 Yosi Gilad
Yosi Gilad Ortal Shimon1,2
Ortal Shimon1,2 David M. Lonard
David M. Lonard Bert W. O’Malley
Bert W. O’Malley- 1Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, United States
- 2CoRegen, Inc., Baylor College of Medicine, Houston, TX, United States
- 3Nuclear Receptor, Transcription and Chromatin Biology Program, Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX, United States
Steroid receptor coactivators (SRCs) are a family of nuclear receptor (NR) coregulators comprised of three evolutionarily related, yet distinct proteins SRC-1, SRC-2 and SRC-3. The SRCs interact with NRs and other transcription factors to regulate their transcriptional activity, impacting a wide variety of physiological and pathological processes. Abnormal expression or dysfunction of SRCs is associated with a diverse range of pathological conditions, including metabolic disorders, genetic and reproductive malfunctioning, impaired embryogenesis and most notably cancer which is the focus of this review. As key integrators of NR and growth factor signaling pathways, SRCs regulate multiple oncogenic programs, particularly in hormone-related malignancies, and thus represent attractive biological targets for cancer treatment. Here we review the evolution of the discovery process for small molecule targeting agents of SRCs and the opportunities they present for cancer therapy.
Introduction
Nuclear receptors (NRs) are a large family of transcription factors (TFs), that is comprised of 48 members in humans. NRs exert their gene-expression regulatory role by directly binding to cis regulatory elements on DNA (De Bosscher et al., 2020; Perissi and Rosenfeld, 2005). Most human NRs transduce their signaling into physiological responses in a hormone-dependent manner. However, several NRs are classified as “orphans”, since they either have no cognate ligand or their ligands have not been identified. To drive transcription, all NRs are dependent on interaction with another family of proteins - nuclear coactivators (Johnson and O’Malley, 2012). Binding of a coactivator to a NR promotes the recruitment of other regulatory proteins and assembly of the transcriptional machinery. SRCs are a family of three paralogous nuclear coactivators that regulate multiple NRs, as well as other TFs that are not dependent on ligand binding, thus having an impact on a wide variety of major physiological processes, including reproduction, development, metabolism and immunity (Xu et al., 1998; Puigserver et al., 1999; Stashi et al., 2014; O’Malley, 2020; Gehin et al., 2002; Mukherjee et al., 2006; Gilad et al., 2022; Gilad et al., 2024). In addition to their importance in maintaining normal physiological functions, all three SRCs are considered oncoproteins, since their overexpression and abnormal functioning are strongly linked to a wide range of malignant diseases, particularly hormone-related cancers. Among the three SRCs, SRC-3 is the most established coactivator in breast cancer (BC); it is usually overexpressed in this disease (Anzick et al., 1997), and its overexpression is strongly associated with poor outcomes (Hudelist et al., 2003; Zhao et al., 2003; Osborne et al., 2003). Beyond BC, SRC-3 has been also associated with multiple other types of cancer, including ovarian (Bautista et al., 1998), endometrial (Kershah et al., 2004), prostate (Gnanapragasam et al., 2001), liver (Wang et al., 2002), pancreatic (Henke et al., 2004), colorectal (Xie et al., 2005), and lung (Cai et al., 2010). Like SRC-3, SRC-1 is also a significant factor in BC, playing key roles in cell migration and metastasis (Qin et al., 2009; Wang et al., 2009) and its increased expression is correlated with poor prognosis and recurrent disease (Fleming et al., 2004). Beyond its role in BC, SRC-1 activity is also associated with prostate, thyroid and endometrial cancers (Walsh et al., 2012). SRC-2 is the most associated member of the SRC family with prostate cancer. It is amplified in close to 10% of primary and 40% of metastatic prostate cancers (Taylor et al., 2010) and its gain of function is correlated with high recurrence of the disease and poor outcomes (Agoulnik et al., 2006; Dasgupta et al., 2015).
Traditional treatment of hormone-related cancers primarily relies on a direct targeting of NRs, to which cancer cells frequently develop resistance. As multifaceted regulatory oncoproteins that orchestrate the activity of NRs, SRCs are considered attractive alternatives to NRs as targets in the treatment of hormone-related cancers. However, SRCs have historically been considered undruggable due to the nature of their biological activity, which relies on protein-protein interactions, their large size and flexible structure, and the lack of a defined ligand activation domain. To circumvent the ‘undruggability’ of the SRCs, early efforts were focused on identifying substances that disrupt the protein-protein interactions between SRCs and NRs, rather than by directly targeting the SRCs themselves (Rodriguez et al., 2004; Mettu et al., 2007; Gunther et al., 2009).
The challenge of directly targeting SRCs has been met by harnessing high throughput (HTP) screening technology as well as specific luciferase-reporter assays designed to detect alterations in SRC transcriptional activity and protein levels. This approach led to the discovery and identification of several groups of small molecules capable of perturbing the biological activity of SRCs. Initially, three naturally occurring small molecule inhibitors (SMIs) were identified, demonstrating that SRCs could be directly inhibited by small-molecule substances (Yan et al., 2014; Wang et al., 2014; Wang et al., 2011). Subsequently, a first-in-class synthetic SRC SMI with improved drug-like properties was discovered (Song et al., 2016). Moreover, this discovery platform, originally designed to identify substances that inhibit SRC activity, also led to the discovery of a group of molecules that stimulate an SRC-driven luciferase signal, classified as SRC small molecule stimulators (SMSs) (Wang et al., 2015). Overall, the discovery of SRC SMIs and SMSs presents a versatile tool-box for targeting these master oncogenes that traditionally were considered elusive biological drug targets. The expanding repertoire of small molecules that can manipulate the activity of the SRCs presents an opportunity to explore new strategies in cancer treatment. In this review we describe the evolution of the discovery process for small molecules that directly target and modulate the activity of SRCs and the potential implications that this presents for cancer treatment.
SRC structure
The SRC family is comprised of three proteins with a molecular weight of ∼160 kDa each that share a high degree of sequence homology and modular similarity (Figure 1). All SRCs contain three conserved structural domains; a basic helix-loop-helix (bHLH)/Per/Arnt/Sim (bHLH-PAS) activation domain at the N-terminus is the most conserved domain among the SRCs that contains a bipartite nuclear localization signal (NLS) and is involved in several protein-protein interactions (Li et al., 2007). The central region contains a NR interaction domain (NRID) comprised of three LXXLL α-helical motifs and flanking sequences around them. The LXXLL motifs (where X represents any amino acid) are required for the ability of the SRCs to interact with NRs (Heery et al., 1997), while the flanking sequences determine NR binding affinity and selectivity (Darimont et al., 1998; Coulthard et al., 2003). The C-terminus of the SRCs contains two activation domains (ADs), AD1 and AD2, which mediate the interactions of the SRCs with other key components of the transcriptional machinery; AD1 recruits the histone acetyltransferases (HATs) p300 and CREB-binding protein (CBP), while AD2 recruits the histone methyltransferases (HMT) coactivator-associated arginine methyltransferase 1 (CARM1) and protein arginine N-methyl transferase 1 (PRMT1) (Johnson and O’Malley, 2012; Lonard et al., 2023). According to their functions, AD1 and AD2 are often referred to as the CBP interacting domain (CID) and the HMT domain respectively. Interestingly, in addition to recruiting HATs through their AD1, there are indications that associate SRC ADs with weak intrinsic HAT activity, although the cellular substrates and the overall impact of this enzymatic activity is not clear (Spencer et al., 1997). Overall, the molecular structure and functional domains of the SRCs allow them to bind to hormone-activated NRs or other TFs, recruit various regulatory proteins, including mediators and RNA polymerase II, and thus promote the formation of an active, multiprotein transcriptional complex.
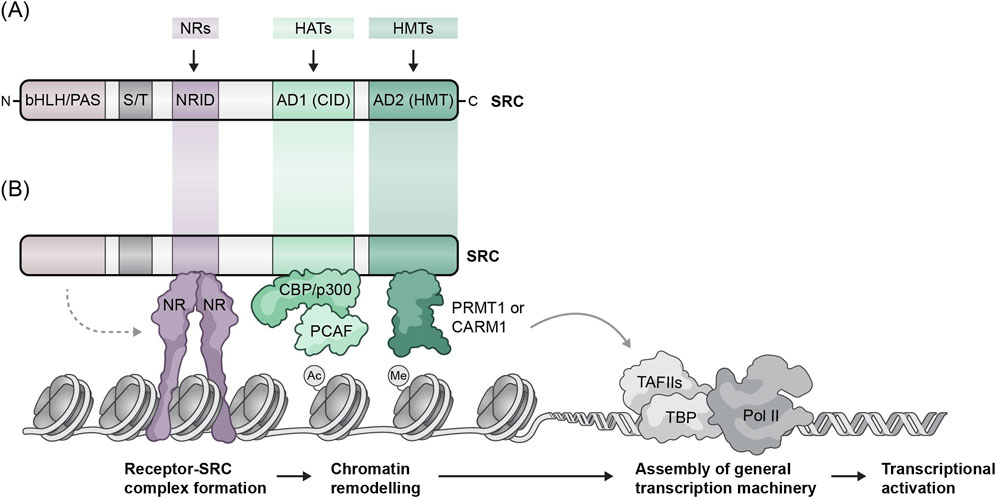
Figure 1. Molecular structure of SRCs and their functional roles in steroid hormone-induced gene expression. (A) Molecular structure of SRC family members. All three SRCs contain five conserved structural domains including a basic helix-loop-helix/period aryl hydrocarbon/simple-minded (bHLH/PAS) domain, a serine/threonine rich (S/T) domain, a nuclear receptor interaction domain (NRID), a CBP interacting domain (CID), and a histone acetyltransferase domain (HAT). (B) The SRCs form a complex with nuclear receptors (NR) in the presence of hormone. The SRCs then directly interact with NRs and subsequently recruit co-coactivator proteins such as CBP/p300 and CARM1 localizing histone acetyltransferase and arginine methyltransferase activity, respectively, to gene promoters and enhancers.
The discovery of naturally occurring SRC inhibitors
Within the broader NR superfamily, steroid hormone receptors (SHRs) represent one of the most important subgroup of NRs to human health and disease, which includes estrogen receptors (ERs), androgen receptor (AR) and progesterone receptor (PR) (Nuclear Receptors Nomenclature Committee, 1999). The transcriptional activity of SHRs under normal physiological conditions is regulated by their hydrophobic small-molecule binding ligands–the steroid hormones. Since SHRs rely on small molecule binding to exert their biological functions and the significance of their activity to human health and disease, they represent important targets for small molecule drug development (Santos et al., 2017). Indeed, many natural and synthetic small molecules that target the ligand binding pocket of SHRs have found their way into the clinic, particularly within the context of hormone-dependent cancers (Ulm et al., 2019). However, despite a substantial contribution of endocrine therapy to cancer treatment, resistance to SHR antagonists in hormone-dependent cancers remains an unmet medical challenge (Musgrove and Sutherland, 2009; Osborne and Schiff, 2011; Watson et al., 2015; Crawford et al., 2018), which provides a strong impetus to develop alternative approaches. Since the SRCs are essential components of SHRs function, manipulating their biological activity represents one such approach. Therefore, the evolving field of SRC small molecule modulators presents an opportunity to implement this strategy. Given that the SRCs are challenging drug targets, early attempts in this direction were focused on targeting the protein-protein interactions of SRCs with SHRs, rather than by direct targeting of the SRCs themselves (Rodriguez et al., 2004; Mettu et al., 2007; Skowron et al., 2019). One such attempt employed a fluorescence resonance energy transfer (FRET)-based assay, which was aimed at identifying small molecules that can disrupt the protein-protein interaction of ERα with an SRC LXXLL NR box motif, resulting in a depository resource of “hit” molecules that are capable of disrupting NR-SRC interactions (PubChem databases AID 629; AID 713). Since this screen was not designed to distinguish between molecules that target the coactivator or SHR, a separate effort was made to unambiguously explore SRC targeting agents. For this purpose a two-steps mini-scale secondary screen of active molecules was performed; 1- The fusion proteins SRC-1A-LUC and SRC-3-LUC (Lonard et al., 2004) were transiently expressed in Hela cells which were then subjected to a screen with hit molecules in order to identify their capability to decrease the steady state levels of exogenous SRCs in a selective manner. 2 - GAL4-responsive luciferase reporter gene (pG5-luc) and expression vectors for GAL4 DBD-SRC-1A (pBIND-SRC-1) or GAL4 DBD-SRC-3 (pBIND-SRC-3) fusion proteins (Lonard et al., 2000) were employed to evaluate the impact of the hit molecules on the intrinsic transcriptional activity of SRC-1 and SRC-3. Using this evaluation method on a cohort of identified SHR-SRC interaction inhibitors, a group of selective SRC SMI compounds was discovered, including gossypol - a naturally occurring polyphenol found in cotton seeds. Since gossypol has been recognized as an anticancer compound with Bcl2 inhibitory activity (Gilbert et al., 1995; Wolter et al., 2006; Huang et al., 2006; Meng et al., 2008), it was selected to demonstrate the concept that SRCs can be targeted directly with small-molecules for therapeutic intervention in cancer. Indeed, gossypol was able to inhibit the proliferation of various cancer cell lines and sensitize them to treatment with other anti-cancer agents (Wang et al., 2011). Co-targeting of SRC family members with other oncogenic proteins/pathways is a valuable tool for overcoming acquired resistance of cancer cells to chemotherapy. Importantly, the anti-proliferative activity of gossypol was selective towards cancer cells and did not alter the levels of other coactivators such as p300 and CARM1. Mechanistically, the anti-cancer activity of gossypol was associated with its binding to the SRC-3 NRID domain and selective reduction of endogenous levels of SRC-1 and SRC-3. Since gossypol is a known inhibitor of Bcl2 on both mRNA and protein levels, and since SRC-3 has several binding sites within Bcl-2 and Bcl-Xl genes (Lanz et al., 2010), it has been suggested that gossypol-mediated disruption of SRC-3 is an upstream event in Bcl2 inhibition. This working model is supported by an observation that depletion of SRC-3 by RNAi brings about decrease in mRNA and protein levels of Bcl2 (Zhou et al., 2005). Overall, identification of gossypol as the first ever direct SRC SMI catalyzed a large-scale discovery campaign to identify additional molecules with such activity. To achieve this, a similar discovery approach was employed: HeLa cells were co-transfected with pG5-luc along with one of the expression vectors encoding the GAL4 DNA binding domain fusion proteins pBIND-SRC-1, pBIND-SRC-2, or pBIND-SRC-3. The transfected cells were then utilized in a HTP screening assay to evaluate the SRC-inhibitory activity of compounds from a chemical library containing 359,484 distinct substances. This enabled the identification of 428, 620 and 621 inhibitors of intrinsic transcriptional activity of SRC-1 [PubChem AID:588,354], SRC-2 [PubChem AID:651,957] and SRC-3 [PubChem AID:588,352] respectively, with gossypol serving as a positive control (Figure 2A). These screens laid the foundation for two separate studies, that were dedicated to a deeper investigation of the primary screen results, which led to the characterization of two distinct naturally-occurring SRC SMIs; In one study, the focus was made on a cluster of molecules that the primary HTP screen identified as potent inhibitors of the intrinsic transcriptional activity of SRCs. All the molecules in this cluster shared a core structure resembling cardiac glycosides, which are known for their anti-cancer potential (Prassas and Diamandis, 2008). Therefore, a secondary evaluation round was performed using MCF-7 cells that were subjected to treatment with a panel of cardiac glycosides, including bufalin - which is originally isolated from Chinese toad parietal glands and has been extensively studied as a potential anti-cancer agent (Ye et al., 2023). Among the tested cardiac glycosides, bufalin most effectively reduced the amount of SRC-3 and SRC-1, and to a lower extent SRC-2, protein levels in MCF-7 cells. Additionally, bufalin significantly inhibited SRC-3 and SRC-1 intrinsic transcriptional activity in HeLa cells at a nanomolar concentration (Wang et al., 2014). The anti-SRC activity of bufalin is reflected in its ability to inhibit cancer cell proliferation and to sensitize cancer cells to other targeted therapies. Moreover, in a xenograft model of an aggressive triple negative breast cancer (TNBC) metastatic tumor, treatment with PEG-PLGA-based bufalin nanoparticles resulted in partial inhibition of tumor progression (Wang et al., 2014). Mechanistically, bufalin does not reduce the mRNA levels of SCR-3 or SRC-1 – it rather brings about a slight increase in them, which classifies it as a post-translational SRC-inhibitor. Bufalin-mediated inhibition of SRCs is associated with its physical interaction with the NRID domain which subjects these proteins to a proteosomal degradation. Interestingly, in contrast to its effect on the protein levels of SRC-1, treatment of mice with bufalin results in increased levels of a lower molecular weight SRC-1 isoform in endometrial lesions (Cho et al., 2018). Intriguingly, upregulation of this SRC-1 isoform–which is a known regulator of endometriosis progression (Cho et al., 2018) - was associated with suppressed growth of endometrial lesions. This phenomenon was attributed to the fact that bufalin-induced SRC-1 isoform overexpression coincided with proteosomal degradation of ERβ. This event led to disruption of the ERβ/SRC-1 isoform axis, which is critical for endometriosis progression. Moreover, overexpression of the SRC-1 isoform and subsequent overstimulation of its transcriptional activity, is associated with cell stress that contributes to restrained endometriotic growth (Han et al., 2012).
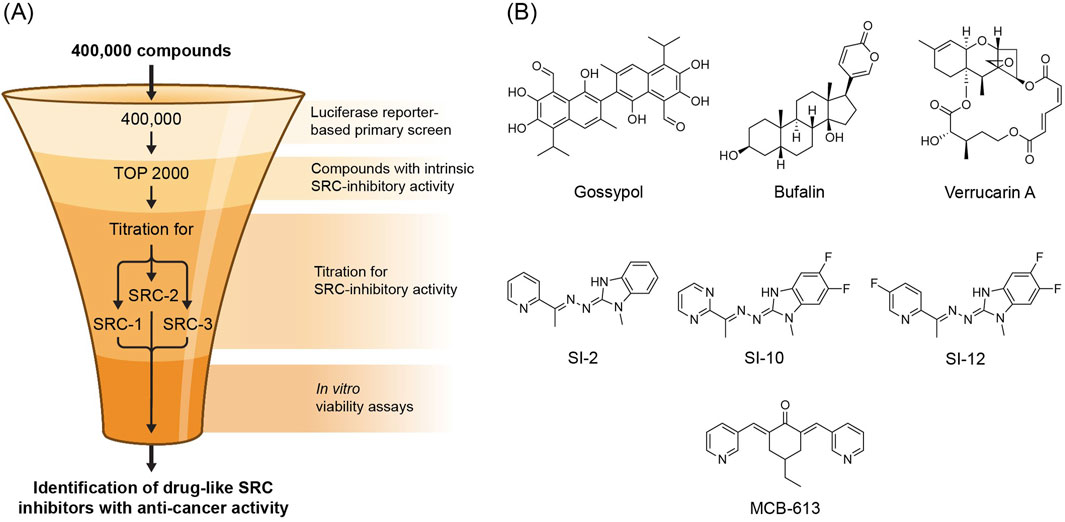
Figure 2. (A) Selection funnel - SRC SMIs discovery HTP screening design. (B) Molecular structures of SRC SMIs and SMS.
In another study verrucarin A, which is structurally distinct from the polyphenol gossypol and cardiac glycosides in general, was characterized as a third class of SRC inhibitors. Verrucarin A is derived from the pathogen fungus Myrothecium and it is structurally related to the trichothecene family of mycotoxins. Like other trichothecenes, Verrucarin A is a cytotoxic molecule with a primary mechanism of action that is associated with protein synthesis inhibition (Jimenez and Vazquez, 1975), albeit other possible mechanisms of action were reported as well (Palanivel et al., 2013; Moon et al., 2013; Jayasooriya et al., 2013). The primary HTP screen indicated that verrucarin A inhibits the intrinsic transcriptional activity of SRCs, leading to a secondary validation which showed that verrucarin A can selectively reduce cellular protein levels of all SRCs, with the most effective inhibitory effect towards SRC-3 (Yan et al., 2014). Like with gossypol and bufalin, verrucarin A-associated inhibition of SRCs results in selective toxicity towards various cancer cell lines, as well as sensitization of cancer cells to treatment with other chemotherapeutic molecules. Unlike gossypol and bufalin, there is no evidence that verrucarin A physically interacts with the SRCs. Therefore, it has been suggested that verrucarin A is an indirect inhibitor of SRCs, that impacts upstream factors which eventually results in destabilization of the SRCs. Overall, luciferase reporter-based HTP screens revealed three distinct classes of naturally occurring SMIs of SRCs (Figure 2B), two of which, gossypol and bufalin, directly bind to SRCs.
The discovery of synthetic molecules that modulate SRC activity
The discoveries of gossypol, bufalin and verrucarin A established the proof-of-principal that the SRCs can be targeted by small-molecules to inhibit cancer cell growth. Moreover, nanoparticle-based formulations and pro-drugs of bufalin improved its bioavailability and effectively inhibited the growth of an aggressive TNBC in vivo (Wang et al., 2014; Song et al., 2015). Encouraged by these findings and by leveraging the data accumulated during the discovery of bufalin and verrucarin A, a new candidate, SI-1, was identified as a potent synthetic SRC SMI. In a search of an optimized version of SI-1, various chemical modifications were performed, while preserving the core structure of the archetype molecule, resulting in a series of SI-1 derivatives. SI-2 (Figure 2B) was identified as the most potent derivative, exhibiting toxicity of 60-fold greater towards cancer cells relative to SI-1 and only requiring nanomolar concentrations to reduce SRC protein levels, whereas SI-1 requires high nanomolar concentrations for the same effect (Song et al., 2016). Furthermore, SI-2 significantly inhibited tumor growth in an in vivo BC tumor model, which was associated with reduced SRC-3 protein levels in the tumor tissue. Interestingly, SI-2-associated reduction in protein levels of SRC-3 did not correlate with SRC-3 mRNA levels, which defines SI-2 as a post-transcriptional inhibitor of SRC-3. Mechanistically, it has been shown that SI-2 inhibits SRC-3 through physical interaction with its NRID domain. Importantly, SI-2 did not possess observable toxicity towards normal cells in vitro or on major organs in vivo. Moreover, cancer cells lacking SRC-3 expression showed much lower sensitivity to treatment with SI-2 compared to their wild type (WT) counterparts. However, a correlation between the expression levels of the other two SRCs and sensitivity to SI-2 has not been evaluated. Nonetheless, the observations mentioned above suggest that SI-2 is a selective SRC SMI with potential applications in cancer therapy.
The introduction of SI-2 as an effective synthetic SRC SMI, with a mechanism of action that involves direct interaction with the SRC NRID domain laid the foundation for the development of other synthetic SMIs with improved drug-like properties. To increase the bio-stability and potency of SI-2, a series of chemical modifications and structure-activity relationship (SAR) exploration, led to the discovery of two analog compounds, SI-10 and SI-12 (Figure 2B), with better metabolic stability and bioavailability compared to SI-2 (Lu et al., 2024). SI-10 and SI-12 both have the core structure of SI-2, that is comprised of a 6-ring heterocycle and benzimidazole-imine moieties. SAR studies revealed that the benzimidazole-imine portion of SI-2 is critical for the preservation of its bioactivity. Therefore, the benzimidazole-imine scaffold was preserved, while introducing two fluorines on the benzene side enhanced metabolic half-life. In the case of SI-12 additional fluorination was introduced into the pyridine ring, while in the case of SI-10 the pyridine heterocycle was substituted with pyrimidine. It has been demonstrated, that like SI-2, the mechanism of action of SI-12 also involves direct binding to SCR-3 (Qin et al., 2021). To shed more light on the mechanism of action of SRC SMIs an in vitro pull-down experiment was performed demonstrating that SI-12 interferes with the formation of the ER-SRC-3/p300 DNA complex (Lu et al., 2024). The disruption of the p300 acetyltransferase recruitment to an active DNA-binding ER complex by SI-12 was associated with significant epigenetic changes in cancer cells as well as a persistent cytotoxic effect of the SI molecules (SI-2 and SI-12) (Lu et al., 2024). These observations suggest that SRC SMIs that bind directly to SRCs prevent the recruitment of other TFs and secondary coregulators, such as p300, that consequently impose cytotoxic epigenetic alterations whose impact on cancer cell survival persists long after the pharmacological pressure of SRC SMI is removed. Overall, SRC SMIs are structurally related molecules with similar impact on SRC activity. SI-1 and its improved version, SI-2, represent the first generation of these synthetic inhibitors of SRCs, while their close structural derivatives, SI-10 and SI-12, represent the second generation with improved drug-like properties, which is reflected in their longer plasma stability and bioavailability.
Like SI-2, SI-10 and SI-12 are selectively toxic towards a variety of cancer cell lines and both are potent tumor growth repressors in vivo, including an ability to repress bone metastases (Lu et al., 2024; Qin et al., 2021). Notably, in vivo treatments with SI-10 and SI-12 required less frequent dosing compared to SI-2, which is attributed to their improved metabolic stability and represents additional drug-like optimization. To expand the therapeutic window and thus enhance the therapeutic applications of the synthetic SRC SMIs, a whole genome screen was performed in an ER + MCF7 cell line to identify drug targets whose inhibition results in an increased sensitivity to SI-12. This screen identified several small-molecule targeted therapies that synergistically enhanced the killing of cancer cells when combined with SI-12. This included DNMT and RhoA inhibitors, which sensitized multiple types of cancer cell lines - beyond MCF-7 cells - to SI-12, demonstrating broad, pan-cancer like efficacy of these drug combinations (Gilad et al., 2021). Overall, the identification of SI-1 and SI-2 as first-generation synthetic SRC SMIs followed by the development of optimized analogs, represent an evolution in the development of small molecule drugs for direct targeting of SRCs, which opens new venues in cancer therapy.
Small molecule-mediated over-stimulation of an oncogene from the SRC family - as a strategy for cancer therapy - represents an alternative approach to small molecule-mediated SRC inhibition. Though it might appear counterintuitive, the underlying hypothesis of such an approach is to put cancer cells out of their homeostatic balance by excessively manipulating the activity of a key transcriptional co-regulator. A few compounds that induce SRC-driven luciferase signal were revealed from the HTP screens that were originally designed to identify SRC inhibitors. Eventually, this led to a detailed characterization of a first-in-class pan-SRC stimulator molecule - MCB-613 (Wang et al., 2015). Like SRC SMIs, MCB-613 directly binds to the SRCs NRID domain, resulting in an enhanced ability of SRCs to interact with other transcriptional activators. Elevated interactions between SRCs and other transcriptional activators result in an overall excess in transcriptional activity. Eventually, an overwhelming MCB-613-induced SRC-hyperactivation converges into a selective toxicity towards cancer cells in vitro and the SRC stimulator’s ability to suppress tumor growth in vivo, through a mechanism that involves excessive generation of reactive oxygen species (ROS) associated with endoplasmic reticulum (ER) stress. Paraptosis was identified as the primary form of cellular death that is induced by MCB-613. Paraptosis is a non-apoptotic form of cell death characterized by lack of DNA fragmentation and presence of massive cytoplasmic vacuoles associated with excessive protein synthesis that ultimately results in elevated levels of ROS, unfolded protein response (UPR) and ER malfunctioning. Hyper-stimulation of an oncogene for killing cancer cells introduces an alternative to the traditional approach of oncogene inhibition, and therefore opens an opportunity for implementing it as a new strategy in cancer treatment (Figure 3).
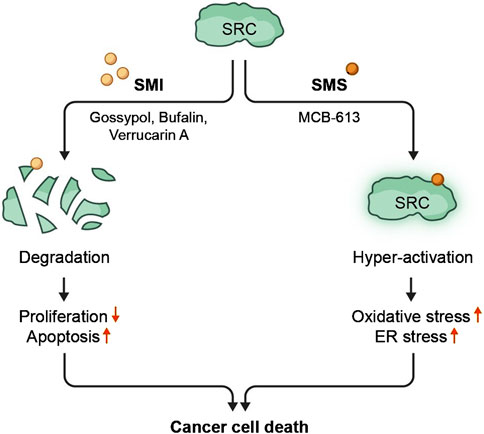
Figure 3. Inhibition or hyperactivation of SRCs with small molecule substances result in cancer cell death. SRC proteins can be targeted by small molecule compounds. Targeting SRC with SMIs results in degradation of SRC proteins, leading to decreased proliferation and increased apoptosis. Targeting SRC with SMSs brings about hyper-activation of SRC transcriptional activities which causes cancer cell-specific cytotoxic stress.
Interestingly, in addition to its direct anti-tumor toxicity, SI-2-mediated SRC-3 inhibition brings about immuno-modulatory effects within the tumor microenvironment (TME). This results in enhanced infiltration of cytotoxic immune cells into the tumor, contributing to overall tumor suppression in immune-intact, but not immune-deficient mice (Han et al., 2022). This observation was further supported by a separate study that showed that a conditional knock out (cKO) of SRC-3 in T regulatory cell (Tregs) results in long-lasting tumor clearance and a vaccine-like tumor rejection effect (Han et al., 2023). Intriguingly, while a SRC SMI exerts immunostimulatory effects, the SRC SMS MCB-613 and its analogs exert an immunosuppressive activity that manifests in post-ischemic tissue protection and repair. This has been exemplified by the establishment of an anti-inflammatory environment and tissue repair in post-myocardial infarction (Mullany et al., 2020), resistance to oxidative stress in cardiac fibroblasts (CFs) (McClendon et al., 2024) and tissue protection following ischemic injury of the brain (McClendon et al., 2022), through mechanisms that involve an activation of anti-inflammatory macrophages, moderation of inflammatory and apoptotic processes such as IL-1b signaling and oxidative stress and induction of antioxidant transcriptional networks. The pro-reparative immuno-suppressive activity of SRC SMSs and anti-tumor immuno-stimulatory activity of SRC SMIs can be regarded as mirror reflections of each other that represent a fascinating theme for expanded investigation. However, the immunomodulatory effects of SRCs and SRC-targeting small molecules constitute a vast topic on their own and are beyond the scope of this review. Readers are encouraged to refer to the references cited above and to our recent reviews on this subject (Gilad et al., 2022; Gilad et al., 2024; Mullany et al., 2021).
Summary and future directions
SRCs regulate the transcriptional activity of NRs and mediate their oncogenic programs. Therefore, since the discovery of the first SRC protein, SRC-1 (Oñate et al., 1995), direct inhibition of SRCs with small molecules was desired, however for a long period of time this was considered unachievable. Thus, strategies designed to effect indirect inhibition of SRCs, such as interference of their interactions with their partner proteins, were developed. The generation of large publicly available depositories of small molecule libraries and the introduction of HTP screening techniques enabled the leveraging of formerly established luciferase-based reporter assays for the discovery of SMIs that directly target the SRCs. Relying on data generated by large-scale HTP screens for the discovery of small molecules that could effectively disrupt SRC-SHR interactions, gossypol was identified as a specific and direct SRC SMI. This discovery provided proof-of-principle that direct inhibition of SRCs with SMIs was possible, thus leading to the design of HTP screens for the identification of additional SRC SMIs. These screens resulted in the discovery and characterization of two additional naturally occurring SRC SMIs, bufalin and verrucarin A, the first synthetic SRC SMI - SI-1 and the SMS–MCB-613. While both naturally occurring substances, bufalin and verrucarin A, inhibit the transcriptional activity of the SRCs and reduce their protein levels, only bufalin was identified as a direct SRC inhibitor that acts through binding to the SRC NRID domain. The structural information received from the discovery of SI-1 served as a starting point and enabled the development of first-generation drug-like synthetic SRC SMI SI-2. Further optimizations of SI-2 led to the discovery of second generation synthetic SRC SMIs SI-10 and SI-12, with improved drug-like characteristics. The first-in-class SRC SMS - MCB-613 was discovered as a ‘byproduct’ of the HTP screens, which were originally designed to discover SRC SMIs. Nonetheless, MCB-613 exhibited effective anti-tumor activity and thus presents an alternative approach for targeting the SRC oncogenes in cancer treatment.
The fact that the SRCs are paralogous proteins with a high degree of structural similarity, likely presents the primary challenge in identifying small molecules that can differentiate between the three family members. Additionally, the SRC small molecule modulators discovered so far, all interact with the NRID domain of SRCs. Consequently, identifying new SRC-small-molecule targeting agents is essential for achieving more specific targeting of each family member. Nonetheless, the discovery of these small-molecule modulators of SRCs represents a major breakthrough and provides a toolkit to overcome the challenge of selectively targeting individual SRC members. Since the SRCs are multifunctional coregulators with a broad impact on human physiology, including roles in systemic physiological processes such as development, metabolism and immunity, interference with their normal functioning is anticipated to result in drug toxicities. Like for other targeted therapies, this presents a significant challenge when considering targeting the SRCs with either SMIs or SMSs for therapeutic purposes.
The introduction of CRISPR-Cas9-based whole-genome screens that includes feasible methods for identifying synthetically lethal anti-cancer drug combinations has significantly accelerated cancer drug discovery (Bock et al., 2022). This is particularly important for understanding and expanding the therapeutic window for pleiotropic drug targets like the SRCs. We have demonstrated that a CRISPR-Cas9 whole-genome screen can be used to identify synergistic anti-cancer effects between SRC SMIs and other anti-cancer agents, allowing for reduced dosages of SRC SMIs which potentiates their utilization with fewer side effects (Gilad et al., 2021). Overall, the discovery of small-molecular modulators that interfere with the biological activity of SRCs through direct interaction with their NRID domain represents a turning point in the therapeutic targeting of this family of coregulators. However, the challenges of selectively targeting specific family members and the risk of major side effects remain as significant barriers that must be overcome for successful clinical applications. With the availability of small molecules that can either stimulate or inhibit SRC activity, alongside the development of CRISPR-Cas9-based high-throughput screens, these challenges are now more readily addressable.
Beyond their potential as anti-cancer therapies, SRC SMIs and SMSs can mediate immunomodulatory effects. This has been demonstrated with SI-2 that can produce an anti-tumor immune response in an immunocompetent mouse tumor model and by SRC SMSs that can promote a reparative environment in tissues subjected to ischemic injury. The surprising ability of SRC small molecule modulators to exert immunomodulatory effects opens new avenues for exploring SRCs as drug targets, beyond the context of their oncogenicity. This is particularly valuable in cancer treatment, where SRC SMIs can act both as direct suppressors of SRC oncogenes and as anti-cancer immunomodulators.
Author contributions
YG: Conceptualization, Writing–original draft, Writing–review and editing. OS: Writing–review and editing. DL: Funding acquisition, Resources, Writing–review and editing. BO’M: Funding acquisition, Resources, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is partly supported by funding from the National Institutes of Health grants R01 HD07857 and R01 HD08188 (BWO) and from the Adrienne Helis Malvin Foundation to DL (DL) and CoRegen Inc. CoRegen Inc was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
The authors acknowledge the graphic design assistance provided by Dr. Sandy R. Westermann (www.scigraphix.com).
Conflict of interest
Author OS was employed by CoRegen, Inc. Authors YG, DL and BO’M were paid consultants by and disclose an equity position in CoRegen, Inc.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agoulnik, I. U., Vaid, A., Nakka, M., Alvarado, M., Bingman, W. E., Erdem, H., et al. (2006). Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 66 (21), 10594–10602. doi:10.1158/0008-5472.CAN-06-1023
Anzick, S. L., Kononen, J., Walker, R. L., Azorsa, D. O., Tanner, M. M., Guan, X.-Y., et al. (1997). AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277 (5328), 965–968. doi:10.1126/science.277.5328.965
Bautista, S., Valles, H., Walker, R. L., Anzick, S., Zeillinger, R., Meltzer, P., et al. (1998). In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clin. cancer Res. official J. Am. Assoc. Cancer Res. 4 (12), 2925–2929.
Bock, C., Datlinger, P., Chardon, F., Coelho, M. A., Dong, M. B., Lawson, K. A., et al. (2022). High-content CRISPR screening. Nat. Rev. Methods Prim. 2 (1), 8. doi:10.1038/s43586-021-00093-4
Cai, D., Shames, D. S., Raso, M. G., Xie, Y., Kim, Y. H., Pollack, J. R., et al. (2010). Steroid receptor coactivator-3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer Res. 70 (16), 6477–6485. doi:10.1158/0008-5472.CAN-10-0005
Cho, Y. J., Lee, J. E., Park, M. J., O’Malley, B. W., and Han, S. J. (2018). Bufalin suppresses endometriosis progression by inducing pyroptosis and apoptosis. J. Endocrinol. 237 (3), 255–269. doi:10.1530/JOE-17-0700
Coulthard, V. H., Matsuda, S., and Heery, D. M. (2003). An extended LXXLL motif sequence determines the nuclear receptor binding specificity of TRAP220. J. Biol. Chem. 278 (13), 10942–10951. doi:10.1074/jbc.M212950200
Crawford, E. D., Schellhammer, P. F., McLeod, D. G., Moul, J. W., Higano, C. S., Shore, N., et al. (2018). Androgen receptor targeted treatments of prostate cancer: 35 years of progress with antiandrogens. J. urology 200 (5), 956–966. doi:10.1016/j.juro.2018.04.083
Darimont, B. D., Wagner, R. L., Apriletti, J. W., Stallcup, M. R., Kushner, P. J., Baxter, J. D., et al. (1998). Structure and specificity of nuclear receptor–coactivator interactions. Genes and Dev. 12 (21), 3343–3356. doi:10.1101/gad.12.21.3343
Dasgupta, S., Putluri, N., Long, W., Zhang, B., Wang, J., Kaushik, A. K., et al. (2015). Coactivator SRC-2–dependent metabolic reprogramming mediates prostate cancer survival and metastasis. J. Clin. investigation 125 (3), 1174–1188. doi:10.1172/JCI76029
De Bosscher, K., Desmet, S. J., Clarisse, D., Estébanez-Perpiña, E., and Brunsveld, L. (2020). Nuclear receptor crosstalk — defining the mechanisms for therapeutic innovation. Nat. Rev. Endocrinol. 16 (7), 363–377. doi:10.1038/s41574-020-0349-5
Fleming, F., Myers, E., Kelly, G., Crotty, T., McDermott, E., O’higgins, N., et al. (2004). Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J. Clin. pathology 57 (10), 1069–1074. doi:10.1136/jcp.2004.016733
Gehin, M., Mark, M., Dennefeld, C., Dierich, A., Gronemeyer, H., and Chambon, P. (2002). The function of TIF2/GRIP1 in mouse reproduction is distinct from those of SRC-1 and p/CIP. Mol. Cell. Biol. 22 (16), 5923–5937. doi:10.1128/mcb.22.16.5923-5937.2002
Gilad, Y., Eliaz, Y., Yu, Y., Dean, A. M., Han, S. J., Qin, L., et al. (2021). A genome-scale CRISPR Cas9 dropout screen identifies synthetically lethal targets in SRC-3 inhibited cancer cells. Commun. Biol. 4 (1), 399. doi:10.1038/s42003-021-01929-1
Gilad, Y., Lonard, D. M., and O’Malley, B. W. (2022). Steroid receptor coactivators – their role in immunity. Front. Immunol. 13, 1079011. doi:10.3389/fimmu.2022.1079011
Gilad, Y., Shimon, O., Han, S. J., Lonard, D. M., and O’Malley, B. W. (2024). Steroid receptor coactivators in Treg and Th17 cell biology and function. Front. Immunol. 15, 1389041. doi:10.3389/fimmu.2024.1389041
Gilbert, N. E., O'Reilly, J. E., Chang, C. G., Lin, Y. C., and Brueggemeier, R. W. (1995). Antiproliferative activity of gossypol and gossypolone on human breast cancer cells. Life Sci. 57 (1), 61–67. doi:10.1016/0024-3205(95)00243-y
Gnanapragasam, V., Leung, H., Pulimood, A., Neal, D., and Robson, C. (2001). Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br. J. cancer 85 (12), 1928–1936. doi:10.1054/bjoc.2001.2179
Gunther, J. R., Du, Y., Rhoden, E., Lewis, I., Revennaugh, B., Moore, T. W., et al. (2009). A set of time-resolved fluorescence resonance energy transfer assays for the discovery of inhibitors of estrogen receptor-coactivator binding. J. Biomol. Screen. 14 (2), 181–193. doi:10.1177/1087057108329349
Han, S. J., Hawkins, S. M., Begum, K., Jung, S. Y., Kovanci, E., Qin, J., et al. (2012). A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat. Med. 18 (7), 1102–1111. doi:10.1038/nm.2826
Han, S. J., Jain, P., Gilad, Y., Xia, Y., Sung, N., Park, M. J., et al. (2023). Steroid receptor coactivator 3 is a key modulator of regulatory T cell–mediated tumor evasion. Proc. Natl. Acad. Sci. 120 (23), e2221707120. doi:10.1073/pnas.2221707120
Han, S. J., Sung, N., Wang, J., O’Malley, B. W., and Lonard, D. M. (2022). Steroid receptor coactivator-3 inhibition generates breast cancer antitumor immune microenvironment. Breast Cancer Res. 24 (1), 73. doi:10.1186/s13058-022-01568-2
Heery, D. M., Kalkhoven, E., Hoare, S., and Parker, M. G. (1997). A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387 (6634), 733–736. doi:10.1038/42750
Henke, R. T., Haddad, B. R., Kim, S. E., Rone, J. D., Mani, A., Jessup, J. M., et al. (2004). Overexpression of the nuclear receptor coactivator AIB1 (SRC-3) during progression of pancreatic adenocarcinoma. Clin. cancer Res. 10 (18), 6134–6142. doi:10.1158/1078-0432.CCR-04-0561
Huang, Y.-W., Wang, L.-S., Chang, H.-L., Ye, W., Dowd, M. K., Wan, P. J., et al. (2006). Molecular mechanisms of (-)-gossypol-induced apoptosis in human prostate cancer cells. Anticancer Res. 26 (3A), 1925–1933.
Hudelist, G., Czerwenka, K., Kubista, E., Marton, E., Pischinger, K., and Singer, C. F. (2003). Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast cancer Res. Treat. 78, 193–204. doi:10.1023/a:1022930710850
Jayasooriya, R. G. P. T., Moon, D.-O., Park, S. R., Choi, Y. H., Asami, Y., Kim, M.-O., et al. (2013). Combined treatment with verrucarin A and tumor necrosis factor-α sensitizes apoptosis by overexpression of nuclear factor-kappaB-mediated Fas. Environ. Toxicol. Pharmacol. 36 (2), 303–310. doi:10.1016/j.etap.2013.04.008
Jimenez, A., and Vazquez, D. (1975). Quantitative binding of antibiotics to ribosomes from a yeast mutant altered on the peptidyl-transferase center. Eur. J. Biochem. 54 (2), 483–492. doi:10.1111/j.1432-1033.1975.tb04160.x
Johnson, A. B., and O’Malley, B. W. (2012). Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol. Cell. Endocrinol. 348 (2), 430–439. doi:10.1016/j.mce.2011.04.021
Kershah, S. M., Desouki, M. M., Koterba, K. L., and Rowan, B. G. (2004). Expression of estrogen receptor coregulators in normal and malignant human endometrium. Gynecol. Oncol. 92 (1), 304–313. doi:10.1016/j.ygyno.2003.10.007
Lanz, R. B., Bulynko, Y., Malovannaya, A., Labhart, P., Wang, L., Li, W., et al. (2010). Global characterization of transcriptional impact of the SRC-3 coregulator. Mol. Endocrinol. 24 (4), 859–872. doi:10.1210/me.2009-0499
Li, C., Wu, R.-C., Amazit, L., Tsai, S. Y., Tsai, M.-J., and O'Malley, B. W. (2007). Specific amino acid residues in the basic helix-loop-helix domain of SRC-3 are essential for its nuclear localization and proteasome-dependent turnover. Mol. Cell. Biol. 27 (4), 1296–1308. doi:10.1128/MCB.00336-06
Lonard, D. M., Nawaz, Z., Smith, C. L., and O'Malley, B. W. (2000). The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol. cell 5 (6), 939–948. doi:10.1016/s1097-2765(00)80259-2
Lonard, D. M., and O’Malley, B. (2023). “Chapter 20 - nuclear receptors and coactivators,” in Genetic steroid disorders. Editor M. I. New Second Edition (San Diego: Academic Press), 373–380.
Lonard, D. M., Tsai, S. Y., and O'Malley, B. W. (2004). Selective estrogen receptor modulators 4-hydroxytamoxifen and raloxifene impact the stability and function of SRC-1 and SRC-3 coactivator proteins. Mol. Cell. Biol. 24 (1), 14–24. doi:10.1128/mcb.24.1.14-24.2004
Lu, D., Chen, J., Qin, L., Bijou, I., Yi, P., Li, F., et al. (2024). Lead compound development of SRC-3 inhibitors with improved pharmacokinetic properties and anticancer efficacy. J. Med. Chem. 67 (7), 5333–5350. doi:10.1021/acs.jmedchem.3c01596
McClendon, L. K., Garcia, R. L., Lazaro, T., Robledo, A., Vasandani, V., Luna, Z. A. E., et al. (2022). A steroid receptor coactivator small molecule “stimulator” attenuates post-stroke ischemic brain injury. Front. Mol. Neurosci. 15, 1055295. doi:10.3389/fnmol.2022.1055295
McClendon, L. K., Lanz, R. B., Panigrahi, A., Gomez, K., Bolt, M. J., Liu, M., et al. (2024). Transcriptional coactivation of NRF2 signaling in cardiac fibroblasts promotes resistance to oxidative stress. J. Mol. Cell. Cardiol. 194, 70–84. doi:10.1016/j.yjmcc.2024.07.001
Meng, Y., Tang, W., Dai, Y., Wu, X., Liu, M., Ji, Q., et al. (2008). Natural BH3 mimetic (-)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol. cancer Ther. 7 (7), 2192–2202. doi:10.1158/1535-7163.MCT-08-0333
Mettu, N. B., Stanley, T. B., Dwyer, M. A., Jansen, M. S., Allen, J. E., Hall, J. M., et al. (2007). The nuclear receptor-coactivator interaction surface as a target for peptide antagonists of the peroxisome proliferator-activated receptors. Mol. Endocrinol. 21 (10), 2361–2377. doi:10.1210/me.2007-0201
Moon, D.-O., Asami, Y., Long, H., Jang, J. H., Bae, E. Y., Kim, B. Y., et al. (2013). Verrucarin A sensitizes TRAIL-induced apoptosis via the upregulation of DR5 in an eIF2α/CHOP-dependent manner. Toxicol. vitro 27 (1), 257–263. doi:10.1016/j.tiv.2012.09.001
Mukherjee, A., Soyal, S. M., Fernandez-Valdivia, R., Gehin, M., Chambon, P., DeMayo, F. J., et al. (2006). Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol. Cell. Biol. 26 (17), 6571–6583. doi:10.1128/MCB.00654-06
Mullany, L. K., Lonard, D. M., and O’Malley, B. W. (2021). Wound healing-related functions of the p160 steroid receptor coactivator family. Endocrinology 162 (3), bqaa232. doi:10.1210/endocr/bqaa232
Mullany, L. K., Rohira, A. D., Leach, J. P., Kim, J. H., Monroe, T. O., Ortiz, A. R., et al. (2020). A steroid receptor coactivator stimulator (MCB-613) attenuates adverse remodeling after myocardial infarction. Proc. Natl. Acad. Sci. 117 (49), 31353–31364. doi:10.1073/pnas.2011614117
Musgrove, E. A., and Sutherland, R. L. (2009). Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 9 (9), 631–643. doi:10.1038/nrc2713
Nuclear Receptors Nomenclature Committee (1999). A unified nomenclature system for the nuclear receptor superfamily. Cell 97, 161–163. doi:10.1016/s0092-8674(00)80726-6
O’Malley, B. W. (2020). SRC-2 Coactivator: a role in human metabolic evolution and disease. Mol. Med. 26, 45. doi:10.1186/s10020-020-00168-0
Oñate, S. A., Tsai, S. Y., Tsai, M.-J., and O'Malley, B. W. (1995). Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270 (5240), 1354–1357. doi:10.1126/science.270.5240.1354
Osborne, C. K., Bardou, V., Hopp, T. A., Chamness, G. C., Hilsenbeck, S. G., Fuqua, S. A., et al. (2003). Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 95 (5), 353–361. doi:10.1093/jnci/95.5.353
Osborne, C. K., and Schiff, R. (2011). Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 62, 233–247. doi:10.1146/annurev-med-070909-182917
Palanivel, K., Kanimozhi, V., Kadalmani, B., and Akbarsha, M. A. (2013). Verrucarin A, a protein synthesis inhibitor, induces growth inhibition and apoptosis in breast cancer cell lines MDA-MB-231 and T47D. Biotechnol. Lett. 35, 1395–1403. doi:10.1007/s10529-013-1238-y
Perissi, V., and Rosenfeld, M. G. (2005). Controlling nuclear receptors: the circular logic of cofactor cycles. Nat. Rev. Mol. cell Biol. 6 (7), 542–554. doi:10.1038/nrm1680
Prassas, I., and Diamandis, E. P. (2008). Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 7 (11), 926–935. doi:10.1038/nrd2682
Puigserver, P., Adelmant, G., Wu, Z., Fan, M., Xu, J., O'Malley, B., et al. (1999). Activation of PPARgamma coactivator-1 through transcription factor docking. Science 286 (5443), 1368–1371. doi:10.1126/science.286.5443.1368
Qin, L., Chen, J., Lu, D., Jain, P., Yu, Y., Cardenas, D., et al. (2021). Development of improved SRC-3 inhibitors as breast cancer therapeutic agents. Endocrine-related cancer 28 (10), 657–670. doi:10.1530/ERC-20-0402
Qin, L., Liu, Z., Chen, H., and Xu, J. (2009). The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 69 (9), 3819–3827. doi:10.1158/0008-5472.CAN-08-4389
Rodriguez, A. L., Tamrazi, A., Collins, M. L., and Katzenellenbogen, J. A. (2004). Design, synthesis, and in vitro biological evaluation of small molecule inhibitors of estrogen receptor alpha coactivator binding. J. Med. Chem. 47 (3), 600–611. doi:10.1021/jm030404c
Santos, R., Ursu, O., Gaulton, A., Bento, A. P., Donadi, R. S., Bologa, C. G., et al. (2017). A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16 (1), 19–34. doi:10.1038/nrd.2016.230
Skowron, K. J., Booker, K., Cheng, C., Creed, S., David, B. P., Lazzara, P. R., et al. (2019). Steroid receptor/coactivator binding inhibitors: an update. Mol. Cell. Endocrinol. 493, 110471. doi:10.1016/j.mce.2019.110471
Song, X., Chen, J., Zhao, M., Zhang, C., Yu, Y., Lonard, D. M., et al. (2016). Development of potent small-molecule inhibitors to drug the undruggable steroid receptor coactivator-3. Proc. Natl. Acad. Sci. 113 (18), 4970–4975. doi:10.1073/pnas.1604274113
Song, X., Zhang, C., Zhao, M., Chen, H., Liu, X., Chen, J., et al. (2015). Steroid receptor coactivator-3 (SRC-3/AIB1) as a novel therapeutic target in triple negative breast cancer and its inhibition with a phospho-bufalin prodrug. PloS one 10 (10), e0140011. doi:10.1371/journal.pone.0140011
Spencer, T. E., Jenster, G., Burcin, M. M., Allis, C. D., Zhou, J., Mizzen, C. A., et al. (1997). Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 389 (6647), 194–198. doi:10.1038/38304
Stashi, E., Lanz, R. B., Mao, J., Michailidis, G., Zhu, B., Kettner, N. M., et al. (2014). SRC-2 is an essential coactivator for orchestrating metabolism and circadian rhythm. Cell Rep. 6 (4), 633–645. doi:10.1016/j.celrep.2014.01.027
Taylor, B. S., Schultz, N., Hieronymus, H., Gopalan, A., Xiao, Y., Carver, B. S., et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer cell 18 (1), 11–22. doi:10.1016/j.ccr.2010.05.026
Ulm, M., Ramesh, A. V., McNamara, K. M., Ponnusamy, S., Sasano, H., and Narayanan, R. (2019). Therapeutic advances in hormone-dependent cancers: focus on prostate, breast and ovarian cancers. Endocr. Connect. 8 (2), R10-R26–R26. doi:10.1530/EC-18-0425
Walsh, C. A., Qin, L., Tien, J.C.-Y., Young, L. S., and Xu, J. (2012). The function of steroid receptor coactivator-1 in normal tissues and cancer. Int. J. Biol. Sci. 8 (4), 470–485. doi:10.7150/ijbs.4125
Wang, L., Yu, Y., Chow, D.-C., Yan, F., Hsu, C.-C., Stossi, F., et al. (2015). Characterization of a steroid receptor coactivator small molecule stimulator that overstimulates cancer cells and leads to cell stress and death. Cancer cell 28 (2), 240–252. doi:10.1016/j.ccell.2015.07.005
Wang, S., Yuan, Y., Liao, L., Kuang, S.-Q., Tien, J.C.-Y., O'Malley, B. W., et al. (2009). Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proc. Natl. Acad. Sci. 106 (1), 151–156. doi:10.1073/pnas.0808703105
Wang, Y., Lonard, D. M., Yu, Y., Chow, D.-C., Palzkill, T. G., and O'Malley, B. W. (2011). Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol. Endocrinol. 25 (12), 2041–2053. doi:10.1210/me.2011-1222
Wang, Y., Lonard, D. M., Yu, Y., Chow, D.-C., Palzkill, T. G., Wang, J., et al. (2014). Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res. 74 (5), 1506–1517. doi:10.1158/0008-5472.CAN-13-2939
Wang, Y., Wu, M. C., Sham, J. S., Zhang, W., Wu, W. Q., and Guan, X. Y. (2002). Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma: a broad survey using high-throughput tissue microarray. Cancer Interdiscip. Int. J. Am. Cancer Soc. 95 (11), 2346–2352. doi:10.1002/cncr.10963
Watson, P. A., Arora, V. K., and Sawyers, C. L. (2015). Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15 (12), 701–711. doi:10.1038/nrc4016
Wolter, K. G., Wang, S. J., Henson, B. S., Wang, S., Griffith, K. A., Kumar, B., et al. (2006). (−)-gossypol inhibits growth and promotes apoptosis of human head and neck squamous cell carcinoma in vivo. Neoplasia 8 (3), 163–172. doi:10.1593/neo.05691
Xie, D., Sham, J. S., Zeng, W.-F., Lin, H.-L., Bi, J., Che, L.-H., et al. (2005). Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum. Pathol. 36 (7), 777–783. doi:10.1016/j.humpath.2005.05.007
Xu, J., Qiu, Y., DeMayo, F. J., Tsai, S. Y., Tsai, M.-J., and O'Malley, B. W. (1998). Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 279 (5358), 1922–1925. doi:10.1126/science.279.5358.1922
Yan, F., Yu, Y., Chow, D.-C., Palzkill, T., Madoux, F., Hodder, P., et al. (2014). Identification of verrucarin a as a potent and selective steroid receptor coactivator-3 small molecule inhibitor. PLoS One 9 (4), e95243. doi:10.1371/journal.pone.0095243
Ye, Q., Zhou, X., Ren, H., Han, F., Lin, R., and Li, J. (2023). An overview of the past decade of bufalin in the treatment of refractory and drug-resistant cancers: current status, challenges, and future perspectives. Front. Pharmacol. 14, 1274336. doi:10.3389/fphar.2023.1274336
Zhao, C., Yasui, K., Lee, C. J., Kurioka, H., Hosokawa, Y., Oka, T., et al. (2003). Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer 98 (1), 18–23. doi:10.1002/cncr.11482
Keywords: Steroid receptor coactivator (SRC), drug discovery, cancer therapy, small molecule drugs (SMDs), drug combinations
Citation: Gilad Y, Shimon O, Lonard DM and O’Malley BW (2024) Targeting steroid receptor coactivators in cancer via small molecule agents. Front. Drug Discov. 4:1514412. doi: 10.3389/fddsv.2024.1514412
Received: 20 October 2024; Accepted: 05 December 2024;
Published: 19 December 2024.
Edited by:
Greco Hernández, National Institute of Cancerology (INCAN), MexicoReviewed by:
Nahuel Peinetti, University of Miami, United StatesRocío García-Becerra, National Autonomous University of Mexico, Mexico
Copyright © 2024 Gilad, Shimon, Lonard and O’Malley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yosi Gilad, eW9zaUBiY20uZWR1; David M. Lonard, ZGxvbmFyZEBiY20uZWR1; Bert W. O’Malley, YmVydG9AYmNtLmVkdQ==