 Roberto Cioeta
Roberto Cioeta Andrea Cossu1
Andrea Cossu1 Emiliano Giovagnoni
Emiliano Giovagnoni Marta Rigoni
Marta Rigoni
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PROTOCOLS article
Front. Drug Saf. Regul. , 09 December 2022
Sec. Substance-Based Medical Devices
Volume 2 - 2022 | https://doi.org/10.3389/fdsfr.2022.992359
This article is part of the Research Topic Natural product research at the crossroad: experimental and clinical approaches in light of the new Regulation on Medical devices (Reg 2017/745/CE), which specifically addresses medical devices made of substances View all 4 articles
The recent passage of European Union (EU) Regulation 2017/745 on medical devices (MDs) has improved the classification of MDs and revised their approval process and the post-marketing evaluation of their safety and effectiveness, promoting transparency and post-marketing oversight in Europe. This new regulation can better ensure patient safety and provide new opportunities for therapeutic innovation. In addition, the new regulations include and define MDs made of substances or of combinations of substances (substance-based MDs: SBMDs). The impressive growth of the MD, including SBMDs, that have been marketed over the recent years has likely been a relevant factor fueling this change. MD regulation requires a major effort from both industry and regulatory bodies to comply with its provisions. Manufacturers should produce sufficient clinical evidence that an MD, under normal conditions of use, provides adequate performance and that the foreseeable risks and frequency of adverse events (AEs) have an acceptable minimum rate, taking into account the benefits provided. We describe how we implemented the post-marketing surveillance (PMS) system of SBMDs to properly deal with post-market monitoring and confirmation of safety and performance, including AEs and the benefit–risk evaluation, as required by the 2017/745 Regulation. The two pillars of this novel system are: 1) passive vigilance, i.e., spontaneous reporting and 2) active post-marketing clinical follow-up (PMCF) activities, which systematically gather, record, and analyze real-world data (RWD) on performance, quality, function, use, tolerability, and safety of an MD, collected through a dedicated, structured web platform. Active PMCF is achieved through a process of generation, validation, and administration of digital questionnaires to all stakeholders, i.e., patients, physicians, both general practitioners and specialists, and pharmacists. The technology, potential use, advantages, and limitations of this large source of RWD are also discussed.
Substance-based medical devices (SBMDs) are an important asset in the European Union (EU) health system. For example, in Italy, for some conditions, such as gastrointestinal functional disorders, SBMD use is almost equal to that of over-the-counter (OTC) medicinal products, and the share of MDs from 2011 to 2022 grew from 9% to 48% in sales value (IQVIA, 2022).
In this context, the recent EU 2017/745 Regulation on MDs (MDR) has substantially increased the obligations of manufacturers to a level almost comparable to that needed for drugs (European Parliament, 2017). Along with improving the classification of MDs and promoting transparency and post-marketing oversight in the EU, the new regulations have reformed the MD approval and post-marketing evaluation, establishing the core relevance of clinical data for demonstrating or confirming MD conformity in relevant general safety and performance requirements (GSPRs).
Clinical data for SBMDs can be generated in several ways. In class III and implantable MDs, clinical investigations should be performed with exceptions envisaged for legacy devices. In the post-market setting, several activities of post-market surveillance (PMS) can be performed to obtain clinical data. PMS includes passive vigilance, based on spontaneous reporting, and post-market clinical follow-up (PMCF) activities (European Parliament, 2017), which can include active real-world (RW) research, such as surveys. Importantly, as per Regulation 2017/745, in the post-market setting, confirmation of GSPRs’ conformity should follow a defined procedure and be updated throughout the entire lifecycle of the MD. In addition, the level of clinical evidence should be appropriate relative to the characteristics of the device and its intended use.
The latter are key points of the new regulation: altogether, they appear to establish the need for manufacturers to implement their own PMS by including PMCF activities, to be planned and carried out to make proactively collected data on the use of a marketed MD available, thus allowing the benefit–risk profile and the acceptability of the identified and possible emerging risks to be continuously confirmed.
In this novel regulatory context, we have developed a dedicated, structured web platform that can actively and systematically gather, record, and analyze relevant RW data (RWD) on the performance, safety, quality, function, and appropriate use of marketed SBMDs, throughout their entire lifetime. RWD for specific SBMDs are continuously obtained through digital surveys of health care professionals (HCPs) and patients. Such a PMCF output, together with passive vigilance, contribute to identify, implement, and monitor any preventive and corrective action when needed. Thus, vigilance became part of an integrated, multidisciplinary, multimedia PMS system, able to provide RWD from relevant populations in a direct and user-friendly way.
The aim of this paper is to describe both the active and passive components of this novel PMS system, as implemented, including the technology used and steps taken to comply with the data protection regulation.
The online RWD platform hosts digital questionnaires specifically designed for patients, physicians, and pharmacists.
This platform (Figure 1) is designed to actively collect digital survey data through a set of procedures able to ensure the individual subject’s protection, data integrity, and reliability. This architecture is very safe, as all the logics and data are stored in the server. The platform is based on a web application structure consisting of the following components: 1) an encrypted database to store data; 2) a web framework page interface; 3) a set of forms to let users’ registration (manufacturer side); and 4) a set of forms to let users choose the product, record that they have bought it, and collect data.
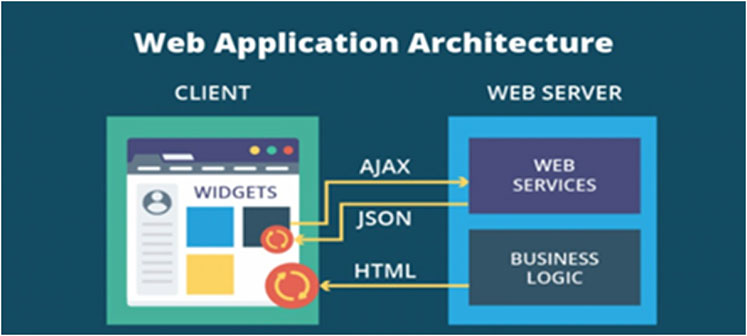
Figure 1. Real-world data platform technology, as implemented. According to a basic web architecture, a server, consisting of web services and business logic (the application), interacts with a client by sending out a HTML page. Each page on the client has separate entities called widgets: elements of interaction in the graphical user interface. By sending AJAX queries to web services, widgets, from the client, can exchange data in the HTML or JSON format. HTML: HyperText Markup Language. AJAX: Asynchronous JavaScript and XML. JSON: JavaScript Object Notation.
The technology provider (Arithmos Srl, Verona-Italy) is ISO27001 certified for all processes and projects. This structure of data collection allows information to be obtained (pseudonymized personal data, separated both logically and physically) from responders using a single sign-on (SSO) system, as well as providing an editor and an export section allowing the administrator to create or manage questionnaires and to extract data (pseudonymized personal data) (Figure 2). The system can manage multiple questionnaire areas depending on the language and country of the users via the SSO.
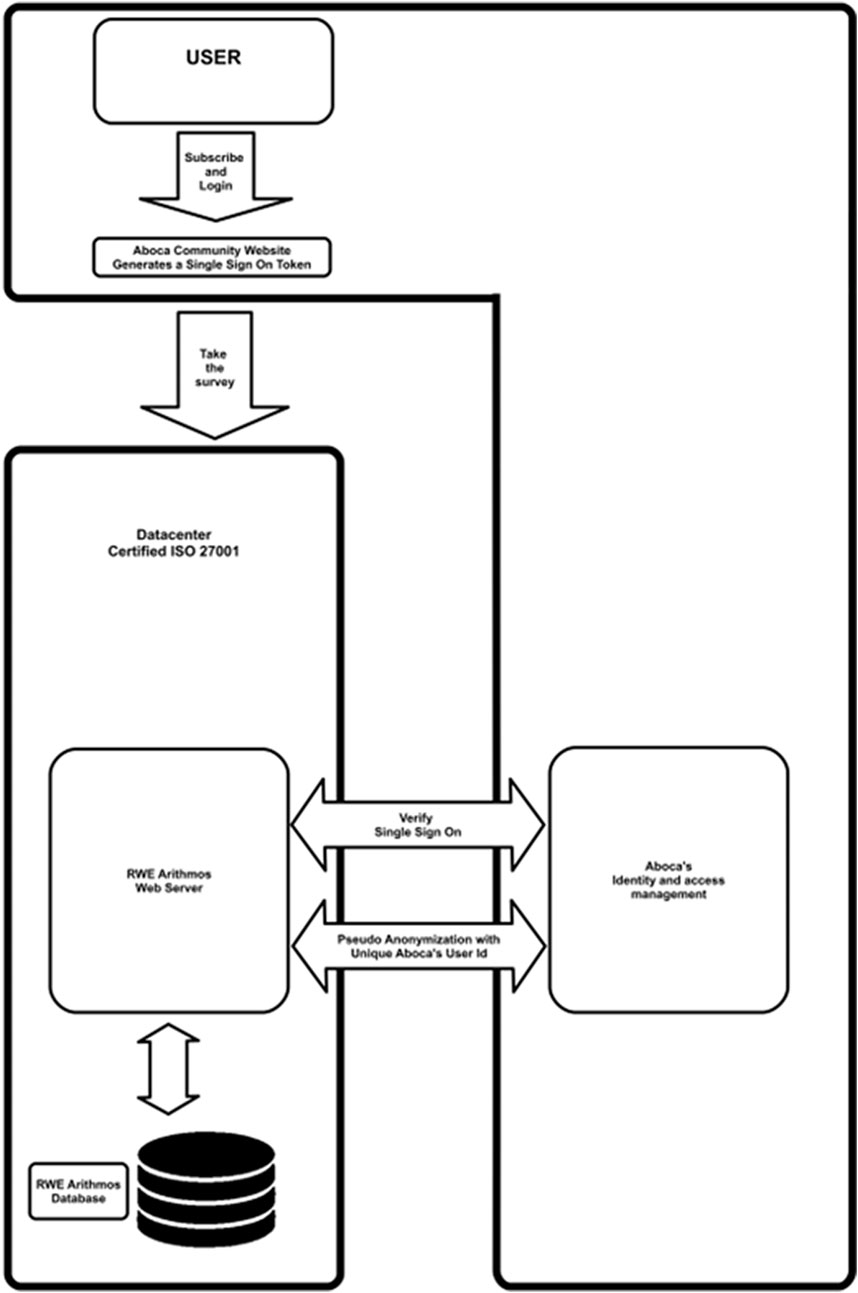
Figure 2. Information flow diagram. Users (consumers, doctors, and pharmacists) will register themselves to the manufacturer’s system by using a unique email. Everyone will access the web framework page interface via the single sign-on (SSO) security system suitably configured. According to EU General Data Protection Regulation, personal data are processed in a lawful, correct, and transparent manner toward the interested party and limited to the purpose of the project.
Through specific questions/fields (e.g., product identifier and proof of purchase), patients are redirected to surveys for the specific product, and HPs to surveys in which aggregate information is collected. In accordance with the EU General Data Protection Regulation (GDPR), data collected via a web framework system are limited to those necessary (name, surname, and email) to complete registration and fill the questionnaires. Before responding to the survey, each customer must sign up into the manufacturer community website (Figure 2) by using a unique email. Access to the web page will be granted via the SSO. Due to the non-anonymous nature of data, starting from 2018, privacy informative is always provided in compliance with current EU Regulations. GDPR requirements for secure data storage of personal information are accomplished by the following steps. A standard authentication approach (username and password) by asp.net is applied for identity verification. For data pseudonymization, the manufacturer sends a user identification (ID) code to the system that is a unique string of 37 alphanumeric characters. The system cannot use this code to access personal registration data (name, surname, or email) that are stored in a separate database. An SSO (based on AES 256 bit) system allows users registered on the manufacturer site to reach the platform in a pseudo-anonymous way (i.e., without the need to re-enter username and password), whereas the ID code will permit to re-map in anytime which ID has inserted questionnaire, if needed (e.g., for legal purposes). A cryptographic mechanism (HTTPS) has been implemented to guarantee the integrity of transmitted data through the web, whereas the database has been encrypted by MS SQL’s standard encryption to limit the potential for unauthorized access to data. Within the Arithmos’s ISO 27001:2013 certified information security management system and manufacturer organization, procedures, and technologies of access control and audit log are in place, as well as procedures and technologies of incident and data breach management, business continuity, and disaster recovery.
The patients can access the platform either using a website link or the QR code, which are both reported on the package of the product. In May 2022, the platform was made available in Germany. Purchase of the product purchase is verified through a batch number and a unique code given on the package, to be entered before the online questionnaire begins.
Reproducibility studies were first performed to measure the precision of the surveys and as indirect measure of questionnaire validity (Lundell et al., 2022). This preliminary assessment was performed for four products; however, the questions were designed in the same way for each cohort (patients, physicians, and pharmacists). Thus, it was considered appropriate to infer that the repeatability results could be extended to each product-specific survey. To assess questionnaire reproducibility, 72 patients, 43 physicians, and 68 pharmacists completed the same questionnaire twice over 20 days. The statistical analysis showed excellent repeatability of the answers in each cohort, with coefficient of agreements, expressed as intraclass correlation coefficient (ICC) of 0.95 (95% confidential interval-CI 0.85–0.96) in 72 patients, of 0.89 (95% CI 0.81–0.94) in 68 pharmacists, and of 0.92 (95% CI 0.84–0.95) in 43 physicians.
Each questionnaire usually consists of approximately 20–25 questions. The questionnaires, for each cohort of participants (patients, physicians, and pharmacists) are developed by clinical experts, in collaboration with the Department of Biomedical, Surgical and Dental Sciences of the University of Milan. All questionnaires focus on the following items: 1) Effectiveness of the SBMD in controlling specific diseases or symptoms; 2) satisfaction level associated with its use; 3) compliance with the suggested dosage and adherence to the marketed indication; 4) quality of life while on treatment; 5) safety and tolerability of the product; 6) identification of unforeseen side-effects or interactions; 7) monitoring known side-effects; 8) identification of possible systematic misuse or off-label use of the MD; and 9) clarity of the information reported on the leaflet (indication, posology and route of administration, and warnings).
Patients are required to confirm that he/she has had experience with the product in the previous year.
Physicians and pharmacists can access the platform through an area specific for HCPs on the manufacturer website. The questions and answer options provided for HCPs are similar to those of patient’s surveys, including, for instance, perceived effectiveness on specific symptoms, pattern of usage, and AEs. Thus, the most relevant effectiveness and safety data are provided by both patients and HCPs.
Questionnaire data are made available for analysis only if all questions are answered.
Almost all questions are given as closed multiple choice questions (MCQ), sometimes providing five options to a statement/question allowing measuring variations in frequency, quality, or magnitude of effects through a five-point Likert verbal scale. Most MCQs require a single answer, and some MCQs allow more than one answer, as specified. Open notes for physicians and pharmacists are provided to collect further detailed information on diseases, symptoms, and treatment regimens. The participants are also requested to report suspected side effects or potential interactions with concomitant treatments, providing potential safety issues directly to the manufacturer’s Vigilance Department at a specific mailbox.
All of the RWD questionnaires obtained for each MD are analyzed by the Department of Biomedical, Surgical and Dental Sciences at the University of Milan, which issues a periodic scientific report. As of June 2022, two complete reports are available for a SBMD, namely, for data obtained over the first 6 and 10 months from the beginning of data collection (Cioeta et al., 2022).
Following the platform’s activation in January 2021, more than 35,000 questionnaires have been completed for the post-market clinical follow-up for 30 different MDs. A table reporting the list of products and the relative number of completed questionnaires obtained so far for each cohort of participants is available in the Supplementary Material S1.
HCP surveys are a central to PMCF activity, allowing information on product performance and safety based on professionals’ experience. Hence, participation in the surveys of this important cohort also relies on the direct recruitment of physicians, performed through manufacturers’ scientific informants, in addition to access to the web platform on their own initiative.
An example of this approach and of the PMS system has been recently published (Cioeta et al., 2022). Beginning in 2017, scientific informants completed nine HCP surveys on six different SBMDs, regarding adult and pediatric formulations in two therapeutic areas: gastrointestinal and respiratory system. The HCP surveys involved a range of hospitals and HCP specializations, such as gastroenterologists, general practitioners, pediatricians, hospital doctors, and medical dieticians, located in Italy and Spain. More than 10,000 completed questionnaires have been acquired and nine clinical reports have been issued.
MD vigilance is part of the PMS system, which collects and analyzes safety information on any AE or special situation (e.g., off-label use, misuse, overdose, pregnancy exposure, etc.) from all potential sources (e.g., health authorities, healthcare professionals, patients, clinical trials, literature, digital media, and so on). The Global Vigilance Department (GVD) collects data related to spontaneous reports by any potential reporter (e.g., HCPs, patients, and health authorities) through various channels, such as company contact details (e.g., institutional website, phone, fax, address, and certified email), company-sponsored digital media, employees, sales agents, scientific representatives, and the local affiliates’ person responsible for vigilance. The data are collected accordingly with internal procedures and the personnel potentially involved in receiving and forwarding any spontaneous report are trained on how reporting the safety data to the GVD. The GVD performs follow-up to collect any available data on the case from the reporter. When business partners are involved (e.g., distributors, importers, and authorized representatives) or a sponsored clinical trial is performed, the process of collection and exchange of safety data is detailed in specific contractual agreements between parties.
Finally, a systematic literature search process is performed to capture any AE on the MDs from published case reports and non-sponsored studies.
Relevant and appropriate data are registered in an ad hoc Oracle Argus safety database, one of the most used pharmacovigilance databases worldwide (Gill et al., 2016). The vigilance system of the SBMDs is similar to a traditional pharmacovigilance system, as the SBMDs and the medicinal products share a therapeutic intended use, pharmaceutical formulations (e.g., tablets, capsules, syrup, drops, spray, micro enema, etc.) and route of administration (oral, topical, rectal, etc.).
The safety database allows each reported case to be registered and evaluated from a clinical and quality standpoint directly in a high-quality standard system, which extracts reliable data that are useful for drawing up periodic aggregate safety reports, such as PMS reports (PMSRs) and periodic safety update reports (PSURs) (art. 85 and 86 Reg. 2017/745), performing statistical analyses for identifying and reporting any potential trend (art. 88 Reg. 2017/745) and generating standard forms (e.g., MIR forms) to submit to the competent authorities any reportable incident (art. 87 Reg. 2017/745).
The 2017/745 MDR has led to a completely new governance in all aspects of the lifecycle of an MD. This profound change was likely prompted by studies on MD approval and post-market evaluation in the EU (Kramer et al., 2012), together with the impressive growth of the MD market and patient use (Giovagnoni, 2022). The new regulation also acknowledges and directly addresses a highly relevant field, i.e., “devices that are composed of substances or of combinations of substances.” Therefore, the new MDR provides framework that, at the same time, guarantees patient safety as well as providing opportunities for innovation and improvement.
In this novel regulatory context, we developed and initiated a new PMS system for properly addressing the post-marketing monitoring and assessment in “conformity with relevant general safety and performance requirements (GSPRs) […] and the evaluation of the undesirable side-effects and of the acceptability of the benefit–risk ratio,” as required by the 2017/745 MDR art. 61 (1), throughout the product lifespan.
A multimodal cross-sectional RWD collection system has been implemented to obtain real-world evidence on safety, tolerability, effectiveness, and patterns of use among physicians, both GPs and specialists, pharmacists, and patients in a continuous flow of updating the benefit–risk evaluation of marketed MDs.
From a regulatory standpoint, the implementation of the PMS system obtained by associating passive vigilance activity with a proactive data collection appears central to compliance with MDR provisions on the centrality of the evaluation, using clinical data in confirming MD conformity with relevant GSPRs. In fact, information on safety and performance from the use of an MD deriving from PMS, in particular PMCF activities, falls within the definition of clinical data under 2017/745 MDR article 2(48). Furthermore, the Medical Device Coordination Group (MDCG) guide document on Regulation 2017/745 regarding clinical evidence needed in post-marketing setting clearly includes High-quality surveys among data sources that can confirm GSPR conformity (MDCG, 2020).
The same document indicates that the hierarchy of clinical evidence, as reported inside, should be understood as a rough indication, as variations may be seen depending on factors such as the specific device’s intended use and source data quality. Thus, according to Regulation 2017/745, MDCG documents underline the necessity of a case-by-case evaluation as to whether the level of evidence is adequate to confirm safety, performance, and the benefit-to-risk profile of a MD. Therefore, while, on the one hand, there is a clear intent to allow a case-by-case evaluation by notified bodies, and the determination of the required level of clinical evidence is left to the manufacturer, on the other hand, RWD, in principle, is indicated as a suitable means of providing sufficient clinical evidence on marketed MDs.
The literature highlighting the strengths of RWD (Concato et al., 2020; Franklin et al., 2021; Concato and Corrigan-Curay, 2022) has been growing in recent decades.
Risk evaluation is of particular relevance for RWD, due to its unique ability to capture all kinds of AEs over a large sample size, including rare AEs. To better identify emergent risks, our RWD platform has been structured to obtain indications by all physicians, both GPs and specialists, in addition to patients and pharmacists. Thus, survey data can provide signals or concerns relative to the tolerability and safety of the SBMDs, prompting further investigation, if necessary.
The analysis of RWD across different cohorts is of great value for the continuous evaluation of the benefit–risk profile of marketed SBMD, especially considering that they are easily accessible over the counter, and in many instances they are used as self-medication.
In fact, moving from the experimental to the real-life context, it is possible to evaluate the benefit–risk ratio of a treatment on larger and less selected populations, which, for instance, may include significant proportions of subjects of extreme ages who have co-morbidities. Furthermore, adequately sized subgroups may be reachable to assess whether the efficacy, tolerability, and safety of a treatment are comparable across different groups of patients (e.g., in children and adults or in pregnant and menopausal women) or, vice versa, to identify subjects who have the highest probability of benefiting from the treatment or those at most risk of side effects.
Consistent with this approach, a first large survey of over 3,000 participants proved the feasibility and the relevance of data from the use of a MD collected through this PMS system (Cioeta et al., 2022).
Some limitations of this system should be acknowledged: as in all web-based systems, one main limitation is that only subjects familiar with the web or having web access and willing to respond will complete the questionnaires. Thus, extremes of ages may be under-represented.
Because the participation in the survey was voluntary, it was not possible to determine the percentage of responders versus those who used the product, so a selection bias is plausible. However, access to large cohorts may counterbalance some of the aforementioned limitations. In conclusion, new MD regulations provide opportunities and challenges for the future research and development of SBMDs, ensuring a higher degree of safety and a continuous monitoring of effectiveness and performance over time. The model that we designed and implemented appears to fulfill the regulatory goals by virtue of a novel, active methodology, in addition to the old PMS procedures. Future reports, and the implementation of this methodology, will allow potential aspects of weakness and improvements to be identified.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
EG: conceptualization, supervision, and review and editing. PM and MR: formal analysis, data curation, and writing—original draft preparation. RC and AC: collaboration in draft preparation, review and editing, and research project management.
This project was funded by Aboca S. p. A that conceived and conducted this research project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fdsfr.2022.992359/full#supplementary-material
Cioeta, C., Muti, P., Rigoni, M., Morlando, L., Siragusa, F., Cossu, A., et al. (2022). Effectiveness and tolerability of poliprotect, a natural mucosal protective agent for gastroesophageal reflux disease and dyspepsia: Surveys from patients, physicians, and pharmacists. Front. Drug Saf. Regul. 2. 969831. doi:10.3389/fdsfr.2022.969831
Concato, J., and Corrigan-Curay, J. (2022). Real-world evidence - where are we now? N. Engl. J. Med. 386 (18), 1680–1682. doi:10.1056/NEJMp2200089
Concato, J., Stein, P., Dal Pan, G. J., Ball, R., and Corrigan-Curay, J. (2020). Randomized, observational, interventional, and real-world-What's in a name? Pharmacoepidemiol. Drug Saf. 29 (11), 1514–1517. doi:10.1002/pds.5123
European Parliament (2017). Regulation (EU) 2017/745 of the European parliament and of the council of 5 april 2017 on medical devices, amending directive 2001/83/EC, regulation (EC) No 178/2002 and regulation (EC) No 1223/2009 and repealing council directives 90/385/EEC and 93/42/EEC. Online: European Union. Available at: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:32017R0745 (Accessed date april, 2022).
Franklin, J. M., Liaw, K. L., Iyasu, S., Critchlow, C. W., and Dreyer, N. A. (2021). Real-world evidence to support regulatory decision making: New or expanded medical product indications. Pharmacoepidemiol. Drug Saf. 30 (6), 685–693. doi:10.1002/pds.5222
Gill, S. K., Christopher, A. F., Gupta, V., and Bansal, P. (2016). Emerging role of bioinformatics tools and software in evolution of clinical research. Perspect. Clin. Res. 7 (3), 115–122. doi:10.4103/2229-3485.184782
Giovagnoni, E. (2022). Substance-based medical devices made of natural substances: An opportunity for therapeutic innovation. Front. Drug Saf. Regul. 2, 998114. doi:10.3389/fdsfr.2022.998114
IQVIA (2022). Multichannel Italy, OTC/SOP and MDs – sell out data mat sept 2022. Durham, North Carolina, United States: IQVIA.
Kramer, D. B., Xu, S., and Kesselheim, A. S. (2012). How does medical device regulation perform in the United States and the European Union? A systematic review. PLoS Med. 9 (7), e1001276. doi:10.1371/journal.pmed.1001276
Lundell, S., Toots, A., Sönnerfors, P., Halvarsson, A., and Wadell, K. (2022). Participatory methods in a digital setting: Experiences from the co-creation of an eHealth tool for people with chronic obstructive pulmonary disease. BMC Med. Inf. Decis. Mak. 22 (1), 68. doi:10.1186/s12911-022-01806-9
MDCG (2020). Regulation (EU) 2017/745: Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC. Online: Medical Device Coordination Group Available at: https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2020_6_guidance_sufficient_clinical_evidence_en_0.pdf (Accessed date april, 2022).
Keywords: EU Regulation 2017/745, substance-based medical devices, post-market surveillance, benefit–risk ratio, real-world data, online platform
Citation: Cioeta R, Cossu A, Giovagnoni E, Rigoni M and Muti P (2022) A new platform for post-marketing surveillance and real-world evidence data collection for substance-based medical devices. Front. Drug Saf. Regul. 2:992359. doi: 10.3389/fdsfr.2022.992359
Received: 12 July 2022; Accepted: 08 November 2022;
Published: 09 December 2022.
Edited by:
Elisabetta Bigagli, University of Florence, ItalyReviewed by:
Laura Hester, Janssen Research and Development, United StatesCopyright © 2022 Cioeta, Cossu, Giovagnoni, Rigoni and Muti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto Cioeta, cmNpb2V0YUBhYm9jYS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.