 Alexandra Gutmann
Alexandra Gutmann Nicole Bobrowski
Nicole Bobrowski Tjarda Jane Roberts
Tjarda Jane Roberts Julian Rüdiger5
Julian Rüdiger5 Thorsten Hoffmann
Thorsten Hoffmann- 1Institute of Inorganic and Analytical Chemistry, Johannes Gutenberg-University Mainz, Mainz, Germany
- 2Institute of Environmental Physics, University of Heidelberg, Heidelberg, Germany
- 3Max Planck Institute for Chemistry, Mainz, Germany
- 4Laboratoire de Physique et de Chimie de l'Environnement et de l'Espace, Université d'Orléans, CNRS UMR7328, Orléans, France
- 5Atmospheric Chemistry, Bayreuth Center of Ecology and Environmental Research, University of Bayreuth, Bayreuth, Germany
Volcanoes are a significant halogen source to the atmosphere. After water, carbon dioxide and sulfur compounds, halogens are often the most abundant gases in volcanic plumes. In the past, less attention was given to the heavy halogens bromine and iodine. However, the discovery of bromine monoxide (BrO) in volcanic plumes led to new interest especially in volcanic bromine chemistry and its impact on atmospheric processes. The BrO detection came along with advances in volcanic remote sensing techniques, in particular, robust DOAS applications and the possibility of continuous measurements by automated instruments located at safe distances from the volcano. As one of the consequences, the volcanic community developed an increased interest to use BrO/SO2 ratios as a potential tracer of volcanic activity. BrO is a secondary volcanic gas, but the only bromine species in volcanic plumes, which has been measured by remote sensing techniques today. For a better understanding on bromine chemistry in volcanic plumes and to gain information on the original amount of emitted bromine by only measuring BrO, additional techniques were developed (alkaline traps, diffusion denuders) and adapted for drone-based sampling to determine further gaseous bromine species (i.e. Br2, HBr, HOBr, interhalogens) at various plume ages. Additionally models of plume-atmospheric chemistry were developed to help the interpretation of field-measurements. Model studies simulating plume conditions indicated that a complex atmospheric chemistry mechanism transforms emitted HBr into BrO and other reactive bromine species such as BrOH, Br2, BrCl, BrONO2 or BrNO2. To reproduce the very rapid formation of BrO observed in volcanic plumes, the volcanic emission input to the (low-temperature) plume chemistry models also needs to consider the high-temperature near-vent plume conditions, as represented by thermodynamic models. The formation of BrO and other reactive bromine species depend not only on the amount of bromine emitted but also on plume mixing processes, relative humidity, and aerosol particle acidity. However, uncertainties remain in the validation of the plume chemistry models by a lack of field-measurements. This review provides a comprehensive summary on volcanic bromine data of the last 15 years achieved from established and cutting edge measurement techniques as well as their treatment and interpretation in recent model experiments. It points out controversially discussed relation of bromine degassing to volcanic activity and puts a light on remaining uncertainties.
Introduction
Volcanic activity is responsible for the formation of a permanent atmosphere on Earth, and volcanic emissions continue to influence atmospheric composition and thereby climate. Thus, even though volcanic activity has acted to promote the development of life it also induces risks and challenges for the terrestrial biosphere (Shaw, 2008). Measurements of volcanic emissions (gas flux, composition) made at the surface provide an indirect means to explore the inaccessible interior of volcanoes and their complex magmatic systems. They can thereby contribute to volcano hazard monitoring and eruption forecasting.
Knowledge about the chemistry occurring in the volcanic plume is required when making inferences about volcanic activity from volcanic gas measurements, particularly in the more chemically-aged downwind plume. Volcanic sulfur emissions become oxidized in the atmosphere to radiatively-active sulfate particles that can cause climate cooling in particular following large eruptions that injected gas into the stratosphere, e.g., Pinatubo 1991. Volcanic sulfate particles in the stratosphere also act as surfaces to activate halogens causing ozone layer depletion (Brasseur and Granier, 1992). More recently, the potentially chemistry-climate importance of volcanic halogen emissions has started to be of increased interest again (von Glasow et al., 2009; Kutterolf et al., 2013, 2015; Cadoux et al., 2015, 2018; Vidal et al., 2016; Klobas et al., 2017). Some eruptions to the stratosphere have been observed to co-inject halogens alongside sulfur (Theys et al., 2009; Hörmann et al., 2013; Carn et al., 2016), leading to volcanic halogen impact on stratospheric ozone and NOX (Lurton et al., 2018). Besides remote sensing observations have been done via in situ sampling (Rose et al., 2006) and analysis of ice cores (Zdanowicz et al., 1999; De Angelis et al., 2003). Volcanic halogen emissions to the troposphere, particularly bromine (but also chlorine and iodine) can become activated through plume atmospheric chemistry that results in tropospheric ozone, NOX, HOX depletion and may convert mercury to a more easily deposited form (Bobrowski et al., 2007; Roberts et al., 2009; von Glasow, 2010; Jourdain et al., 2016). The plume chemical activation of volcanic halogen emissions, especially bromine, is the focus of this review.
In general, volcanic gas emissions consist of: 50–90% H2O, 1–40% CO2, 1–25% SO2, and trace species such as ca. 0.5% H2 and 0.03% CO (Gerlach, 2004; Textor et al., 2004). For arc volcanoes, halogen emissions are represented by about 0.84% HCl, 0.061% HF, 0.0025% HBr (Gerlach, 2004), with trace-level HI emissions. Global average bromine degassing fluxes from arc volcanoes are estimated to be 5 ± 15 Gg/year HBr (Pyle and Mather, 2009), based on collated data on Br/S ratios in volcanic emissions combined with assuming an annual arc-related SO2 flux of 15 Tg/a. However, this may underestimate total volcanic bromine emissions, given the most recent estimate of the global SO2 flux from passively degassing volcanoes is 23 ± 2 Tg/a (Carn et al., 2017), and the new identification of bromine-rich volcanic sources (e.g., Nyiragongo, Democratic Republic of the Congo, Bobrowski et al., 2015).
Previous reviews on volcanic halogens have focused on petrological approaches (e.g., Devine et al., 1984; Wallace, 2005) and volcanic degassing fluxes of halogens (Pyle and Mather, 2009) or their general cycling and distribution in the earth system (Webster et al., 2018). Platt and Bobrowski (2015) summarized observations of reactive bromine species mainly by spectroscopic technologies. Several reviews have also been published about the origin and impact of volcanic halocarbons as a minor halogen emission (Schwandner et al., 2004, 2013; Frische et al., 2006). Lately developments of new measurement techniques, collection and interpretation of data sets (by both, new and old techniques), and approach by model applications moved forward the knowledge related to bromine transformations in volcanic plumes. To our knowledge no comprehensive summary and overview of this still relatively new and fast advancing field has been done today. Here we conflate all recent approaches to draw an overall image about the state of the art by pointing out commonly confirmed statements, inconsistencies and general knowledge gaps suggesting further work. We focus on inorganic bromine compounds within passively degassing volcanic plumes entering the troposphere, for which the datasets and plume model studies are most comprehensive, but including a brief discussion of other halogens (Cl, I).
This review starts with an overview of existing measurement techniques for the determination of both speciated and total bromine in volcanic plumes. This is followed by a discussion of numerical modeling of plume bromine chemistry. Then a summary of volcanic bromine measurements is presented. An overview is given of the chemical reaction mechanisms that activate bromine (as well as chlorine and iodine) in volcanic plumes, detailing the roles of both low and high-temperature chemistry, photochemical and multi-phase reactions, aerosol/humidity, and impacts and feedbacks of ozone, NOX, and mercury. This conceptual framework is then used to discuss reported field-studies and trends in the collated volcanic bromine datasets. The review concludes by describing the current knowledge gaps, particularly in relation to the use of bromine as monitoring parameter for forecasting eruptions, and highlights recent developments that provide new opportunities in this field.
Methods Used to Investigate Bromine in Volcanic Plumes
Measurement Techniques
Volcanoes present a challenging context for both instruments and scientists seeking to measure volcanic plume gases in remote, inaccessible areas and during hazardous volcanic eruptions. For continuous and safe observations remote sensing techniques are often the instruments of choice. However, not all species can be measured by remote sensing. Also, in situ and direct sampling techniques can enable measurements closer to the emission source and thus capture degassed species at an earlier point of plume age. Making measurements very near-to-source is essential for the case of highly reactive species such as bromine: it allows to investigate as “pure” volcanic signal as possible, opening up the possibility to study the chemical transformation processes as a function of distance (and travel time) downwind, as the volcanic emissions progressively mix with the surrounding atmosphere.
In Situ – Application of Instruments in the Plume
Earlier determinations of volcanic halogens were realized by condensate sampling in volcanic fumaroles (Oana, 1962; Sugiura et al., 1963; Goff and McMurtry, 2000; Amachi et al., 2001) and by the use of passive alkaline traps (Noguchi and Kamiya, 1963; Goff et al., 1998; Witter et al., 2004). Today, actively pumped alkaline traps are typically applied. The main idea is to retain and enrich acid compounds within strong alkaline solutions (e.g., 1–4 M NaOH) when air is pumped through the substrate. Due to their light weight and easy handling, “filter-packs” (FP) are still the most commonly applied alkaline traps to sample volcanic plume gases. Different kinds of filters (cellulose, Nylon, Teflon) have been used with several mainly alkaline coatings (e.g., 1M NaHCO3) and glycerol to improve the absorption efficiency. Nevertheless, saturation of filters or evaporation can readily occur. Limitations of the sampling efficiency were pointed out especially for saturated filter packs leading to elevated halogen/sulfur ratios due to distinct acidity (e.g., SO2 and HF) and therefore different retention in the alkaline solution (Martin et al., 2010; Sawyer et al., 2011; Wittmer et al., 2014). To avoid or at least be aware of saturation effects, several filters are usually mounted in series. If the last filter of a pack yields < 10% of the total amount of the species of interest, a quantitative trapping of the compounds is assumed (Lazrus et al., 1976; Sedlacek et al., 1984). If particle filters in front of filter packs are installed (i.e., to avoid uptake of aerosol and ash particles), possible interactions of gases with the particle-loaded filter have also to be considered. For volcanic bromine, HBr is assumed to be efficiently collected, but uncertainties remain whether species such as HOBr, Br2, BrCl, BrNO2, and BrONO2 are trapped quantitatively by alkaline impregnated filters (Kitto et al., 1988).
Using these techniques extensive data sets of volcanic plume composition were established, however, in the case of halogens most of them focus on chlorine and fluorine and do not take bromine and iodine into account. One of the early plume studies on heavy halogens was carried out by Aiuppa et al. (2005) who assumed to determine the total molar amount of emitted bromine species (Brtotal) on alkaline filters since bromine on particulate filters were negligible and no BrO was measurable with DOAS at the same location.
Uncoated filters can be used for the determination of particulate bromine, although few observations have been reported (Sturges and Shaw, 1993; Zelenski et al., 2013). Size-resolved analysis is enabled when cascade impactors are used. Here, the particles are deposited on several filter membranes according to their size. In comparison to alkaline filter sampling packs (sampling volume about 120 L; Aiuppa, 2009), particle analysis typically requires much higher sampling volumes ranging from 2,000 up to 10,000 L (Erebus, Antarctica: Ilyinskaya et al., 2010; about cascade impactors: Marple et al., 1991; Ma and Kim, 2008).
For avoiding saturation during sampling, active liquid alkaline traps were developed, which solve acid gas species in alkaline solutions by bubbling volcanic gases through the solution. To enhance the efficiency in Drechsel bottles (based on gas washing bottles named after Edmund Drechsel, DB) frits are used to decrease bubble size and therefore increase the relative surface of introduced gas samples to the alkaline solution (Liotta et al., 2012; Wittmer et al., 2014). The same aim of increasing the surface between the solution and introduced gas is used by filling a glass tube with glass rings (so-called Raschig rings; Raschig, 1914) and adding alkaline solution which is able to cover all the surface by rotating the tube (the so-called Raschig tube; RT, Levin et al., 1980). The enhanced efficiency allows a higher volume flow-rate that yields in increased sample concentrations and facilitates analyses close to detection limits for a specified sampling time or alternatively sampling time in the field can be reduced, in cases where a reduced detection limit is not necessary. Since it is assumed that the dominant fraction of bromine compounds are acidic and are therefore dissolved in the alkaline sampling solution the results are assumed to represent the Brtotal. Today, all those alkaline trap samples are usually analyzed by IC and ICP-MS (see overview in Wittmer et al., 2014). An alternative are neutron activation analysis, but often much more expensive and comparison studies showed similar results between the methods (Strellis et al., 1996; Wittmer, 2012). Wittmer et al. (2014) characterized the sampling efficiency of various alkaline traps. In general, good agreements for filter packs, Drechsel bottles, and Raschig Tubes were found.
Alkaline sampling techniques do not allow speciation (i.e., the determination of individual halogen species). Moreover, alkaline traps cannot distinguish between gaseous and particulate phase. To provide a more in-depth halogen-speciation, gas diffusion denuders take advantage of the different diffusion coefficients of gaseous compounds and particles. The tube-shaped denuder systems are applied with a coated inner surface. Gas molecules diffuse to coated denuder walls, which represent an ideal sink for the gas molecules of interest and thus are retained and enriched in the coating. In contrast, due to the much smaller diffusion coefficient, the aerosol particles pass the denuder. Using various coatings, which undergo species specific derivatization of analytes, enables selective analysis of bromine species (Huang and Hoffmann, 2008, 2009; Rüdiger et al., 2017).
In situ techniques applicable for bromine determination are summarized in Table 1. Beside ground-based applications, drone–based sampling units proved to be a valuable tool for in situ volcanic gas measurements of difficult-to-access crater emissions as well as aged plumes downwind from the emission source that have become lofted away from the surface (e.g., McGonigle et al., 2008; Rüdiger et al., 2018b).
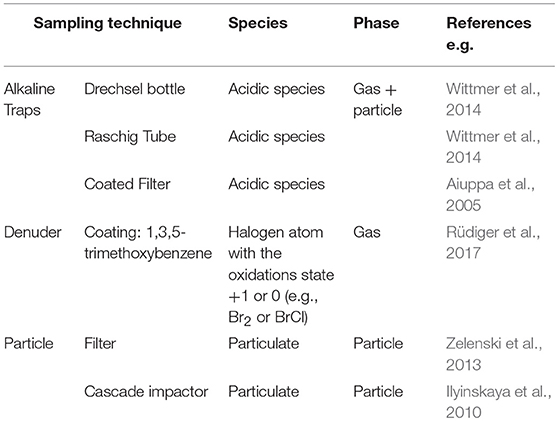
Table 1. Overview in situ plume bromine sampling techniques.
Remote Sensing
Especially during explosive episodes, remote sensing techniques become an indispensable tool for safe monitoring of volcanoes. Optical remote sensing techniques, using spectroscopic approaches from ultra-violet to thermal infrared, measure integrated gas concentrations through cross-sections of the plume. An overview of typical applied remote sensing techniques is given e.g., in McGonigle and Oppenheimer (2003) and Platt et al. (2018).
Restrictions for remote sensing techniques might occur due to high and/or variable atmospheric backgrounds for some gases (mainly for H2O and CO2) and since remote sensing techniques are sensitive for species with differential absorption structures, accessible compounds are limited to the wavelength range of the particular spectrometer and light source. Fourier transform infrared (FTIR) technique has been used for spectroscopic halogen observations of HCl and HF (e.g., Mori and Notsu, 1997; Oppenheimer et al., 1998). However, due to “small” absorption cross-sections, low abundances and limited spectral resolution, portable FTIR instruments have not been able to detect HBr.
In volcanic environments, BrO is the only bromine species that has been determined by remote sensing. Satellites, portable and continuously installed Differential Optical Absorption Spectroscopy (DOAS) instruments, are used to investigate BrO in volcanic plumes. DOAS instruments collect light (from scattered skylight, direct solar light or artificial lamps) that has passed through the volcanic plume. Light collected by a telescope is coupled into a spectrometer often by using an optical fiber and is then dispersed by a grating inside the spectrometer. Signal intensity is quantified via a CCD array or photomultiplier device. The most commonly used instrument are the so-called passive DOAS instruments, which use scattered skylight as a light source. The plume absorption spectrum is compared with an additional spectrum taken outside the plume under nearly identical conditions to correct influences of atmospheric background absorptions and the solar spectral structure (Fraunhofer lines). Today portable Multi-Axis-DOAS (MAX-DOAS) instruments are a widespread instrument in volcanic plume investigations yielding many observations of BrO alongside SO2. Details are described in e.g., Hönninger et al. (2004), Bobrowski et al. (2007), Bobrowski and Platt (2007), and Gliß et al. (2015). Zenith-sky pointing DOAS instruments can be deployed whilst traversing underneath the plume to yield total column amounts across a transect, which in combination with wind-speed estimates provides total SO2 and BrO fluxes. Traverses have been undertaken by foot, car, ship, and even by UAVs in inaccessible areas (e.g., McGonigle et al., 2002; Galle et al., 2003; López et al., 2013; Rüdiger et al., 2018b).
Imaging of plumes with SO2-cameras (see e.g., Mori and Burton, 2006; Kern et al., 2015; McGonigle et al., 2017) enables to spatially map complex plumes at high time resolution (<1 s). Imaging of volcanic plume BrO has been achieved using Imaging DOAS (IDOAS). Spatial resolution is generated by spatial and spectrally resolved vertical columns applying a two dimensional detector and by horizontally scanning gaining the second spatial dimension (Lohberger et al., 2004; Louban et al., 2009). However, the temporal resolution to record a single image by this method is in the order of minutes, much slower than the fast (seconds) plume chemical and physical (dilution) processes. The BrO image obtained is only an average. Ongoing development of Fabry Perot Interferometers (FPI; Kern et al., 2010; Kuhn et al., 2018) may enable fast imaging of BrO in the future with a temporal resolution similar to SO2 cameras (<1 s).
Beside portable instruments, instrument networks for permanent and automatic monitoring of several volcanoes by DOAS instruments have been created [NOVAC, (Galle et al., 2010); FLAME, (Burton et al., 2009), FLYSPEC, (Businger et al., 2015)]. Typically instruments are installed 5–10 km downwind of the crater observing plumes with an age of about 10–30 min (Galle et al., 2010). Further satellite ultraviolet instruments like GOME-2 (Global Ozone Monitoring Experiment), OMI (Ozone Monitoring Instrument) and the recently launched TROPOMI (Tropospheric Monitoring Instrument) enable BrO (and SO2) monitoring in the troposphere and stratosphere on a global scale. Main differences between satellite instruments are spatial resolution and overpass time (for details see e.g., Theys et al., 2011; Carn et al., 2013; Hörmann et al., 2013). Systematic analysis of GOME-2 satellite observations of volcanic BrO are presented from volcanic eruptions entering the troposphere (Heue et al., 2011; Rix et al., 2012), and upper troposphere–lower stratosphere region (Theys et al., 2011; Hörmann et al., 2013). Due to the spatial resolution (100's km, reaching 10's km for the new TROPOMI instrument), observations are typically available for somewhat aged eruption plumes. Interpretation of such data is made more complicated because total bromine measurements are often not available and for commonly investigated plume ages >day, SO2 might not be a reliable conservative plume tracer anymore. We focus our discussion of volcanic bromine speciation on ground based and airborne measurements made on the volcano flank (relatively close to the emission source) by remote sensing (DOAS), in situ methods, and associated numerical modeling studies.
Model Application
The volcanic plume is a diverse environment with dynamic variability in temperature, humidity, aerosol loading, and chemistry. Because of this, modelers typically employ separate techniques to describe phenomena at the vent (high-temperatures) and at ambient atmospheric conditions downwind (low temperatures). Numerical models of volcanic plume chemistry have been developed and applied at both high-temperature and low-temperatures. Comparison of model simulations to plume observations provides a wider context to improve our understanding of bromine evolution in the plume and to forecast impacts of bromine chemistry. Calculations identified that volcanic gases undergo high-temperature interactions followed by low-temperature reactions in the downwind plume that has been mixed with ambient air and cooled to ambient temperatures (Gerlach, 2004; Bobrowski et al., 2007; Roberts et al., 2009).
Model Initialization – High-Temperature Near-Vent Conditions, Dilution, and Aerosols
Following the discovery of BrO in a volcanic plume (Bobrowski et al., 2003) thermodynamic equilibrium models were applied at high-temperature to forecast bromine speciation. However, these were not able to simulate bromine monoxide to sulfur dioxide (BrO/SO2) ratios (10−4 mol/mol) observed downwind from the crater (Gerlach, 2004). Rather, the predicted BrO/SO2 ratios at the vent (10−8) are consistent with the observation that BrO is often not detectable at the crater rim (see also section Dependence on Plume Age–Temporal Evolution).
Numerical models of atmospheric chemistry were then adapted to volcanic plume environments and/or newly developed and applied to forecast the plume halogen chemistry at ambient-temperatures. These model simulations were able to reproduce measurement results (e.g., BrO/SO2 downwind from the crater). Three key aspects to the initialization of atmospheric chemistry models of volcanic plume halogen chemistry are: (1) the high-temperature near-vent conditions, (2) dilution (that controls entrainment of background air), and (3) volcanic aerosols.
For near vent conditions, it is assumed that near the emission source volcanic gases mix with atmospheric gases and undergo high-temperature chemistry. If reaction kinetics proceed faster than plume dilution a thermodynamic equilibrium may be assumed. This equilibrium systems depend on pressure, temperature (magmatic and ambient) and oxygen fugacity (P-T-fO2) conditions and have been applied to calculate the composition of the “effective source region” (Gerlach and Nordlie, 1975; Gerlach, 2004; Martin et al., 2006; Bobrowski et al., 2007). One of the most common used thermodynamic equilibrium models is HSC Chemistry (Outokumpu Tech, Finland). A free-energy minimization algorithm is used to calculate molecular equilibrium compositions of gas mixtures. The estimated model input data are based on crater rim or fumarole measurements and include temperature and pressure as well as a starting elemental plume composition. This is based on mixing the observed composition of volcanic gases with air (N2, O2, Ar), for which the proportion of mixing between atmospheric and magmatic gases (VA:VM) must be assumed. Thermodynamic modeling of the hot plume-air mixing predicts that a VA:VM>0 ambient air oxidizes gases like H2S and H2 and produces small quantities of reactive halogen species such as Br, BrO, Cl and ClO, as well as relatively high quantities of HOX (hydrogen oxide radicals) and NOX (NO, NO2 and combinations) (Gerlach, 2004; Martin et al., 2006). Thermodynamic modeling also predicts the high-temperature formation of SO3 that is a precursor to sulfate aerosols (see also section Bromine Explosion). The output from the thermodynamic models is then used as input (“the effective source region”) to atmospheric chemistry models that simulate halogen processes in the downwind plume at ambient temperatures. The high-temperature composition of the near vent plume is crucial for the initialization of low-temperature kinetic models: the radicals generated accelerating the onset (“kick-start”) of halogen cycling at ambient temperatures, hence can affect plume evolution downwind from the crater.
However, the high-temperature near-vent conditions (“effective source region”) remain a source of uncertainty in the initialization of low-temperature models of the plume halogen chemistry. The high-temperature modeling involves several unverified assumptions since high-temperature conditions are hard to constrain by direct measurements. For example, VA:VM in the near-vent plume exerts a strong influence on the predicted composition but is somewhat arbitrarily chosen: early studies used 40:60, and later on 15:85, 10:90 and 5:95. For some processes assumptions of the thermodynamic equilibrium seem to be inappropriate [e.g., re-equilibration of H2S (Martin et al., 2009), H2 (Aiuppa et al., 2011) or formation of NOX from background N2 that is likely kinetics limited due to the high bond strength for N2 (Martin et al., 2012a)]. At VA:VM = 40:60, high levels of Cl radicals are predicted to form, that subsequently act to rapidly destroy H2S in the atmospheric chemistry models. However, measurements by Aiuppa et al. (2007) did not show a decrease of H2S as a function of distance to the emission source (0.1–1 km). Measurements of HO2 and OH are very rare even though they play an important role in the atmospheric chemistry/halogen reaction cycles. OH radicals can be measured by very high-resolution active DOAS instruments (Platt et al., 1988) or by laser-induced fluorescence (LIF). Indirect detection of HO2 is possible by titrating HO2 with NO to form OH, which can be detected by LIF. Subtraction of the measured OH background then yields HO2 (Hard et al., 1984; Brune et al., 1995). However, the power requirements and large size of such measurement techniques for OH and HO2 generally limit their use on the volcano, restricting their potential applicability to aircraft measurements of the (typically more aged) plume. One of those examples is the airplane measurements of the eruptive plume of Mt Hekla, Iceland in 2000 (Hunton et al., 2005). No measurements are available, to the authors' knowledge, in the proximity of volcanic emission sources.
When the plume reaches the crater rim, more ambient air is entrained and the plume cools to ambient temperatures. The background air contains oxidants (HOX, NOX, O3) whose entrainment into the plume affects the plume chemistry. Therefore, important controls on the halogen evolution in the downwind plume are the rate or extent of dilution and nature of the background atmosphere. Beside latitude, longitude and altitude dependent parameters (e.g., temperature, time of the day) more polluted and NOX-rich atmosphere may need to be considered for some locations. In contrast to emissions entering cleaner free tropospheric conditions like for many high altitude volcanoes in the Andes, impacts of other environmental emissions on the plume chemistry have to be considered close to large cities or for instance, for volcanic emissions entering the planetary boundary layer like it is the case for Stromboli in the Mediterranean, or Masaya in Nicaragua. Dilution of the downwind plume can be represented by simple dispersion parameterizations such as Pasquill-Gifford or can be adapted to reproduce reported plume dilution rates.
Aerosol particles in volcanic plumes are highly acidic, often sulfate-rich, and include both primary aerosol (directly emitted) and secondary aerosols (e.g., formed from the atmospheric oxidation of volcanic SO2). Volcanic aerosols can be measured by in situ offline time-averaged sampling using filter packs (Allen et al., 2002; Mather et al., 2003) or in situ real-time sampling by optical particle counters (Allen et al., 2006) and by remote sensing with sun-photometers (Watson and Oppenheimer, 2000; Roberts et al., 2018). The role of aerosol in partitioning between the gas phase and particles is considered by equilibrium models AIM (Aerosol Inorganic Model; Wexler and Clegg, 2002; Martin et al., 2012b) or MOCCA (Model of Chemistry in Clouds and Aerosols in MISTRA; Sander and Crutzen, 1996; Vogt et al., 1996; von Glasow, 2000). Aerosols catalyze heterogeneous reactions that have a crucial impact on plume halogen chemistry (see section Bromine Explosion). This is demonstrated by model simulations that can only reproduce downwind observations of BrO/SO2 when volcanic aerosol is included. Reactions of halogens on volcanic aerosol are thus a key element in the atmospheric models of volcanic plume halogen chemistry outlined below.
Modeling Plume Chemistry – Low-Temperature Kinetic Models
In the downwind plume low-temperature kinetic models investigate plume chemistry. Mainly two low-temperature kinetic 0/1D models have been developed: MISTRA (simulates an advected column of air) and PlumeChem (in expanding box or multi-grid box modes). Recently also the regional 3D model CCAT-BRAMS was applied to study volcanic halogen chemistry. All these models take into account detailed atmospheric chemistry schemes including reactive halogens (bromine and chlorine) with gas-phase photolytic and gas-aerosol reactions. These kinetic models of volcanic plume chemistry at ambient temperatures used an “effective emissions source” (Bobrowski et al., 2007) in their inputs based on thermodynamic equilibrium models (HSC) representations of the near-vent high-temperature chemistry.
The numerical model MISTRA simulates a column of atmosphere that is divided into multiple layers. This column is moving over a volcano where the plume is emitted as a puff into few adjacent model layers at a given height. This column of air is then advected downwind according to an assumed wind speed (Bobrowski et al., 2007), whilst the plume puff disperses vertically in the column. Horizontal dilution is taken into account with a simple parameterization, assuming that the horizontal evolution of the plume corresponds to a Gaussian plume. MISTRA was originally developed for microphysics in the marine boundary layer including a hydrodynamic part, a radiation code and a parameterization of the turbulence by means of a first-order. The turbulence closure was based on the model of Mellor and Yamada (1982) and the meteorological core was developed by Bott et al. (1996). von Glasow (2000) extended MISTRA with a module that describes chemical reactions of the gas phase, aerosol particles and cloud droplets (this extension was based on MOCCA). So far, collision and coalescence of aerosol and new particle formation are not considered in the model. The one-dimensional MISTRA has been applied in volcanic plumes with focus on halogen mechanisms by Aiuppa et al. (2007), Bobrowski et al. (2007), von Glasow (2010), Bobrowski et al. (2015), and Surl et al. (2015).
PlumeChem is a 0D/1D box model that also uses a Lagrangian-type approach to follow a volcanic plume puff with time or distance downwind according to an assumed wind-speed (Roberts et al., 2009, 2014b, 2018; Kelly et al., 2013). The model simulates chemical reactions in the plume as it disperses and entrains background air. This approach is similar to that applied to modeling the chemistry of ship-plumes (Song, 2003). In the single grid box mode, plume dispersion is simulated by a single grid box (an ellipse) that expands as background atmospheric air is entrained into it. In the multiple grid box mode, the plume is described by a series of chemical boxes in a row, i.e., plume chemistry is spatially resolved horizontally. Plume dispersion is represented in the vertical direction by box-growth, whereas it is simulated in the horizontal direction by mixing between adjacent boxes. The plume dispersion is modeled assuming a Gaussian plume dispersion and parameters are given by Pasquill-Gifford parameterizations, or can be user-defined to approximate actual observed dispersion of plume tracers (Kelly et al., 2013). The PlumeChem model includes detailed volcanic plume halogen atmospheric chemistry similar to MISTRA, but the gas-aerosol reactions as treated using reactive uptake coefficients on an aerosol surface area rather than directly simulating the aqueous-phase chemistry. This reduces the computational requirements of the model, and is the approach also used in 3D models as discussed below. Considered model parameters are the chemical composition of model initialization, the aerosol loading and plume dispersion.
Recently a non-hydrostatic mesoscale atmospheric chemistry model, CCATT-BRAMS, was developed to simulate volcanic plume halogen chemistry and regional impacts in a Eulerian-type study (Jourdain et al., 2016). The BRAMS model has been used for studies of planetary boundary layer, operational weather forecasting and climate and includes parameterizations of physical processes such as surface–air exchanges, turbulence, convection, radiation and cloud microphysics that are particularly well adapted for tropics. Coupling to CCATT enables the transport, chemical evolution, emission and deposition of molecules and aerosol particles (Freitas et al., 2009; Longo et al., 2013). The CCATT preprocessor was adapted to include volcanic emissions, and the chemistry scheme developed to include volcanic halogens. Two-way nested grids are used to capture plume processes at the small-scale (<1 km) with larger grids simulating the tropospheric impacts of the dispersed plume at the regional scale (>1,000 km). A similar development has also been undertaken to include volcanic halogens in the WRFChem model (Surl et al., 2014).
First steps have also been made to introduce volcanic halogens and their tropospheric plume chemistry into global models. A major issue is the coarse resolution of global models (typically 100–200 km). This causes the volcanic emissions to be too rapidly diluted compared to reality. Global models, therefore, cannot resolve the volcanic plume halogen processes that occur at much smaller scales (km), particularly in young plumes/near-to-source. Grellier et al. (2014) proposed a sub-grid-scale parametrization approach to represent the near-source halogen chemistry plume processes in a global tropospheric model. Future work should further develop the volcanic plume halogen chemistry in such approaches.
The comparison of the currently available model results with one another and with measured observations is usually difficult because starting conditions vary between models due to consideration of different data arrays and volcanoes in model applications. In general, total emissions and their composition may vary immensely over just a few days or between different volcanoes. Furthermore, measurement data sets at different distances downwind are usually not taken simultaneously. The model parameter space is vast. Sensitivity studies show how variability in initial plume compositions (e.g., variable HBr emissions, plume composition, mixing or high-temperature interactions of the young plume in the source region) affect downwind BrO formation (e.g., Roberts et al., 2014b).
Current Understanding
Overview of BrO/SO2 and Br/S Field-Measurements
Upon the discovery of BrO in the volcanic plume of Soufriere Hills, Montserrat (Bobrowski et al., 2003) detection of BrO in many volcanic plumes globally shows that BrO formation is a common process in volcanic plumes including different settings of arc, rift and hotspot volcanoes as shown below.
Nevertheless, data sets are still very limited regarding the geographical distribution and temporal variability of bromine emissions. Figure 1 and the Supplementary Table 1 summarize all available volcanic plume bromine data since 2003. Earlier results are already summarized elsewhere (e.g., Gerlach, 2004). Figure 1 gives an overview on particulate and gaseous bromine to sulfur (Br/S) and BrO/SO2 ratios, across different volcanological settings (arc, hotspot and rift).
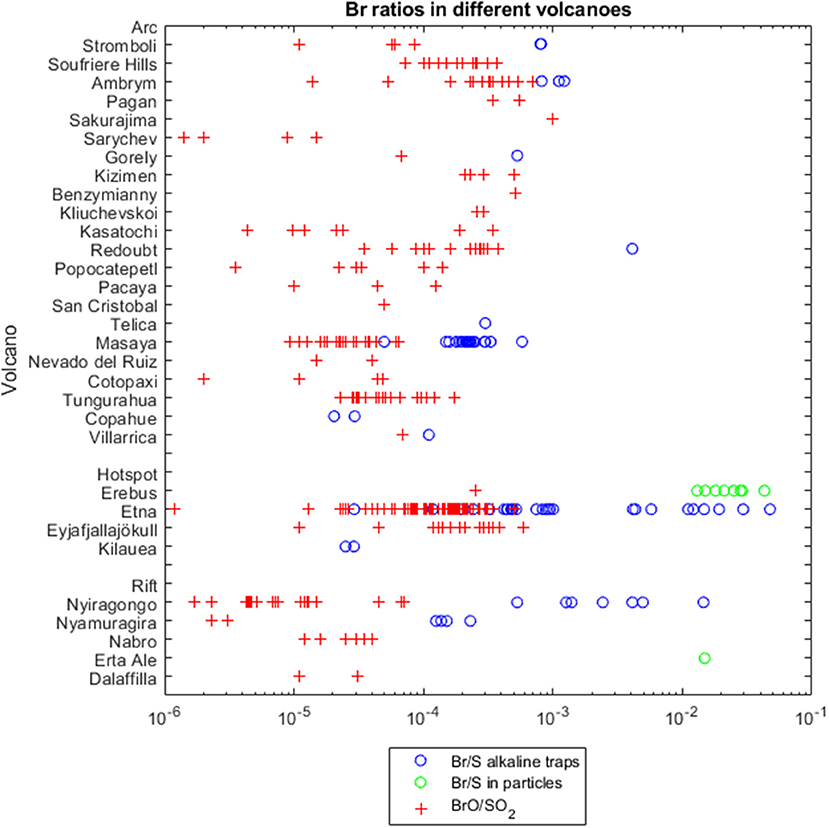
Figure 1. Volcanoes and their bromine emissions. In blue circles Br/S ratios of total amounts measured by alkaline traps, in red crosses BrO/SO2 ratios measured by DOAS instruments, in green circles Br/S ratios of total amounts in particles measured by particle filters. Data from Supplementary Table 1, x-axis follows a logarithmic scale. Variability may be due to magma types and volcanic (tectonic) environments, volcanic activity, distance to the source and chemical aging, or measurement uncertainties (see section Overview of BrO/SO2 and Br/S Field-Measurements).
Bromine species are typically reported relative to sulfur, to enable distinction between plume chemistry and dilution effects. Assuming slow in-plume oxidation, SO2 will act as a plume tracer on the relatively short observation timescales (typically minutes to hours) of measurements on the volcano flank. Indeed, volcanic plume SO2 can often be considered to be conserved over the short distance between “source” and “downwind” observation points on the volcano flank because the acidity of the volcanic aerosol in the plume, prevents the SO2 uptake, and because SO2 concentrations are in excess of most oxidants (McGonigle, 2004; von Glasow et al., 2009; Galeazzo et al., 2018). Variations in bromine/sulfur observed at just one fixed measurement point might be a result from changes in emission composition. But the assumption of SO2 as a plume tracer may not always be valid for measurements of very dilute or aged plumes (in the order of days), e.g., as may be detected by satellites.
The Br/S emissions in alkaline-trap samples are assumed to represent total bromine, Brtotal. Figure 1 shows that Br/S values (displayed as blue circles) are generally higher than BrO/SO2 ratios (red crosses) measured by DOAS instruments. This is consistent with the partial conversion of emitted bromine into BrO, Particulate measurements tend to report higher Br/S values (green dots) in comparison to gas ratios or gas-particle mixtures. Increased Br/S ratios in particles may demonstrate enhanced bromine partitioning into particles compared to sulfur, or unusually low sulfate for the few volcanoes studied e.g., Erebus, Antarctica (Ilyinskaya et al., 2010).
Several factors may explain the variability in Figure 1, including:
- Degassing derived from different magma types and volcanic (tectonic) environments e.g., deep or shallower sources associated with melt generation and degassing systems or crustal fluids (Pyle and Mather, 2009; Mather et al., 2012) as discussed below.
- Different states of volcanic activity such as eruptive or quiescent degassing (section Forecasting–Correlating Volcanic Plume Composition to Volcanic Activity).
- Different distances to the source and therefore different extents to which bromine has been chemically transformed into BrO as a function of plume age (section BrO Evolution in Volcanic Plumes).
- Also, processes such as scrubbing of water-soluble compounds may impact Br/S (Symonds et al., 2001; Textor, 2003).
- Uncertainties in individual measuring techniques (section Measurement Techniques), as discussed below.
Considering subaerial actively degassing volcanoes, arc volcanoes are the most abundant tectonic setting and therefore provide the majority of data. The volcanic Br/S shows a very large variation over five orders of magnitude. This is probably primarily due to variation in bromine abundance of the arc volcano plumes.
Hotspot volcanoes have typically been described as halogen-poor (e.g., Pyle and Mather, 2009). However, we find that the available hotspot volcanoes fall in the same range of the data available for arc zones (Figure 1). Note that the representation of the samples formerly and still up to now is not statistically relevant. Hawaii falls at the very low end (about 0.3 × 10−4) but Mt Etna, Sicily, Italy is at the high end of Br/S ratios (up to 0.05). There may be influence of recycled oceanic crust as a halogens source in some hotspot cases, e.g., Eyjafjallajökull, Iceland (Moune et al., 2012).
Intriguingly, the few samples taken from rift volcanoes tend to show lower BrO/SO2 ratios, but no clear differences for Br/S ratios in comparison to arc and hotspot volcanoes.
Within the arc volcano category, the bromine abundances of southern American volcanoes seem to lay at the lower end of the Br/S range. Maximal BrO/SO2 values for South American volcanoes are below the mean of other available arc volcanic values, with the exception of Tungurahua (max. 1.7 × 10−4). Lowest global Br/S values since 2003 were found for Copahue's plume (0.20 × 10−4 and 0.29 × 10−4). Although for Japanese arc volcanoes Gerlach (2004) cited also low Br/S ratios (Tokachi 0.25 × 10−4, Kudryavy 7.8 × 10−4), BrO/SO2 obervations by Lee et al. (2005) at Sakurajima are a bit in contrast to those earlier determined values in Japan. However, some caution must be applied to the comparisons regarding the methods, because most of the observations of the South American volcanoes are ground-based, in contrast, many observations of the Kamchatka arc (Kizimen, Benzymianny, Kliuchevskoi) were gained by satellite measurements of potentially more aged plume.
Uncertainties in individual measuring techniques were discussed in section Measurement Techniques. Measurements of trace bromine emissions can be particularly challenging close to the detection limit. Also, high gas concentrations can limit the accuracy of the results e.g., due to saturation of alkaline filter packs (see section In Situ–Application of Instruments in the Plume). In Figure 1 the high Br/S ratios of up to 475 × 10−4 measured on Etna, Sicily, Italy in 2005 (Martin et al., 2008) are significantly greater than Br/S observations from January to October 2004 where all data points are below 100 × 10−4 (Aiuppa et al., 2005), as well as Br/S on Etna reported by Oppenheimer et al. (2006) and Wittmer et al. (2014). Whilst change in volcanic activity could be one explanation, oversaturated filter samples might be a conceivable explanation of the exceptional high Br/S ratios by Martin et al. (2008).
Plume Chemistry and the Formation of Reactive Bromine
Bromine Explosion
Since observations show BrO is not directly emitted from the volcano (see section Dependence on Plume Age–Temporal Evolution), model studies were undertaken to explain the measured BrO content and its formation process.
Thermodynamic studies predict that halogens are mainly degassed as hydrogen halides, e.g., HBr (Symonds et al., 1994). Gerlach (2004) calculated that BrO in volcanic plumes must originate from the enhancement of reactive halogen species by oxidation of HBr and HCl through autocatalytic cycles involving heterogeneous chemistry. Numerical models of volcanic plume atmospheric chemistry were then developed to explain the measured BrO content and its formation process. Volcanic BrO formation is currently assumed to occur through a bromine reaction cycle, the so-called “bromine explosion,” that has been previously identified in the polar and marine boundary layers and above salt plains (Barrie et al., 1988; von Glasow and Crutzen, 2003; Saiz-Lopez and von Glasow, 2012).
The most relevant gas phase reaction of HBr is the formation of Br radicals with OH radicals (R1; von Glasow, 2010).
High-temperature chemistry is suggested to accelerate the start of low-temperature cycles by elevated concentrations of HOX derived from high-temperature chemistry in the near-vent plume that enhance the initial formation of Br radicals via R1 (Oppenheimer et al., 2006; Martin et al., 2009; Roberts et al., 2009; von Glasow, 2010). Therefore, small amounts of BrO (R2) and subsequent formation of HOBr (R3) are already expected to be formed in the high temperature zone as calculated by thermodynamical equilibrium models.
However, those gas phase reactions alone cannot explain the measured abundance of volcanic BrO. Calculations predict that heterogeneous reactions enhance BrO formation compared to gas phase oxidation. Indeed, plume model simulations that do not include volcanic aerosols cannot reproduce the downwind BrO. The key reaction is the reactive uptake of HOBr (reacting with aqueous HBr in the acidic aerosol) to form the reactive halogen Br2 (R4).
Br2 has low solubility in the aqueous acidic sulfate particles and thus partitions into the gas phase where it is photolyzed into Br radicals (R5).
Overall, this repeated sequence R2–R5 each time doubles BrO concentrations and is therefore known as the “bromine explosion” (Figure 2). Due to its autocatalytic nature, it can produce a very rapid increase in BrO concentrations.
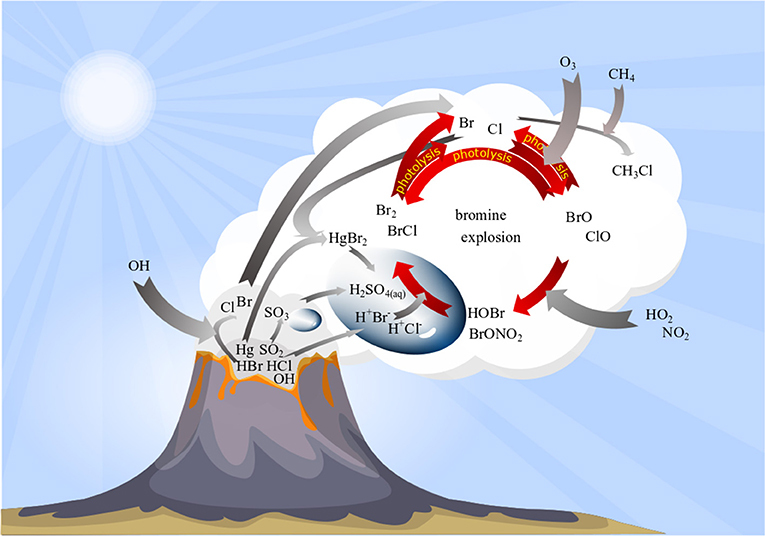
Figure 2. Schematic overview on bromine plume chemistry. Volcanic emissions (dark gray cloud) are processed in high-temperature reactions forming minor amounts of halogen radicals (light gray cloud). Bromine radicals “kick-start” an autocatalytic cycle (“bromine explosion”) that converts emitted HBr into BrO via multistep reactions including gas and particle phase and leads to O3 depletion (for details see section bromine explosion).
The sequence can be terminated by the loss of Br due to its conversion back to HBr (R6–R7; von Glasow and Crutzen, 2003; Roberts et al., 2009).
In the following, we discuss the importance of photochemistry, the role of aerosols, the production of HNO2 (von Glasow, 2010) or HNO3 (Roberts et al., 2009) predicted by models, the depletion of tropospheric ozone (section Causes and Effects of O3 Depletion), interhalogen, chlorine and iodine plume chemistry (section Interhalogen, Chlorine, and Iodine Plume Chemistry), and conversion of mercury into a more toxic and easily deposited form (von Glasow, 2010; von Glasow et al., 2015) in relation to reactive bromine formation in volcanic plumes (section Mercury in Volcanic Plumes).
Importance of Photochemistry
Photochemistry plays a key role for doubling Br radicals in the autocatalytic cycle of the bromine explosion (R5). BrO measurements made round–the–clock at Masaya volcano, Nicaragua, confirmed that photolytic reactions are necessary for the formation of BrO (Kern et al., 2009). Kern et al. (2009) observed significant BrO during daytime whereas BrO was below the detection limit during night. Other measurements were performed at Mt Etna, Sicily, Italy in the early morning during and shortly after sunrise (Gliß et al., 2015; Butz et al., 2017). The data shows an increase of the BrO/SO2 ratio within the first hour of increasing sunlight and a constant ratio afterward.
Influence of aerosol or humidity
Regarding volcanic halogen emission estimates, the non-reactive uptake of HCl and HBr onto sulfate particles should be considered as it may overprint magmatic HX/SO2 gas ratios. Significant uptake could lead to underestimation of volcanic halogen emissions if the particle phase is not taken into account alongside the gas phase (Pyle and Mather, 2009).
Aerosols provide surface area promoting heterogeneous reactions within or on the particles and therefore have a key role in volcanic BrO formation (R4), the cycling, and speciation of bromine in volcanic plumes. The volcanic plume environment is abundant in particles, including both primary and secondary aerosols (see also section Model Initialization–High-Temperature Near-Vent Conditions, Dilution, and Aerosols). Primary sulfate is emitted at up to about 1% (mol/mol) of the volcanic SO2 emission. One mechanism for primary sulfate formation is the high-temperature formation of SO3 radicals in the near-vent plume, that upon cooling are converted into H2SO4 in the presence of water vapor. Secondary sulfate aerosol particles are formed from the oxidation of SO2 to H2SO4 as the plume disperses into the background atmosphere. Oxidation can occur by gas-phase reaction with OH, and aqueous-phase reaction with ozone, H2O2, and O2 with transition metal ions. In volcanic plumes, catalysis by metallic ions might significantly enhance oxidation of SO2 with O2 (Galeazzo et al., 2018). H2SO4 is highly hygroscopic and can create aqueous H2SO4(aq) droplets via homogeneous nucleation or condense onto existing accumulation mode particles (Seinfeld et al., 1998; Mather et al., 2006; Martin et al., 2012b; Galeazzo et al., 2018).
The surface area of hygroscopic sulfate-rich particles is a strong function of humidity and temperature conditions. However, as the abundant amounts of aerosol provide a substantial surface area for reactions, the formation of BrO may not be limited by aerosol availability, particularly near-downwind. Bobrowski and Giuffrida's studies (Bobrowski and Giuffrida, 2012) at 6 km downwind did not find influences on the BrO/SO2 ratio due to seasons, relative humidity, and wind velocities. Although higher relative humidity and aerosol with high sulfuric acid concentrations are proposed to increase the probabilities and rates of halogen reactions, they seem not to be a real limiting factor in volcanic plumes. However, Dinger et al. (2018) identified correlations between observed BrO/SO2 molar ratios and relative humidity (correlation coefficient 33%) by investigating periodic patterns in the BrO/SO2 ratio of Cotopaxi's volcanic plume.
Nitrogen compounds
There is still a great deal of uncertainty as to whether nitrogen compounds NOY (NOX and oxidation products including organic nitrogen compounds) are emitted or formed in volcanic emissions and which products are built by the interaction of bromine and nitrogen compounds in volcanic plumes. In polluted environments, NOX might be present in the atmosphere and enter volcanic plumes upon dilution. Mather et al. (2004a) observed nitric acid at the volcano crater rim but proposed not a direct emission of nitric acid. Rather NO is predicted to be formed thermally by equilibrium models at near vent conditions from atmospheric N2 and O2 (Gerlach, 2004; Martin et al., 2006). It is assumed that in the volcanic plume at ambient temperature NO will be rapidly oxidized with O3 to NO2 (von Glasow, 2010). Further oxidation pathways (R10–R11 below), or reaction of NO2 with OH may then produce HNO3.
von Glasow (2000) suggested a reaction of Br radicals with volcanic NO2 immediately after plume release of BrNO2 (R8) and due to photolysis a lifetime of about 150 s (R9). Since large amounts of NO2 are proposed in the plume the continuous reformation and accumulation of BrNO2 is assumed. This reduces the amount of bromine available to form BrO.
M is a chemically inert collisional partner.
In contrast, Roberts et al. (2014b) suggest the volcanic plume BrNO2 lifetime will be short. Roberts et al. (2009) suggest rather a formation of BrONO2 due to the reaction of BrO with NO2 (R10). BrONO2 rapidly reacts heterogeneous on volcanic aerosols producing HOBr that may immediately react via R4, forming Br2 and nitric acid (R11). Under high aerosol conditions, BrONO2 can thereby promote the “bromine explosion,”
However, the importance of these chemical pathways depends on the abundance of NOX in volcanic plumes. As mentioned above a key question is whether NOX is generated from volcanoes or simply being entrained into the plume by dilution. Although the formation of NOX is predicted by equilibrium models, a high-temperature kinetic model study found that the time-scale to produce significant quantities of thermal NOX formation is too slow in the near-vent plumes (Martin et al., 2012a). However, several observations of elevated NOX and NOY in volcanic plume have been reported: an overview is given in Martin et al. (2012a). For example, aircraft measurements were able to identify volcanogenic HNO3 and HO2NO2 (Oppenheimer et al., 2010). However, this study did not observe any NOX in the downwind plume. They assume that near-source NOX was quickly oxidized to HNO3 and HO2NO2. HNO3 has been reported in several volcanic plumes (e.g., Mt Etna, Sicily, Italy; Voigt et al., 2014), Masaya, Nicaragua; Mather et al., 2004b). However, Martin et al. (2010) did not detect any HNO3 at Masaya in 2009. For some eruptions (particularly explosive), volcanic-induced lightning could be a source of NOX in parts of the plume. For volcanic plumes entering polluted background atmospheres, the entrained air can be a source of NOX (and NOY) as the plume dilutes. Overall there remain uncertainties in the sources of volcanic plume NOY, and hence the role of coupled nitrogen-bromine chemistry in volcanic plumes and its importance for Br-speciation.
Mercury in volcanic plumes
Mercury is emitted from volcanoes, but is also present in the background atmosphere, predominantly in the inert form Hg(0). It is believed that reactive bromine (as well as chlorine, see section Interhalogen, Chlorine, and Iodine Plume Chemistry) species might promote conversion from relatively inert Hg(0) (emitted as well as atmospheric mercury) into more reactive and toxic oxidized mercury [Hg(II), R12–R13], although observations of volcanic mercury transformations are few and somewhat inconclusive to date. Oxidized mercury is soluble and partitions into aerosol particles so can be more easily deposited near the volcano (R14).
There are uncertainties in mercury chemistry in volcanic plume reactions, notably whether a back-reaction can reform inert Hg(0) from oxidized Hg(II), possibly involving photolysis, SO2 and particles. Mercury concentrations are generally thought to be at sufficiently trace-levels that the mercury chemistry does not impact overall Br-speciation (Bagnato et al., 2007; von Glasow, 2010).
Causes and Effects of O3 Depletion
Models predict the depletion of tropospheric ozone destroyed by the reaction sequence R2–R5 each cycle (e.g. Bobrowski et al., 2007; Roberts et al., 2009; von Glasow et al., 2009; Kelly et al., 2013; Surl et al., 2015). In addition, ozone loss occurs through a second cycle between the self-reactions of BrO (R15, R16). This rapid interconversion causes ozone loss in the plume at high halogen concentrations, and so can be particularly important in destroying ozone in the near-downwind (concentrated) volcanic plume.
Note also that the reactions R15 and R16 will tend to reduce the abundance of BrO at high halogen concentrations and therefore can bring additional non-linearities to the chemistry of volcanic BrO formation. Determination of ozone in volcanic plumes by UV absorption is difficult due to high SO2 concentrations (see Table 2). As both O3 and SO2 broadly absorb UV radiation, two or more simultaneous observations at different wavelengths are required to deconvolute SO2 from O3 by UV absorbance alone. Because many UV absorbance instruments employ a UV line source (frequently Hg vapor, 253.7 nm), additional treatments are needed to correct for SO2 coabsorption. In those cases to avoid SO2 interferences a CrO3 scrubber is necessary to remove SO2 from the inflow to the instrument (e.g., Schumann et al., 2011; Surl et al., 2015). Dual channel ozone instruments also yield accurate results in the presence of SO2. These detect ozone by taking the difference between a “measurement” cell and “background” cell (with in-line ozone scrubber), therefore any SO2 interference cancels out (Vance et al., 2010). In cases of large ozone depletion at low SO2 the interference may be estimated using field-observations of SO2 and subtracted in data post-processing (Kelly et al., 2013). Electrochemical concentration cells (ECCs) sense ozone as it reacts with a diluted solution of potassium iodide to produce a weak electrical current proportional to the ozone concentration of the sampled air. A negative cross-sensitivity (observed ozone destruction) on mole-per-mole-basis may be caused due to SO2 (Vömel and Diaz, 2010). However, no interferences in volcanic applications are known with chemiluminescence-based techniques that measure O3 by sensing light output from the reaction of O3 with ethylene, although an overread of O3 (few %) at high humidity may occur (Vance et al., 2010). Recently an Aeroqual instrument, WO3 semiconductor, was used for ozone measurements, for which the most important cross-sensitivity in volcanic plumes is that of H2S (Roberts, 2018). Time-averaged ozone measurements have also been made using diffusion tube passive sampling devices (Vance et al., 2010). These are filled with an adsorbing cartridge that has a highly specific irreversible chemical affinity to ozone (e.g., the “ozonolysis” of double bonds to form an aldehyde). In the lab, hydrazine is added to form an acid that can be detected via absorption.
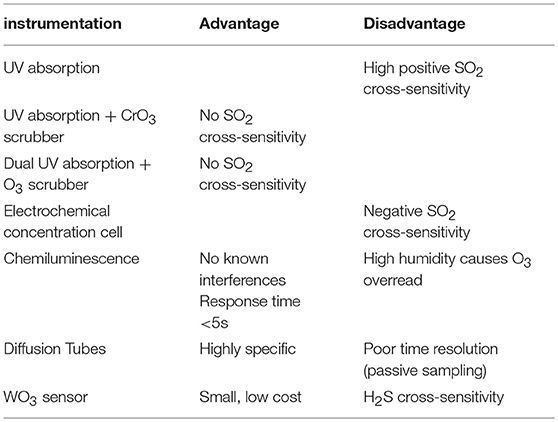
Table 2. Ozone measurement devices and their advantages and disadvantages for application in volcanic plumes.
Ozone loss due to heterogeneous reactions in the presence of halogens has already been observed in several tropospheric volcanic plumes (see Table 3, for a discussion of halogen impacts on ozone in the stratosphere see section Introduction). Ozone in Kilauea's plumes was reported close to ambient levels in crater-rim emissions (Halema “uma”u) and only slightly depleted in the 10 km downwind plume (Pu‘u ‘Ō‘ō) (Roberts, 2018). This is consistent with Kilauea being a low halogen emitter. Large ozone depletions (several 10's%) were observed in the plumes of higher halogen emitters such as Mt Etna, Sicily, Italy and Mt Redoubt, Alaska (see Table 3). In general, the largest O3 depletion was observed in the core of the plume and was correlated with volcanic gases (e.g. Kelly et al., 2013), with a maximum deficit of −30 ppbv and an average O3 deficit reaching −12 ppbv at 20–40 min plume age. That is consistent with model simulations that reproduced ozone depletion in the core of the plume (Kelly et al., 2013). Observational and modeling results suggest that the conversion of volcanic Br emissions into reactive bromine are key controls in the depletion of tropospheric ozone downwind from the volcano. If ozone becomes entirely depleted in the plume this might in turn limit the partitioning of reactive bromine into BrO in the near-source (concentrated) plume, e.g., as suggested for conditions of extreme emissions and/or low background ozone concentrations (Jourdain et al., 2016). However, full depletion of ozone in a volcanic plume has only been observed in two measurements at Augustine in 1976 (Vance et al., 2010). Note that humidity might have an influence and is not considered here and that Hegg et al. (1976) reported a calibration accuracy of ±5% when the chemiluminscence technique was compared to ozone detection via potassium iodide. Reactive bromine is the principal cause of ozone depletion in volcanic plumes, but models predict that the interaction of BrO with reactive chlorine (or iodine) can augment the ozone loss.
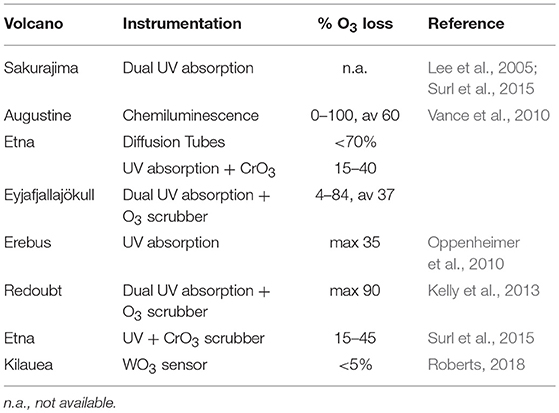
Table 3. Overview of sampling campaigns on O3 depletion in volcanic plumes.
Interhalogen, Chlorine, and Iodine Plume Chemistry
Chlorine
The bromine explosion can ultimately also lead to the production of reactive chlorine. First, studies of Roberts et al. (2009) using the AIM model (Wexler and Clegg, 2002) on gas-particle-partitioning for mean arc volcanic compositions (Gerlach, 2004) reported that Br2 is the favored product in heterogeneous reactions (R4) for typical volcanic Br/Cl emission compositions, according to aqueous-phase equilibria (R18). However, depletion of plume HBr through the bromine explosion can significantly reduce HBr/HCl ratios (100–1,000 times lower than arc mean composition). Under these conditions, the reactive uptake of HOBr can lead to the formation of BrCl (R17) instead of Br2 in R4.
Subsequent partitioning of BrCl (and Br2) into the gas phase and photolysis produces Cl alongside Br radicals (R19) that can react with ozone (R20) and contribute to ozone loss (see also R5 and R2).
The reaction of ClO with BrO can lead to OClO (R21, Bobrowski and Platt, 2007; Gliß et al., 2015; Roberts et al., 2018).
OClO/SO2 has been reported to increase from below detection limit at the vent up to (3–6) × 10−5 at Mt. Etna, Sicily, Italy [BrO/SO2: (1–5) × 10−4; Bobrowski et al., 2007; General et al., 2015; Gliß et al., 2015] and (4–6) × 10−4 at Soufriere Hills, Montserrat [BrO/SO2: (1–4) × 10−4; Donovan et al., 2014]. Compared to bromine activation <1% HCl is converted into reactive chlorine, according to both models and observations (Gliß et al., 2015). The few field-data are too limited to determine further controls on OClO formation, although the increasing OClO/SO2 is consistent with an in-plume formation pathway.
Recently, models have been able to reproduce volcanic OClO observations (Roberts et al., 2018), according to the HBr depletion and BrCl formation pathway. Models predict that bromine emissions and humidity can both impact OClO formation rates. For higher bromine emissions HBr may not become depleted immediately by the bromine explosion, leading to a later onset of BrCl formation and thus delayed rise in OClO/SO2. Modeled OClO/SO2 formation near downwind is enhanced at greater relative humidity because this enhances the aerosol surface that supports the heterogeneous reactions, leading to a more rapid bromine cycling and faster depletion of HBr and enabling BrCl formation (Roberts et al., 2018).
Kern and Lyons (2018) report observations of OClO (with BrO) at the plume edge, whilst only BrO is observed at the plume center. The measurements are very consistent with the horizontal spatial distribution of BrO-OClO predicted by Roberts et al. (2018). Kern and Lyons (2018) propose that measurements of OClO alongside BrO can help determine the degree of depletion of Br(aq) (and HBr) and therefore guide interpretation of BrO observations in terms of total bromine emission.
Importantly, the fact that Br2 (or BrCl when HBr is depleted) are the preferential products released from the reactive uptake of HOX (X = Br, Cl) and the fast reaction of Cl with methane (R22) prevents a self-amplifying cycle for chlorine oxides. Thus, HCl cannot become depleted by plume reactive halogen chemistry (unlike HBr).
Iodine
Only a few studies are available that report about iodine in tropospheric volcanic plumes, although iodine chemistry is well-studied in marine environments (Saiz-Lopez and von Glasow, 2012 and references therein). Iodine is released as a trace emission in tropospheric volcanic plumes. For instance a total iodine to sulfur ratio (I/S) has been observed between (4–7) × 10−6 detected with filter packs at Etna, Sicily, Italy in 2004 (Aiuppa et al., 2005), (7–22) × 10−6 and (4–90) × 10−6 at Etna and Stromboli, Mediterranean with alkaline traps in 2010–2012 (Wittmer et al., 2014), (3–5) × 10−6 with the Raschig Tube at Nyamuragira, in the Virunga Mountains of the Democratic Republic of the Congo in 2015 (Bobrowski et al., 2017) and 2 × 10−5 and 5.8 × 10−6 at Masaya and Telica, both Nicaragua respectively with filter packs in 2006 (Witt et al., 2008). Bobrowski et al. (2017) reported that the total iodine already comprises 8–18% non-hydrogen iodide measured at the crater rim of the pit crater at Nyamuragira. A major eruption of Kasatochi, Aleutian Islands 2008 enabled rare detection of IO in the volcanic plume in the stratosphere by satellite (Schönhardt et al., 2017). We highlight that the ozone depletion potential of iodine may have significant ozone-depletion impacts even as a trace emission. Iodine has an alpha factor (chemical effectiveness relative to chlorine for global ozone destruction, globally and annually averaged) of 150–300. Compared to bromine (alpha factor ca. 60), iodine can affect ozone levels even more effectively (World Meteorological Organization, 2014 and references therein).
Like the other halogens volcanic iodine is thought to be emitted mainly as hydrogen iodide with subsequent radical formation near the vent in a similar manner to bromine (R1, see section Bromine Explosion). IO is formed out of iodine radicals reacting with ozone (R23).
The reaction of IO with another halogen oxide (XO, with X = Br, I) leads to OIO formation (R24). OIO can form higher iodine oxides which are precursors in particle formation and thus affect the atmospheric radiation balance (Hoffmann et al., 2001).
Reactions with HOI (formed analogously to R3) form interhalogens (R25, XI with X = Br, Cl) that are released to the gas phase. Interhalogens can be photolyzed (analogous to R10) leading to halogen radicals that are involved in the catalytic cycles of halogens through plume chemistry as outlined above.
Further details are given in von Glasow and Crutzen (2003) and literature therein.
BrO Evolution in Volcanic Plumes
Dependence on Plume Age – Temporal Evolution
Observations of BrO/SO2 ratios as a function of distance from the emission source allow to trace chemical transformations of volcanic bromine (under the condition that SO2 is conserved). BrO was reported to be not detectable at crater rims but is formed inside the atmosphere-volcanic gas plume mixture over very short time-scales (minutes). The development of BrO/SO2 with distance downwind is shown in Figure 3, based on results of Bobrowski and Giuffrida (2012) and Platt and Bobrowski (2015) complemented by further measurement series (derived from the Supplementary Table 1). Each data series is normalized to the maximum BrO/SO2 observed, and averages were taken for measurements at the same distance within a given data series. All data arrays confirm a general rising trend. Close to the emission source BrO/SO2-ratios are low. Excluding Ambrym, Vanuatu BrO/SO2-ratios seem to reach a relatively stable level within 5–10 km from the source. The near-constant levels then appear to decrease in some cases, for example Etna, Sicily, Italy 2004, Nyiragongo, in the Virunga Mountains of the Democratic Republic of the Congo 2004 but not for other data series e.g., Etna 2005 and Ambrym 2007.
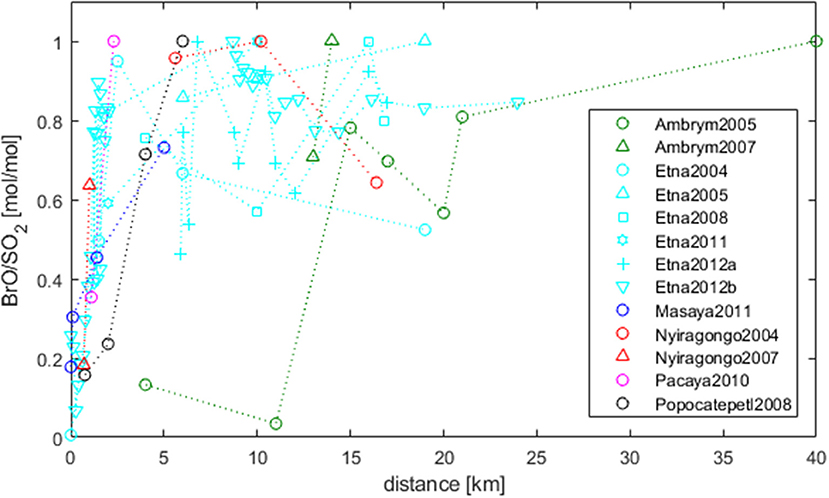
Figure 3. Development of BrO/SO2-ratios (normalized) in the plume as a function of distance from the emission source [extended from Bobrowski and Giuffrida (2012) and Platt and Bobrowski (2015), using datasets in Supplementary Table 1 and references therein]. For data-series with the same distance averages are formed. BrO is not detectable at crater rims but typically rises to a near stable level within 5–10 km (for details see section Dependence on Plume Age–Temporal Evolution).
The development of BrO is a function of plume age (and therefore depends as well on the distance); however, the distance-time interconversion depends on wind-speeds that are specific to each data-series. This means BrO formation, even if taking place at the same plume age, might be measured at a different distance. Mixing between the plume and air is also a function of distance or time downwind, and is suggested by models to strongly influence halogen speciation, hence BrO/SO2. Indeed, whilst all volcanic plume model studies predict the initial rise in BrO/SO2, the occurrence of a subsequent plateau or decline depends strongly on the downwind plume-air mixing (Roberts et al., 2014b). For large gas emission fluxes like at Ambrym a lower proportion of air may initially mix into the plume. This could reduce BrO/SO2 close to the source, as is found for both models and observations (Bani et al., 2009; Jourdain et al., 2016). However, this aspect still needs to be further investigated using measurement data.
Spatial Distribution in the Cross-Section of the Plume
Beside distribution of BrO along the plume-direction (as a function of plume age, dependent on distance from the emission source), measurement and model studies have also investigated the vertical or horizontal distribution of BrO and OClO in the cross-section of the plume.
Vertical profiles were calculated from two-dimensional BrO and SO2 distributions measured by imaging DOAS instruments (Bobrowski et al., 2007; Louban et al., 2009; General et al., 2015). Gliß et al. (2015) and recently Kern and Lyons (2018) investigated the vertical BrO/SO2 distribution by using a 1 dimensional scanner unit. Bobrowski et al. (2007) found highest BrO/SO2 values at the edges of the plume (3 × 10−4) compared to lower values in the center (2 × 10−4) and reproduced this trend with their model data.
Jourdain et al. (2016) discussed favored BrO formation for the edges of a volcanic plume more quantitatively, finding general agreement between model and measurement data in the trend in BrO/SO2. While at the edges BrO reached up to 60% of the total bromine species, in the core BrO remains below 30%. Modeling predicts that Br radicals are the abundant species in the core. An explanation might be the entrainment of O3-rich ambient air at the plume edges to promote BrO formation, but limited transport of tropospheric O3 and HOX radicals toward the plume center (see section Causes and Effects of O3 Depletion). However model studies show that plumes without ozone depletion in the core can also exhibit greater BrO/SO2 (and OClO/SO2) at the plume edge (Roberts et al., 2018). This is due to the interconversion reactions between reactive bromine species, in particular Br, BrO (section Causes and Effects of O3 Depletion), that enhances BrO/Br in the more dilute edge compared to the core of the near-downwind plume.
BrO as a Fraction of Total Bromine Under Different Plume Conditions
The conversion of HBr and the formation of BrO compared with total SO2 flux, as well as the depletion of ozone have been reported in few measurement observations (see section Causes and Effects of O3 Depletion) and have been investigated with model studies. Roberts et al. (2014b) showed in studies of Etna's plume HBr fully converted within tens of minutes with HBr/SO2 ratios of 7.4 × 10−4. While HBr/SO2 ratios of 24 × 10−4 a conversion of only about 50% was determined within an hour of simulation (both with 20 kg/s SO2). Bani et al. (2009) observed an increased and faster BrO/SO2 formation dependent on the distance by comparing data with SO2 flux below 100 kg/s and above 100 kg/s at Ambrym, Vanuatu in 2005 and 2007. Although it has to be noted that no influence of bromine emissions can be investigated in this case due to the lack of simultaneous HBr measurements.
The impact of total (Br flux) and relative (Br/S) bromine emissions on BrO development can be investigated using simultaneous measurements of Br/S and BrO/SO2, which enables the calculation of BrO as a fraction of total bromine (BrO%). Near-simultaneous Br/S and BrO/SO2 data sets from Supplementary Table 1 have been used to calculate the BrO% fraction as a function of the distance from the emission source in Figure 4. The dependence of BrO% with distance shows a positive dependency (similar as for Figure 3) further supporting the assumption that increased BrO/SO2 is caused by the formation of BrO. The particularly low BrO% for Redoubt, Alaska might be explained by the fact that the measurements of Br/S and BrO/SO2 were not quite simultaneous (which could introduce non-linearities if the bromine emission changed).
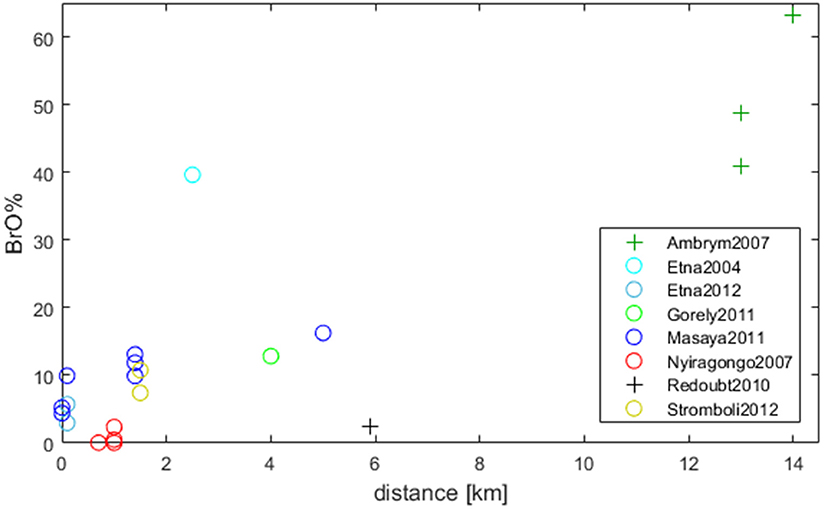
Figure 4. The BrO% fraction is shown dependent on the distance to the emission source. BrO% describes (BrO/SO2)/(Br/S). The trend (also observed in Figure 3) leads to the assumption that increased BrO/SO2 is caused by the formation of BrO. Circles (o): simultaneous taken data-sets; Plus (+): non-simultaneous data sets but have been compared in other publications (Ambrym: Bani et al., 2009; Allard et al., 2016; Redoubt: Kelly et al., 2013). For further details see Supplementary Table 1 and section BrO as a Fraction of Total Bromine Under Different Plume Conditions.
The highest BrO% between 40 and 65% was observed at Ambrym, Vanuatu in 2007 (SO2 flux of 47 kg/s). These datasets were also obtained at the furthest distance downwind (12–14 km) i.e., greater time-evolution to form BrO. Note that BrO/SO2 were measured in August 2007 (Bani et al., 2009) while Br/S results derive from October 2007 (Allard et al., 2016). Jourdain et al. (2016) performed model simulations of the Ambrym plume, comparing to BrO/SO2 measurements in January 2005 (Bani et al., 2009). The SO2 flux was significant higher in 2005 (average 218 kg/s) than in 2007. The model predicted up to 60% BrO% at 200 km distance to the emission source at the edge of the plume (HBr/SO2 6.87 × 10−4). In the core only below 30% BrO% is observed.
A high BrO fraction of about 40% was observed at Etna, Sicily, Italy in 2004 with Br/S 4.8 × 10−4 and an SO2 flux of 11 kg/s (Oppenheimer et al., 2006). Note that measurements on Etna in 2012 (Wittmer et al., 2014) resulting in 3 and 6% at similar Br/S ratios of 8.8 × 10−4 and 4.2 × 10−4 respectively occurred at plume ages < 1 min. While 40% were reached by observations of Oppenheimer et al. (2006) at 2–3 km (3–4 min estimated plume age), the model results by Roberts et al. (2014b) mentioned above with HBr/SO2 7.4 × 10−4 (SO2 20 kg/s) resulted in approximately 25% at 5 min plume age. This comparison lacks of the fact that we do not know if the model set-up would agree with Oppenheimer et al.'s plume measurement parameters. It is worth highlighting that the value of 40% BrO% found for the Etna 2004 measurements is rather high compared to other datasets around this distance downwind (Figure 4).
Model studies of Bobrowski et al. (2015) compared Etna's plume (bromine flux 22 g/s, maximum BrO/SO2 2.1 × 10−4) with slightly greater bromine flux of Nyiragongo's plume (in the Virunga Mountains of the Democratic Republic of the Congo, bromine flux 30 g/s, maximum BrO/SO2 0.7 × 10−4). Simulations predicted similar BrO columns in both cases but differ in relative BrO formation. Even though Nyiragongo has greater Br/S ratios in its volcanic plume than Etna, observations of the relative conversion to BrO downwind show smaller values (BrO%: 0–2%) in the aged plume. For Nyiragongo 2007 model predictions fit with measurements, if a plume age of 1–2 min is assumed.
Comparison of Nyiragongo with studies by Kelly et al. (2013) at Mt Redoubt results in a slightly higher conversion of HBr (although low compared to Etna) in Mt Redoubt's plume. Even though Br/S emissions of 41 × 10−4 are smaller than in some cases of Nyiragongo (note also the SO2 flux of 4.3 kg/s is lower, resulting in also lower quantities of bromine compared to Nyiragongo as well as Etna). Model simulations that reproduced the observed ozone loss in the plume of Mt. Redoubt predicted only 30% of emitted HBr to be converted into reactive forms. This may be due to the rather dilute Redoubt plume because conversion of HBr into reactive bromine via the autocatalytic bromine explosion strongly depends on abundances of bromine and aerosols. BrO formation in this case seems to be rather low compared to other data sets mentioned above (see Figure 4). Note that model parameters and Br/S results are based on measurements at Mt Redoubt in June 2010 but BrO/SO2 observations were made in August 2010. The observed BrO/SO2 of 1 × 10−4 in August is rather low (BrO% of 2.4%) compared to the June model results (maximum BrO/SO2: 6 × 10−4, BrO%: 14%). For comparison, GOME-2 observations of Mt. Redoubt plume in the end of May found 2.5 × 10−4 BrO/SO2 (Hörmann et al., 2013, see Supplementary Table 1). Also, the ozone depletion of August (up to 70% below ambient air at 19 min estimated plume age) is lower than observed in June (up to 90% below ambient air at 38 min estimated plume age). Since the rapid conversion of HBr to form BrO leads to ozone consumption (see section Causes and Effects of O3 Depletion), the differences in observed ozone loss over June-August imply that BrO/SO2 was likely higher in June 2010 than reported in August 2010. A possible explanation is that the bromine emission decreased over these months, or that changes in the plume chemistry led to a reduced conversion of emitted bromine into BrO. Unfortunately, this hypothesis cannot easily be tested as Br/S was not monitored throughout.
To study the relationship between emission fluxes and BrO fractions there are mainly three mandatory tasks: (1) Simultaneous measurements of Br/S and BrO/SO2. (2) The determination of bromine fluxes or at least SO2 fluxes so Br flux can be calculated from Br/S ratios. (3) Observations of BrO should be made at a certain distance/plume age from the emission source. As discussed before (section Dependence on Plume Age–Temporal Evolution) for comparison of BrO results in plumes measurements should be used, which reached the BrO/SO2 “plateau” after ~5–10 min (2,5–10 km, depending on wind speed and plume condition). All three tasks are more or less realized by only two data sets for Etna in 2004 (Oppenheimer et al., 2006) and Gorely in 2011 (Aiuppa et al., 2012; Bobrowski et al., 2012; Platt and Bobrowski, 2015). Remarkable is the fact that Etna's plume (Br/S: 4.8 × 10−4, SO2: 11 kg/s) results in a significant higher HBr to BrO conversion of ~40% (BrO% 40%) than in Gorely's plume that had a slightly lower SO2 flux (9.3 kg/s) and slightly higher Br/S ratio (5.3 × 10−4, i.e., resulting in similar Br flux) resulting in ~13% BrO%.
All low-temperature kinetic models pointed out that the absolute amount of total gas emission, the total emitted bromine (also chlorine) and the initial formation of HOX or halogen radicals in the effective source region, and the role of plume-air mixing all seem to be exert strong controls on BrO formation. However, these parameters are not well constrained in the models, nor for the intercompared field-observations, due to lack of datasets. For the intercomparison of volcanoes, the overall atmospheric setting where volcanoes are located at should also be noted. Etna is in a more polluted region, while Nyiragongo and Redoubt release plumes in more remote, clean atmospheres. Therefore, there might be a difference in the ambient ozone and NOX levels. It is also possible that volcanic plume halogen chemistry might be influenced by marine sources of halogens and aerosols. Finally, temporal variations due to meteorological or/and volcanological conditions may introduce variability in the datasets used for intercomparisons (noting Br/S and BrO/SO2 measurements are difficult to perform exactly simultaneously as they are made at different locations on the volcano).
The challenge to interpreting BrO observations is highlighted by Cadoux et al. (2018). GOME-2 observations of Merapi's eruption in Indonesia 2010 resulted in extremely low BrO/SO2 ratios (maximum 8 × 10−6; Hörmann et al., 2013), much lower than expected based on petrological estimates of the halogen/sulfur emission. Cadoux et al. (2018) discussed that BrO formation could be limited by the restriction of photochemical driven reactions caused by the ash-rich paroxysmal phase and the absence of UV light during the eruption in the night (the BrO was detected by GOME-2 in the following day). According to Cadoux et al. (2018) other possibilities include: (a) scavenging of bromine in the troposphere (e.g., adsorption of bromine on ash), (b) kinetic factors lead to favored S degassing from the magma, (c) brine saturation leading to Br uptake when magma rises, and (d) changes in Br partitioning caused by other volatile species in the magma. To this, we add the important role of plume chemistry in converting emitted HBr into BrO, as emphasized in this review.
Forecasting – Correlating Volcanic Plume Composition to Volcanic Activity
The characterization of volcanic gases can give insights about processes inside volcanoes. Volcanic gases are solved in magma at depth under high pressure. When magma is rising, decompression, diffusion, and coalescence lead to bubble formation and growth and therefore the release of gases. The depth at which gases start to escape can be different with different types of magma. The release of gases has an influence on the rising magma due to their influence on density, compressibility and flow properties. Therefore, magma degassing is a crucial control of eruption dynamics (Malinconico, 1979; Carroll and Holloway, 1994; Sparks, 2003; Moussallam et al., 2016). A strong influence is already known for the most abundant emitted gases like H2O and CO2. Nevertheless, even trace gases like fluorine are thought to control the viscosity of magmas with effects on crystallization-fractionation paths, controlling degassing paths and therefore the eruptive behavior (Giordano et al., 2004).
H2O is the most abundant emitted species, but water vapor emissions are difficult to determine due to high and very variable atmospheric background. Further, emitted water vapor is difficult to interpret because it does not always purely originate from magma but might derive also from hydrothermal systems or in some cases even from incorporated local seawater. Nevertheless, recent studies show the potential to determine water vapor in volcanic plumes and renew the hope to use them as eruption precursors in the future (Moor et al., 2016a,b; Kern et al., 2017).
Monitoring of CO2 by remote sensing instruments is difficult as well. Non-volcanic CO2 sources and high and variable atmospheric background concentrations may lead to variability in volcanic CO2/SO2 measurements (Burton et al., 2000). Today, CO2/SO2 ratios need usually to be determined by in situ sampling, however these are spatially limited and sampling points are not always representative and are frequently destroyed during eruptive periods. CO2 is exsolved at relatively large depth and SO2 is released at much shallower levels. Degassing of the halogens Cl and F occurs often at even lower pressure, closer to the Earth's surface (Figure 5; Spilliaert et al., 2006; Burton et al., 2007; Aiuppa, 2009). Observations of decreasing CO2/SO2 ratios and increasing Cl/SO2 ratios could therefore be interpreted as gases derived from magma moving to shallower depth or magma from already exhausted reservoirs.
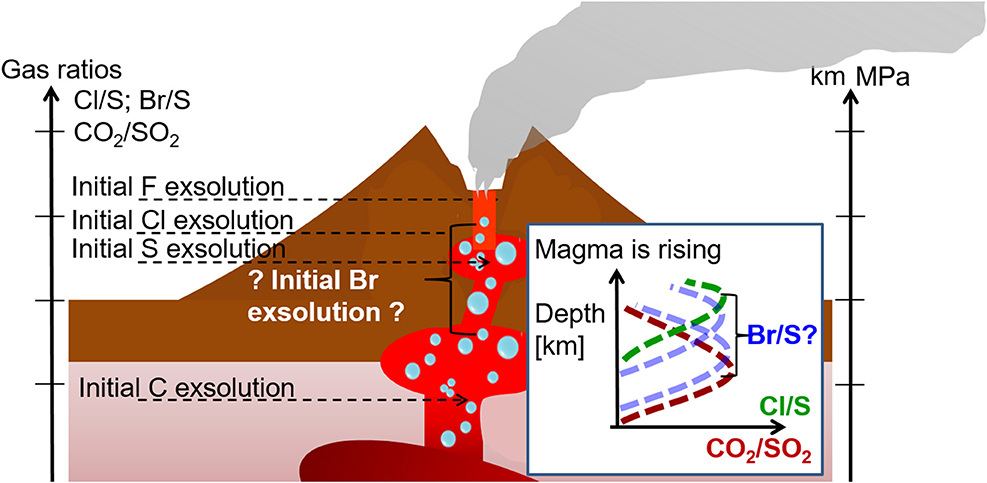
Figure 5. Magma degassing model for volcanic gases. Carbon is held to exsolve at depth, S, Cl, F follow at lower pressures closer to the surface. Diagram illustrates changes of observed gas ratios if magma is rising. Whether bromine partitioning occurs at higher or lower pressure than sulfur is not clarified and part of current scientific discussions (for details see section Forecasting–Correlating Volcanic Plume Composition to Volcanic Activity).
Although bromine is not known to have a large influence on eruption dynamics, bromine degassing could be nevertheless a good indicator for rising magma. In contrast to CO2 and H2O, SO2 and bromine species have negligible background concentrations and clearly derive from magma degassing. The fact that SO2 and BrO are measurable simultaneously by remote sensing instruments facilitates its applicability for monitoring volcanic activity.
Aiuppa et al. (2005) correlated Br/S ratios from filter pack samples to Etna's activity in 2004 observing increasing Br/S as well as Cl(F)/S prior to an eruption, followed by a rapid decrease. In contrast to the Br/S observation of Aiuppa et al. (2005), several authors observed lower BrO/SO2 ratios during or also prior to eruptive activities in comparison to observations made during quiescent degassing or after eruptive activity phases. For instance as reported by Bobrowski and Giuffrida (2012) during eruptive and non-eruptive periods of Etna, Sicily, Italy from 2006 to 2009, Lübcke et al. (2014) at Nevado del Ruiz, Colombia from 2009 to 2013, Dinger et al. (2018) and Hidalgo et al. (2018) at Cotopaxi, Ecuador 2008, Warnach et al. (in review) at Tungurahua, Ecuador 2008–2014. The time lag between gas composition change and start of eruptive activities differs between the various studies and volcanoes; however, the general trend seems to be similar. Conversely, the discussion of BrO-mediated ozone depletion at Mt Redoubt, Alaska (see section BrO as a Fraction of Total Bromine Under Different Plume Conditions) points toward a possible decrease in bromine emission with assured activity.
In contrast to most chlorine and fluorine studies, recent investigations observe a correlation between BrO/SO2 ratios and CO2/SO2 ratios (Bobrowski et al., 2015, 2017) leading to the assumption that bromine exsolution might be deeper than sulfur. Only recently studies on partitioning and/or knowledge of the magma movement through other geophysical or geochemical data start to deliver a basis for the interpretation of available field data (Cadoux et al., 2018). With increased degassing a change of the gas composition from S-rich to Cl-Br-rich is predicted, favoring the hypothesis of a relatively shallow degassing of bromine in comparison to sulfur that goes along with measurement results of Aiuppa et al. (2005). However, both possibilities are discussed–shallow and deep degassing of bromine (Figure 5).
Summary of Main Findings
For the interpretation of BrO data regarding volcanic activity, it is essential to have a detailed understanding of the ongoing chemistry, to be able to recalculate total bromine emissions. There is a temporal and spatial heterogeneous distribution of the BrO concentration in volcanic plumes (see sections Dependence on Plume Age–Temporal Evolution and Spatial Distribution in the Cross-Section of the Plume). Current model and field studies demonstrate that most of the BrO is not directly emitted and describe its formation through low-temperature, kinetically controlled photochemical and multiphase reactions and mixing processes with the surrounding atmosphere in the downwind plume (see section Plume Chemistry and the Formation of Reactive Bromine, summary in Figure 2).
Common main findings are:
- BrO/Brtotal rises rapidly within the first minutes of plume age but usually stabilizes and then decreases again further downwind (section Dependence on Plume Age–Temporal Evolution).
- Low-temperature “bromine explosion” chemistry mechanism is able to explain BrO abundances in the downwind plume. But elevated concentrations of radicals e.g., HOX and/or NOX derived from high-temperature near-vent chemistry in the initial plume seem necessary to accelerate the start to enhance low-temperature BrO formation downwind in order to reach observed magnitudes of BrO (section Plume Chemistry and the Formation of Reactive Bromine).
- The autocatalytic cycles forming BrO are driven by reaction cycles involving aerosol- and gas-phase and photolysis reactions and affected by O3 entrainment, leading to spatial heterogeneity in plume BrO/SO2 as seen in vertical plume profiles (sections Spatial Distribution in the Cross-Section of the Plume).
- The products from heterogeneous halogen reactions are controlled by aqueous-phase equilibria that produce Br2 (hence BrO) in volcanic plume environment. Formation of BrCl (hence OClO) occurs upon very low levels of HBr as a consequence of the bromine explosion (section Interhalogen, Chlorine, and Iodine Plume Chemistry).
- Studies of the near-source plume chemistry point to non-linearities in HBr conversion into BrO depending on the bromine content of the emissions, plume-air mixing and other factors.
Limitations
Today, the knowledge on bromine plume chemistry is limited mainly by measurement restrictions. Model studies that have been performed in relation to particular data arrays can generally reproduce the available (limited) observations but operate in a vast parameter space that is currently not well constrained. Discrepancies exist in the predicted bromine speciation between the two available model (MISTRA and PlumeChem) probably as a result from differences in simulation algorithms, but also the wide volcanological and meteorological parameter space to which they have been applied. A general takeaway of obtained findings on one volcano cannot necessarily be applied to other volcanoes (e.g., comparison of Etna, Sicily, Italy and Nyiragongo, in the Virunga Mountains of the Democratic Republic of the Congo in Bobrowski et al., 2017). The high-temperature near-vent plume compositions are crucial for initializing low-temperature models that simulate the downwind plume chemistry. Near-source chemistry is usually investigated with high-temperature thermodynamic models. Since the hot near-vent plume cannot easily be observed by measurements, there are still uncertainties in parameterizations. The often assumed thermodynamically equilibrium conditions might not hold true (see section Model Initialization–High-Temperature Near-Vent Conditions, Dilution and Aerosols), therefore, a better characterization of the near-source volcanic plume chemistry is an essential step for understanding the evolution of volcanic plumes in the atmosphere. Even though the “bromine explosion” chemistry mechanism is relatively well-understood although still not fully characterized for volcanic plume environments there remain also some uncertainties in the low-temperature modeling for example in the parameterization of the gas-aerosol reactions (Roberts et al., 2014a), and the role of NOX in volcanic plumes. A major issue, however, is that available datasets are typically too limited to fully constrain the model initialization such that some parameters have to be assumed, and only very limited data exist for model evaluation.
The growing amount of data sets and detailed model studies help to improve hypotheses. However, most collected data sets are incomplete toward bromine speciation and development in the plume. Future data sampling is recommended to be taken as comprehensive as possible. Therefore, three main points need to be considered: (1) Simultaneous measurements of the emissions-plume chemistry and physics are needed as both emissions and plume dynamics can vary rapidly with time. (2) Measurements of BrO for volcanic activity monitoring should be systematically undertaken at a certain plume age also because the influence of plume age on bromine chemistry needs to be taken into account to enable the interpretation of the data in the context of volcanic activity. (3) Additional parameters like ozone, aerosol particle composition, and size distribution as well as meteorological conditions, and constraints on gas flux and plume dilution/mixing into the background atmosphere are mandatory. Evidently, many restrictions reduce the availability of those data sets. For most of the bromine species as well as key chemical compounds such as OH and HO2 no applicable methods are available for definite speciation. Recent developments have started to provide complement measurements for reactive bromine species, as outlined below. However, data sets are still rare. This activity demands a large pool of instruments, involved people, and logistical resources. Application of UAV systems is revealed as a useful tool for plume investigation but is still limited in its scope due to the often high altitude of volcanoes or other environmental effects.
Recent Developments – Opportunities and Challenges
Development of measuring techniques to add further knowledge on bromine speciation and quantification of other involved species such as OH and bromine speciation is needed to evaluate model predictions of plume chemistry. To pursue this target Rüdiger et al. (2017) presented a new in situ method for the determination of gaseous reactive bromine species in volcanic plumes (see section In Situ–Application of Instruments in the Plume). Applying active alkaline traps (Raschig Tubes) can simultaneously determine Brtotal. So far, data sets are very rare. The method has been applied at Etna, Sicily, Italy 2015 (Rüdiger et al., 2017), Nyamuragira, in the Virunga Mountains of the Democratic Republic of the Congo 2015 (Bobrowski et al., 2017), Stromboli, Mediterranean 2016 (Rüdiger et al., 2018b) and Masaya, Nicaragua 2016 (Rüdiger et al., in preparation). The proof of principle of ground-based sampling sets in 2015's campaigns resulted in averaged reactive bromine to Brtotal ratios of 29 ±17% at Etna (plume age < 1 min) and 25% ±9% at Nyamuragira. In 2016 applicability of UAV-based sampling at Stromboli was undertaken and resulted in an average reactive bromine to SO2 ratio of (5.1 ±4.2) × 10−4 for the very young plume. Measurement results regarding reactive bromine do not allow an ideal and entire description of bromine plume evolution yet, because spatial information in the plume is still very limited (distances mainly at 100–200 m from emission sources and at several craters in the case of Etna). Additional methods for determination of other bromine species such as HBr in the gas phase are under development (Gutmann et al., 2018) and may complement bromine speciation soon.
Development of Fabry Perot Interferometers (FPI, Kern et al., 2010; Kuhn et al., 2018) may allow faster imaging of BrO in the future, than current DOAS application. This can be particularly valuable to separate plume chemistry (as a function of dilution, plume age etc.) from temporal variability in bromine emissions. Permanently installed DOAS instruments (e.g., NOVAC network) now provide the potential for long-term BrO/SO2 time series on a daily basis, at many volcanoes worldwide. Whilst these datasets are regularly being exploited for SO2, so far only a few of these datasets have been analyzed for BrO, but the number is increasing. As our understanding of volcanic plume bromine chemistry improves, we may be able to better interpret these observations in terms of volcanological and meteorological conditions, to yield a valuable resource for daily bromine emissions monitoring. Recently, Kern and Lyons (2018) demonstrate the use of co-measured OClO to help better interpret volcanic BrO.
Atmospheric simulation chamber studies show promise to discover further details of volcanic plume chemistry by mimicking the plume gas and particle conditions in a controlled environment. In those experiments chemical reactions are continuously observed in real-time by a multi-instrument setup, measuring BrO, O3, NOX and further compounds as well as meteorological parameters to constrain their effect on the bromine activation (Rüdiger et al., 2018a).
To provide better constraints for the initial composition of the emissions-plume (“effective source region”) there are efforts to improve the understanding of high-temperature chemistry in the transient near-vent plume. A major limitation to existing thermodynamic models is the assumption of chemical equilibrium. Development of new chemical-kinetics approaches is being undertaken to more accurately reproduce the transient (hot, rapidly diluting and cooling) near-vent plume (Roberts et al., in review). This effort will improve the representation of the high-temperature near-vent chemistry as needed for initialization of the low-temperature atmospheric models of volcanic BrO.
Recent advances introducing the mechanisms developed from 1D/box models into 3D models deliver a more comprehensive understanding of volcanic halogen chemistry as plumes disperse more widely into the atmosphere, enabling the quantification of impacts on tropospheric composition and the potential for halogens to be transported into the stratosphere. Mesoscale models with nested grids enable the simulation of plume processes and impacts from local up to regional scales (Surl et al., 2014; Jourdain et al., 2016). Advances have also been made toward developing parameterizations to include volcanic plume halogen processes in global models (Grellier et al., 2014). Whilst the box/1D models are an essential tool to probe the chemistry of volcanic plumes at the plume-scale and for comparison and to aid interpretation of near-downwind plume observations, regional and global models are needed to assess more fully impacts and to compare to satellite observations of BrO in more aged/evolved plumes. In particular, the recently launched TROPOMI satellite instrument will enable to measure BrO globally at higher spatial resolutions than previously possible.
Advances in in situ and remote sensing field-observations from the crater to flank, and by satellite, and development of a chain of numerical models from local (0D/1D) to regional and global (3D) scales, as well as laboratory experiments, will together address current knowledge gaps in volcanic plume bromine chemistry. Anticipated future benefits include the potential for widespread use of bromine monitoring as part of volcano hazard forecasting and a quantitative assessment of volcanic bromine impacts on the atmosphere over regional-global scales.
Author Contributions
AG and NB collected research results. AG processed data collections. AG, NB, and TR wrote the paper with input from all authors. JR and TH contributed to the final version of the manuscript. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Research Center VAMOS (Volcanoes and Atmosphere in Magmatic Open Systems) at the Johannes Gutenberg-Universität Mainz. NB and JR thank the Deutsche Forschungsgemeinschaft DFG for financial support in the frame of the the project BO 3611/2-1/HE5214/8-1. TR acknowledges the Orleans Labex VOLTAIRE (VOLatils-Terre Atmosphère Interactions–Ressources et Environnement, ANR-10-LABX-100-0) and ANR Projet de Recherche Collaborative VOLC-HALCLIM (Volcanic Halogens: from Deep Earth to Atmospheric Impacts, ANR-18-CE01-0018-01). We thank the reviewers for their detailed comments and constructive suggestions, which helped to significantly improve the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2018.00213/full#supplementary-material
References
Aiuppa, A. (2009). Degassing of halogens from basaltic volcanism: insights from volcanic gas observations. Chem. Geol. 263, 99–109. doi: 10.1016/j.chemgeo.2008.08.022
Aiuppa, A., Federico, C., Franco, A., Giudice, G., Gurrieri, S., Inguaggiato, S., et al. (2005). Emission of bromine and iodine from Mount Etna volcano. Geochem. Geophys. Geosyst. 6:Q08008. doi: 10.1029/2005GC000965
Aiuppa, A., Franco, A., von Glasow, R., Allen, A. G., D'Alessandro, W., Mather, T. A., et al. (2007). The tropospheric processing of acidic gases and hydrogen sulphide in volcanic gas plumes as inferred from field and model investigations. Atmos. Chem. Phys. 7, 1441–1450. doi: 10.5194/acp-7-1441-2007
Aiuppa, A., Giudice, G., Liuzzo, M., Tamburello, G., Allard, P., Calabrese, S., et al. (2012). First volatile inventory for Gorely volcano, Kamchatka. Geophys. Res. Lett. 39:L06307. doi: 10.1029/2012GL051177
Aiuppa, A., Shinohara, H., Tamburello, G., Giudice, G., Liuzzo, M., and Moretti, R. (2011). Hydrogen in the gas plume of an open-vent volcano, Mount Etna, Italy. J. Geophys. Res. Solid Earth 116:1861. doi: 10.1029/2011JB008461
Allard, P., Aiuppa, A., Bani, P., Métrich, N., Bertagnini, A., Gauthier, P.-J., et al. (2016). Prodigious emission rates and magma degassing budget of major, trace and radioactive volatile species from Ambrym basaltic volcano, Vanuatu island Arc. J. Volcanol. Geotherm. Res. 322, 119–143. doi: 10.1016/j.jvolgeores.2015.10.004
Allen, A. G., Mather, T. A., McGonigle, A. J. S., Aiuppa, A., Delmelle, P., Davison, B., et al. (2006). Sources, size distribution, and downwind grounding of aerosols from Mount Etna. J. Geophys. Res. Atmos. 111:D10302. doi: 10.1029/2005JD006015
Allen, A. G., Oppenheimer, C., Ferm, M., Baxter, P. J., Horrocks, L. A., Galle, B., et al. (2002). Primary sulfate aerosol and associated emissions from Masaya Volcano, Nicaragua. J. Geophys. Res. Atmos. 107, ACH 5-1-ACH 5-8. doi: 10.1029/2002JD002120
Amachi, S., Muramatsu, Y., and Kamagata, Y. (2001). Radioanalytical determination of biogenic volatile iodine emitted from aqueous environmental samples. J. Radioanal. Nucl. Chem. 246, 337–341. doi: 10.1023/A:1006786826998
Bagnato, E., Aiuppa, A., Parello, F., Calabrese, S., D'Alessandro, W., Mather, T. A., et al. (2007). Degassing of gaseous (elemental and reactive) and particulate mercury from Mount Etna volcano (Southern Italy). Atmos. Environ. 41, 7377–7388. doi: 10.1016/j.atmosenv.2007.05.060
Bani, P., Oppenheimer, C., Tsanev, V. I., Carn, S. A., Cronin, S. J., Crimp, R., et al. (2009). Surge in sulphur and halogen degassing from Ambrym volcano, Vanuatu. Bull. Volcanol. 71, 1159–1168. doi: 10.1007/s00445-009-0293-7
Barrie, L. A., Bottenheim, J. W., Schnell, R. C., Crutzen, P. J., and Rasmussen, R. A. (1988). Ozone destruction and photochemical reactions at polar sunrise in the lower Arctic atmosphere. Nature 334, 138–141. doi: 10.1038/334138a0
Bobrowski, N., and Giuffrida, G. (2012). Bromine monoxide/sulphur dioxide ratios in relation to volcanological observations at Mt. Etna. Solid Earth 3, 433–445. doi: 10.5194/se-3-433-2012
Bobrowski, N., Giuffrida, G. B., Arellano, S., Yalire, M., Liotta, M., Brusca, L., et al. (2017). Plume composition and volatile flux of Nyamulagira volcano, Democratic Republic of Congo, during birth and evolution of the lava lake, 2014–2015. Bull. Volcanol. 79:B09207. doi: 10.1007/s00445-017-1174-0
Bobrowski, N., Hönninger, G., Galle, B., and Platt, U. (2003). Detection of bromine monoxide in a volcanic plume. Nature 423, 273–276. doi: 10.1038/nature01625
Bobrowski, N., and Platt, U. (2007). SO2/BrO ratios studied in five volcanic plumes. J. Volcanol. Geotherm. Res. 166, 147–160. doi: 10.1016/j.jvolgeores.2007.07.003
Bobrowski, N., Vogel, L., Platt, U., Arellano, S., Galle, B., Hansteen, T., et al. (2012). “Bromine monoxide evolution in early plumes of Mutnovsky and Gorely (Kamchatka, Russia),” in EGU General Assembly 2012, held 22-27 April, 2012 in (Vienna), 6556.
Bobrowski, N., von Glasow, R., Aiuppa, A., Inguaggiato, S., Louban, I., Ibrahim, O. W., et al. (2007). Reactive halogen chemistry in volcanic plumes. J. Geophys. Res. 112:D06311. doi: 10.1029/2006JD007206
Bobrowski, N., von Glasow, R., Giuffrida, G. B., Tedesco, D., Aiuppa, A., Yalire, M., et al. (2015). Gas emission strength and evolution of the molar ratio of BrO/SO2 in the plume of Nyiragongo in comparison to Etna. J. Geophys. Res. Atmos. 120, 277–291. doi: 10.1002/2013JD021069
Bott, A., Trautmann, T., and Zdunkowski, W. (1996). A numerical model of the cloud-topped planetary boundary-layer: radiation, turbulence and spectral microphysics in marine stratus. Q. J. R. Meteorol. Soc. 122, 635–667. doi: 10.1002/qj.49712253105
Brasseur, G., and Granier, C. (1992). Mount pinatubo aerosols, chlorofluorocarbons, and ozone depletion. Science 257, 1239–1242. doi: 10.1126/science.257.5074.1239
Brune, W. H., Stevens, P. S., and Mather, J. H. (1995). Measuring OH and HO2 in the troposphere by laser-induced fluorescence at low pressure. J. Atmos. Sci. 52, 3328–3336. doi: 10.1175/1520-0469(1995)052<3328:MOAHIT>2.0.CO;2
Burton, M. R., Caltabiano, T., Murè, F., Salerno, G., and Randazzo, D. (2009). SO2 flux from Stromboli during the 2007 eruption: results from the FLAME network and traverse measurements. J. Volcanol. Geotherm. Res. 182, 214–220. doi: 10.1016/j.jvolgeores.2008.11.025
Burton, M. R., Mader, H. M., and Polacci, M. (2007). The role of gas percolation in quiescent degassing of persistently active basaltic volcanoes. Earth Planet. Sci. Lett. 264, 46–60. doi: 10.1016/j.epsl.2007.08.028
Burton, M. R., Oppenheimer, C., Horrocks, L. A., and Francis, P. W. (2000). Remote sensing of CO2 and H2O emission rates from Masaya volcano, Nicaragua. Geology 28, 915. doi: 10.1130/0091-7613(2000)28<915:Rsocah>2.0.Co;2
Businger, S., Huff, R., Pattantyus, A., Horton, K., Sutton, A. J., Elias, T., et al. (2015). Observing and Forecasting Vog Dispersion from Kilauea Volcano, Hawaii. Bull. Am. Meteorol. Soc. 96, 1667–1686. doi: 10.1175/BAMS-D-14-00150.1
Butz, A., Dinger, A. S., Bobrowski, N., Kostinek, J., Fieber, L., Fischerkeller, C., et al. (2017). Remote sensing of volcanic CO2, HF, HCl, SO2, and BrO in the downwind plume of Mt. Etna. Atmos. Meas. Tech. 10, 1–14. doi: 10.5194/amt-10-1-2017
Cadoux, A., Iacono-Marziano, G., Scaillet, B., Aiuppa, A., Mather, T. A., Pyle, D. M., et al. (2018). The role of melt composition on aqueous fluid vs. silicate melt partitioning of bromine in magmas. Earth Planet. Sci. Lett. 498, 450–463. doi: 10.1016/j.epsl.2018.06.038
Cadoux, A., Scaillet, B., Bekki, S., Oppenheimer, C., and Druitt, T. H. (2015). Stratospheric Ozone destruction by the Bronze-Age Minoan eruption (Santorini Volcano, Greece). Sci. Rep. 5:12243. doi: 10.1038/srep12243
Carn, S. A., Clarisse, L., and Prata, A. J. (2016). Multi-decadal satellite measurements of global volcanic degassing. J. Volcanol. Geotherm. Res. 311, 99–134. doi: 10.1016/j.jvolgeores.2016.01.002
Carn, S. A., Fioletov, V. E., Mclinden, C. A., Li, C., and Krotkov, N. A. (2017). A decade of global volcanic SO2 emissions measured from space. Sci. Rep. 7:44095. doi: 10.1038/srep44095
Carn, S. A., Krotkov, N. A., Yang, K., and Krueger, A. J. (2013). “Measuring global volcanic degassing with the Ozone Monitoring Instrument (OMI),” in Remote Sensing of Volcanoes and Volcanic Processes: Integrating Observation and Modelling, eds D. M. Pyle, T. A. Mather, and J. Biggs (McLean: Va. GeoScienceWorld), 229–257.
Carroll, M. R., and Holloway, J. R. (eds.). (1994). Volatiles in Magmas. Washington, DC: Mineralogical Society of America.
De Angelis, M., Simões, J., Bonnaveira, H., Taupin, J.-D., and Delmas, R. J. (2003). Volcanic eruptions recorded in the Illimani ice core (Bolivia): 1918–1998 and Tambora periods. Atmos. Chem. Phys. 3, 1725–1741. doi: 10.5194/acp-3-1725-2003
Devine, J. D., Sigurdsson, H., Davis, A. N., and Self, S. (1984). Estimates of sulfur and chlorine yield to the atmosphere from volcanic eruptions and potential climatic effects. J. Geophys. Res. Solid Earth 89, 6309–6325. doi: 10.1029/JB089iB07p06309
Dinger, F., Bobrowski, N., Warnach, S., Bredemeyer, S., Hidalgo, S., Arellano, S., et al. (2018). Periodicity in the BrO/SO2; molar ratios in the volcanic gas plume of Cotopaxi and its correlation with the Earth tides during the eruption in 2015. Solid Earth 9, 247–266. doi: 10.5194/se-9-247-2018
Donovan, A., Tsanev, V., Oppenheimer, C., and Edmonds, M. (2014). Reactive halogens (BrO and OClO) detected in the plume of Soufrière Hills Volcano during an eruption hiatus. Geochem. Geophys. Geosyst. 15, 3346–3363. doi: 10.1002/2014GC005419
Freitas, S. R., Longo, K. M., Silva Dias, M. A. F., Chatfield, R., Silva Dias, P., Artaxo, P., et al. (2009). The Coupled Aerosol and Tracer Transport model to the Brazilian developments on the Regional Atmospheric Modeling System (CATT-BRAMS)–Part 1: model description and evaluation. Atmos. Chem. Phys. 9, 2843–2861. doi: 10.5194/acp-9-2843-2009
Frische, M., Garofalo, K., Hansteen, T. H., Borchers, R., and Harnisch, J. (2006). The origin of stable halogenated compounds in volcanic gases. Environ. Sci. Pollut. Res. 13, 406–413. doi: 10.1065/espr2006.01.291
Galeazzo, T., Bekki, S., Martin, E., Savarino, J., and Arnold, S. R. (2018). Photochemical box-modelling of volcanic SO2 oxidation: Isotopic constraints. Atmos. Chem. Phys. Discuss. 1–38. doi: 10.5194/acp-2018-381
Galle, B., Johansson, M., Rivera, C., Zhang, Y., Kihlman, M., Kern, C., et al. (2010). Network for observation of volcanic and atmospheric change (NOVAC)—A global network for volcanic gas monitoring: network layout and instrument description. J. Geophys. Res. 115:151. doi: 10.1029/2009JD011823
Galle, B., Oppenheimer, C., Geyer, A., McGonigle, A. J. S., Edmonds, M., and Horrocks, L. (2003). A miniaturised ultraviolet spectrometer for remote sensing of SO2 fluxes: a new tool for volcano surveillance. J. Volcanol. Geotherm. Res. 119, 241–254. doi: 10.1016/S0377-0273(02)00356-6
General, S., Bobrowski, N., Pöhler, D., Weber, K., Fischer, C., and Platt, U. (2015). Airborne I-DOAS measurements at Mt. Etna: BrO and OClO evolution in the plume. J. Volcanol. Geotherm. Res. 300, 175–186. doi: 10.1016/j.jvolgeores.2014.05.012
Gerlach, T. M. (2004). Volcanic sources of tropospheric ozone-depleting trace gases. Geochem. Geophys. Geosyst. 5:Q09007. doi: 10.1029/2004GC000747
Gerlach, T. M., and Nordlie, B. E. (1975). The C-O-H-S gaseous system; Part II, Temperature, atomic composition, and molecular equilibria in volcanic gases. Am. J. Sci. 275, 377–394. doi: 10.2475/ajs.275.4.377
Giordano, D., Romano, C., Dingwell, D. B., Poe, B., and Behrens, H. (2004). The combined effects of water and fluorine on the viscosity of silicic magmas. Geochim. Cosmochim. Acta 68, 5159–5168. doi: 10.1016/j.gca.2004.08.012
Gliß, J., Bobrowski, N., Vogel, L., Pöhler, D., and Platt, U. (2015). OClO and BrO observations in the volcanic plume of Mt. Etna – implications on the chemistry of chlorine and bromine species in volcanic plumes. Atmos. Chem. Phys. 15, 5659–5681. doi: 10.5194/acp-15-5659-2015
Goff, F., Janik, C. J., Delgado, H., Werner, C., Counce, D., Stimac, J. A., et al. (1998). Geochemical surveillance of magmatic volatiles at Popocatèpetl volcano, Mexico. Geol. Soc. Am. Bull. 110, 695. doi: 10.1130/0016-7606(1998)110<0695:GSOMVA>2.3.CO;2
Goff, F., and McMurtry, G. M. (2000). Tritium and stable isotopes of magmatic waters. J. Volcanol. Geotherm. Res. 97, 347–396. doi: 10.1016/S0377-0273(99)00177-8
Grellier, L., Marécal, V., Josse, B., Hamer, P. D., Roberts, T. J., Aiuppa, A., et al. (2014). Towards a representation of halogen chemistry within volcanic plumes in a chemistry transport model. Geosci. Model Dev. Discuss. 7, 2581–2650. doi: 10.5194/gmdd-7-2581-2014
Gutmann, A., Bobrowski, N., Liotta, M., Rüdiger, J., and Hoffmann, T. (2018). “Bromine Chemistry in volcanic plumes – Development of in-situ denuder sampling techniques for hydrogen bromine,” in EGU General Assembly 2018, held 8-13 April, 2018 (Vienna), 17096.
Hard, T. M., O'Brien, R. J., Chan, C. Y., and Mehrabzadeh, A. A. (1984). Tropospheric free radical determination by fluorescence assay with gas expansion. Environ. Sci. Technol. 18, 768–777. doi: 10.1021/es00128a009
Hegg, D. A., Hobbs, P. V., and Radke, L. F. (1976). Reactions of Nitrogen Oxides and Sulfur in Power Plant Plumes. Techcnical Report, EPRI EA-270, Electric Power Research Institute, Palo Alto, CA. Available online at: http://carg.atmos.washington.edu/sys/research/archive/react_no_o_so2_power.pdf
Heue, K.-P., Brenninkmeijer, C. A. M., Baker, A. K., Rauthe-Schöch, A., Walter, D., Wagner, T., et al. (2011). SO2 and BrO observation in the plume of the Eyjafjallajökull volcano 2010: CARIBIC and GOME-2 retrievals. Atmos. Chem. Phys. 11, 2973–2989. doi: 10.5194/acp-11-2973-2011
Hidalgo, S., Battaglia, J., Arellano, S., Sierra, D., Bernard, B., Parra, R., et al. (2018). Evolution of the 2015 cotopaxi eruption revealed by combined geochemical & seismic observations. Geochem. Geophys. Geosyst. 19, 2087–2108. doi: 10.1029/2018GC007514
Hoffmann, T., O'Dowd, C. D., and Seinfeld, J. H. (2001). Iodine oxide homogeneous nucleation: an explanation for coastal new particle production. Geophys. Res. Lett. 28, 1949–1952. doi: 10.1029/2000GL012399
Hönninger, G., von Friedeburg, C., and Platt, U. (2004). Multi axis differential optical absorption spectroscopy (MAX-DOAS). Atmos. Chem. Phys. 4, 231–254. doi: 10.5194/acp-4-231-2004
Hörmann, C., Sihler, H., Bobrowski, N., Beirle, S., Penning de Vries, M., Platt, U., et al. (2013). Systematic investigation of bromine monoxide in volcanic plumes from space by using the GOME-2 instrument. Atmos. Chem. Phys. 13, 4749–4781. doi: 10.5194/acp-13-4749-2013
Huang, R.-J., and Hoffmann, T. (2008). A denuder-impinger system with in situ derivatization followed by gas chromatography-mass spectrometry for the determination of gaseous iodine-containing halogen species. J. Chromatogr. A 1210, 135–141. doi: 10.1016/j.chroma.2008.08.003
Huang, R.-J., and Hoffmann, T. (2009). Development of a coupled diffusion denuder system combined with gas chromatography/mass spectrometry for the separation and quantification of molecular iodine and the activated iodine compounds iodine monochloride and hypoiodous acid in the marine atmosphere. Anal. Chem. 81, 1777–1783. doi: 10.1021/ac801839v
Hunton, D. E., Viggiano, A. A., Miller, T. M., Ballenthin, J. O., Reeves, J. M., Wilson, J. C., et al. (2005). In-situ aircraft observations of the 2000 Mt. Hekla volcanic cloud: Composition and chemical evolution in the Arctic lower stratosphere. J. Volcanol. Geotherm. Res. 145, 23–34. doi: 10.1016/j.jvolgeores.2005.01.005
Ilyinskaya, E., Oppenheimer, C., Mather, T. A., Martin, R. S., and Kyle, P. R. (2010). Size-resolved chemical composition of aerosol emitted by Erebus volcano, Antarctica. Geochem. Geophys. Geosyst. 11:Q03017. doi: 10.1029/2009GC002855
Jourdain, L., Roberts, T. J., Pirre, M., and Josse, B. (2016). Modeling the reactive halogen plume from Ambrym and its impact on the troposphere with the CCATT-BRAMS mesoscale model. Atmos. Chem. Phys. 16, 12099–12125. doi: 10.5194/acp-16-12099-2016
Kelly, P. J., Kern, C., Roberts, T. J., Lopez, T., Werner, C., and Aiuppa, A. (2013). Rapid chemical evolution of tropospheric volcanic emissions from Redoubt Volcano, Alaska, based on observations of ozone and halogen-containing gases. J. Volcanol. Geotherm. Res. 259, 317–333. doi: 10.1016/j.jvolgeores.2012.04.023
Kern, C., Kick, F., Lübcke, P., Vogel, L., Wöhrbach, M., and Platt, U. (2010). Theoretical description of functionality, applications, and limitations of SO2 cameras for the remote sensing of volcanic plumes. Atmos. Meas. Tech. 3, 733–749. doi: 10.5194/amt-3-733-2010
Kern, C., Lübcke, P., Bobrowski, N., Campion, R., Mori, T., Smekens, J.-F., et al. (2015). Intercomparison of SO 2 camera systems for imaging volcanic gas plumes. J. Volcanol. Geotherm. Res. 300, 22–36. doi: 10.1016/j.jvolgeores.2014.08.026
Kern, C., and Lyons, J. J. (2018). Spatial distribution of halogen oxides in the plume of Mount Pagan volcano, Mariana Islands. Geophys. Res. Lett. 45, 9588–9596. doi: 10.1029/2018GL079245
Kern, C., Masias, P., Apaza, F., Reath, K. A., and Platt, U. (2017). Remote measurement of high preeruptive water vapor emissions at Sabancaya volcano by passive differential optical absorption spectroscopy. J. Geophys. Res. Solid Earth 122, 3540–3564. doi: 10.1002/2017JB014020
Kern, C., Sihler, H., Vogel, L., Rivera, C., Herrera, M., and Platt, U. (2009). Halogen oxide measurements at Masaya Volcano, Nicaragua using active long path differential optical absorption spectroscopy. Bull. Volcanol. 71, 659–670. doi: 10.1007/s00445-008-0252-8
Kitto, M. E., Anderson, D. L., and Zoller, W. H. (1988). Simultaneous collection of particles and gases followed by multielement analysis using nuclear techniques. J. Atmos. Chem. 7, 241–259. doi: 10.1007/BF00130932
Klobas, J. E., Wilmouth, D. M., Weisenstein, D. K., Anderson, J. G., and Salawitch, R. J. (2017). Ozone depletion following future volcanic eruptions. Geophys. Res. Lett. 44, 7490–7499. doi: 10.1002/2017GL073972
Kuhn, J., Platt, U., Bobrowski, N., and Wagner, T. (2018). Towards imaging of atmospheric trace gases using Fabry Perot Interferometer Correlation Spectroscopy in the UV and visible spectral range. Atmos. Meas. Tech. Discuss. 1–19. doi: 10.5194/amt-2018-350
Kutterolf, S., Hansteen, T. H., Appel, K., Freundt, A., Krüger, K., Pérez, W., et al. (2013). Combined bromine and chlorine release from large explosive volcanic eruptions: a threat to stratospheric ozone? Geology 41, 707–710. doi: 10.1130/G34044.1
Kutterolf, S., Hansteen, T. H., Freundt, A., Wehrmann, H., Appel, K., Krüger, K., et al. (2015). Bromine and chlorine emissions from Plinian eruptions along the Central American Volcanic Arc: from source to atmosphere. Earth Planet. Sci. Lett. 429, 234–246. doi: 10.1016/j.epsl.2015.07.064
Lazrus, A. L., Gandrud, B. W., Woodard, R. N., and Sedlacek, W. A. (1976). Direct measurements of stratospheric chlorine and bromine. J. Geophys. Res. 81, 1067–1070. doi: 10.1029/JC081i006p01067
Lee, C., Kim, Y. J., Tanimoto, H., Bobrowski, N., Platt, U., Mori, T., et al. (2005). High ClO and ozone depletion observed in the plume of Sakurajima volcano, Japan. Geophys. Res. Lett. 32:450. doi: 10.1029/2005GL023785
Levin, I., Münnich, K. O., and Weiss, W. (1980). The Effect of Anthropogenic CO2 and 14C Sources on the Distribution of 14C in the Atmosphere. Radiocarbon 22, 379–391. doi: 10.1017/S003382220000967X
Liotta, M., Rizzo, A., Paonita, A., Caracausi, A., and Martelli, M. (2012). Sulfur isotopic compositions of fumarolic and plume gases at Mount Etna (Italy) and inferences on their magmatic source. Geochem. Geophys. Geosyst. 13:1861. doi: 10.1029/2012GC004118
Lohberger, F., Hönninger, G., and Platt, U. (2004). Ground-based imaging differential optical absorption spectroscopy of atmospheric gases. Appl. Opt. 43:4711. doi: 10.1364/AO.43.004711
Longo, K. M., Freitas, S. R., Pirre, M., Marécal, V., Rodrigues, L. F., Panetta, J., et al. (2013). The Chemistry CATT-BRAMS model (CCATT-BRAMS 4.5): a regional atmospheric model system for integrated air quality and weather forecasting and research. Geosci. Model Dev. 6, 1389–1405. doi: 10.5194/gmd-6-1389-2013
López, T., Ushakov, S., Izbekov, P., Tassi, F., Cahill, C., Neill, O., et al. (2013). Constraints on magma processes, subsurface conditions, and total volatile flux at Bezymianny Volcano in 2007–2010 from direct and remote volcanic gas measurements. J. Volcanol. Geotherm. Res. 263, 92–107. doi: 10.1016/j.jvolgeores.2012.10.015
Louban, I., Bobrowski, N., Rouwet, D., Inguaggiato, S., and Platt, U. (2009). Imaging DOAS for volcanological applications. Bull. Volcanol. 71, 753–765. doi: 10.1007/s00445-008-0262-6
Lübcke, P., Bobrowski, N., Arellano, S., Galle, B., Garzón, G., Vogel, L., et al. (2014). BrO/SO2 molar ratios from scanning DOAS measurements in the NOVAC network. Solid Earth 5, 409–424. doi: 10.5194/se-5-409-2014
Lurton, T., Jégou, F., Berthet, G., Renard, J.-B., Clarisse, L., Schmidt, A., et al. (2018). Model simulations of the chemical and aerosol microphysical evolution of the Sarychev Peak 2009 eruption cloud compared to in situ and satellite observations. Atmos. Chem. Phys. 18, 3223–3247. doi: 10.5194/acp-18-3223-2018
Ma, C.-J., and Kim, K.-H. (2008). A combination of size-resolved particle samplers and XRF microprobe technique for single particle study. Atmos. Environ. 42, 7022–7026. doi: 10.1016/j.atmosenv.2008.04.045
Malinconico, L. L. (1979). Fluctuations in SO2 emission during recent eruptions of Etna. Nature 278, 43–45. doi: 10.1038/278043a0
Marple, V. A., Rubow, K. L., and Behm, S. M. (1991). A microorifice uniform deposit impactor (MOUDI): description, calibration, and use. Aerosol Sci. Technol. 14, 434–446. doi: 10.1080/02786829108959504
Martin, R. S., Ilyinskaya, E., and Oppenheimer, C. (2012a). The enigma of reactive nitrogen in volcanic emissions. Geochim. Cosmochim. Acta 95, 93–105. doi: 10.1016/j.gca.2012.07.027
Martin, R. S., Mather, T. A., and Pyle, D. M. (2006). High-temperature mixtures of magmatic and atmospheric gases. Geochem. Geophys. Geosyst. 7:Q04006. doi: 10.1029/2005GC001186
Martin, R. S., Mather, T. A., Pyle, D. M., Power, M., Allen, A. G., Aiuppa, A., et al. (2008). Composition-resolved size distributions of volcanic aerosols in the Mt. Etna plumes. J. Geophys. Res. 113:D17211. doi: 10.1029/2007JD009648
Martin, R. S., Roberts, T. J., Mather, T. A., and Pyle, D. M. (2009). The implications of H2S and H2 kinetic stability in high-T mixtures of magmatic and atmospheric gases for the production of oxidized trace species (e.g., BrO and NOx). Chem. Geol. 263, 143–150. doi: 10.1016/j.chemgeo.2008.12.028
Martin, R. S., Sawyer, G. M., Spampinato, L., Salerno, G. G., Ramirez, C., Ilyinskaya, E., et al. (2010). A total volatile inventory for Masaya Volcano, Nicaragua. J. Geophys. Res. 115:L02610. doi: 10.1029/2010JB007480
Martin, R. S., Wheeler, J. C., Ilyinskaya, E., Braban, C. F., and Oppenheimer, C. (2012b). The uptake of halogen (HF, HCl, HBr and HI) and nitric (HNO3) acids into acidic sulphate particles in quiescent volcanic plumes. Chem. Geol. 296-297, 19–25. doi: 10.1016/j.chemgeo.2011.12.013
Mather, T. A., Allen, A. G., Davison, B. M., Pyle, D. M., Oppenheimer, C., and McGonigle, A. J. S. (2004a). Nitric acid from volcanoes. Earth Planet. Sci. Lett. 218, 17–30. doi: 10.1016/S0012-821X(03)00640-X
Mather, T. A., Allen, A. G., Oppenheimer, C., Pyle, D. M., and McGonigle, A. J. S. (2003). Size-resolved characterisation of soluble ions in the particles in the tropospheric plume of masaya volcano, nicaragua: origins and plume processing. J. Atmos. Chem. 46, 207–237. doi: 10.1023/A:1026327502060
Mather, T. A., Pyle, D. M., and Allen, A. G. (2004b). Volcanic source for fixed nitrogen in the early Earth's atmosphere. Geology 32:905. doi: 10.1130/G20679.1
Mather, T. A., Pyle, D. M., Tsanev, V. I., McGonigle, A. J. S., Oppenheimer, C., and Allen, A. G. (2006). A reassessment of current volcanic emissions from the Central American arc with specific examples from Nicaragua. J. Volcanol. Geotherm. Res. 149, 297–311. doi: 10.1016/j.jvolgeores.2005.07.021
Mather, T. A., Witt, M. L. I., Pyle, D. M., Quayle, B. M., Aiuppa, A., Bagnato, E., et al. (2012). Halogens and trace metal emissions from the ongoing 2008 summit eruption of Kilauea volcano, Hawai‘i. Geochim. Cosmochim. Acta 83, 292–323. doi: 10.1016/j.gca.2011.11.029
McGonigle, A. J. S. (2004). SO2 depletion in tropospheric volcanic plumes. Geophys. Res. Lett. 31:L13201. doi: 10.1029/2004GL019990
McGonigle, A. J. S., Aiuppa, A., Giudice, G., Tamburello, G., Hodson, A. J., and Gurrieri, S. (2008). Unmanned aerial vehicle measurements of volcanic carbon dioxide fluxes. Geophys. Res. Lett. 35:L06303. doi: 10.1029/2007GL032508
McGonigle, A. J. S., and Oppenheimer, C. (2003). “Optical sensing of volcanic gas and aerosol emissions,” in Volcanic Degassing, eds C. Oppenheimer, D. M. Pyle, and J. Barclay (London: Geological Society of London), 149–168.
McGonigle, A. J. S., Oppenheimer, C., Galle, B., Mather, T. A., and Pyle, D. M. (2002). Walking traverse and scanning DOAS measurements of volcanic gas emission rates. Geophys. Res. Lett. 29, 46-1–46-4. doi: 10.1029/2002GL015827
McGonigle, A. J. S., Pering, T. D., Wilkes, T. C., Tamburello, G., D'Aleo, R., Bitetto, M., et al. (2017). Ultraviolet imaging of volcanic plumes: a new paradigm in volcanology. Geosciences 7:68. doi: 10.20944/preprints201707.0076.v1
Mellor, G. L., and Yamada, T. (1982). Development of a turbulence closure model for geophysical fluid problems. Rev. Geophys. 20:851. doi: 10.1029/RG020i004p00851
Moor, J. M. d., Aiuppa, A., Avard, G., Wehrmann, H., Dunbar, N., Muller, C., et al. (2016a). Turmoil at Turrialba Volcano (Costa Rica): degassing and eruptive processes inferred from high-frequency gas monitoring. J. Geophys. Res. Solid Earth 121, 5761–5775. doi: 10.1002/2016JB013150
Moor, J. M. d., Aiuppa, A., Pacheco, J., Avard, G., Kern, C., Liuzzo, M., et al. (2016b). Short-period volcanic gas precursors to phreatic eruptions: insights from Poás Volcano, Costa Rica. Earth Planet. Sci. Lett. 442, 218–227. doi: 10.1016/j.epsl.2016.02.056
Mori, T., and Burton, M. (2006). The SO 2 camera: a simple, fast and cheap method for ground-based imaging of SO2 in volcanic plumes. Geophys. Res. Lett. 33:329. doi: 10.1029/2006GL027916
Mori, T., and Notsu, K. (1997). Remote CO, COS, CO2, SO2, HCl detection and temperature estimation of volcanic gas. Geophys. Res. Lett. 24, 2047–2050. doi: 10.1029/97GL52058
Moune, S., Sigmarsson, O., Schiano, P., Thordarson, T., and Keiding, J. K. (2012). Melt inclusion constraints on the magma source of Eyjafjallajökull 2010 flank eruption. J. Geophys. Res. Solid Earth 117:1485. doi: 10.1029/2011JB008718
Moussallam, Y., Edmonds, M., Scaillet, B., Peters, N., Gennaro, E., Sides, I., et al. (2016). The impact of degassing on the oxidation state of basaltic magmas: a case study of Kilauea volcano. Earth Planet. Sci. Lett. 450, 317–325. doi: 10.1016/j.epsl.2016.06.031
Noguchi, K., and Kamiya, H. (1963). Prediction of volcanic eruption by measuring the chemical composition and amounts of gases. Bull. Volcanol. 26, 367–378. doi: 10.1007/BF02597298
Oana, S. (1962). Volcanic gases and sublimates from Showashinzan. Bull. Volcanol. 24, 49–57. doi: 10.1007/BF02599328
Oppenheimer, C., Francis, P., Burton, M., Maciejewski, A. J. H., and Boardman, L. (1998). Remote measurement of volcanic gases by Fourier transform infrared spectroscopy. Appl. Phys. B Lasers Opt. 67, 505–515. doi: 10.1007/s003400050536
Oppenheimer, C., Kyle, P., Eisele, F., Crawford, J., Huey, G., Tanner, D., et al. (2010). Atmospheric chemistry of an Antarctic volcanic plume. J. Geophys. Res. 115, 5473. doi: 10.1029/2009JD011910
Oppenheimer, C., Tsanev, V. I., Braban, C. F., Cox, R. A., Adams, J. W., Aiuppa, A., et al. (2006). BrO formation in volcanic plumes. Geochim. Cosmochim. Acta 70, 2935–2941. doi: 10.1016/j.gca.2006.04.001
Platt, U., and Bobrowski, N. (2015). “Quantification of volcanic reactive halogen emissions,” in Volcanism and Global Environmental Change, eds A. Schmidt, K. E. Fristad, and L. T. Elkins-Tanton (Cambridge: Cambridge University Press), 115–132.
Platt, U., Bobrowski, N., and Butz, A. (2018). Ground-based remote sensing and imaging of volcanic gases and quantitative determination of multi-species emission fluxes. Geosciences 8:44. doi: 10.3390/geosciences8020044
Platt, U., Rateike, M., Junkermann, W., Rudolph, J., and Ehhalt, D. H. (1988). New tropospheric OH measurements. J. Geophys. Res. 93:5159. doi: 10.1029/JD093iD05p05159
Pyle, D. M., and Mather, T. A. (2009). Halogens in igneous processes and their fluxes to the atmosphere and oceans from volcanic activity: a review. Chem. Geol. 263, 110–121. doi: 10.1016/j.chemgeo.2008.11.013
Rix, M., Valks, P., Hao, N., Loyola, D., Schlager, H., Huntrieser, H., et al. (2012). Volcanic SO2, BrO and plume height estimations using GOME-2 satellite measurements during the eruption of Eyjafjallajökull in May 2010. J. Geophys. Res. Atmos. 117, 147. doi: 10.1029/2011JD016718
Roberts, T. (2018). Ozone Depletion in Tropospheric Volcanic Plumes: From Halogen-Poor to Halogen-Rich Emissions. Geosciences 8, 68. doi: 10.3390/geosciences8020068
Roberts, T. J., Braban, C. F., Martin, R. S., Oppenheimer, C., Adams, J. W., Cox, R. A., et al. (2009). Modelling reactive halogen formation and ozone depletion in volcanic plumes. Chem. Geol. 263, 151–163. doi: 10.1016/j.chemgeo.2008.11.012
Roberts, T. J., Jourdain, L., Griffiths, P. T., and Pirre, M. (2014a). Re-evaluating the reactive uptake of HOBr in the troposphere with implications for the marine boundary layer and volcanic plumes. Atmos. Chem. Phys. 14, 11185–11199. doi: 10.5194/acp-14-11185-2014
Roberts, T. J., Martin, R. S., and Jourdain, L. (2014b). Reactive bromine chemistry in Mount Etna's volcanic plume: the influence of total Br, high-temperature processing, aerosol loading and plume–air mixing. Atmos. Chem. Phys. 14, 11201–11219. doi: 10.5194/acp-14-11201-2014
Roberts, T. J., Vignelles, D., Liuzzo, M., Giudice, G., Aiuppa, A., Coltelli, M., et al. (2018). The primary volcanic aerosol emission from Mt Etna: size-resolved particles with SO2 and role in plume reactive halogen chemistry. Geochim. Cosmochim. Acta 222, 74–93. doi: 10.1016/j.gca.2017.09.040
Rose, W. I., Millard, G. A., Mather, T. A., Hunton, D. E., Anderson, B., Oppenheimer, C., et al. (2006). Atmospheric chemistry of a 33–34 hour old volcanic cloud from Hekla Volcano (Iceland): insights from direct sampling and the application of chemical box modeling. J. Geophys. Res. 111, Q08008. doi: 10.1029/2005JD006872
Rüdiger, J., Bobrowski, N., Liotta, M., and Hoffmann, T. (2017). Development and application of a sampling method for the determination of reactive halogen species in volcanic gas emissions. Anal. Bioanal. Chem. 409, 5975–5985. doi: 10.1007/s00216-017-0525-1
Rüdiger, J., Schmitt, S., Pitton, D., Tirpitz, J.-L., Gutmann, A., Gutierrez, X., et al. (2018a). “HALVIRE: HALogen activation in Volcanic plumes In Reaction chamber Experiments,” in EGU General Assembly 2018, held 8-13 April, 2018 (Vienna), 14827.
Rüdiger, J., Tirpitz, J.-L., Moor, J. M., de Bobrowski, N., Gutmann, A., Liuzzo, M., et al. (2018b). Implementation of electrochemical, optical and denuder-based sensors and sampling techniques on UAV for volcanic gas measurements: examples from Masaya, Turrialba and Stromboli volcanoes. Atmos. Meas. Tech. 11, 2441–2457. doi: 10.5194/amt-11-2441-2018
Saiz-Lopez, A., and von Glasow, R. (2012). Reactive halogen chemistry in the troposphere. Chem. Soc. Rev. 41, 6448–6472. doi: 10.1039/c2cs35208g
Sander, R., and Crutzen, P. J. (1996). Model study indicating halogen activation and ozone destruction in polluted air masses transported to the sea. J. Geophys. Res. Atmos. 101, 9121–9138. doi: 10.1029/95JD03793
Sawyer, G. M., Salerno, G. G., Le Blond, J. S., Martin, R. S., Spampinato, L., Roberts, T. J., et al. (2011). Gas and aerosol emissions from Villarrica volcano, Chile. J. Volcanol. Geotherm. Res. 203, 62–75. doi: 10.1016/j.jvolgeores.2011.04.003
Schönhardt, A., Richter, A., Theys, N., and Burrows, J. P. (2017). Space-based observation of volcanic iodine monoxide. Atmos. Chem. Phys. 17, 4857–4870. doi: 10.5194/acp-17-4857-2017
Schumann, U., Weinzierl, B., Reitebuch, O., Schlager, H., Minikin, A., Forster, C., et al. (2011). Airborne observations of the Eyjafjalla volcano ash cloud over Europe during air space closure in April and May 2010. Atmos. Chem. Phys. 11, 2245–2279. doi: 10.5194/acp-11-2245-2011
Schwandner, F. M., Seward, T. M., Gize, A. P., Hall, K., and Dietrich, V. J. (2013). Halocarbons and other trace heteroatomic organic compounds in volcanic gases from Vulcano (Aeolian Islands, Italy). Geochim. Cosmochim. Acta 101, 191–221. doi: 10.1016/j.gca.2012.10.004
Schwandner, F. M., Seward, T. M., Gize, A. P., Hall, P. A., and Dietrich, V. J. (2004). Diffuse emission of organic trace gases from the flank and crater of a quiescent active volcano (Vulcano, Aeolian Islands, Italy). J. Geophys. Res. Atmos. 109. doi: 10.1029/2003JD003890
Sedlacek, W. A., Lazrus, A. L., and Gandrud, B. W. (1984). Measurements of stratospheric bromine. J. Geophys. Res. 89, 4821. doi: 10.1029/JD089iD03p04821
Seinfeld, J. H., Pandis, S. N., and Noone, K. (1998). Atmospheric chemistry and physics: from air pollution to climate change. Phys. Today 51, 88–90. doi: 10.1063/1.882420
Shaw, G. H. (2008). Earth's atmosphere–Hadean to early Proterozoic. Chem. Erde 68, 235–264. doi: 10.1016/j.chemer.2008.05.001
Song, C. H. (2003). Dispersion and chemical evolution of ship plumes in the marine boundary layer: Investigation of O 3 /NO y /HO x chemistry. J. Geophys. Res. 108:215. doi: 10.1029/2002JD002216
Sparks, R. S. J. (2003). “Dynamics of magma degassing,” in Volcanic Degassing, eds C. Oppenheimer, D. M. Pyle, and J. Barclay (London: Geological Society of London), 5–22.
Spilliaert, N., Metrich, N., and Allard, P. (2006). S–Cl–F degassing pattern of water-rich alkali basalt: modelling and relationship with eruption styles on Mount Etna volcano. Earth Planet. Sci. Lett. 248, 772–786. doi: 10.1016/j.epsl.2006.06.031
Strellis, D. A., Hwang, H. H., Anderson, T. F., and Landsberger, S. (1996). A comparative study of IC, ICP-AES, and NAA measurements on chlorine, bromine, and sodium in natural waters. J. Radioanal. Nucl. Chem. 211, 473–484. doi: 10.1007/BF02039713
Sturges, W. T., and Shaw, G. E. (1993). Halogens in aerosols in Central Alaska. Atmos. Environ. A 27, 2969–2977. doi: 10.1016/0960-1686(93)90329-W
Sugiura, T., Mizutani, Y., and Oana, S. (1963). Fluorine, chlorine, bromine and iodine in volcanic gases. J. Earth Sci. Nagoya Univ. 11, 272–278.
Surl, L., Donohoue, D., Aiuppa, A., Bobrowski, N., and von Glasow, R. (2015). Quantification of the depletion of ozone in the plume of Mount Etna. Atmos. Chem. Phys. 15, 2613–2628. doi: 10.5194/acp-15-2613-2015
Surl, L., Donohoue, D., and von Glasow, R. (2014). “Modelling the regional impact of volcanic bromine using WRF-Chem,” in EGU General Assembly 2014, held 27 April-2 May, 2014 (Vienna), 1509.
Symonds, R. B., Gerlach, T. M., and Reed, M. H. (2001). Magmatic gas scrubbing: implications for volcano monitoring. J. Volcanol. Geotherm. Res. 108, 303–341. doi: 10.1016/S0377-0273(00)00292-4
Symonds, R. B., Rose, W. I., Bluth, G. J. S., and Gerlach, T. M. (1994). Volcanic-gas studies; methods, results, and applications. Rev. Mineral. Geochem. 30, 1–66.
Textor, C. (2003). Injection of gases into the stratosphere by explosive volcanic eruptions. J. Geophys. Res. 108:647. doi: 10.1029/2002JD002987
Textor, C., Graf, H.-F., Timmreck, C., and Robock, A.(eds.). (2004). Emissions from volcanoes: Emissions of Atmospheric Trace Compounds. Dordrecht: Springer Netherlands.
Theys, N., van Roozendael, M., Dils, B., Hendrick, F., Hao, N., and de Mazière, M. (2009). First satellite detection of volcanic bromine monoxide emission after the Kasatochi eruption. Geophys. Res. Lett. 36:L03809. doi: 10.1029/2008GL036552
Theys, N., van Roozendael, M., Hendrick, F., Yang, X., Smedt, I., de, Richter, A., et al. (2011). Global observations of tropospheric BrO columns using GOME-2 satellite data. Atmos. Chem. Phys. 11, 1791–1811. doi: 10.5194/acp-11-1791-2011
Vance, A., McGonigle, A. J. S., Aiuppa, A., Stith, J. L., Turnbull, K., and von Glasow, R. (2010). Ozone depletion in tropospheric volcanic plumes. Geophys. Res. Lett. 37:L22802. doi: 10.1029/2010GL044997
Vidal, C. M., Métrich, N., Komorowski, J.-C., Pratomo, I., Michel, A., Kartadinata, N., et al. (2016). The 1257 Samalas eruption (Lombok, Indonesia): the single greatest stratospheric gas release of the Common Era. Sci. Rep. 6:34868. doi: 10.1038/srep34868
Vogt, R., Crutzen, P. J., and Sander, R. (1996). A mechanism for halogen release from sea-salt aerosol in the remote marine boundary layer. Nature 383, 327–330. doi: 10.1038/383327a0
Voigt, C., Jessberger, P., Jurkat, T., Kaufmann, S., Baumann, R., Schlager, H., et al. (2014). Evolution of CO2, SO2, HCl, and HNO3 in the volcanic plumes from Etna. Geophys. Res. Lett. 41, 2196–2203. doi: 10.1002/2013GL058974
Vömel, H., and Diaz, K. (2010). Ozone sonde cell current measurements and implications for observations of near-zero ozone concentrations in the tropical upper troposphere. Atmos. Meas. Tech. 3, 495–505. doi: 10.5194/amt-3-495-2010
von Glasow, R. (2000). Modeling the Gas and Aqueous Phase Chemistry of the Marine Boundary Layer. Dissertation. Mainz: Johannes Gutenberg-Universität, Physik.
von Glasow, R. (2010). Atmospheric chemistry in volcanic plumes. Proc. Natl. Acad. Sci. U.S.A. 107, 6594–6599. doi: 10.1073/pnas.0913164107
von Glasow, R., Bobrowski, N., and Kern, C. (2009). The effects of volcanic eruptions on atmospheric chemistry. Chem. Geol. 263, 131–142. doi: 10.1016/j.chemgeo.2008.08.020
von Glasow, R., and Crutzen, P. J. (2003). “Tropospheric halogen chemistry,” in Treatise on Geochemistry, eds H. D. Holland and K. K. Turekian (Oxford: Elsevier), 1–67.
von Glasow, R., Donohoue, D., Bobrowski, N., Witt, M., and Mather, T. (2015). “Reactive plume chemistry and links to mercury deposition at Masaya volcano, Nicaragua,” in EGU General Assembly 2015, held 12-17 April, 2015 (Vienna), 6134.
Wallace, P. J. (2005). Volatiles in subduction zone magmas: concentrations and fluxes based on melt inclusion and volcanic gas data. J. Volcanol. Geotherm. Res. 140, 217–240. doi: 10.1016/j.jvolgeores.2004.07.023
Watson, I. M., and Oppenheimer, C. (2000). Particle size distributions of Mount Etna's aerosol plume constrained by Sun photometry. J. Geophys. Res. Atmos. 105, 9823–9829. doi: 10.1029/2000JD900042
Webster, J. D., Baker, D. R., and Aiuppa, A. (2018). “Halogens in mafic and intermediate-silica content magmas,” in The Role of Halogens in Terrestrial and Extraterrestrial Geochemical Processes, eds D. E. Harlov and L. Aranovich (Cham: Springer International Publishing), 307–430.
Wexler, A. S., and Clegg, S. L. (2002). Atmospheric aerosol models for systems including the ions H+, NH, Na+, SO, NO, Cl−, Br−, and H2O. J. Geophys. Res. 107:745. doi: 10.1029/2001JD000451
Witt, M. L. I., Mather, T. A., Pyle, D. M., Aiuppa, A., Bagnato, E., and Tsanev, V. I. (2008). Mercury and halogen emissions from Masaya and Telica volcanoes, Nicaragua. J. Geophys. Res. 113, Q08008. doi: 10.1029/2007JB005401
Witter, J. B., Kress, V. C., Delmelle, P., and Stix, J. (2004). Volatile degassing, petrology, and magma dynamics of the Villarrica Lava Lake, Southern Chile. J. Volcanol. Geotherm. Res. 134:303–337. doi: 10.1016/j.jvolgeores.2004.03.002
Wittmer, J. (2012). Development of an Active Alkaline Trap to Determine Acidic Gas Ratios in Volcanic Plumes: Sampling Technique and Analytical Methods. Diploma Thesis. Heidelberg University Library: University of Heidelberg.
Wittmer, J., Bobrowski, N., Liotta, M., Giuffrida, G., Calabrese, S., and Platt, U. (2014). Active alkaline traps to determine acidic-gas ratios in volcanic plumes: sampling techniques and analytical methods. Geochem. Geophys. Geosyst. 15, 2797–2820. doi: 10.1002/2013GC005133
World Meteorological Organization (2014). Scientific Assessment of Ozone Depletion: 2014, Global Ozone Research and Monitoring Project-Report No. 55, 1.50.
Zdanowicz, C. M., Zielinski, G. A., and Germani, M. S. (1999). Mount mazama eruption: calendrical age verified and atmospheric impact assessed. Geology 27:621. doi: 10.1130/0091-7613(1999)027<0621:MMECAV>2.3.CO;2
Keywords: volcanic halogen emissions, volcanic plumes, plume chemistry, bromine explosion, bromine speciation, gas monitoring, troposphere
Citation: Gutmann A, Bobrowski N, Roberts TJ, Rüdiger J and Hoffmann T (2018) Advances in Bromine Speciation in Volcanic Plumes. Front. Earth Sci. 6:213. doi: 10.3389/feart.2018.00213
Received: 30 June 2018; Accepted: 05 November 2018;
Published: 30 November 2018.
Edited by:
Andrew McGonigle, University of Sheffield, United KingdomReviewed by:
Eisuke Fujita, National Research Institute for Earth Science and Disaster Prevention, JapanJ. Eric Klobas, Harvard University, United States
Copyright © 2018 Gutmann, Bobrowski, Roberts, Rüdiger and Hoffmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandra Gutmann, Z3V0bWFubkB1bmktbWFpbnouZGU=
Nicole Bobrowski, bmJvYnJvd3NAaXVwLnVuaS1oZWlkZWxiZXJnLmRl