 Priyanka Rani
Priyanka Rani Pranabesh Mandal
Pranabesh Mandal Bikash Kumar Rajak
Bikash Kumar Rajak Durg Vijay Singh
Durg Vijay Singh- Molecular Modelling and Computer Aided Drug Discovery Laboratory, Department of Bioinformatics Central University of South Bihar, School of Earth Biological and Environmental Sciences, Gaya, India
Permeability-glycoprotein (P-gp) belongs to the ABS transporter protein family, with a high expression rate in cancerous cells. The substrate/inhibitors of the protein are structurally diverse, with no lucid mechanism of inhibition. There are two schools of thought on the inhibition mechanism: (i) P-gp inhibitors bind to the huge hydrophobic cavity between two Trans-Membrane Domains (TMDs), supported by ample literary proof and (ii) P-gp inhibitors bind to the vicinity of Nucleotide-Binding Sites (NBSs). Structural biologists have presented several experimental and theoretical structures of P-gp with bound nucleotides and inhibitors to explain the same. However, the available experimental P-gp structures are insufficient to address the catalytic transition path of mammalian P-gp in detail, thus the dynamics and mechanism by which drugs are effluxed is still unknown. Targeted Molecular Dynamics (targeted MD) could be used to minutely analyse and explore the catalytic transition inward open (IO) to outward open (OO) and relaxation path (OO to IO). Finally, analysis of targeted MD trajectory may help to explore different conformational states of Pg-p (reaction coordinate of catalytic transition/relaxation), efflux of compounds aided by the dynamics of Nucleotide Binding Domains/NBDs (ATP coupled process) and TMDs (peristalsis-like movement pushes the bound molecule). This review presents an understanding of the catalytic transition and dynamics of protein which provides insights at the efflux of chemotherapeutic drug using in cancer treatment.
1 Introduction
ABC transporter proteins are among the essential membrane transporters responsible for reduced bioavailability and multi-drug resistance (MDR), representing a captivating target for intrusion. Permeability-glycoprotein (P-gp) or human MDR1/ABCB1 protein is the extensively used transporter protein (Ambudkar et al., 1999). P-gp is abundantly present in the kidney, adrenal gland, colon, testis endothelial cells, liver, brain, respiratory mucosa, placenta, and other tissues as well. However, in solid tumour tissues, expression of P-gp is abundant in non-small cell lung carcinoma and neuroblastoma of the breast, pancreas, colon, adrenal, liver, renal, and ovaries.
As formerly mentioned, P-gp expression has been linked to reduced bioavailability of substrates resulting in poor clinical outcomes in several pathological conditions. It reduces the bioavailability of clinically essential molecules, mainly hydrophobic, and acts as a ‘hydrophobic’ vacuum cleaner (Sharom, 2011a). However, how ABC effluxes its substrate is still not very lucid. Furthermore, several hypotheses of the transport mechanism of ABC transporter have been critically analyzed by various research groups (Tripathi et al., 2015). This review focuses on Inward Opern (IO) and Outward Open (OO) states of P-gp structure, the structural arrangement of different domains, the use of targeted Molecular Dynamics (targeted MD) to explore the dynamics of Trans-membrane Helices (THs) and Trans-Membrane Domains (TMDs), and different conformational states/catalytic transition states of p-gp protein during catalysis.
Bacterial P-gp is encoded in two peptides (e.g., PDB ID: 2HYD) (Dawson and Locher, 2006) compared to single peptides in mice and humans. Mouse P-gp has ∼1,280 residues, organized into two homologous, pseudo-symmetrical domains joined by a linker of ∼76 residues (633–709) (Nuti and Rao, 2002). The basic domain organization of ABCC1 (MRP1), ABCB1 (P-gp), and ABCG2 (BCRP) transporters are shown in Figure 1. The MDR1 protein has two TMDs with six THs; respectively in each domain with two Nucleotide Binding Domains/NBDs and an additional Trans-Membrane Domain/TMD named as TMD0 consisting of five THs (Bera et al., 2018), while the ABCG2 (BCRP) family has only one NBD and TMD with six THs. While ABCB1 (P-gp/MDR1) family of transporter consist of two TMDs, each consisting of six Trans-membrane Helices (THs) that provide a passageway for the cargo, and two cytoplasmic NBDs that binds and hydrolyze ATP (Sharom, 2011a) (Figure 2). The NBDs of ABC proteins contain three highly conserved regions namely: the Walker A and Walker B motifs (common in many ATP-binding proteins) and the Walker C (Signature of ABC transporter proteins) motif (Holland and Blight, 1999). The X-ray crystal structures of several isolated nucleotide binding subunits of bacterial ABC transporter proteins reveal an interdigitated head-to-tail arrangement, also acknowledged as a sandwich dimer (Eckford and Sharom, 2009). It has been also noted that the two ATP molecules bind at the interface of the sandwich dimer, which interacts with the Walker A and B motifs of one NBD and C motif of the partner domain.
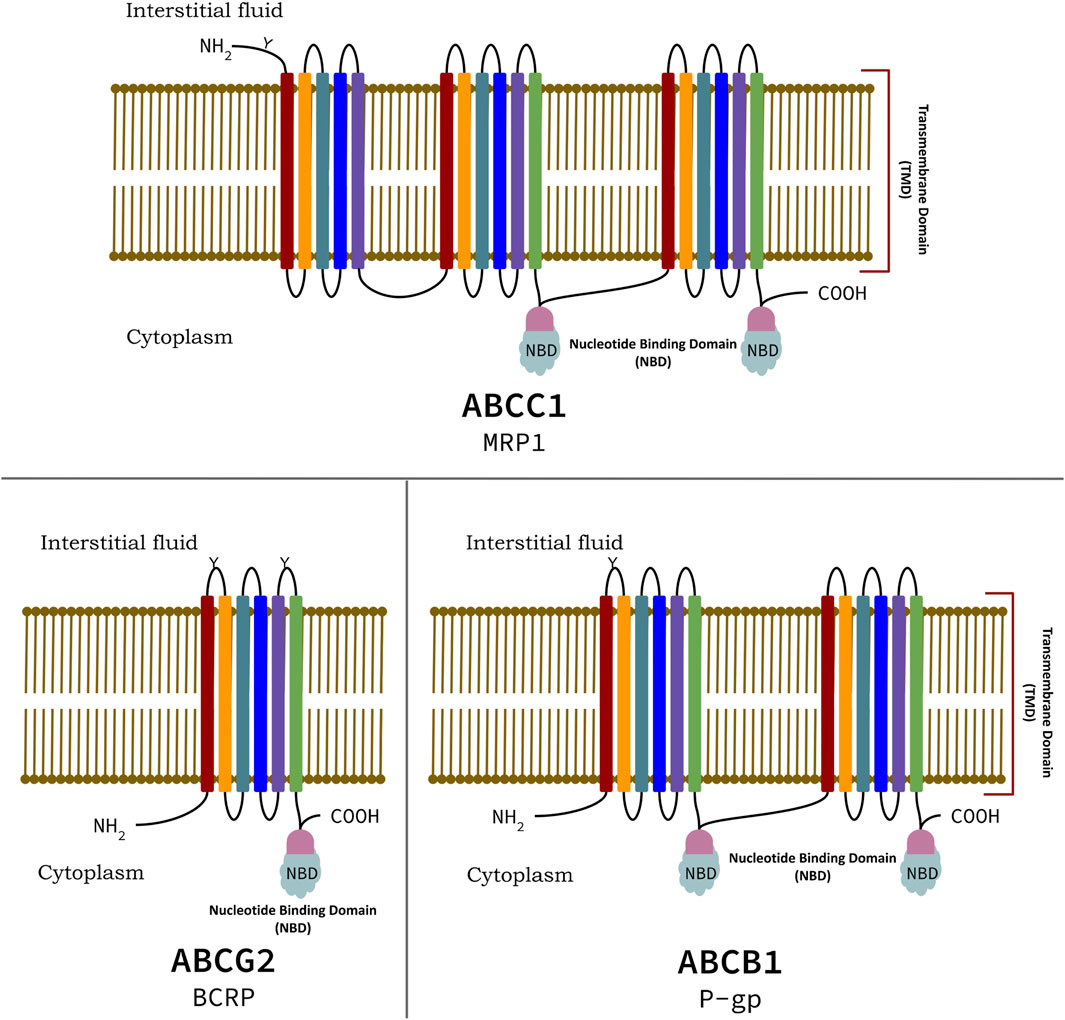
Figure 1. Domain organization of highly expressive ABC transporter proteins of ABCC1, ABCB1, and ABCG2 family, conferring Multiple Drug Resistance (MDR).

Figure 2. Multiple sequence alignment of Nucleotide Binding Domains (NBDs) of different organisms: Homo sapiens (P08183), Gorilla gorilla (G3S959), Nomascus leucogenys (A0A2I3GNI3), Sapajus apella (A0A6J3FUE0), Canis lupus familiaris (A0A5F4BVH2) and Lynx canadensis (A0A667GS53), showing high dissimilarities among them.
P-gp, plays a significant role in cancer treatment as well, particularly in the development of resistance against chemotherapeutic drugs (Alfarouk et al., 2015). It is a well-established fact that P-gp is a membrane protein that acts as an efflux pump/vacuum cleaner which actively transports a wide range of drugs out of cancerous cells, reducing the intracellular concentration of drugs, resulting in low susceptibility towards chemotherapeutic agent cytotoxic effects. Moreover, P-gp is overexpressed in cancerous cells which enhances the efflux of several chemotherapeutic drugs leading to accelerating problem of Multiple Drug Resistance (MDR) rendering treatment management (Assaraf et al., 2019). One among many strategies to overcome P-gp mediated drug resistance includes the incorporation of P-gp inhibitors with chemotherapy to hinder the efflux activity of P-gp to enhance the concentration of drugs for more effectiveness (Zhang et al., 2021). Researchers are additionally working on ways to develop chemotherapeutic drugs that are not suitable substrates for P-gp but have potential activity concerning cancer treatment. To actively induce this idea, it is very important to understand the structure and mechanism of P-gp which has been discussed in this review, along with possible approaches and techniques required to understand the function of P-gp.
2 Substrate, modulator, and inhibitors of permeability-glycoprotein
Over decades, many small molecules such as clinically used drugs, natural products, chemotherapeutic agents, fluorescent dyes, and linear and cyclic peptides (Sharom, 2008; Eckford and Sharom, 2009) have been identified as P-gp substrates (Table 1) and inhibitors (Table 2). Over a while, many molecules have been reported to be P-gp inhibitors, belonging to varied groups and generations of compounds. Some of them act as modulators for anti-cancer drugs as well. However, the question arises: “How does P-gp accommodate these diverse substrates and inhibitors?” There are two schools of thought about the binding of P-gp inhibitors: First, P-gp inhibitors bind at the substrate-binding domain, which is located at TMD/TMD interfaces (Pleban et al., 2005; Aller et al., 2009). The second states that P-gp inhibitors interact at the NBDs, thereby competing with ATP binding (Brewer et al., 2014; Tripathi et al., 2015). Studies have been reported that, there are inhibitor compounds that affect nucleotide (ATP) binding to the P-gp causing the hindrance in efflux (Brewer et al., 2014). Moreover, it has also been reported that phospholipid in bilayer membrane composition do affect the thermal unfolding of P-gp, altering the basal ATPase (Clay et al., 2015). Our group (Singh et al., 2013) reinforce both of these theories, i.e., the third generation of inhibitor; flavonoid (Abdallah et al., 2015) and steroid (Orlowski et al., 1996) binds at NBDs. In contrast, the sizable macrocyclic molecule may bind at the TMD domain of P-gp and compete with the binding of P-gp substrates.

Table 1. Small molecules identified to be interacting with Permeability-glycoprotein.
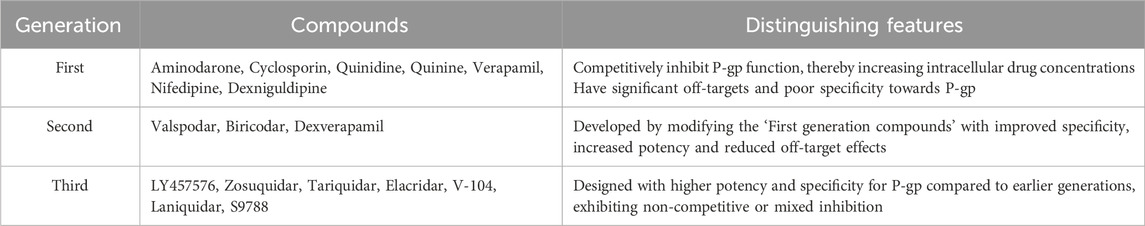
Table 2. Different class of compounds with inhibiting/modulating activity against Permeability-glycoprotein (Bansal et al., 2009; Boumendjel et al., 2009; Singh et al., 2013).
Apart from the formerly mentioned two schools of thought, P-gp inhibitors might be binding somewhere at structural transition states (NBDs/TMDs) of P-gp. Different structural transition states of NBDs/TMDs of P-gp may answer substrate and inhibitor recognition promiscuity through this transporter and hence it is of utmost importance to study the mechanism of catalytic transition of P-gp and its different conformational states.
3 Conserveness in nucleotide binding domain (NBDs) and trans-membrane domains (TMDs) in permeability-glycoprotein
Multiple Sequence Alignment (MSA) has been performed using online server Clustal Omega (https://www.ebi.ac.uk/jdispatcher/msa/clustalo) at its default settings, to estimate the conserveness in different helices and domains of P-gp. As discussed formerly, P-gp belongs to the transporter protein subfamily ABCB, responsible for transporting many substrates and drugs in and out of the cell. It is well perceived that P-gp has two NBD that bind ATP, which further participates in the drug effluxing mechanism through ATP hydrolysis (Sharom, 2011b). So, it is worth presenting the MSA of NBD to reasonably infer the evident changes in the sequence as evolution has proceeded. Visual depictions of the alignment as in Figure 2 illustrate conserveness in the NBD domain of P-gp in various organisms with a few mutation events that have appeared at different amino acids, during evolution in protein segments of different organisms.
THs of human P-gp have also been aligned (Figure 3) with each other. Different helices have different movements to ease the transportation of substrates through the membrane. Each helix has a different amino acid sequence to allow movement and flexibility. Variation in sequence of THs is quite visible through MSA, and the hydrophobic region formed by the hydrophobic amino acid residues cannot be denied. Later the sequence has been evaluated based on the arrangement of amino acids in 3D coordinates.

Figure 3. Multiple sequence alignment of twelve Trans-membrane Helices (THs) of human P-gp showing high dissimilarity.
It has been unearthed that several sets of amino acid residues of THs responsible for binding drugs are present in a specific 3D arrangement (hydrophobic inner core), making it possible for compounds to be transported through membranes even after mutation at the binding site as visible MSA of Figure 3. Thus, proving the presence of multiple sites in the TMD interface, it provides numerous possibilities, including substrate diversity, which explains the broad substrate specificity also known as poly-specificity of P-gp. Sequence variability of THs admittedly facilitates fine-tuning between the substrate and binding sites (primary or secondary site) for effective binding, facilitating the accommodation of different molecules on multiple sites, thereby explaining the phenomena of poly-specificity of P-gps and other ABC transporters.
4 Structural organisation of human P-gp: arrangement of loop, helices, and motifs
The molecular structure of ABC transporters has been determined from various organisms in IO and OO states (Figure 2). The first X-ray crystal structures (PDB ID: 3G5U) of mouse P-gp in an IO conformation were reported in 2009 (Aller et al., 2009). A drug-binding pocket within the protein’s trans-membrane sections has yielded many crystal structures, both with and without bound cyclic peptide substrates. These structures have been identified in the absence of nucleotides. Earlier, a atomistic resolution of 3.4 Å crystal structure of C. elegans ABC transporter protein in an IO state (PDB ID: 4F4C), without bound substrate and nucleotide was reported in 2001 (Jin et al., 2012). Another structure of mouse P-gp (PDB ID: 4M2S) with resolution 3.4Å was also solved (Li et al., 2014). In 2009, researchers started to focus their interest on P-gp however, besides chimeric structures, there is no reliable structure yet available for human P-gp.
Human P-gp (ABCB1/MDR1) consists of single peptide of 1,280 amino acids and has an approximate weight of 170 kDa (Sharom, 2011a). It has a domain arrangement of TMD1-NBD1-TMD2-NBD2, which forms two pseudo-symmetrical halves linked by a residue chain of approximately 76 amino acids (from 633 to 709), known as the linker as shown in Figure 4 (Nuti and Rao, 2002). The approximate human P-gp linker region starts from G633 and expands upto 76 residues downstream. It has been reported that the linker regulates the inherent substrate specificity of P-gp and also maintains the tightness of coupling between the ATP hydrolase reaction and substrate recognition (Nuti and Rao, 2002). Each of the halves has one TMD and one NBD (Higgins and Linton, 2004). Both TMDs has six THs linked by intercellular helices (Coupler Helices) and extracellular loops. P-gp has four coupling helices/intracellular helices (CH1, CH2, CH3, CH4) as listed in Table 3, that mediate the cross-talk between TMDs and NBDs through interaction with residues of NBDs. The CH1, CH4 interact with NBD1, and CH1 is located in between TH2 and TH3, while CH4 is located in between TH10 and TH11. CH2 is present in between of TH4 and TH5, while a CH3 is located at the end of TH8 and begins TH9 and interacts with NBD2. All these CHs serve as physical relays that carry a conformational shift from NBD to TMD (Bansal et al., 2009; Boumendjel et al., 2009) as presented in Figure 4. The length, position of CHs and residues of NBDs adjacent to each CHs (4Å) are shown in Figure 4; Table 3 (Loo and Clarke, 2013; Loo and Clarke, 2014). Each NBD has an ATP binding pocket. Walker A, Walker B, H loop, Q loop, A loop of one NBD, D loop, and ABC signature motif (Walker C) of other NBD forms one ATP binding pocket (Sauna and Ambudkar, 2007; Pan and Aller, 2015). The positions of Walker A, Walker B, Walker C, A loop, Q loop, H loop, and D loop have been mentioned in Table 4; Figure 5.
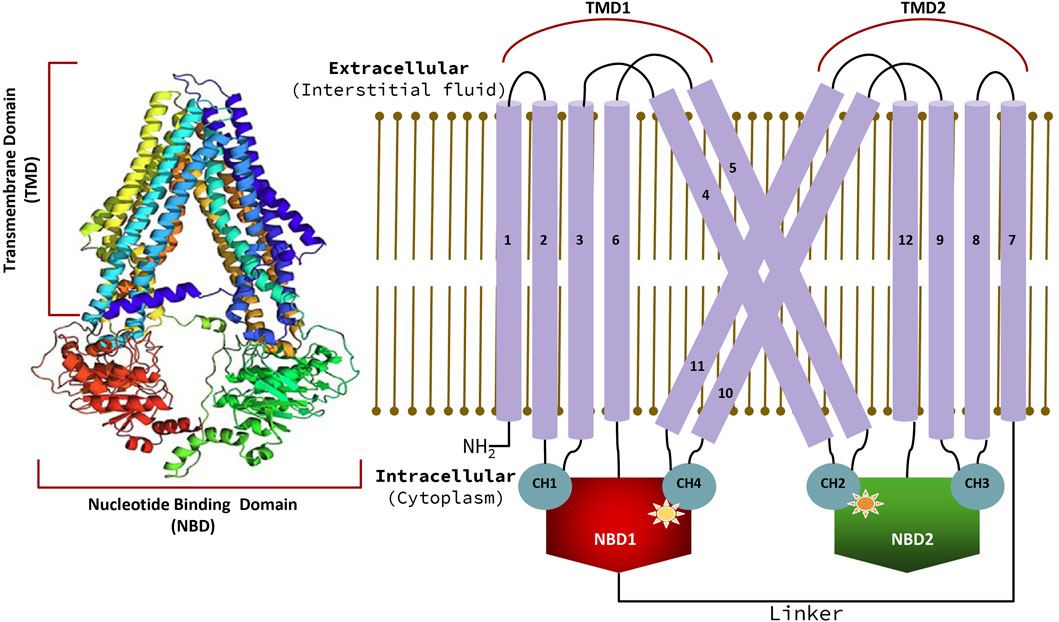
Figure 4. Model of human “ABC transporter protein” with uniprot ID. P08183, along with the arrangement of Trans-Membrane Domains (TMDs) and Nucleotide Binding Domains (NBDs) in human P-gp with detailed positioning of Trans-membrane Helices (THs) interlinking/coupler helices (CHs) that connects THs to NBDs.
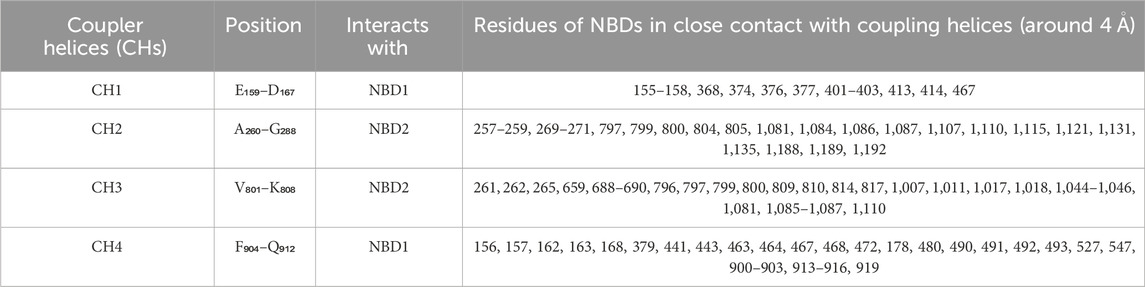
Table 3. Position of coupler helices/interlinking helices in protein sequence and residues from Nucleotide Binding Domain (NBD) interacting with coupling helices (around 4 Ǻ).
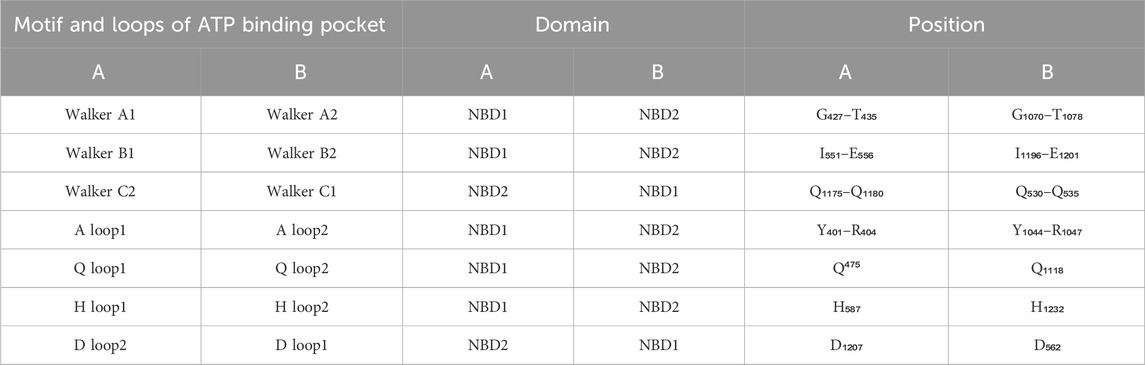
Table 4. Motifs and loops at ATP binding domain and their relative position in the sequence.
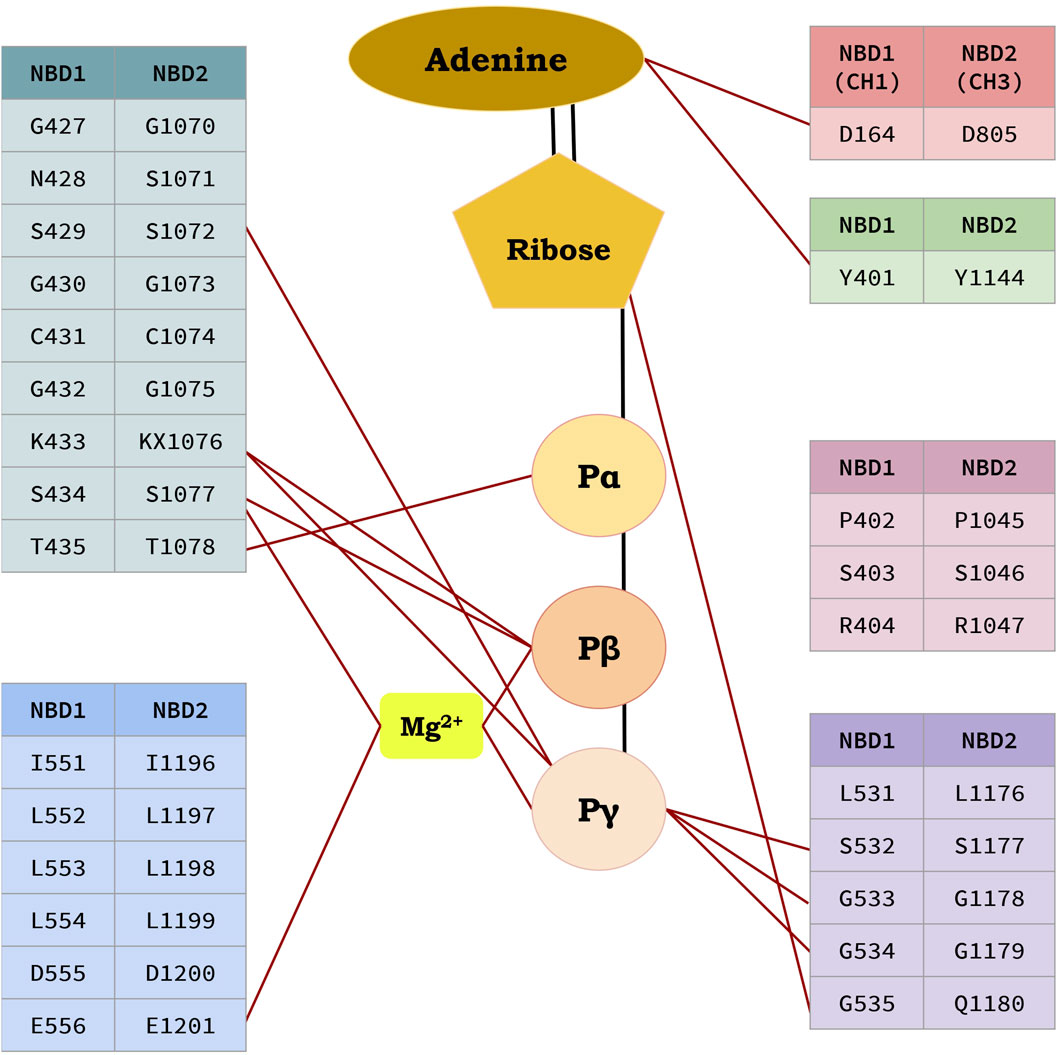
Figure 5. Residues interacting with ATP molecules at the ATP binding site in Nucleotide Binding Domains (NBDs).
5 Structure of human permeability-glycoprotein: determined through various computational approach
Despite the many P-gp structures solved, no high-resolution crystallographic structure is available for human P-gp. Several attempts have been made to model human P-gp in IO as well as OO conformation, to represent the two conformational states of the protein. Recently, an IO confirmation electron microscopic structure of P-gp (PDB ID: 6QEX) has been reported (Alam et al., 2019). Due to a lack of clear structure with good resolution in the database, structural biologists have moved towards the theoretical structure of human P-gp. It could be designed using either homology modelling, fold recognition/threading, ab initio (if the homologous structure is unavailable and the protein fold is unique), or a combination of two techniques. Homologous structures of human P-gp are available except for the linker region. Thus, either homology modelling, fold recognition, or a combination of both could be used to establish a theoretical model. A modelled and validated structure of human P-gp RefSeq sequence NP_001335875.1 has been shown in Figure 2, with elaborated and detailed annotations as shown in Tables 3–5; Figure 6 (Sayers et al., 2010; Bera et al., 2018).
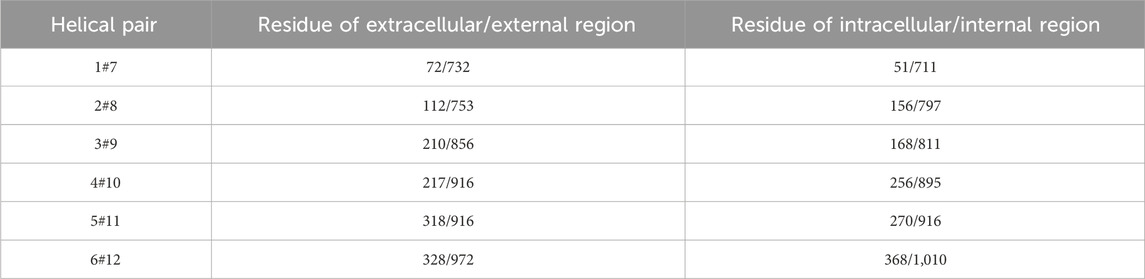
Table 5. Selected amino acid residue pair for computing dynamic movement of respective Trans-membrane Helices (THs) and their Trans-Membrane Domains (TMDs) (Wise, 2012).
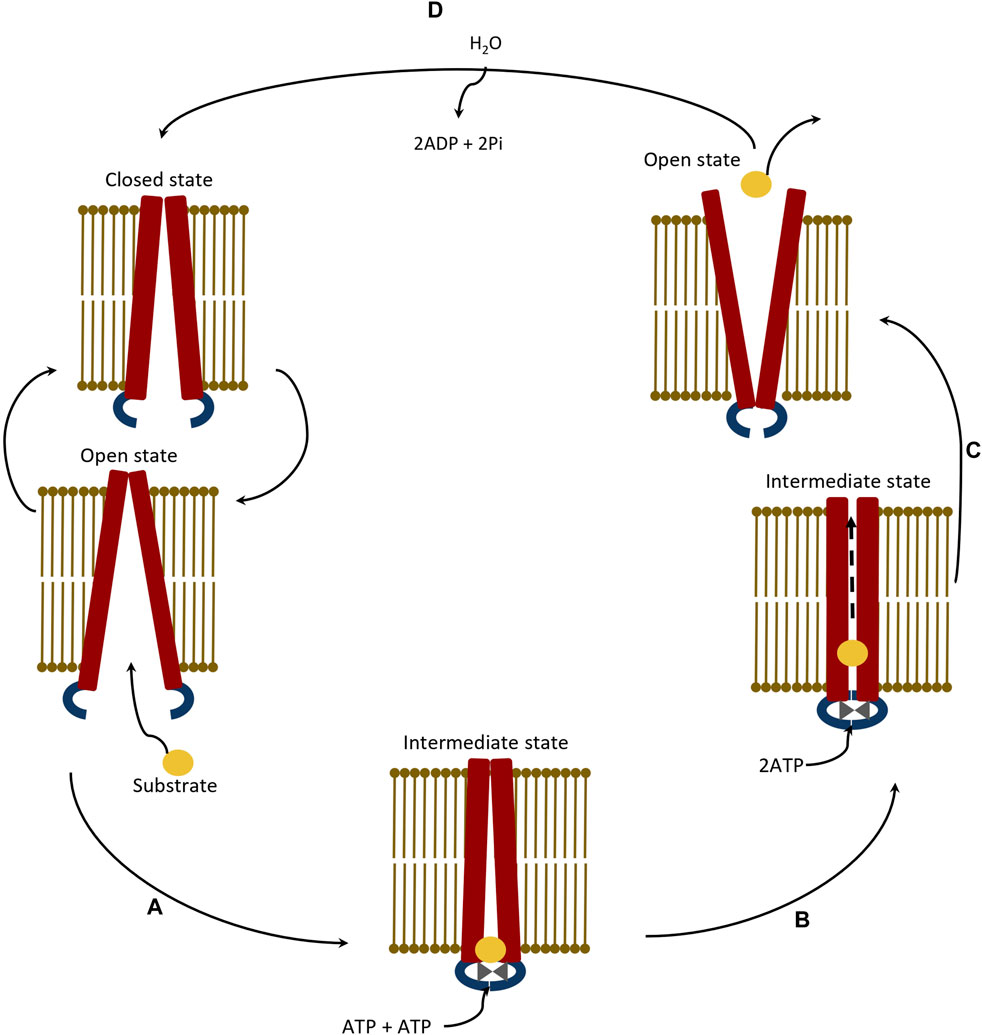
Figure 6. Schematic representation of catalytic transition stepwise pathway of ABC transporter protein showing efflux process of a compatible substrate. (A) Binding of substrate at Nucleotide Binding Domains (NBDs) in Inward Open (IO) state of protein, (B) Transport of bound substrate through Trans-membrane Helices (THs) of Trans-Membrane Domains (TMDs) of protein, (C) Efflux of substrate through Outward Open (IO) conformation of protein, and (D) Relaxation of protein at Inward Open (IO) state.
Since the P-gp protein shows sequence variability and functional (substrate and inhibitor recognition) diversity, it is difficult to model P-gp using the homology modelling method. Thus, it would be wise to model the structure using theoretical methods (threading or ab initio modelling) to create average IO and OO structure frames with greater clarity, followed by its Molecular Dynamics (MD) simulation to determine its structural stability, as discussed in the next section.
6 Importance of molecular dynamics simulation to explore conformational states of permeability-glycoprotein
Studying structural stability in protein (no steric clashes or inappropriate geometries) is of utmost importance before taking it further for complex analysis. Structural stability could be achieved by performing short conventional molecular dynamics simulations, and stability could be determined via various analyses (basic analysis: RMSD, RMSF, Rg, H-bond analysis etc. as well as advanced analysis: protein-ligand interaction analysis through MM-PBSA/MM-GBSA, PCA, free energy landscape, energy maps etc.) of simulation trajectories. These analyses are necessary to explore the dynamics of protein during simulation, and to understand the conformational changes in a protein which are important to carry out its biological functioning a large-scale molecular dynamics simulation or enhanced conformational sampling approaches may be useful (Mandal et al., 2023). They also aid in identifying stable and flexible regions of the simulated protein structure, relating to the characterize conformational changes, which crucial in context to protein function.
Similarly, P-gp mechanism and drug efflux process could be explored by employing molecular dynamics simulations, which has been a subject of active study in recent years. In the case of Pg-p dynamic movement in all the domains, especially in TMDs (mimics peristalsis-like movement) has been reported during its catalytic transition of effluxing molecules out of the cell (Korkhov et al., 2012; Manolaridis et al., 2018). Another study reported difference in the interaction pattern of molecules with substrate or modulator activity towards P-gp, as observed throughout simulation. The difference in compound could be correlated to the conformational changes of protein attained after small molecule interaction, that may trigger initial step of efflux. It has been observed that modulators tend to have higher number of non-bonded interactions (hydrophobic) when compared with P-gp substrate molecules. Thus, it has been proposed that the signal triggering the efflux could be interconnected to the presence/entrance of molecule in the internal cavity of P-gp (Ferreira et al., 2012). A different study talks about the energetic being the driving force for transport of molecules like verapamil and doxorubicin. Electrostatic repulsion (powered by positively charged resides) initially greatly contributes in verapamil transport and in later the stage transport in driven by hydrophobic interaction (Wang and Sun, 2020). Several studies with special emphasized on THs revealed that the trans-membrane region forms active sites for varied molecules comprising of hydrophobic residues which contributes in ligand binding to P-gp, explaining the reason for binding of hydrophobic drugs. It is also revealed that rigid compounds are less venerable as compared to flexible compounds when it comes at being P-gp substrate, as they are incompetent to modify their conformation to interact with binding site. These facts combined indicates that lipophilicity is a crucial criteria for designing of anticancer drugs (Prajapati et al., 2013). In a recent study researchers have reported natural P-gp inhibitors with potential study, based on their molecular docking outcomes, promising the innovative approach of computational study could be a cost-effect by-pass of conventional time inhibitive screening method (Dey et al., 2024). Such studies exhibits the utility of computational biology and molecular dynamics to elucidate the structural dynamics correlating to mechanism of P-gp, presenting the research community with valuable insight about drug efflux, multidrug resistance, pointers in potential drug development targeting P-gp to treat diseases like cancer.
7 Conformational changes in nucleotide binding and trans-membrane domains
As catalytic transition proceeds in P-gp and it goes from IO→OO, changes in the position of domains (NBDs and TMDs) are observed. The coordinated movement of these THs (Table 6) plays a major role in conformational shift of the P-gp structure once ATP hydrolysis has occurred. By monitoring the changes in the position of the domain, conformational changes could be traced. It is inconvenient to trace changes in whole domains, though this slight hindrance can be surpassed by picking up a relevant and essential residue that could be monitored in the domain. As reported by Wise, J.G., and coworkers in 2012, the catalytically critical glutamyl residues 556 and 1,201 present in the two NBDs are thought to be involved in the activation of the catalytic water of hydrolysis at the catalytic sites. As the simulation proceeds from IO→OO conformation, the separation distance in these residues decreases and afterwards remains relatively constant. These changes and movements in P-gp correlate with the rotation of the two NBDs relative to each other. Simultaneously, these conformational changes also affect the drug-binding domain, which undergoes a catalytic transition from fully IO→OO.
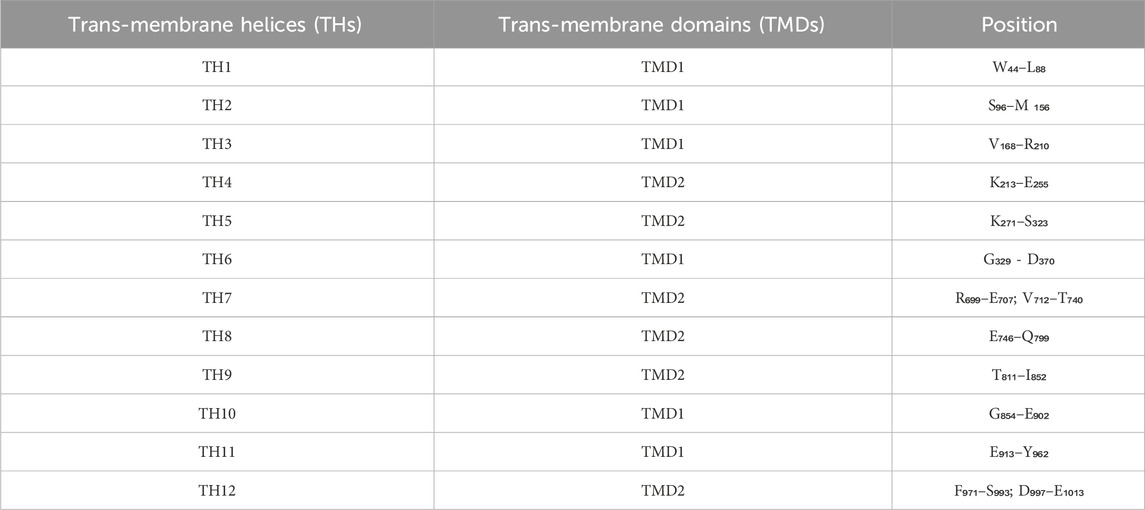
Table 6. Position of 12 Trans-membrane Helices (THs) in protein sequence of human Permeability-glycoprotein.
Direct involvement of Q-loop in NBD-TMD interaction/communication is well known as reported in earlier works (Lawson et al., 2008). Q-loop consists of a conserved glutamine residue followed by an 8-9 residue segment (Table 5). It is highly flexible and probably undergoes conformational changes during the catalytic transition from IO→OO. This loop or the conserved glutamine residue has been monitored to overt knowledge of the mechanism of P-gp catalytic transition. There has also been a report regarding the conformational switching of Q-loop, which causes communication between the substrate and catalytic site (TMDs and NBDs) in ABC transporter proteins (Jones and George, 2002).
Similarly, the dynamics of TMD have been monitored by computing the relative position of selected intracellular and extracellular amino acid residues. Drug molecules are effluxed through a channel/passage built by two trans-membrane domains, each having six THs (Wilkens, 2015). Catalytic transition powered by ATP hydrolysis triggers the conformational change in the helices of both TMDs and dynamic movement allows translocation of the substrate from intracellular to extracellular space (Hollenstein et al., 2007) (Table 5). As mentioned earlier, the whole domain cannot be monitored for movements; thus, a few residues from each TMD are selected based on their importance in functional aspects. The occurring changes during the catalytic transition (Table 5) can be monitored. Wise JG has reported significant dynamic movement occurring in the helices that form the drug-binding sites of P-gp, during catalytic transition. Opening of the drug binding sites to the extracellular space of the cell occurs as the ADP-Vi transition state approaches. The twisting of the NBDs as they fully take part in the catalytic transition, coupled with the helical movements of TMDs, leads to the opening of the drug’s two efflux sites (Wise, 2012).
8 The conformational state of permeability-glycoprotein may explain the promiscuity of substrate/inhibitor recognition
P-gp is known to recognize and efflux a variety of chemical compounds (also named the hydrophobic vacuum cleaner of the cell) of varied nature, including amphipathic anti-cancer-agents as mentioned in earlier sections as well (Ambudkar et al., 1999; Ambudkar et al., 2006; Szakács et al., 2006). It is also tiring to predict the mechanism of P-gp, and whether it adapts the same conformation when binding to its substrate, modulator, and inhibitors in addition to that, it is also difficult to decipher whether its schematic activity is dependent upon the size, shape, or other substrate properties such as their interaction feature. The curiosity of researchers has led to the enlightenment of substantially valuable knowledge regarding this specific property, i.e., the poly-specificity of P-gp. It was believed that P-gp exhibited a large conical cavity in its trans-membrane region consisting of a pocket with multiple binding sites (Loo et al., 2003; Aller et al., 2009). Two different cyclic peptides (QZ59-RRR and QZ59-SSS) bind at the same binding pocket but at different sites (Aller et al., 2009).
Another team of researchers reported the possibility of P-gp containing at least two transport-active drug binding sites: (i.) the H-site having specificity for Hoechst 33342 and colchicine, and (ii.) the R-site, as it binds to rhodamine 123 and anthracyclines (Shapiro and Ling, 1997). Later, a third binding site with allosteric properties was reported with an affinity for prazosin and progesterone (Shapiro et al., 1999). In a radioligand study, it has been revealed that P-gp accommodates more than two drug-binding sites (Martin et al., 2000), precisely indicating the seven varied sites (Safa, 2004). Hence, suggesting the multiple binding sites of P-gp protein to accommodate various drugs. However, data about whether each drug has a single or numerous binding sites is still scarce. A few compounds have been shown to have one active site, while some have an affinity toward more than one active site. In one of the recent investigations published in 2013, the possibility of a molecule having multiple binding sites has been clarified. It has also been reported that human P-gp has multiple binding sites or a molecule/substrate, and each site is analogously capable of transporting them. P-gp can also bind the compounds at the secondary sites, and when its primary binding sites are chemically modified, it eases the transportation. The existence of multiple diverse binding sites for varied compounds makes it a very efficient transporter (Chufan et al., 2013).
While conducting an investigation using site-directed mutagenesis of ABC transporters, including P-gp, they have multiple catalytic sites for each compound that should be considered. The central drug-cavity has several similar binding sites for drug binding, guaranteeing the binding of the compound followed by its transport, despite mutations at the primary drug-binding site. It has been reported that a few sets of important amino acid residues that are responsible for the interaction and binding of drugs are present in a specific 3D arrangement, thus making it possible for compounds to be transported (Chakraborty et al., 2012). This provides multiple possibilities, including chemical flexibility, which explains the broad substrate specificity aka poly-specificity of P-gp. Structural flexibility admittedly facilitates fine-tuning between the substrate and binding sites (primary or secondary site) for effective binding, facilitating the accommodation of different molecules on multiple sites, thereby explaining the phenomena of poly-specificity for P-gp and other ABC transporters. The last step of the process is the efflux of its substrate. This process is of aid to cells to efflux xenobiotics, though P-gp also effluxes essential therapeutic molecules along with xenobiotics without discrimination which becomes a problem in treatment.
Furthermore, the compiled knowledge about structural details and mechanism of catalytic transition of P-gp and its inhibitors has boosted the research for development of new potential inhibitor molecules with specific as well as broad activity surpassing the problem of efflux caused due to MDR and over-expression of P-gp, as discussed in the succeeding section.
9 Recent advances in permeability-glycoprotein inhibitor design
The area of inhibitor designing against P-gp is of great interest and a recent advance in its inhibitor design is focused on improving the potency, specificity and pharmacokinetic properties of P-gp inhibitors. The neoteric advancements are interdisciplinary approach intervening biotechnology, chemistry and bioinformatics, some of them are discussed below.
a) Structure-Based Drug Design (SBDD): The structural information about P-gp obtained through X-ray crystallography, cryo-electron microscopy or predicted computational models enables researchers to rationally design P-gp inhibitors via means of Structure-Based Drug Design (SBDD), encompassing the knowledge about compound binding sites and respective interacting residues of P-gp at different conformational states, which guides the development of more potent and selective inhibitors (Palmeira et al., 2012).
b) Ligand-Bases Drug Designing (LBDD): Another approach could be High Throughput Virtual Screening (HTVS) of small molecules to identify the potential active compound (Database screening) or starting point compound (Fragment-Based Drug Design aka FBDD) for inhibitor design through de novo or combinatorial chemistry approach followed by their optimization. Moreover, design of covalent P-gp inhibitors are also being excavated with an aim to find molecules with improved efficacy and selectivity as covalent inhibitors forms a more stable bond with their interacting proteins when compared to other inhibitors. The knowledge about the “Structure Activity Relationship (SAR)” of inhibitor molecule with the protein would further enrich the knowledge about interacting residues aiding in inhibitor development (Zhao et al., 2023) through “Quantitative Structure Activity Relationship (QSAR).” This ensures maximised target effect i.e., selectivity of chemotherapeutic drugs. Multitargeted inhibitors are also being explored to overcome resistance mechanism by inhibiting P-gp as well as other proteins involved in MDR.
c) Natural Product-Derived Inhibitors: Natural products and their derivatives has also been explored for their potential P-gp inhibitor activity (Dewanjee et al., 2017; Kumar and Jaitak, 2019), with an effort to optimize their pharmacological properties through structural modifications and analog synthesis as mentioned above (Dinić et al., 2018).
d) Nanotechnology-Based Approaches: Recent advancement in nanotechnology offers innovative strategies for development and delivery of P-gp inhibitor-loaded nanoparticles or nanocarriers, which improves the selective delivery of inhibitors specifically to cancer cells thereby minimizing the systemic toxicity caused by overdosing of chemotherapeutic drugs (Yadav et al., 2022).
e) Prodrug Strategies: Use of prodrugs (inactive compounds that undergo activation through enzymatic or chemical to release the active drug) could enhance the bioavailability cellular uptake as well as tissue distribution of active P-gp inhibitors, that would in turn potentially overcome the issues relating to poor pharmacokinetic properties (absorption, distribution and efflux) of traditional inhibitors (Guillemard and Uri Saragovi, 2004; Jain et al., 2004; Tanino et al., 2007; Di et al., 2009).
As discussed earlier, the mechanisms of P-gp inhibition has a significant role in drug development with special emphasis at the issue of MDR in cancer treatment. P-gp inhibition mechanisms impact drug development and clinical practice by enhancing drug delivery and reversal of drug resistance, which is achieved by increased intracellular accumulation of chemotherapeutic drugs to attain drug concentration to therapeutic level. Studies have revealed that Zosuquidar (LY335979) is a third-generation P-gp inhibitor when combined with standard chemotherapeutic regimes, has the ability to overcome P-gp-mediated resistance by improving the response rates and survival outcomes in patients with refractory or relapsed in acute myeloid leukemia (AML) (Pote and Gacche, 2023). Another such example is Tariquidar (XR9576) which is also a potent third-generation P-gp inhibitor with the ability to enhance the efficacy of anticancer drugs such as paclitaxel and doxorubicin (Akhtar et al., 2011; Zhang et al., 2016). In clinical practice, other P-gp inhibitors like verapamil and cyclosporine have also been used in combination with standard chemotherapeutics in order to improve response rates in patients suffering with solid tumors, leukemia and lymphoma. A study has reported that a combination of cyclosporine A and anthracyclines effectively increases response rates in patients with relapsed/refractory lymphomas (Albadari et al., 2024). In another study pharmacokinetic interaction of ketoconazole (antifungal compound) has proved to be increasing plasma concentration of P-gp drugs (digoxin and loperamide) which in turn enhcnces therapeutic effect of drugs (jia et al., 2002). Besides above mentioned strategies involved in development of P-gp inhibitors other conventional techniques are also still in practice, although these are some of the promising advances to overcome the hurdle of MDR in cancer treatment with improved efficacy of therapeutic drugs.
Overall, the mechanisms of P-gp inhibition has a significant impact on drug development and clinical practices as mentioned above. By targeting P-gp-mediated drug efflux, P-gp inhibitors offer promising strategies to enhance drug delivery, overcome drug resistance, and improve treatment outcomes in patients with MDR cancers and other conditions.
10 Final remarks
P-gp is one of the most studied proteins among all ABC transporters. However, the mechanism of substrate and inhibitor binding, catalytic transition, and efflux still needs to be unearthed. The high expression of P-gp has been directly linked to the reduced bioavailability of clinically essential drugs (Wise, 2012). Structurally diverse sets of P-gp inhibitors, substrates and modulators have been identified and reported in the past few years. However, several aspects remain unanswered: “How does P-gp accommodate the diverse sets of substrates and inhibitors?” P-gp amino acid sequences of various species have shown conserveness in the critically important structural segments, thereby maintaining the biological functioning of P-gp. From the first mammalian X-ray crystal structure of P-gp in IO conformation, structural biologists started to focus their interest in P-gp, and later, many other structures were deposited. However, human P-gp in its IO or OO states still needs to be crystallized, further enhancing our understanding. The uniqueness of human P-gp is the presence of a linker (∼76 amino acid residue long) region responsible for linking the pseudo-symmetrical domains (TMD1-NBD1/TMD2-NBD2) with each other. Due to the lack of reliable experimentally reported structures of human P-gp, researchers have moved towards creating a theoretical model structure.
Further to pursue catalytic transition (IO→OO) and relaxation (OO → IO), studies of the protein holding great hidden treasure about how it accommodates the varied range of compounds within its niche molecular dynamics/targeted molecular dynamics simulation techniques have been adopted by researchers. It will reveal the possible intermediate states (structural frames) and their energies, which will help to understand minute conformational changes occurring during the transition. Different structural states may help explain the dynamics of NBDs and TMDs substrate promiscuity, and inhibitor binding in the NBDs/TMDs. Moreover, the structural frames/states may be utilized for better inhibitor design to tackle the problem of MDR and the rampant efflux of chemotherapeutic drugs used during cancer treatment.
Author contributions
PR: Writing–original draft, Writing–review and editing, Data curation, Investigation. PM: Writing–original draft, Writing–review and editing, Data curation. BR: Writing–original draft, Writing–review and editing. DS: Supervision, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors are mightly thankful to Central University of South Bihar, Gaya.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
P-gp, permeability-glycoprotein; IO, inward opening; OO, outward opening; NBSs, nucleotide binding sites; TMD, trans-membrane domain; CT, catalytic transition; NBD, nucleotide binding domain.
References
Abdallah, H. M., Al-Abd, A. M., El-Dine, R. S., and El-Halawany, A. M. (2015). P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: a review. J. Adv. Res. 6 (1), 45–62. doi:10.1016/j.jare.2014.11.008
Akhtar, N., Ahad, A., Khar, R. K., Jaggi, M., Aqil, M., Iqbal, Z., et al. (2011). The emerging role of P-glycoprotein inhibitors in drug delivery: a patent review. Expert Opin. Ther. Pat. 21 (4), 561–576. doi:10.1517/13543776.2011.561784
Alam, A., Kowal, J., Broude, E., Roninson, I., and Locher, K. P. (2019). Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 363 (6428), 753–756. doi:10.1126/science.aav7102
Albadari, N., Xie, Y., and Li, W. (2024). Deciphering treatment resistance in metastatic colorectal cancer: roles of drug transports, EGFR mutations, and HGF/c-MET signaling. Front. Pharmacol. 14, 1340401. doi:10.3389/fphar.2023.1340401
Alfarouk, K. O., Stock, C. M., Taylor, S., Walsh, M., Muddathir, A. K., Verduzco, D., et al. (2015). Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int. 15 (1), 71. doi:10.1186/s12935-015-0221-1
Aller, S. G., Yu, J., Ward, A., Weng, Y., Chittaboina, S., Zhuo, R., et al. (2009). Structure of P-glycoprotein reveals a molecular Basis for poly-specific drug binding. Science 323 (5922), 1718–1722. doi:10.1126/science.1168750
Ambudkar, S. V., Dey, S., Hrycyna, C. A., Ramachandra, M., Pastan, I., and Gottesman, M. M. (1999). Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39 (1), 361–398. doi:10.1146/annurev.pharmtox.39.1.361
Ambudkar, S. V., Kim, I. W., and Sauna, Z. E. (2006). The power of the pump: mechanisms of action of P-glycoprotein (ABCB1). Eur. J. Pharm. Sci. 27 (5), 392–400. doi:10.1016/j.ejps.2005.10.010
Assaraf, Y. G., Brozovic, A., Gonçalves, A. C., Jurkovicova, D., Linē, A., Machuqueiro, M., et al. (2019). The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updat. 46, 100645. doi:10.1016/j.drup.2019.100645
Bansal, T., Jaggi, M., Khar, R., and Talegaonkar, S. (2009). Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. J. Pharm. Pharm. Sci. 12 (1), 46–78. doi:10.18433/j3rc77
Bera, K., Rani, P., Kishor, G., Agarwal, S., Kumar, A., and Singh, D. V. (2018). Structural elucidation of transmembrane domain zero (TMD0) of EcdL: a multidrug resistance-associated protein (MRP) family of ATP-binding cassette transporter protein revealed by atomistic simulation. J. Biomol. Struct. Dyn. 36 (11), 2938–2950. doi:10.1080/07391102.2017.1372311
Boumendjel, A., Boutonnat, J., and Robert, J. (2009). ABC transporters and multidrug resistance. 1st ed. Wiley. Available from: https://onlinelibrary.wiley.com/doi/book/10.1002/9780470495131.
Breedveld, P., Beijnen, J. H., and Schellens, J. H. (2006). Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol. Sci. 27 (1), 17–24. doi:10.1016/j.tips.2005.11.009
Brewer, F. K., Follit, C. A., Vogel, P. D., and Wise, J. G. (2014). In silico screening for inhibitors of p-glycoprotein that target the nucleotide binding domains. Mol. Pharmacol. 86 (6), 716–726. doi:10.1124/mol.114.095414
Chakraborty, S., Asgeirsson, B., and Rao, B. J. (2012). A measure of the broad substrate specificity of enzymes based on ‘duplicate’catalytic residues. PloS One. 7 (11), e49313. doi:10.1371/journal.pone.0049313
Chufan, E. E., Kapoor, K., Sim, H. M., Singh, S., Talele, T. T., Durell, S. R., et al. (2013). Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1). PloS one 8 (12), e82463. doi:10.1371/journal.pone.0082463
Clay, A. T., Lu, P., and Sharom, F. J. (2015). Interaction of the P-glycoprotein multidrug transporter with Sterols. Biochemistry 54 (43), 6586–6597. doi:10.1021/acs.biochem.5b00904
Dantzig, A. H., de Alwis, D. P., and Burgess, M. (2003). Considerations in the design and development of transport inhibitors as adjuncts to drug therapy. Adv. drug Deliv. Rev. 55 (1), 133–150. doi:10.1016/s0169-409x(02)00175-8
Dawson, R. J., and Locher, K. P. (2006). Structure of a bacterial multidrug ABC transporter. Nature 443 (7108), 180–185. doi:10.1038/nature05155
Dewanjee, S., Dua T, K., Bhattacharjee, N., Das, A., Gangopadhyay, M., Khanra, R., et al. (2017). Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules 22 (6), 871. doi:10.3390/molecules22060871
Dey, A., Li, R., Larzat, N., Idoipe, J. B., Kati, A., and Sharma, A. (2024). Investigating natural inhibitors of Permeability-glycoprotein (P-gp) liver transporter via molecular docking simulation for Hepatocellular carcinoma therapy. Commun. Comput. Inf. Sci. 2030, 81–93. doi:10.1007/978-3-031-53731-8_7
Di, L., Kerns, E. H., and Carter, G. T. (2009). Drug-like property concepts in pharmaceutical design. Curr. Pharm. Des. 15 (19), 2184–2194. doi:10.2174/138161209788682479
Dinić, J., Podolski-Renić, A., Jeremić, M., and Pešić, M. (2018). Potential of natural-based anticancer compounds for P-glycoprotein inhibition. Curr. Pharm. Des. 24 (36), 4334–4354. doi:10.2174/1381612825666190112164211
Eckford, P. D. W., and Sharom, F. J. (2009). ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 109 (7), 2989–3011. doi:10.1021/cr9000226
Ferreira, R. J., Ferreira, M. J. U., and Dos Santos, DJVA (2012). Insights on P-Glycoprotein’s efflux mechanism obtained by molecular dynamics simulations. J. Chem. Theory Comput. 8 (6), 1853–1864. doi:10.1021/ct300083m
Guillemard, V., and Uri Saragovi, H. (2004). Prodrug chemotherapeutics bypass p-glycoprotein resistance and kill tumors in vivo with high efficacy and target-dependent selectivity. Oncogene 23 (20), 3613–3621. doi:10.1038/sj.onc.1207463
Higgins, C. F., and Linton, K. J. (2004). The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11 (10), 918–926. doi:10.1038/nsmb836
Holland, I. B., and Blight, M. A. (1999). ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol. 293 (2), 381–399. doi:10.1006/jmbi.1999.2993
Hollenstein, K., Dawson, R. J., and Locher, K. P. (2007). Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 17 (4), 412–418. doi:10.1016/j.sbi.2007.07.003
Jain, R., Majumdar, S., Nashed, Y., Pal, D., and Mitra, A. K. (2004). Circumventing P-Glycoprotein-Mediated cellular efflux of Quinidine by prodrug Derivatization. Mol. Pharm. 1 (4), 290–299. doi:10.1021/mp049952s
jia, W. E., Lew, K., Casciano, C. N., Clement, R. P., and Johnson, W. W. (2002). Interaction of common Azole antifungals with P glycoprotein. Antimicrob. Agents Chemother. 46 (1), 160–165. doi:10.1128/aac.46.1.160-165.2002
Jin, M. S., Oldham, M. L., Zhang, Q., and Chen, J. (2012). Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490 (7421), 566–569. doi:10.1038/nature11448
Jones, P. M., and George, A. M. (2002). Mechanism of ABC transporters: a molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc. Natl. Acad. Sci. U. S. A. 99 (20), 12639–12644. doi:10.1073/pnas.152439599
Korkhov, V. M., Mireku, S. A., and Locher, K. P. (2012). Structure of AMP-PNP-bound vitamin B12 transporter BtuCD–F. Nature 490 (7420), 367–372. doi:10.1038/nature11442
Kumar, A., and Jaitak, V. (2019). Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 176, 268–291. doi:10.1016/j.ejmech.2019.05.027
Lawson, J., O’Mara, M. L., and Kerr, I. D. (2008). Structure-based interpretation of the mutagenesis database for the nucleotide binding domains of P-glycoprotein. Biochimica Biophysica Acta (BBA)-Biomembranes. 1778 (2), 376–391. doi:10.1016/j.bbamem.2007.10.021
Li, J., Jaimes, K. F., and Aller, S. G. (2014). Refined structures of mouse P-glycoprotein. Protein Sci. 23 (1), 34–46. doi:10.1002/pro.2387
Loo, T. W., Bartlett, M. C., and Clarke, D. M. (2003). Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 278 (41), 39706–39710. doi:10.1074/jbc.M308559200
Loo, T. W., and Clarke, D. M. (2013). Drug Rescue Distinguishes between different structural models of human P-glycoprotein. Biochemistry 52 (41), 7167–7169. doi:10.1021/bi401269m
Loo, T. W., and Clarke, D. M. (2014). Locking intracellular helices 2 and 3 together inactivates human P-glycoprotein. J. Biol. Chem. 289 (1), 229–236. doi:10.1074/jbc.M113.527804
Mandal, P., Rani, P., Chandra, G., and Singh, D. V. (2023). Flap sub-domain dynamics of serine-threonine phosphatase (Stp1) of Staphylococcus aureus: an accelerated molecular dynamics simulation study. J. Biomol. Struct. Dyn. 41 (13), 6413–6421. doi:10.1080/07391102.2022.2107575
Manolaridis, I., Jackson, S. M., Taylor, N. M., Kowal, J., Stahlberg, H., and Locher, K. P. (2018). Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 563 (7731), 426–430. doi:10.1038/s41586-018-0680-3
Martin, C., Berridge, G., Higgins, C. F., Mistry, P., Charlton, P., and Callaghan, R. (2000). Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol. 58 (3), 624–632. doi:10.1124/mol.58.3.624
Nuti, S. L., and Rao, U. S. (2002). Proteolytic cleavage of the linker region of the human P-glycoprotein modulates its ATPase function. J. Biol. Chem. 277 (33), 29417–29423. doi:10.1074/jbc.M204054200
Orlowski, S., Mir, L. M., Belehradek, J., and Garrigos, M. (1996). Effects of steroids and verapamil on P-glycoprotein ATPase activity: progesterone, desoxycorticosterone, corticosterone and verapamil are mutually non-exclusive modulators. Biochem. J. 317 (2), 515–522. doi:10.1042/bj3170515
Palmeira, A., Sousa, E., Helena Vasconcelos, M., Pinto, M., and X Fernandes, M. (2012). Structure and ligand-based design of P-glycoprotein inhibitors: a historical perspective. Curr. Pharm. Des. 18 (27), 4197–4214. doi:10.2174/138161212802430530
Pan, L., and Aller, S. G. (2015). Equilibrated atomic models of outward-facing P-glycoprotein and effect of ATP binding on structural dynamics. Sci. Rep. 5 (1), 7880. doi:10.1038/srep07880
Pleban, K., Kopp, S., Csaszar, E., Peer, M., Hrebicek, T., Rizzi, A., et al. (2005). P-glycoprotein substrate binding domains are located at the transmembrane domain/transmembrane domain interfaces: a combined photoaffinity labeling-protein homology modeling approach. Mol. Pharmacol. 67 (2), 365–374. doi:10.1124/mol.104.006973
Pote, M. S., and Gacche, R. N. (2023). ATP-binding cassette efflux transporters and MDR in cancer. Drug Discov. Today 28 (5), 103537. doi:10.1016/j.drudis.2023.103537
Prajapati, R., Singh, U., Patil, A., Khomane, K. S., Bagul, P., Bansal, A. K., et al. (2013). In silico model for P-glycoprotein substrate prediction: insights from molecular dynamics and in vitro studies. J. Comput. Aided Mol. Des. 27 (4), 347–363. doi:10.1007/s10822-013-9650-x
Safa, A. R. (2004). Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr. Med. Chemistry-Anti-Cancer Agents. 4 (1), 1–17. doi:10.2174/1568011043482142
Sanchez-Covarrubias, L., Slosky, L. M., Thompson, B. J., Zhang, Y., Laracuente, M. L., DeMarco, K. M., et al. (2014). P-glycoprotein modulates morphine uptake into the CNS: a role for the non-steroidal anti-inflammatory drug diclofenac. PloS one 9 (2), e88516. doi:10.1371/journal.pone.0088516
Sauna, Z. E., and Ambudkar, S. V. (2007). About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. cancer Ther. 6 (1), 13–23. doi:10.1158/1535-7163.MCT-06-0155
Sayers, E. W., Barrett, T., Benson, D. A., Bolton, E., Bryant, S. H., Canese, K., et al. (2010). Database resources of the national center for biotechnology information. Nucleic acids Res. 39 (Suppl. l_1), D38–D51. doi:10.1093/nar/gkq1172
Shapiro, A. B., Fox, K., Lam, P., and Ling, V. (1999). Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone: Evidence for a third drug-binding site. Eur. J. Biochem. 259 (3), 841–850. doi:10.1046/j.1432-1327.1999.00098.x
Shapiro, A. B., and Ling, V. (1997). Positively Cooperative sites for drug transport by P-glycoprotein with Distinct drug Specificities. Eur. J. Biochem. 250 (1), 130–137. doi:10.1111/j.1432-1033.1997.00130.x
Sharom, F. J. (2008). ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 9 (1), 105–127. doi:10.2217/14622416.9.1.105
Sharom, F. J. (2011a). The P-glycoprotein multidrug transporter. Essays Biochem. 50, 161–178. doi:10.1042/bse0500161
Sharom, F. J. (2011b). The P-glycoprotein multidrug transporter. Essays Biochem. 50, 161–178. doi:10.1042/bse0500161
Shebley, M., Liu, J., Kavetskaia, O., Sydor, J., de Morais, S. M., Fischer, V., et al. (2017). Mechanisms and predictions of drug-drug interactions of the hepatitis C virus three direct-acting antiviral regimen: paritaprevir/ritonavir, ombitasvir, and dasabuvir. Drug Metabolism Dispos. 45 (7), 755–764. doi:10.1124/dmd.116.074518
Silva, V., Gil-Martins, E., Silva, B., Rocha-Pereira, C., Sousa, M. E., Remião, F., et al. (2021). Xanthones as P-glycoprotein modulators and their impact on drug bioavailability. Expert Opin. Drug Metabolism Toxicol. 17 (4), 441–482. doi:10.1080/17425255.2021.1861247
Singh, D. V., Godbole, M. M., and Misra, K. (2013). A plausible explanation for enhanced bioavailability of P-gp substrates in presence of piperine: simulation for next generation of P-gp inhibitors. J. Mol. Model 19 (1), 227–238. doi:10.1007/s00894-012-1535-8
Szakács, G., Paterson, J. K., Ludwig, J. A., Booth-Genthe, C., and Gottesman, M. M. (2006). Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 5 (3), 219–234. doi:10.1038/nrd1984
Tanino, T., Nawa, A., Kondo, E., Kikkawa, F., Daikoku, T., Tsurumi, T., et al. (2007). Paclitaxel-2′-Ethylcarbonate prodrug can Circumvent P-glycoprotein-mediated cellular efflux to increase drug Cytotoxicity. Pharm. Res. 24 (3), 555–565. doi:10.1007/s11095-006-9171-6
Tripathi, A., Singh, D. V., Kesharwani, R. K., and Misra, K. (2015). P-glycoprotein: a critical comparison of models depicting mechanism of drug efflux and role of modulators. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 85, 359–375. doi:10.1007/s40011-014-0405-9
Wang, L., and Sun, Y. (2020). Efflux mechanism and pathway of verapamil pumping by human P-glycoprotein. Archives Biochem. biophysics 696, 108675. doi:10.1016/j.abb.2020.108675
Warrington, J. S., and Shaw, L. M. (2005). Pharmacogenetic differences and drug–drug interactions in immunosuppressive therapy. Expert Opin. Drug Metabolism Toxicol. 1 (3), 487–503. doi:10.1517/17425255.1.3.487
Wessler, J. D., Grip, L. T., Mendell, J., and Giugliano, R. P. (2013). The P-glycoprotein transport System and Cardiovascular drugs. J. Am. Coll. Cardiol. 61 (25), 2495–2502. doi:10.1016/j.jacc.2013.02.058
Wilkens, S. (2015). Structure and mechanism of ABC transporters. F1000prime Rep. 7, 14. doi:10.12703/P7-14
Wise, J. G. (2012). Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry 51 (25), 5125–5141. doi:10.1021/bi300299z
Yadav, P., Ambudkar, S. V., and Rajendra Prasad, N. (2022). Emerging nanotechnology-based therapeutics to combat multidrug-resistant cancer. J. Nanobiotechnol 20 (1), 423. doi:10.1186/s12951-022-01626-z
Zhang, H., Xu, H., Ashby, C. R., Assaraf, Y. G., Chen, Z., and Liu, H. (2021). Chemical molecular-based approach to overcome multidrug resistance in cancer by targeting P-glycoprotein (P-gp). Med. Res. Rev. 41 (1), 525–555. doi:10.1002/med.21739
Zhang, Y., Sriraman, S. K., Kenny, H. A., Luther, E., Torchilin, V., and Lengyel, E. (2016). Reversal of chemoresistance in ovarian cancer by co-delivery of a P-glycoprotein inhibitor and paclitaxel in a liposomal platform. Mol. cancer Ther. 15 (10), 2282–2293. doi:10.1158/1535-7163.MCT-15-0986
Keywords: ABC transporter, homology modelling, targeted molecular dynamics, catalytic transition, dynamics of NBDs and TMDs, P-gp inhibitors and substrates, multiple drug resistance (MDR), efflux
Citation: Rani P, Mandal P, Rajak BK and Singh DV (2024) A review on dynamics of permeability-glycoprotein in efflux of chemotherapeutic drugs. Front. Drug Discov. 4:1363364. doi: 10.3389/fddsv.2024.1363364
Received: 30 December 2023; Accepted: 27 March 2024;
Published: 11 April 2024.
Edited by:
Rajesh Kumar Pathak, Chung-Ang University, Republic of KoreaReviewed by:
Muktesh Chandra, Marwadi University, IndiaRohit Shukla, Jaypee University of Information Technology, India
Shubham Pant, Central Electrochemical Research Institute (CSIR), India
Akshay Singh, Indian Council of Agricultural Research (ICAR), India
Copyright © 2024 Rani, Mandal, Rajak and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Durg Vijay Singh, ZHVyZ3ZpamF5c2luZ2hAY3ViLmFjLmlu, ZHZzYmlvdGVjaEBnbWFpbC5jb20=