 Maria Grazia Leone
Maria Grazia Leone- Directorate General of Medical Devices and Pharmaceutical Services, Ministry of Health, Rome, Italy
Regulation (EU) 2017/745 provides for new provisions for medical devices made of substances as well as specific classification rules and requirements. The demarcation line between medical devices composed of substances and medicinal products is not always easy to define. The recent publication by the European Commission of the guidance on borderline between medical devices and medicinal products under the Regulation (EU) 2017/745 on medical devices, MDCG 2022-5, addresses some important issues in this topic. This article will discuss some controversial aspects in this field in order to clarify the product qualification process.
Introduction
Regulation (EU) 2017/745 fully applicable since May 2021, has introduced several novelties in the medical device sector; among these medical devices (MD) composed of substances deserve a special mention. The definition of MD reported in the Regulation (EU) 2017/745 (MDR), states that the use for which medicinal products (MP) and MD are intended are very similar, the only difference is given by the mechanism of action being pharmacological, immunological or metabolic for MP (Table 1). MDs composed of substances have a medical purpose and are available in a presentation similar to MPs, e.g., eye drops, creams, syrups, capsules, nevertheless they do not fall within the definition of MP as the mechanism of action accounting for the principal intended action is non-pharmacological, -immunological or -metabolic, but physical (e.g., mechanical action, physical barrier, lubrication, etc.).
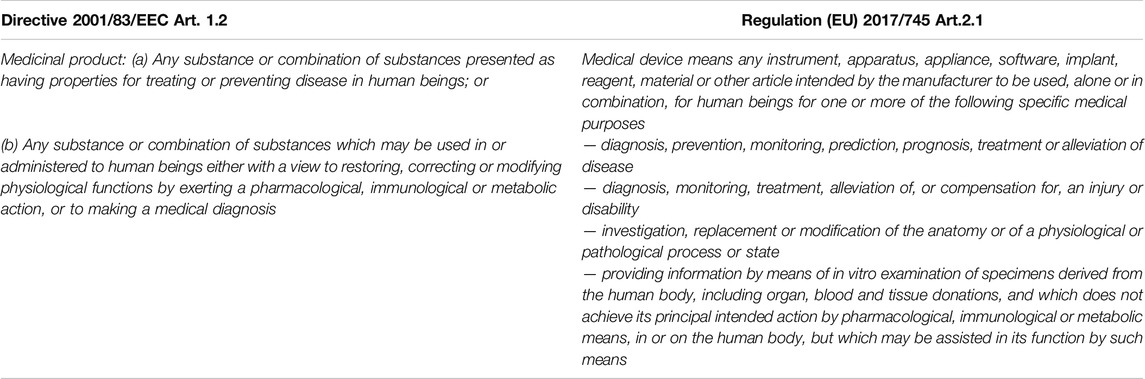
Table 1. Definition of MP and MD.
In this context the qualification of a product is not always immediate, so that at the European level long discussions occur on the so-called borderline products, which by their nature do not clearly belong to a specific sector and for whom it is therefore difficult to define to which regulatory framework they apply.
It is important to underline that the topics covered in this article will have great relevance for the future of substance-based MDs.
Evolution of MDs Made of Substances
In the last 20 years, since the entry into force of Directive 93/42/EEC, many substance-based products have been CE marked as MDs, even if these kind of products were likely not in mind of the legislator when drafting the directive. At that time the term “material” reported in the English version of Directive 93/42/EEC in the definition of MD was translated in some languages, including Italian, as “substance” opening the possibility to this category of products made of substances with a mode of action non-pharmacological, immunological or metabolic, entering the market as MDs in compliance with the provisions of the legislation. For this reason, the above-mentioned directive lacked specific requirements and classification rules for this kind of products. In force of Directive 93/42/EEC, and despite not fitting properly, the rule applied for the definition of the risk class for MDs made of substances was mainly rule 5 (Annex IX), relating to invasive devices in natural orifices. The risk class for this rule was based on the duration of use, criterion considered insufficient to address the safety of MDs made of substances.
Regulation (EU) 2017/745: Further Steps Forward
The MDR has introduced an important innovation for substance-based MDs (Marletta, 2020), definitively legitimizing their qualification as MDs. During the drafting of MDR, in the discussions at the European Council some competent authorities were in favor of maintaining this category of devices within the scope of the MD regulatory framework, while other would have them merged into the MP sector. The final version of the MDR foresees specific requirements and a specific classification rule that take into account the concerns regarding the safety of these products, as highlighted in recital 59.
In the context of the previous regulatory regime, discussions at European level were mainly focused on the lack of adequate provisions in terms of managing the risk related to the invasiveness and potential toxicity of these devices, as well as the lack of a suitable classification rule reflecting their risk level. In order to obtain a suitable risk-based classification of devices composed of substances, specific classification rules were introduced taking into account the site where the device performs its action in or on the human body, whether it is introduced or applied, and whether a systemic absorption of the substances occurs. Based on these considerations, the legislator introduced within the MDR an ad hoc classification rule, rule 21 (Annex VIII), as well as specific essential requirements.
Rule 21 takes into consideration the specific risks for substance-based medical devices, precisely in relation to whether they are intended to be introduced into the human body (through an orifice or to be applied to the skin) and, above all, considering that the substances are absorbed by the human body or locally dispersed in it. The risk class depends on the route of administration and possible systemic absorption.
Devices that are composed of substances or of combinations of substances that are intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed by or locally dispersed in the human body are classified as:
• class III if they, or their products of metabolism, are systemically absorbed by the human body in order to achieve the intended purpose;
• class III if they achieve their intended purpose in the stomach or lower gastrointestinal tract and they, or their products of metabolism, are systemically absorbed by the human body;
• class IIa if they are applied to the skin or if they are applied in the nasal or oral cavity as far as the pharynx, and achieve their intended purpose on those cavities; and
• class IIb in all other cases.
Unlike the previous regulatory framework, MDs made of substances can no longer be classified as class I, therefore all the conformity assessment procedures provide for the intervention of a notified body.
Moreover, for the first time the concept of local dispersion is reported into the MDR and defined in the guidance MDCG 2021–24 as “the condition whereby substances remain in a specific site without being distributed throughout the body through the blood and/or lymphatic system”.
Guidance on Borderline Between Medical Devices and Medicinal Products Under MDR (MDCG 2022–5).
The recently published guideline has undergone a long review process. It is a very important document in the context of the qualification of borderline products.
A first question may arise concerning what substances are allowed in MDs made of substances since the term “substance” is not defined in the MDR, although the guideline MDCG 2022-5, refers to the definition reported in Directive 2001/83/EC. For substances of herbal origin, the definition includes micro-organisms, plants, parts of plants, vegetable secretions, extracts. A critical aspect is defining “substance” a complex mixture like those derived from herbal products, for which since it could be not easy identifying the mode of action, new methods should be developed in order to qualify these products (Bilia et al., 2021).
As previously reported, it is important to underline that the intended use of a MD (diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of a disease, etc.) may recall the one of a medicinal product, however the mechanism of action, responsible for the therapeutic effect, determines the difference between the two.
It is important to distinguish between the mechanism of action based on the interaction between the substance or substances and the body (refer to the text below), and the effect which is the result produced by the mechanism of action (Racchi and Govoni, 2020). Two products may have the same therapeutic effect (e.g., treatment of gastric acid hypersecretion), obtained by different mechanisms of action (H2 receptor antagonism or barrier mechanism).
It is also important to distinguish between the principal mechanism of action, accounting for the principal intended use, and other mechanisms of action, if any, not accounting for the intended use. In general, the pharmacological mechanism of action is expressed through an interaction with biological macromolecules, generally proteins. Examples include the binding to specific protein receptors, enzymes, etc. which can be physiologically present in the patient’s body, in pathogenic microorganisms, in neoplastic cells, etc. The immunological and metabolic mechanisms of action are two specific aspects of the pharmacological one, as the type of interaction is the same, but it occurs at the level of the immune or metabolic systems respectively.
The pharmacological mode of action includes two sequential steps, which are evident, in the definition of both the previous guidance, MEDDEV 2.1/3 rev.3 and the updated version:
1 The first step is an interaction (“Pharmacological means” is understood as an interaction between a substance or its metabolites and a constituent of the human body),
2 the second step (signal transduction) is an event triggered by the interaction characterized by a conformational change (“which results in initiation, enhancement, reduction or blockade of physiological or pathological functions”).
To provide a clear explanation, some definitions reported above are better elucidated:
• Interaction: binding to a body component or organisms or pathogens within or on the body.
• Event: signal transduction pathway (production of G proteins, ion channel opening, enzymatic activity, lysis of membranes, etc.), in the definition: initiation, enhancement, reduction or blockade.
• Therapeutic effect: principal intended action.
Within the pharmacological mode of action, the interaction, by itself, is not sufficient to determine the therapeutic effect. The effect is mediated by the subsequent event triggered by the interaction (e.g., signal transduction pathway as reported in classical pharmacology). On the other end, the MD’s mode of action is based on a simple interaction sufficient to determine a therapeutic effect. The mode of action is physical, mechanical, etc. and the interaction by itself generally determines the effect without triggering a subsequent event (e.g., signal transduction pathway).
The interactions reported in the examples of MPs are pharmacological, since in all cases they result in a subsequent event. These interactions are targeted to a specific site of action taking into consideration that an interaction with a different site of action would not lead to the same event (Table 2).
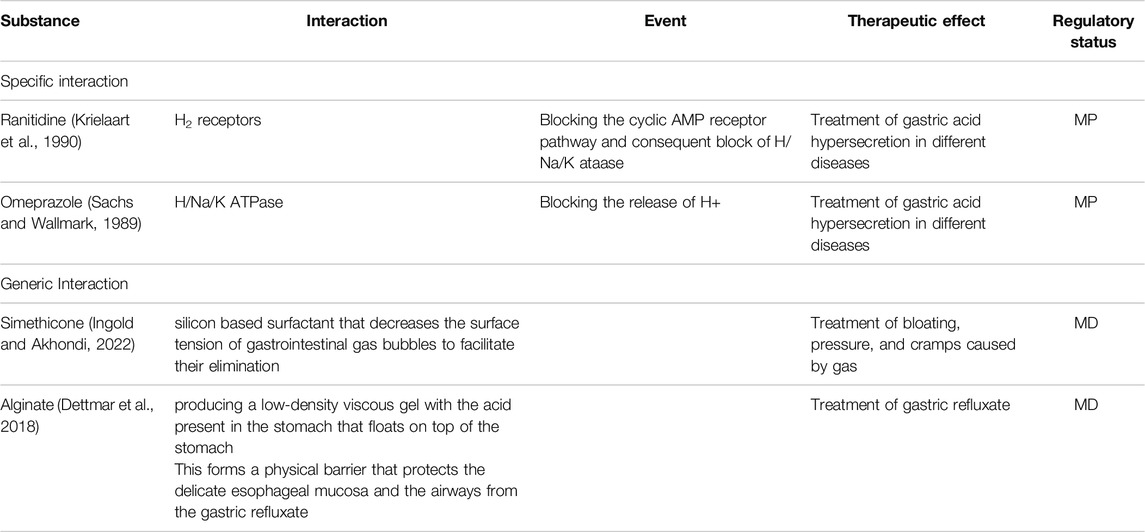
Table 2. Examples of different types of interactions.
MDs Composed of Substances of Herbal Origin
When borderline products contain herbals, the qualification is even more difficult. Complex mixtures of herbal origin have multiple mechanisms of action and it is not always clear how to identify the principal one to properly qualify the product. The legislator has already faced this issue; in fact, Directive 2001/83/EC as amended by Directive 2004/24/EC foresees a simplified registration for traditional herbal medicinal products (THMP). Due to the difficulties of identifying the mode of action, the Summary of product characteristic (SmPC), in section 5 Pharmacological properties, 5.1 Pharmacodynamic may not foresee its description, as reported in Article 16c (1) (a) (iii) Directive 2001/83/EC as amended: “Not required as per of Directive 2001/83/EC”.
THMP monographs published by the European Medicines Agency (EMA), specify the intended use and the route of administration (excluding the parenteral one) for products whose traditional use has been recognized for at least 15 years in the European Union and for 30 outside of it. The above mentioned directive foresees that the national competent authority cannot require additional data to assess the safety and the traditional use of these products.
Based on these considerations, the level of scientific evidences (i.e., safety and efficacy) required for THMPs is much less compared with those required for MDs composed of substances foreseen by the MDR (refer to the section below).
Data Required by the Regulation (EU) 2017/745 for Substance-Based MDs.
MDR focus the attention to the safety and performance of MDs made of substances: specific general safety and performance requirements have been set in the Annex I section 12.2 (General Safety and Performance Requirements). MDs composed of substances shall comply, where applicable and in a manner limited to the aspects not covered by the MDR, with the relevant requirements laid down in Annex I of Directive 2001/83/EC for the evaluation of absorption, distribution, metabolism, excretion, local tolerance, toxicity, interaction with other devices, medicinal products or other substances and potential for adverse reactions. The technical documentation to be submitted is shown in Annex IX, Chapter II paragraph 5.4.
As a further novelty, for MDs that are absorbed systemically in order to achieve their intended use (first indent of rule 21), the MDR foresees a consultation procedure. The notified body shall seek a scientific opinion from one of the competent authorities for MPs or EMA (Regulation (EC) 726/2004), on the above reported aspects. The scientific opinion of the competent authority for MPs, released within 150 days, albeit not binding, is included in the conformity assessment of the notified body concerning the device. The notified body shall give due consideration to the views expressed in the scientific opinion when making its decision, and shall convey its final decision to the consulted MPs competent authority.
Furthermore, Annex II paragraph 6.2. c “Additional information necessary for specific cases” detailed the information, including test design, complete test or study protocols, methods of data analysis, and data summaries and test conclusions, regarding studies in relation to:
• absorption, distribution, metabolism and excretion;
• possible interactions of those substances, or of their products of metabolism in the human body, with other devices, medicinal products or other substances, considering the target population, and its associated medical conditions;
• local tolerance; and
• toxicity, including single-dose toxicity, repeat-dose toxicity, genotoxicity, carcinogenicity, and reproductive and developmental toxicity, as applicable depending on the level and nature of exposure to the device.
In the absence of such studies, an adequate justification shall be provided.
Discussion
The qualification of a product as MD is under the responsibility of the manufacturer, in consultation with the notified body, and consequently it should take up the charge of the burden of complying with the relevant regulatory requirements. Data contained in the technical file should be solid, scientifically valid and correspond to the state of the art.
In order to ensure consistent decisions in all member states, the Commission can decide, on a case-by-case basis, on its own initiative or following a motivated request of a member state, and after consulting the Medical Device Coordination Group (MDCG), whether or not a specific product, category or group of products falls within the scope of the MDR.
In taking the decision on the regulatory status of borderline products with MPs, human cells and tissues, biocides or food products, the Commission should consult (Art. 4 MDR) the EMA, the European Chemicals Agency (ECHA) and the European Food Safety Authority (EFSA), respectively.
The belief that a product, qualified as MP was safer than a product qualified as MD, has been overtaken by the provisions of the MDR which requires, for MDs composed of substance, data comparable to the ones listed in Annex I of Directive 2001/83/EC. Moreover, in the case of MDs composed of herbal substances, data requested by the MDR are even more extensive than those requested for THMP. On the other hand, the MP regulatory framework, in this specific context, is not appropriate to foster innovation, since the EMA monographs are based on well-established use data. New paradigms should be developed to solve these issues in order to promote innovation in this area.
Both on the pharmacological and the toxicological side there is the need of new methods and paradigms to deal with the complexity derived from the introduction of innovative products in particular if derived from herbal extracts.
In conclusion, the MDR raises the standard of evidences required to demonstrate safety and performance for MDs composed of substances with a positive impact on the health of citizens.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bilia, A. R., Corazziari, E. S., Govoni, S., Mugelli, A., and Racchi, M. (2021). Medical Devices Made of Substances: Possible Innovation and Opportunities for Complex Natural Products. Planta Med. 87, 1110–1116. doi:10.1055/a-1511-8558
Dettmar, P. W., Gil-Gonzalez, D., Fisher, J., Flint, L., Rainforth, D., Moreno-Herrera, A., et al. (2018). A Comparative Study on the Raft Chemical Properties of Various Alginate Antacid Raft-Forming Products. Drug Dev. Industrial Pharm. 44 (1), 30–39. doi:10.1080/03639045.2017.1371737
Ingold, C. J., and Akhondi, H. (2022). “Simethicone,” in StatPearls (Treasure Island (FL)). [Internet].
Krielaart, M. J., Veenstra, D. M., and van Buuren, K. J. (1990). Mechanism of Action of H2-Antagonists on Histamine- or Dimaprit-Stimulated H2-Receptors of Spontaneously Beating guinea-pig Atrium. Agents Actions 31 (1-2), 23–35.
Marletta, M. (2020). The New Regulation 2017/745: an Opportunity for Innovation. Pharm. Adv. 01, 13–15. doi:10.36118/pharmadvances.01.2020.03s
Racchi, M., and Govoni, S. (2020). The Concept of Non-pharmacological Mechanism of Action in Medical Devices Made of Substances in Practice: what Pharmacology Can Do to Promote the Scientific Implementation of the European Medical Device Regulation. Pharmadvances 01s, 4–12. doi:10.36118/pharmadvances.01.2020.02s
Keywords: regulation UE 2017/745, medical devices, substances, borderline products, medicinal products, pharmacological mechanism of action
Citation: Leone MG (2022) Medical Devices Made of Substances: A New Challenge. Front. Drug Saf. Regul. 2:952013. doi: 10.3389/fdsfr.2022.952013
Received: 24 May 2022; Accepted: 13 June 2022;
Published: 04 July 2022.
Edited by:
Alessandro Mugelli, University of Florence, ItalyCopyright © 2022 Leone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Grazia Leone, bWcubGVvbmVAc2FuaXRhLml0