
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Drug Discov. , 05 March 2024
Sec. In silico Methods and Artificial Intelligence for Drug Discovery
Volume 4 - 2024 | https://doi.org/10.3389/fddsv.2024.1339697
This article is part of the Research Topic Drug Discovery and Development Explained: Introductory Notes for the General Public View all 11 articles
 Astrid Musnier1
Astrid Musnier1 Christophe Dumet1Saheli Mitra1Adrien Verdier1Raouf Keskes1Augustin Chassine1Yann Jullian1Mélanie Cortes1Yannick Corde1Zakaria Omahdi1Vincent Puard1Thomas Bourquard1†
Christophe Dumet1Saheli Mitra1Adrien Verdier1Raouf Keskes1Augustin Chassine1Yann Jullian1Mélanie Cortes1Yannick Corde1Zakaria Omahdi1Vincent Puard1Thomas Bourquard1† Anne Poupon1,2*†
Anne Poupon1,2*†As in all sectors of science and industry, artificial intelligence (AI) is meant to have a high impact in the discovery of antibodies in the coming years. Antibody discovery was traditionally conducted through a succession of experimental steps: animal immunization, screening of relevant clones, in vitro testing, affinity maturation, in vivo testing in animal models, then different steps of humanization and maturation generating the candidate that will be tested in clinical trials. This scheme suffers from different flaws, rendering the whole process very risky, with an attrition rate over 95%. The rise of in silico methods, among which AI, has been gradually proven to reliably guide different experimental steps with more robust processes. They are now capable of covering the whole discovery process. Amongst the players in this new field, the company MAbSilico proposes an in silico pipeline allowing to design antibody sequences in a few days, already humanized and optimized for affinity and developability, considerably de-risking and accelerating the discovery process.
Antibodies have long been irreplaceable tools for research. They have more recently emerged as powerful drugs, allowing considerably higher specificity than traditional chemicals, and offering new treatment options in a growing number of pathologies (Lu et al., 2020). Thanks to their half-lives, antibody drugs also have long-lasting effects as compared to small-molecules, rendering them more adapted to chronic pathologies. Many new formats derived from antibodies have been designed allowing to exploit their exquisite specificity (Vega et al., 2022). Antibody-drug conjugates can be used to bring chemical drugs to the precise location where their action is to take place, which is particularly useful for chemotherapies involving very toxic molecules (Jin et al., 2022). Using an antibody to bring the chemotherapy to the tumor allows to increase the doses, rendering cancer therapies more efficient, and decreasing the side-effects. T-cells expressing chimeric antigen receptors (CAR-T cells), recognize their target cells through antibody-like receptors (Mehrabadi et al., 2022), for example, binding to markers of cancer, and destroy them. Bispecifics recognize two different targets and can, for example, activate immune cells in the micro-tumoral environment (Bejarano et al., 2021).
As currently performed, antibody discovery starts by immunizing animals: the target is injected into an animal (mostly mice or rabbits), together with an immune booster. The immune system of the animal reacts by producing antibodies against this molecule. The second step is the screening, which consists in finding, amongst the antibodies of the animal, those binding to the target of interest. Successive rounds of selections, mainly based on refined binding assays (in vitro cross-species binding, paralogs binding) and both in vitro and in vivo functional assays, are applied to downsize the number of initial hit molecules and to identify the final “leads”, resulting in the well-known funnel-shaped process of antibody discovery (Hoover et al., 2021). These successive elimination steps are highly empirical, and depend more on the scalability of wet-lab techniques than on the importance of the information provided. Epitope mapping is a very good example of that. Antibody/antigen complex is highly useful for further engineering, and mandatory for IP protection, but requires time-consuming and low throughput methods, like the gold-standards X-ray crystallography or NMR. As a result, epitope mapping is carried out very late in the cycle, as a check prior patenting, whereas it would have made much sense at the very beginning of the project, as a decision-making element (Bauer et al., 2023).
After these first selections, only a few leads actually display the suitable physico-chemical properties to be qualified as candidate molecules that could be moved to preclinical and clinical trials, and ultimately become therapeutics. Maturation steps are hence often engaged to optimize the affinity and the developability properties (low immunogenicity, solubility at high concentrations, manufacturability at large scale). Sequences are herein modified, meaning that the number of molecules to test is increased back, and that the final molecules are, strictly speaking, different from the originally characterized ones. A new round of validations aiming at requalifying the matured molecule is hence necessary, hoping that the biological properties are retained along the process.
Artificial intelligence methods are gradually replacing all these experimental steps, lowering the attrition rate and shortening the whole process. This technological transition happened a decade earlier for small chemical molecules, but the complexity of biologics prevented any transfer of technology from one area to the other and specific methods had to be designed. Here are described some of the AI-based innovations dedicated at antibody discovery.
Artificial intelligence, theorized by Alan Turing in the 50 s, was born with the description of genetic algorithms by J.H. Holland in 1975 (Holland, 1992). However, computers were not powerful enough for these methods to be useful, and the real takeoff happened 15 years later with the publication of David Golberg Genetic Algorithms in Search, Optimization, and Machine Learning (Golberg, 1989). AI methods have considerably diversified and can be divided in two main categories: machine-learning and knowledge-based methods (Figure 1). Machine-learning methods are, by far, the most used, among which neural networks. There are again many categories within neural networks, the most popular being deep-learning. Once a model has been trained or learned, for example, using a neural network, it allows to either evaluate examples not present in the training stage, or even generate new ones (generative AI). Language models are another popular application of AI which bloomed after the arrival of the iconic transformer paper “Attention is all you need” in 2017 (Vaswani et al., 2017). The model, often a deep neural network, is fed with a corpus of texts, and it learns the meaning of word ensembles in a context. This type of model has been generalized to many types of objects (apart from texts), such as images, molecules, etc.
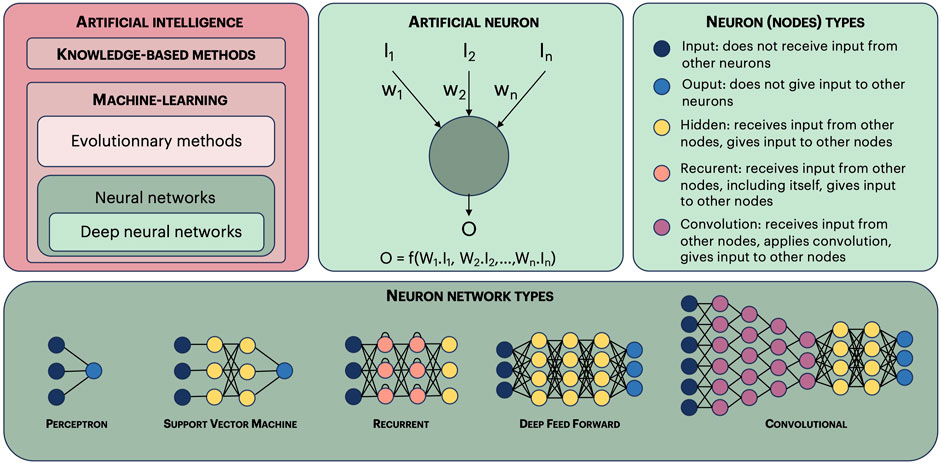
FIGURE 1. Artificial intelligence methods. Artificial intelligence methods can be divided in two main categories: knowledge-based methods and machine-learning methods. Machine-learning methods can be further divided in evolutionary methods and neural networks. This last category contains deep-learning methods. An artificial neuron (or node) receives input values (I1, I2, In), and computes an output value O, using a function and weights (w1, w2, wn). Learning consists in optimizing these weights using input values for which the output value is known (learning set). The nodes are classified in five main categories. There are many types of neuron networks, we show here only the most common ones. Finally, a neuron network is qualified as “deep”, allowing to make deep-learning, if it has three or more layers of hidden nodes.
A very important aspect of machine-learning methods in general, is that they need to be trained on a set of data called learning set. The result of a machine-learning campaign certainly depends as much on the quality of this learning dataset than on the detailed implementation. Many databases related to antibodies have emerged these last years (Khetan et al., 2022), that can be used to train machine-learning methods. However, most of these databases have been themselves built using automated methods and are lacking one or the other essential pieces of information like affinity, aggregation parameters, or the epitope. One crucial piece of information is the pairing between heavy and light chains, which is missing in all the large databases. For this reason, we have developed our own database, which contains more than 80,000 well-characterized antibodies: heavy and light chain pairing, but also epitopes, affinities, in vitro and in vivo data, cross-species reactivity, etc. This database is accessible through a software platform, MAbFactory1.
The first area in which AI has been used in the context of antibody discovery has been epitope and paratope prediction, which consists in predicting the regions of each protein (the region on the antibody side is called paratope and the region on the antigen side is called epitope) involved in their interaction. Whereas initial trials at tackling this problem only allowed to predict linear epitopes (which represent only 10% of antibody epitopes (Rubinstein et al., 2008)), gradual introduction of more complex algorithms, such as docking and machine-learning trained scoring functions allowed to reach useful accuracy levels (Zeng et al., 2023), such as epitope3D (da Silva et al., 2022), RosettaDock (Lyskov and Gray, 2008) or MAbTope (Bourquard et al., 2018; Tahir et al., 2021). MAbTope is docking-based and uses a coarse-grained formalism, which requires only the antibody sequences and allows high-throughput epitope mapping. It allows identifying a correct epitope region in more than 80% of cases. This method has been successfully applied to many examples (Kizlik-Masson et al., 2017; Ashraf et al., 2019; Neiveyans et al., 2019; Granel et al., 2020; Trilleaud et al., 2021; Vayne et al., 2021; Ugamraj et al., 2022), including when the crystal structure of the target is unknown, and a 3D homology model has to be built.
Whether working from immune animal or from already established antibody banks, the first set of hits is mainly selected on the recombinant target using classical biology approaches based on hybridomas or display technologies, either in bacteriophages or yeasts (Köhler and Milstein, 1975; Clackson et al., 1991). High affinity is the main success criterion. This approach has three major limitations: i/many leads displaying sub-optimal affinity or less represented molecules are below the threshold of such approaches and are de facto excluded from the selection, and ii/the epitope cannot be selected, meaning that selected hits binding to different places on the target molecule. Experimentally determining the epitopes of these hits, or at least knowing which ones are in competition (epitope binning) is far from trivial. The third limitation is due to the process used for transferring the animal immune repertoire to either bacteriophages or yeasts. Heavy and light chain pairing is not maintained, and the resulting antibodies are largely non-natural.
More recently, single B cells technologies have greatly improved the process of this initial clone selection (Pedrioli and Oxenius, 2021). Instead of building a bank from the immune repertoire, the B-cells of the animal, which each express a unique antibody in their membrane, can be directly selected on their affinity for the target using single-cell technologies. The antibodies coded by the retained B-cells can then be sequenced individually, resulting in natural-paired sequences. However, this technology is also relying on high affinity selection, and leads displaying sub-optimal affinity or less represented molecules are again eliminated. Moreover, even within a few thousand clones, experimentally characterization remains a problem.
Today, no published in silico method allows find leads against a selected target while fully exploring the sequential space of a natural repertoire, diverse both on the frameworks and CDRs (∼109–1012 in diversity). State-of-the-art methods still require a seed antibody to guide the search. Deep-learning language models have had nice successes in finding novel and better leads in very large artificial library of CDR-degenerated parental antibodies, paving the way to future extension to antibody repertoires (Liu et al., 2020; Mason et al., 2021; Saka et al., 2021; Bachas et al., 2022). Examples are mentioned in the affinity maturation section.
The first step in antibody characterization is often affinity evaluation, since the experimental technologies allow reasonably high-throughput as compared to other in vitro assays. Rough but large-scale evaluation is often performed in ELISA, while more precise but low throughput evaluation is performed in SPR to provide the ground-truth KD. However, these technologies require the production of both antibody and antigen, limiting the number of clones that can be evaluated. Affinity prediction from the sequences and structures of antibody and antigen would therefore allow the evaluation of much larger ensembles. Many computational methods have been proposed for this task, and benchmarks collected, but the models still have limited efficacy (Guest et al., 2021). Moreover, many of these methods rely on the knowledge of the accurate structural assembly of antibody and target, which is generally not available, and certainly not for very large collections of antibodies.
Antibodies obtained either through immunization or by screening existing antibody banks, often have insufficient affinities. Experimental methods to enhance affinity rely on random mutagenesis, usually restricted to the CDRs, and require intensive wet-lab labor. Deep-learning language models proved themselves successful at finding better binders than a parental antibody. Using the same principle as the experimental approach, language models start by building a library of the parental antibody which CDR residues are degenerated and substituted in all 20 or selected amino-acids. But the theoretical diversity to explore, even considering only the CDRs, remains largely beyond the interrogation by any wet-lab or computational means, and maturation methods are constrained to consider only a few mutated positions. As a matter of dimension, considering the CDRH3 is 10 aa-long on average, testing its full theoretical mutational space only raises the library to 1020. Bachas and Mason (Bachas et al., 2022) used degenerated Trastuzumab libraries, cloned either in bacteriophages or hybridomas, and used their binding to a fluorescent HER2 (in FACS) to train models which allowed them to retrieve better binders than the parentals. They included up to 3 mutations on respectively 10 and 17 positions. Saka et al. (Saka et al., 2021) and Liu et al. (Liu et al., 2020) created degenerated libraries of an anti-kinurenin and an anti-VEGF-A (Rabinizumab), respectively, and trained a directed evolution-based model from the enriched sequences along panning rounds. The major limitation of such models, beside the restricted number of mutations, is that they are learnt on a given antibody-antigen pair, and that the resulting training set is not target-agnostic. The whole procedure is not applicable to the next target.
With the improvement of structure determination methods, rational design of mutants has significantly increased the success rate of affinity maturation (Li et al., 2023). Although rational design leads to testing a much lower number of mutants than random mutagenesis, it also requires to have precise structural data, which is in itself a difficult task. To tackle this problem, many computational methods allowing affinity prediction of mutants have appeared recently (Li et al., 2023) with various success rates. RosettaAntibodyDesign (Adolf-Bryfogle et al., 2018) is one of the most successful.
One parameter often underestimated during antibody discovery is off-target binding. Indeed, if selectivity for the target is commonly verified by evaluating the absence of binding to close homologs, binding to unrelated proteins is usually not addressed, or very late in the discovery process. Yet, there is now ample evidence that this phenomenon, called cross-reactivity, is far from anecdotal, as it can lead to auto-immune diseases (Cusick et al., 2012), and is most-probably also responsible for some failures in clinical trials (Lecerf et al., 2019; Cunningham et al., 2021; Loberg et al., 2021). However, cross-reactivity can also be an advantage, as in the case of rituximab. Indeed, rituximab not only binds its cognate target CD20, but also the sphingomyelin-phosphodiesterase-acid-like-3b (SMPDL-3b), and offers a treatment option for follicular segment glomerulosclerosis (FSGS). Some experimental methods exist to evaluate cross-reactivity, like tissue cross reactivity, or protein arrays, but are lengthy and expensive. MAbSilico has developed a computational method allowing to predict off-target binding with good accuracy (Musnier et al., 2022). In this method, both sequence and predicted 2D structure of antibodies are used to encode the CDRs of the antibodies. These encodings can then be compared using a specific score, based on the similarity of itemsets (Egho et al., 2015). This method allowed us predicting that 238D2 (Jähnichen et al., 2010), an anti-CXCR4 antibody, also binds hemagglutinin, and 6 human proteins. We were able to experimentally validate these predictions (Musnier et al., 2022). Using this method and our database of more than 80.000 antibodies having known targets, we are able to identify off-targets as soon as the sequences are known, and this does not require the knowledge of the antigen’s 3D structure.
The last step of antibody discovery is the evaluation of developability. The term developability generally covers different aspects: (1) immunogenicity: will this antibody elicit immune reaction when injected into human? (2) Producibility: will this antibody have high production yields in bioproduction? (3) Aggregation: will it be possible to make high concentration solution, or will the antibody aggregate? The methods and databases developed to date, are largely reviewed in (Khetan et al., 2022). Briefly, for example, prediction of immunogenicity is largely based on humanness scores, such as the OASis score (Prihoda et al., 2022). These scores evaluate how close the antibody of interest is to known human sequences, and are correlated with the levels of anti-drug antibodies (ADA) observed in clinical trials. Optimization of one antibody’s immunogenicity starts with its humanization, which consists in modifying patterns to go back to the closest human germline. MAbSilico’s CDR similarity measure (see above) allows to performed humanization. In fact, since it can identify the human antibody having the most similar CDRs, it can be considered that the frameworks of the retrieved human antibody constitute an optimal scaffold to support the CDRs. The CDRs of the animal antibody can then be grafted into the human frameworks, leading to a fully human candidate.
More general evaluation of developability can be obtained through the Therapeutic Antibody Profiler (TAP) tool (Raybould et al., 2019). This method allows to anticipate expression or aggregation issues of antibodies based on characteristics such as CDRH3 length, hydrophobicity within the CDRs or canonical forms. Gentiluomo et al. (Gentiluomo et al., 2019) use interpretable neural networks to successfully predict aggregation, together with melting temperature. Hou et al. (Hou et al., 2020) have developed the SOLart software, which uses both sequence and structure, and is based on a random-forest algorithm.
Producibility prediction seems to be an even more difficult challenge. Different studies show a correlation between the production titer and the stability of the antibody (Goldenzweig et al., 2016; Jain et al., 2017), especially the melting temperature and solubility. Harmalkar et al. (Harmalkar et al., 2023) use pre-trained language models and convolutional neural networks to predict melting temperature. Avoiding antibodies predicted to have low melting temperature or poor solubility is thus desirable, but is not a guarantee of good production titers.
De novo antibody design holds the hope of being able to generate a highly affine, soluble, non-immunogenic, and epitope-directed antibody starting only from the name of the target. It implies mastering, at least, affinity prediction, structural characterization, and developability assessment. Solutions aiming at solving each pitfall are developed, as mentioned above, but they are still used individually along the funnel-shaped process dictated by the classical biological pipeline. Chaining them all together, in a virtuous circle, is certainly one key to success.
The whole design process must start by creating candidates, either randomly which would imply subsequent rational selection, or rationally, by “walking” on the target structure. Language-based approaches were expected to fulfill the first approach at high throughput but they are, as described above, still highly limited on the antibody diversity that can be injected in the computations (Liu et al., 2020; Mason et al., 2021; Saka et al., 2021; Lim et al., 2022). The approach proposed by Aguilar Rangel et al. (Aguilar Rangel et al., 2022) is based on a structural approach, computing CDR and epitope peptide complementarity. Authors show that the method can design de novo CDR peptides, which can then be grafted into nanobodies binding to three different targets (human serum albumin, SARS-CoV-2 spike protein, and trypsin), although with limited affinities. The method proposed by Anishchenko et al. (Anishchenko et al., 2021), which computes a structural evaluation of randomly generated and modeled peptides, proved accurate for protein design, but has not yet been applied to antibodies.
At MAbSilico, we have designed our own algorithms for the different steps, which allowed us to finally chain them all, unlocking our ability to de novo design antibodies. Our new in silico pipeline is target-agnostic and epitope-driven, and was successful at designing binders against the immune checkpoint inhibitor TIGIT (T-cell immunoreceptor with Ig and ITIM domains, unpublished) and against the Receptor-Binding Domain of SARS-CoV-2 (data presented at the Antibody Engineering and Therapeutics 2023; Amsterdam). In the latter project, thousands of paired VH/VL sequences were obtained from COVID-19 vaccinated patients, modeled and selected against chosen epitopes of the RBD. We identified 5 candidates, displaying nM and sub-nM affinities, and cross-neutralizing several viral strains (pre- and post- Omicron lineage emergence). Our method was successfully scaled. In fact, starting from a collection of 4.25 × 1012 VH/VL pairs (artificially reconstituted from 1.7 × 106 VH, and 2.5 × 106 VL sequences obtained by NGS of a human scFv library), 16 VHs and 22 VLs were predicted as affine binders on a specified epitope of TIGIT. Amongst the 352 possible pairings, 94% were binding in an ELISA assay, and after developability optimization, the best binder had sub-nanomolar affinity in BLI. We were also able to de novo design binders against a GPCR, whose 3D structure has not yet been determined and for which we built several homology models. This demonstrates that our method does not require an experimental structure of the target.
In silico methods are being developed to replace or support antibody selection and their molecular characterization and optimization. As shown Figure 2, AI-based methods covering one step of the classical funnel-like discovery pipeline are undoubtedly useful, but they do not change the global shaping of discovery.
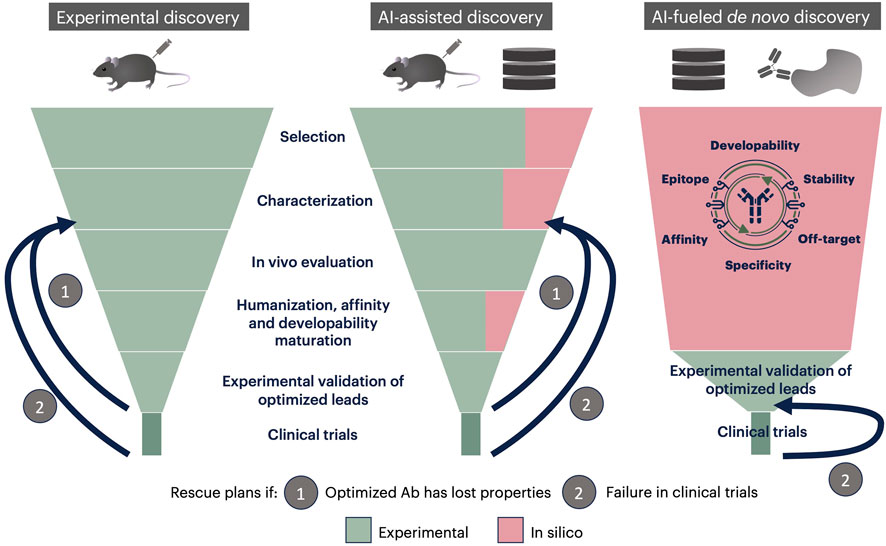
FIGURE 2. Classical, AI-assisted and AI-fueled de novo discovery pipelines. In the classical discovery pipeline (left), initial candidates (hundreds to thousands) are selected within the immune repertoire of an immunized animal, in vitro characterized (a few tens are retained) then in vivo evaluated, resulting in a few leads. These leads are humanized and optimized for affinity and developability. The resulting antibodies are evaluated in vitro and in vivo to verify that activity has been maintained. One of those is then selected for clinical trials. If antibodies have lost their activity during humanization and optimization (rescue plan 1), or if the chosen candidate fails in clinical trials (rescue plan 2), new candidates have to be selected within those characterized in vitro, and later steps gone through again. In the AI-assisted discovery pipeline (center), selection, characterization and optimization steps are partially conducted in silico, which accelerates the process, without changing its general organization. AI-based selection procedures allow to start from databases rather than physical antibody banks. In the AI-fueled de novo discovery pipeline, starting point is a database. Moreover, some technologies, such as those developed by MAbSilico, allow choosing the targeted epitope at the beginning. In this pipeline, all the antibodies of the initial database are fully evaluated in parallel, resulting in a few hundred well qualified, humanized and optimized antibodies. These candidates are then evaluated in vitro and in vivo to choose one lead for clinical trials. If this candidate fails, the next one can be chosen, without the need to repeat the whole process.
De novo AI-fueled methodologies, such as the one developed by MAbSilico allow to generate a few tens to a few hundred well-qualified leads, which are predicted to have high affinity, low off-target binding and good developability (Figure 2). These candidates can then be tested in vitro and in vivo, without the need to optimize or humanize them before clinical trials, which eliminates the risk of losing activity in the process. The chances of success are consequently much higher than in the classical process. Finally, the initial in silico step only takes up to 21 days, considerably shortening the process, and drastically abating the costs as the number of biological assays needed is decreased and the chances of success increased.
Among all characterization steps, the prediction of one antibody’s biological function remains the least amenable to in silico prediction, as the molecular mechanisms involved are either not fully understood, or highly complex and target-specific. Targeting a precise epitope can partially circumvent this issue. For example, targeting the interaction region of a ligand on its receptor will in most cases inhibit the action of the ligand. However, antibodies having the same epitope can have different functions as illustrated by Zaitseva et al. (Zaitseva et al., 2023). These authors have generated different variants of an anti-Fn14 (fibroblast growth factor (FGF)-inducible 14) antibody, and show that, despite all binding the same epitope, they have different biological functions.
AM: Writing–original draft, Writing–review and editing, Conceptualization, Data curation, Investigation, Project administration, Validation. CD: Writing–review and editing, Conceptualization, Data curation, Formal Analysis, Investigation, Software, Visualization. SM: Writing–review and editing, Formal Analysis, Investigation, Methodology, Software. AV: Writing–review and editing, Formal Analysis, Investigation, Methodology, Software, Visualization. RK: Writing–review and editing, Formal Analysis, Investigation, Methodology, Software. AC: Writing–review and editing, Data curation, Resources, Software, Supervision, Visualization. YJ: Writing–review and editing, Formal Analysis, Investigation, Methodology, Software. MC: Writing–review and editing, Investigation, Validation. YC: Writing–review and editing, Investigation, Validation. ZO: Writing–review and editing, Investigation, Validation. VP: Writing–review and editing, Conceptualization, Project administration, Resources. TB: Supervision, Writing–review and editing, Conceptualization, Formal Analysis, Investigation, Methodology, Project administration, Software. AP: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Supervision, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Authors AC, YJ, MC, YC, ZO, VP, TB, and AP were employed by MAbSilico.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1https://app-publicdemo-mabfactory-97288.azurewebsites.net/
Adolf-Bryfogle, J., Kalyuzhniy, O., Kubitz, M., Weitzner, B. D., Hu, X., Adachi, Y., et al. (2018). RosettaAntibodyDesign (RAbD): a general framework for computational antibody design. PLOS Comput. Biol. 14, e1006112. doi:10.1371/journal.pcbi.1006112
Aguilar Rangel, M., Bedwell, A., Costanzi, E., Taylor, R. J., Russo, R., Bernardes, G. J. L., et al. (2022). Fragment-based computational design of antibodies targeting structured epitopes. Sci. Adv. 8, eabp9540. doi:10.1126/sciadv.abp9540
Anishchenko, I., Pellock, S. J., Chidyausiku, T. M., Ramelot, T. A., Ovchinnikov, S., Hao, J., et al. (2021). De novo protein design by deep network hallucination. Nature 600, 547–552. doi:10.1038/s41586-021-04184-w
Ashraf, Y., Mansouri, H., Laurent-Matha, V., Alcaraz, L. B., Roger, P., Guiu, S., et al. (2019). Immunotherapy of triple-negative breast cancer with cathepsin D-targeting antibodies. J. Immunother. cancer 7, 29. doi:10.1186/s40425-019-0498-z
Bachas, S., Rakocevic, G., Spencer, D., Sastry, A. V., Haile, R., Sutton, J. M., et al. (2022). Antibody optimization enabled by artificial intelligence predictions of binding affinity and naturalness. arXiv. doi:10.1101/2022.08.16.504181
Bauer, J., Rajagopal, N., Gupta, P., Gupta, P., Nixon, A. E., and Kumar, S. (2023). How can we discover developable antibody-based biotherapeutics? Front. Mol. Biosci. 10, 1221626. doi:10.3389/fmolb.2023.1221626
Bejarano, L., Jordāo, M. J. C., and Joyce, J. A. (2021). Therapeutic targeting of the tumor microenvironment. Cancer Discov. 11, 933–959. doi:10.1158/2159-8290.CD-20-1808
Bourquard, T., Musnier, A., Puard, V., Tahir, S., Ayoub, M. A., Jullian, Y., et al. (2018). MAbTope: a method for improved epitope mapping. J. Immunol. 201, 3096–3105. doi:10.4049/jimmunol.1701722
Clackson, T., Hoogenboom, H. R., Griffiths, A. D., and Winter, G. (1991). Making antibody fragments using phage display libraries. Nature 352, 624–628. doi:10.1038/352624a0
Cunningham, O., Scott, M., Zhou, Z. S., and Finlay, W. J. J. (2021). Polyreactivity and polyspecificity in therapeutic antibody development: risk factors for failure in preclinical and clinical development campaigns. mAbs 13, 1999195. doi:10.1080/19420862.2021.1999195
Cusick, M. F., Libbey, J. E., and Fujinami, R. S. (2012). Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 42, 102–111. doi:10.1007/s12016-011-8294-7
da Silva, B. M., Myung, Y., Ascher, D. B., and Pires, D. E. V. (2022). epitope3D: a machine learning method for conformational B-cell epitope prediction. Briefings Bioinforma. 23, bbab423. doi:10.1093/bib/bbab423
Egho, E., Raïssi, C., Calders, T., Jay, N., and Napoli, A. (2015). On measuring similarity for sequences of itemsets. Data Min. Knowl. Disc 29, 732–764. doi:10.1007/s10618-014-0362-1
Gentiluomo, L., Roessner, D., Augustijn, D., Svilenov, H., Kulakova, A., Mahapatra, S., et al. (2019). Application of interpretable artificial neural networks to early monoclonal antibodies development. Eur. J. Pharm. Biopharm. 141, 81–89. doi:10.1016/j.ejpb.2019.05.017
Golberg, D. E. (1989). Genetic algorithms in search, optimization, and machine learning. Reading, MA, USA: Addison Welssey Publishing company.
Goldenzweig, A., Goldsmith, M., Hill, S. E., Gertman, O., Laurino, P., Ashani, Y., et al. (2016). Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell 63, 337–346. doi:10.1016/j.molcel.2016.06.012
Granel, J., Lemoine, R., Morello, E., Gallais, Y., Mariot, J., Drapeau, M., et al. (2020). 4C3 human monoclonal antibody: a proof of concept for non-pathogenic proteinase 3 anti-neutrophil cytoplasmic antibodies in granulomatosis with polyangiitis. Front. Immunol. 11, 573040. doi:10.3389/fimmu.2020.573040
Guest, J. D., Vreven, T., Zhou, J., Moal, I., Jeliazkov, J. R., Gray, J. J., et al. (2021). An expanded benchmark for antibody-antigen docking and affinity prediction reveals insights into antibody recognition determinants. Structure 29, 606–621.e5. doi:10.1016/j.str.2021.01.005
Harmalkar, A., Rao, R., Richard Xie, Y., Honer, J., Deisting, W., Anlahr, J., et al. (2023). Toward generalizable prediction of antibody thermostability using machine learning on sequence and structure features. mAbs 15, 2163584. doi:10.1080/19420862.2022.2163584
Holland, J. H. (1992). Adaptation in natural and artificial systems: an introductory analysis with applications to biology, control, and artificial intelligence. Cambridge, MA: MIT press. Available at: https://books.google.fr/books?hl=fr&lr=&id=5EgGaBkwvWcC&oi=fnd&pg=PR7&dq=J.+H.+Holland,+Adaptation+In+Natural+And+Artificial+Systems&ots=mJoq2YKqxq&sig=fAjjZ6AOubm3-FNuSNT_Tf-CHfU (Accessed October 25, 2023).
Hoover, J. M., Prinslow, E. G., Teigler, J. E., Truppo, M. D., and La Porte, S. L. (2021). Therapeutic antibody discovery. Remingt. Twenty-third Ed. 2021, 417–436. doi:10.1016/B978-0-12-820007-0.00023-4
Hou, Q., Kwasigroch, J. M., Rooman, M., and Pucci, F. (2020). SOLart: a structure-based method to predict protein solubility and aggregation. Bioinformatics 36, 1445–1452. doi:10.1093/bioinformatics/btz773
Jähnichen, S., Blanchetot, C., Maussang, D., Gonzalez-Pajuelo, M., Chow, K. Y., Bosch, L., et al. (2010). CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. 107, 20565–20570. doi:10.1073/pnas.1012865107
Jain, T., Sun, T., Durand, S., Hall, A., Houston, N. R., Nett, J. H., et al. (2017). Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. 114, 944–949. doi:10.1073/pnas.1616408114
Jin, S., Sun, Y., Liang, X., Gu, X., Ning, J., Xu, Y., et al. (2022). Emerging new therapeutic antibody derivatives for cancer treatment. Sig Transduct. Target Ther. 7, 39–28. doi:10.1038/s41392-021-00868-x
Khetan, R., Curtis, R., Deane, C. M., Hadsund, J. T., Kar, U., Krawczyk, K., et al. (2022). Current advances in biopharmaceutical informatics: guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. mAbs 14, 2020082. doi:10.1080/19420862.2021.2020082
Kizlik-Masson, C., Vayne, C., McKenzie, S. E., Poupon, A., Zhou, Y., Champier, G., et al. (2017). 5B9, a monoclonal antiplatelet factor 4/heparin IgG with a human Fc fragment that mimics heparin-induced thrombocytopenia antibodies. J. Thromb. Haemost. 15, 2065–2075. doi:10.1111/jth.13786
Köhler, G., and Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497. doi:10.1038/256495a0
Lecerf, M., Kanyavuz, A., Lacroix-Desmazes, S., and Dimitrov, J. D. (2019). Sequence features of variable region determining physicochemical properties and polyreactivity of therapeutic antibodies. Mol. Immunol. 112, 338–346. doi:10.1016/j.molimm.2019.06.012
Li, J., Kang, G., Wang, J., Yuan, H., Wu, Y., Meng, S., et al. (2023). Affinity maturation of antibody fragments: a review encompassing the development from random approaches to computational rational optimization. Int. J. Biol. Macromol. 247, 125733. doi:10.1016/j.ijbiomac.2023.125733
Lim, Y. W., Adler, A. S., and Johnson, D. S. (2022). Predicting antibody binders and generating synthetic antibodies using deep learning. mAbs 14, 2069075. doi:10.1080/19420862.2022.2069075
Liu, G., Zeng, H., Mueller, J., Carter, B., Wang, Z., Schilz, J., et al. (2020). Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics 36, 2126–2133. doi:10.1093/bioinformatics/btz895
Loberg, L. I., Chhaya, M., Ibraghimov, A., Tarcsa, E., Striebinger, A., Popp, A., et al. (2021). Off-target binding of an anti-amyloid beta monoclonal antibody to platelet factor 4 causes acute and chronic toxicity in cynomolgus monkeys. mAbs 13, 1887628. doi:10.1080/19420862.2021.1887628
Lu, R.-M., Hwang, Y.-C., Liu, I.-J., Lee, C.-C., Tsai, H.-Z., Li, H.-J., et al. (2020). Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 27, 1. doi:10.1186/s12929-019-0592-z
Lyskov, S., and Gray, J. J. (2008). The RosettaDock server for local protein–protein docking. Nucleic Acids Res. 36, W233–W238. doi:10.1093/nar/gkn216
Mason, D. M., Friedensohn, S., Weber, C. R., Jordi, C., Wagner, B., Meng, S. M., et al. (2021). Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat. Biomed. Eng. 5, 600–612. doi:10.1038/s41551-021-00699-9
Mehrabadi, A. Z., Ranjbar, R., Farzanehpour, M., Shahriary, A., Dorostkar, R., Hamidinejad, M. A., et al. (2022). Therapeutic potential of CAR T cell in malignancies: a scoping review. Biomed. Pharmacother. 146, 112512. doi:10.1016/j.biopha.2021.112512
Musnier, A., Bourquard, T., Vallet, A., Mathias, L., Bruneau, G., Ayoub, M. A., et al. (2022). A New in silico antibody similarity measure both identifies large sets of epitope binders with distinct CDRs and accurately predicts off-target reactivity. Int. J. Mol. Sci. 23, 9765. doi:10.3390/ijms23179765
Neiveyans, M., Melhem, R., Arnoult, C., Bourquard, T., Jarlier, M., Busson, M., et al. (2019). A recycling anti-transferrin receptor-1 monoclonal antibody as an efficient therapy for erythroleukemia through target up-regulation and antibody-dependent cytotoxic effector functions. mAbs 11, 593–605. doi:10.1080/19420862.2018.1564510
Pedrioli, A., and Oxenius, A. (2021). Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 42, 1143–1158. doi:10.1016/j.it.2021.10.008
Prihoda, D., Maamary, J., Waight, A., Juan, V., Fayadat-Dilman, L., Svozil, D., et al. (2022). BioPhi: a platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning. mAbs 14, 2020203. doi:10.1080/19420862.2021.2020203
Raybould, M. I. J., Marks, C., Krawczyk, K., Taddese, B., Nowak, J., Lewis, A. P., et al. (2019). Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. 116, 4025–4030. doi:10.1073/pnas.1810576116
Rubinstein, N. D., Mayrose, I., Halperin, D., Yekutieli, D., Gershoni, J. M., and Pupko, T. (2008). Computational characterization of B-cell epitopes. Mol. Immunol. 45, 3477–3489. doi:10.1016/j.molimm.2007.10.016
Saka, K., Kakuzaki, T., Metsugi, S., Kashiwagi, D., Yoshida, K., Wada, M., et al. (2021). Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci. Rep. 11, 5852. doi:10.1038/s41598-021-85274-7
Tahir, S., Bourquard, T., Musnier, A., Jullian, Y., Corde, Y., Omahdi, Z., et al. (2021). Accurate determination of epitope for antibodies with unknown 3D structures. mAbs 13, 1961349. doi:10.1080/19420862.2021.1961349
Trilleaud, C., Gauttier, V., Biteau, K., Girault, I., Belarif, L., Mary, C., et al. (2021). Agonist anti-ChemR23 mAb reduces tissue neutrophil accumulation and triggers chronic inflammation resolution. Sci. Adv. 7, eabd1453. doi:10.1126/sciadv.abd1453
Ugamraj, H. S., Dang, K., Ouisse, L.-H., Buelow, B., Chini, E. N., Castello, G., et al. (2022). TNB-738, a biparatopic antibody, boosts intracellular NAD+ by inhibiting CD38 ecto-enzyme activity. mAbs 14, 2095949. doi:10.1080/19420862.2022.2095949
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., et al. (2017). “Attention is all you need,” in Advances in neural information processing systems (Red Hook, NY: Curran Associates, Inc.). Available at: https://proceedings.neurips.cc/paper_files/paper/2017/hash/3f5ee243547dee91fbd053c1c4a845aa-Abstract.html (Accessed November 16, 2023).
Vayne, C., Nguyen, T.-H., Rollin, J., Charuel, N., Poupon, A., Pouplard, C., et al. (2021). Characterization of new monoclonal PF4-specific antibodies as useful tools for studies on typical and autoimmune heparin-induced thrombocytopenia. Thromb. Haemost. 121, 322–331. doi:10.1055/s-0040-1717078
Vega, F. A., Berraondo, P., and Galluzzi, L. (2022). New antibody formats. Cambridge, MA: Academic Press. Available at: https://books.google.fr/books?hl=fr&lr=&id=Er5mEAAAQBAJ&oi=fnd&pg=PP1&dq=new+antibody+formats+review&ots=a2FNJEpNaQ&sig=XVPDRssdpQUnv3IlkV7jtase8NQ (Accessed January 9, 2024).
Zaitseva, O., Hoffmann, A., Löst, M., Anany, M. A., Zhang, T., Kucka, K., et al. (2023). Antibody-based soluble and membrane-bound TWEAK mimicking agonists with FcγR-independent activity. Front. Immunol. 14, 1194610. doi:10.3389/fimmu.2023.1194610
Keywords: antibody, artificial intelligence, discovery, manufacturability, affinity
Citation: Musnier A, Dumet C, Mitra S, Verdier A, Keskes R, Chassine A, Jullian Y, Cortes M, Corde Y, Omahdi Z, Puard V, Bourquard T and Poupon A (2024) Applying artificial intelligence to accelerate and de-risk antibody discovery. Front. Drug Discov. 4:1339697. doi: 10.3389/fddsv.2024.1339697
Received: 16 November 2023; Accepted: 13 February 2024;
Published: 05 March 2024.
Edited by:
Bruno Villoutreix, Hôpital Robert Debré, FranceReviewed by:
Taisiia Feoktistova, Merck, United StatesCopyright © 2024 Musnier, Dumet, Mitra, Verdier, Keskes, Chassine, Jullian, Cortes, Corde, Omahdi, Puard, Bourquard and Poupon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne Poupon, YW5uZS5wb3Vwb25AbWFic2lsaWNvLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.