 Alina Mostovaya
Alina Mostovaya Jeffrey A. Hawkes
Jeffrey A. Hawkes Thorsten Dittmar
Thorsten Dittmar Lars J. Tranvik
Lars J. Tranvik
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Earth Sci. , 18 December 2017
Sec. Biogeoscience
Volume 5 - 2017 | https://doi.org/10.3389/feart.2017.00106
Lakes in the boreal region have been recognized as the biogeochemical hotspots, yet many questions regarding the regulators of organic matter processing in these systems remain open. Molecular composition can be an important determinant of dissolved organic matter (DOM) fate in freshwater systems, but many aspects of this relationship remain unclear due to the complexity of DOM and its interactions in the natural environment. Here, we combine ultrahigh resolution mass spectrometry (FT-ICR-MS) with kinetic modeling of decay of >1,300 individual DOM molecular formulae identified by mass spectrometry, to evaluate the role of specific molecular characteristics in decomposition of lake water DOM. Our data is derived from a 4 months microbial decomposition experiment, carried out on water from three Swedish lakes, with the set-up including natural lake water, as well as the lake water pretreated with UV light. The relative decay rate of every molecular formula was estimated by fitting a single exponential model to the change in FT-ICR-MS signal intensities over decomposition time. We found a continuous range of exponential decay coefficients (kexp) within different groups of compounds and show that for highly unsaturated and phenolic compounds the distribution of kexp was shifted toward the lowest values. Contrary to this general trend, plant-derived polyphenols and polycondensed aromatics were on average more reactive than compounds with an intermediate aromaticity. The decay rate of aromatic compounds increased with increasing nominal oxidation state of carbon, and molecular mass in some cases showed an inverse relationship with kexp in the UV-manipulated treatment. Further, we observe an increase in formulae-specific kexp as a result of the UV pretreatment. General trends in reactivity identified among major compound groups emphasize the importance of the intrinsic controllers of lake water DOM decay. However, we additionally indicate that each compound group contained a wide spectrum of reactivities, suggesting that high resolution is needed to further ascertain the complex reasons behind DOM reactivity in lake water.
Dissolved organic matter (DOM) is the quantitatively most abundant pool of organic carbon in the oceans and in most inland waters (Thurman, 1985). It is an important vector of energy and nutrients from terrestrial to aquatic systems, shaping communities and ecosystems (Jansson et al., 2007), and is a precursor of a substantial sediment carbon sink (von Wachenfeldt and Tranvik, 2008) as well as a large source of atmospheric carbon dioxide (Cole et al., 2007; Raymond et al., 2013). Accordingly, there is a considerable interest in identifying the main drivers of DOM decomposition in freshwater systems. Boreal lakes draw especially close attention in this regard as they receive and actively process increasingly large amounts of terrigenous organic matter (Sobek et al., 2003; Algesten et al., 2004; Tranvik et al., 2009). Microbial and photochemical mineralization of this organic matter contributes to the flux of greenhouse gases from lakes to the atmosphere (Duarte and Prairie, 2005; Raymond et al., 2013). Moreover, the boreal and arctic regions have the highest abundance and area of lake water bodies on Earth (Verpoorter et al., 2014), and the terrestrial export and concentrations of dissolved organic carbon in the boreal region are predicted to increase with elevated temperatures (Weyhenmeyer and Karlsson, 2009).
There is increasing evidence that the degradation of organic matter in soils is constrained largely by extrinsic, environmental factors, rather than intrinsic properties of the organic matter itself (Kleber, 2010; Schmidt et al., 2011). However, in aquatic environment intrinsic factors have been identified as important controllers of DOM fate (Kellerman et al., 2015). A number of studies suggested a link between the reactivity of DOM and its specific properties, e.g., elemental ratios (Sun et al., 1997; D'Andrilli et al., 2015), nominal oxidation state of carbon (Kattner et al., 2011; LaRowe and Van Cappellen, 2011), apparent molecular weight (Meyer, 1986; Amon and Benner, 1994), actual molecular weight (Kim et al., 2006), and aliphatic vs. aromatic content (Hopkinson et al., 1998). Notably, the ideas about the exact relationship between DOM properties and degradability are sometimes conflicting, such as in case of nominal oxidation state of carbon and molecular weight.
Commonly, to establish a link between the reactivity and properties of DOM, a comparison is made between two and three contrasting DOM fractions (Tranvik, 1990; Amon and Benner, 1996; Guillemette et al., 2013) or a limited number of reactivity pools (Hopkinson et al., 2002; Chen and Jaffé, 2016). Recent works emphasize that DOM in freshwater systems has rather a continuum of reactivity (Vähätalo et al., 2010; Sierra et al., 2011; Koehler et al., 2012; Mostovaya et al., 2016, 2017) and thus, examining the whole range of DOM constituents could be beneficial in search for connections between the properties and reactivity of DOM. Characterizing a multitude of compounds within DOM with both molecular characteristics and a specific decay rate could help to comprehensively assess major intrinsic drivers of DOM decay. In recent decades Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) has emerged as a powerful tool for resolving the composition of DOM on the level of individual molecular formulae (Marshall, 2000). Moreover, the signal intensity corresponding to each formula is in principal linearly proportional to the concentration of the corresponding isomeric mixture of compounds, as long as the sample matrix is similar (Lechtenfeld et al., 2014; Seidel et al., 2015). Therefore, following the loss of formula-specific intensity during decomposition can allow assessment of the relative rates of each individual molecular mass' decay (Mostovaya et al., 2017). Acquired individual decay coefficients can be further related to specific molecular properties, such as degree of compound saturation, carbon oxidation state, and molecular mass.
The ideas outlined above allowed us to break down the degradation of bulk DOM to behavior of a multitude of individual molecular masses, and relate the differing reactivity to their averaged structural properties. To do this, we performed a 120-day DOM decomposition experiment, using waters from three boreal lakes. One of the two experimental treatments was preexposed to UV light to accelerate the subsequent microbial decomposition (Lindell et al., 1995; Wetzel et al., 1995; Moran and Zepp, 1997). Based on the time series of intensities, we derived apparent exponential decay coefficients kexp for every molecular formula detected with FT-ICR-MS. We then explored the relationship between molecular properties of a large number (>1,300) of DOM constituents and the apparent decay rate coefficient kexp, including photochemical effects on subsequent DOM decomposition.
Three humic brown-water lakes (DOC concentration range 21.2–23.7 mg C L−1; Specific UV absorbance at 254 nm (SUVA254) range 3.3–4.1 L mg C−1 m−1), Lumpen, Ramsjön, and Övre Långsjön, were sampled in a forested area of east-central Sweden in October 2013. The catchments of the lakes are dominated by boreal forest (~20% of peat in the catchment of the Lake Lumpen) and underlain by the calcareous moraine and granitic bedrock. Mean annual precipitation in this area is 600–700 mm and mean annual temperature is 4–6°C (1961–1990, Swedish Meteorological and Hydrological Institute; http://www.smhi.se/klimatdata).
The water was collected from the surface (oxic epilimnion, 0−0.5 m depth), filtered through 0.2 μm membrane filters (Supor, Pall) and stored in the dark at 4°C. Portions of filtered water were irradiated with UV light (300−400 nm range) for 144 h, and about 30% of initial DOC was photochemically mineralized as a result (see also Mostovaya et al., 2016). Thus, the subsequent dark decomposition experiment was provided with treatments containing non-manipulated and UV-manipulated lake water.
The experimental setup consisted of non-manipulated and UV-manipulated lake water treatments, duplicated for each lake (12 experiments in total). The DOM incubations were conducted on 0.2 μm filtered water, inoculated with a 64 μm plankton net filtered aliquot from the respective lake (5% of the volume), with addition of inorganic nutrients (480 μg N L−1 as KNO3 and 100 μg P L−1 as Na2HPO4). Lake water was incubated in pre-acid washed (10% HCl), pre-combusted (4 h at 550°C) 40 ml glass vials sealed with PTFE-lined silicone septa, at 20°C and in the dark. All vials were submersed in water to minimize the gas exchange, and incubations lasted for 120 days. The length of the experiment was motivated by the fact that humic DOM can decay very slowly (e.g., Koehler et al., 2012). At 12 different time points of the experiment individual vials were sacrificed for measuring the DOC concentration and taking the samples for mass spectrometry analysis. The DOC concentration was measured on a Sievers 900 TOC Analyzer (General Electric Analytical Instruments). Water for mass spectrometry analysis was stored in pre-combusted (4 h at 450°C) 2 ml glass vials with PTFE-lined silicone septa at 4°C. Prior to usage, the septa and screw caps were pre-soaked in methanol for 24 h and repeatedly rinsed with MilliQ water (Millipore).
Mass spectrometry analysis was performed with a 15-T Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR-MS; Solarix, Bruker Daltonics) with an electrospray ionization source operating in negative mode. Lake water samples were diluted with ultrapure water to a concentration of 10 mg C L−1 and mixed with methanol (HPLC grade, Sigma-Aldrich) in a proportion of 2:1 (final concentration 6.7 mg C L−1). The samples were injected without further treatment, i.e., without performing the solid phase extraction, at 360 μL h−1. Mass spectra were collected over 400 scans, with ion accumulation time of 0.5 s, and within a range of 150–2,000 m/z. Each sample was internally calibrated with a reference mass list generated from North Equatorial Pacific Intermediate Water (NEqPIW) (Green et al., 2014) using the Bruker Daltonics Data Analysis software package. Molecular formulae containing the elements C, H, O, N, S, and P were assigned to peaks with a signal to noise ratio greater than four and based on the following criteria: O ≤ C; O > (2P + S); N ≤ 4; S ≤ 4 and P ≤ 1. The S-, P-, and N > 1-containing formulae did not show any specific behavior (see section Formula-Specific Apparent Decay Coefficient kexp) and, since higher errors are associated with the assignments containing S, P, and N > 1, these formulae were excluded from further analysis to avoid excessive complexity. Unassigned peaks were disregarded, as they were mainly noise or isotopologue (e.g., 13C) peaks. Known contaminant peaks and peaks found in procedural blanks, as well as the peaks with unusually high intensity, were removed from the dataset. For each lake, only peaks found in both replicates of each experimental treatment were considered. For each decomposition experiment, time series of total intensity were compared to the corresponding bulk DOC loss (Figure S1). This was done to check the consistency of the data, as we expected linear relationship between total intensity and bulk DOC. One of the replicate incubations (lake Övre Långsjön, non-manipulated water, replicate 2) and four individual samples from different incubations fell out of the linear trend, possibly due to the short-term instrument or software problems, (Figure S1) and, thus, were removed from the subsequent analysis. Further, data from each lake-treatment-replicate combination (eleven in total, since one incubation time series was removed) was processed separately. A detection limit was applied based on dynamic range. The dynamic range of each individual sample was calculated as a ratio of maximum to minimum peak intensity found in the sample. Detection limit was then calculated for each sample, by dividing maximum intensity in this sample by the lowest dynamic range found among the samples. Intensities below the detection limit were removed. This left in consideration 1,654−2,045 formulae across the range of samples.
In each sample the intensities were normalized to the total intensity and multiplied to account for dilution and to estimate the equivalent intensity in the undiluted sample. In other words, the samples were normalized internally for total intensity and then externally to account for DOC concentration. This was done to correctly estimate the relative change in concentration over the course of incubation (Lechtenfeld et al., 2014; Seidel et al., 2015; Hawkes et al., 2016b; Mostovaya et al., 2017). The signal intensity we refer to further in the text (e.g., in the section Formula-Specific Apparent Decay Coefficient kexp) corresponds to DOC normalized intensity in undiluted sample.
Double bond equivalence (DBE), reflecting the degree of unsaturation, was calculated for each formula as DBE = 1 + ½(2C – H + N) and modified aromaticity index (AImod), reflecting the existence of aromatic and condensed aromatic structures, was calculated as AImod = [1 + C − ½O − ½(N + H)]/(C − ½O − N), where C, H, O, and N refer to a number of respective atoms per molecule. The formulae for DBE and AImod are adjusted from (Koch and Dittmar, 2006, 2016) as S- and P-containing formulae were not included in our analysis.
Molecular data was visualized in van Krevelen diagrams, i.e., the plots of formula-specific H/C vs. O/C ratios (Kim et al., 2003). Compound groups were assigned based on AImod and H/C ratio, with AImod > 0.66 indicating the presence of combustion derived polycondensed aromatics (Koch and Dittmar, 2006), 0.50 < AImod ≤ 0.66 delineating plant polyphenols and condensed aromatics with few aliphatic side chains (Šantl-Temkiv et al., 2013; Kellerman et al., 2015), AImod ≤0.5 & H/C < 1.5 indicating highly unsaturated and phenolic compounds (Šantl-Temkiv et al., 2013), and H/C ≥ 1 indicating aliphatic compounds (Riedel et al., 2016). We also calculated nominal oxidation state of carbon (NOSC) as NOSC = 4−[(4C+H−3N−2O)/C]. This formula is modified from Riedel et al. (2012), since S-containing compounds were not included in our analysis.
For each formula, a single-exponential model (Olson, 1963) was fit into the 12-point time series of relative change in signal intensity following the equation
where kexp is an apparent exponential decay coefficient (day−1) and It/I0 is the relative change in intensity over time (Mostovaya et al., 2017). The choice of the model is rooted in the theory of reactivity continuum (Boudreau and Ruddick, 1991), which emerges as a realistic model of organic matter decay (Vähätalo et al., 2010; Koehler and Tranvik, 2015; Mostovaya et al., 2016) and assumes that each individual reactive type within bulk DOM degrades following the single-exponential kinetics. For the purpose of model fitting, only formulae that were detected on at least 8 days of the 12-point time series were considered. The model fitting was conducted using generalized least squares modeling, and the goodness of model fit was described with normalized root-mean-square error (NRMSE) (Mostovaya et al., 2017). The time series containing a lot of noise were not considered in the subsequent analysis. The threshold for noise was established at NRMSE = 35.8%–a 95% quantile of the NRMSE distribution calculated from a series of repeated measurements of DOM NEqPIW standard (Green et al., 2014) of steady concentration (15 mg C L−1). Between 89.7 and 97.3% of the formulae in the experimental datasets met this criterion. Since the context of our study was DOM decomposition, we were interested in the molecular formulae that exhibited net decay, and, hence, only used the formulae with positive kexp in the subsequent analysis. It should be remembered that the apparent exponential decay coefficient kexp represents the net of simultaneous loss and formation of the corresponding formula. The formulae demonstrating the net decay constituted the majority in our datasets (76.1−98.1% of all formulae with accepted NRMSE). The formulae demonstrating the net accumulation (Figure S2) are not discussed further.
The relationship of kexp with molecular characteristics, including the H/C and O/C ratios, DBE, AImod, NOSC, and molecular mass were approximated using generalized additive models (GAM) based on cubic splines (R function stat_smooth in package ggplot2). The model fitting, and all calculations were performed using R (version 3.3.2, R Development Core team, 2016).
We focused on two groups of formulae from the comparison between the non-manipulated and UV-manipulated experimental treatments: (1) shared formulae, found in both treatments and (2) the formulae found only in UV treatment, i.e., corresponding to photoproduced molecules. For the shared formulae, apparent decay coefficients kexp were compared between the treatments. To explore possible similarities in chemical characteristics and reactivity of photoproduced molecules, the corresponding formulae were plotted in van Krevelen space with apparent decay coefficients kexp as a third, color-coded dimension.
Between 1,308 and 1,820 formulae could be assigned an apparent decay coefficient kexp in each treatment (see Formula-Specific Apparent Decay Coefficient kexp in Methods). To compare the distribution of kexp we considered the following groups of compounds: aliphatics, highly unsaturated and phenolic compounds, plant polyphenols, and polycondensed aromatics. The values of kexp varied from 4.3 × 10−8 to 0.02 day−1. While most of kexp in this range were found within each group of compounds, the exact distribution of kexp, i.e., the prevalence of lower or higher kexp, differed between the groups (Figures 1, 2, Figures S3, S4).
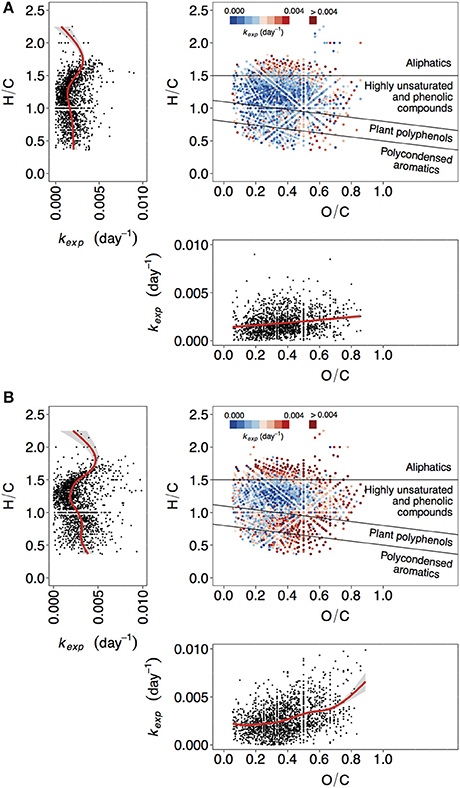
Figure 1. Van Krevelen plots depicting molecular formulae color-coded by value of the decay coefficient kexp. Two additional panels isolate relationship of kexp with H/C (left), and O/C (bottom) ratios. Red lines correspond to fitted generalized additive models (GAM). (A) Lake Ramsjön, non-manipulated treatment, (B) Lake Ramsjön, UV-manipulated treatment. One of the duplicate treatments is shown in each case (two other lakes and all the duplicates are shown in the Figure S3).
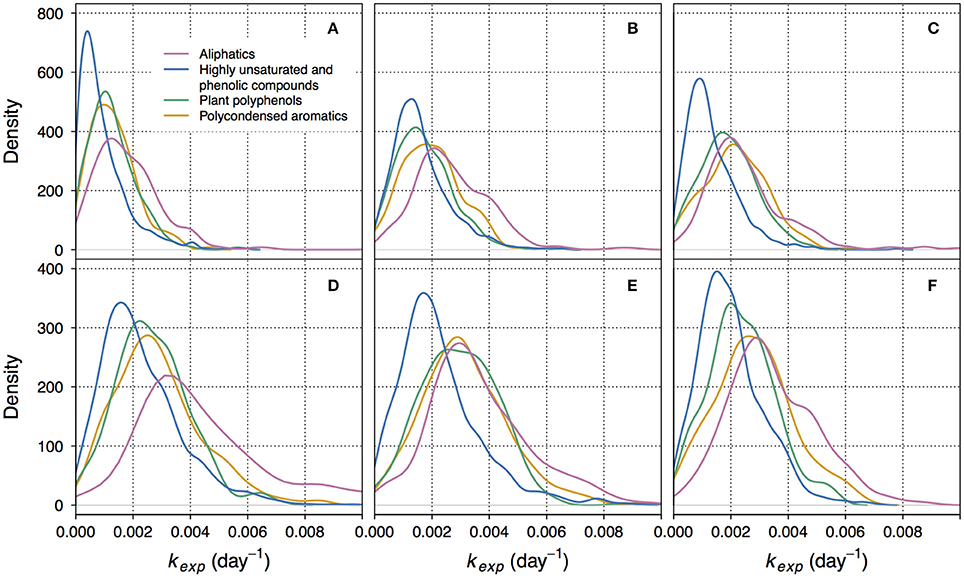
Figure 2. Kernel density plots showing the distributions of decay coefficients kexp within different groups of compounds. Here, kernel density function is a non-parametrically estimated probability density function of kexp. The integral of each function over the range of corresponding kexp equals 1. (A–C) correspond to non-manipulated treatments of lake Lumpen, Ramsjön, and Övre Långsjön, respectively. (D–F) correspond to the UV-manipulated treatments of the same lakes. Note that the Y-axis scale is different for different treatments. One of the duplicate treatments is shown (data from the second duplicate is shown in the Figure S4).
The lowest kexp were mostly associated with highly unsaturated and phenolic compounds (Figures 1, 2, Figures S3, S4). This group is ambiguous with respect to aromaticity–molecules may contain aromatic rings, or may simply have a high density of double bonds in a different configuration (Koch and Dittmar, 2006). Given that the DOM of boreal lakes originates largely from terrestrial vascular plants (Thurman, 1985; Hessen and Tranvik, 1998), a reasonable assumption can be made that most of the highly unsaturated and phenolic compounds found in our experiment are lignin-derived.
In non-manipulated treatments, representing the natural lake water, the share of slowly decaying formulae was also substantial within the groups of polyphenols and polycondensed aromatics (Figures 1A, 2A,B, Figures S3, S4). In one of the lakes (Figure 2C), and in UV-manipulated treatment (Figures 2E,F) these groups were almost as reactive as aliphatics.
Relatively low reactivity of vascular plant-derived components of DOM has been reported previously (Benner et al., 1987; Moran and Hodson, 1994; Benner and Kaiser, 2011). However, we do not suggest that the low reactivity of lignin-derived and polyphenolic compounds is ubiquitous. Our results showed the presence of high apparent decay rates within each group of compounds (Figure 2, Figure S4). Accordingly, fast decomposition of lignin phenols was recently documented for Amazon River DOM (Ward et al., 2013). The fact that some fraction of phenolic compounds is likely to readily participate in decomposition at the timescales of several months was also previously reported (Mostovaya et al., 2016).
The reactivity of aliphatic compounds was predominantly high (Figures 1, 2, Figures S3, S4), which agrees with existing concepts of OM lability (Sun et al., 1997; D'Andrilli et al., 2015). Studies have shown that labile fractions of DOM are often of algal origin (e.g., Guillemette et al., 2013), and there is evidence that algal DOM is to a large extent aliphatic (H/C > 1.5) (Mangal et al., 2016). Yet, we do not imply that aliphatic compounds in our study are exclusively of algal origin, because they may also be components of fresh terrestrial DOM of increased lability. In the study evaluating the persistence of DOM in boreal lakes Kellerman et al. (2015) found that aliphatic compounds were abundant in lakes with higher water residence time. While, at first, this seems to contradict our results, it should be considered that persistence is not necessarily caused by the resistance to degradation and sedimentation, but can be also a result of the constant replenishment. This might be especially relevant for algal DOM, as the conditions for photosynthesis improve in more transparent waters of the lakes with higher water residence time.
It should be reemphasized that kexp formed a continuous distribution within each group of compounds, with both high and low kexp present (Figure 2, Figure S4). The average behavior of each compound group was, hence, determined by the character of the distribution and whether it was shifted toward lower or higher values of kexp. This gives further support to the idea that overall DOM decomposition kinetics emerges from the decay kinetics of multitude of individual constituents (Mostovaya et al., 2017). The presence of a wide range of reactivities within each compound group, however, signifies that molecular formulae with different bulk properties can be equally reactive. This likely reflects the complexity of structural constraints on reactivity and suggests that higher structural resolution is needed to further ascertain the reasons behind DOM reactivity in lake water.
Our experiment characterized the aerobic decomposition of DOM typically present in boreal humic lakes, not limited by nutrients. While it is not possible to say whether the observed trends in compound reactivity would be retrievable from the other aquatic settings, they might be characteristic of other lakes with high terrestrial DOM input. At the same time, it has been shown before that the freshness of terrestrial DOM can influence the rates of microbial DOM utilization (Berggren et al., 2010a,b). This exemplifies that the exact chemical composition might be a more universal predictor of decay patterns than the degree of allochthony. With the increasing number of studies using the ultrahigh resolution mass spectrometry to understand the molecular drivers of DOM reactivity in a variety of systems (Gonsior et al., 2013; Sleighter et al., 2014; D'Andrilli et al., 2015; Kellerman et al., 2015; Hawkes et al., 2016b; Riedel et al., 2016; Kamjunke et al., 2017) a better understanding of the intrinsic controls of aquatic DOM reactivity becomes increasingly possible to achieve.
We examined the relationship of kexp with compounds' saturation (H/C), modified aromaticity index (AImod), double bond equivalency (DBE), molecular mass, oxygen content (O/C ratio), and nominal oxidation state of carbon (NOSC). Despite the large variability in the data, fitting the GAM models allowed us to identify general trends (Figures 1, 3, Figures S3, S5).
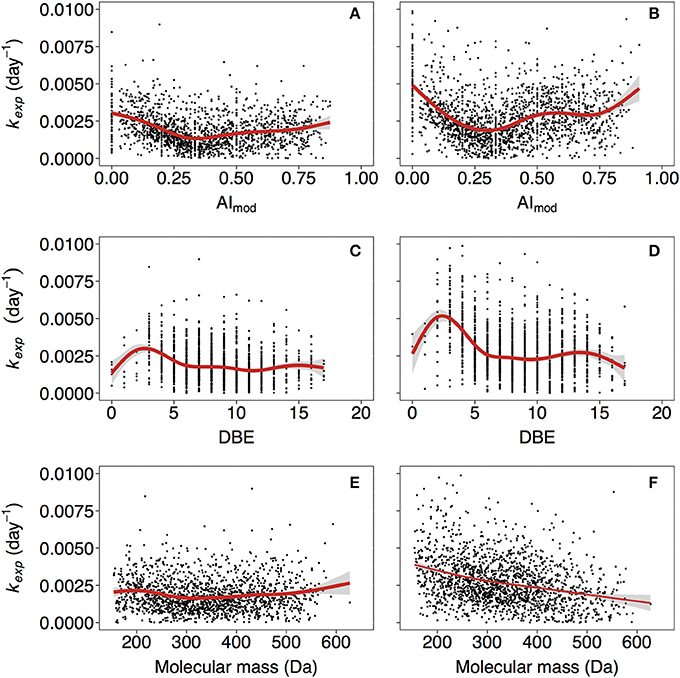
Figure 3. Relationships of kexp with modified aromaticity index (AImod; A,B), double bond equivalence (DBE; C,D) and molecular mass (E,F). (A,C,E, and B,D,F) correspond to non-manipulated and UV-manipulated treatment, respectively. Red lines correspond to fitted generalized additive models (GAM). The data is shown for one of the duplicates of lake Ramsjön (data for the second duplicate and two other lakes is shown in the Figure S5).
The reactivity of aromatic compounds (H/C~below 1.1) was on average higher than reactivity in the “highly unsaturated” region (H/C~between 1.1 and 1.5) (Figure 1, Figure S3), and AImod showed curved relationship with kexp: the kexp first decreased with increasing AImod, but at AImod between 0.25 and 0.33 began to increase again (Figures 3A,B, Figure S5). As was pointed out above, highly unsaturated compounds may contain aromatic structures but their content is assumed to be lower than in the groups of plant polyphenols and polycondensed aromatics. A high contribution of aromatic structures is typical for the colored DOM of humic lakes, and traditionally considered to be associated with low reactivity (Tranvik, 1998). Hence, at the first sight our findings seem surprising. However, substantial evidence has accumulated that this material is not as persistent as often assumed (Tranvik, 1992), and in boreal lakes colored DOM disappears often faster than bulk DOM (Koehler et al., 2012; Weyhenmeyer et al., 2012). Likewise, FT-ICR-MS analysis of DOM from a large number of lakes (Kellerman et al., 2015) showed that the prevalence of aromatic compounds decreases with increasing water residence time of lakes, suggesting that they are selectively removed. The concept of universally low reactivity of aromatic DOM has been further challenged as higher-than-previously-thought lability of lignin-derived components was described in a variety of systems (Holmes et al., 2008; Spencer et al., 2008; Ward et al., 2013). The variability in reactivity observed for aromatic DOM reflects the fact that compounds that belong to a large common group (e.g., aromatics, lignin) can be extremely diverse with respect to exact structure and chemical properties.
Decay rate tended to be slightly higher for compounds with lowest DBE (Figures 3C,D, Figure S5), but generally DBE did not have a strong effect on kexp.
We did not find strong indications of the effect of molecular mass on kexp in the non-manipulated treatment (Figure 3E, Figure S5), and in the UV treatment reactivity tended to decrease with increasing mass in some cases (Figure 3F, Figure S5). It should be noted that our data covered the DOM mass range of ~150−600 Da. Other studies considered apparent masses up to 1−10 kDa (Tranvik, 1990; Amon and Benner, 1996), acquired using ultrafiltration techniques, and showed that bacteria preferentially utilize substrates of higher apparent molecular weight. A systematic comparison of apparent (i.e., assessed on the basis of separation techniques such as ultrafiltration) vs. actual (i.e., assessed based on high resolution methods targeting individual molecules) molecular weight across lakes showed that the patterns of the two are not systematically related (Kellerman, 2015). Our results indicate that in natural lake water actual molecular mass might not be important for reactivity, as opposed to other characteristics, such as compound class and, to a certain degree, nominal oxidation state of carbon (see below). The possible reasons for emerging relationship between molecular mass and reactivity in the UV treatment are discussed in section Treatment Effects.
Apparent decay coefficient kexp was positively related to oxygen content, i.e., O/C ratio (Figure 1, Figure S3), and tended to be lowest at intermediate levels of NOSC (Figure 4, Figure S6). Figure 4 and Figure S6 demonstrate that exact relationships of kexp with NOSC can differ between the groups of compounds. For all lakes, treatments, and replicates, reactivity of plant polyphenols increases nearly linearly with increasing NOSC (Figure 4, Figure S6). For other compound classes relationship of kexp with NOSC was less straightforward, but demonstrated a similar tendency in many cases (Figure 4, Figure S6).
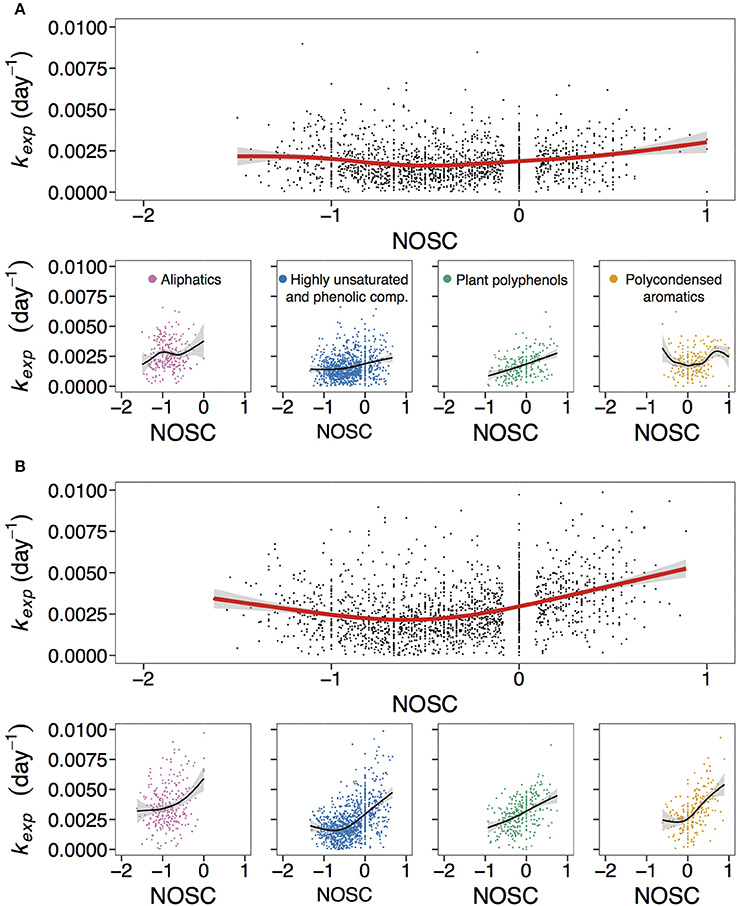
Figure 4. Overall and compound group-specific relationship of kexp with nominal oxidation state of carbon (NOSC). (A,B) correspond to non-manipulated and UV-manipulated treatment, respectively. Red and black lines correspond to fitted generalized additive models (GAM). The data is shown for one of the duplicates of lake Ramsjön (data for the second duplicate and two other lakes is shown in the Figure S6). Color-coded are aliphatics (pink), highly unsaturated and phenolic compounds (blue), plant polyphenols (green), and polycondensed aromatics (yellow).
There is no absolute agreement in the literature on the exact relationship between oxygen content and reactivity of OM. Negative relationship between O/C and bioavailability of DOM (assessed as bacterial growth) has been reported by Sun et al. (1997). In contrast, based on the results of high resolution mass spectrometry analysis, Kim et al. (2006) concluded that biodegradation selectively removed compounds with higher O/C. However, there are also studies that did not find a strong connection between O/C and bioavailability of humic DOM (Hunt et al., 2000), or a direct correlation between NOSC and degradation rate coefficient of DOM compounds (Lechtenfeld et al., 2014).
There is evidence for a positive relationship between the degree of oxidation and apparent radiocarbon age of marine DOM (Lechtenfeld et al., 2014). It seems logical to assume that, unlike more reduced substrates, oxidized DOM has low energetic value for DOM decomposers (Perdue, 2009; Kattner et al., 2011). LaRowe and Van Cappellen (2011) challenge this concept and argue that removal of electrons from a substrate is thermodynamically more favorable at higher NOSC. Yet, the authors note that thermodynamic predictions may not match the observations due to complexity of factors affecting decomposition. Accordingly, in a conceptual model of DOM degradation in the oceans Williams (1999) proposes that there is a general mismatch between the susceptibility of DOM components toward microbial degradation and Gibb's free energy, i.e., apparently energetically favorable substrates appear to be recalcitrant, and vice versa.
Our results are corroborated by the findings of Kellerman et al. (2015) in the study of molecular composition of freshwater DOM across 109 Swedish lakes. This study demonstrated that degradation processes preferentially remove aromatic oxidized terrestrial compounds in lakes with longer water residence times. Considering how different our approach is from Kellerman et al. (2015), the similarity in the findings is striking, further emphasizing that terrestrially derived, predominantly aromatic DOM is a dynamic component within the organic matter field. There is also a possibility that both in our experiments and in the study by Kellerman et al. (2015) formulae with higher NOSC were partially lost through non-biological processes (e.g., aggregation and flocculation). It has been shown that compounds with higher NOSC are preferentially precipitated by Fe and Al salts (Riedel et al., 2012; Lavonen, 2015). Studies on the flocculation of DOM in lakes suggest that allochthonous colored DOM is a dominant precursor of organic matter settling onto the sediment (von Wachenfeldt and Tranvik, 2008; von Wachenfeldt et al., 2009). Although we lack the data to adequately assess the importance of non-biological pathways for our experiment, relationship between NOSC and non-biological DOM removal could be an interesting topic for the future studies.
All trends of kexp with molecular parameters were consistent between different lakes and replicates (Figures S3–S6 in the supplement, complementing the figures displayed in the main text). Hence, our results provide additional support to the idea that patterns of DOM utilization are in principle predictable from the intrinsic properties of DOM (Seitzinger et al., 2005; Kellerman et al., 2015), but we also acknowledge that there is high variability in reactivity which limits the potential to predict it from broad molecular properties, such as element ratios. The variability in our data could be due to the fact that each molecular formula describes a large number of potential isomers (Hertkorn et al., 2007; Zark et al., 2017), and the variations in specific structure likely translate into variations in decay behavior. Despite this limitation, our method was able to capture the general trends in reactivity of different DOM groups, including the effects of UV pretreatment on distribution of apparent decay coefficients kexp.
Between 87.7 and 97.9% of formulae found in non-manipulated treatments were also detected in the UV-manipulated treatments. Interestingly, the reactivity (kexp) of these shared compounds was often higher in the UV-manipulated treatment (Figure 5, Figure S7). Despite a general increase in reactivity, also indicated by the shift toward higher values of kexp in all compound groups (Figure 2, Figure S4), the recalcitrant core in the region of highly unsaturated compounds persisted in the UV-manipulated treatment (Figures 1B, 2, Figures S3, S4).
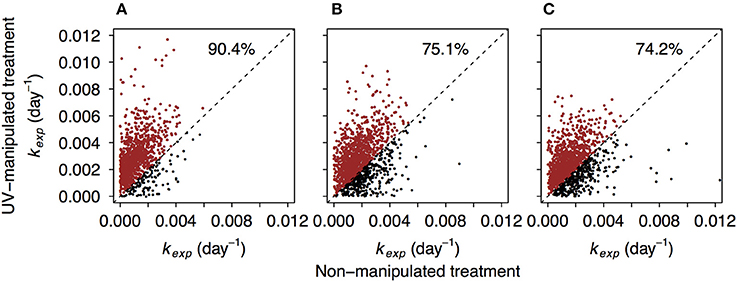
Figure 5. Comparison of decay coefficients kexp of the formulae shared by non-manipulated and UV-manipulated experimental treatments. Dashed line refers to hypothetical 1:1 relationship. The data points above the 1:1 line are depicted in red. Percentages refer to the share of data points above the 1:1 line. (A–C) correspond to lake Lumpen, Ramsjön, and Övre Langsjön, respectively. One of the experimental duplicates is shown in each case (data from the second duplicate is shown in the Figure S7).
A possible reason for higher DOM reactivity in the UV treatment is that the microbial community was stimulated by labile compounds produced as a result of DOM exposure to UV, and this triggered an ability to decompose formerly “recalcitrant” compounds. This phenomenon, known as the priming effect, is commonly described from terrestrial ecosystems (Fontaine et al., 2007; Kuzyakov, 2010). However, an increasing number of studies in aquatic systems have failed to corroborate its existence in aquatic ecosystems (Bengtsson et al., 2014; Catalán et al., 2015; Dorado-García et al., 2016). The main mechanisms of priming in soils are thought to involve nutrient limitations, co-metabolism, and higher enzyme production (Blagodatskaya and Kuzyakov, 2008). All our experiments were carried out with the addition of inorganic nutrients, therefore microbes should not have been limited by the availability of specific elements. Therefore, it seems unlikely that nutrient priming is responsible for increase in compounds decomposition in UV-manipulated treatment.
DOM degradation kinetics can also depend on the concentration of the individual DOM constituents (Dittmar, 2015). For example, it was proposed that the millennium-scale stability of DOM in the deep ocean is due to extreme dilution of the individual DOM compounds, and possibly not due to structural constraints (Arrieta et al., 2015). In our experiment, initial concentration of DOC was 30% lower in the UV treatment than in the non-manipulated treatment, yet decomposition in the UV treatment proceeded faster. Previous studies also showed that overall degradation and kinetics in freshwater systems are not constrained by DOC concentration within the concentration range studied here (Eiler et al., 2003; Koehler et al., 2012). Hence, changes in structural properties of DOM are most likely to be responsible for the differences in decay rates between the treatments.
We considered that UV treatment may lead to broad compositional or structural changes of the organic matter, such as loss of aromaticity (Stubbins et al., 2010) or loss of carboxylic acid functionality (Hawkes et al., 2016b) but these types of chemical change were not apparent from average H/C or O/C ratios (respectively), and loss of intensity of individual molecular masses did not show clear patterns in these respects (Figures S8, S9). It is still possible that individual isomers were affected but such changes, but not on a broad scale. As was mentioned above, each molecular formula encompasses a large number of structural isomers (Zark et al., 2017). Therefore, while the majority of formulae identified in the UV treatment were identical to those in the non-manipulated treatment, the isomeric mixture of structures underlying each molecular formulae could differ between the treatments.
Another possible explanation for different reactivity of the same compounds in the two treatments might be that specific molecules were to a different extent entangled in colloidal or supramolecular structures. For example, soil-derived humic DOM can exist in the form of supramolecular complexes where molecules are stabilized by weak hydrophobic interactions and hydrogen bonds (Conte and Piccolo, 1999; Piccolo, 2001). Multiple studies indicated the decrease in apparent molecular weight of DOM as a result of UV exposure (Helms et al., 2008, 2014), suggesting a disruption of multiple bonds within aggregated and colloidal DOM structures.
Electrospray ionization can disperse loosely, non-covalently bound structures (Yamashita and Fenn, 1984). Accordingly, the formulae identified by FT-ICR-MS would carry no information on whether corresponding compounds were previously part of a supramolecular complex. It is possible that UV light cleaved the larger complexes into more available fragments, no longer protected from decomposition by multiple weak bonds. Supramolecular complexes of high apparent molecular weight (measured with size exclusion chromatography) can be associated with plant polyphenols and polycondensed aromatics (Kellerman, 2015). In our study, these groups of compounds increased in reactivity in the UV-manipulated treatment, which suggests that supramolecular structure could have been at least partly responsible for lower reactivity of polyphenols in non-manipulated treatment.
In the UV treatment kexp in some cases decreased with increasing molecular mass (Figure 3F, Figure S5)–the relationship that was not observed in non-manipulated treatment (Figures 3E, Figure S5). Pretreatment with UV light likely diminished the effects of steric hindrance as molecular aggregates were broken into smaller fragments, and relationship between structure of molecules and kexp became more apparent. Notably, in the UV treatment relationship of kexp with molecular characteristics had more pronounced character (steeper curves of the fitted GAMs) in many cases (Figures 1, 3, 4, Figures S3, S5, S6).
Photoproduced compounds, identified as formulae unique to the UV treatment, appeared among all compound groups and covered the whole range of apparent decay coefficients kexp (Figure 6, Figure S10). This shows that products of photoreactions are not restricted to the aliphatic region where many described photoproducts belong (Moran and Zepp, 1997) and are not necessarily highly reactive. Instead, some photoproducts can persist and act as a slowly degrading substrate on a timescale of several months.
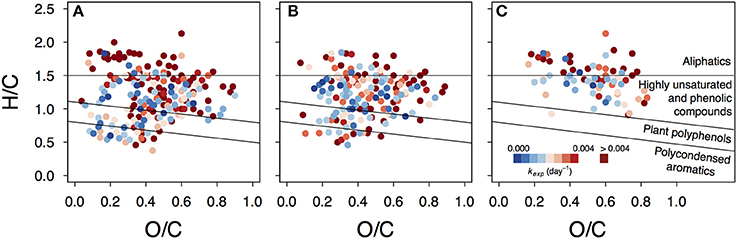
Figure 6. Van Krevelen plots depicting variability in decay rates of photoproduced compounds. Presented are data from lake Lumpen (A), Ramsjön (B) and Övre Langsjön (C). One of the experimental duplicates is shown in each case (data from the second duplicate is shown in the Figure S10).
It should be mentioned that the most reactive compounds (such as low molecular weight carboxylic acids) were not addressed in our study, as they can disappear within a few hours. It should also be taken into account that many photoproducts tend to end up below the detection limit or are inefficiently ionized (Stubbins et al., 2010; Raeke et al., 2016) and therefore might fall outside our analytical window.
While phototransformations of lake DOM has been extensively studied before (e.g., Bertilsson and Tranvik, 2000; Moran et al., 2000; Stubbins et al., 2010; Gonsior et al., 2013), the exact contribution of photoreactions to lake carbon budgets has been rarely assessed. The existing studies on small boreal lakes suggest that sunlight-induced CO2 emission constitutes <10% of the mean annual CO2 emission from these lakes (Koehler et al., 2014; Groeneveld et al., 2015). This fact can be related to relatively low mean annual irradiance in boreal latitudes and strong light attenuation in brown humic waters. The analysis of hundreds of freshwater systems in boreal Canada, however, showed that the increased terrestrial influence increases photodegradability of DOM (Lapierre et al., 2013), hence the contribution of sunlight-induced CO2 emissions from lakes might increase with changing climate and increased terrestrial runoff. The carbon budgets (e.g., Koehler et al., 2014; Groeneveld et al., 2015) do not account for the effects of phototransformations on DOM bioreactivity and proneness to coagulation, as those are especially difficult to evaluate quantitatively. We here show that a complete loss of 30% of DOM after the UV exposure coincided with phototransformations that result in a systematic increase in compound loss rates trough biological and, possibly, abiotic removal (see also Mostovaya et al., 2016).
Our study presents a direct analysis of the relationship between molecular properties and degradation kinetics of lake water DOM. Using ultrahigh resolution mass spectrometry and kinetic modeling we were able to describe the decay of individual molecular formulae with exponential decay coefficients kexp. We show that each group of compounds is characterized by a continuous range of decay coefficients kexp, and the distribution of kexp is shifted toward lower or higher values, depending on compound group. The lowest reactivity was attributed to the group of highly unsaturated and phenolic compounds, likely dominated by the products of vascular plant decay. The reactivity of compounds with higher aromatic content (plant polyphenols and polycondensed aromatics) was on average higher, and aliphatic compounds had the highest decay rates. Nominal oxidation state of carbon appeared to play a role in reactivity of DOM, with the higher NOSC often related to higher reactivity. The relatively high reactivity of aromatic and oxidized compounds is an important observation that may challenge common assumptions about the relationship between composition and decay of freshwater DOM. Molecular mass did not have a pronounced effect on reactivity, except for the DOM in the UV treatment where kexp in some cases decreased with increasing mass. Based on the comparison of non-manipulated and UV-manipulated DOM, we propose that reactivity of molecules within DOM may be constrained by the formation of supramolecular complexes, e.g., through multiple hydrogen bonds, hydrophobic interactions, and steric hindrance.
Finally, we demonstrate that high resolution mass spectrometry can be used to assess how decay of organic matter is distributed among the numerous compounds that make up the DOM, and has the capacity to reveal the impact of factors such as solar UV exposure on the detailed dynamics of DOM. With further development and increasing access (Hawkes et al., 2016a) to high resolution techniques for DOM characterization, we foresee substantial progress in the understanding of how the dynamics of natural organic matter depends on molecular characteristics.
AM and LT designed the DOM decomposition experiment. The experiment was performed by AM. Molecular analyses were performed by AM and JH under the supervision of TD. Kinetic modeling was performed by AM. Graphics were produced by AM. The manuscript was written under the lead of AM, with the contribution of JH, TD, and LT.
The study was financed by the Swedish Research Council (grant 2011-3475-88773-67), by the Nordforsk (DOMQUA project 60501), and by the Knut and Alice Wallenberg Foundation (grant KAW 2013.0091).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Karólína Einarsdóttir and Jan Johansson for laboratory assistance; Katrin Klaproth and Helena Osterholz for help with the processing of mass spectrometry data; Birgit Koehler for assistance with R code and modeling.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2017.00106/full#supplementary-material
Algesten, G., Sobek, S., Bergström, A.-K., Ågren, A., Tranvik, L. J., and Jansson, M. (2004). Role of lakes for organic carbon cycling in the boreal zone. Glob. Chang. Biol. 10, 141–147. doi: 10.1111/j.1365-2486.2003.00721.x
Amon, R. M. W., and Benner, R. (1994). Rapid cycling of high-molecular-weight dissolved organic matter in the ocean. Nature 369, 549–552. doi: 10.1038/369549a0
Amon, R. M. W., and Benner, R. (1996). Bacterial utilization of different size classes of dissolved organic matter. Limnol. Oceanogr. 41, 41–51. doi: 10.4319/lo.1996.41.1.0041
Arrieta, J. M., Mayol, E., Hansman, R. L., Herndl, G. J., Dittmar, T., and Duarte, C. M. (2015). Dilution limits dissolved organic carbon utilization in the deep ocean. Science 348, 331–333. doi: 10.1126/science.1258955
Bengtsson, M. M., Wagner, K., Burns, N. R., Herberg, E. R., Wanek, W., Kaplan, L. A., et al. (2014). No evidence of aquatic priming effects in hyporheic zone microcosms. Sci. Rep. 4:5187. doi: 10.1038/srep05187
Benner, R., Fogel, M. L., Sprague, E. K., and Hodson, R. E. (1987). Depletion of 13C in lignin and its implication for stable carbon isotope studies. Nature 329, 708–710. doi: 10.1038/329708a0
Benner, R., and Kaiser, K. (2011). Biological and photochemical transformations of amino acids and lignin phenols in riverine dissolved organic matter. Biogeochemistry 102, 209–222. doi: 10.1007/s10533-010-9435-4
Berggren, M., Laudon, H., Haei, M., Ström, L., and Jansson, M. (2010a). Efficient aquatic bacterial metabolism of dissolved low-molecular-weight compounds from terrestrial sources. ISME J. 4, 408–416. doi: 10.1038/ismej.2009.120
Berggren, M., Ström, L., Laudon, H., Karlsson, J., Jonsson, A., Giesler, R., et al. (2010b). Lake secondary production fueled by rapid transfer of low molecular weight organic carbon from terrestrial sources to aquatic consumers. Ecol. Lett. 13, 870–880. doi: 10.1111/j.1461-0248.2010.01483.x
Bertilsson, S., and Tranvik, L. J. (2000). Photochemical transformation of dissolved organic matter in lakes. Limnol. Oceanogr. 45, 753–762. doi: 10.4319/lo.2000.45.4.0753
Blagodatskaya, E., and Kuzyakov, Y. (2008). Mechanisms of real and apparent priming effects and their dependence on soil microbial biomass and community structure: critical review. Biol. Fertil. Soils 45, 115–131. doi: 10.1007/s00374-008-0334-y
Boudreau, B. P., and Ruddick, B. R. (1991). On a reactive continuum representation of organic matter diagenesis. Am. J. Sci. 291, 507–538. doi: 10.2475/ajs.291.5.507
Catalán, N., Kellerman, A. M., Peter, H., Carmona, F., and Tranvik, L. J. (2015). Absence of a priming effect on dissolved organic carbon degradation in lake water. Limnol. Oceanogr. 60, 159–168. doi: 10.1002/lno.10016
Chen, M., and Jaffé, R. (2016). Quantitative assessment of photo- and bio-reactivity of chromophoric and fluorescent dissolved organic matter from biomass and soil leachates and from surface waters in a subtropical wetland. Biogeochemistry 129, 273–289. doi: 10.1007/s10533-016-0231-7
Cole, J. J., Prairie, Y. T., Caraco, N. F., McDowell, W. H., Tranvik, L. J., Striegl, R. G., et al. (2007). Plumbing the global carbon cycle: integrating inland waters into the terrestrial carbon budget. Ecosystems 10, 171–184. doi: 10.1007/s10021-006-9013-8
Conte, P., and Piccolo, A. (1999). Conformational arrangement of dissolved humic substances. Influence of solution composition on association of humic molecules. Environ. Sci. Technol. 33, 1682–1690. doi: 10.1021/es9808604
D'Andrilli, J. D., Cooper, W. T., Foreman, C. M., and Marshall, A. G. (2015). An ultrahigh-resolution mass spectrometry index to estimate natural organic matter lability. Rapid Commun. Mass Spectrom. 29, 2385–2401. doi: 10.1002/rcm.7400
Dittmar, T. (2015). “Reasons behind the long-term stability of dissolved organic matter,” in Biogeochemistry of Marine Dissolved Organic Matter, eds D. A. Hansell and C. A. Carlson (London: Elsevier), 369–388.
Dorado-García, I., Syväranta, J., Devlin, S. P., Medina-Sánchez, J. M., and Jones, R. I. (2016). Experimental assessment of a possible microbial priming effect in a humic boreal lake. Aquat. Sci. 78, 191–202. doi: 10.1007/s00027-015-0425-4
Duarte, C. M., and Prairie, Y. T. (2005). Prevalence of heterotrophy and atmospheric CO2 emissions from aquatic ecosystems. Ecosystems 8, 862–870. doi: 10.1007/s10021-005-0177-4
Eiler, A., Langenheder, S., Bertisson, S., and Tranvik, L. J. (2003). Heterotrophic bacterial growth efficiency and community structure at different natural organic carbon concentrations. Appl. Environ. Microbiol. 69, 3701–3709. doi: 10.1128/AEM.69.7.3701-3709.2003
Fontaine, S., Barot, S., Barré, P., Bdioui, N., Mary, B., and Rumpel, C. (2007). Stability of organic carbon in deep soil layers controlled by fresh carbon supply. Nature 450, 277–280. doi: 10.1038/nature06275
Gonsior, M., Schmitt-Kopplin, P., and Bastviken, D. (2013). Depth-dependent molecular composition and photo-reactivity of dissolved organic matter in a boreal lake under winter and summer conditions. Biogeosciences 10, 6945–6956. doi: 10.5194/bg-10-6945-2013
Green, N. W., Perdue, E. M., Aiken, G. R., Butler, K. D., Chen, H., Dittmar, T., et al. (2014). An intercomparison of three methods for the large-scale isolation of oceanic dissolved organic matter. Mar. Chem. 161, 14–19. doi: 10.1016/j.marchem.2014.01.012
Groeneveld, M. M., Tranvik, L. J., and Koehler, B. (2015). Photochemical mineralisation in a humic boreal lake: temporal variability and contribution to carbon dioxide production. Biogeosci. Discuss. 12, 17125–17152. doi: 10.5194/bgd-12-17125-2015
Guillemette, F., McCallister, S. L., and del Giorgio, P. A. (2013). Differentiating the degradation dynamics of algal and terrestrial carbon within complex natural dissolved organic carbon in temperate lakes. J. Geophys. Res. Biogeosci. 118, 963–973. doi: 10.1002/jgrg.20077
Hawkes, J. A., Dittmar, T., Patriarca, C., Tranvik, L. J., and Bergquist, J. (2016a). Evaluation of the Orbitrap mass spectrometer for the molecular fingerprinting analysis of natural dissolved organic matter. Anal. Chem. 88, 7698–7704. doi: 10.1021/acs.analchem.6b01624
Hawkes, J. A., Hansen, C. T., Goldhammer, T., Bach, W., and Dittmar, T. (2016b). Molecular alteration of marine dissolved organic matter under experimental hydrothermal conditions. Geochim. Cosmochim. Acta 175, 68–85. doi: 10.1016/j.gca.2015.11.025
Helms, J. R., Mao, J., Stubbins, A., Schmidt-Rohr, K., Spencer, R. G. M., Hernes, P. J., et al. (2014). Loss of optical and molecular indicators of terrigenous dissolved organic matter during long-term photobleaching. Aquat. Sci. 76, 353–373. doi: 10.1007/s00027-014-0340-0
Helms, J. R., Stubbins, A., Ritchie, J. D., Minor, E. C., Kieber, D. J., and Mopper, K. (2008). Absorption spectral slopes and slope ratios as indicators of molecular weight, source, and photobleaching of chromophoric dissolved organic matter. Limnol. Oceanogr. 53, 955–969. doi: 10.4319/lo.2008.53.3.0955
Hertkorn, N., Ruecker, C., Meringer, M., Gugisch, R., Frommberger, M., Perdue, E. M., et al. (2007). High-precision frequency measurements: indispensable tools at the core of the molecular-level analysis of complex systems. Anal. Bioanal. Chem. 389, 1311–1327. doi: 10.1007/s00216-007-1577-4
Hessen, D., and Tranvik, L. (eds.). (1998). Aquatic Humic Substances: Ecology and Biogeochemistry. Berlin; Heidelberg: Springer.
Holmes, R. M., McClelland, J. W., Raymond, P. A., Frazer, B. B., Peterson, B. J., and Stieglitz, M. (2008). Lability of DOC transported by Alaskan rivers to the Arctic Ocean. Geophys. Res. Lett. 35, 3–7. doi: 10.1029/2007GL032837
Hopkinson, C. S., Buffam, I., Hobbie, J., Vallino, J., Perdue, M., Eversmeyer, B., et al. (1998). Terrestial imputs of organic matter to coastal ecosystems: an intercomparison of chemical characteristics and bioavailability. Biogeochemistry 43, 221–234. doi: 10.1023/A:1006016030299
Hopkinson, C. S., Vallino, J. J., and Nolin, A. (2002). Decomposition of dissolved organic matter from the continental margin. Deep Sea Res. II 49, 4461–4478. doi: 10.1016/S0967-0645(02)00125-X
Hunt, A. P., Parry, J. D., and Hamilton-Taylor, J. (2000). Further evidence of elemental composition as an indicator of the bioavailability of humic substances to bacteria. Limnol. Oceanogr. 45, 237–241. doi: 10.4319/lo.2000.45.1.02376
Jansson, M., Persson, L., De Roos, A. M., Jones, R. I., and Tranvik, L. J. (2007). Terrestrial carbon and intraspecific size-variation shape lake ecosystems. Trends Ecol. Evol. 22, 316–322. doi: 10.1016/j.tree.2007.02.015
Kamjunke, N., Tümpling, W., Von Hertkorn, N., Harir, M., Schmitt-kopplin, P., Norf, H., et al. (2017). A new approach for evaluating transformations of dissolved organic matter (DOM) via high-resolution mass spectrometry and relating it to bacterial activity. Water Res. 123, 513–523. doi: 10.1016/j.watres.2017.07.008
Kattner, G., Simon, M., and Koch, B. P. (2011). “Molecular characterization of dissolved organic matter and constraints for prokaryotic utilization,” in Microbial Carbon Pump in the Ocean, eds S. S. N. Jiao and F. Azam (Washington, DC: Science/AAAS), 60–61.
Kellerman, A. M. (2015). Molecular-Level Dissolved Organic Matter Dynamics in Lakes. Doctoral thesis, Uppsala University, Uppsala.
Kellerman, A. M., Kothawala, D. N., Dittmar, T., and Tranvik, L. J. (2015). Persistence of dissolved organic matter in lakes related to its molecular characteristics. Nat. Geosci. 8, 454–457. doi: 10.1038/ngeo2440
Kim, S., Kaplan, L. A., and Hatcher, P. G. (2006). Biodegradable dissolved organic matter in a temperate and a tropical stream determined from ultra-high resolution mass spectrometry. Limnol. Oceanogr. 51, 1054–1063. doi: 10.4319/lo.2006.51.2.1054
Kim, S., Kramer, R. W., and Hatcher, P. G. (2003). Graphical method for analysis of ultrahigh-resolution broadband mass spectra of natural organic matter, the van Krevelen diagram. Anal. Chem. 75, 5336–5344. doi: 10.1021/ac034415p
Kleber, M. (2010). What is recalcitrant soil organic matter? Environ. Chem. 7, 320–332. doi: 10.1071/EN10006
Koch, B. P., and Dittmar, T. (2006). From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun. Mass Spectrom. 20, 926–932. doi: 10.1002/rcm.2386
Koch, B. P., and Dittmar, T. (2016). Erratum: from mass to structure: an aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun. Mass Spectrom. 30, 250. doi: 10.1002/rcm.7433
Koehler, B., Landelius, T., Weyhenmeyer, G. A., Machida, N., and Tranvik, L. J. (2014). Sunlight-induced carbon dioxide emissions from inland waters. Glob. Biogeochem. Cylces 28, 696–711. doi: 10.1002/2014GB004850
Koehler, B., and Tranvik, L. J. (2015). Reactivity continuum modeling of leaf, root, and wood decomposition across biomes. J. Geophys. Res. Biogeosci. 120, 1196–1214. doi: 10.1002/2015JG002908
Koehler, B., Von Wachenfeldt, E., Kothawala, D., and Tranvik, L. J. (2012). Reactivity continuum of dissolved organic carbon decomposition in lake water. J. Geophys. Res. Biogeosci. 117, 1–15. doi: 10.1029/2011JG001793
Kuzyakov, Y. (2010). Priming effects: interactions between living and dead organic matter. Soil Biol. Biochem. 42, 1363–1371. doi: 10.1016/j.soilbio.2010.04.003
Lapierre, J.-F., Guillemette, F., Berggren, M., and Del Giorgio, P. A. (2013). Increases in terrestrially derived carbon stimulate organic carbon processing and CO2 emissions in boreal aquatic ecosystems. Nat. Commun. 4:2972. doi: 10.1038/ncomms3972
LaRowe, D. E., and Van Cappellen, P. (2011). Degradation of natural organic matter: a thermodynamic analysis. Geochim. Cosmochim. Acta 75, 2030–2042. doi: 10.1016/j.gca.2011.01.020
Lavonen, E. (2015). Tracking Changes in Dissolved Natural Organic Matter Composition: Evaluating Drinking Water Production using Optical Molecular Level Tools. Doctoral thesis, Swedish University of Agricultural Sciences, Uppsala.
Lechtenfeld, O. J., Kattner, G., Flerus, R., McCallister, S. L., Schmitt-Kopplin, P., and Koch, B. P. (2014). Molecular transformation and degradation of refractory dissolved organic matter in the Atlantic and Southern Ocean. Geochim. Cosmochim. Acta 126, 321–337. doi: 10.1016/j.gca.2013.11.009
Lindell, M. J., Granéli, W., and Tranvik, L. J. (1995). Enhanced bacterial growth in response to photochemical transformation of dissolved organic matter. Limnol. Oceanogr. 40, 195–199. doi: 10.4319/lo.1995.40.1.0195
Mangal, V., Stock, N. L., and Guéguen, C. (2016). Molecular characterization of phytoplankton dissolved organic matter (DOM) and sulfur components using high resolution Orbitrap mass spectrometry. Anal. Bioanal. Chem. 408, 1891–1900. doi: 10.1007/s00216-015-9295-9
Marshall, A. G. (2000). Milestones in Fourier transform ion cyclotron resonance mass spectrometry technique development. Int. J. Mass Spectrom. 200, 331–356. doi: 10.1016/S1387-3806(00)00324-9
Meyer, J. L. (1986). Dissolved organic carbon dynamics in two subtropical blackwater rivers. Arch. für Hydrobiol. 108, 119–134.
Moran, M. A., and Hodson, R. E. (1994). Dissolved humic substances of vascular plant origin in a coastal marine environment. Limnol. Oceanogr. 39, 762–771. doi: 10.4319/lo.1994.39.4.0762
Moran, M. A., Sheldon, W. M., and Zepp, R. G. (2000). Carbon loss and optical property changes during long-term photochemical and biological degradation of estuarine dissolved organic matter. Limnol. Oceanogr. 45, 1254–1264. doi: 10.4319/lo.2000.45.6.1254
Moran, M. A., and Zepp, R. G. (1997). Role of photoreactions in the formation of biologically labile compounds from dissolved organic matter. Limnol. Oceanogr. 42, 1307–1316. doi: 10.4319/lo.1997.42.6.1307
Mostovaya, A., Hawkes, J. A., Koehler, B., Dittmar, T., and Tranvik, L. J. (2017). The emergence of the reactivity continuum of organic matter from kinetics of a multitude of individual molecular constituents. Environ. Sci. Technol. 51, 11571–11579. doi: 10.1021/acs.est.7b02876
Mostovaya, A., Koehler, B., Guillemette, F., Brunberg, A.-K., and Tranvik, L. J. (2016). Effects of compositional changes on reactivity continuum and decomposition kinetics of lake dissolved organic matter. J. Geophys. Res. Biogeosci. 121, 1733–1746. doi: 10.1002/2016JG003359
Olson, J. S. (1963). Energy storage and the balance of producers and decomposers in ecological systems. Ecology 44, 322–331. doi: 10.2307/1932179
Perdue, E. M. (2009). “Natural organic matter,” in Encyclopedia of Inland Waters, ed G. Likens (Oxford: Academic Press), 806–819.
Piccolo, A. (2001). The supramolecular structure of humic substances. Soil Sci. 166, 810–832. doi: 10.1097/00010694-200111000-00007
R Development Core team, (2016). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org/
Raeke, J., Lechtenfeld, O. J., Wagner, M., Herzsprung, P., and Reemtsma, T. (2016). Selectivity of solid phase extraction of freshwater dissolved organic matter and its effect on ultrahigh resolution mass spectra. Environ. Sci. Process. Impacts 18, 918–927. doi: 10.1039/C6EM00200E
Raymond, P. A., Hartmann, J., Lauerwald, R., Sobek, S., McDonald, C., Hoover, M., et al. (2013). Global carbon dioxide emissions from inland waters. Nature 503, 355–359. doi: 10.1038/nature12760
Riedel, T., Biester, H., and Dittmar, T. (2012). Molecular fractionation of dissolved organic matter with metal salts. Environ. Sci. Technol. 46, 4419–4426. doi: 10.1021/es203901u
Riedel, T., Zark, M., Vähätalo, A. V., Niggemann, J., Spencer, R. G. M., Hernes, P. J., et al. (2016). Molecular signatures of biogeochemical transformations in dissolved organic matter from ten world rivers. Front. Earth Sci. 4:85. doi: 10.3389/feart.2016.00085
Schmidt, M. W. I., Torn, M. S., Abiven, S., Dittmar, T., Guggenberger, G., Janssens, I. A., et al. (2011). Persistence of soil organic matter as an ecosystem property. Nature 478, 49–56. doi: 10.1038/nature10386
Seidel, M., Yager, P. L., Ward, N. D., Carpenter, E. J., Gomes, H. R., Krusche, A. V., et al. (2015). Molecular-level changes of dissolved organic matter along the Amazon River-to-ocean continuum. Mar. Chem. 177, 218–231. doi: 10.1016/j.marchem.2015.06.019
Seitzinger, S. P., Hartnett, H., Lauck, R., Mazurek, M., Minegishi, T., Spyres, G., et al. (2005). Molecular-level chemical characterization and bioavailability of dissolved organic matter in stream water using electrospray-ionization mass spectrometry. Limnol. Oceanogr. 50, 1–12. doi: 10.4319/lo.2005.50.1.0001
Sierra, C. A., Harmon, M. E., and Perakis, S. S. (2011). Decomposition of heterogeneous organic matter and its long-term stabilization in soils. Ecol. Monogr. 81, 619–634. doi: 10.1890/11-0811.1
Sleighter, R. L., Cory, R. M., Kaplan, L. A., Abdulla, H. A. N., and Hatcher, P. G. (2014). A coupled geochemical and biogeochemical approach to characterize the bioreactivity of dissolved organic matter from a headwater stream. J. Geophys. Res. Biogeosci. 119, 1520–1537. doi: 10.1002/2013JG002600
Sobek, S., Algesten, G., Bergström, A. K., Jansson, M., and Tranvik, L. J. (2003). The catchment and climate regulation of pCO2 in boreal lakes. Glob. Chang. Biol. 9, 630–641. doi: 10.1046/j.1365-2486.2003.00619.x
Spencer, R. G. M., Aiken, G. R., Wickland, K. P., Striegl, R. G., and Hernes, P. J. (2008). Seasonal and spatial variability in dissolved organic matter quantity and composition from the Yukon River basin, Alaska. Glob. Biogeochem. Cycles 22, 1–13. doi: 10.1029/2008GB003231
Stubbins, A., Spencer, R. G. M., Chen, H., Hatcher, P. G., Mopper, K., Hernes, P. J., et al. (2010). Illuminated darkness: molecular signatures of Congo River dissolved organic matter and its photochemical alteration as revealed by ultrahigh precision mass spectrometry. Limnol. Oceanogr. 55, 1467–1477. doi: 10.4319/lo.2010.55.4.1467
Sun, L., Perdue, E. M., Meyer, J. L., and Weis, J. (1997). Use of elemental composition to predict bioavailability of dissolved organic matter in a Georgia river. Limnol. Oceanogr. 42, 714–721. doi: 10.4319/lo.1997.42.4.0714
Šantl-Temkiv, T., Finster, K., Dittmar, T., Hansen, B. M., Thyrhaug, R., Nielsen, N. W., et al. (2013). Hailstones: a window into the microbial and chemical inventory of a storm cloud. PLoS ONE 8:e53550. doi: 10.1371/journal.pone.0053550
Thurman, E. M. (1985). Developments in Biogeochemistry: Organic Geochemistry of Natural Waters. Dordrecht: Martinus Nijhoff/Dr. W. Junk Publishers.
Tranvik, L. J. (1990). Bacterioplankton growth on fractions of dissolved organic carbon of different molecular weights from humic and clear waters. Appl. Environ. Microbiol. 56, 1672–1677.
Tranvik, L. J. (1992). Allochthonous dissolved organic matter as an energy source for pelagic bacteria and the concept of the microbial loop. Hydrobiologia 229, 107–114. doi: 10.1007/BF00006994
Tranvik, L. J. (1998). “Degradation of dissolved organic matter in humic waters by bacteria,” in Aquatic Humic Substances, eds D. Hessen and L. Tranvik (Berlin; Heidelberg: Springer), 259–283.
Tranvik, L. J., Downing, J. A., Cotner, J. B., Loiselle, S. A., Striegl, R. G., Ballatore, T. J., et al. (2009). Lakes and reservoirs as regulators of carbon cycling and climate. Limnol. Oceanogr. 54, 2298–2314. doi: 10.4319/lo.2009.54.6_part_2.2298
Vähätalo, A. V., Aarnos, H., and Mäntyniemi, S. (2010). Biodegradability continuum and biodegradation kinetics of natural organic matter described by the beta distribution. Biogeochemistry 100, 227–240. doi: 10.1007/s10533-010-9419-4
Verpoorter, C., Kutser, T., Seekell, D. A., and Tranvik, L. J. (2014). A global inventory of lakes based on high-resolution satellite imagery. Geophys. Res. Lett. 41, 6396–6402. doi: 10.1002/2014GL060641
von Wachenfeldt, E., Bastviken, D., and Tranvik, L. J. (2009). Microbially induced flocculation of allochthonous dissolved organic carbon in lakes. Limnol. Oceanogr. 54, 1811–1818. doi: 10.4319/lo.2009.54.5.1811
von Wachenfeldt, E., and Tranvik, L. J. (2008). Sedimentation in boreal lakes—the role of flocculation of allochthonous dissolved organic matter in the water column. Ecosystems 11, 803–814. doi: 10.1007/s10021-008-9162-z
Ward, N. D., Keil, R. G., Medeiros, P. M., Brito, D. C., Cunha, A. C., Dittmar, T., et al. (2013). Degradation of terrestrially derived macromolecules in the Amazon River. Nat. Geosci. 6, 530–533. doi: 10.1038/ngeo1817
Wetzel, R. G., Hatcher, P. G., and Bianchi, T. S. (1995). Natural photolysis by ultraviolet irradiance of recalcitrant dissolved organic matter to simple substrates for rapid bacterial metabolism. Limnol. Oceanogr. 40, 1369–1380. doi: 10.4319/lo.1995.40.8.1369
Weyhenmeyer, G. A., Fröberg, M., Karltun, E., Khalili, M., Kothawala, D., Temnerud, J., et al. (2012). Selective decay of terrestrial organic carbon during transport from land to sea. Glob. Chang. Biol. 18, 349–355. doi: 10.1111/j.1365-2486.2011.02544.x
Weyhenmeyer, G. A., and Karlsson, J. (2009). Nonlinear response of dissolved organic carbon concentrations in boreal lakes to increasing temperatures. Limnol. Oceanogr. 54, 2513–2519. doi: 10.4319/lo.2009.54.6_part_2.2513
Williams, P. J. L. B. (1999). “Heterotrophic bacteria and the dynamics of dissolved organic material,” in Microbial Ecology of the Oceans, 1st Edn., ed D. J. Kirchman (New York, NY: Wiley-Liss), 153–199.
Yamashita, M., and Fenn, J. B. (1984). Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 88, 4451–4459. doi: 10.1021/j150664a002
Keywords: dissolved organic matter, reactivity, ultrahigh resolution mass spectrometry, decomposition kinetics, phototransformations, saturation, nominal oxidation state of carbon, supramolecular complexes
Citation: Mostovaya A, Hawkes JA, Dittmar T and Tranvik LJ (2017) Molecular Determinants of Dissolved Organic Matter Reactivity in Lake Water. Front. Earth Sci. 5:106. doi: 10.3389/feart.2017.00106
Received: 12 June 2017; Accepted: 30 November 2017;
Published: 18 December 2017.
Edited by:
Sandra Arndt, University of Bristol, United KingdomReviewed by:
Norbert Kamjunke, Helmholtz-Zentrum für Umweltforschung (UFZ), GermanyCopyright © 2017 Mostovaya, Hawkes, Dittmar and Tranvik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alina Mostovaya, YWxpbmEubW9zdG92YXlhQGViYy51dS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.