
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
POLICY AND PRACTICE REVIEWS article
Front. Drug Saf. Regul. , 15 December 2022
Sec. Substance-Based Medical Devices
Volume 2 - 2022 | https://doi.org/10.3389/fdsfr.2022.1089965
This article is part of the Research Topic Treatment of Comorbidities of Asthma and Its Safety View all 5 articles
 Sara Manellari1
Sara Manellari1 Maria Grazia Leone2
Maria Grazia Leone2 Antonella Casiraghi1
Antonella Casiraghi1 Chiara Grazia Milena Gennari1
Chiara Grazia Milena Gennari1 Paola Minghetti1*
Paola Minghetti1*European legislation provides that each product used for healthcare purposes be regulated according to specific directives and regulations based on its intended use and mechanism of action. However, qualification issues may arise for medical devices, their accessories, and medicinal products. This is the case with gases for spirometry, which support spirometers in measuring patients’ pulmonary capacities. This article discusses criticisms connected to their proper regulatory qualification, detailing why they should be more properly qualified as accessories of medical devices instead of as medicinal products or medical devices.
In health settings, many products used for healthcare purposes (medical or not) may fall under different regulatory categories according to their characteristics, mechanisms of action, intended uses, and claims. They may be qualified as medicinal products, medical devices, accessories of medical devices, food supplements, or cosmetics. Even if each of these categories is ruled by specific European directives and regulations, sometimes borderline and classification issues arise. This can happen, for instance, when the mechanism of action of a product is uncertain or is not univocally identified, as with medical devices composed of substances (Scaglione et al., 2021). It may not be easy to establish the principal mode of action on which its qualification as a medicinal product or a medical device depends. Medicinal products and medical devices, including their accessories, are regulated by Directive 2001/83/EC (MPD) and Regulation (EU) 2017/745 (MDR), respectively (European Parliament and Council, 2001; European Parliament and Council, 2017). Nevertheless, it is sometimes difficult to qualify a borderline product. European guidelines are published with the aim of interpreting legislative provisions, even if they are not legally binding. Borderline products include the gases used for spirometry, a diagnostic test for cardiopulmonary diseases. These gases are used together with spirometers to make diagnosis on the pulmonary capacity of patients (Wanger et al., 2005; Flesh and Dine, 2012; Vaz Fragoso et al., 2017). Notably, the intended action of gases for spirometry only contributes to making a diagnosis. More precisely, the spirometer is intended to make a diagnosis, measuring the gas volume that fills out the lungs. In this light, such gases have been historically qualified as medical devices in some European countries (e.g., Italy) since they are used with diagnostic devices for medical purposes. This manuscript aims to discuss the regulatory status of gases used for spirometry in the EU. It analyzes legislative provisions for medicinal products, medical devices, and accessories, highlighting the intended purposes and mechanisms of action of each category of products to determine which one best suits.
The current EU regulatory framework for healthcare products has been designed with the objective of protecting citizens’ health. Therefore, the higher the risks for the users, the more restrictive the provisions of the regulatory framework for placing that product on the market. The different regulatory qualifications for a good have relevant impacts on the provisions a manufacturer must fulfil to place it on the market. For example, the requirements for medicinal products and medical devices are more stringent than those for food supplements, since the first are developed to fulfil a specific medical purpose and must demonstrate their efficacy and safety through solid scientific data.
Art. 2(1) of Regulation (EU) 2017/745 (MDR) (European Parliament and Council, 2017) defines medical devices as “instruments, apparatus, appliances, software, implants, reagents, materials, or other articles intended by the manufacturer to be used, alone or in combination, for human beings for medical purposes such as a) diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease; b) diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability; c) investigation, replacement or modification of the anatomy or a physiological or pathological process or state; d) providing information through in vitro examination of specimens derived from the human body, including organ, blood and tissue donations; and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.”
On the other hand, in agreement with MPD (Art. 1.2), a medicinal product is “a substance, or a combination of substances presented as having properties for treating or preventing disease in human beings or which may be used in or administered to human beings either to restore, correct or modify physiological functions by exerting a pharmacological, immunological or metabolic action or to making a medical diagnosis” (European Parliament and Council, 2001).
Although the medical intended use of medicinal products and medical devices is clear in both definitions, a remarkable difference exists between them in their mechanism of action. Unlike medicinal products, the principal intended action of medical devices shall not be based on a pharmacological, immunological, or metabolic mechanism but on physical or mechanical means. Typically, the latter includes mechanical action, physical barriers such as films, lubrication, heat transfer, radiation, ultrasound, replacement of or support to organs or body functions, hydration or dehydration, and pH modification (Medical Device Coordination Group, 2022). Nevertheless, in vivo diagnostic agents, although they do not have pharmacological, immunological, or metabolic modes of action, are qualified as medicinal products by MPD.
It is often difficult to clearly distinguish between medicinal products and medical devices due to the complex mixture of components (e.g., herbal products) or a lack of consensus on the nature of the primary mechanism of action. In this light, the most recent MDCG 2022-5 guidance on borderline products defines pharmacological, immunological, and metabolic means to help manufacturers and notified bodies perform a proper assessment (Medical Device Coordination Group, 2022). For example, pharmacological means is “an interaction typically at a molecular level between a substance or its metabolites and a constituent of the human body (e.g., cell membranes, intracellular structures, RNA, DNA, proteins, components of extracellular matrix, components of blood and components of body fluids) which results in initiation, enhancement, reduction or blockade of physiological functions or pathological processes” (Medical Device Coordination Group, 2022). Therefore, the pharmacological mechanism of action shall be established based on two pillars: 1) the existence of an interaction between the molecule of the substance and a cellular constituent of the human body, and 2) that such interaction shall induce a direct response, initiating, enhancing, reducing, or blocking a physiological function or a pathological process (Leone, 2022).
Notably, Art. 2(1) of MDR does not preclude the possibility that, in parallel to the primary mechanism of action, a medical device may include substances acting by pharmacological, immunological, or metabolic means (European Parliament and Council, 2017). However, the manufacturer shall demonstrate that such substances have an ancillary action contributing to the primary physical or mechanical mechanism of action. Therefore, for the qualification of a product, the primary mechanism of action shall be considered. The manufacturer should describe the intended use and the principal mechanism of action of the product in technical files and in labelling and instructions for use, and should support their claims with current scientific data, according to all information and requirements laid down in the annexes of the Regulation (European Parliament and Council, 2017).
Directive 93/42/EEC first distinguished between “medical device” and “accessory” (European Parliament and Council, 1993). The MDR defines an accessory of a medical device as: “an article which, whilst not being itself a medical device, is intended by its manufacturer to be used together with one or several particular medical device(s) to specifically enable the medical device(s) to be used in accordance with its/their intended purpose(s) or to specifically and directly assist the medical functionality of the medical device(s) in terms of its/their intended purpose(s)” [Art. 2(2) of MDR]. The term “article” used in Art. 2(2) seems quite broad for distinguishing clearly which kinds of products can be classified as accessories. In this context, the MDCG guidance 2022-5 reports several examples of accessories of medical devices (Medical Device Coordination Group, 2022): contact lens care products (rinsing and hydrating solutions, including those which aid the insertion and/or wearing of contact lenses without therapeutic claim), lubricants specifically intended for use together with medical devices (e.g., for gloves, endoscopes, condoms), skin barrier powders and pastes, or other skin care products specifically intended for use with ostomy bags, gases used to drive cryoprobes and surgical tools, and ultrasound gels. Although this list is not exhaustive, and MDCG documents are not legally binding, it underlines how far the meaning of “article” in the accessory definition—which includes instruments, apparatus, implants, reagents, or substances—is from the regulatory perspective.
Doubtless, accessories shall not act by pharmacological, immunological, or metabolic means as medical devices do. Apart from this, the European regulatory framework does not characterize accessories in terms of their mechanism of action. However, the definition in Art. 2(2) of MDR suggests that the medical purpose of an accessory is strictly connected to the medical device with which it is used, regardless of its mechanism of action. The US regulatory framework defines the functions of an accessory in more detail. The Federal Food, Drug, and Cosmetic Act (FD&C Act) classifies accessories of medical devices similarly to the EU framework; indeed, they shall follow the same provisions of medical devices (United States Code, 2022). However, FDA guidance defines an accessory as a “finished device that is intended to support, supplement, and/or augment the performance of one or more parent devices” (Food and Drug Administration, 2017). Therefore, with respect to the “used together” of Art. 2(2) of MDR (European Parliament and Council, 2017), the FDA defines more precisely the functions of an accessory. A “support” function means that the accessory enables or facilitates a medical device to perform according to its intended use. Instead, an accessory can supplement the performance of a parent device by adding a new function or a new way of using it, without changing the intended use of the parent device. Finally, an accessory can augment the performance of a parent device by enabling it to perform its intended use more safely or effectively (Food and Drug Administration, 2017).
Focusing on the provisions that manufacturers must follow to CE-mark their products, the European legislator has established that accessories must be ruled and classified by the same provisions as medical devices. Indeed, the Directive, firstly—and the MDR, more recently—should apply to both medical devices and their accessories; in other words, accessories are subjected to the conformity assessment procedure as medical devices from a regulatory perspective. Moreover, MEDDEV 2.1/1 stated that classification as a “device” or an “accessory” had no practical consequence because these were considered as medical devices in their own right (European Commission, 1994). The guideline emphasized that “a product could only become an accessory to a medical device if the manufacturer of such a product established an intended use in conjunction with one or several medical devices.”
In line with Directive 93/42/EEC, MDR confirms this provision as reported in the text: “accessories for a medical device shall be classified in their own right separately from the device with which they are used and the application of the classification rules shall be governed by the intended purpose of the devices (medical devices or accessories)” (European Parliament and Council, 2017). However, while accessories are different from medical devices, Art. 1(4) states that they are subjected to all provisions that apply to medical devices, including general safety and performance requirements, information supplied with the device, clinical investigations, technical documentation, and CE mark conformity assessment.
Products are regulated either by the MDR or by the MPD, but not both, and the procedures of these regulatory regimes do not apply cumulatively (Medical Device Coordination Group, 2022). After a case-by-case assessment, “in cases of doubt, where, taking into account all its characteristics, a product may fall within the definition of a “medicinal product” and within the definition of a product covered by other Community legislation (e.g., MDR), the provisions of Directive 2001/83/EC shall apply” [Art. 2(2) of MPD] (European Parliament and Council, 2001). An EU Court judgement in 2012 took this concept up again and added that within the same Member State, a product, which, while not identical to another product qualified as a medicinal product, but with an identical substance and the same mode of action, cannot, in principle, be marketed as a medical device, unless it demonstrates to have a specific characteristic relevant for the purposes of a medical device (Court, 2013). However, the Court also stated that the qualification of a product in one Member State as a medical device bearing a CE marking, in accordance with Directive 93/42/EEC (now repealed by MDR), does not preclude the competent authorities of another Member State from qualifying the same product, based on its pharmacological, immunological or metabolic action, as a medicinal product within the meaning of Art. 1(2)(b) of MPD (European Parliament and Council, 2001). In cases of doubts, National Competent Authorities (NCAs) should refer to the existing regulatory sources (MDR, MDCG guidance, EU Commission documents) or activate the so-called “Helsinki Procedure” (Medical Device Coordination Group, 2021). The Helsinki Procedure states that if the NCA is not able to solve the qualification issue by referring to the regulatory sources available, it can engage other NCAs to reach a consensus on a specific issue. Released after the Medical Device Competent Authorities Meeting in Helsinki in October 2002, the procedure was reviewed in 2021 to improve the harmonization of product qualification and classification among EU Member States.
Regarding the distinction between medical devices and accessories, it is noteworthy that the same MDR provides that “without prejudice to Article 2(2) of Directive 2001/83/EC, upon a duly substantiated request of a Member State, the Commission shall, after consulting the Medical Device Coordination Group (MDCG), by means of implementing acts, determine whether or not a specific product, or category or group of products, falls within the definitions of medical device or accessory for a medical device” [Art. 4(1) of MDR] (European Parliament and Council, 2017).
Gases have long been used in the medical field, for preventing, diagnosing, and treating diseases. According to their intended use and mechanism of action, they may fall under several legislations. If gases are used with a medical scope and exert a pharmacological, immunological, or metabolic action, they are classified as medicinal products. Examples of medical gases are those intended for use in anesthesia and inhalation therapy (e.g., oxygen, medical air supplied in containers) (Medical Device Coordination Group, 2022). If gases have a medical application and the primary mechanism of action is physical or mechanical, they are considered to be medical devices. Carbon dioxide (CO2) is the most used gas for insufflation during laparoscopic surgery because it increases operative space and visualization for surgeons (Park et al., 2012). CO2’s principal mode of action is physical because it allows the formation of a space inside an organ or tissue by temporarily modifying its anatomical structure (i.e., surgical cavity). This concept has also been confirmed by MDCG guidance 2022-5, which states in a note (paragraph 1.2.6.3) concerning gases to be considered medicinal products that the same gas “intended exclusively for minimal access surgery with a physical mode of action (e.g., insufflation) would be a medical device” (Medical Device Coordination Group, 2022).
Gases like oxygen, nitrogen, helium, acetylene, argon, methane, carbon monoxide, and neon are used in spirometry (Wanger et al., 2005; Flesh and Dine, 2012; Vaz Fragoso et al., 2017). Spirometry is one of the most readily available and useful tests of pulmonary function. After a maximal inhalation, spirometry devices (spirometers) measure the volume of exhaled gas at specific time points during complete exhalation by force. Spirometry is a key diagnostic test for asthma and chronic obstructive pulmonary disease; moreover, it is indicated to diagnose and monitor several other pathological cardio-pulmonary conditions (Wanger et al., 2005; Flesch and Dine, 2012; Vaz Fragoso et al., 2017). The test is performed using an instrument called spirometer, a computerized machine connected via a cable to a mouthpiece inserted between the teeth of the person undergoing the examination. Spirometers are classified as medical devices since they have a medical purpose, which is diagnosis by detecting pulmonary diseases or by monitoring lung function, and do not achieve their intended use by pharmacological, immunological, or metabolic means.
It could also be argued that gases should be classified as medical devices or medicinal products since they are used with diagnostic devices and are involved in that medical use. In this light, it is noteworthy that the intended or claimed purpose of gases for spirometry to make a diagnosis is only indirect. More precisely, the spirometer is intended to make a diagnosis, while the gases are intended to support the device in measuring lung volumes. There is no doubt that their principal mode of action is not pharmacological, immunological, or metabolic, since the gases fill empty spaces already present in the lungs without altering their anatomical structure (i.e., physical mode of action). If there may be a pharmacological interaction between gases and other biological components, it would be an ancillary mechanism of action but not the principal one, and this cannot identify them as medicinal products.
For these reasons, gases for spirometry may be classified as accessories of medical devices because, according to the definition given by MDR, they are not themselves medical devices but may be intended by their manufacturer to be used together with spirometers in agreement with Art. 2(2) MDR (European Parliament and Council, 2017). Even if the FDA provisions are not valid in Europe, they are a useful point of reference. As mentioned above, in the USA, the gases in question would be considered accessories because they participate in a diagnosis by supporting the performance of the medical devices (spirometers) and do not achieve their intended use by chemical or metabolic means. In the end, classifying these gases as accessories does not affect the protection of public health because accessories are regulated as medical devices by the MDR. They are classified according to a risk assessment, their manufacturers must fulfil all provisions established by the European law and, overall, their safety level is the same as for medical devices (Figure 1).
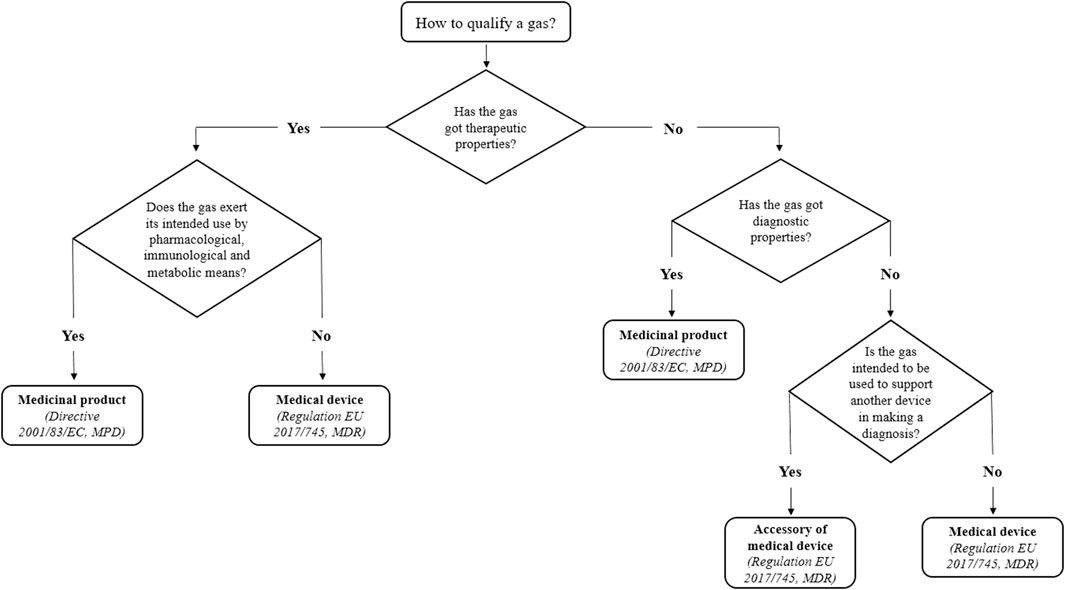
Figure 1. Decisional flowchart for the qualification of a gas.
The new MDCG guidance 2022-5 clarifies the border between medical devices and medicinal products used for diagnostic purposes and gives examples (Medical Device Coordination Group, 2022). Diagnosis is defined as “the process of investigation of the anatomy, morphology, the condition, or the functions of the human body irrespective if these are physiological or pathological, and subsequent interpretation of this information with a view to determining possible abnormalities. Investigation can include visualization, detection, or measurement.” Interesting considerations emerge from the aforementioned guideline regarding both a product’s mechanism of action and its intended purpose. Based on the definition of a medicinal product according to article 1(2)(b) MPD, a substance or combination of substances administered to make a medical diagnosis should be classified as a medicinal product independently of its mechanism of action. Indeed, it is required that only “substances which may be used in or administered to human beings with a view to restoring, correcting or modifying physiological functions” shall have a pharmacological, immunological, or metabolic action; on the contrary, this is not explicitly required for substances which make medical diagnoses. Thus, substances used for diagnostic purposes—namely, X-ray contrast media, NMR enhancing agents, SPECT- and PET-radiopharmaceuticals, fluorescein strips for diagnostic purposes, radioactive tracers, and substances for tumor identification—are medicinal products, regardless of their mechanism of action.
However, the MDCG guidance 2022-5 provided clarification on the meaning of “making a medical diagnosis.” If the intended purpose of the product is to simply visualize an anatomical structure without determining possible abnormalities (e.g., colorant used to mark the location of the site of a surgical procedure, markers placed or implanted for radiation therapy, fluorescein strips intended for the adjustment of contact lenses), then it is not regarded as diagnostic and the product could be qualified as a medical device or as an accessory to a medical device. On the contrary, if the product is used with the aim of distinguishing between healthy and pathological tissues, then it would be considered diagnostic and would be classified as a medicinal product. Among the examples given for medicinal products are gases for in vivo diagnostic purposes, including lung function and tests (e.g., carbon dioxide for vascular diagnostic purposes). However, the same guideline, in the aforementioned note, specifies that it is not simply the mechanism of action of a gas that determines whether it would be qualified as a medicinal product or a medical device. A gas intended to be used in anesthesia and inhalation therapy is a medicinal product, but the same gas intended exclusively for minimal access surgery with a physical mode of action (e.g., insufflation) would be a medical device. Gases intended to be used to drive cryoprobes and surgical tools are given as examples of accessories.
Gases used in clinical settings are sometimes difficult to qualify. The new MDCG guidance 2022-5 guideline on borderline products qualifies these gases as medicinal products a priori because they are considered substances with diagnostic purposes, which are different from simple visualization of an anatomical structure without aiming to determine abnormalities. However, gases used for spirometry do not have a primary intended medical use as diagnostics because spirometers play this role and, for this reason, they could be qualified as medical devices. Gases merely enable spirometers to be used in accordance with their intended purpose. Moreover, the mechanism of action of gases consists in filling a space of an existing cavity (i.e., lungs) without modifying the anatomical structure. Additionally, any pharmacological interaction with the human body would be an unwanted ancillary mechanism of action but not the principal one. In this sense, different nontoxic gases could be indifferently used in accordance with the spirometer characteristics. Considering these factors, the qualification of gases for spirometry as accessories of medical devices seems preferable. Since there are no other provisions in the MDR about the medical purpose and the mechanism of action of accessories, gases for spirometry may fall completely under this regulatory category of products. This guarantees a high level of safety and performance since accessories follow the same provisions for the CE mark laid down in the MDR for medical devices. Again, their qualification as medicinal products may have negative long-term impacts for European citizens. On the one hand, the switch from MDR to MPD may dramatically affect the economic sustainability of manufacturers, and it may result in an elevated risk of product withdrawal from the market with unpredictable consequences for patients’ access to diagnosis. On the other hand, since these gases currently fall under different regulatory statuses among Member States, this switch to medicinal products may contribute to the development of non-harmonized interpretations of the same regulatory framework at the European level. For example, as observed in the case of other well-established products that have been qualified as medicinal products after MPD entered in force (Decristoforo et al., 2017; Bonertz et al., 2018), gases for spirometry may be maintained on the market on the basis of Art. 5 of MPD, a procedure that does not require the evaluation of a Common Technical Document (CTD) (European Parliament and Council, 2001). Regardless of the approach adopted by the NCAs, a strong heterogeneity among Member States in terms of market access and required data authorization cannot be excluded.
Spirometry is a crucial test for diagnosing cardiopulmonary diseases, thus it is necessary that all products used for it (e.g., gases, spirometers) be properly qualified in order to guarantee reliable results that impact patients’ health. As discussed in this article, gases used in combination with spirometers can be qualified as accessories since they do not make diagnosis by themselves but rather support medical devices (i.e., spirometers) in making it. In other words, gases alone are not able to make the diagnosis, while the spirometers do, using different types of gas mixtures.
SM: conceptualization, writing—review and editing. ML: supervision and critical revision of the manuscript. AC: conceptualization, writing—review and editing. CG: conceptualization, writing—review and editing. PM: supervision.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bonertz, A., Roberts, G. C., Hoefnagel, M., Timon, M., Slater, J. E., Rabin, R. L., et al. (2018). Challenges in the implementation of eaaci guidelines on allergen immunotherapy: A global perspective on the regulation of allergen products. Allergy 73, 64–76. doi:10.1111/all.13266
Court, E. U. (2013). Judgment C-109/12 - laboratoires Lyocentre v Lääkealan turvallisuus– ja kehittämiskeskus, Sosiaali– ja terveysalan lupa– ja valvontavirasto. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:62012CJ0109&from=IT (accessed July 22, 2022).
Decristoforo, C., Penuelas, I., Patt, M., and Todde, S. (2017). European regulations for the introduction of novel radiopharmaceuticals in the clinical setting. Q. J. Nucl. Med. Mol. Imaging 61, 135–144. doi:10.23736/S1824-4785.17.02965-X
European Commission (Ec), (1994). MEDDEV 2.1/1, guidelines relating to the application of: The council directive 90/385/EEC on active implantable medical devices and the council directive 93/42/EEC on medical devices. Available at: https://ec.europa.eu/docsroom/documents/10278/attachments/1/translations (accessed July 22, 2022).
European Parliament and Council (2017). Regulation (EU) 2017/745 of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC 2017Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:02017R0745-20170505&from=EN (accessed July 22, 2022).
European Parliament and Council (2001). Directive 2001/83/EC of 6 November 2001 on the Community code relating to medicinal products for human use 2001. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:02001L0083-20190726&from=EN (accessed July 22, 2022).
European Parliament and Council (2001). Directive 93/42/EEC of 14 June 1993 concerning medical devices 1993. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:01993L0042-20071011&from=EN (accessed July 22, 2022).
Flesch, J. D., and Dine, C. J. (2012). Lung volumes: Measurement, clinical use, and coding. Chest 142, 506–510. doi:10.1378/chest.11-2964
Food and Drug Administration (Fda), (2017). Medical device accessories – describing accessories and classification pathways. Silver Spring, MD: Guidance for Industry and Food and Drug Administration Staff.
Leone, M. G. (2022). Medical devices made of substances: A new challenge. Front. Drug Saf. Regul. 2, 1–5. doi:10.3389/fdsfr.2022.952013
Medical Device Coordination Group (MDCG) (2022). MDCG 2022-5, Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical devices.
Medical Devices Coordination Group (2021). Exchange of information between medical device competent authorities on borderline and classification cases - Helsinki Procedure 2021 2021. Bruxelles.
Park, E. Y., Kwon, J-Y., and Kim, K. J. (2012). Carbon dioxide embolism during laparoscopic surgery. Yonsei Med. J. 53, 459–466. doi:10.3349/ymj.2012.53.3.459
Scaglione, F., Musazzi, U. M., and Minghetti, P. (2021). Considerations on D-mannose mechanism of action and consequent classification of marketed healthcare products. Front. Pharmacol. 12, 636377–7. doi:10.3389/fphar.2021.636377
United States Code (2022). Title 21 chapter 9, federal food, Drug, and cosmetic act. Washinghton, DC.
Vaz Fragoso, C. A., Cain, H. C., Casaburi, R., Lee, P. J., Iannone, L., Leo-Summers, L. S., et al. (2017). Spirometry, static lung volumes, and diffusing capacity. Respir. Care 62, 1137–1147. doi:10.4187/respcare.05515
Keywords: regulation EU 2017/745, MDCG 2022-5, medical devices, borderline products, accessories of medical devices, regulatory qualification, gases for spirometry
Citation: Manellari S, Leone MG, Casiraghi A, Gennari CGM and Minghetti P (2022) Medicinal products, medical devices, or accessories of medical devices: How to qualify gases for spirometry?. Front. Drug Saf. Regul. 2:1089965. doi: 10.3389/fdsfr.2022.1089965
Received: 04 November 2022; Accepted: 28 November 2022;
Published: 15 December 2022.
Edited by:
Alessandro Mugelli, University of Florence, ItalyReviewed by:
Serghei Covantsev, S. P. Botkin Clinical Hospital, RussiaCopyright © 2022 Manellari, Leone, Casiraghi, Gennari and Minghetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Minghetti, cGFvbGEubWluZ2hldHRpQHVuaW1pLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.