 Utkarsha Naithani
Utkarsha Naithani Vandana Guleria
Vandana Guleria
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Drug Discov. , 05 April 2024
Sec. In silico Methods and Artificial Intelligence for Drug Discovery
Volume 4 - 2024 | https://doi.org/10.3389/fddsv.2024.1362456
This article is part of the Research Topic Enhancing Drug Discovery Through Structure-Based Design and Computational Techniques View all 3 articles
In the drug discovery and development, the identification of leadcompoundsplaysa crucial role in the quest for novel therapeutic agents. Leadcompounds are the initial molecules that show promising pharmacological activity againsta specific target and serve as the foundation for drug development. Integrativecomputational approaches have emerged as powerful tools in expediting this complex andresource-intensive process. They enable the efficient screening of vast chemical librariesand the rational design of potential drug candidates, significantly accelerating the drugdiscoverypipeline. This review paper explores the multi-layered landscape of integrative computationalmethodologies employed in lead compound discovery and evaluation. These approaches include various techniques, including molecular modelling, cheminformatics, structure-based drug design (SBDD), high-throughput screening, molecular dynamics simulations, ADMET (absorption, distribution, metabolism, excretion, and toxicity) prediction, anddrug-target interaction analysis. By revealing the critical role ofintegrative computational methods, this review highlights their potential to transformdrug discovery into a more efficient, cost-effective, and target-focused endeavour, ultimately paving the way for the development of innovative therapeutic agents to addressa multitude of medical challenges.
In the ever-evolving landscape of drug development, the quest for new and potent treatments stands as an imperative challenge. To tackle this challenge, the combination of computational techniques into the drug discovery process is a good example of innovation that has the potential to drastically alter the course of lead compound development. Computational techniques are an outstanding example of innovation, providing unique benefits beyond conventional methods and enabling lead compound identification to occur more quickly and accurately than ever before (Tiwari and Singh, 2022). Finding new treatments has always been difficult due to the complexity and size of the chemical space. But the emergence of integrative computational methods has relieved this bottleneck and brought about a revolutionary new phase in drug design. These techniques use data analytics, molecular modelling, and algorithms (Outeiral et al., 2021) to predict pharmacokinetic characteristics, decipher intricate molecular interactions, and expedite the laborious lead compound discovery process (Prieto-Martínez et al., 2019).
Although conventional techniques for discovering new drugs have been essential (Leveridge et al., 2018), the pharmaceutical industry has adopted integrative computational methodologies as a revolutionary answer because of their high attrition rates, resource intensity, and time limits (Berdigaliyev and Aljofan, 2020). One of the main benefits of integrative computational techniques is their capacity to address the large chemical space more efficiently. Researchers are given with a large pool of potential candidates in their search for lead compounds, and navigating this vast environment quickly and precisely is necessary. Utilising computing capacity, integrative techniques sort through large chemical libraries to find molecules that have the best chance of succeeding. This speeds up the preliminary phases of lead generation, enabling a more concentrated and targeted strategy.
The collaborative synergy between experimental validations and computer models goes beyond lead compound discovery. The significance of assessing these substances pharmacokinetic and safety profiles is acknowledged by integrative approaches. The computational framework’s integration of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) predictions enhances our comprehension of a lead compound’s path through the biological system (Mohs and Greig, 2017). The pharmaceutical industry is using a growing number of integrative computational approaches, making real-world case studies a more valuable source of success (Wu et al., 2023). These studies present examples of how the intelligent fusion of technology and experimentation has resulted in the discovery and creation of new treatments. The versatility and impact of integrative techniques are highlighted by the successful uses in a range of therapeutic domains, including infectious diseases and cancer treatment.
Globally, the integrative approach to drug discovery is having a revolutionary effect and changing the landscape of healthcare. This cooperative approach, which combines knowledge from various scientific fields, accelerates the creation of new and efficient treatments. Through the effective identification of targeted treatments and customization of interventions based on individual characteristics, the integrative approach more effectively addresses diseases with worldwide prevalence. Its importance in global health emergencies is highlighted by its ability to react quickly to newly emerging infectious diseases, as demonstrated by the COVID-19 pandemic and other incidents. Furthermore, by creating treatments that work in a variety of genetic and socioeconomic contexts, the strategy advances health equity. Simplifying clinical trials ensures that life-saving medications are approved more quickly and can be administered to patients all over the world. Because integrative research is collaborative in nature, it promotes global collaboration and knowledge sharing that advances our collective understanding of diseases and treatment approaches. All things considered, the integrative approach is a driving force behind positive, profound changes in healthcare that promise better outcomes and increased access to cutting-edge treatments for people all over the world. Despite the fact that integrated computational techniques present a promising future, difficulties and constraints must be recognized. Among the challenges faced by researchers include the precision of computer forecasts, the quality and quantity of data intake, and the need for substantial computational resources (Sliwoski et al., 2014). It is imperative that these issues be resolved to improve and advance integrative drug discovery methodologies. This review digs into the complex intersections of technology, biology, and chemistry by undertaking a comprehensive analysis of integrative computational methodologies for the detection and assessment of lead compounds in drug creation. As the field continues to evolve, the integration of computational methods is poised to play an increasingly pivotal role in shaping the future of drug creation and design (Nicolaou, 2014). These methods overcome the drawbacks of conventional approaches by fusing modern technology with well-established experimental protocols in a way that presents a dynamic and comprehensive view.
Drug discovery used to be a laboriousand costly procedure. Empirical observations, unpredictability, and time-consuming procedures for screening natural compounds were major components of drug discovery. Traditional screening methods involve the manual testing of large numbers of compounds, a process that is both time-consuming and expensive. This approach limits the number of compounds that can be explored, slowing down the drug discovery process (Khan et al., 2021). A thorough understanding of the molecular interactions between compounds and their targets is not possible with empirical screening techniques. It is difficult to accurately predict the behaviour of the compounds in complex biological systems due to this lack of mechanistic insight. Traditional methods often neglect factors such as a compound’s pharmacokinetics and bioavailability (Berdigaliyev and Aljofan, 2020). These are critical for determining how a drug is absorbed, distributed, metabolized, and excreted in the body, influencing its overall efficacy and safety. Many successful drugs discovered using traditional methods were often a result of coincidence rather than a systematic approach. This reliance on chance discoveries is inefficient and may not be sustainable for addressing complex diseases. As our understanding of ethical and safety standards has evolved, traditional empirical screening methods face increased scrutiny for their reliance on animal testing and potential safety risks that may not be apparent until later stages of development.
The field was revolutionised by the introduction of genomics, high-throughput screening, and molecular biology in the late 20th century. Structural biology, bioinformatics, and computational techniques are all used in modern drug discovery to speed up target identification and lead optimisation. The influence of traditional medical knowledge has shaped the evolution of modern medicine over-time (Raviña, 2011). The medical science narrative is always being shaped by the wisdom of past civilizations, influencing anything from surgical methods to philosophical ideas. The integration of historical perspectives and modern techniques results in a comprehensive strategy that recognises the complex relationship between tradition and innovation in the quest for wellbeing (Singh et al., 2022). Within the drug development process, traditional drug discovery lays the groundwork for lead discovery, in which the identification and optimisation of possible lead compounds are guided by the knowledge of biological targets and the insights obtained from comprehensive screening.
In the field of drug development, a chemical entity exhibiting potential therapeutic characteristics against a particular biological target or disease is known as a lead compound. It is the foremost step in the creation of new drugs and provides the framework for additional optimization aimed at improving their pharmacokinetic, safety, and effectiveness characteristics. A critical phase in the process is identifying a lead drug, which entails screening a variety of synthetic and natural compounds to identify one with potential biological activity. Lead compounds are excellent choices for additional development because of their qualities (Cheng et al., 2007). They frequently show selectivity and affinity for a given target, like an enzyme or protein linked to a disease. Various techniques are used to efficiently sift through compound libraries and identify potential leads. Some of them are:
• High Throughput Screening (HTS)—High Throughput Screening (Zhu and Cuozzo, 2009) is a widely used lead discovery method that involves the rapid testing of large compound libraries for their ability to interact with a target of interest. Automated systems allow the screening of thousands to millions of compounds, assessing their biological activity. This approach is characterized by its speed and efficiency, enabling the identification of lead compounds with potential therapeutic effects. However, it also poses challenges related to false positives and the need for robust validation. HTS often lacks detailed information about the mechanism of action and may identify compounds with non-specific binding.
• Fragment-Based Screening—Fragment-based screening involves testing smaller, low molecular weight compounds (fragments) for their binding affinity to a target. This approach focuses on identifying key fragments that can be built upon to create more potent lead compounds. Although it requires more detailed structural information and sophisticated methods such as X-ray crystallography or NMR spectroscopy, fragment-based screening offers the advantage of exploring a broader chemical space and often results in leads with improved binding affinities. Fragment-based approaches are particularly suitable for challenging targets that may be difficult to address with traditional screening methods. Fragments help identify key binding sites (hotspots) on the target, guiding the design of compounds with optimal interactions.
• Affinity-Based Techniques -Affinity-based techniques aim to identify lead compounds based on their specific interactions with the target molecule. This includes techniques like surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), and bio-layer interferometry (BLI). These methods measure the binding affinity, kinetics, and thermodynamics of interactions between molecules, providing valuable insights into the strength and nature of the binding. Affinity-based techniques are particularly useful for understanding the binding kinetics of lead compounds, helping prioritize candidates with optimal drug-like properties (Bergsdorf and Ottl, 2010).
Effective drug development depends on reliable data availability, where extensive information on biological sequences, chemical compounds, and pertinent discoveries from scientific publications are arranged in a methodical manner across several databases. Hundreds of biological databases appear every year in this rapidly expanding field (Song et al., 2009). The integration of database resources plays a transformative role, facilitating the exploration and optimization of potential lead compounds. These databases, encompassing a broad spectrum of chemical and biological information, are instrumental in enhancing the efficiency of lead identification processes. Among the essential resources, PubChem, ChEMBL, the Protein Data Bank (PDB), and structural databases such as Cambridge Structural Database (CSD) stand out for their invaluable contributions (Galperin, 2008).
• PubChem offers a comprehensive platform for accessing a wide array of chemical compounds and their associated biological activities. This database is a cornerstone for high throughput screening (HTS) initiatives, enabling the swift identification of compounds with potential therapeutic benefits.
• ChEMBL stands as a meticulously curated repository of bioactive molecules with detailed records of their effects on drug targets. It significantly aids in both HTS and affinity-based screening techniques by providing crucial insights into the biological efficacy of compounds, thus facilitating the selection of promising lead candidates.
• The Protein Data Bank (PDB) supplies extensive structural data on proteins and nucleic acids, which is pivotal for the success of fragment-based screening approaches. Access to detailed molecular structures allows for the precise engineering and refinement of lead compounds, ensuring their optimal interaction with target biomolecules.
• Cambridge Structural Database (CSD), specifically catering to the realm of crystallographic data, enriches the structural analysis further. It enables researchers to delve deep into the three-dimensional configurations of small molecules, supporting the rational design of lead compounds based on structural compatibility with biological targets.
• DrugBank stands out as a vital resource in the pharmaceutical industry. This comprises more than 3,243 experimental medications, 123 FDA-approved biotech pharmaceuticals, 71 nutraceuticals, and more than 1,350 small molecule drugs (Knox et al., 2023). DrugBank’s extensive dataset, which it has carefully selected, makes it easier to comprehend the properties, modes of action, and possible side effects of drugs. As a result, it can be applied to make well-informed decisions from target identification through clinical trials, among other phases of drug development.
• ChemDB greatly expands the variety of small molecules available to researchers by housing a collection of about 5 million commercially available chemicals. This large collection encourages originality and innovation in drug development efforts by offering a wide chemical area for investigation. ChemDB provides a wide range of chemicals, enabling researchers to investigate new chemical entities and find possible medicinal agents.
• Another notable database is ZINsC, which aims to offer a publicly accessible library of substances that may be purchased. ZINC supports well-informed decision-making in the early phases of drug development with its 20,089,615 3D structures annotated with physiologically important parameters including molecular weight, estimated Log P, and the number of rotatable bonds. This wealth of annotated information helps researchers filter and prioritize compounds based on their physicochemical properties, streamlining the process of hit identification.
Incorporating these database resources into the lead discovery phase not only streamlines the search for and optimization of lead compounds but also significantly propels forward the drug development process. By leveraging the extensive data these databases provide, researchers can tailor their screening strategies more effectively, prioritize compounds with higher therapeutic potentials, and unveil novel interactions for drug development. This integrative approach marks a pivotal advancement in the quest for new and efficacious therapeutic agents.
Once a lead compound is identified, medicinal chemists and researchers work to optimize its structure through a process known as lead optimization (Jorgensen, 2009). This involves modifying the substance to maximize desired effects while enhancing potency, selectivity, and other pharmacological qualities. Hydrogen bonding, hydrophobic interactions, van der Waals forces, and other non-bonded interactions are all included in its investigation and optimisation. The molecular recognition between the lead compound as well as the target site is shaped by these interactions, which consequently affects the drug candidate’s overall efficacy and specificity. They are crucial for forming precise and complementary interactions between the drug and the target, influencing the binding affinity and stabilizing the drug-target complex (Arya et al., 2021). In lead optimization, hydrophobic interactions are significant for enhancing the stability of the drug-target complex. By promoting the burial of hydrophobic molecules in the protein’s hydrophobic pockets, these interactions contribute to the overall binding energy, influencing the compound’s binding affinity and specificity. However, lead optimisation goes beyond simple structural adjustments. Let’s look at various approaches used in lead optimization (de Souza Neto et al., 2020).
• SAR (Structure-Activity Relationship) Studies—In Structure-Activity Relationship (SAR) studies, scientists systematically vary substituent on a molecule to observe how these changes influence its biological activity. This involves introducing different chemical groups or alterations to specific parts of the molecule. The goal is to understand the relationship between the structure of the compound and its activity against the target, such as a disease-related protein or enzyme. Once the impact of these substituent variations is understood, rational modifications are made based on the observed SAR. In other words, researchers make informed adjustments to the molecular structure, strategically choosing modifications that enhance the desired biological activity. This iterative process helps optimize the lead compound by fine-tuning its structure for improved potency, selectivity, and other relevant properties in drug development (Temml and Kutil, 2021).
• Pharmacokinetic Enhancements—In the realm of pharmacokinetic enhancements, lead optimization involves strategic modifications to address Absorption, Distribution, Metabolism, and Excretion (ADME) (Ferreira and Andricopulo, 2019). By strategically modifying the structure of a drug candidate, scientists can improve its Absorption (how well it enters the bloodstream), Distribution (how it travels throughout the body), Metabolism (how it is broken down), and Excretion (how it is eliminated). For example, improving a drug’s water solubility can enhance its absorption from the gut. Similarly, modifying functional groups susceptible to metabolism can slow down its breakdown and extend its action time. Optimizing ADME properties leads to improved bioavailability, enhanced drug efficacy, and potentially reduced dosing frequency, all contributing to a more effective and patient-friendly drug candidate (Kar and Leszczynski, 2020).
• Solubility Optimization—During lead optimisation, medicinal chemists strive to optimise the compound’s solubility by adding polar groups to its structure. This calculated addition improves the compound’s solubility by increasing its affinity for water. Enhanced solubility consequently makes the drug candidate easier to formulate, less difficult to administer, and more easily absorbed overall. This sophisticated strategy guarantees that the lead compound is efficiently delivered for the best possible clinical impact in addition to being therapeutically potent.
• Hybrid Molecule Design—Hybrid molecule design is a novel approach to lead optimisation in which scientists combine specific characteristics from different lead compounds. By combining the advantages of several leads, this novel strategy aims to produce hybrid molecules with enhanced activity and other desired characteristics. Medicinal chemists seek to improve the balance of efficacy, selectivity, and other pharmacological properties by carefully combining essential structural components. In the optimisation process, this calculated combination helps to produce more effective and adaptable drug candidates.
Computational approaches in lead discovery have become a game-changing paradigm in the dynamic field of drug development, revolutionising conventional approaches and speeding up the detection of promising lead compounds with therapeutic efficacy (Sliwoski et al., 2014). As compared to the traditional reliance only on empirical methods, this revolutionary shift utilises the computational power of sophisticated tools and algorithms. Here, we dive deep into the different computational techniques that have completely changed the lead discovery landscape.
• Quantitative Structure-Activity Relationship (QSAR) Modeling: QSAR models are pivotal for SAR studies as they quantitatively correlate chemical structures with biological activity or chemical reactivity, providing insights into how structural variations influence molecular behaviour. QSAR models can predict various ADME properties such as oral bioavailability, plasma protein binding, and potential toxicities, including hepatotoxicity and cardiotoxicity. ADMET predictions aid in the optimization of drug pharmacokinetics, ensuring adequate bioavailability, suitable half-life, and appropriate distribution to target tissues (Azzam, 2023). QSAR modelling ensures a multifaceted approach to predictive modelling, facilitating the identification and optimization of promising drug candidates. To evaluate the built models’ dependability and predictive capacity, they go through a rigorous validation process. The model’s good generalisation to novel compounds and situations is ensured by validation. Common criteria include using diverse test sets, employing cross-validation techniques, and assessing statistical metrics like sensitivity, specificity, and accuracy. Diverse test sets validate the model’s ability to generalize to different compounds and biological contexts. In order to forecast important pharmacokinetic characteristics of possible treatment candidates, validated models are utilised (Azzam, 2023). These characteristics include the way the drug enters the bloodstream, how it is distributed throughout the body, how it is broken down, how it is excreted, and how it is eliminated. Additionally, candidate compounds’ possible toxicological effects are predicted using computational models. The lead compounds are prioritised and optimised by researchers based on the predicted ADMET profiles (Ferreira and Andricopulo, 2019). The compounds that exhibit favourable pharmacokinetic i.e., Validated pharmacokinetic models offer insights into crucial aspects of a drug’s journey within the body. Predictions encompass drug entry into the bloodstream (absorption), distribution to various tissues, breakdown (metabolism), excretion from the body, and elimination. Absorption models estimate how efficiently a drug enters the bloodstream, influencing its bioavailability. Distribution predictions consider the drug’s movement between blood and tissues, impacting its concentration at the target site. Metabolism models evaluate the enzymatic transformation of the drug, affecting its activity and duration in the body. Excretion models assess the removal of the drug or its metabolites from the body, predominantly through urine or faeces. Elimination predictions encompass the overall clearance of the drug, reflecting the rate at which it is removed from the bloodstream. A holistic understanding of these pharmacokinetic characteristics aids in optimizing drug formulations, dosages, and administration schedules, contributing to the development of safer and more effective therapeutic interventions. ADMET modeling thus provides a comprehensive tool to anticipate a drug’s behavior within the body, informing critical decisions in the drug discovery and development process.
• Molecular Docking and Simulation: Molecular docking can be used to visualize how two or more molecules (or parts thereof) might be combined to form a hybrid molecule with desired properties. It helps in understanding the binding affinities and interaction mechanisms of potential hybrid molecules with their targets. Docking simulations can also support SAR studies by identifying critical interactions between the molecule and the target, guiding structural modifications to enhance activity or selectivity (Temml and Kutil, 2021). Advanced Docking Algorithms, such as AutoDock Vina, GOLD, and Glide, offer refined approaches for predicting how small molecules, like drugs, bind to a receptor (Figure 1) or enzyme. They employ various search algorithms (e.g., genetic algorithms, Monte Carlo simulation) to explore possible binding modes and use scoring functions to predict the binding affinity.
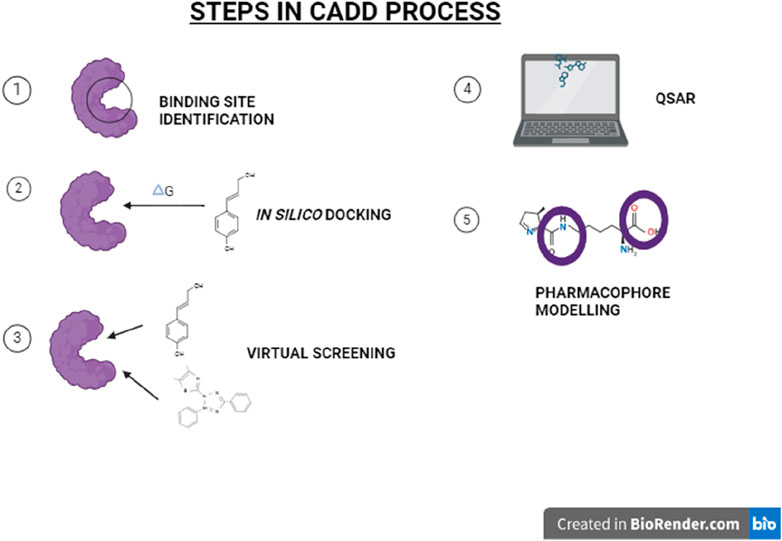
Figure 1. Major steps in CADD (Computer-Aided Drug Design).
These two steps pertain to scoring schemes and sampling techniques, respectively.
Posing: Predicting the possible binding conformations of the ligand in the receptor’s binding site. This involves exploring various orientations and conformations that the ligand can adopt within the binding pocket.
Scoring: Calculating the binding affinity of the ligand for each predicted pose. This involves estimating the energy of interaction between the ligand and the receptor.
Molecular docking relies heavily on scoring functions, which reduce the complex interactions between the ligand and the receptor to a single docking score, a numerical value. Researchers can compare various binding poses and determine the most likely binding mode using this score. There are three main scoring schemes, and each has advantages and disadvantages of its own (Torres et al., 2019).
Typical scoring functions that are used are:
Force field-based—In molecular docking, force field-based scoring functions are an effective tool that provide a thorough and physically based method of assessing the binding affinity between a ligand and its target receptor. By taking into account a variety of physical forces and interactions, these functions estimate the energy of the two molecule’s interaction and offer important insights into the underlying binding mechanisms. Hence, by using a force field, the strengths of the electrostatic interactions and vanderwaals between each atom in the two molecules in the complex are added up to estimate the affinities. It is also common to include the intra molecular energies (also known as strain energy) of the two binding partners (Stanzione et al., 2021). The totality of two energies—the energy of the receptor-ligand interaction and the internal ligand energy (such as steric strain brought on by binding)—is typically measured by molecular mechanics force fields.
Empirical scoring functions—Empirical scoring functions incorporate information from experimentally determined ligand-receptor complexes and learn from previous data sets. By serving as archives of known binding affinities, these training sets enable the scoring function to identify traits and attributes linked to robust binding interactions. Empirical scoring functions can therefore identify structural components and molecular motifs that aid in binding (Li et al., 2019).
Knowledge-based scoring functions—These calculate the binding affinity by utilising data on established protein-protein interactions. Through inference from this abundance of data, these scoring functions provide insights into the variables affecting binding affinity. This method allows for the incorporation of a variety of interaction patterns, it is particularly helpful when working with targets where there is an abundance of experimental data.
Combining these methods improves binding affinity predictions’ durability and advances our knowledge of molecular recognition events in the context of drug discovery. Researchers can more accurately and thoroughly navigate the complexities of ligand-receptor interactions when a variety of scoring functions are integrated, which is consistent with the interdisciplinary nature of computational biology (Kitchen et al., 2004).
• Virtual screening: In the process of finding new drugs, a computational method called virtual screening (VS) is used to select promising candidates from vast chemical libraries (McInnes, 2007). The goal of virtual screening is to forecast a ligand’s (molecule’s) ability to bind to a particular target protein, or receptor. The form and chemical characteristics of the ligand and the receptor among various factors, are considered when making this prediction. Suppose you are trying to find a key that will fit a certain lock. The target protein is represented by the lock, and you can select from a wide range of keys, which are possible therapeutic candidates. Before you ever try any of the keys in person, you can use virtual screening to evaluate each one digitally and ascertain which ones are the mostlikely to fit the lock (Srinivas Reddy et al., 2007).
• Molecular Dynamics Simulations: MD simulations allow for the observation of the dynamic behavior of atoms and molecules over time. By applying Newton’s laws of motion to atoms and molecules, MD simulations can predict the movement of each atom in a system, providing detailed insights into the structural dynamics, conformational changes, and interaction energies of biological molecules in a simulated physiological environment (Hollingsworth and Dror, 2018). MD simulations can provide the dynamic context within which binding processes occur, including the exploration of binding modes, conformational changes upon ligand binding, and the evaluation of entropic contributions to binding affinity.
The process’s initial step is choosing a biological target, which is frequently a protein connected to a disease pathway. The choice is guided by the understanding of the target’s role in the pathology. Molecular docking and virtual screening are used to find possible ligands from chemical libraries prior to MD. The selection of lead-like compounds is guided by these techniques, which predict binding affinities. A thorough preparation process is applied to the chosen ligand-protein complex, which includes the addition of water molecules to produce a solvated system. For an accurate description of intermolecular interactions, force fields are selected. In order to alleviate steric clashes and arrive at an energetically favourable simulation starting point, the system is subjected to energy minimization. After that, there are phases of equilibration that enable the system to stabilise by modifying the pressure and temperature. This stage resembles natural physiological settings (Hollingsworth and Dror, 2018). The production MD simulation starts and tracks the atoms trajectory over time. The dynamic interactions are captured by the simulation, providing information on stability, structural alterations, and ligand binding. Trajectory analysis, done after simulation, breaks down the complicated data produced. Important metrics that reveal the dynamics of the ligand-protein complex include RMSF (Root Mean Square Fluctuation) and RMSD (Root Mean Square Deviation) (Adelusi et al., 2022). The computational insights are validated through the integration of experimental data with MD predictions. The behaviour of lead compounds is better understood through this iterative process (Kairys et al., 2019; Salo-Ahen et al., 2020).
• Cheminformatics and Molecular Descriptors: The use of cheminformatics involves analyzing molecular descriptors and fingerprints to understand and predict the activity of chemical compounds, supporting SAR analysis. Molecular descriptors related to polarity, molecular volume, and hydrogen bonding can be analyzed to predict and optimize the solubility of compounds (Green and Segall, 2013). An open-source toolkit called RDKit provides researchers with many features, such ascalculating molecular descriptors and searching substructures. Each of these instruments work together to provide the effective manipulation and evaluation of chemical data that is important for lead detection (Begam and Kumar, 2012).
R, a statistical programming language and software environment, has applications in cheminformatics data processing. DataWarrior, an open-source program, aids in the display and interpretation of data in cheminformatics. These tools help researchers analyse the chemical and biological data necessary to drive discovery by enabling them to glean insightful information from vast volumes of data (Sander et al., 2015).
• Machine Learning and Data Mining: The foundation of ML in drug discovery rests on the comprehensive collection of diverse datasets. Molecular structures, biological activities, and relevant information converge to form datasets that reflect the intricacies of the drug development landscape. The quality and representativeness of these datasets become pivotal for subsequent ML endeavours (Lavecchia, 2015). Using mathematical descriptors, the unprocessed molecular structures are transformed into numerical representations. Important molecular details including structural characteristics, physicochemical attributes, and molecular fingerprints are captured by these descriptors. A quantitative framework is established in this step for further machine learning analysis (Lavecchia, 2015).
Not all descriptors are created equal. ML uses careful selection procedures to set out on a mission to find the optimal subset of variables (Carracedo-Reboredo et al., 2021). The most informative descriptors are found using feature selection methods, which can vary from statistical analyses to model-driven approaches. The goal of this step is to lessen the possibility ofoverfitting while increasing model efficiency. When ML models have a more refined set of descriptors, they start the training process. Using different algorithms, the model discovers complex relationships and patterns in thedata, iteratively adjusting parameters to reduce the discrepancy between expected and actual results (Cano et al., 2017). Machine learning-based drug discovery is an iterative procedure. Iterative refinement and optimisation of models are guided by insights into strengths and limitations discovered during validation. Scientists adjust variables, investigate substitute algorithms, and incorporate supplementary information to improve forecast precision and applicability. The integration of ML predictions with experimental data is crucial as it establishes a dynamic feedback loop. This integration serves as a roadmap for future improvement in addition to validating predictions in an actual setting. The interplay between experimental validation and computational insights improves the dependability of drug discovery procedures (Kolluri et al., 2022).
• AI-powered De Novo Design: Artificial Intelligence (AI), especially generative models like Generative Adversarial Networks (GANs) (Barigye et al., 2020) and Variational Autoencoders (VAEs), are used to design new molecules with desired properties from scratch. This technique helps in generating novel compounds that are optimized for potency, selectivity, and pharmacokinetic profiles (Gupta et al., 2021; Jiménez-Luna et al., 2021).
• Pharmacophore Modeling: Pharmacophore modeling identifies the essential features responsible for a drug’s biological activity. It can guide the design of hybrid molecules by combining the active features of different pharmacophores. It helps in understanding the structural requirements for activity, aiding in the rational design of molecules with enhanced efficacy.
Computational approaches in drug discovery, while powerful, are not without limitations. Quantitative Structure-Activity Relationship (QSAR) modeling faces challenges related to data quality and oversimplification of biological interactions. Molecular docking and simulations demand high computational resources and accuracy in input structures, with limitations in representing solvent effects. Molecular dynamics simulations are computationally expensive and sensitive to initial conditions. Cheminformatics and molecular descriptors depend on data quality and may struggle with subtle structural variations. Machine learning, though powerful, risks overfitting and may lack interpretability. Pharmacophore modeling is highly dependent on accurate input structures and may oversimplify ligand-receptor interactions. Integrating diverse data sources poses challenges due to heterogeneity, and over-reliance on computational predictions without experimental validation can lead to false positives.
The discovery of lead compounds has been transformed by computational methods, which provide creative answers to problems in drug development. This section looks at case studies that show how computational methods can be successfully combined to identify potential lead compounds.
Case study 1 -This case study explores the search for novel antimalarial drugs by looking into the molecular docking and antimalarial evaluation of novel N-(4-aminobenzoyl)-L-glutamic acid conjugated 1,3,5-triazine derivatives, exploring their potential as Pf-DHFR inhibitors. In order to do this, Molinspiration cheminformatics and Biovia Discovery Studio (DS) 2020 were used to conduct molecular modelling studies on 120 designed compounds. Moreover, the toxicity of the compounds that passed screening was assessed using the TOPKAT module. Using the Pf-DHFR-TS protein (PDB IDs 1J3I and 1J3K), protein–ligand docking was examined using the CDOCKER docking technology. A conventional and microwave-assisted nucleophilic substitution reaction was used to create these compounds, and a range of physicochemical and spectroscopic techniques were employed to characterise them. With an IC50 value of 9.54 μg mL−1, Df3 exhibited the highest antimalarial activity among the ten compounds tested against the chloroquine-resistant (Dd2) strain. Moreover, molecular dynamics (MD) simulation studies and the estimation of MM-PBSA-based free binding energies of docked complexes with 1J3I and 1J3K were conducted. The discovery of a novel class of Pf-DHFR inhibitors can be accomplished using this hybrid scaffold (Adhikari et al., 2022).
Case study 2—This study investigates the effects of 137 antimalarial and antihuman African trypanosomiasis compounds on three distinct in vitro assays (Trypanosoma brucei rhodesiense (T.b.r.), Plasmodium falciparum (P.f.), and cytotoxicity-L6 cells) using bis(2-aminoimidazolines), bisguanidinediphenyls, and polyamines. For the ligands under consideration, ΔTm values were also looked at when they were available. Based on structural similarity, eight DNA–ligand complexes and one DNA structure without a ligand were chosen from the Protein Data Bank (PDB). At the theoretical level of B3LYP/6-31G, geometry optimisation was performed for all the ligands under consideration. These molecules were docked at the minor groove of nine different DNA crystal structures using the AutoDock4 tool. We found that the majority of the ligands under consideration interact with the residues of DT20, DA6, DT8, and DT19. Molecular dynamics simulations, generalised born surface area calculations, and Poisson Boltzmann surface area calculations in molecular mechanics suggest that docked ligands exhibit little deviation in the minor groove of DNA until 10 ns simulation. Generally, the docked poses are stable. For T.b.r., P. f., C-L6, and ΔTm values, effective and statistically significant quantitative structure–activity relationship models were created. Every model that is generated undergoes both internal and external validation. Based on the developed models, we predicted a few ligands with significant IC50 values against P. f. The development of novel, highly effective antimalarial and antihuman African trypanosomal drugs may be aided by these findings (Gahtori et al., 2020).
Case study 3—Stepping into the field of possible therapeutic interventions against SARS-CoV-2, this case study investigates the carvedilol’s in silico molecular docking with the virus’s target proteins, illuminating the complex interactions that could offer hints for new treatment approaches. They first used computational methods to test the binding affinities of carvedilol with putative SARS-CoV-2 target proteins, such as RdRp, 3CL/Mpro, NSP13 helicase, NSP2, PLpro, RBD, ACE2, and the complex of RBD-ACE2, in order to explore the possible mechanisms of carvedilol against SARS-CoV-2 infection. Using the RdRp crystal structure (PDB ID:6XQB, resolution of 3.40 Å, Chain A), performed blind molecular docking with carvedilol and its metabolites in order to identify the best protein–ligand binding complex with the highest binding affinity. The optimal binding sites of candidate compounds with target proteins were screened using the molecular docking method.
The results of the in silico molecular docking demonstrated that the docking score between carvedilol and the viral protein RdRp was higher (−10.0 kcal/mol) than the scores of the other proteins that were tested, such as 3CL/Mpro (−7.2 kcal/mol), NSP13 Helicase (−7.4 kcal/mol), NSP2 (−7.5 kcal/mol), PLpro (−7.9 kcal/mol), RBD (−6.8 kcal/mol), ACE2 (−8.0 kcal/mol), and the complex of RBD-ACE2 (−8.1 kcal/mol). This implies that RdRp might be a prime candidate for carvedilol as a therapeutic target to treat SARS-CoV-2 infection (Zhang et al., 2023).
Integrative computational techniques have emerged as essential tools in the field of lead chemical identification for drug design, transforming the traditional drug development environment. The success of these techniques is due to their ability to seamlessly integrate disparate computational methodologies, moving the field toward more efficient and focused drug development. Virtual screening and molecular docking investigations stand out as essential approaches, allowing for the precise prediction of ligand-protein interactions. These technologies, which make use of software like as AutoDock and Glide, speed up the discovery of prospective lead compounds by realistically mimicking binding interactions (Macalino et al., 2015).
Quantitative structure-activity relationship (QSAR) modelling, which employs machine learning algorithms to forecast the biological activity of substances based on their chemical structures, is another crucial component of integrative techniques. This computer modelling allows for a logical and systematic study of chemical space, which aids in the discovery of molecules with superior pharmacological characteristics. Molecular dynamics simulations, aided by sophisticated software such as GROMACS and AMBER, offer significant understanding of the dynamic behaviour of biomolecular systems throughout time. These models are helpful for determining the stability and conformational changes of ligand-protein complexes, which is important for lead compound selection.
Cheminformatics and data mining are critical in the age of big data, when enormous chemical databases must be browsed effectively to uncover possible leads. Virtual screening based on ligands and structures, combined with machine learning algorithms, provides a comprehensive filtering process that allows compounds to be prioritized for experimental validation (Lin et al., 2020). Network pharmacology integrates computational biology, cheminformatics, and systems biology to create a more comprehensive approach to drug development. This technique finds lead compounds that affect many targets while maximizing therapeutic efficacy by taking into account the extensive network of molecular pathways.
Integrative computational techniques that focus on the discovery of smaller molecular fragments aid in fragment-based drug creation. MOE and GOLD are tools that aid in the methodical assembly of fragments into bigger, more powerful lead compounds. Artificial intelligence (AI) has lately taken centre stage in drug development, especially deep learning algorithms that forecast molecular actions with extraordinary precision. Integrating AI technologies, like as neural networks, with conventional methodologies improves the precision of forecasting bioactivity and toxicity profiles, driving lead optimization (Katsila et al., 2016). Furthermore, integrated computational techniques contribute greatly to medication repurposing efforts by comparing existing pharmacological databases against potential therapeutic targets. This method expedites the discovery of lead compounds with known safety profiles, therefore speeding up the medication development process. Hybrid techniques that combine diverse computational methodologies demonstrate the synergistic power of integrative tactics. Combining virtual screening with experimental high-throughput screening, for example, improves lead discovery and validation efficiency. Integrative computational techniques in personalized medicine consider individual differences in genetics, lifestyle, and illness. Tailoring lead chemical discoveries to unique patient profiles boosts the chance of therapy effectiveness and supports a paradigm shift toward more patient-centric therapeutic treatments (Ou-Yang et al., 2012).
Integrative computational techniques are at the forefront of transforming lead chemical discovery and drug design, providing significant prospects for speeding up the drug development pipeline. But along with these advantages come a number of restrictions and difficulties that must be carefully taken into account in order to use these approaches effectively. The availability of varied, accurate, and thorough biological and chemical data is critical to the success of integrative techniques. Incomplete or incorrect datasets might jeopardize the accuracy of forecasts, highlighting the importance of improved data gathering procedures and improved data-sharing norms within the scientific community. The computational complexity inherent in integrative techniques is a substantial impediment. As these methods amalgamate data from various sources, the algorithms and models become increasingly complicated. This complexity demands substantial computational resources, and as the number of integrated parameters and datasets grows, processing times can extend, potentially hindering the efficiency of the drug discovery pipeline. Overcoming this challenge necessitates ongoing advancements in computational infrastructure and algorithmic efficiency to handle the intricacies of integrative analyses.
The biological complexity adds another level of complication. Biological systems function on various scales, with complex interactions at the molecular, cellular, and organismal levels. To effectively discover lead drugs, integrative techniques must deal with the complexities of protein-protein interactions, pathway crosstalk, and cell-specific responses. Inadequate knowledge of these biological complexities might hamper prediction precision, underscoring the importance of continuing study to improve our understanding of biological processes. Navigating the chemical space is a daunting task. To efficiently explore the immensity of chemical space, which represents the infinite possible chemical compounds, creative algorithms and methodologies are required. To sift through the huge quantity of possible candidates and identify compounds with desired drug-like characteristics, advanced computational algorithms are required. This difficulty emphasises how new strategies must be developed in order to expedite the search for lead compounds with the best pharmacological profiles and to explore chemical space more quickly.
The future landscape of integrated computational techniques for lead compound identification and drug design is set for dramatic shifts. The combination of multi-omics data, artificial intelligence, and quantum computing ushers in a new era in drug discovery. Utilising advanced machine learning methods, specifically deep learning, can improve the accuracy of drug-target interaction and toxicity profile predictions. Quantum computing, with its extraordinary computational capability, has the potential to rapidly unravel the complexity of molecular interactions. As we progress toward customized treatment, these integrative techniques will adapt to individual variances by taking genetics, lifestyle, and environmental variables into account. Network pharmacology will take centre stage, revealing the interrelated mechanisms underlying illnesses. Virtual screening will grow more complex, allowing for the faster discovery of possible lead compounds, while in silico trials will allow for the simulation of various patient reactions prior to clinical trials. Explainable AI will address prediction transparency, while open science projects will promote collaborative research. Ethical concerns and appropriate AI practices will be of the utmost importance in guaranteeing the ethical use of patient data. Finally, education and training will equip researchers to traverse these changing approaches, moving the profession toward new, ethical, and successful drug development paradigms.
The use of computational approaches has transformed drug development, ushering in a new era of increased efficiency and more understanding. Deep learning, in particular, allows us to anticipate medication interactions and comprehend their processes with unparalleled precision. The impending arrival of quantum computing offers considerably more processing power. In the future, these approaches have the potential to catapult us into the realm of customized treatment, where individual differences are methodically examined. The interrelated nature of illnesses in the human body is being revealed via network pharmacology. Virtual screening and computer-based trials are emerging as ways to speed up drug development while reducing expenses. Nonetheless, ethical concerns about data exploitation must be addressed with vigour. It is critical to balance innovation with ethical data handling. In essence, the future of drug design is being charted by the dynamic interplay of computational prowess and biological insight, heralding a new era of targeted, efficient, and personalized therapeutics.
UN: Formal Analysis, Supervision, Writing–original draft, Writing–review and editing. VG: Supervision, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adelusi, T. I., Oyedele, A.-Q. K., Boyenle, I. D., Ogunlana, A. T., Adeyemi, R. O., Ukachi, C. D., et al. (2022). Molecular modeling in drug discovery. Inf. Med. Unlocked 29, 100880. doi:10.1016/j.imu.2022.100880
Adhikari, N., Choudhury, A. A. K., Shakya, A., Ghosh, S. K., Patgiri, S. J., Singh, U. P., et al. (2022). Molecular docking and antimalarial evaluation of novel N-(4-aminobenzoyl)-l-glutamic acid conjugated 1,3,5-triazine derivatives as Pf-DHFR inhibitors. 3 Biotech. 12, 347. doi:10.1007/s13205-022-03400-2
Arya, H., and Coumar, M. S. (2021). “Chapter 4 - lead identification and optimization,” in The design & development of novel drugs and vaccines. Editors T. K. Bhatt,, and S. Nimesh (New York: Academic Press), 31–63.
Azzam, K. A. L. (2023). SwissADME and pkCSM webservers predictors: an integrated online platform for accurate and comprehensive predictions for in silico ADME/T properties of artemisinin and its derivatives. Kompleksnoe Ispolʹzovanie Mineralʹnogo syrʹâ/Complex Use Mineral Resources/Mineraldik Shikisattardy Keshendi Paidalanu 325, 14–21. doi:10.31643/2023/6445.13
Barigye, S. J., García de la Vega, J. M., and Perez-Castillo, Y. (2020). Generative adversarial networks (GANs) based synthetic sampling for predictive modeling. Mol. Inf. 39, e2000086. doi:10.1002/minf.202000086
Begam, B. F., and Kumar, J. S. (2012). A study on cheminformatics and its applications on modern drug discovery. Procedia Eng. 38, 1264–1275. doi:10.1016/j.proeng.2012.06.156
Berdigaliyev, N., and Aljofan, M. (2020). An overview of drug discovery and development. Future Med. Chem. 12, 939–947. doi:10.4155/fmc-2019-0307
Bergsdorf, C., and Ottl, J. (2010). Affinity-based screening techniques: their impact and benefit to increase the number of high quality leads. Expert Opin. Drug Discov. 5, 1095–1107. doi:10.1517/17460441.2010.524641
Cano, G., Garcia-Rodriguez, J., Garcia-Garcia, A., Perez-Sanchez, H., Benediktsson, J. A., Thapa, A., et al. (2017). Automatic selection of molecular descriptors using random forest: application to drug discovery. Expert Syst. Appl. 72, 151–159. doi:10.1016/j.eswa.2016.12.008
Carracedo-Reboredo, P., Liñares-Blanco, J., Rodríguez-Fernández, N., Cedrón, F., Novoa, F. J., Carballal, A., et al. (2021). A review on machine learning approaches and trends in drug discovery. Comput. Struct. Biotechnol. J. 19, 4538–4558. doi:10.1016/j.csbj.2021.08.011
Cheng, K.-C., Korfmacher, W. A., White, R. E., and Njoroge, F. G. (2007). Lead optimization in discovery drug metabolism and pharmacokinetics/case study: the hepatitis C virus (HCV) protease inhibitor SCH 503034. Perspect. Med. Chem. 1, 1177391X0700100–9. doi:10.1177/1177391x0700100001
de Souza Neto, L. R., Moreira-Filho, J. T., Neves, B. J., Maidana, R. L. B. R., Guimarães, A. C. R., Furnham, N., et al. (2020). In silico strategies to support fragment-to-lead optimization in drug discovery. Front. Chem. 8, 93. doi:10.3389/fchem.2020.00093
Ferreira, L. L. G., and Andricopulo, A. D. (2019). ADMET modeling approaches in drug discovery. Drug Discov. Today 24, 1157–1165. doi:10.1016/j.drudis.2019.03.015
Gahtori, J., Pant, S., and Srivastava, H. K. (2020). Modeling antimalarial and antihuman African trypanosomiasis compounds: a ligand- and structure-based approaches. Mol. Divers 24, 1107–1124. doi:10.1007/s11030-019-10015-y
Galperin, M. Y. (2008). The molecular biology database collection: 2008 update. Nucleic Acids Res. 36, D2–D4. doi:10.1093/nar/gkm1037
Green, D. V. S., and Segall, M. (2013). “CHEMOINFORMATICS IN LEAD OPTIMIZATION,” in Chemoinformatics for drug discovery (China: Wiley), 149–178.
Gupta, R., Srivastava, D., Sahu, M., Tiwari, S., Ambasta, R. K., and Kumar, P. (2021). Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol. Divers 25, 1315–1360. doi:10.1007/s11030-021-10217-3
Hollingsworth, S. A., and Dror, R. O. (2018). Molecular dynamics simulation for all. Neuron 99, 1129–1143. doi:10.1016/j.neuron.2018.08.011
Jiménez-Luna, J., Grisoni, F., Weskamp, N., and Schneider, G. (2021). Artificial intelligence in drug discovery: recent advances and future perspectives. Expert Opin. Drug Discov. 16, 949–959. doi:10.1080/17460441.2021.1909567
Jorgensen, W. L. (2009). Efficient drug lead discovery and optimization. Acc. Chem. Res. 42, 724–733. doi:10.1021/ar800236t
Kairys, V., Baranauskiene, L., Kazlauskiene, M., Matulis, D., and Kazlauskas, E. (2019). Binding affinity in drug design: experimental and computational techniques. Expert Opin. Drug Discov. 14, 755–768. doi:10.1080/17460441.2019.1623202
Kar, S., and Leszczynski, J. (2020). Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin. Drug Discov. 15, 1473–1487. doi:10.1080/17460441.2020.1798926
Katsila, T., Spyroulias, G. A., Patrinos, G. P., and Matsoukas, M.-T. (2016). Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 14, 177–184. doi:10.1016/j.csbj.2016.04.004
Khan, S. R., Al Rijjal, D., Piro, A., and Wheeler, M. B. (2021). Integration of AI and traditional medicine in drug discovery. Drug Discov. Today 26, 982–992. doi:10.1016/j.drudis.2021.01.008
Kitchen, D. B., Decornez, H., Furr, J. R., and Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 3, 935–949. doi:10.1038/nrd1549
Knox, C., Wilson, M., Klinger, C. M., Franklin, M., Oler, E., Wilson, A., et al. (2023). DrugBank 6.0: the DrugBank knowledgebase for 2024. Nucleic Acids Res. 52, D1265–D1275. doi:10.1093/nar/gkad976
Kolluri, S., Lin, J., Liu, R., Zhang, Y., and Zhang, W. (2022). Machine learning and artificial intelligence in pharmaceutical research and development: a review. AAPS J. 24, 19. doi:10.1208/s12248-021-00644-3
Lavecchia, A. (2015). Machine-learning approaches in drug discovery: methods and applications. Drug Discov. Today 20, 318–331. doi:10.1016/j.drudis.2014.10.012
Leveridge, M., Chung, C.-W., Gross, J. W., Phelps, C. B., and Green, D. (2018). Integration of lead discovery tactics and the evolution of the lead discovery toolbox. SLAS Discov. 23, 881–897. doi:10.1177/2472555218778503
Li, J., Fu, A., and Zhang, L. (2019). An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdiscip. Sci. 11, 320–328. doi:10.1007/s12539-019-00327-w
Lin, X., Li, X., and Lin, X. (2020). A review on applications of computational methods in drug screening and design. Molecules 25, 1375. doi:10.3390/molecules25061375
Macalino, S. J. Y., Gosu, V., Hong, S., and Choi, S. (2015). Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 38, 1686–1701. doi:10.1007/s12272-015-0640-5
McInnes, C. (2007). Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 11, 494–502. doi:10.1016/j.cbpa.2007.08.033
Mohs, R. C., and Greig, N. H. (2017). Drug discovery and development: role of basic biological research. Alzheimer’s Dementia Transl. Res. Clin. Interventions 3, 651–657. doi:10.1016/j.trci.2017.10.005
Nicolaou, K. C. (2014). Advancing the drug discovery and development process. Angew. Chem. 126, 9280–9292. doi:10.1002/ange.201404761
Outeiral, C., Strahm, M., Shi, J., Morris, G. M., Benjamin, S. C., and Deane, C. M. (2021). The prospects of quantum computing in computational molecular biology. WIREs Comput. Mol. Sci. 11. doi:10.1002/wcms.1481
Ou-Yang, S., Lu, J., Kong, X., Liang, Z., Luo, C., and Jiang, H. (2012). Computational drug discovery. Acta Pharmacol. Sin. 33, 1131–1140. doi:10.1038/aps.2012.109
Prieto-Martínez, F. D., López-López, E., Eurídice Juárez-Mercado, K., and Medina-Franco, J. L. (2019). Computational drug design methods—current and future perspectives. In: In Silico drug design. Germany, Elsevier, 2019: 19–44.
Raviña, E. (2011). The evolution of drug discovery: from traditional medicines to modern drugs. Wiley-VCH.
Salo-Ahen, O. M. H., Alanko, I., Bhadane, R., Bonvin, A. M. J. J., Honorato, R. V., Hossain, S., et al. (2020). Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 9, 71. doi:10.3390/pr9010071
Sander, T., Freyss, J., von Korff, M., and Rufener, C. (2015). DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 55, 460–473. doi:10.1021/ci500588j
Singh, D. B., Pathak, R. K., and Rai, D. (2022). From traditional herbal medicine to rational drug discovery: strategies, challenges, and future perspectives. Rev. Bras. Farmacogn. 32, 147–159. doi:10.1007/s43450-022-00235-z
Sliwoski, G., Kothiwale, S., Meiler, J., and Lowe, E. W. (2014). Computational methods in drug discovery. Pharmacol. Rev. 66, 334–395. doi:10.1124/pr.112.007336
Song, C. M., Lim, S. J., and Tong, J. C. (2009). Recent advances in computer-aided drug design. Brief. Bioinform 10, 579–591. doi:10.1093/bib/bbp023
Srinivas Reddy, A., Priyadarshini Pati, S., Praveen Kumar, P., Pradeep, H. N., and Narahari Sastry, G. (2007). Virtual screening in drug discovery - a computational perspective. Curr. Protein Pept. Sci. 8, 329–351. doi:10.2174/138920307781369427
Stanzione, F., Giangreco, I., and Cole, J. C. (2021). Use of molecular docking computational tools in drug discovery.
Temml, V., and Kutil, Z. (2021). Structure-based molecular modeling in SAR analysis and lead optimization. Comput. Struct. Biotechnol. J. 19, 1431–1444. doi:10.1016/j.csbj.2021.02.018
Tiwari, A., and Singh, S. (2022). “Computational approaches in drug designing,” in Bioinformatics (Germany: Elsevier), 207–217.
Torres, P. H. M., Sodero, A. C. R., Jofily, P., and Silva-Jr, F. P. (2019). Key topics in molecular docking for drug design. Int. J. Mol. Sci. 20, 4574. doi:10.3390/ijms20184574
Wu, D., Sanghavi, M., Kollipara, S., Ahmed, T., Saini, A. K., and Heimbach, T. (2023). Physiologically based pharmacokinetics modeling in biopharmaceutics: case studies for establishing the bioequivalence safe space for innovator and generic drugs. Pharm. Res. 40, 337–357. doi:10.1007/s11095-022-03319-6
Zhang, C., Liu, J., Sui, Y., Liu, S., and Yang, M. (2023). In silico drug repurposing carvedilol and its metabolites against SARS-CoV-2 infection using molecular docking and molecular dynamic simulation approaches. Sci. Rep. 13, 21404. doi:10.1038/s41598-023-48398-6
Keywords: drug design, integrative computational approaches, ADMET, molecularmodelling, artificial intelligence in drug discovery
Citation: Naithani U and Guleria V (2024) Integrative computational approaches for discovery and evaluation of lead compound for drug design. Front. Drug Discov. 4:1362456. doi: 10.3389/fddsv.2024.1362456
Received: 28 December 2023; Accepted: 25 March 2024;
Published: 05 April 2024.
Edited by:
Rajesh Kumar Pathak, Chung-Ang University, Republic of KoreaReviewed by:
Jyoti Kant Choudhari, Maulana Azad National Institute of Technology, IndiaCopyright © 2024 Naithani and Guleria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vandana Guleria, dmFuZGFuYUBzaG9vbGluaXVuaXZlcnNpdHkuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.