 Madison Ambrose
Madison Ambrose Jeremy Lee
Jeremy Lee Aleem Syed
Aleem Syed Zamal Ahmed
Zamal Ahmed Guang Peng
Guang Peng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Drug Discov., 29 January 2025
Sec. Anti-Cancer Drugs
Volume 5 - 2025 | https://doi.org/10.3389/fddsv.2025.1520734
Increased research attention has been brought to non-enzymatic protein targeting agents as a new and effective strategy for advancing cancer treatment. To discover this class of new anticancer drugs, two molecular approaches targeting the non-enzymatic activities of proteins have shown promising experimental, preclinical, and clinical results. In the first approach, selective agents known as PROteolysis-TArgeting Chimeras (PROTACs) employ innate endogenous protein degradation machinery in cells to proteolyze the targeted protein. The combination of the highly selective PROTACs and exploitation of cellular protein degradation pathways provides the opportunity to treat diseases that were previously deemed incurable due to lack of enzymatic activities of the targeted proteins. The second approach targets protein-protein interactions (PPIs) as an alternative non-enzymatic route that alters the functional activities of protein complexes and thus significantly influence cancer cell fitness and survival. To efficiently identify potential chemical leads for these approaches, high-throughput screening (HTS) has been extremely valuable due to its ability to quickly screen large libraries of compounds. In this review paper, we will provide an overview of developing anti-cancer agents targeting non-enzymatic activities of proteins and the potential clinical impact of this new class of inhibitors.
Traditional cancer treatments, including chemotherapy, surgical intervention, and radiation, all have significant drawbacks; these approaches lack specificity, produce undesirable side effects, and negatively impact healthy tissues in the process. To resolve these complications and improve effectiveness, cancer therapy research initiatives have shifted towards developing highly specific drugs that target individual molecular identifiers, allowing for the selective elimination of cancer cells. The first steps towards achieving this goal focused on identifying proteins that exhibit either altered enzymatic activity or altered enzymatic expression within cancer cells. Once identified, small molecule inhibitors were designed to bind at or near the active site of the enzyme. A diverse array of inhibitors that target kinase activity have shown positive results in clinical practice. The first U.S. Food & Drug Administration (FDA) approved drug that used this approach was Imatinib (Gleevec), a Bcr-Abl kinase inhibitor that successfully treated Philadelphia chromosome-positive chronic myelogenous leukemia (Farmer et al., 2005; Bryant et al., 2005). This development served as the springboard for future drugs, with approximately over 60 kinase-inhibiting drugs receiving FDA approval as of 2023 (Table 1). In addition to kinase activity, enzymatic activity involving poly (ADP-ribose) polymerase-1 (PARP1) has also served as an effective target for developing inhibitors, particularly for BRCA-mutated cancer cells.
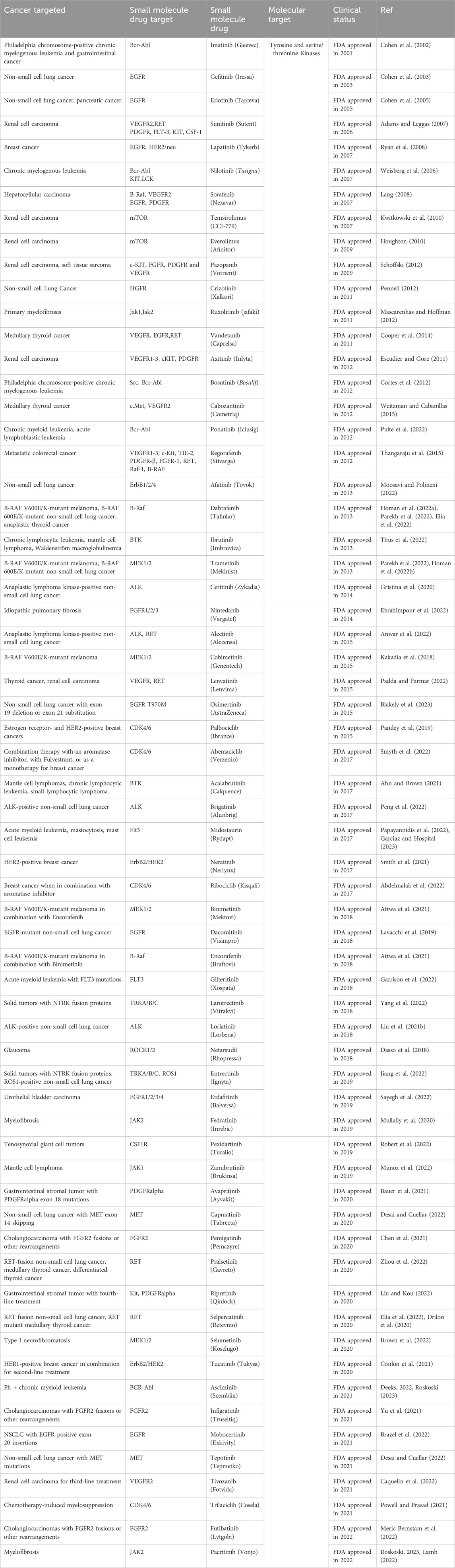
Table 1. List of small molecule inhibitors targeting enzymatic activity.
While enzymatic targeting proteins have been successful in blocking the enzymatic domain of proteins involved in cancer survival, many proteins are complex and have both enzymatic and non-enzymatic domains. For instance, the majority of enzymes include both a catalytic domain where the respective substrate undergoes a chemical reaction as well as a regulatory domain where allosteric regulators, signaling molecules, and scaffold proteins bind to control protein activity (Sun et al., 2022). These proteins cannot be exclusively neutralized by an enzymatic targeting protein (Farley et al., 2024). However, non-enzymatic targeting proteins such as PROTACs provide the opportunity to inhibit these target proteins due to their ability to capitalize on the hosts protein degradation system via the ubiquitin-proteasome system. Specifically, PROTACs promote the ubiquitination of the target protein by recruiting the E3 ubiquitin ligase to the target protein (Sun et al., 2022). In addition, PROTACs have the unique function to be reused and continuously promote this interaction, increasing the selectivity and clinical efficacy of the drug. One example of PROTACs being able to block complex, multifunctional proteins is with the zeste homolog 2 (EZH2) within the PRC2 complex. EZH2 is a highly oncogenic protein that inhibits the expression of 200 tumor suppressor genes. However, Liu et al. had significant success in designing an EZH2 PROTAC that could completely block the oncogenic activity of EZH2 (Liu Z. et al., 2021). Another promising PROTAC includes AU-15330. AU-15330 is a SMARCA2/4 degrader that utilizes both a bait and ligand moiety that is currently in clinical trials for the treatment of prostate cancer (He et al., 2024). These alongside the numerous PROTACs that are in development demonstrate the advantages of this approach in targeting complex proteins that have both enzymatic and non-enzymatic domain for the treatment of diverse cancers.
While there have been many successful small inhibitors that target the enzymatic activity of influential cancer proteins, small inhibitors that target the non-enzymatic activity of proteins have also been identified. These include drugs that mark proteins for degradation as well as drugs that inhibit or stabilize protein-protein interactions (PPIs) (Table 2). Due to the selectivity, these approaches have displayed promising results for the treatment of diseases that were previously viewed as untreatable and should further be explored in the upcoming years. In this article, we will describe these new, non-enzymatic approaches for cancer treatment that utilize protein degradation and PPIs interfering methods.
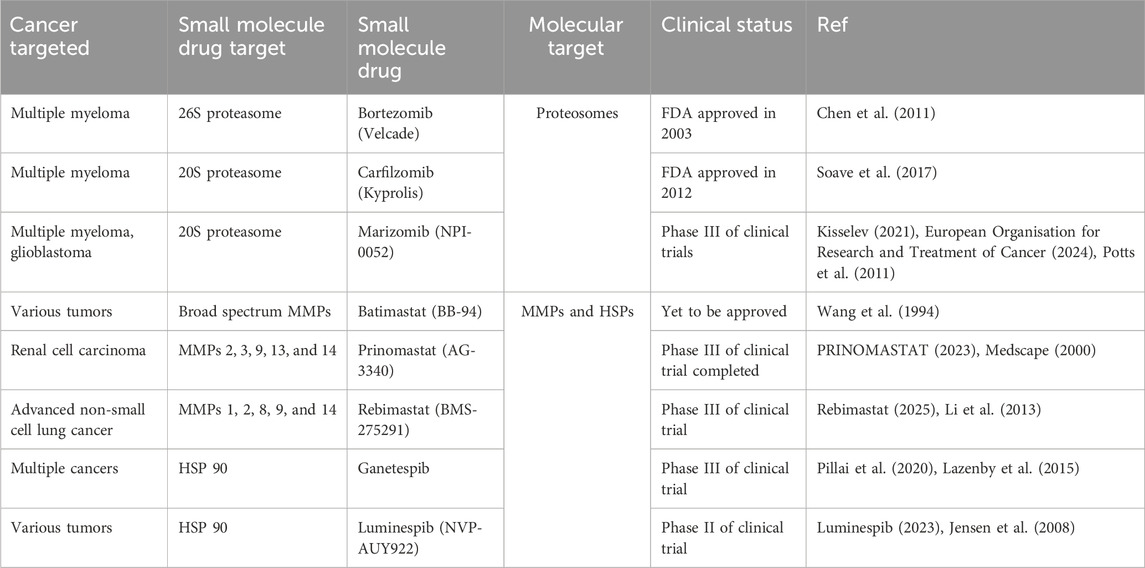
Table 2. List of small molecule inhibitors targeting non-enzymatic activity.
Targeted protein degradation is an area that has gained increased interest in recent years, with many pharmaceutical companies investing in small inhibitor drugs that use targeted protein degradation machinery. Targeted protein degradation works by utilizing the cell’s endogenous machinery to discard the cell of damaged or unwanted proteins (Lai and Crews, 2017; Varshavsky, 2005). Thus, by developing drugs that selectively mark cancerous proteins for degradation, this will allow the cell to eliminate improper protein faster than without the supplemental intervention.
In the early 1900s, it was assumed that intracellular proteins lived for long periods of time. However, in the 1980s, two different experiments made discoveries that complemented each other. The first experiment, which was conducted in the lab of Avram Hershko, discovered that some proteins were degraded by cellular machinery (Varshavsky, 2005; Ciehanover et al., 1978; Hershko et al., 1980). This mechanism, later known as protein degradation, worked by adding a protein known as ubiquitin to the protein of interest (Figure 1A). This ubiquitin-protein complex would then be degraded by an ATP-dependent protease. The experiment labeled the enzymes that conjugated the protein and ubiquitin together as E1, E2, and E3 (Hershko et al., 1983). The ATP-dependent protease that was discovered here, is now commonly known as the 26S proteasome (Baumeister et al., 1998). The second experiment, which was conducted at a lab at the Massachusetts Institute of Technology (MIT), discovered that the ubiquitin system functioned as the main perpetrator for protein degradation in living cells; furthermore, that it played roles in cell cycle regulation, protein synthesis, transcriptional regulation, stress response, DNA repair, and cell viability (Finley et al., 1984; Varshavsky, 1997; Pickart, 2004). Therefore, it is clear that protein degradation serves as a critical function in protecting the body by eliminating damaged or misfolded proteins, recycling of amino acids, regulating cellular metabolism, and generating of active proteins (Goldberg, 2003).
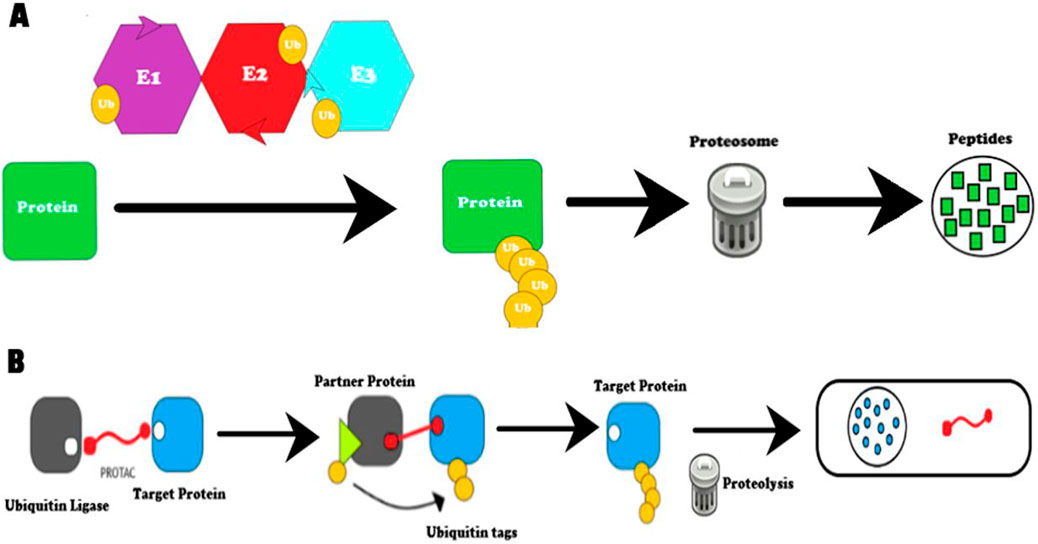
Figure 1. Diagram of protein degradation methods. (A) Protein degradation by the ubiquitination pathway. Ubiquitin ligases E1, E2, and E3 help carry out ubiquitination by tagging the target protein. The protein tagged with ubiquitin is degraded by the proteasome and converted to peptides. (B) Protein degradation by proteolysis-targeting chimeras (PROTACS). PROTACS work by recruiting ubiquitin ligase to a target protein. First, the ubiquitin ligase and target protein are brought in close proximity to each other by the PROTAC molecule due to its dual binding ability. Second, the E3 ligase, with the help of E1 and E2, ubiquitinates the target protein. This modification is a signal for endogenous proteasomal machinery to act on the target protein and degrade the target protein (Farmer et al., 2005).
In 1999, a heat shock protein 90 (HSP90) inhibiting drug know as17-AGG became one of the first protein degradation inducing drugs to enter FDA clinical trials (Kim et al., 2009). HSP90 molecular chaperones are a class of proteins that help prevent nascent or misfolded proteins from aggregating in cells (Zuehlke and Johnson, 2010). This was an important target since researchers had previously discovered that cancer cells upregulated HSP90 to multiply faster and increase survival pathways (Moulick et al., 2011). Another reason being that HSP90 in cancer cells was found to be more sensitive to HSP90 inhibitors. This is due to the fact that since tumor cells contain HSP90 in their activated conformation, cancer cells have a higher affinity for the inhibitors when compared to normal cells (Kamal et al., 2003).
It was then proposed that HSP90 inhibitors were capable of reducing tumor sizes as well as delaying or completely stopping tumor progression. HSP90 inhibitors function by targeting the ATP-binding domain of the HSP90 chaperone, which leads to the breakdown of the HSP90 target proteins (Prodromou et al., 1997). While there are 39 ongoing clinical trials for inhibitors of HSP90, FDA has not approved of any of them (Zhang et al., 2022). The FDA cannot approve of these inhibitors because of their poor in vivo properties, structural toxicity, and the broad range of HSP90 target proteins (Taldone et al., 2008; McClellan et al., 2007). However, two FDA approved drugs, Panobinostat and Irsogladine, have secondary HSP90 inhibitory functions (Zhang et al., 2022). Panobinostat is primarily a histone deacetylase (HDAC) inhibitor. When it is combined with Bortezomib and dexamethasone to treat recurrent multiple myeloma, Panobinostat hyperacetylates HSP90, inhibiting its function. Irsogladine is a phosphodiesterase inhibitor and inhibits HSP90 by disrupting its folding machinery. Nevertheless, inhibitors of HSP90 have provided researchers with a positive step in the direction for further protein degradation experiments.
Since these experiments were conducted, researchers have continued to explore the potential applications of induced protein degradation, specifically a drug that could directly degrade improper cancerous proteins. For the purpose of this article, we will focus on PROTACs as TPDs; however, other emerging TPD techniques that are promising include lysosome-targeting chimeras (LYTACs), asialoglycoprotein receptor (ASGPR) targeting chimera (ATAC), bispecific atamer chimera (BIAC), antibody-based PROTAC (AbTAC), glueTAC, autophagosome-tethering compound (ATTEC), autophagy-targeting chimera (AUTAC), and chaperone-mediated autophagy-based degraders (Banik et al., 2020; Ahn et al., 2021; Zhao et al., 2022; Li et al., 2020; Takahashi and Arimoto, 2020; Kirchner et al., 2019).
PROTACs are a class of drugs have been the topic of investigation for more than 20 years, with several currently in clinical trials (Bekes et al., 2022). In this unconventional approach, PROTACs destroy the target protein by ubiquitination (Figure 1B) (Sun et al., 2019). By eliminating the target, PROTACs could potentially overcome the resistance faced by other therapeutic treatments such as chemotherapy. In addition, PROTACs are unconventional since they can bind to areas that other drugs cannot access. Specifically, due to their ability to induce protein degradation, PROTACs have the potential to provide access to shallow binding regions that other drugs cannot get access to. This opens up the possibility to attack diseases that have been accepted as “undruggable,” such as the myc proto-oncogene, or the Tau protein that is distorted in Alzheimer’s disease (Scudellari, 2019).
Early proof-of-concept studies provided promising theoretical proof of PROTACs. This was due to the fact that molecules that employed similar methods were effective in inhibiting protein activity (Cromm and Crews, 2017). However, experimentally, early PROTACs showed poor results due to low cell permeability, sensitivity of E3 recruiting phosphorylation, and micromolar potency (Buckley et al., 2012). To improve the PROTAC technology, E3 recognition was the first hurdle to tackle. To do so, the switch from a phosphatase sensitive degron to an oxygen-dependent degron was made. This was achieved through the use of the Von Hippel-Landau (VHL) protein which can recognize a hydroxylated proline sequence (Hon et al., 2002). The VHL recruiting motif was deemed successful since it was able to degrade AR-GFP and FKBP12-GFP during in vitro cell experiments (Diehl and Ciulli, 2022). Since then, it has been confirmed that hydroxylated proline sequence can degrade several proteins that are influential in disease progression. These proteins include ER alpha, aryl hydrocarbon receptor, Smad3, Tau, Akt, and the X-protein of the hepatitis B virus (Cromm and Crews, 2017).
Despite the success, there were still issues of potency in micromolar range due to low permeability. To overcome this, the peptidomimetic VHL ligand 1 was added and this resulted in a significantly improved the binding affinity with VHL (Kd = 185 nM) (Buckley et al., 2012). The ability for VHL ligand 1 to bind to VHL was identified by using crystal structure intermediates, fragment-based screening, and computer simulations (Van Molle et al., 2012). It was also determined that the VHL ligand 1 had a R-hydroxyproline core which was crucial for the interaction with VHL. The effectiveness of VHL ligand 1 was proven in 2015 by the degradation of target fusion proteins composed of HaloTag-GFP (Buckley et al., 2015). In this experiment, HaloPROTAC was able to bind to VHL and subsequently degrade the HaloTag-GFP target protein in cell based assays, displaying the dependency of HaloPROTAC protein degradation on VHL binding.
Another major development in PROTACs was the discovery of their catalytic function. In the experiment by Bondeson et al., they developed a PROTAC that was able to decrease target protein levels by roughly 90%, even at nanomolar concentrations (Bondeson et al., 2015). Bondeson et al. then designed a PROTAC to target RIPK2, a serine-threonine kinase that functions as a mediator for innate immune signaling (Bondeson et al., 2015) (reference). When the RIPK2 and PROTAC interacted, the RIPK2 could recruit other kinases which increased the speed of ubiquitination. This experiment illustrated that PROTACs could function catalytically by inducing an enzyme cascade, which in turn accelerates protein degradation.
In order to have reversible activation of PROTACs, the photoPROTAC was designed. The photoPROTAC is composed of a PROTAC with the addition of ortho-F4-azobenzene linkers between two ligands of the target protein (warhead ligands) (Pfaff et al., 2019; Bondeson et al., 2018). In the cis-azobenzene form, the photoPROTAC is inactive and highly stable but can be activated to the trans-azobenzene form when exposed to visible light. Subsequently, when exposed to light outside of the visible light range, it reverts back to the cis-azobenzene form, becoming inactive. Pfaff et al. confirmed this concept by measuring BRD degradation within Ramos cells of varying concentrations and visible light wavelengths. Therefore, by being able to control degradation activity, the results of this experiment illustrate that PROTACs have the ability for spatiotemporal control.
Due to the continuous efforts to enhance PROTAC technology, PROTACs have the potential to be an alternative drug discovery approach to compliment traditional cancer therapies and to combat treatment resistance. Indeed, as of 2023, there are seventeen PROTACs in clinical trials encompassing treatment for a wide range of cancer types (Table 3).
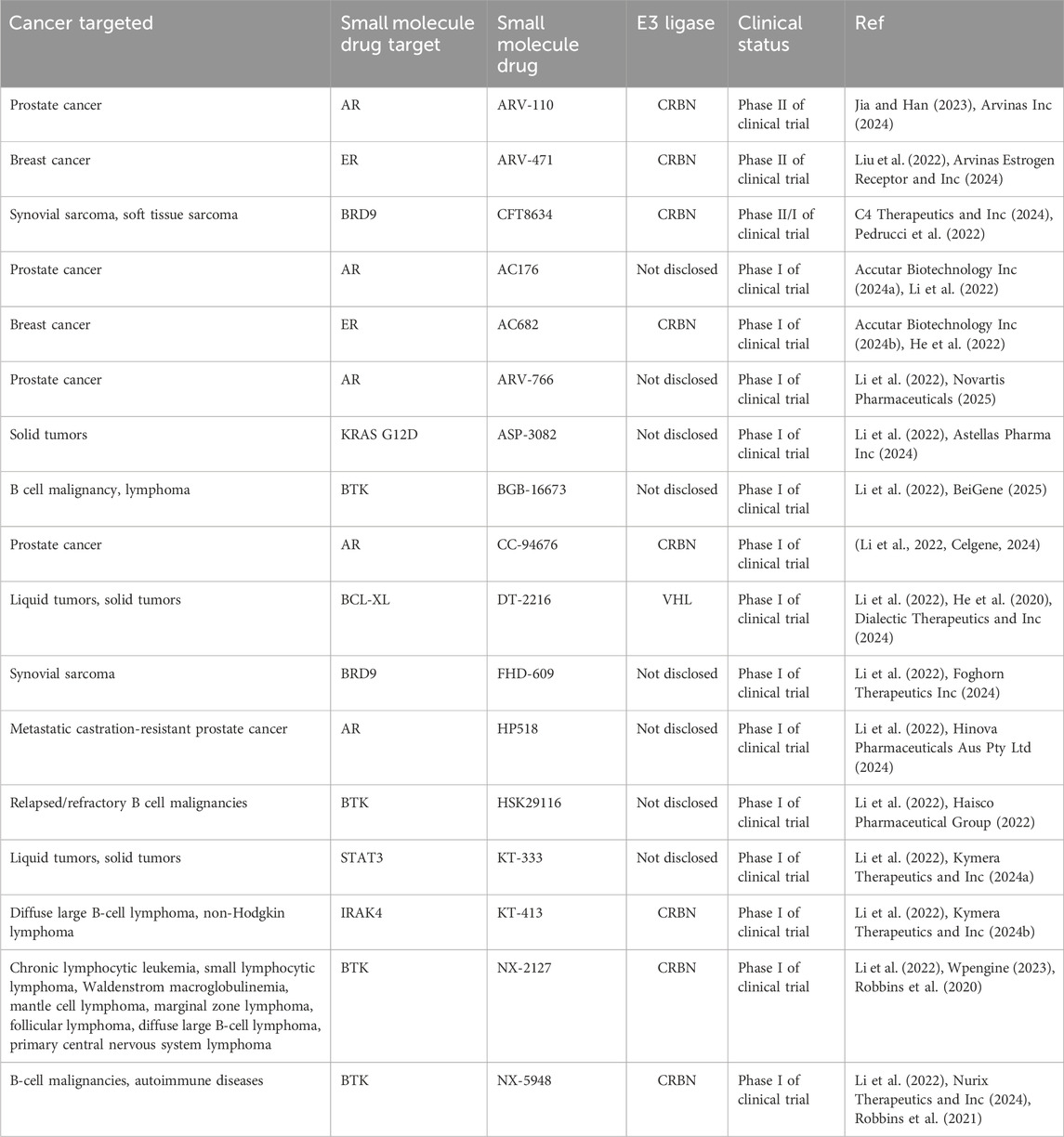
Table 3. PROTACs testing in clinical trials.
Protein degradation is an endogenous recycling mechanism that allows cells to dispose of unwanted proteins, in addition to the aforementioned roles it plays in maintaining overall cell health. Due to its regulatory role in cell cycle, it is of interest to investigate how protein degradation inducers could influence the p53/MDM2 pathway, which is frequently mutated in cancer cells.
The tumor suppressor protein, p53, plays an important role in cell proliferation and cell death. The function of p53 is to act as a regulator of the cell cycle and as a signal for apoptosis when physiological stress of damage occurs. Specifically, p53 will signal for apoptosis when oncogenic stress arises to prevent further proliferation (Vousden and Lu, 2002). Other functions of p53 include DNA repair, senescence, and angiogenesis.
p53 has to be heavily regulated because constant activation would result in excessive apoptosis, leading to accelerated bodily aging (Shi and Gu, 2012). One way p53 is regulated is by having a quick half-life (Rotter, 1983). However, when p53 is mutated in cancer cells, it has a dramatically longer half-life which allows for the constant proliferation of the mutated cancer cells. Unfortunately, around 50% of all cancers involve the loss of anti-tumor function from p53 or suffer from other defects in the signaling pathways that try to alert p53, resulting in cancer cell survival (Vousden and Lu, 2002; Olivier et al., 2010).
In regard to the p53/MDM2 pathway, p53 functions as a transcriptional regulator, modulating the expression of Mouse double minute 2 homolog (MDM2) for p53’s own regulation. MDM2 is an oncoprotein that functions to suppress p53 by binding directly to the protein. When MDM2 binds to p53, it acts as an E3 ligase to promote the degradation of p53 (Freedman et al., 1999; Brooks and Gu, 2006). Therefore, MDM2 works as a negative autoregulatory feedback loop to p53 (Kulikov et al., 2010). There are many ways the p53/MDM2 pathway can be altered in cancer cells to promote cancer survival. One avenue being an increased expression of the p53 mutant proteins which results in the inability to signal for MDM2 expression, preventing p53 degradation and instead, cancer cell proliferation (Sionoc et al., 2024; Blagosklonny, 2000).
There have been many attempts to resolve mutations in this pathway, one proposal was by interfering with MDM2 and p53 interactions by introducing a PPI inhibitor which is described in detail later. Overall, this mechanism aimed to inhibit MDM2 which would lead to cell cycle arrest or apoptosis in cancer cells. While this approach is promising with many ongoing clinical trials, PROTACs have the potential to address the shortcoming seen in MDM2 inhibitors (Wang and Chen, 2022). Since PROTACs serve as E3 ligase recruiters, instead of inhibiting MDM2, it can ubiquitinate MDM2 for its subsequent degradation (Han et al., 2022); therefore, producing similar effects to the MDM2 inhibitors. PROTACs that have been designed to target MDM2 include MD-224, WB214, and TW-32. MD-224 in particular had a respectable performance with reports of its ability to rapidly degrade MDM2 in leukemia cells as well as inhibit growth of leukemia cells with wildtype p53 (Li et al., 2019). Secondly, since MDM2 and PROTACs both act as E3 ligases, PROTACs can ubiquitinate p53 in a similar manner to MDM2. Thus, PROTACs can decrease mutated p53 levels, resulting in reduced, or even halted, cancer cell proliferation.
Through the combination of continuously improving technology and applications of PROTACs in the p53/MDM2 pathway, PROTACs have a wide range of treatment applications, displaying that future efforts that need to be made in the area of protein degradation.
Another exciting area for non-enzymatic drug development involves interfering with PPIs. PPIs are understood as the intentional physical contact between two or more protein molecules due to hydrophobic interaction, hydrogen bonding, or salt-bridges through electrostatic interactions. When two proteins interact directly with each other, their function changes distinctly. PPI networks have shown a recurrent theme in healthy organisms. When mutations, different gene expressions, or diseases occur the PPI interaction networks become altered and disrupt cellular functions (Chatr-Aryamontri et al., 2008; Yeger-Lotem and Sharan, 2015). More precisely, experiments related to PPIs illustrate how signal flow within the networks can be altered by mutations or diseases such as cancer, and therefore shows a path where PPI interfering drugs could be used to reestablish functional homeostasis at molecular, cellular, and tissue levels (Goncearenco et al., 2017).
Conditions for PPI should be explained before any further context is given. Physical contact in PPI is specific, not all proteins that collide with neighboring proteins will interact. Another aspect to note is that the contact between two proteins is neither static nor permanent. Proteins will constantly be modified as well as undergo conformational changes, turnover, and assembly. Few proteins will stay stable since they make up complex, macromolecular protein structures and cellular machines, such as ATP synthase or cytochrome oxidase. Lastly, interactions depend on the type of cell, its stage of development, its stage in the cell cycle, environmental variables, structural alterations, presence of cofactors, and potential other binding partners (De Las Rivas and Fontanillo, 2010). In conclusion, PPIs have to be intentional, and the interaction interface must be specific.
Researchers have commonly approached PPIs with two alternative experiment types: binary and co-complex. The binary approach is a technique that measures direct physical interactions between protein pairs. The common method is to apply the binary method to yeast two-hybrid (Y2H) that utilizes a “bait” and “prey” protein to analyze the transcription of a reporter gene (Suter et al., 2008). The “bait” protein is modified to include a DNA binding domain while the “prey” is modified to include an activation domain for the transcription factor. When the “prey” protein has bound to the “bait” protein, the transcription factor is reconstituted, and transcription of the reporter gene can proceed. When successful in the interaction, a reporter gene is able to produce a color or promote cell growth, both of which can be measured to determine the outcome. Y2H assay has been the favored methodology in PPI research since the majority of PPIs publications are now based on Y2H data. Co-complex functions similarly except that co-complex methods are able to be applied directly or indirectly between the protein pairs (Yu et al., 2008). The co-complex approach is to use tandem affinity purification coupled to mass spectrometry (TAP-MS) (Berggard et al., 2007).
Not all protein complexes can be targeted by small molecules. To discover the protein complexes that would work, Goncearenco et al. evaluated the superposition of preexisting protein-small molecule and protein-protein binding interfaces in structural complexes (Goncearenco et al., 2017). If the binding modes between the two protein-small molecule or protein-protein complexes overlapped, these PPIs were in contention to be druggable. Goncearenco et al. then used Inferred Biomolecule Interaction Server (IBIS) to locate similar binding sites found in homologous proteins. Similarity between homologous proteins was determined by their sequence and structural conservation. The usage of IBIS found that between MDM2 and small molecules, there were three conserved binding sites. The usage of IBIS also illustrated that there was a conserved binding site cluster between MDM2 and p53.
Cancer research efforts have started to design drugs that inhibit PPIs (Figure 2A; Table 4). Petta et al. looked at inhibiting the interaction between MDM2 and p53 because in certain cancers, MDM2 overexpression has been associated with reduced treatment response and poor clinical prognosis (Petta et al., 2016). In light of this, it was suggested that by inhibiting HDM2 (MDM2 in mice) E3 ligase activity, this would cause p53 dependent cell cycle activity to stop, or cause apoptosis in p53-positive stressed cancer cells (Chene, 2003; Davydov et al., 2004).
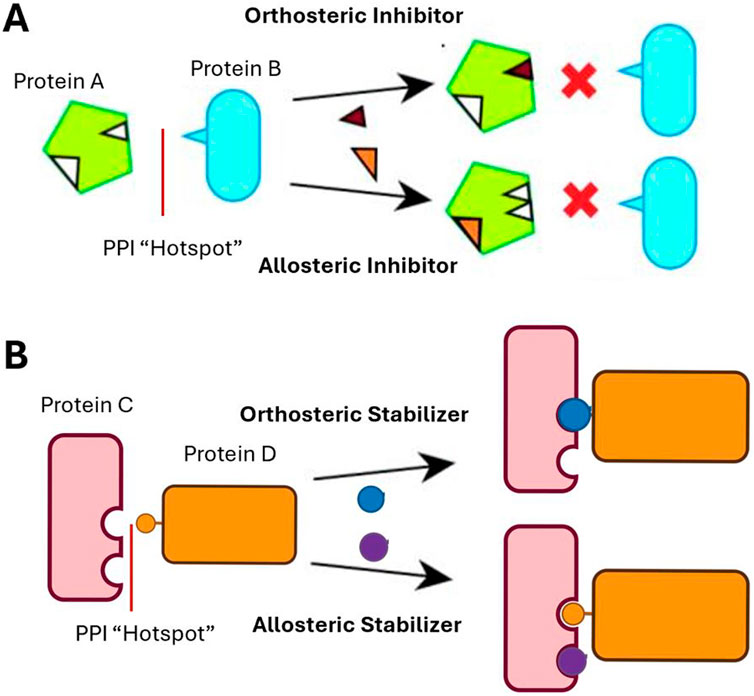
Figure 2. (A) PPI Function–Inhibition. The orthosteric inhibitor binds directly to the interaction area between the two proteins, preventing interactions. The allosteric inhibitor binds to another area, outside of the PPI surface, creating a change in protein shape that inhibits the interaction. (B) PPI Function - Stabilization. The orthosteric stabilizer binds directly to the interaction area between the two proteins, stabilizing the interaction. The allosteric stabilizer binds to an area outside of the PPI surface, creating a change in protein shape, stabilizing the interaction.
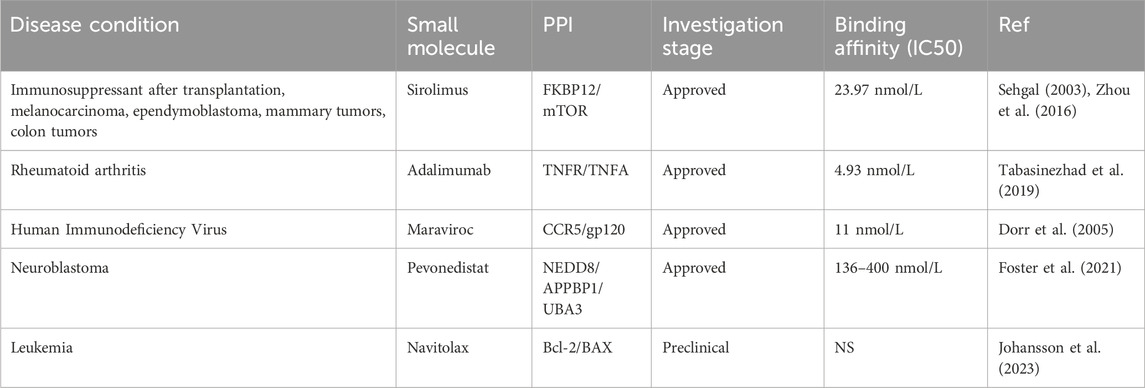
Table 4. List of PPI inhibitors.
A HTS assay was employed to screen small-molecule inhibitors for ubiquitin ligases (E3s). E3 was chosen because this enzyme allows for the conjugation of ubiquitin to the protein that is being targeted through an isopeptide connection (Pickart, 2001). The HTS assay measured the autoubiquitination of RING domain of E3 HDM2 as a read-out (Vousden, 2002). A library that was comprised of 100,000 compounds was screened and potential targets were selected based on the extent of inhibition (e.g., if there was at least 50% inhibition in the wells of the HTS assay) (Davydov et al., 2004). This experiment was able to successfully identify several compounds that were consistently specific for small-molecule HDM2 inhibitors. Therefore, this shows the potential for developing many HDM2 inhibitors for cancer treatment. In addition to identifying HDM2 specific inhibitors, some of the compounds were able to inhibit or activate other E3 ligases. Overall, this study opened up the possibility to discover several other compounds that can be used as PPI inhibitors for cancer treatment.
The first MDM2-p53 inhibitor to enter clinical trials was RG-7112 in 2013 (Vu et al., 2013). Since, there have been many that have enter clinical trials, with the most promising being Alrizomadlin (APG-115) (Ascentage Pharma Group Inc, 2022). Alrizomadlin was granted a fast track designation by the FDA in September 2021 for the treatment of melanoma, and in July 2021, it completed Phase 1 of clinical trials for the treatment of advanced solid tumors or lymphoma (Karlovitch, 2021). Furthermore, Alrizomadlin in combination with a programed death-1 (PD-1) inhibitor, toripalimab, is currently in Phase 1 for liposarcoma and Phase 2 for advanced solid tumors (Ascentage Pharma Group, 2024). Through this approach of altering the abnormal p53/MDM2 pathway, Alrizomadlin works as a MDM2 inhibitor to increase the overexpression of p53 and p21 to induce apoptosis in cancer cells. Interfering with the p53/MDM2 PPI to restore p53 anti-tumor functions shows the potential of how other influential PPIs can be interfered with to treat many cancer types. Ultimately, this approach has limitless opportunities for cancer treatment that need to be investigated further.
Each day, cells undergo replicative stress which results in damage to their respective genomes. Cells resolve these lesions and preserve genome stability through the use of DDR machinery. DDR machinery involves, but is not limited to, homologous recombination (HR), non-homologous end joining (NHEJ), alternative end-joining (A-EJ), nucleotide excision repair (NER), mismatch repair (MMR), and base excision repair (BER) (Ye et al., 2021). While these mechanisms are advantageous for the maintenance of health cells, eventually, the healthy cells reach a point at which excessive DNA damage that cannot be repaired (Wang, 2001). When this occurs, the healthy cells undergo apoptosis. However, in cancer cells, DDR genes become alerted and apoptosis does not occur (Ye et al., 2021). This results in increased genome instability and prolonged cancer cell survival. There are over 450 genes that encode the proteins that are utilized within DDR machinery (McPherson and Korzhnev, 2021). Thus, this provides researchers with numerous opportunities to use PPIs as a form of cancer treatment. Two examples of non-enzymatic PPIs being used to interfere with DDR machinery as a cancer treatment strategy include Mitoxantrone and Peposertib. Mitoxantrone is a HR cancer treatment drug that has been FDA approved for the treatment of acute myeloid leukemia, prostate cancer, and multiple sclerosis (Fox, 2004). In particular, Mitoxantrone targets the RPA:RAD52 PPI which is a key interaction for annealing single stranded DNA (Al-Mugotir et al., 2021; Liang et al., 2024). Peposertib is a medication that capitalizes on the NHEJ mechanism and is currently in FDA clinical trials for the treatment of head tumors, neck tumors, rectal cancer, and HPV-associated cancers (Samuels et al., 2024; Romesser et al., 2024; Gordhandas et al., 2022). Peposertib specifically targets the DNA dependent protein kinase (DNA:PK) interactions which is a crucial aspect of NHEJ. By inhibiting the RPA:RAD52 and DNA:PK interactions, single stranded and double stranded DNA breaks are not amended and cancer cells are forced into apoptosis. Due to the prevalence of non-enzymatic PPIs within the DDR machinery, there are vast amounts of interactions that can be explored by researchers in the future, demonstrating the true potential of PPIs as cancer treatment options.
In the recent years, another class of epigenetic target called bromodomain and extra terminal domain (BET) proteins have been discovered. BET proteins consist of four types of proteins: bromodomain testis-specific protein (BRDT), Brd2, Brd3, and Brd4. The function of BET proteins is to promote and inhibit transcription, as well as aid cell cycle regulation (Ali et al., 2022). BRDT regulates the cell cycle in germ cells, Brd2 and Brd3 are able to initiate transcription by activating the promoter of regulatory genes, and Brd4 functions to elongate transcription by activating promoters and transcription (Ali et al., 2022; LeRoy et al., 2008). Brd4 is also a mitotic bookmark and cell cycle regulator. One of the reasons why inhibitors of BET proteins have received attention is due to their overexpression in a wide range of tumor types (Segura et al., 2013). In addition, since they consist of four types of proteins, this proposes numerous avenues for PPI inhibition.
To prevent BET protein interactions, several novel BET protein inhibitors that use the PPI approach have recently been developed. Various degrees of anti-tumor activity have been found with BET PPI inhibitors and new treatment options are being investigated by combining these PPIs with either regulators or small protein inhibitors (Belkina and Denis, 2012). BET PPI inhibitors work by causing cell cycle arrest and transcriptional repression. Prior to testing, one of the anticipated issues of these inhibitors was that BET PPI inhibitors would cause general transcriptional repression, damaging the individual’s healthy tissue. However, preclinical models proved that BET PPI inhibitors did not affect normal tissues and only targeted tumor cells. This was as a result of the BET PPI inhibitors having preference for binding to super enhancers, which are more prominent in tumor cells than healthy tissues (Pott and Lieb, 2015; Chapuy et al., 2013).
As of now, results from early clinical data have shown mixed results for BET PPI inhibitors as a monotherapy. More specifically, the results indicated positive treatments within a matter of weeks for some individuals, while other showed progress after 8 months (Stathis et al., 2016). Thus, BET PPI inhibitors as a monotherapy have not been proven to have a dependable outcome; however, the 35 ongoing or completed clinical trials hope to change that in the near future (Sarnik et al., 2021). With this, preclinical data has shown promising results when BET PPI inhibitors are combined with other agents. This appears to be because BET PPI inhibitors have been able to overcome resistance to a single target agent or collaboration with other epigenetic agents for immune checkpoint inhibitors (Doroshow et al., 2017).
PPIs are a broad target to tackle since they have diverse and complex qualities. For example, PPI size can range from 4 amino acids to thousands of angstroms long, and each is characterized by its dynamics, binding affinities, number of proteins in the complex (Wade et al., 2004). Nevertheless, HTS programs have been adapted to find PPI targets which have allowed for a more rapid discovery of PPI interfaces (Taylor et al., 2018). Two HTS programs to note are the Alpha-Lisa and split-GFP programs which specializes in PPIs (Beaudet et al., 2008; Feng et al., 2017).
Since the PPIs of interest for cancer treatment frequently have weak binding affinities or large, shallow interfaces, fundamental HTS was not able to initially screen complex PPIs (Smith and Gestwicki, 2012). This was due to many reasons, with of the primarily ones being that most of the PPI interfaces are located within protein complexes that have three or more different components, making them difficult to access through HTS. In attempts to identify how complex PPIs can be analyzed by HTS, the molecular chaperone, heat shock protein 70 (Hsp70), served as a model system due to its complex and tightly regulated structure. Hsp70 is a well analyzed multiprotein complex that is highly regulated by its co-chaperones: nucleotide exchange factors (NEFs), BAGs, and J-domain proteins (also known as Hsp40s or DNAJs) (Chiappori et al., 2016). Hsp70 is made of an 44ka ATPase N-terminal nucleotide-binding domain (NBD), a 18kD C-terminal substrate-binding domain (SBD), and a 10kD C-terminal domain (CTD) (Sharma and Masison, 2009). When ATP enters the NBD, J-domain proteins stimulate ATP hydrolysis and NEFs promote ADP release. The combination of these co-chaperones has been shown to increase the steady-state ATP hydrolysis by approximately 200-fold, one reason why it is a prominent model system for analyzing complex PPIs (Sharma and Masison, 2009; McCarty et al., 1995). In addition, the NEFs interact with Hsp70 over a large, multi-subdomain area and the J-proteins weakly interact with Hsp70 over a large, buried area that is highly polar (Xu et al., 2008; Ahmad et al., 2011). Ultimately, if HTS was able to screen for the complex Hsp70, the potential to identify compounds for complex PPIs for cancer treatment existed.
In experiment by Taylor et al., Hsp70 was used in an HTS assay and had two positive results, highlighting that HTS can be used for complex PPIs (Taylor et al., 2018). In the HTS assay, Taylor et al. used human Hsp70 with co-chaperones BAG2 and DNAJA2 to recreate the ternary protein structure. This complex was then screened by 100,000 different compounds that inhibit co-chaperone-stimulated ATPase activity using HTS (Taylor et al., 2018; Miyata et al., 2010). Two compounds were able to deemed successful, Compound F and Compound R. Explicitly, Compound R was able to inhibit the Hsp70-BAG2 interaction and Compound F was able to inhibit the Hsp70-DNAJ interaction, blocking approximately 80% of steady-state turnover (Taylor et al., 2018; Bonomo et al., 2010; Chang et al., 2011). Thus, HTS has shown a promising start to towards identifying complex PPIs inhibitors and this approach should be further explored.
Another approach to drug design is to stabilize PPIs instead of inhibiting their activity (Figure 2B). One function for small molecule PPI stabilizers is to use allosteric stabilizers to associate with one of the proteins within the complex (Petta et al., 2016) (Figure 2B). In doing so, the allosteric stabilizers increase the binding affinity of both proteins involved in the PPI. Similarly, the other function for small molecule PPI stabilizers to increase binding affinity is by binding to the interfacial surface of the protein complex. Therefore, by increasing binding affinity of the PPIs, the complex is more stable and less likely to dissociate.
PPI stabilization is used in the clinic as an anti-cancer treatment by disrupting the cell cycle. For the cell cycle to occur properly, microtubules function to transfer material within the cell while also influencing cell anatomy through shortening and lengthening of individual microtubules (Downing, 2000). Microtubules are made by protein heterodimers of alpha and beta tubulin. To fulfill all of these tasks, the microtubules must be able to rearrange regularly by consistent polymerization and depolymerization (Inoue and Sato, 1967). Because microtubules contribute such a significant role in cell growth and cell division, microtubules have served as one of the best cancer targets for researchers to work with PPI stabilizers (Mukhtar et al., 2014). In the late 1900s, the U.S. Department of Agriculture (USDA) and the National Cancer Institute (NCI) worked together on a program to identify natural compounds with anti-cancer activity (Weaver, 2014; Orr et al., 2003). This involved screening 15,000 different species of plants and 115,000 plant extracts. Of these, the bark of a Pacific yew tree sample, Taxus brevifoliai, that was collected by USDA botanist Arthur Barclay, was found to have cytotoxic properties. This anti-cancer agent, later known as Paclitaxel, was shown positive results in disrupting the microtubules. The Taxus brevifoliai-derived Paclitaxel was able to disrupt the breakdown of microtubules because of its high affinity for a hydrophobic pocket of polymerized tubulin that is located on the beta subunit of the microtubule structure (Orr et al., 2003; Nogales et al., 1995). Paclitaxel binds to the microtubule and stabilizes the structure in an allosteric manner, preventing the cell cycle from progressing (Thiel et al., 2012; Rohena and Mooberry, 2014). Therefore, this prevents cancer cells from proliferating and thus serves as a treatment through PPI stabilization. By 1994, the FDA had approved of the ingredients derived from Taxus brevifoliai for the treatment of breast cancer and since, it has been used in the treatment of colorectal, bladder, lung, and ovarian cancer as well as Kaposi’s sarcoma (National Cancer Institute, 2023; Zhu and Chen, 2019).
Another example, and a more novel target class that involves PPI stabilization, is the 14-3-3 proteins. 14-3-3 proteins are a family of homologous proteins comprised of seven isoforms. Most of these proteins are expressed in the brain and have been found in the cerebrospinal fluid of patients with various neurological disorders (Berg et al., 2003). Some of these neurological disorders include, but are not limited to, Parkinson’s disease, Alzheimer’s disease, Creutzfeldt-Jakob disease, lissencephaly, schizophrenia, and bipolar disorder (Foote and Zhou, 2012). Because 14-3-3 proteins are capable of binding to more than 100 different binding sites, research has focused on looking into 14-3-3 proteins as a potential new PPI pharmacological intervention by either stabilization or inhibition.
A more focused interaction partner that has been experimented on is the interaction between 14-3-3 proteins and its interaction partner, TASK3. TASK3 is a pore-domain potassium channel that contains a conserved C-terminal of five amino acids (Rajan et al., 2002). TASK3 is prominently expressed in the central nervous system and is involved in cell cycle management, apoptosis, cell signaling, and regulation of protein kinases (Rajan et al., 2002). Potassium channels are critical since they are involved the signal pathways that manage cell death and proliferation (Kunzelmann, 2005; Lang et al., 2005). When TASK3 is inhibited, potassium channels cannot function properly and data has shown that there is decreased cell proliferation in embryonic stem cells, hepatocarcinoma cells, breast and prostate cancer cells, and melanoma cells when this occurs (Bittner et al., 2010). Unbalanced control of TASK3 has been shown to increase the rates of inflammation, epilepsy, and cancer. Consequently, stabilization of 14-3-3 proteins and TASK3 interactions is an increasing area of interest for PPI interference.
Inhibition of estrogen receptor alpha (ER alpha) binding to 14-3-3 proteins also has shown promising results to combat breast cancer. A current breast cancer treatment is to block estrogen production through anti-estrogens or aromatase competitive inhibitors that compete for hormone binding (De Vries-van Leeuwen et al., 2013). Due to varying resistances, there is a need to find alternative treatments for breast cancer. One small molecule that has been able to inhibit ER alpha binding to 14-3-3 proteins is Fuscicoccin (Stevers et al., 2018). Fusicoccin can stabilize the 14-3-3/ER alpha site by binding directly to the interface rim. This alternative method of using Fusicoccin can diminish estradiol-mediated ER dimerization, limit ER binding to chromatin, and downstream gene activation, all resulting in decreased cell proliferation (De Vries-van Leeuwen et al., 2013).
Immunosuppression is the partial or complete repression of the body’s immune system. Often times, drugs will be deliberately used to suppress the immune system in order to prepare for transplantations, such as bone marrow or other organs. Immunosuppression is important in order to prevent the body’s natural system from rejecting the donor tissue. Rapamycin (sirolimus) and FK506 (tacrolimus) are two PPI stabilizing molecules that interact directly with FKBP12 immunophilin (Vellanki et al., 2020) (Table 5). These molecules are well-known in the clinic as immunosuppressants. Rapamycin and FK506 share a common mechanism of action, which is to stabilize FKBP12/protein phosphatase calcineurin and FKBP12/mTOR interactions, respectively. Stabilization of these domains allows temporary control of cell signaling. Additionally, these domains are able to control cell growth, induce G protein-coupled receptor (GPCR) signaling, and transfer proteins to either the nucleus or plasma membrane (Coutinho-Budd et al., 2013). Experiments have proven that the resulting Rapamycin/FKBP12 interaction produces a toxic complex that interferes with intracellular signaling during G1 of interphase, therefore impacting cell proliferation (Sabers et al., 1995). Lastly, a small molecular known as Mizoribine is also currently being studied in combination with 14-3-3η and glucocorticoid receptors for its potential to enhance immunosuppression (Takahashi et al., 2000). While these PPI drugs are not being directly used as cancer therapies, their applications within cancer treatment can significantly benefit treatment patient outcomes.
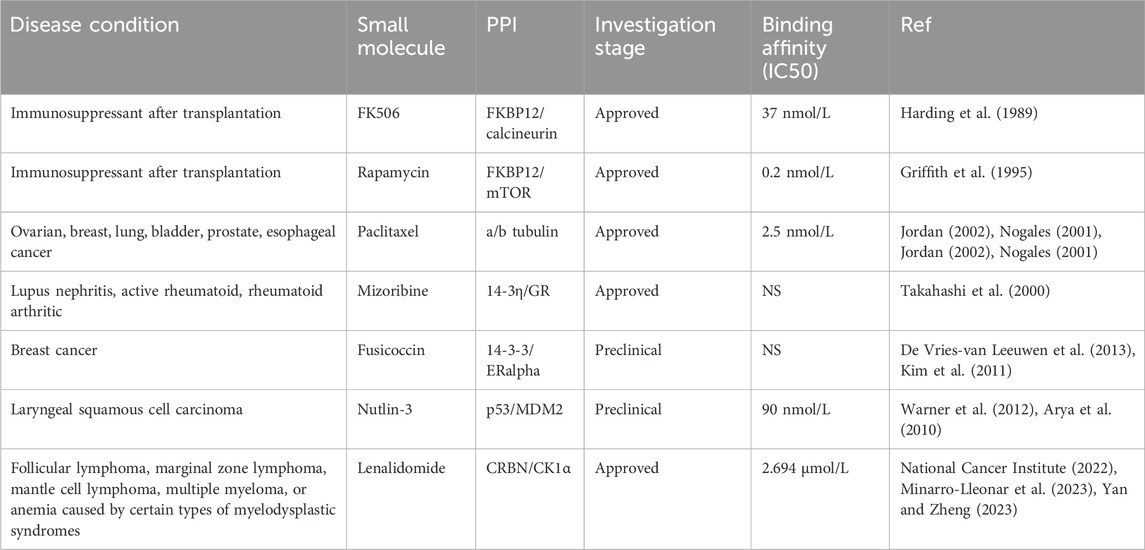
Table 5. List of PPI stabilizers.
While PPIs interfering and protein degrading drugs have displayed promising results, there are challenges of using these approaches. For instance, it is difficult to develop PPI interfering drugs since the majority of the protein-protein interfaces are flat and lack pockets for PPI modulators to bind to (Jubb et al., 2017; Lu et al., 2020). A solution that has been proposed is to predict an area of DNA that is likely to mutate (hot spot) that the PPI interfering drug can bind to. Predicting hotspots can be identified in silico, by either 1) analyzing how the binding sites have been conserved evolutionarily or 2) by scanning of PPI interfaces and determining if certain amino acid substitutions would alter the binding affinity (Goncearenco et al., 2017). Two software programs that have been created to assist in locating hot spots are MutaBind2 and Hot-Region. Mutabind utilizes molecular mechanics force fields, statistical potentials, and fast side-chain optimization algorithms to determine the impact of either protein variants or mutations on the binding affinity of the protein complex (MutaBind2, 2024; Li et al., 2016). In addition to determining the binding affinity, Mutabind provides a map, structural model, and determines the deleterious effect of a mutation on the protein along with a confidence interval of the generated prediction. HotRegion, on the other hand, utilizes residue network topology (the study of geometric properties and spatial relations) and a statistical pairwise contact energy function to determine the impact of mutations on the protein (PRISM, 2024). By using Mutabind and HotRegion, this allows researchers to digitally identify how mutations can alter protein structure and create binding pockets for PPI interfering drugs to interact with.
Although protein degradation and PPI based treatments are highly specific, like all cancer therapies, the possibility of developing drug resistance exists (Zheng, 2017). A potential solution to prevent tumor drug resistance is to design the drug to be able to continue functioning when the target proteins undergo extensive selection in the tumor (Goncearenco et al., 2017). Extensive clonal selection allows the tumor cells to maintain the necessary interactions between the target proteins, but eliminate the drug binding site, causing drug resistance. If this can be overcome, drug resistance can be reduced. One inhibitor that has attempted to overcome drug resistance is the MDM2 inhibitor, Nutlin-3. Nutlin-3 exhibits anti-cancer effects, even in cells that do not express functional p53 (Kumamoto et al., 2008). In addition, other p53/MDM2 PPI inhibitors that are currently in clinical trials, as well as emerging peptide inhibitors, have resisted the extensive tumor clonal selection that leads to drug resistance. Overall, there are a few shortcomings in these novel approaches, however, they are effectively being addressed and still hold promising results.
Alternatives for the current treatments of cancer such as radiation therapy, chemotherapy, and surgery have been at the center of investigation to reduce the damage of healthy cells. As a result, efforts are being made to look towards novel therapeutic approaches through the concepts of targeted protein degradation and PPI interference. These concepts have led to the development of inhibitors that exclusively target cancer cells, with several receiving FDA approval and many in clinical trials. Protein degradation continues to show its promising potential as a new cancer treatment, specifically in regard to PROTACs. Furthermore, PPI interfering drugs continue to display significant results within the development of p53/MDM2 inhibitors, BET protein inhibitors, and as immunosuppressants. With the assistance from HTS, protein degradation and PPI interfering drugs have progressed immensely, and targets have been discovered. In the future, we suggest that these avenues should continue to be investigated and that BET protein inhibitors should be combined with other agents due to their promising preclinical results. Overall, protein degradation and PPI interfering drugs promise better cancer treatment options than the current approaches and should be further explored.
MA: Writing–original draft, Writing–review and editing. JL: Writing–original draft, Writing–review and editing. AS: Writing–original draft, Writing–review and editing. ZA: Writing–original draft, Writing–review and editing. GP: Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by NCI Cancer Center Support Grant CA016672 to The University of Texas MD Anderson Cancer Center and NIH R01 grant 2CA181663 to G.P.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelmalak, M., Singh, R., Anwer, M., Ivanchenko, P., Randhawa, A., Ahmed, M., et al. (2022). The renaissance of CDK inhibitors in breast cancer therapy: an update on clinical trials and therapy resistance. Cancers (Basel) 14 (21), 5388. doi:10.3390/cancers14215388
Accutar Biotechnology Inc (2024a). A study of AC176 for the treatment of metastatic castration resistant prostate cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT05241613.
Accutar Biotechnology Inc (2024b). A study of AC682 for the treatment of locally advanced or metastatic ER+ breast cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT05080842.
Adams, V. R., and Leggas, M. (2007). Sunitinib malate for the treatment of metastatic renal cell carcinoma and gastrointestinal stromal tumors. Clin. Ther. 29 (7), 1338–1353. doi:10.1016/j.clinthera.2007.07.022
Ahmad, A., Bhattacharya, A., McDonald, R. A., Cordes, M., Ellington, B., Bertelsen, E. B., et al. (2011). Heat shock protein 70 kDa chaperone/DnaJ cochaperone complex employs an unusual dynamic interface. Proc. Natl. Acad. Sci. U. S. A. 108 (47), 18966–18971. doi:10.1073/pnas.1111220108
Ahn, G., Banik, S. M., Miller, C. L., Riley, N. M., Cochran, J. R., and Bertozzi, C. R. (2021). LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 17 (9), 937–946. doi:10.1038/s41589-021-00770-1
Ahn, I. E., and Brown, J. R. (2021). Targeting bruton's tyrosine kinase in CLL. Front. Immunol. 12, 687458. doi:10.3389/fimmu.2021.687458
Ali, H. A., Li, Y., Bilal, A. H. M., Qin, T., Yuan, Z., and Zhao, W. (2022). A comprehensive review of BET protein biochemistry, physiology, and pathological roles. Front. Pharmacol. 13, 818891. doi:10.3389/fphar.2022.818891
Al-Mugotir, M., Lovelace, J. J., George, J., Bessho, M., Pal, D., Struble, L., et al. (2021). Selective killing of homologous recombination-deficient cancer cell lines by inhibitors of the RPA:RAD52 protein-protein interaction. PLoS One 16 (3), e0248941. doi:10.1371/journal.pone.0248941
Anwar, K., Nguyen, L., Nagasaka, M., Ou, I., and Chan, A. (2022). Overview of drug-drug interactions between ritonavir-boosted nirmatrelvir (paxlovid) and targeted therapy and supportive care for lung cancer. JTO Clin. Res. Rep. 4 (2), 100452. doi:10.1016/j.jtocrr.2022.100452
Arvinas Estrogen Receptor, Inc. (2024). A phase 1/2 trial of ARV-471 alone and in combination with palbociclib (IBRANCE®) in patients with ER+/HER2- locally advanced or metastatic breast cancer (mBC). Available at: https://clinicaltrials.gov/ct2/show/NCT04072952.
Arvinas Inc. (2024). Trial of ARV-110 in patients with metastatic castration resistant prostate cancer (mCRPC). Available at: https://clinicaltrials.gov/ct2/show/NCT03888612.
Arya, A. K., El-Fert, A., Devling, T., Eccles, R. M., Aslam, M. A., Rubbi, C. P., et al. (2010). Nutlin-3, the small-molecule inhibitor of MDM2, promotes senescence and radiosensitises laryngeal carcinoma cells harbouring wild-type p53. Br. J. Cancer 103 (2), 186–195. doi:10.1038/sj.bjc.6605739
Ascentage Pharma Group (2024). APG-115 in combination with PD-1 inhibitor in patients with advanced liposarcoma or advanced solid tumors. Available at: https://clinicaltrials.gov/ct2/show/NCT04785196.
Ascentage Pharma Group Inc. (2022). APG-115 in patients with advanced solid tumors or lymphomas (APG-115). Available at: https://clinicaltrials.gov/ct2/show/NCT02935907.
Astellas Pharma Inc (2024). A study of ASP3082 in adults with previously treated solid tumors. Available at: https://clinicaltrials.gov/ct2/show/NCT05382559.
Attwa, M. W., Darwish, H. W., Al-Shakliah, N. S., and Kadi, A. A. (2021). A validated LC-MS/MS assay for the simultaneous quantification of the FDA-approved anticancer mixture (encorafenib and binimetinib): metabolic stability estimation. Molecules 26 (9), 2717. doi:10.3390/molecules26092717
Banik, S. M., Pedram, K., Wisnovsky, S., Ahn, G., Riley, N. M., and Bertozzi, C. R. (2020). Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584 (7820), 291–297. doi:10.1038/s41586-020-2545-9
Bauer, S., George, S., von Mehren, M., and Heinrich, M. C. (2021). Early and next-generation KIT/PDGFRA kinase inhibitors and the future of treatment for advanced gastrointestinal stromal tumor. Front. Oncol. 11, 672500. doi:10.3389/fonc.2021.672500
Baumeister, W., Walz, J., Zuhl, F., and Seemuller, E. (1998). The proteasome: paradigm of a self-compartmentalizing protease. Cell 92 (3), 367–380. doi:10.1016/s0092-8674(00)80929-0
Beaudet, L., Rodriguez-Suarez, R., Venne, M.-H., Caron, M., Bédard, J., Brechler, V., et al. (2008). AlphaLISA immunoassays: the no-wash alternative to ELISAs for research and drug discovery. Nat. Methods 5 (12), an8–an9. doi:10.1038/nmeth.f.230
BeiGene (2025). A phase 1 dose-escalation and expansion study of BGB-16673 in patients with B-cell malignancies. Available at: https://clinicaltrials.gov/ct2/show/NCT05006716.
Bekes, M., Langley, D. R., and Crews, C. M. (2022). PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21 (3), 181–200. doi:10.1038/s41573-021-00371-6
Belkina, A. C., and Denis, G. V. (2012). BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 12 (7), 465–477. doi:10.1038/nrc3256
Berg, D., Holzmann, C., and Riess, O. (2003). 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 4 (9), 752–762. doi:10.1038/nrn1197
Berggard, T., Linse, S., and James, P. (2007). Methods for the detection and analysis of protein-protein interactions. Proteomics 7 (16), 2833–2842. doi:10.1002/pmic.200700131
Bittner, S., Budde, T., Wiendl, H., and Meuth, S. G. (2010). From the background to the spotlight: TASK channels in pathological conditions. Brain Pathol. 20 (6), 999–1009. doi:10.1111/j.1750-3639.2010.00407.x
Blagosklonny, M. V. (2000). p53 from complexity to simplicity: mutant p53 stabilization, gain-of-function, and dominant-negative effect. FASEB J. 14 (13), 1901–1907. doi:10.1096/fj.99-1078rev
Blakely, C. M., Weder, W., Bubendorf, L., He, J., Majem, M., Shyr, Y., et al. (2023). Primary endpoints to assess the efficacy of novel therapeutic approaches in epidermal growth factor receptor-mutated, surgically resectable non-small cell lung cancer: a review. Lung Cancer 177, 59–72. doi:10.1016/j.lungcan.2023.01.002
Bondeson, D. P., Mares, A., Smith, I. E., Ko, E., Campos, S., Miah, A. H., et al. (2015). Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11 (8), 611–617. doi:10.1038/nchembio.1858
Bondeson, D. P., Smith, B. E., Burslem, G. M., Buhimschi, A. D., Hines, J., Jaime-Figueroa, S., et al. (2018). Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 25 (1), 78–87 e5. doi:10.1016/j.chembiol.2017.09.010
Bonomo, J., Welsh, J. P., Manthiram, K., and Swartz, J. R. (2010). Comparing the functional properties of the Hsp70 chaperones, DnaK and BiP. Biophys. Chem. 149 (1-2), 58–66. doi:10.1016/j.bpc.2010.04.001
Brazel, D., Kroening, G., and Nagasaka, M. (2022). Non-small cell lung cancer with EGFR or HER2 exon 20 insertion mutations: diagnosis and treatment options. BioDrugs 36 (6), 717–729. doi:10.1007/s40259-022-00556-4
Brooks, C. L., and Gu, W. (2006). p53 ubiquitination: mdm2 and beyond. Mol. Cell 21 (3), 307–315. doi:10.1016/j.molcel.2006.01.020
Brown, R. M., Farouk Sait, S., Dunn, G., Sullivan, A., Bruckert, B., and Sun, D. (2022). Integrated drug mining reveals actionable strategies inhibiting plexiform neurofibromas. Brain Sci. 12 (6), 720. doi:10.3390/brainsci12060720
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 (7035), 913–917. doi:10.1038/nature03443
Buckley, D. L., Raina, K., Darricarrere, N., Hines, J., Gustafson, J. L., Smith, I. E., et al. (2015). HaloPROTACS: use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem. Biol. 10 (8), 1831–1837. doi:10.1021/acschembio.5b00442
Buckley, D. L., Van Molle, I., Gareiss, P. C., Tae, H. S., Michel, J., Noblin, D. J., et al. (2012). Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc. 134 (10), 4465–4468. doi:10.1021/ja209924v
C4 Therapeutics, Inc. (2024). A study to assess the safety and tolerability of CFT8634 in locally advanced or metastatic SMARCB1-perturbed cancers, including synovial sarcoma and SMARCB1-null tumors. Available at: https://clinicaltrials.gov/ct2/show/NCT05355753.
Caquelin, L., Gewily, M., Mottais, W., Tebaldi, C., Laviolle, B., Naudet, F., et al. (2022). Tivozanib in renal cell carcinoma: a systematic review of the evidence and its dissemination in the scientific literature. BMC Cancer 22 (1), 381. doi:10.1186/s12885-022-09475-7
Celgene (2024). Study to evaluate the safety and tolerability of CC-94676 in participants with metastatic castration-resistant prostate cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT04428788.
Chang, L., Miyata, Y., Ung, P. M., Bertelsen, E. B., McQuade, T. J., Carlson, H. A., et al. (2011). Chemical screens against a reconstituted multiprotein complex: myricetin blocks DnaJ regulation of DnaK through an allosteric mechanism. Chem. Biol. 18 (2), 210–221. doi:10.1016/j.chembiol.2010.12.010
Chapuy, B., McKeown, M. R., Lin, C. Y., Monti, S., Roemer, M. G., Qi, J., et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24 (6), 777–790. doi:10.1016/j.ccr.2013.11.003
Chatr-Aryamontri, A., Ceol, A., Licata, L., and Cesareni, G. (2008). Protein interactions: integration leads to belief. Trends Biochem. Sci. 33 (6), 241–243. doi:10.1016/j.tibs.2008.04.002
Chen, D., Frezza, M., Schmitt, S., Kanwar, J., and Dou, Q. P. (2011). Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets 11 (3), 239–253. doi:10.2174/156800911794519752
Chen, L., Zhang, Y., Yin, L., Cai, B., Huang, P., Li, X., et al. (2021). Fibroblast growth factor receptor fusions in cancer: opportunities and challenges. J. Exp. Clin. Cancer Res. 40 (1), 345. doi:10.1186/s13046-021-02156-6
Chene, P. (2003). Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat. Rev. Cancer 3 (2), 102–109. doi:10.1038/nrc991
Chiappori, F., Merelli, I., Milanesi, L., Colombo, G., and Morra, G. (2016). An atomistic view of Hsp70 allosteric crosstalk: from the nucleotide to the substrate binding domain and back. Sci. Rep. 6, 23474. doi:10.1038/srep23474
Ciehanover, A., Hod, Y., and Hershko, A. (1978). A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 81 (4), 1100–1105. doi:10.1016/0006-291x(78)91249-4
Cohen, M. H., Johnson, J. R., Chen, Y. F., Sridhara, R., and Pazdur, R. (2005). FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist 10 (7), 461–466. doi:10.1634/theoncologist.10-7-461
Cohen, M. H., Williams, G., Johnson, J. R., Duan, J., Gobburu, J., Rahman, A., et al. (2002). Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin. Cancer Res. 8 (5), 935–942.
Cohen, M. H., Williams, G. A., Sridhara, R., Chen, G., and Pazdur, R. (2003). FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist 8 (4), 303–306. doi:10.1634/theoncologist.8-4-303
Conlon, N. T., Kooijman, J. J., van Gerwen, S. J. C., Mulder, W. R., Zaman, G. J. R., Diala, I., et al. (2021). Comparative analysis of drug response and gene profiling of HER2-targeted tyrosine kinase inhibitors. Br. J. Cancer 124 (7), 1249–1259. doi:10.1038/s41416-020-01257-x
Cooper, M. R., Yi, S. Y., Alghamdi, W., Shaheen, D. J., and Steinberg, M. (2014). Vandetanib for the treatment of medullary thyroid carcinoma. Ann. Pharmacother. 48 (3), 387–394. doi:10.1177/1060028013512791
Cortes, J. E., Kim, D. W., Kantarjian, H. M., Brummendorf, T. H., Dyagil, I., Griskevicius, L., et al. (2012). Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J. Clin. Oncol. 30 (28), 3486–3492. doi:10.1200/JCO.2011.38.7522
Coutinho-Budd, J. C., Snider, S. B., Fitzpatrick, B. J., Rittiner, J. E., and Zylka, M. J. (2013). Biological constraints limit the use of rapamycin-inducible FKBP12-Inp54p for depleting PIP2 in dorsal root ganglia neurons. J. Negat. Results Biomed. 12, 13. doi:10.1186/1477-5751-12-13
Cromm, P. M., and Crews, C. M. (2017). Targeted protein degradation: from chemical biology to drug discovery. Cell Chem. Biol. 24 (9), 1181–1190. doi:10.1016/j.chembiol.2017.05.024
Dasso, L., Al-Khaled, T., Sonty, S., and Aref, A. A. (2018). Profile of netarsudil ophthalmic solution and its potential in the treatment of open-angle glaucoma: evidence to date. Clin. Ophthalmol. 12, 1939–1944. doi:10.2147/OPTH.S154001
Davydov, I. V., Woods, D., Safiran, Y. J., Oberoi, P., Fearnhead, H. O., Fang, S., et al. (2004). Assay for ubiquitin ligase activity: high-throughput screen for inhibitors of HDM2. J. Biomol. Screen 9 (8), 695–703. doi:10.1177/1087057104267956
De Las Rivas, J., and Fontanillo, C. (2010). Protein-protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 6 (6), e1000807. doi:10.1371/journal.pcbi.1000807
Desai, A., and Cuellar, S. (2022). The current landscape for METex14 skipping mutations in non-small cell lung cancer. J. Adv. Pract. Oncol. 13 (5), 539–544. doi:10.6004/jadpro.2022.13.5.8
De Vries-van Leeuwen, I. J., da Costa Pereira, D., Flach, K. D., Piersma, S. R., Haase, C., Bier, D., et al. (2013). Interaction of 14-3-3 proteins with the estrogen receptor alpha F domain provides a drug target interface. Proc. Natl. Acad. Sci. U. S. A. 110 (22), 8894–8899. doi:10.1073/pnas.1220809110
Dialectic Therapeutics, Inc (2024). A study of DT2216 in relapsed/refractory malignancies. Available at: https://clinicaltrials.gov/ct2/show/NCT04886622.
Diehl, C. J., and Ciulli, A. (2022). Discovery of small molecule ligands for the von Hippel-Lindau (VHL) E3 ligase and their use as inhibitors and PROTAC degraders. Chem. Soc. Rev. 51 (19), 8216–8257. doi:10.1039/d2cs00387b
Doroshow, D. B., Eder, J. P., and LoRusso, P. M. (2017). BET inhibitors: a novel epigenetic approach. Ann. Oncol. 28 (8), 1776–1787. doi:10.1093/annonc/mdx157
Dorr, P., Westby, M., Dobbs, S., Griffin, P., Irvine, B., Macartney, M., et al. (2005). Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49 (11), 4721–4732. doi:10.1128/AAC.49.11.4721-4732.2005
Downing, K. H. (2000). Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu. Rev. Cell Dev. Biol. 16, 89–111. doi:10.1146/annurev.cellbio.16.1.89
Drilon, A., Oxnard, G. R., Tan, D. S. W., Loong, H. H. F., Johnson, M., Gainor, J., et al. (2020). Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N. Engl. J. Med. 383 (9), 813–824. doi:10.1056/NEJMoa2005653
Ebrahimpour, A., Ahir, M., Wang, M., Jegga, A. G., Bonnen, M. D., Eissa, N. T., et al. (2022). Combination of esomeprazole and pirfenidone enhances antifibrotic efficacy in vitro and in a mouse model of TGFβ-induced lung fibrosis. Sci. Rep. 12 (1), 20668. doi:10.1038/s41598-022-24985-x
Elia, G., Patrizio, A., Ragusa, F., Paparo, S. R., Mazzi, V., Balestri, E., et al. (2022). Molecular features of aggressive thyroid cancer. Front. Oncol. 12, 1099280. doi:10.3389/fonc.2022.1099280
Escudier, B., and Gore, M. (2011). Axitinib for the management of metastatic renal cell carcinoma. Drugs R. D. 11 (2), 113–126. doi:10.2165/11591240-000000000-00000
European Organisation for Research and Treatment of Cancer (2024). A phase III trial of with marizomib in patients with newly diagnosed glioblastoma (MIRAGE). Available at: https://clinicaltrials.gov/ct2/show/NCT03345095.
Farley, K., Bhattacharya, S., Cleland, J., Chandran, P., and Wu, J. (2024). The targeted protein degradation landscape. Nat. Rev. Drug Discov. doi:10.1038/d41573-024-00187-0
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434 (7035), 917–921. doi:10.1038/nature03445
Feng, S., Sekine, S., Pessino, V., Li, H., Leonetti, M. D., and Huang, B. (2017). Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun. 8 (1), 370. doi:10.1038/s41467-017-00494-8
Finley, D., Ciechanover, A., and Varshavsky, A. (1984). Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85. Cell 37 (1), 43–55. doi:10.1016/0092-8674(84)90299-x
Foghorn Therapeutics Inc. (2024). FHD-609 in subjects with advanced synovial sarcoma or advanced SMARCB1-loss tumors. Available at: https://clinicaltrials.gov/ct2/show/NCT04965753.
Foote, M., and Zhou, Y. (2012). 14-3-3 proteins in neurological disorders. Int. J. Biochem. Mol. Biol. 3 (2), 152–164.
Foster, J. H., Barbieri, E., Zhang, L., Scorsone, K. A., Moreno-Smith, M., Zage, P., et al. (2021). The anti-tumor activity of the NEDD8 inhibitor pevonedistat in neuroblastoma. Int. J. Mol. Sci. 22 (12), 6565. doi:10.3390/ijms22126565
Fox, E. J. (2004). Mechanism of action of mitoxantrone. Neurology 63 (12 Suppl. 6), S15–S18. doi:10.1212/wnl.63.12_suppl_6.s15
Freedman, D. A., Wu, L., and Levine, A. J. (1999). Functions of the MDM2 oncoprotein. Cell Mol. Life Sci. 55 (1), 96–107. doi:10.1007/s000180050273
Garciaz, S., and Hospital, M. A. (2023). FMS-like tyrosine kinase 3 inhibitors in the treatment of acute myeloid leukemia: an update on the emerging evidence and safety profile. Onco Targets Ther. 16, 31–45. doi:10.2147/OTT.S236740
Garrison, D. A., Jin, Y., Talebi, Z., Hu, S., Sparreboom, A., Baker, S. D., et al. (2022). Itraconazole-induced increases in gilteritinib exposure are mediated by CYP3A and OATP1B. Molecules 27 (20), 6815. doi:10.3390/molecules27206815
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426 (6968), 895–899. doi:10.1038/nature02263
Goncearenco, A., Li, M., Simonetti, F. L., Shoemaker, B. A., and Panchenko, A. R. (2017). Exploring protein-protein interactions as drug targets for anti-cancer therapy with in silico workflows. Methods Mol. Biol. 1647, 221–236. doi:10.1007/978-1-4939-7201-2_15
Gordhandas, S. B., Manning-Geist, B., Henson, C., Iyer, G., Gardner, G. J., Sonoda, Y., et al. (2022). Pre-clinical activity of the oral DNA-PK inhibitor, peposertib (M3814), combined with radiation in xenograft models of cervical cancer. Sci. Rep. 12 (1), 974. doi:10.1038/s41598-021-04618-5
Griffith, J. P., Kim, J. L., Kim, E. E., Sintchak, M. D., Thomson, J. A., Fitzgibbon, M. J., et al. (1995). X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 82 (3), 507–522. doi:10.1016/0092-8674(95)90439-5
Gristina, V., La Mantia, M., Iacono, F., Galvano, A., Russo, A., and Bazan, V. (2020). The emerging therapeutic landscape of ALK inhibitors in non-small cell lung cancer. Pharm. (Basel) 13 (12), 474. doi:10.3390/ph13120474
Haisco Pharmaceutical Group (2022). A study of HSK29116 in adults with relapsed/refractory B-cell malignancies. Available at: https://clinicaltrials.gov/ct2/show/NCT05252364.
Han, X., Wei, W., and Sun, Y. (2022). PROTAC degraders with ligands recruiting MDM2 E3 ubiquitin ligase: an updated perspective. Acta Mater Med. 1 (2), 244–259. doi:10.15212/amm-2022-0010
Harding, M. W., Galat, A., Uehling, D. E., and Schreiber, S. L. (1989). A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 341 (6244), 758–760. doi:10.1038/341758a0
He, M., Cao, C., Ni, Z., Liu, Y., Song, P., Hao, S., et al. (2022). PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target Ther. 7 (1), 181. doi:10.1038/s41392-022-00999-9
He, T., Cheng, C., Qiao, Y., Cho, H., Young, E., Mannan, R., et al. (2024). Development of an orally bioavailable mSWI/SNF ATPase degrader and acquired mechanisms of resistance in prostate cancer. bioRxiv 121, e2322563121. doi:10.1073/pnas.2322563121
He, Y., Koch, R., Budamagunta, V., Zhang, P., Zhang, X., Khan, S., et al. (2020). DT2216-a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J. Hematol. Oncol. 13 (1), 95. doi:10.1186/s13045-020-00928-9
Hershko, A., Ciechanover, A., Heller, H., Haas, A. L., and Rose, I. A. (1980). Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl. Acad. Sci. U. S. A. 77 (4), 1783–1786. doi:10.1073/pnas.77.4.1783
Hershko, A., Heller, H., Elias, S., and Ciechanover, A. (1983). Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258 (13), 8206–8214. doi:10.1016/s0021-9258(20)82050-x
Hinova Pharmaceuticals Aus Pty Ltd (2024). A study to assess the safety, pharmacokinetics, and anti-tumor activity of oral HP518 in patients with metastatic castration-resistant prostate cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT05252364.
Homan, M., Warrier, G., Lao, C. D., Yentz, S., Kraft, S., and Fecher, L. A. (2022a). Treatment related toxicities with combination BRAF and MEK inhibitor therapy in resected stage III melanoma. Frontiers. 12, 855794. doi:10.3389/fonc.2022.855794
Homan, M., Warrier, G., Lao, C. D., Yentz, S., Kraft, S., and Fecher, L. A. (2022b). Treatment related toxicities with combination BRAF and MEK inhibitor therapy in resected stage III melanoma. Front. Oncol. 12, 855794. doi:10.3389/fonc.2022.855794
Hon, W. C., Wilson, M. I., Harlos, K., Claridge, T. D., Schofield, C. J., Pugh, C. W., et al. (2002). Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 417 (6892), 975–978. doi:10.1038/nature00767
Houghton, P. J. (2010). Everolimus. Clin. Cancer Res. 16 (5), 1368–1372. doi:10.1158/1078-0432.CCR-09-1314
Inoue, S., and Sato, H. (1967). Cell motility by labile association of molecules. J. Gen. Physiol. 50 (6), 259–292. doi:10.1085/jgp.50.6.259
Jensen, M. R., Schoepfer, J., Radimerski, T., Massey, A., Guy, C. T., Brueggen, J., et al. (2008). NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 10 (2), R33. doi:10.1186/bcr1996
Jia, X., and Han, X. (2023). Targeting androgen receptor degradation with PROTACs from bench to bedside. Biomed. Pharmacother. 158, 114112. doi:10.1016/j.biopha.2022.114112
Jiang, Q., Li, M., Li, H., and Chen, L. (2022). Entrectinib, a new multi-target inhibitor for cancer therapy. Biomed. Pharmacother. 150, 112974. doi:10.1016/j.biopha.2022.112974
Johansson, K. B., Zimmerman, M. S., Dmytrenko, I. V., Gao, F., and Link, D. C. (2023). Idasanutlin and navitoclax induce synergistic apoptotic cell death in T-cell acute lymphoblastic leukemia. Leukemia 37 (12), 2356–2366. doi:10.1038/s41375-023-02057-x
Jordan, M. A. (2002). Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anticancer Agents 2 (1), 1–17. doi:10.2174/1568011023354290
Jubb, H. C., Pandurangan, A. P., Turner, M. A., Ochoa-Montano, B., Blundell, T. L., and Ascher, D. B. (2017). Mutations at protein-protein interfaces: small changes over big surfaces have large impacts on human health. Prog. Biophys. Mol. Biol. 128, 3–13. doi:10.1016/j.pbiomolbio.2016.10.002
Kakadia, S., Yarlagadda, N., Awad, R., Kundranda, M., Niu, J., Naraev, B., et al. (2018). Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 11, 7095–7107. doi:10.2147/OTT.S182721
Kamal, A., Thao, L., Sensintaffar, J., Zhang, L., Boehm, M. F., Fritz, L. C., et al. (2003). A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425 (6956), 407–410. doi:10.1038/nature01913
Karlovitch, S. (2021). Alrizomadlin lands FDA fast track designation for melanoma: targeted oncology. Available at: https://www.targetedonc.com/view/alrizomadlin-lands-fda-fast-track-designation-for-melanoma.
Kim, Y., Kim, H., Jang, S. W., and Ko, J. (2011). The role of 14-3-3β in transcriptional activation of estrogen receptor α and its involvement in proliferation of breast cancer cells. Biochem. Biophys. Res. Commun. 414 (1), 199–204. doi:10.1016/j.bbrc.2011.09.056
Kim, Y. S., Alarcon, S. V., Lee, S., Lee, M. J., Giaccone, G., Neckers, L., et al. (2009). Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem. 9 (15), 1479–1492. doi:10.2174/156802609789895728
Kirchner, P., Bourdenx, M., Madrigal-Matute, J., Tiano, S., Diaz, A., Bartholdy, B. A., et al. (2019). Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 17 (5), e3000301. doi:10.1371/journal.pbio.3000301
Kisselev, A. F. (2021). Site-specific proteasome inhibitors. Biomolecules 12 (1), 54. doi:10.3390/biom12010054
Kulikov, R., Letienne, J., Kaur, M., Grossman, S. R., Arts, J., and Blattner, C. (2010). Mdm2 facilitates the association of p53 with the proteasome. Proc. Natl. Acad. Sci. U. S. A. 107 (22), 10038–10043. doi:10.1073/pnas.0911716107
Kumamoto, K., Spillare, E. A., Fujita, K., Horikawa, I., Yamashita, T., Appella, E., et al. (2008). Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res. 68 (9), 3193–3203. doi:10.1158/0008-5472.CAN-07-2780
Kunzelmann, K. (2005). Ion channels and cancer. J. Membr. Biol. 205 (3), 159–173. doi:10.1007/s00232-005-0781-4
Kwitkowski, V. E., Prowell, T. M., Ibrahim, A., Farrell, A. T., Justice, R., Mitchell, S. S., et al. (2010). FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 15 (4), 428–435. doi:10.1634/theoncologist.2009-0178
Kymera Therapeutics, Inc. (2024a). Safety, PK, PD, clinical activity of KT-333 in adult patients with refractory lymphoma, large granular lymphocytic leukemia, solid tumors. Available at: https://clinicaltrials.gov/ct2/show/NCT05225584.
Kymera Therapeutics, Inc. (2024b). Safety, PK/PD, and clinical activity of KT-413 in adult patients with relapsed or refractory B-cell NHL. Available at: https://clinicaltrials.gov/ct2/show/NCT05233033.
Lai, A. C., and Crews, C. M. (2017). Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16 (2), 101–114. doi:10.1038/nrd.2016.211
Lang, F., Foller, M., Lang, K. S., Lang, P. A., Ritter, M., Gulbins, E., et al. (2005). Ion channels in cell proliferation and apoptotic cell death. J. Membr. Biol. 205 (3), 147–157. doi:10.1007/s00232-005-0780-5
Lang, L. (2008). FDA approves sorafenib for patients with inoperable liver cancer. Gastroenterology 134 (2), 379. doi:10.1053/j.gastro.2007.12.037
Lavacchi, D., Mazzoni, F., and Giaccone, G. (2019). Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): current perspectives. Drug Des. Devel Ther. 13, 3187–3198. doi:10.2147/DDDT.S194231
Lazenby, M., Hills, R., Burnett, A. K., and Zabkiewicz, J. (2015). The HSP90 inhibitor ganetespib: a potential effective agent for Acute Myeloid Leukemia in combination with cytarabine. Leuk. Res. 39 (6), 617–624. doi:10.1016/j.leukres.2015.03.016
LeRoy, G., Rickards, B., and Flint, S. J. (2008). The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 30 (1), 51–60. doi:10.1016/j.molcel.2008.01.018
Li, M., Simonetti, F. L., Goncearenco, A., and Panchenko, A. R. (2016). MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 44 (W1), W494–W501. doi:10.1093/nar/gkw374
Li, R., Liu, M., Yang, Z., Li, J., Gao, Y., and Tan, R. (2022). Proteolysis-targeting chimeras (PROTACs) in cancer therapy: present and future. Molecules 27 (24), 8828. doi:10.3390/molecules27248828
Li, W., Saji, S., Sato, F., Noda, M., and Toi, M. (2013). Potential clinical applications of matrix metalloproteinase inhibitors and their future prospects. Int. J. Biol. Markers. 28 (2), 117–130. doi:10.5301/jbm.5000026
Li, Y., Yang, J., Aguilar, A., McEachern, D., Przybranowski, S., Liu, L., et al. (2019). Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 62 (2), 448–466. doi:10.1021/acs.jmedchem.8b00909
Li, Z., Zhu, C., Ding, Y., Fei, Y., and Lu, B. (2020). ATTEC: a potential new approach to target proteinopathies. Autophagy 16 (1), 185–187. doi:10.1080/15548627.2019.1688556
Liang, C. C., Greenhough, L. A., Masino, L., Maslen, S., Bajrami, I., Tuppi, M., et al. (2024). Mechanism of single-stranded DNA annealing by RAD52-RPA complex. Nature 629 (8012), 697–703. doi:10.1038/s41586-024-07347-7
Liu, B., and Kou, Y. (2022). Fourth-line rescue treatment ripretinib of advanced small intestine gastrointestinal stromal tumors who achieved partial response: a case report. J. Gastrointest. Oncol. 13 (3), 1505–1513. doi:10.21037/jgo-22-534
Liu, T., Merguerian, M. D., Rowe, S. P., Pratilas, C. A., Chen, A. R., and Ladle, B. H. (2021b). Exceptional response to the ALK and ROS1 inhibitor lorlatinib and subsequent mechanism of resistance in relapsed ALK F1174L-mutated neuroblastoma. Cold Spring Harb. Mol. Case Stud. 7 (4), a006064. doi:10.1101/mcs.a006064
Liu, Z., Hu, M., Yang, Y., Du, C., Zhou, H., Liu, C., et al. (2022). An overview of PROTACs: a promising drug discovery paradigm. Mol. Biomed. 3 (1), 46. doi:10.1186/s43556-022-00112-0
Liu, Z., Hu, X., Wang, Q., Wu, X., Zhang, Q., Wei, W., et al. (2021a). Design and synthesis of EZH2-based PROTACs to degrade the PRC2 complex for targeting the noncatalytic activity of EZH2. J. Med. Chem. 64 (5), 2829–2848. doi:10.1021/acs.jmedchem.0c02234
Lu, H., Zhou, Q., He, J., Jiang, Z., Peng, C., Tong, R., et al. (2020). Recent advances in the development of protein-protein interactions modulators: mechanisms and clinical trials. Signal Transduct. Target Ther. 5 (1), 213. doi:10.1038/s41392-020-00315-3
Luminespib (2023). Luminespib. Available at: https://www.sciencedirect.com/topics/medicine-and-dentistry/luminespib.
Mascarenhas, J., and Hoffman, R. (2012). Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis. Clin. Cancer Res. 18 (11), 3008–3014. doi:10.1158/1078-0432.CCR-11-3145
McCarty, J. S., Buchberger, A., Reinstein, J., and Bukau, B. (1995). The role of ATP in the functional cycle of the DnaK chaperone system. J. Mol. Biol. 249 (1), 126–137. doi:10.1006/jmbi.1995.0284
McClellan, A. J., Xia, Y., Deutschbauer, A. M., Davis, R. W., Gerstein, M., and Frydman, J. (2007). Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131 (1), 121–135. doi:10.1016/j.cell.2007.07.036
McPherson, K. S., and Korzhnev, D. M. (2021). Targeting protein-protein interactions in the DNA damage response pathways for cancer chemotherapy. RSC Chem. Biol. 2 (4), 1167–1195. doi:10.1039/d1cb00101a
Medscape (2000). Phase III trials of prinomastat in advanced cancers discontinued but multiple phase II trials continue. Available at: https://www.medscape.com/viewarticle/412097.
Meric-Bernstam, F., Bahleda, R., Hierro, C., Sanson, M., Bridgewater, J., Arkenau, H. T., et al. (2022). Futibatinib, an irreversible FGFR1-4 inhibitor, in patients with advanced solid tumors harboring FGF/FGFR aberrations: a phase I dose-expansion study. Cancer Discov. 12 (2), 402–415. doi:10.1158/2159-8290.CD-21-0697
Minarro-Lleonar, M., Bertran-Mostazo, A., Duro, J., Barril, X., and Juarez-Jimenez, J. (2023). Lenalidomide stabilizes protein-protein complexes by turning labile intermolecular H-bonds into robust interactions. J. Med. Chem. 66 (9), 6037–6046. doi:10.1021/acs.jmedchem.2c01692
Miyata, Y., Chang, L., Bainor, A., McQuade, T. J., Walczak, C. P., Zhang, Y., et al. (2010). High-throughput screen for Escherichia coli heat shock protein 70 (Hsp70/DnaK): ATPase assay in low volume by exploiting energy transfer. J. Biomol. Screen 15 (10), 1211–1219. doi:10.1177/1087057110380571
Moulick, K., Ahn, J. H., Zong, H., Rodina, A., Cerchietti, L., Gomes DaGama, E. M., et al. (2011). Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 7 (11), 818–826. doi:10.1038/nchembio.670
Mukhtar, E., Adhami, V. M., and Mukhtar, H. (2014). Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 13 (2), 275–284. doi:10.1158/1535-7163.MCT-13-0791
Mullally, A., Hood, J., Harrison, C., and Mesa, R. (2020). Fedratinib in myelofibrosis. Blood Adv. 4 (8), 1792–1800. doi:10.1182/bloodadvances.2019000954
Munoz, J., Wang, Y., Jain, P., and Wang, M. (2022). Zanubrutinib in lymphoproliferative disorders: a comprehensive review. Ther. Adv. Hematol. 13, 20406207221093980. doi:10.1177/20406207221093980
MutaBind2 (2024). MutaBind2. Available at: https://lilab.jysw.suda.edu.cn/research/mutabind2/.
National Cancer Institute (2022). Lenalidomide. Bethesda, MA: National Cancer Institute. Available at: https://www.cancer.gov/about-cancer/treatment/drugs/lenalidomide.
National Cancer Institute (2023). Success story: taxol. Bethesda, MA: National Cancer Institute. Available at: https://dtp.cancer.gov/timeline/noflash/success_stories/s2_taxol.htm#:∼:text=1-,Dr.,the%20segregation%20of%20the%20chromosomes.
Nogales, E. (2001). Structural insight into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 30, 397–420. doi:10.1146/annurev.biophys.30.1.397
Nogales, E., Wolf, S. G., Khan, I. A., Luduena, R. F., and Downing, K. H. (1995). Structure of tubulin at 6.5 A and location of the taxol-binding site. Nature 375 (6530), 424–427. doi:10.1038/375424a0
Novartis Pharmaceuticals (2025). A study of ARV-766 given by mouth in men with metastatic castration-resistant prostate cancer who have progressed on prior approved systemic therapies. Available at: https://clinicaltrials.gov/ct2/show/NCT05067140.
Nurix Therapeutics, Inc (2024). A study of NX-5948 in adults with relapsed/refractory B-cell malignancies. Available at: https://clinicaltrials.gov/ct2/show/NCT05131022.
Olivier, M., Hollstein, M., and Hainaut, P. (2010). TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2 (1), a001008. doi:10.1101/cshperspect.a001008
Orr, G. A., Verdier-Pinard, P., McDaid, H., and Horwitz, S. B. (2003). Mechanisms of Taxol resistance related to microtubules. Oncogene 22 (47), 7280–7295. doi:10.1038/sj.onc.1206934
Pandey, K., An, H. J., Kim, S. K., Lee, S. A., Kim, S., Lim, S. M., et al. (2019). Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: a review. Int. J. Cancer 145 (5), 1179–1188. doi:10.1002/ijc.32020
Papayannidis, C., Federico, V., Fianchi, L., Pregno, P., Pugliese, N., Romano, A., et al. (2022). Treatment of advanced systemic mastocytosis with midostaurin: practical guidance for optimal therapy and management. Mediterr. J. Hematol. Infect. Dis. 14 (1), e2022073. doi:10.4084/MJHID.2022.073
Parekh, P. R., Botting, G. M., Thurber, D. B., Boruszczak, M., Murphy, W., and Bertenshaw, G. P. (2022). Predictive biomarkers for response to trametinib in non-small cell lung cancer. Tumour Biol. 44 (1), 249–267. doi:10.3233/TUB-220009
Pedrucci, F., Pappalardo, C., Marzaro, G., Ferri, N., Ferlin, A., and De Toni, L. (2022). Proteolysis targeting chimeric molecules: tuning molecular strategies for a clinically sound listening. Int. J. Mol. Sci. 23 (12), 6630. doi:10.3390/ijms23126630
Peng, L., Zhu, L., Sun, Y., Stebbing, J., Selvaggi, G., Zhang, Y., et al. (2022). Targeting ALK rearrangements in NSCLC: current state of the art. Front. Oncol. 12, 863461. doi:10.3389/fonc.2022.863461
Pennell, N. A. (2012). Treating ALK-positive lung cancer in the weeks after the FDA approval of crizotinib. Am. J. Manag. Care 18 (5 Spec No. 2), SP84–7.
Petta, I., Lievens, S., Libert, C., Tavernier, J., and De Bosscher, K. (2016). Modulation of protein-protein interactions for the development of novel therapeutics. Mol. Ther. 24 (4), 707–718. doi:10.1038/mt.2015.214
Pfaff, P., Samarasinghe, K. T. G., Crews, C. M., and Carreira, E. M. (2019). Reversible spatiotemporal control of induced protein degradation by bistable PhotoPROTACs. ACS Cent. Sci. 5 (10), 1682–1690. doi:10.1021/acscentsci.9b00713
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi:10.1146/annurev.biochem.70.1.503
Pickart, C. M. (2004). Back to the future with ubiquitin. Cell 116 (2), 181–190. doi:10.1016/s0092-8674(03)01074-2
Pillai, R. N., Fennell, D. A., Kovcin, V., Ciuleanu, T. E., Ramlau, R., Kowalski, D., et al. (2020). Randomized phase III study of ganetespib, a heat shock protein 90 inhibitor, with docetaxel versus docetaxel in advanced non-small-cell lung cancer (GALAXY-2). J. Clin. Oncol. 38 (6), 613–622. doi:10.1200/JCO.19.00816
Pott, S., and Lieb, J. D. (2015). What are super-enhancers? Nat. Genet. 47 (1), 8–12. doi:10.1038/ng.3167
Potts, B. C., Albitar, M. X., Anderson, K. C., Baritaki, S., Berkers, C., Bonavida, B., et al. (2011). Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 11 (3), 254–284. doi:10.2174/156800911794519716
Powell, K., and Prasad, V. (2021). Concerning FDA approval of trilaciclib (Cosela) in extensive-stage small-cell lung cancer. Transl. Oncol. 14 (11), 101206. doi:10.1016/j.tranon.2021.101206
PRINOMASTAT (2023). PRINOMASTAT. Available at: https://drugs.ncats.io/drug/10T6626FRK.
PRISM (2024). PRISM. Available at: http://prism.ccbb.ku.edu.tr/hotregion.
Prodromou, C., Roe, S. M., O'Brien, R., Ladbury, J. E., Piper, P. W., and Pearl, L. H. (1997). Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90 (1), 65–75. doi:10.1016/s0092-8674(00)80314-1
Pulte, E. D., Chen, H., Price, L. S. L., Gudi, R., Li, H., Okusanya, O. O., et al. (2022). FDA approval summary: revised indication and dosing regimen for ponatinib based on the results of the OPTIC trial. Oncologist 27 (2), 149–157. doi:10.1093/oncolo/oyab040
Rajan, S., Preisig-Muller, R., Wischmeyer, E., Nehring, R., Hanley, P. J., Renigunta, V., et al. (2002). Interaction with 14-3-3 proteins promotes functional expression of the potassium channels TASK-1 and TASK-3. J. Physiol. 545 (1), 13–26. doi:10.1113/jphysiol.2002.027052
Rebimastat (2025). Rebimastat. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/Rebimastat.
Robbins, D. W., Kelly, A., Tan, M., McIntosh, J., Wu, J., Konst, Z., et al. (2020). Nx-2127, a degrader of BTK and IMiD neosubstrates, for the treatment of B-cell malignancies. Blood 136, 34. doi:10.1182/blood-2020-141461
Robbins, D. W., Noviski, M., Rountree, R., Tan, M., Brathaban, N., Ingallinera, T., et al. (2021). Nx-5948, a selective degrader of BTK with activity in preclinical models of hematologic and brain malignancies. Blood 138, 2251. doi:10.1182/blood-2021-147473
Robert, M., Farese, H., and Miossec, P. (2022). Update on tenosynovial giant cell tumor, an inflammatory arthritis with neoplastic features. Front. Immunol. 13, 820046. doi:10.3389/fimmu.2022.820046
Rohena, C. C., and Mooberry, S. L. (2014). Recent progress with microtubule stabilizers: new compounds, binding modes and cellular activities. Nat. Prod. Rep. 31 (3), 335–355. doi:10.1039/c3np70092e
Romesser, P. B., Capdevila, J., Garcia-Carbonero, R., Philip, T., Fernandez Martos, C., Tuli, R., et al. (2024). A phase ib study of the DNA-PK inhibitor peposertib combined with neoadjuvant chemoradiation in patients with locally advanced rectal cancer. Clin. Cancer Res. 30 (4), 695–702. doi:10.1158/1078-0432.CCR-23-1129
Roskoski, R. (2023). Properties of FDA-approved small molecule protein kinase inhibitors: a 2023 update. Pharmacol. Res. 187, 106552. doi:10.1016/j.phrs.2022.106552
Rotter, V. (1983). p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells. Proc. Natl. Acad. Sci. U. S. A. 80 (9), 2613–2617. doi:10.1073/pnas.80.9.2613
Ryan, Q., Ibrahim, A., Cohen, M. H., Johnson, J., Ko, C. W., Sridhara, R., et al. (2008). FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 13 (10), 1114–1119. doi:10.1634/theoncologist.2008-0816
Sabers, C. J., Martin, M. M., Brunn, G. J., Williams, J. M., Dumont, F. J., Wiederrecht, G., et al. (1995). Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 270 (2), 815–822. doi:10.1074/jbc.270.2.815
Samuels, M., Falkenius, J., Bar-Ad, V., Dunst, J., van Triest, B., Yachnin, J., et al. (2024). A phase 1 study of the DNA-PK inhibitor peposertib in combination with radiation therapy with or without cisplatin in patients with advanced head and neck tumors. Int. J. Radiat. Oncol. Biol. Phys. 118 (3), 743–756. doi:10.1016/j.ijrobp.2023.09.024
Sarnik, J., Poplawski, T., and Tokarz, P. (2021). BET proteins as attractive targets for cancer therapeutics. Int. J. Mol. Sci. 22 (20), 11102. doi:10.3390/ijms222011102
Sayegh, N., Tripathi, N., Agarwal, N., and Swami, U. (2022). Clinical evidence and selecting patients for treatment with erdafitinib in advanced urothelial carcinoma. Onco Targets Ther. 15, 1047–1055. doi:10.2147/OTT.S318332
Schoffski, P. (2012). Pazopanib in the treatment of soft tissue sarcoma. Expert Rev. Anticancer Ther. 12 (6), 711–723. doi:10.1586/era.12.41
Scudellari, M. (2019). Protein-slaying drugs could be the next blockbuster therapies. Nature 567 (7748), 298–300. doi:10.1038/d41586-019-00879-3
Segura, M. F., Fontanals-Cirera, B., Gaziel-Sovran, A., Guijarro, M. V., Hanniford, D., Zhang, G., et al. (2013). BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 73 (20), 6264–6276. doi:10.1158/0008-5472.CAN-13-0122-T
Sehgal, S. N. (2003). Sirolimus: its discovery, biological properties, and mechanism of action. Transpl. Proc. 35 (3 Suppl. l), 7S–14S. doi:10.1016/s0041-1345(03)00211-2
Sharma, D., and Masison, D. C. (2009). Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept. Lett. 16 (6), 571–581. doi:10.2174/092986609788490230
Shi, D., and Gu, W. (2012). Dual roles of MDM2 in the regulation of p53: ubiquitination dependent and ubiquitination independent mechanisms of MDM2 repression of p53 activity. Genes Cancer 3 (3-4), 240–248. doi:10.1177/1947601912455199
Sionoc, R. V., Hayon, I. L., and Haupt, Y. (2024). The regulation of p53 growth suppression - madame curie bioscience database - NCBI bookshelf. Available at: https://www.ncbi.nlm.nih.gov/books/NBK6412/.
Smith, A. E., Ferraro, E., Safonov, A., Morales, C. B., Lahuerta, E. J. A., Li, Q., et al. (2021). HER2 + breast cancers evade anti-HER2 therapy via a switch in driver pathway. Nat. Commun. 12 (1), 6667. doi:10.1038/s41467-021-27093-y
Smith, M. C., and Gestwicki, J. E. (2012). Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev. Mol. Med. 14, e16. doi:10.1017/erm.2012.10
Smyth, E. N., Beyrer, J., Saverno, K. R., Hadden, E., Abedtash, H., DeLuca, A., et al. (2022). Real-world patient characteristics, utilization patterns, and outcomes of US patients with HR+, HER2- metastatic breast cancer treated with abemaciclib. Drugs Real World Outcomes 9 (4), 681–693. doi:10.1007/s40801-022-00327-1
Soave, C. L., Guerin, T., Liu, J., and Dou, Q. P. (2017). Targeting the ubiquitin-proteasome system for cancer treatment: discovering novel inhibitors from nature and drug repurposing. Cancer Metastasis Rev. 36 (4), 717–736. doi:10.1007/s10555-017-9705-x
Stathis, A., Zucca, E., Bekradda, M., Gomez-Roca, C., Delord, J. P., de La Motte Rouge, T., et al. (2016). Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor otx015/MK-8628. Cancer Discov. 6 (5), 492–500. doi:10.1158/2159-8290.CD-15-1335
Stevers, L. M., Sijbesma, E., Botta, M., MacKintosh, C., Obsil, T., Landrieu, I., et al. (2018). Modulators of 14-3-3 protein-protein interactions. J. Med. Chem. 61 (9), 3755–3778. doi:10.1021/acs.jmedchem.7b00574
Sun, D., Zhang, J., Dong, G., He, S., and Sheng, C. (2022). Blocking non-enzymatic functions by PROTAC-mediated targeted protein degradation. J. Med. Chem. 65 (21), 14276–14288. doi:10.1021/acs.jmedchem.2c01159
Sun, X., Gao, H., Yang, Y., He, M., Wu, Y., Song, Y., et al. (2019). PROTACs: great opportunities for academia and industry. Signal Transduct. Target Ther. 4, 64. doi:10.1038/s41392-019-0101-6
Suter, B., Kittanakom, S., and Stagljar, I. (2008). Two-hybrid technologies in proteomics research. Curr. Opin. Biotechnol. 19 (4), 316–323. doi:10.1016/j.copbio.2008.06.005
Tabasinezhad, M., Mahboudi, F., Wenzel, W., Rahimi, H., Walther, T. H., Blattner, C., et al. (2019). The transient production of anti-TNF-α antibody Adalimumab and a comparison of its characterization to the biosimilar Cinorra. Protein Expr. Purif. 155, 59–65. doi:10.1016/j.pep.2018.11.006
Takahashi, D., and Arimoto, H. (2020). Targeting selective autophagy by AUTAC degraders. Autophagy 16 (4), 765–766. doi:10.1080/15548627.2020.1718362
Takahashi, S., Wakui, H., Gustafsson, J. A., Zilliacus, J., and Itoh, H. (2000). Functional interaction of the immunosuppressant mizoribine with the 14-3-3 protein. Biochem. Biophys. Res. Commun. 274 (1), 87–92. doi:10.1006/bbrc.2000.3104
Taldone, T., Gozman, A., Maharaj, R., and Chiosis, G. (2008). Targeting Hsp90: small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 8 (4), 370–374. doi:10.1016/j.coph.2008.06.015
Taylor, I. R., Dunyak, B. M., Komiyama, T., Shao, H., Ran, X., Assimon, V. A., et al. (2018). High-throughput screen for inhibitors of protein-protein interactions in a reconstituted heat shock protein 70 (Hsp70) complex. J. Biol. Chem. 293 (11), 4014–4025. doi:10.1074/jbc.RA117.001575
Thangaraju, P., Singh, H., and Chakrabarti, A. (2015). Regorafenib: a novel tyrosine kinase inhibitor: a brief review of its therapeutic potential in the treatment of metastatic colorectal carcinoma and advanced gastrointestinal stromal tumors. Indian J. Cancer 52 (3), 257–260. doi:10.4103/0019-509X.176690
Thiel, P., Kaiser, M., and Ottmann, C. (2012). Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery? Angew. Chem. Int. Ed. Engl. 51 (9), 2012–2018. doi:10.1002/anie.201107616
Thus, Y. J., De Rooij, M. F. M., Beijersbergen, R. L., and Spaargaren, M. (2022). An unbiased CRISPR-cas9 screening method for the identification of positive and negative regulatory proteins of cell adhesion. Bio Protoc. 12 (21), e4545. doi:10.21769/BioProtoc.4545
Van Molle, I., Thomann, A., Buckley, D. L., So, E. C., Lang, S., Crews, C. M., et al. (2012). Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1α protein-protein interface. Chem. Biol. 19 (10), 1300–1312. doi:10.1016/j.chembiol.2012.08.015
Varshavsky, A. (1997). The ubiquitin system. Trends Biochem. Sci. 22 (10), 383–387. doi:10.1016/s0968-0004(97)01122-5
Varshavsky, A. (2005). Regulated protein degradation. Trends Biochem. Sci. 30 (6), 283–286. doi:10.1016/j.tibs.2005.04.005
Vellanki, S., Garcia, A. E., and Lee, S. C. (2020). Interactions of FK506 and rapamycin with FK506 binding protein 12 in opportunistic human fungal pathogens. Front. Mol. Biosci. 7, 588913. doi:10.3389/fmolb.2020.588913
Vousden, K. H. (2002). Activation of the p53 tumor suppressor protein. Biochim. Biophys. Acta 1602 (1), 47–59. doi:10.1016/s0304-419x(02)00035-5
Vousden, K. H., and Lu, X. (2002). Live or let die: the cell's response to p53. Nat. Rev. Cancer 2 (8), 594–604. doi:10.1038/nrc864
Vu, B., Wovkulich, P., Pizzolato, G., Lovey, A., Ding, Q., Jiang, N., et al. (2013). Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4 (5), 466–469. doi:10.1021/ml4000657
Wade, M., Mendez, J., Coussens, N. P., Arkin, M. R., and Glicksman, M. A. (2004). “Inhibition of protein-protein interactions: cell-based assays,” in Assay guidance manual. Editors S. Markossian, A. Grossman, K. Brimacombe, M. Arkin, D. Auld, and C. Austin, (Bethesda (MD).
Wang, J. Y. (2001). DNA damage and apoptosis. Cell Death Differ. 8 (11), 1047–1048. doi:10.1038/sj.cdd.4400938
Wang, S., and Chen, F. E. (2022). Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. Eur. J. Med. Chem. 236, 114334. doi:10.1016/j.ejmech.2022.114334
Wang, X., Fu, X., Brown, P. D., Crimmin, M. J., and Hoffman, R. M. (1994). Matrix metalloproteinase inhibitor BB-94 (batimastat) inhibits human colon tumor growth and spread in a patient-like orthotopic model in nude mice. Cancer Res. 54 (17), 4726–4728.
Warner, W. A., Sanchez, R., Dawoodian, A., Li, E., and Momand, J. (2012). Identification of FDA-approved drugs that computationally bind to MDM2. Chem. Biol. Drug Des. 80 (4), 631–637. doi:10.1111/j.1747-0285.2012.01428.x
Weaver, B. A. (2014). How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 25 (18), 2677–2681. doi:10.1091/mbc.E14-04-0916
Weisberg, E., Manley, P., Mestan, J., Cowan-Jacob, S., Ray, A., and Griffin, J. D. (2006). AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br. J. Cancer 94 (12), 1765–1769. doi:10.1038/sj.bjc.6603170
Weitzman, S. P., and Cabanillas, M. E. (2015). The treatment landscape in thyroid cancer: a focus on cabozantinib. Cancer Manag. Res. 7, 265–278. doi:10.2147/CMAR.S68373
Wpengine (2023). A study of NX-2127 in adults with relapsed/refractory B-cell malignancies. Available at: https://clin.larvol.com/trial-detail/NCT05233033.
Xu, Z., Page, R. C., Gomes, M. M., Kohli, E., Nix, J. C., Herr, A. B., et al. (2008). Structural basis of nucleotide exchange and client binding by the Hsp70 cochaperone Bag2. Nat. Struct. Mol. Biol. 15 (12), 1309–1317. doi:10.1038/nsmb.1518
Yan, J., and Zheng, Z. (2023). Discovery of highly potent CRBN ligands and insight into their binding mode through molecular docking and molecular dynamics simulations. ChemMedChem. 18 (5), e202200573. doi:10.1002/cmdc.202200573
Yang, J. C. H., Brose, M. S., Castro, G., Kim, E. S., Lassen, U. N., Leyvraz, S., et al. (2022). Rationale and design of ON-TRK: a novel prospective non-interventional study in patients with TRK fusion cancer treated with larotrectinib. BMC Cancer 22 (1), 625. doi:10.1186/s12885-022-09687-x
Ye, Z., Shi, Y., Lees-Miller, S. P., and Tainer, J. A. (2021). Function and molecular mechanism of the DNA damage response in immunity and cancer immunotherapy. Front. Immunol. 12, 797880. doi:10.3389/fimmu.2021.797880
Yeger-Lotem, E., and Sharan, R. (2015). Human protein interaction networks across tissues and diseases. Front. Genet. 6, 257. doi:10.3389/fgene.2015.00257
Yu, H., Braun, P., Yildirim, M. A., Lemmens, I., Venkatesan, K., Sahalie, J., et al. (2008). High-quality binary protein interaction map of the yeast interactome network. Science. 322 (5898), 104–110. doi:10.1126/science.1158684
Yu, J., Mahipal, A., and Kim, R. (2021). Targeted therapy for advanced or metastatic cholangiocarcinoma: focus on the clinical potential of infigratinib. Onco Targets Ther. 14, 5145–5160. doi:10.2147/OTT.S272208
Zhang, J., Li, H., Liu, Y., Zhao, K., Wei, S., Sugarman, E. T., et al. (2022). Targeting HSP90 as a novel therapy for cancer: mechanistic insights and translational relevance. Cells 11 (18), 2778. doi:10.3390/cells11182778
Zhao, L., Zhao, J., Zhong, K., Tong, A., and Jia, D. (2022). Targeted protein degradation: mechanisms, strategies and application. Signal Transduct. Target Ther. 7 (1), 113. doi:10.1038/s41392-022-00966-4
Zheng, H. C. (2017). The molecular mechanisms of chemoresistance in cancers. Oncotarget 8 (35), 59950–59964. doi:10.18632/oncotarget.19048
Zhou, L., Li, J., Zhang, X., Xu, Z., Yan, Y., and Hu, K. (2022). An integrative pan cancer analysis of RET aberrations and their potential clinical implications. Sci. Rep. 12 (1), 13913. doi:10.1038/s41598-022-17791-y
Zhou, Y., Zhao, R. H., Tseng, K. F., Li, K. P., Lu, Z. G., Liu, Y., et al. (2016). Sirolimus induces apoptosis and reverses multidrug resistance in human osteosarcoma cells in vitro via increasing microRNA-34b expression. Acta Pharmacol. Sin. 37 (4), 519–529. doi:10.1038/aps.2015.153
Zhu, L., and Chen, L. (2019). Progress in research on paclitaxel and tumor immunotherapy. Cell Mol. Biol. Lett. 24, 40. doi:10.1186/s11658-019-0164-y
Keywords: protein-protein interaction (PPI), proteolysis targeting chimera (PROTAC), non-enzymatic protein targeting agents, targeted protein degradation (TPD), DNA damage repair (DDR), P53/MDM2 pathway, BET protein inhibitor, immunosupperssion
Citation: Ambrose M, Lee J, Syed A, Ahmed Z and Peng G (2025) Non-enzymatic protein targeting agents as a promising strategy for cancer treatment. Front. Drug Discov. 5:1520734. doi: 10.3389/fddsv.2025.1520734
Received: 05 November 2024; Accepted: 08 January 2025;
Published: 29 January 2025.
Edited by:
Hoang V. Le, National Institute on Drug Abuse (NIH), United StatesReviewed by:
Shang Su, Louisiana State University, United StatesCopyright © 2025 Ambrose, Lee, Syed, Ahmed and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang Peng, Z3BlbmdAbWRhbmRlcnNvbi5vcmc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.