 Stéphane Giraud
Stéphane Giraud
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Drug Discov., 25 January 2024
Sec. Technologies and Strategies to Enable Drug Discovery
Volume 4 - 2024 | https://doi.org/10.3389/fddsv.2024.1342866
This article is part of the Research TopicDrug Discovery and Development Explained: Introductory Notes for the General PublicView all 11 articles
In the quest for the discovery of new therapies, the identification of the initial active molecules is a major challenge. Although significant progress in chemistry and biology has been made in recent years, the process remains difficult. In this mini-review, we will explain the major approaches and experimental methods that can be used to identify these molecules. Two main approaches are described, target-based and phenotypic-based and a focus is made on some high throughput technologies and biophysical methods.
Despite scientific and methodological advances made over the last 20 years, identifying and developing new therapies remain long and costly processes (Wouters et al., 2020). Indeed, the marketing and distribution to the general public of small molecules, antibodies or therapeutic proteins take several years; for instance, a small chemical molecule is commercially available after 12–15 years. As the duration of certain developmental phases, such as clinical trials, can hardly be reduced, a great effort has been made to spend less time on the first steps of the process (pre-discovery, drug discovery and preclinical). New biophysical, biochemical, biological and in silico technologies have emerged to accelerate the discovery stage.
In this review we will focus on small organic compounds; vaccines, cellular therapy, therapeutic antibodies and other biologics will not be addressed. We will particularly discuss the main methods for identifying the first bioactive small molecules, also known as “hits”. The definition of a hit can vary across the scientific community, but in this article a hit will be considered as a molecule whose activity is confirmed in one or several primary biological and/or biophysical assays. These hits will then be optimized through an iterative cycle involving biology, biophysics, chemistry and AI-based methods (Vemula et al., 2023) to obtain a new drug, displaying a high efficacy and a low or even no toxicity.
The identification of new small organic molecules-based therapies requires a set of molecules to be tested and a robust validated assay. These molecules can be obtained in different ways, which will not be detailed herein, however here are some commons sources:
(1) Natural products: nature has always been a source of valuable bioactive molecules. Natural products and their derived molecules have been used since ancient times to treat diseases. These molecules are found in plants, microbes, aquatic organisms, animals, fungi and insects. Many drugs, such as antibiotics and anticancer agents, were originally derived from natural sources. (Newman and Cragg, 2020; Naeem et al., 2022).
(2) Synthetic compounds: pharmaceutical companies and research institutions often maintain libraries of synthetic chemical compounds. Combinatorial and parallel chemistry have been used to generate thousands of molecules by systematically varying chemical structures. Today more rational approaches are used to design and synthesized specific molecules intended to inhibit particular targets like kinases, ion channels, GPCR or biological mechanisms like protein-protein interactions or DNA methylation. Artificial intelligence and machine learning algorithms are also used to identify potential drug candidates by analyzing huge datasets and predicting the biological activity of molecules. (Yu and MacKerell, 2017).
(3) Repurposing of existing drugs: sometimes existing drugs that were developed for one indication can be repurposed for treating different diseases. This approach emerged in the early 1990s and has been proven to be a viable alternative to the identification of new drugs. (Gns et al., 2019).
(4) Drug design: this rational methodology consists in designing potential active compounds, i.e., compounds that bind to a particular target, based on structural data of the target or based on data of the ligand. Many computational techniques have recently emerged that help researchers identify innovative compounds. (Hoffer et al., 2018; Singh et al., 2020).
To identify new hits, a screening strategy (or method) must be adopted. A set of specific assays must be carried out to identify and optimize potential drugs that then become drug candidates for clinical trials. Two major kinds of approaches exist (Figure 1), namely, those that require the identification of a target and validation of the relationship between that target and a particular disease–called target-based approaches–and those that work in a target agnostic fashion known as phenotypic approaches. The latter consist in observing the effect(s) of a new potential therapy at the level of cells or whole organisms. Phenotypic approaches require an experimental model as close as possible of the pathology and symptoms observed in human.
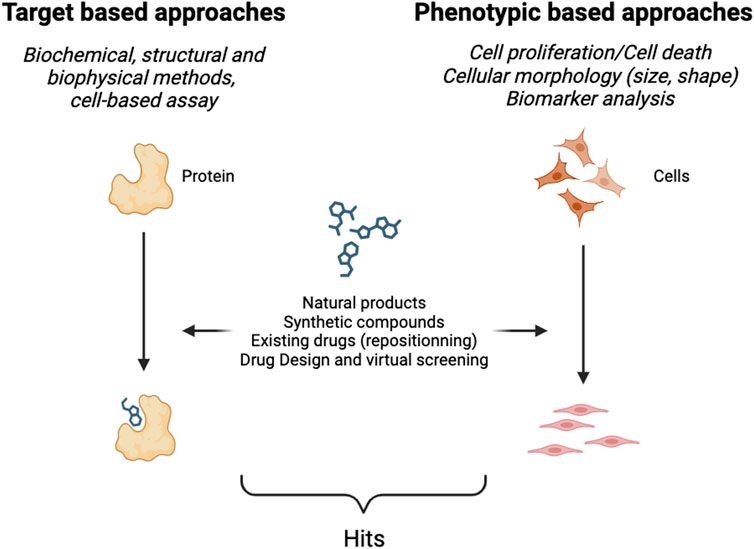
FIGURE 1. Schematic representation of hit identification.
Target-based screening relies primarily on the identification of a disease-relevant target; typically, for example, proteins and nucleic acids. This type of screening can be performed in vitro using biochemical and biophysical methods, or in cellulo using cellular models to assess the activity of the compound towards the target in a cellular context. The assays developed to perform the screening are designed either to measure the interaction between a potential drug and the chosen target, or the ability of a drug to modulate a cellular function through its interaction with the target. The aim of these methods is to develop an assay that produces a detectable signal in order to visualize, primarily through the emission of light (in the visible or fluorescence spectra), the activity of a given compound towards the target. The development of an assay is not trivial and can take time as it must be sensitive, reliable and reproducible enough to provide comparative results when thousands of compounds are screened.
High Throughput Screening consists in the screening of large libraries of compounds (from thousands to sometime millions) in order to identify hits (Blay et al., 2020). This approach is based on the automation of biological and biophysical assays that can be miniaturized and must be a highly sensitive method to identify active compounds. At its inception, HTS screening campaigns were carried out using 96-well plates, now screens are conducted in 384-well or 1,536-well plates of the same dimension as their predecessor. This means that the number of reactions that can be performed in parallel has significantly increased over the last 2 decades, and that the time needed to screen large libraries has thus been considerably reduced. However, it still takes several weeks to months to complete a screen, and the typical hit rate is around 1%. The success of an HTS screening largely depends on the design of the assay and the achieved statistical performance. The robustness of the assay can be assessed using a statistical parameter like the Z’ score (Zhang, 1999). This parameter has been widely used to determine the suitability of an HTS assay but other parameters like the distribution of standard deviation have been described (Hanley, 2019)
HTS was the gold standard in the 1990s and gave good results. Today it is used alongside other approaches, like structure-based drug design or other computational techniques (Macarrón and Hertzberg, 2011). In order to be robust and not too expensive, an HTS assay should not comprise too many steps. To that effect, several homogeneous-phase assays have been developed, in which all reagents are mixed and no washing step is required. Among the homogeneous-phase techniques, HTRF (Homogeneous Time Resolved Fluorescence) is widely used (Gotoh et al., 2010; Shin et al., 2023). This technique is based on the transfer of energy between two fluorophores, a donor and an acceptor. This transfer occurs when both fluorophores are in close proximity, resulting in a measurable fluorescent signal. This kind of methodology is used for a number of applications, such as the detection of protein-protein interactions (each fluorophore being linked to one of the proteins), enzymatic activities or receptor binding. Another technology named ALPHAscreen (Amplified Luminescent Proximity Homogenous Assay) is also based on a signal obtained when two entities are in close proximity or linked. In this assay the donor and the acceptor are microbeads that are brought together by the molecular interaction of the binding partners that are linked to these beads. Fluorescence polarization which measures the rate of rotation of a fluorescent-labeled ligand is also a powerful method to identify hits and to obtain information during the optimization process (Lea and Simeonov, 2011; Hua et al., 2023).
Cell-based assays also played a crucial role in the identification and validation of bioactive compounds serving as versatile tools to assess cellular responses to various stimuli and compounds. These assays use living cells to investigate drug efficacy on cell viability, proliferation or specific cellular functions. These assays differ from phenotypic screening in terms of complexity of the readouts. While phenotypic assays generally involve the simultaneous analysis of multiple cellular parameters (shape, size, surface, biomarkers of specific pathways), cell-based assays focus on a single parameter. One notable example is the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay, where the reduction of a yellow tetrazolium salt to purple formazan product by metabolically active cells is measured, providing an indirect assessment of cell viability.
Specific target-based screens like protein-protein interactions can also be conducted using cell-based assays. For example, bimolecular complementation assays, also known as PCA (Protein-fragments Complementation assays) have been developed in the last decade (Kodama and Hu, 2012; Sharma and Anand, 2016; Bellón-Echeverría et al., 2018). In these assays, a fluorescent reporter protein is divided into two non-functional fragments. Each fragment is then fused to two proteins of interest. When these proteins are in close proximity, the split fragments of the reporter protein reassemble, restoring its functionality and resulting in a measurable signal. One of the limitations of the system is that complex reassembly is irreversible; hence, more dynamic systems have recently been developed (Tebo and Gautier, 2019).
Protein-protein interactions within cells can also be monitored, for instance using energy transfer as in FRET (Fluorescence Resonance Energy Transfer) (Song et al., 2011) or BRET (Bioluminescence Resonance Energy Transfer) (Machleidt et al., 2015; Cho and Dalby, 2021). Both methodologies rely on the transfer of energy between a donor and an acceptor, one being a bioluminescent protein in the case of BRET. The major advantage of cell-based assays is that it addresses the activity of a candidate compound in a specific cellular context. If an effect is detected this means that the compound is able to cross the cellular membrane and to reach a target. Additionally, if cells tolerate the compound, this is a first indication that the compound is not toxic for the cell.
Structural an biophysical methods are now systematically integrated into the hit identification and validation process, as well as in the subsequent steps of candidate molecule optimization. Their use for hit identification dates back to the 1990s, via Nuclear Magnetic Resonance (NMR) and X-ray crystallography coupled with computational analyses. Since then, several techniques have been developed or adapted particularly in terms of throughput, and they have become complementary to biochemical or cellular biology methods fostering a positive selection of the compounds. These technologies have provided scientists with important information for the development of compounds, such as evidence that the compound binds to the target, the kinetics of the binding, the affinity (measurement of the strength) of the binding, or thermodynamic parameters. In addition, these techniques can also help to identify the binding mode and the binding pocket of molecules. This information is essential for developing a molecule with the right mode of action. Indeed, as an example an enzyme can be inhibited by a molecule that binds in the active site or at a distal site (allosteric inhibition).
A wide range of techniques is now available and their use in drug discovery has been reviewed elsewhere (Renaud et al., 2016; Holdgate and Bergsdorf, 2021). A majority of these methods focus on analyzing isolated targets, which implies producing and purifying the target, albeit more recent methods can now be performed using cellular extracts. Some of them require labeling of the target with a fluorochrome or use a native unmodified target, the ultimate goal for all these techniques being to demonstrate that a candidate compound binds to the target.
Among the most frequently employed methods, we can mention:
(i) Calorimetry techniques (like Isothermal Titration Calorimetry (ITC) or Differential Scanning Calorimetry (DSC) provides thermodynamic data about the protein-ligand complex. For example, ITC measures the consumption or generation of heat when a compound binds to the protein (Falconer et al., 2021). (ii) Temperature-related intensity change that measures the modification of fluorescence intensity of a fluorochrome when the target and the compound are bound (Jerabek-Willemsen et al., 2014). (iii) NMR that relies on the behavior of certain atomic nuclei when placed in a strong magnetic field and exposed to a specific frequency of radiofrequency radiation (Shimada et al., 2019). (iv) Surface plasmon resonance detects changes in the refractive index near a metal surface. (v) Mass spectrometry that determines the mass-to-charge ratio of ions (Gavriilidou et al., 2022) (vi) X-ray diffraction that measures the diffraction angles and changes of intensities of X-rays can be applicable to crystals (X-ray crystallography) (Maveyraud and Mourey, 2020) but also to proteins in solution like enzymes (Byer et al., 2023) (vii) cryo-electron microscopy (cryo-EM) is a powerful technique used for imaging macromolecules at near atomic resolution. This technique is now complementary to NMR or X-ray diffraction in small molecule drug design ((Vénien-Bryan et al., 2017; Renaud et al., 2018)
These techniques all rely on high-standard equipment, and depending on the method employed the throughput can vary from a few compounds a day to a few compounds a week or month. These methodologies are part of the drug discovery process from the early phases to the selection of the preclinical candidate therapy.
Historically, the discovery of medicines relied on phenotypic approaches, however with the advent of genomics in the 1980s and the sequencing of the human genome in 2001, these approaches were neglected. Nevertheless, over the last decade there has been renewed interest in phenotypic approaches, as they are valuable at identifying novel therapeutic agents (Ege et al., 2021; Vincent et al., 2022). One of the advantages of phenotypic assays is that they explore a broader spectrum of biological responses than target-based approaches, elucidating complex biological pathways and uncovering unforeseen interactions, thus offering a holistic perspective of the potential effect of a new agent. Technological advances have played a pivotal role in boosting phenotypic screening. Assay miniaturization, development of high-throughput screening platforms (gathering automated equipment to rapidly test a huge quantity of samples rapidly), automated imaging (microscopy technology) and data analysis systems have opened new avenues to perform phenotypic analyses. One such technique is fluorescence imaging, which enables scientists to visualize and quantify various biological processes at the cellular and subcellular levels. This technique uses fluorescent probes, markers or genetically-encoded fluorescent proteins to highlight specific cellular structures, proteins, or functional activities. High content screening (a combination of powerful imaging tools and biochemical/molecular biology assays) captures dynamic cellular events in real-time; for instance, the monitoring of processes like cell migration, proliferation, modification of the cellular morphology (shape, size…) and cell death. Additionally, it allows the concomitant assessment of multiparametric data including protein localization, analysis of subcellular organelles or responses to external stimuli. Numerous approved therapies for cardiovascular diseases, viral infections, neurodegenerative disorders and cancers originate from phenotypic screening (Blay et al., 2020). Despite, many advantages that have led to the identification of innovative therapies that could not have been identified without this approach, phenotypic screening has one major drawback–this global approach makes it difficult to decipher the molecular mechanisms of action of a drug and to identify its target(s), both necessary to optimize the potency of a drug and for its further development in the clinic.
Drug discovery is a long and challenging process which involves various fields of expertise. A crucial step of the development of a new small organic-based therapy is the identification of hits. Target-based, phenotypic-based and biophysical methods can be employed throughout the process to identify these hits and to participate in the optimization and development process of a new promising therapy. Despite significant advances in scientific and technological methods, the identification and development of new therapies remain arduous and resource-intensive. The journey from identifying bioactive molecules to developing a marketable drug involves an intricate interplay of biology, biophysics, chemistry and cutting-edge technologies. As science continues to advance, the hope is to streamline this process, making drug discovery more efficient and accessible for the benefit of patients.
SG: Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The author received funding from the Synergy Lyon Cancer foundation, the Centre Léon Bérard, the Lyrican+ (grant INCa-DGOS-INSERM-ITMO cancer_18003) and the Institut Convergence Plascan (Grant Number ANR-17-CONV-0002).
The author thanks reviewers for their comments and suggestions on the manuscript and Brigitte Manship for her help in proofreading the manuscript. Figure 1 was Created with BioRender.com.
SG is co-founder of TheraPPI Bioscience, a spin-off of his host academic institutions.
The handling editor BOV declared a shared affiliation with the author SG at the time of the review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bellón-Echeverría, I., Carralot, J.-P., Del Rosario, A. A., Kueng, S., Mauser, H., Schmid, G., et al. (2018). MultiBacMam Bimolecular Fluorescence Complementation (BiFC) tool-kit identifies new small-molecule inhibitors of the CDK5-p25 protein-protein interaction (PPI). Sci. Rep. 8, 5083. doi:10.1038/s41598-018-23516-x
Blay, V., Tolani, B., Ho, S. P., and Arkin, M. R. (2020). High-Throughput Screening: today’s biochemical and cell-based approaches. Drug Discov. Today 25, 1807–1821. doi:10.1016/j.drudis.2020.07.024
Byer, A. S., Pei, X., Patterson, M. G., and Ando, N. (2023). Small-angle X-ray scattering studies of enzymes. Curr. Opin. Chem. Biol. 72, 102232. doi:10.1016/j.cbpa.2022.102232
Cho, E. J., and Dalby, K. N. (2021). Luminescence energy transfer–based screening and target engagement approaches for chemical biology and drug discovery. SLAS Discov. 26, 984–994. doi:10.1177/24725552211036056
Ege, N., Bouguenina, H., Tatari, M., and Chopra, R. (2021). Phenotypic screening with target identification and validation in the discovery and development of E3 ligase modulators. Cell Chem. Biol. 28, 283–299. doi:10.1016/j.chembiol.2021.02.011
Falconer, R. J., Schuur, B., and Mittermaier, A. K. (2021). Applications of isothermal titration calorimetry in pure and applied research from 2016 to 2020. J. Mol. Recognit. 34, e2901. doi:10.1002/jmr.2901
Gavriilidou, A. F. M., Sokratous, K., Yen, H.-Y., and De Colibus, L. (2022). High-throughput native mass spectrometry screening in drug discovery. Front. Mol. Biosci. 9, 837901. doi:10.3389/fmolb.2022.837901
Gns, H. S., Gr, S., Murahari, M., and Krishnamurthy, M. (2019). An update on Drug Repurposing: Re-written saga of the drug’s fate. Biomed. Pharmacother. 110, 700–716. doi:10.1016/j.biopha.2018.11.127
Gotoh, Y., Nagata, H., Kase, H., Shimonishi, M., and Ido, M. (2010). A homogeneous time-resolved fluorescence-based high-throughput screening system for discovery of inhibitors of IKKbeta-NEMO interaction. Anal. Biochem. 405, 19–27. doi:10.1016/j.ab.2010.05.028
Hanley, Q. S. (2019). The distribution of standard deviations applied to high throughput screening. Sci. Rep. 9, 1268. doi:10.1038/s41598-018-36722-4
Hoffer, L., Muller, C., Roche, P., and Morelli, X. (2018). Chemistry-driven hit-to-lead optimization guided by structure-based approaches. Mol. Inf. 37, 1800059. doi:10.1002/minf.201800059
Holdgate, G. A., and Bergsdorf, C. (2021). Applications of biophysics in early drug discovery. SLAS Discov. 26, 945–946. doi:10.1177/24725552211035123
Hua, L., Wang, D., Wang, K., Wang, Y., Gu, J., Zhang, Q., et al. (2023). Design of tracers in fluorescence polarization assay for extensive application in small molecule drug discovery. J. Med. Chem. 66, 10934–10958. doi:10.1021/acs.jmedchem.3c00881
Jerabek-Willemsen, M., André, T., Wanner, R., Roth, H. M., Duhr, S., Baaske, P., et al. (2014). MicroScale thermophoresis: interaction analysis and beyond. J. Mol. Struct. 1077, 101–113. doi:10.1016/j.molstruc.2014.03.009
Kodama, Y., and Hu, C.-D. (2012). Bimolecular fluorescence complementation (BiFC): a 5-year update and future perspectives. BioTechniques 53, 285–298. doi:10.2144/000113943
Lea, W. A., and Simeonov, A. (2011). Fluorescence polarization assays in small molecule screening. Expert Opin. Drug Discov. 6, 17–32. doi:10.1517/17460441.2011.537322
Macarrón, R., and Hertzberg, R. P. (2011). Design and implementation of high throughput screening assays. Mol. Biotechnol. 47, 270–285. doi:10.1007/s12033-010-9335-9
Machleidt, T., Woodroofe, C. C., Schwinn, M. K., Méndez, J., Robers, M. B., Zimmerman, K., et al. (2015). NanoBRET—a novel BRET platform for the analysis of protein–protein interactions. ACS Chem. Biol. 10, 1797–1804. doi:10.1021/acschembio.5b00143
Maveyraud, L., and Mourey, L. (2020). Protein X-ray crystallography and drug discovery. Molecules 25, 1030. doi:10.3390/molecules25051030
Naeem, A., Hu, P., Yang, M., Zhang, J., Liu, Y., Zhu, W., et al. (2022). Natural products as anticancer agents: current status and future perspectives. Molecules 27, 8367. doi:10.3390/molecules27238367
Newman, D. J., and Cragg, G. M. (2020). Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 83, 770–803. doi:10.1021/acs.jnatprod.9b01285
Renaud, J.-P., Chari, A., Ciferri, C., Liu, W., Rémigy, H.-W., Stark, H., et al. (2018). Cryo-EM in drug discovery: achievements, limitations and prospects. Nat. Rev. Drug Discov. 17, 471–492. doi:10.1038/nrd.2018.77
Renaud, J.-P., Chung, C., Danielson, U. H., Egner, U., Hennig, M., Hubbard, R. E., et al. (2016). Biophysics in drug discovery: impact, challenges and opportunities. Nat. Rev. Drug Discov. 15, 679–698. doi:10.1038/nrd.2016.123
Sharma, H., and Anand, B. (2016). Fluorescence bimolecular complementation enables facile detection of ribosome assembly defects in Escherichia coli. RNA Biol. 13, 872–882. doi:10.1080/15476286.2016.1207037
Shimada, I., Ueda, T., Kofuku, Y., Eddy, M. T., and Wüthrich, K. (2019). GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 18, 59–82. doi:10.1038/nrd.2018.180
Shin, Y. H., Kim, D.-E., Yu, K. L., Park, C. M., Kim, H. G., Kim, K.-C., et al. (2023). A novel time-resolved fluorescence resonance energy transfer assay for the discovery of small-molecule inhibitors of HIV-1 tat-regulated transcription. Int. J. Mol. Sci. 24, 9139. doi:10.3390/ijms24119139
Singh, N., Chaput, L., and Villoutreix, B. O. (2020). Virtual screening web servers: designing chemical probes and drug candidates in the cyberspace. Brief. Bioinform. 22, 1790–1818. doi:10.1093/bib/bbaa034
Song, Y., Madahar, V., and Liao, J. (2011). Development of FRET assay into quantitative and high-throughput screening technology platforms for protein–protein interactions. Ann. Biomed. Eng. 39, 1224–1234. doi:10.1007/s10439-010-0225-x
Tebo, A. G., and Gautier, A. (2019). A split fluorescent reporter with rapid and reversible complementation. Nat. Commun. 10, 2822. doi:10.1038/s41467-019-10855-0
Vemula, D., Jayasurya, P., Sushmitha, V., Kumar, Y. N., and Bhandari, V. (2023). CADD, AI and ML in drug discovery: a comprehensive review. Eur. J. Pharm. Sci. 181, 106324. doi:10.1016/j.ejps.2022.106324
Vénien-Bryan, C., Li, Z., Vuillard, L., and Boutin, J. A. (2017). Cryo-electron microscopy and X-ray crystallography: complementary approaches to structural biology and drug discovery. Acta Crystallogr. Sect. F. Struct. Biol. Commun. 73, 174–183. doi:10.1107/S2053230X17003740
Vincent, F., Nueda, A., Lee, J., Schenone, M., Prunotto, M., and Mercola, M. (2022). Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat. Rev. Drug Discov. 21, 899–914. doi:10.1038/s41573-022-00472-w
Wouters, O. J., McKee, M., and Luyten, J. (2020). Estimated research and development investment needed to bring a new medicine to market, 2009-2018. JAMA 323, 844–853. doi:10.1001/jama.2020.1166
Yu, W., and MacKerell, A. D. (2017). “Computer-aided drug design methods,” in Antibiotics, methods in molecular biology. Editor P. Sass (New York, NY: Springer New York), 85–106. doi:10.1007/978-1-4939-6634-9_5
Keywords: drug discovery, small organic compounds, screening methods, HTS, biophysics
Citation: Giraud S (2024) Identification of first active compounds in drug discovery. how to proceed?. Front. Drug Discov. 4:1342866. doi: 10.3389/fddsv.2024.1342866
Received: 22 November 2023; Accepted: 05 January 2024;
Published: 25 January 2024.
Edited by:
Bruno Villoutreix, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Antoine Gedeon, Sorbonne Université, FranceCopyright © 2024 Giraud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stéphane Giraud, c3RlcGhhbmUuZ2lyYXVkQGx5b24udW5pY2FuY2VyLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.