 Luca Santuari1
Luca Santuari1 Ioannis Xenarios
Ioannis Xenarios Bulak Arpat
Bulak Arpat
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Drug Discov., 03 September 2024
Sec. In silico Methods and Artificial Intelligence for Drug Discovery
Volume 4 - 2024 | https://doi.org/10.3389/fddsv.2024.1447867
Antibodies represent the largest class of biotherapeutics thanks to their high target specificity, binding affinity and versatility. Recent breakthroughs in Artificial Intelligence (AI) have enabled information-rich in silico representations of antibodies, accurate prediction of antibody structure from sequence, and the generation of novel antibodies tailored to specific characteristics to optimize for developability properties. Here we summarize state-of-the-art methods for antibody analysis. This valuable resource will serve as a reference for the application of AI methods to the analysis of antibody sequencing datasets.
Antibodies are the largest class of biotherapeutics, with a projected market size of US$ 300 Billion by 2025 (Lu et al., 2020). They are used for treating cancer, autoimmune and infectious diseases (Lu et al., 2020; Weiner et al., 2010; Chan and Carter, 2010), as they can be designed to recognize any antigen at high specificity and binding affinity. Antibody discovery is traditionally performed with directed evolution using experimental assays such as hybridoma or phage display (Lu et al., 2020). Although well-established, these methods remain costly, time-consuming and prone to fail due to experimental challenges.
The introduction of Next-Generation Sequencing (NGS) for antibody screening in place of random colony picking has enabled to cover a much larger sequence diversity, a wider binding affinity range, and isolate sequences that target distinct epitopes (Spoendlin et al., 2023). Short read sequencing is limited to a single chain, either heavy (VH) and light chain (VL), while long reads can obtain paired information of both chains, increasing our understanding of inter-chain residue dependencies (Burbach and Briney, 2024).
Recently, Artificial Intelligence (AI) has experienced accelerated progress, particularly in the fields of Deep Learning (DL) and Natural Language Processing (NLP), and biology has been greatly benefited from it (Khakzad et al., 2023; Graves et al., 2020; Nam Kim et al., 2024; Bender and Cortés-Ciriano, 2021; Bender and Cortes-Ciriano, 2021; Kim et al., 2023). A notable example is the model AlphaFold2 for structural biology (Jumper et al., 2021), which brought sequence-based protein structure prediction close to experimental accuracy.
The success of the Transformer architecture (Vaswani et al., 2023) in NLP has led to the creation of Large Language Models (LLM), statistical models trained on large collections of texts to capture semantic similarity among words in the form of vector representations, called embeddings, without relying on expensive and hard to obtain labels. Embeddings are very versatile, with applications that include text classification and generation. In biology, LLMs trained on curated databases of millions of protein sequences [UniProt (UniProt Consortium et al., 2023), UniRef (Suzek et al., 2007) and BFD (Jumper et al., 2021; Steinegger and Söding, 2018)] were shown to be able to learn secondary and tertiary structural information from sequence (Ahmed et al., 2022; Lin et al., 2023) and can be used to predict protein function. More recently LLMs have been trained on databases of antibody sequences, such as the Observed Antibody Space (OAS), leading to the creation of antibody-specific language models (ALMs) (Leem et al., 2022; Ruffolo et al., 2023; Prihoda et al., 2022; Olsen et al., 2022).
Despite the availability of these models, bringing an antibody from discovery to the patient remains challenging. Once a candidate antibody has been found, it must be optimized to match the properties of therapeutic antibodies, grouped under the term of developability. A consensus is lacking in the literature for which properties are part of developability (Habib et al., 2023; Raybould et al., 2024; Fernández-Quintero et al., 2023; Khetan et al., 2022; Zhang et al., 2023; Evers et al., 2023). Some of these properties are humanization, prediction of solubility and aggregation, for which several ML methods have been proposed (Prihoda et al., 2022; Parkinson and Wang, 2024; Pujols et al., 2022).
The type of license associated with a ML model (code, weights and training data) plays a key role in the choice of integration into industrial applications. A commercially permissive license favors rapid prototyping in research and development within an industrial setting and rapid, cost-free integration into a product. In this review, we indicate the license type associated to the methods presented, in the hope that this resource will serve as a reference to accelerate the adoption of these models in industry.
Several reviews have been published that discuss ML applications to antibody discovery and development (Graves et al., 2020; Nam Kim et al., 2024; Kim et al., 2023). These reviews are focused on giving an academic perspective on the field. Our review stands out not only for its breadth, by providing a comprehensive, up-to-date overview of the state-of-the-art AI methods and resources for antibody sequence, structure and developability, but also for the particular focus on practical considerations in regard to product integration, such as licenses.
Providing a comprehensive benchmark for these methods is outside the scope of this review, and would require testing against specific benchmark datasets like ProteinGym (Notin et al., 2023) for general protein language models and FlAb (Chungyoun et al., 2024) for antibody language models.
This review is divided into three parts (Figure 1). The first part covers recent applications of LLMs to protein (PLM) and antibody (ALM) sequences. The second part focuses on folding models, models that can predict protein structure from sequence, and inverse folding models, models that can identify the sequences a specific structure can fold into. The third part covers ML methods that can be used to optimize developability properties. Finally, we conclude with some remarks and perspectives.
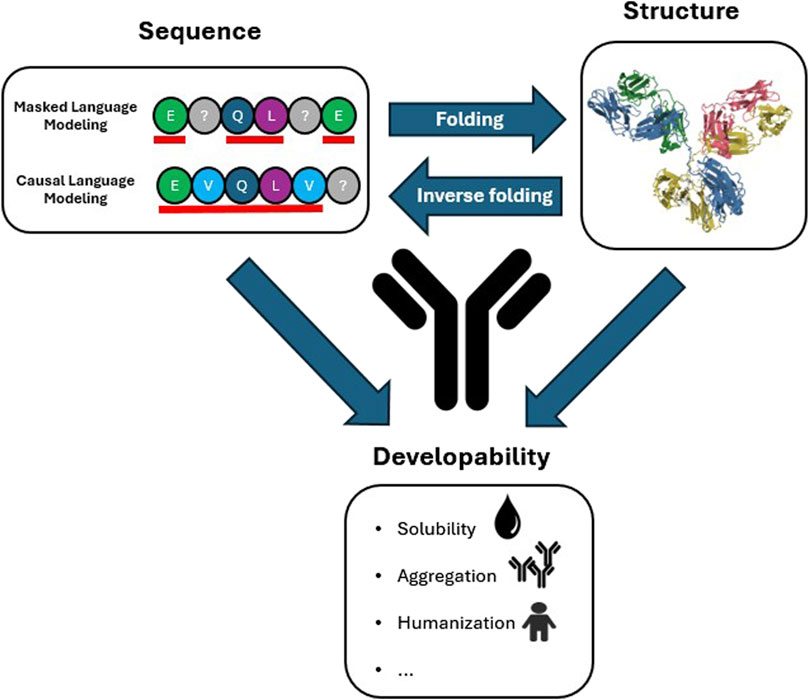
Figure 1. Antibody sequence and structure information in relation to developability properties. Masked (contextual, BERT-like) and Causal (autoregressive, GPT-like) Language Modeling prediction strategies are highlighted. The residues underlined in red are the residues that are used to train the model to predict the masked residues (Masked) and next residue (Causal) indicated with a grey question mark. The representative antibody structure is the structure of immunoglobulin (PDB:1IGY). Arrows indicate the information flow from sequence to structure (folding models), from structure to sequence (inverse folding models) and that developability properties are determined by both antibody sequence and structure.
The field of NLP was revolutionized by the introduction of the Transformer (Vaswani et al., 2017), a DL architecture that was able to achieve unprecedented accuracy in understanding and generation of written and spoken languages, programming languages, images and videos (Islam et al., 2023). At the core of the Transformer is the attention layer, a neural network layer inspired by cognitive attention, the human ability to focus on important signals and exclude irrelevant information. Through attention the model learns the relative importance all parts of the input sequence (tokens) have with respect to each other. This is used to generate a vector representation of each token in the sequence (embedding) that can be leveraged for specific tasks. Training is performed with either a Masked Language Modelling (MLM) objective, the prediction of a randomly chosen subset of masked tokens, a Causal Language Modelling (CLM) objective, the prediction of the next token based on the preceding tokens, or both (Figure 1).
When trained on large collections of protein sequences [UniProt (UniProt Consortium et al., 2023), UniRef (Suzek et al., 2007) and BFD (Steinegger and Söding, 2018)] as protein Language Models (PLMs) (Table 1), these models capture information on evolutionary constraints, secondary and tertiary structures (Ahmed et al., 2022; Lin et al., 2023; Rives et al., 2021). For a comprehensive overview of NLP applied to the protein sequence domain we refer to the available reviews (Ofer et al., 2021; Valentini et al., 2023; Dounas et al., 2024).
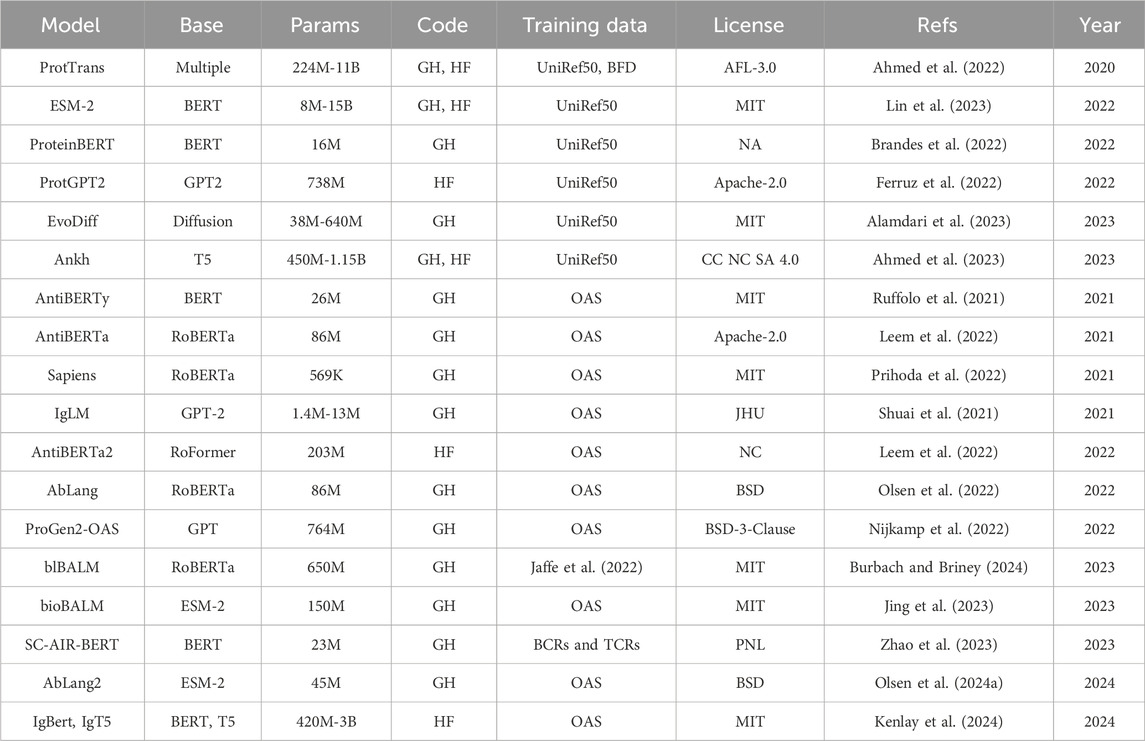
Table 1. Specification for general protein language models (top) and antibody-specific language models (bottom). Base, base model architecture; Params, number of trainable parameters; Code, model and code availability; Training data, dataset used for training; License, release license; Refs, references; Year, year of first release. ProtTrans is a collection of models with base architecture Transformer-XL (Dai et al., 2019), XLNet (Yang et al., 2020), BERT (Jacob et al., 2019), Albert (Lan et al., 2019), Electra (Clark et al., 2020), T5 (Raffel et al., 2023). For disambiguation we refer to Baseline Antibody Language Model (BALM) as blBALM and to Bio-inspired Antibody Language Model (BALM) as bioBALM. GH: GitHub. HF: HuggingFace.
The concept of PLMs was later applied to antibody sequences, resulting in Antibody Language Models (ALM) (Table 1). Most of these models have been trained on unpaired data, either a single model including both chain types (AntiBERTa (Leem et al., 2022), AntiBERTy (Ruffolo et al., 2023), IgLM (Shuai et al., 2021), BALM-unpaired (Burbach and Briney, 2024), Bio-inspired Antibody Language Model (Jing et al., 2023)) or with chain-specific models [Sapiens (Prihoda et al., 2022), AbLang (Olsen et al., 2022)]. Other models, such as BALM-paired (Burbach and Briney, 2024), ESM2-paired (Burbach and Briney, 2024), SC-AIR-BERT (Zhao et al., 2023), and AbLang2 (Olsen et al., 2024a), make use of paired sequence information to capture inter-chain residue dependencies. Applications of these models include paratope prediction [AntiBERTa (Leem et al., 2022)), humanization (Sapiens (Prihoda et al., 2022)], sequence completion [AbLang (Olsen et al., 2022)] and generation conditioned on species and chain type [AntiBERTy (Ruffolo et al., 2023), IgLM (Shuai et al., 2021)]. pAbT5 (Simon et al., 2023) stands out as it is tasked to predict one chain type starting from the other.
AlphaFold2 (Jumper et al., 2021) led to impressive improvements in the accuracy of protein sequence-to-structure prediction. One of major bottlenecks for the runtime of AlphaFold2 is the need to construct a Multiple Sequence Alignment (MSA) from the input sequence. Recently, structural information learned with PLMs has been leveraged to substitute the MSA dependency leading to the release of sequence-only models (ESMFold (Lin et al., 2023), BALMFold (Jing et al., 2023), OmegaFold (Wu Ruidong et al., 2022), HelixFold-Single (Fang et al., 2023) and EMBER3D (Weissenow et al., 2022)). Barret and coauthors (Barrett et al., 2022) compared an AlphaFold architecture using either only MSA or sequence (MonoFold) or both inputs together (PolyFold) and showed that the two input modes are complementary to each other, although using MSA has still higher performance. The reliance of AlphaFold2 on the MSA is also reflected in the lower accuracy when predicting the structure of orphan and de novo proteins. Both RGN2 (Chowdhury et al., 2021) and trRosettaX-Single (Wang et al.) have been proposed to address this limitation. More recently several models have been published addressing the structure prediction at the atomic level [Protpardelle (Chu et al., 2023), EquiFold (Lee et al., 2022), RoseTTAFold All-Atom (Krishna et al., 2024)] instead of the amino acid level, opening up new possibilities for modelling protein complexes with DNA, RNA, and small molecules. This is also the focus of AlphaFold3 (Abramson et al., 2024).
The prediction of antibody structure carries additional challenges with respect to other proteins. The Complementary Determining Regions (CDRs) responsible for the binding with the antigen are the most variable and therefore difficult to predict, especially the CDR3 region of the heavy chain (HCDR3). Several models have been published to address these challenges [ABlooper (Brennan et al., 2022), IgFold (Ruffolo et al., 2023), EquiFold (Lee et al., 2022), DeepAB (Ruffolo et al., 2022), ABodyBuilder2 (Brennan et al., 2023)]. ABodyBuilder2 is part of the ImmuneBuilder (Brennan et al., 2023) suite and has better performance with respect to ABlooper, IgFold, EquiFold and AlphaFold-Multimer (Evans et al., 2021), specifically for the prediction of the HCDR3 loops, achieving a RMSD of 2.81 Å. This improvement was achieved by using an ensemble of four models built on the structure module of AlphaFold-Multimer followed by refinement with OpenMM and pdbfixer. tFold-Ab (Wu Jiaxiang et al., 2022) first computes single chain structure predictions using the PLM ProtXLNet (Ahmed et al., 2022) and then predicts the multimer conformation of the heavy and light chains using a simplified version of the Evoformer module of AlphaFold that takes single sequence in input. However, the availability of this method only as a web-server hinders the possibility to assess its performance with respect to available benchmarks.
DL has been recently applied to the inverse folding problem, that is the problem of determining which sequences can fold into a predefined structure. This is especially useful in the context of protein and antibody design. For instance, the structure for a particular antibody sequence can be first derived with folding models, further optimized in structure space for developability properties and then converted back into sequence format for experimental validation. Inverse folding models for general proteins include ESM-IF1 (Hsu et al., 2022), KW-Design (Gao et al., 2024), ProRefiner (Zhou et al., 2023), GraDe_IF (Yi et al., 2023), ProteinMPNN (Dauparas et al., 2023) and SeqPredNN (Adriaan Lategan et al., 2023). Inverse folding methods specifically designed for antibodies are AntiFold (Haraldson Høie et al., 2024), AbMPNN (Dreyer et al., 2023), IgDesign (Shanehsazzadeh et al., 2023) and DiscoTope-3.0 (Haraldson Høie et al., 2024). AntiFold is a version of ESM-IF1 fine-tuned on experimental and predicted antibody structures.
Table 2 summarizes the information of the models mentioned in this section with the respective licenses.
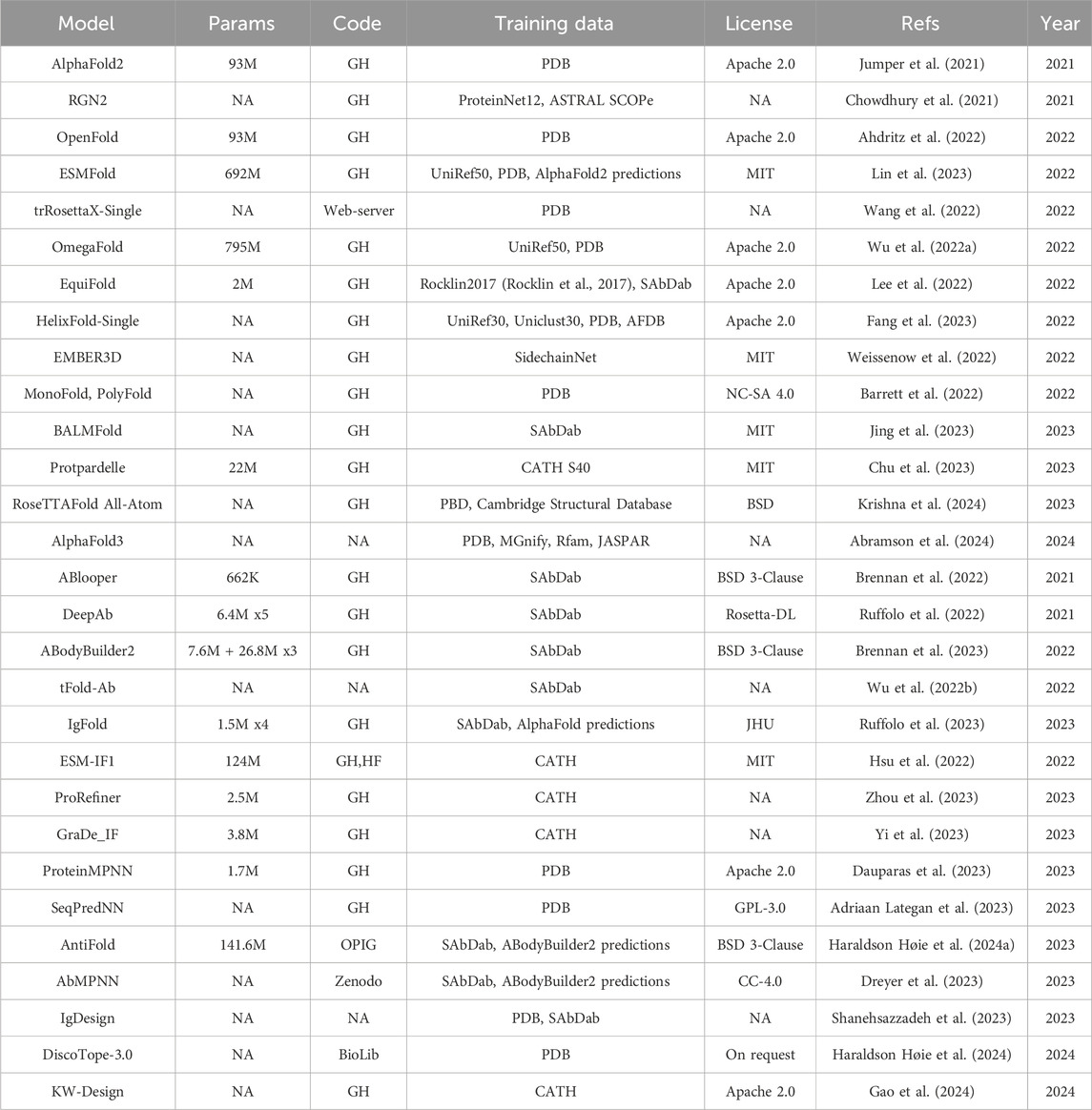
Table 2. Specification for general protein folding models (top), antibody-specific folding models (middle) and inverse folding models (bottom). DeepAb and ABodyBuilder2 are ensemble of models. The table follows the same structure as Table 1, apart from the Base column that does not apply here. The models for which we could not determine the number of parameters are indicated as NA in the Params column. GH: GitHub. HF: HuggingFace.
Screening for a high affinity antibody is only the first step in the antibody development process. To match the characteristics of therapeutic antibodies, the selected antibody must be further optimized to adhere to the properties of therapeutic antibodies (developability) (Fernández-Quintero et al., 2023; Khetan et al., 2022). Raybould and coauthors (Raybould et al., 2024) developed the Therapeutic Antibody Profile, a webserver used to evaluate antibody developability as including immunogenicity, solubility, specificity, stability, manufacturability, and storability. They focused on five metrics calculated from the CDRs based on total length, surface hydrophobicity, positive and negative charge of surface patches, and net charge of VH and VL chains.
Habib and coauthors (Habib et al., 2023) have compiled a list of 40 sequence- and 46 structure-based developability parameters (DP). They showed that sequence DPs are better predictors than structure DPs in single DP ablation experiments using Multiple Linear Regression (MLR) layer, especially when using sequence-based embeddings generated with the PLM ESM-1v as features. This reflects the fact that ESM-1v has direct access to the sequence information it is trained on and only learns structure information indirectly from sequence.
For the scope of this review, we will focus on ML methods for humanization and prediction of solubility, Methods for aggregation predictions are not mentioned, and viscosity. Humanization is the process of lowering the risk of immunogenicity by increasing the human-like content of the antibody sequence while maintaining binding affinity (Carter and Rajpal, 2022). Solubility, aggregation, and viscosity are properties that determine if an antibody will perform well in a solution. These are properties that are important for sub-cutaneous delivery, which represent an attractive alternative with respect to intra-venous delivery because of ease and speed of administration (Viola et al., 2018) but require maximizing the dose (Jiskoot et al., 2022). If an antibody has been selected in a screening procedure that uses the naive immune repertoire of a non-human species, when administered to a patient it can elicit an immunogenic response, whereby Anti-Drug Antibodies (ADAs) are raised against the engineered antibody by the immune system of the host. Humanization is the process by which non-human residues lying outside of the epitope-binding regions are iteratively swapped with human-like residues to increase the humanness of the antibody while retaining its binding affinity. Several ML methods have been presented to address humanization. Hu-mAb (Marks et al., 2021) uses a set of V gene type-specific Random Forest (RF) models to iteratively select the top scoring single-site mutation in the framework region based on a humanness score until it reaches a target score.
BioPhi (Prihoda et al., 2022) is a platform for humanness evaluation and humanization. Humanness is evaluated using OASis, a database of 9-mers (k-mers of nine residues) constructed from over 188 million sequences from 231 human subjects comprising 26 studies. Humanization is performed with Sapiens, which comprises two chain-specific ALMs, one for the heavy and one for the light chain, trained using a MLM objective on 20 million heavy chain human sequences from 38 OAS studies from 2011 to 2017, and 19 million light chain human sequences from 14 OAS studies from 2011 to 2017. The Sapiens network returns per-position posterior probabilities for all 20 amino acids conditioned on the input sequence that are used to introduce humanizing mutations. BioPhi includes an interface (Designer) that allows to select which of the suggested mutations the user would like to introduce. The software is available as a web-server or with a command line interface for processing set of sequences.
An alternative method not based on DL is AntPack (Parkinson and Wang, 2024). First, the authors developed a new antibody numbering method that is much faster than existing methods. Then they fitted a gaussian mixture model on the numbered antibody sequences using 60 million heavy and 70 million light sequences from the cAbRep database (Guo et al., 2019). The authors originally trained the model on human sequences of the OAS dataset and by inspecting the sequences of the training set that were responsible of giving unusually high probability to mouse sequences were able to identify more than 7,000 sequences that had been incorrectly labelled as both mouse and human.
Therapeutic antibodies are often produced and utilized at high concentration, so they require high solubility and low aggregation. Several methods have been proposed to predict solubility from sequence-based and structure-based features. SOLart (Hou et al., 2020) is a Random Forest model trained on a combination of 52 sequence-based and structure-based features. In a comparison with 9 SOTA methods using a dataset of experimentally determined and modelled structures of S. cerevisiae it was able to achieve a Pearson correlation of 0.65. Language models have been recently employed for binary prediction of soluble versus non soluble proteins, either by using fine-tuning or by training only the last classification layer of the neural network. NetSolP (Thumuluri et al., 2022), an ensemble of fine-tuned ESM1b models, achieves a performance comparable to the version of ESM with MSA input on datasets of proteins expressed in E. coli that were assessed for solubility.
Another favorable property related to developability for antibodies to perform well in a solution is low viscosity. Rai et al. (2023) proposed PfAbNet-viscosity, a 3D convolutional neural network to predict viscosity from antibody structures trained under a low training data regime. The authors used data augmentation to try to mitigate the limitations of working with few antibodies for training. PfAbNet-viscosity outperformed two SOTA models, Sharma (Sharma et al., 2014) and Surface Charge Model (SCM) (Agrawal et al., 2015).
The field of antibody discovery and development is experiencing an acceleration thanks to the successes of DL in protein structure prediction and representation learning from protein sequences. Here we focused on the applicability of these methods and resources in an industrial setting, especially with respect to the possibility of integrating these methods into commercial products. In Table 3 we highlighted a selection of protein and antibody-specific models for sequence, folding, inverse folding and developability (humanization). Our choice is based on our assessment of usability and license considerations.

Table 3. A selection of models with highlighted strengths and limitations. General protein models (top) and antibody-specific models (bottom).
To evaluate the performance of these models, a benchmark has been recently proposed, Fitness Landscape for Antibodies (FLAb), that covers six properties of therapeutic antibodies: expression, thermostability, immunogenicity, aggregation, polyreactivity and binding affinity (Chungyoun et al., 2024). The models considered in the study include decoder-only generative models trained with next token prediction [ProGen2 (Nijkamp et al., 2022), IgLM (Shuai et al., 2021) and ProtGPT2 (Ferruz et al., 2022)], encoder-only models for representation learning [AntiBERTy (Ruffolo et al., 2023)], inverse folding models [ProteinMPNN (Dauparas et al., 2023), ESM-IF (Hsu et al., 2022)] and a physics-based model [Rosetta (Koehler Leman et al., 2020)]. The authors showed that none of the models outperformed all the other models for all the tasks, underscoring the challenges in the development and application of these models for specific tasks.
Additional insights on how to improve these models will come from extending these benchmarks to different models, including PLMs like ESM-2 fine-tuned on antibody sequence data. With the increased availability of predicted antibody structures the current direction in the field is to integrate structural information in ALMs, with AntiBERTa2 (Barton et al., 2024) being an example. Antibody datasets, such as OAS, suffer from a germline content bias that can prevent the model from suggesting mutations that are further away from the germline sequence space. AbLang2 (Olsen et al., 2024b) is a recent model that addresses this bias, where the authors trained the model to focus more on non-germline residues using focal loss instead of cross-entropy loss to handle the class imbalance of germline versus non-germline residues in the training data.
The availability of models specifically developed for antibody structure prediction and inverse folding models allows to address the developability optimization problem both in sequence and structure space. This is particularly important as optimization in sequence space appears to be more constrained than in structure space (as mentioned in the developability cartography study (Habib et al., 2023)).
Humanization is the process that is best addressed by current developability methods, while other methods aimed at predicting solubility and viscosity suffers from the limited availability of experimental data for training and have been exploring less the application of language models.
These are exciting times for antibody discovery and development with AI that is being leveraged as a catalyst to accelerate and de-risk drug development on many fronts. We are starting to see how the process of bringing a drug from discovery to pre-clinical and clinical trials can be shortened and how the costs of this process can be reduced. The next few years will continue to see a fast-paced development and integration of these methods and resources in industrial applications, with the goal of ensuring that a newly found treatment can arrive faster to the patients.
LS: Writing–original draft, Writing–review and editing, Investigation. MB: Investigation, Writing–original draft. IX: Conceptualization, Writing–review and editing. BA: Conceptualization, Supervision, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Authors LS, MB, IX, and BA were employed by JSR Life Sciences.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500. doi:10.1038/s41586-024-07487-w
Adriaan Lategan, F., Schreiber, C., and Patterton, H. G. (2023). SeqPredNN: a neural network that generates protein sequences that fold into specified tertiary structures. BMC Bioinforma. 24 (1), 373. doi:10.1186/s12859-023-05498-4
Agrawal, N. J., Helk, B., Kumar, S., Mody, N., Sathish, H. A., Samra, H. S., et al. (2015). Computational tool for the early screening of monoclonal antibodies for their viscosities. mAbs 8 (1), 43–48. doi:10.1080/19420862.2015.1099773
Ahdritz, G., Bouatta, N., Floristean, C., Kadyan, S., Xia, Q., Gerecke, W., et al. (2022). OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. BioRxiv, Preprint. (Accessed November 22, 2022). doi:10.1038/s41592-024-02272-z
Ahmed, E., Essam, H., Salah-Eldin, W., Moustafa, W., Mohamed, E., Rochereau, C., et al. (2023). Ankh: optimized Protein Language model unlocks general-purpose modelling. preprint, Bioinformatics. (Accessed January 16 2023). doi:10.1101/2023.01.16.524265
Ahmed, E., Heinzinger, M., Dallago, C., Rehawi, G., Wang, Yu, Jones, L., et al. (2022). ProtTrans: toward understanding the language of Life through self-supervised learning. IEEE Trans. Pattern Analysis Mach. Intell. 44 (10), 7112–7127. doi:10.1109/TPAMI.2021.3095381
Alamdari, S., Thakkar, N., Van Den Berg, R., Lu, A. X., Fusi, N., Amini, A. P., et al. (2023). Protein generation with evolutionary diffusion: sequence is all you need. BioRxiv. (Accessed September 12, 2023). doi:10.1101/2023.09.11.556673
Barrett, T. D., Villegas-Morcillo, A., Robinson, L., Gaujac, B., Adméte, D., Saquand, E., et al. (2022). ManyFold: an efficient and flexible library for training and validating protein folding models. Bioinformatics 39 (1). doi:10.1093/bioinformatics/btac773
Barton, J., Jacob, D. G., and Leem, J. (2024). Enhancing Antibody Language Models with structural information. BioRxiv. (Accessed January 04, 2024). doi:10.1101/2023.12.12.569610
Bender, A., and Cortes-Ciriano, I. (2021). Artificial intelligence in drug discovery: what is realistic, what are illusions? Part 2: a discussion of chemical and biological data. Drug Discov. Today 26 (4), 1040–1052. doi:10.1016/j.drudis.2020.11.037
Bender, A., and Cortés-Ciriano, I. (2021). Artificial intelligence in drug discovery: what is realistic, what are illusions? Part 1: ways to make an impact, and why we are not there yet. Drug Discov. Today 26 (2), 511–524. doi:10.1016/j.drudis.2020.12.009
Brandes, N., Ofer, D., Peleg, Y., Rappoport, N., and Linial, M. (2022). ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics 38 (8), 2102–2110. doi:10.1093/bioinformatics/btac020
Brennan, A., Georges, G., Bujotzek, A., and Deane, C. M. (2022). ABlooper: fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinformatics 38 (7), 1877–1880. doi:10.1093/bioinformatics/btac016
Brennan, A., Wong, W.Ki, Boyles, F., Georges, G., Bujotzek, A., and ImmuneBuilder, C. M. D. (2023). ImmuneBuilder: deep-Learning models for predicting the structures of immune proteins. Commun. Biol. 6 (1), 575. doi:10.1038/s42003-023-04927-7
Burbach, S. M., and Briney, B. (2024). Improving antibody language models with native pairing. Patterns (N Y). 5 (5), 100967. doi:10.1016/j.patter.2024.100967
Carter, P. J., and Rajpal, A. (2022). Designing antibodies as therapeutics. Cell 185 (15), 2789–2805. doi:10.1016/j.cell.2022.05.029
Chan, A. C., and Carter, P. J. (2010). Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 10 (5), 301–316. doi:10.1038/nri2761
Chowdhury, R., Bouatta, N., Biswas, S., Rochereau, C., Church, G. M., Sorger, P. K., et al. (2021). Single-sequence protein structure prediction using language models from deep learning. preprint. BioRxiv. (Accessed August 04, 2021). doi:10.1038/s41587-022-01432-w
Chu, A. E., Cheng, L., El Nesr, G., Xu, M., and Huang, P.-S. (2023). An all-atom protein generative model. BioRxiv. (Accessed May 25, 2023). doi:10.1101/2023.05.24.542194
Chungyoun, M., Ruffolo, J., and Gray, J. (2024). FLAb: benchmarking deep learning methods for antibody fitness prediction. preprint. BioRxiv. (Accessed January 15, 2024). doi:10.1101/2024.01.13.575504
Clark, K., Luong, M.-T., Quoc, V.Le, and Christopher, D. (2020). Manning. ELECTRA: pre-training text encoders as discriminators rather than generators. arXiv:2003.10555. doi:10.48550/arXiv.2003.10555
Dai, Z., Yang, Z., Yang, Y., Carbonell, J., Le, Q. V., and Salakhutdinov, R. (2019). Transformer-XL: attentive Language Models beyond a fixed-length context. arXiv:1901.02860, 1. doi:10.48550/arXiv.1901.02860
Dauparas, J., Anishchenko, I., Bennett, N., Bai, H., Ragotte, R. J., Milles, L. F., et al. (2023). Robust deep learning based protein sequence design using ProteinMPNN. Sci. 378 (6615), 49–56. doi:10.1126/science.add2187
Dounas, A., Cotet, T.-S., and Yermanos, A. (2024). Learning immune receptor representations with protein language models. arXiv. doi:10.48550/arXiv.2402.03823
Dreyer, F. A., Cutting, D., Schneider, C., Kenlay, H., and Deane, C. M. (2023). Inverse folding for antibody sequence design using deep learning. arXiv. doi:10.3389/fmolb.2023.1237621
Evans, R., O’Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T., et al. (2021). Protein complex prediction with AlphaFold-Multimer. BioRxiv. (Accessed March 10, 2022). doi:10.1101/2021.10.04.463034
Evers, A., Malhotra, S., and Sood, V. D. (2023). In silico approaches to deliver better antibodies by design – the past in The present and the future. arXiv. doi:10.48550/arXiv.2305.07488
Fang, X., Wang, F., Liu, L., He, J., Lin, D., Xiang, Y., et al. (2023). A method for multiple-sequence-alignment-free protein structure prediction using a protein language model. Nat. Mach. Intell. 5 (10), 1087–1096. doi:10.1038/s42256-023-00721-6
Fernández-Quintero, M. L., Ljungars, A., Waibl, F., Greiff, V., Andersen, J. T., Gjølberg, T. T., et al. (2023). Assessing developability early in the discovery process for novel biologics. mAbs 15 (1), 2171248. doi:10.1080/19420862.2023.2171248
Ferruz, N., Schmidt, S., and Höcker, B. (2022). ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13 (1), 4348. doi:10.1038/s41467-022-32007-7
Gao, Z., Tan, C., Chen, X., Zhang, Y., Xia, J., Li, S., et al. (2024). KW-DESIGN: pushing the limit of protein design via knowledge refinement. arXiv. doi:10.48550/arXiv.2305.15151
Graves, J., Jacob, B., Priego, E., Makkapati, N., Parish, S., Medellin, B., et al. (2020). A review of deep learning methods for antibodies. Antibodies 9 (2), 12. doi:10.3390/antib9020012
Guo, Y., Chen, K., Kwong, P. D., Shapiro, L., and Sheng, Z. (2019). cAb-rep: a database of curated antibody repertoires for exploring antibody diversity and predicting antibody prevalence. Front. Immunol. 10, 2365. doi:10.3389/fimmu.2019.02365
Habib, B., Smorodina, E., Pariset, M., Zhong, J., Akbar, R., Chernigovskaya, M., et al. (2023). Biophysical cartography of the native and human-engineered antibody landscapes quantifies the plasticity of antibody developability. BioRxiv. (Accessed December 13, 2023). doi:10.1101/2023.10.26.563958
Haraldson Høie, M., Gade, F. S., Johansen, J. M., Würtzen, C., Winther, O., Nielsen, M., et al. (2024). DiscoTope-3.0: improved B-cell epitope prediction using inverse folding latent representations. Front. Immunol. 15, 1322712. doi:10.3389/fimmu.2024.1322712
Haraldson Høie, M., Hummer, A. M., Olsen, T. H., Nielsen, M., and Deane, C. M. (2024a). AntiFold: improved antibody structure design using inverse folding. arXiv. doi:10.48550/arXiv.2405.03370
Hou, Q., Marc Kwasigroch, J., Rooman, M., and Pucci, F. (2020). SOLart: a structure-based method to predict protein solubility and aggregation. Bioinformatics 36 (5), 1445–1452. doi:10.1093/bioinformatics/btz773
Hsu, C., Verkuil, R., Liu, J., Lin, Z., Hie, B., Sercu, T., et al. (2022). Learning inverse folding from millions of predicted structures. BioRxiv. (Accessed September 06, 2022). doi:10.1101/2022.04.10.487779
Islam, S., Elmekki, H., Ahmed, E., Bentahar, J., Drawel, N., Rjoub, G., et al. (2023). A comprehensive survey on applications of transformers for deep learning tasks. arXiv. doi:10.48550/arXiv.2306.07303
Jacob, D., Chang, M.-W., Lee, K., and Toutanova, K. (2019). BERT: pre-training of deep bidirectional transformers for language understanding. arXiv. doi:10.48550/arXiv.1810.04805
Jaffe, D. B., Shahi, P., Adams, B. A., Chrisman, A. M., Finnegan, P. M., Raman, N., et al. (2022). Functional antibodies exhibit light chain coherence. Nature 611 (7935), 352–357. doi:10.1038/s41586-022-05371-z
Jing, H., Gao, Z., Xu, S., Shen, T., Peng, Z., He, S., et al. (2023). Accurate prediction of antibody function and structure using bio-inspired Antibody Language model. Preprint. Bioinformatics. 25 (4), bbae245. doi:10.1093/bib/bbae245
Jiskoot, W., Hawe, A., Menzen, T., Volkin, D. B., and Crommelin, D. J. A. (2022). Ongoing challenges to develop high concentration monoclonal antibody-based formulations for subcutaneous administration: quo vadis? J. Pharm. Sci. 111 (4), 861–867. doi:10.1016/j.xphs.2021.11.008
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Kenlay, H., Dreyer, F. A., Kovaltsuk, A., Miketa, D., Douglas, P., and Deane, C. M. (2024). Large scale paired antibody language models. arXiv. doi:10.48550/arXiv.2403.17889
Khakzad, H., Igashov, I., Schneuing, A., Goverde, C., Bronstein, M., and Correia, B. (2023). A new age in protein design empowered by deep learning. Cell Syst. 14 (11), 925–939. doi:10.1016/j.cels.2023.10.006
Khetan, R., Curtis, R., Deane, C. M., Hadsund, J. T., Kar, U., Krawczyk, K., et al. (2022). Current advances in biopharmaceutical informatics: guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. mAbs 14 (1), 2020082. doi:10.1080/19420862.2021.2020082
Kim, J., McFee, M., Fang, Q., Abdin, O., and Kim, P. M. (2023). Computational and artificial intelligence-based methods for antibody development. Trends Pharmacol. Sci. 44 (3), 175–189. doi:10.1016/j.tips.2022.12.005
Koehler Leman, J., Weitzner, B. D., Lewis, S. M., Adolf-Bryfogle, J., Alam, N., Alford, R. F., et al. (2020). Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17 (7), 665–680. doi:10.1038/s41592-020-0848-2
Krishna, R., Wang, J., Ahern, W., Sturmfels, P., Kalvet, I., Lee, G. R., et al. (2024). Generalized biomolecular modeling and design with RoseTTAFold all-atom. Sci. 384 (6693), eadl2528. doi:10.1126/science.adl2528
Lan, Z., Chen, M., Goodman, S., Gimpel, K., Sharma, P., and Soricut, R. (2019). ALBERT: a lite BERT for self-supervised learning of language representations. arXiv. doi:10.48550/arXiv.1909.11942
Lee, J. H., Yadollahpour, P., Watkins, A., Frey, N. C., Leaver-Fay, A., Ra, S., et al. (2022). Protein structure prediction with a novel coarse-grained structure representation. Preprint. BioRxiv. (Accessed October 08, 2022). doi:10.1101/2022.10.07.511322
Leem, J., Mitchell, L. S., Farmery, J. H. R., Barton, J., and Galson, J. D. (2022). Deciphering the language of antibodies using self-supervised learning. Patterns 3 (7), 100513. doi:10.1016/j.patter.2022.100513
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., et al. (2023). Evolutionary-scale prediction of atomic level protein structure with a language model. Sci. 379 (6637), 1123–1130.
Lu, R.-M., Hwang, Y.-C., Liu, I.-Ju, Lee, C.-C., Tsai, H.-Z., Li, H.-J., et al. (2020). Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 27 (1), 1. doi:10.1186/s12929-019-0592-z
Marks, C., Hummer, A. M., Chin, M., and Deane, C. M. (2021). Humanization of antibodies using a machine learning approach on large-scale repertoire data. Bioinform. 37 (22), 4041–4047.
Nam Kim, D., McNaughton, A. D., and Kumar, N. (2024). Leveraging artificial intelligence to expedite antibody design and enhance antibody–antigen interactions. Bioengineering 11 (2), 185. doi:10.3390/bioengineering11020185
Nijkamp, E., Jeffrey, R., Weinstein, E. N., Naik, N., and Ali, M. (2022). ProGen2: exploring the boundaries of Protein Language Models. arXiv. doi:10.48550/arXiv.2206.13517
Notin, P., Kollasch, A. W., Ritter, D., van Niekerk, L., Paul, S., Spinner, H., et al. (2023). ProteinGym: large-scale benchmarks for protein fitness prediction and design. BioRxiv. doi:10.1101/2023.12.07.570727
Ofer, D., Brandes, N., and Linial, M. (2021). The language of proteins: NLP, machine learning and protein sequences. Comput. Struct. Biotechnol. J. 19, 1750–1758. doi:10.1016/j.csbj.2021.03.022
Olsen, T. H., Moal, I. H., and Deane, C. M. (2024a). Addressing the antibody germline bias and its effect on language models for improved antibody design. BioRxiv. (Accessed February 07, 2024). doi:10.1101/2024.02.02.578678
Olsen, T. H., Moal, I. H., and Deane, C. M. (2024b). Addressing the antibody germline bias and its effect on language models for improved antibody design. bioRxiv. Available at: https://www.biorxiv.org/content/early/2024/02/07/2024.02.02.578678.full.pdf. doi:10.1101/2024.02.02.578678
Olsen, T. H., Moal, I. H., and Deane, C. M. (2022). AbLang: an antibody language model for completing antibody sequences. Bioinforma. Adv. 2 (1), vbac046. doi:10.1093/bioadv/vbac046
Parkinson, J., and Wang, W. (2024). For antibody sequence generative modeling, mixture models may be all you need. Bioinformatics 40, btae278. doi:10.1093/bioinformatics/btae278
Prihoda, D., Maamary, J., Waight, A., Juan, V., Fayadat-Dilman, L., Svozil, D., et al. (2022). BioPhi: a platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning. mAbs 14 (1), 2020203. doi:10.1080/19420862.2021.2020203
Pujols, J., Iglesias, V., Santos, J., Kuriata, A., and Ventura, S. (2022). A3D 2.0 update for the prediction and optimization of protein solubility. Methods Mol Biol. 2406, 65–84. doi:10.1007/978-1-0716-1859-2_3
Raffel, C., Shazeer, N., Roberts, A., Lee, K., Narang, S., Matena, M., et al. (2023). Exploring the limits of transfer learning with a unified text-to-text transformer. arXiv.
Rai, B. K., Apgar, J. R., and Bennett, E. M. (2023). Low-data interpretable deep learning prediction of antibody viscosity using a biophysically meaningful representation. Sci. Rep. 13 (1), 2917. doi:10.1038/s41598-023-28841-4
Raybould, M. I. J., Turnbull, O. M., Suter, A., Guloglu, B., and Deane, C. M. (2024). Contextualising the developability risk of antibodies with lambda light chains using enhanced therapeutic antibody profiling. Commun. Biol. 7 (1), 62. doi:10.1038/s42003-023-05744-8
Rives, A., Meier, J., Sercu, T., Goyal, S., Lin, Z., Liu, J., et al. (2021). Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. 118 (15), e2016239118. doi:10.1073/pnas.2016239118
Rocklin, G. J., Chidyausiku, T. M., Goreshnik, I., Ford, A., Houliston, S., Lemak, A., et al. (2017). Global analysis of protein folding using massively parallel design, synthesis, and testing. Science 357 (6347), 168–175. doi:10.1126/science.aan0693
Ruffolo, J. A., Chu, L.-S., Mahajan, S. P., and Gray, J. J. (2023). Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Nat. Commun. 14 (1), 2389. doi:10.1038/s41467-023-38063-x
Ruffolo, J. A., Gray, J. J., and Sulam, J. (2021). Deciphering antibody affinity maturation with language models and weakly supervised learning. arXiv. doi:10.48550/arXiv.2112.07782
Ruffolo, J. A., Sulam, J., and Gray, J. J. (2022). Antibody structure prediction using interpretable deep learning. Patterns 3 (2), 100406. doi:10.1016/j.patter.2021.100406
Shanehsazzadeh, A., Alverio, J., Kasun, G., Levine, S., Khan, J. A., Chung, C., et al. (2023). In vitro validated antibody design against multiple therapeutic antigens using generative inverse folding. BioRxiv. Absci Corporation, 570889. doi:10.1101/2023.12.08.570889
Sharma, V. K., Patapoff, T. W., Kabakoff, B., Pai, S., Hilario, E., Zhang, B., et al. (2014). In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc. Natl. Acad. Sci. U. S. A. 111 (52), 18601–18606. doi:10.1073/pnas.1421779112
Shuai, R. W., Ruffolo, J. A., and Gray, J. J. (2021). Generative language modeling for antibody design. BioRxiv. (Accessed December 20, 2022). doi:10.1101/2021.12.13.472419
Simon, K. S. C., and Wei, K. Y. (2023). Generative antibody design for complementary chain pairing sequences through encoder-decoder Language Model. arXiv. doi:10.48550/arXiv.2301.02748
Spoendlin, F. C., Abanades, B., Raybould, M. I. J., Wong, W.Ki, Georges, G., and Deane, C. M. (2023). Improved computational epitope profiling using structural models identifies a broader diversity of antibodies that bind to the same epitope. Front. Mol. Biosci. 10, 1237621. doi:10.3389/fmolb.2023.1237621
Steinegger, M., and Söding, J. (2018). Clustering huge protein sequence sets in linear time. Nat. Commun. 9 (1), 2542. doi:10.1038/s41467-018-04964-5
Suzek, B. E., Huang, H., McGarvey, P., Mazumder, R., and Wu, C. H. (2007). UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23 (10), 1282–1288. doi:10.1093/bioinformatics/btm098
Thumuluri, V., Martiny, H.-M., Armenteros, J. J. A., Salomon, J., Nielsen, H., and Johansen, A. R. (2022). NetSolP: predicting protein solubility in Escherichia coli using language models. Bioinformatics 38 (4), 941–946. doi:10.1093/bioinformatics/btab801
UniProt Consortium, T., Bateman, A., Martin, M.-J., Orchard, S., Magrane, M., Ahmad, S., et al. (2023). UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51 (D1), D523–D531. doi:10.1093/nar/gkac1052
Valentini, G., Malchiodi, D., Gliozzo, J., Mesiti, M., Soto-Gomez, M., Cabri, A., et al. (2023). The promises of large language models for protein design and modeling. Front. Bioinforma. 3, 1304099. doi:10.3389/fbinf.2023.1304099
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., et al. (2017). Attention is all you need. arXiv:1706.03762. doi:10.48550/arXiv.1706.03762
Viola, M., Sequeira, J., Seiça, R., Veiga, F., Serra, J., Santos, A. C., et al. (2018). Subcutaneous delivery of monoclonal antibodies: how do we get there? J. Control. Release 286, 301–314. doi:10.1016/j.jconrel.2018.08.001
Wang, W., Peng, Z., and Yang, J. (2022). Single-sequence protein structure prediction using supervised transformer protein language models. Nat. Comput. Sci. 2, 804–814. doi:10.1038/s43588-022-00373-3
Weiner, L. M., Surana, R., and Wang, S. (2010). Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 10 (5), 317–327. doi:10.1038/nri2744
Weissenow, K., Heinzinger, M., Steinegger, M., and Rost, B. (2022). Ultra-fast protein structure prediction to capture effects of sequence variation in mutation movies. BioRxiv. (Accessed November 16, 2022). doi:10.1101/2022.11.14.516473
Wu, J., Wu, F., Jiang, B., Liu, W., and Zhao, P. (2022b). tFold-ab: fast and accurate antibody structure prediction without sequence homologs. BioRxiv. (Accessed November 13, 2022). doi:10.1101/2022.11.10.515918
Wu, R., Ding, F., Wang, R., Shen, R., Zhang, X., Luo, S., et al. (2022a). High-resolution de novo structure prediction from primary sequence. BioRxiv. (Accessed July 22, 2022). doi:10.1101/2022.07.21.500999
Yang, Z., Dai, Z., Yang, Y., Carbonell, J., Salakhutdinov, R., Quoc, V., et al. (2020). Generalized autoregressive pretraining for language understanding. arXiv. doi:10.48550/arXiv.1906.08237
Yi, K., Zhou, B., Shen, Y., Liò, P., and Wang, Yu G. (2023). Graph denoising diffusion for inverse protein folding. arXiv. doi:10.48550/arXiv.2306.16819
Zhang, W., Wang, H., Feng, N., Li, Y., Gu, J., and Wang, Z. (2023). Developability assessment at early-stage discovery to enable development of antibody-derived therapeutics. Antib. Ther. 6 (1), 13–29. doi:10.1093/abt/tbac029
Zhao, Yu, Su, X., Zhang, W., Mai, S., Xu, Z., Qin, C., et al. (2023). SC-AIR-BERT: a pre-trained single-cell model for predicting the antigen-binding specificity of the adaptive immune receptor. Briefings Bioinforma. 24 (4), bbad191. doi:10.1093/bib/bbad191
Keywords: LLM, ALM (antibody language model), developability, inverse folding, deep learning, artificial intelligence
Citation: Santuari L, Bachmann Salvy M, Xenarios I and Arpat B (2024) AI-accelerated therapeutic antibody development: practical insights. Front. Drug Discov. 4:1447867. doi: 10.3389/fddsv.2024.1447867
Received: 12 June 2024; Accepted: 12 August 2024;
Published: 03 September 2024.
Edited by:
Ferdinand Molnár, Nazarbayev University, KazakhstanReviewed by:
Markus Heinonen, Aalto University, FinlandCopyright © 2024 Santuari, Bachmann Salvy, Xenarios and Arpat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bulak Arpat, YmFycGF0QGpzcmxpZmVzY2llbmNlcy5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.