 Lusine Tonoyan1,2
Lusine Tonoyan1,2 Arno G. Siraki2*
Arno G. Siraki2*- 1Applied Pharmaceutical Innovation, Edmonton, AB, Canada
- 2Faculty of Pharmacy and Pharmaceutical Sciences, College of Health Sciences, University of Alberta, Edmonton, AB, Canada
Machine learning (ML) in toxicological sciences is growing exponentially, which presents unprecedented opportunities and brings up important considerations for using ML in this field. This review discusses supervised, unsupervised, and reinforcement learning and their applications to toxicology. The application of the scientific method is central to the development of a ML model. These steps involve defining the ML problem, constructing the dataset, transforming the data and feature selection, choosing and training a ML model, validation, and prediction. The need for rigorous models is becoming more of a requirement due to the vast number of chemicals and their interaction with biota. Large datasets make this task possible, though selecting databases with overlapping chemical spaces, amongst other things, is an important consideration. Predicting toxicity through machine learning can have significant societal impacts, including enhancements in assessing risks, determining clinical toxicities, evaluating carcinogenic properties, and detecting harmful side effects of medications. We provide a concise overview of the current state of this topic, focusing on the potential benefits and challenges related to the availability of extensive datasets, the methodologies for analyzing these datasets, and the ethical implications involved in applying such models.
1 Introduction to machine learning
Machine learning (ML) has developed into a powerful tool in toxicological sciences, revolutionizing how we analyze and interpret complex datasets. The fundamental concepts of ML are explored here in its applications to toxicology, with attention to specific areas regarding drug toxicity. ML has undoubtedly impacted the research landscape, and the number of publications in the area of ML is growing exponentially (Figure 1). It is not practical to summarize the literature due to this level of activity; however, this review intends to provide foundational knowledge and applications of certain critical aspects of ML algorithms and how they are used in specific areas of toxicology.

FIGURE 1. The exponential growth of publications in ML and drug toxicity. This graph is based on a scopus.com search (January 2024): “machine learning” and “toxicity” for all fields.
1.1 What is artificial intelligence and machine learning?
Artificial Intelligence (AI) is a branch of computer science dedicated to creating systems capable of tasks that normally require human intelligence. Machine Learning (ML), is a subset of AI, which involves developing algorithms that allow machines to learn and make decisions from data (Figures 2, 5).
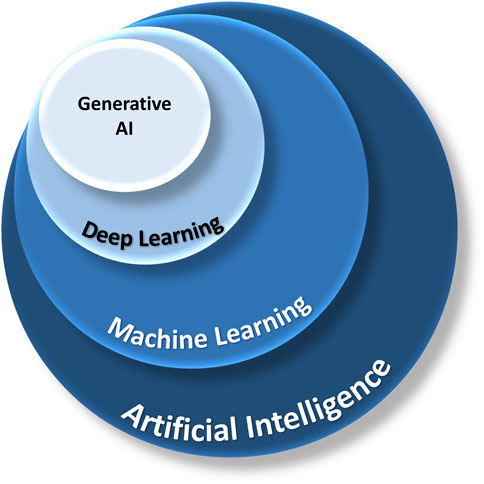
FIGURE 2. The relationship between AI, ML, deep learning, and generative AI. AI is a broad field of study, and ML is a branch of AI that focuses on algorithms mimicking human learning. Deep learning utilizes multiple layers of neural networks. Generative AI can synthesize new content based on existing data from deep learning.
1.2 Main types of machine learning algorithms
Machine learning encompasses a variety of algorithms, falling into three primary categories: supervised learning, unsupervised learning, and reinforcement learning (Ribba et al., 2020). Each type addresses distinct learning objectives and is tailored to specific applications in toxicology (Figure 3). Although these algorithms are different, they can be used to manage the same questions, with the difference being prediction capabilities.
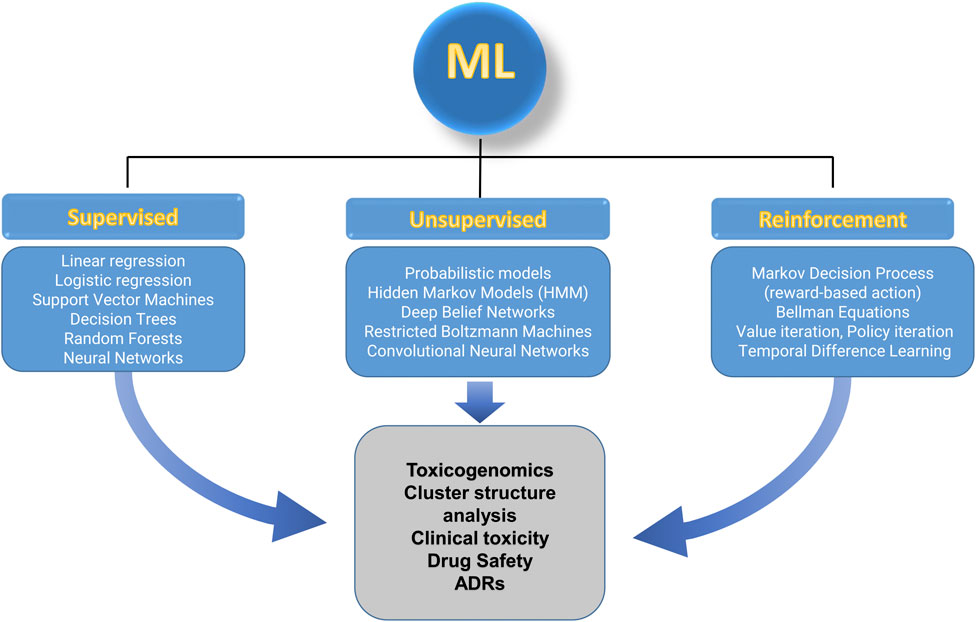
FIGURE 3. Different algorithms of ML, and applications to toxicology.
1.3 Machine learning model representation
The intersection of ML and drug toxicology holds immense promise. By leveraging machine learning, researchers can analyze vast datasets to predict and understand adverse drug reactions, aiding in developing safer pharmaceuticals.
As with all pathways of scientific inquiry, the first steps are critical to choosing the right approach. In Figure 4, we present a workflow for ML for scientific questions, toxicological ones in this case. We begin to “define an ML problem” at the beginning of the research workflow. This step aims to define the unsolved problem or research question, which differs from other approaches because of its potential mathematical complexity. This usually involves finding patterns, features, and classifications that ultimately lead to predictions. ML requires a large amount of data, that must be reliable. The next step is data collection. Data (i.e., categorical, nominal, or numerical) can be annotated with labels intended to represent accurate answers, although it should be mentioned that these labels, often considered gold-standard, may be subject to human error or bias. Notably, the data must contain parameters relevant to addressing the ML problem. ML requires 1) training data, 2) testing data, and 3) validation data. There is no single “right” answer to the amount of data required for ML. Ideally, one should collect about 1,000 samples. For most “average” problems, you need to have 10,000–100,000 samples, although examples in the literature have been published with <1,000 data points (the layers therein can enhance the usefulness of the data). For “hard” problems like machine translation, high dimensional data generation, or anything requiring deep learning, one should aim for 100,000–1,000,000 samples. Equally crucial to data collection is data transformation or data cleanup. Clean up the data involves removing repeats, filling in or imputing missing values, reformatting for compatibility, fixing outliers, grouping sparse classes, in essence, data cleansing. This also involves converting categorical or nominal data to numeric data, one-hot encoding (converting categorical variables to binary), normalizing skewed data, and range scale, i.e., data transformation. Feature engineering involves selecting relevant features and discarding irrelevant data through methods based on statistical analysis or expert guidance. Once the data is ready, one can choose and train a model. This step will entail selecting the approaches as mentioned earlier, where supervised, unsupervised, or reinforcement models can be applied. Testing and validating ensures that the ML model is not using too few parameters (under fitting) or too many parameters (overfitting). The problem of overfitting (when too many parameters are used) can be prevented by 1) external validation datasets, 2) K-fold cross-validation, 3) leave-one-out methods, and 4) permutation testing or the combinations of all four methods. Data splitting is an integral part of validation. The data set can be divided into two groups (the holdout method), where 2/3 (66% of the data) is the training data, and 1/3 (33% of the data) is the testing data, which is “held out” and used to test the model. The rule in model training is that the testing data must remain unseen during the training phase. One can further divide the training data into smaller parts or “folds” to avoid training bias. Studies that report on the validation of their model will typically report on assessing models based on confusion matrix or ROC curves (Rashidi et al., 2023; Valero-Carreras et al., 2023). For more details on evaluation metrics, such as accuracy, precision, and mean squared error (MSE), refer to a recent review (Sinha et al., 2023). Ideally, once the model is validated, it is ready for use and application. This may result in an end-user interface, webpage, or other resource. However, it should be kept in mind that models can be continually improved and that the workflow presented in Figure 4 is an iterative process that can be continually tweaked to minimize error and maximize accuracy in prediction. Lastly, It is important to recall and apply the fundamental framework in ML (Mitchell, 1997). A computer program (algorithm) learns from experience (E) concerning tasks (T) and a performance measure (P), where its performance at tasks measured by P improves with experience (Figure 5).
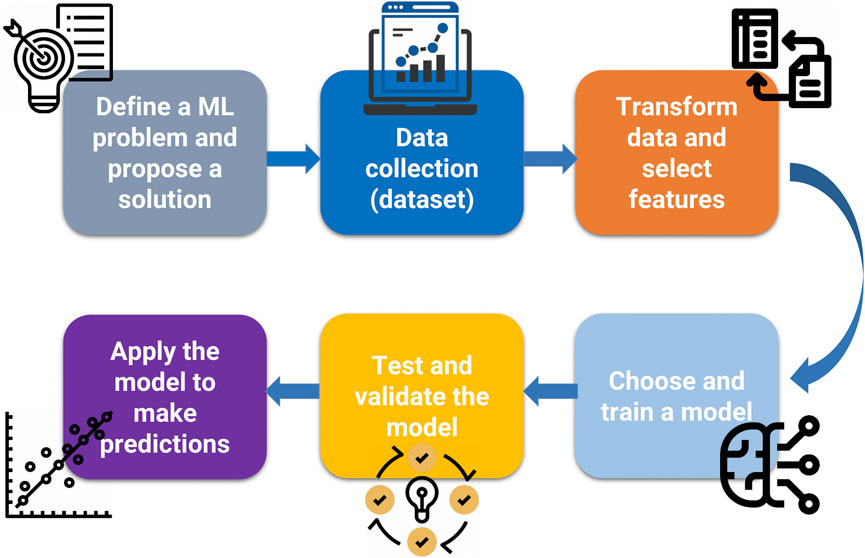
FIGURE 4. The generalized workflow for ML application. The first step requires the definition of the unsolved problem. Dataset collection is highly dependent the availability or acquisition of reliable data. Robust models require significant data points for training, testing and validation. Data transformation is a critical step to ensure the “cleanliness” of data. Machine learning algorithms, such as decision trees/random forests, hidden Markov models, and artificial neural networks, are selected for model building only after the data set has been curated and cleaned.
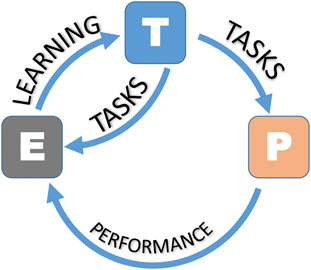
FIGURE 5. The ML framework (as per Mitchell). The task (T) is what the ML model is designed to do or the problem it is supposed to solve. The experience (E) refers to the data that the ML model uses to learn and make predictions. The performance measure (P) is a metric or evaluation criterion that quantifies how well the machine learning system is performing the task, T, based on Mitchell (1997).
An important point to emphasize is the “all-metrics” approach, where models and algorithms are evaluated based on a wide range of performance metrics. The latter is important in considering a comprehensive view of a model’s performance. However, there are some key consideration in order to make a holistic conclusion for performance. For instance, a surface-level analysis with greater focus numerical readouts rather than mechanistic information can be misleading. The value of certain metrics is also important to consider, as not all metrics are of equal value. The latter can also lead to overfitting (Figure 6), where a real-world test of an arguably perfect model may fail. Thus, keeping in mind the practical applications, potential biases, ethical implications, and general application of the model are more tangible criteria in performance evaluation (Duffull and Isbister, 2022; Muntean and Militaru, 2023).
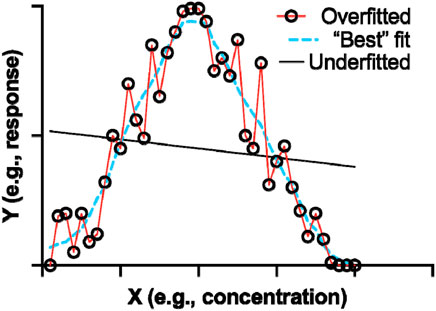
FIGURE 6. A graphical representation of fitting. Hypothetical data was plotted on an XY plot in GraphPad Prism 9. The “Best” fit should form a prediction based on appropriate training set parameters.
1.4 Machine learning applications in toxicology
Machine learning has numerous applications in toxicology, ranging from predicting the toxicity of chemicals to identifying new drug targets. We have focused on selected areas in toxicology, particularly human drug toxicology. Guidance on other literature is provided as appropriate.
1.4.1 Chemoinformatics, bioinformatics and structure-toxicity relationships
Machine learning is crucial in bridging bioinformatics with chemoinformatics and quantitative structure-toxicity relationships modelling, facilitating the prediction of chemical properties, biological readouts, and toxicity. Chemoinformatics involves the acquisition or collection of chemical information and can be considered a combination of information technology and chemistry (Lo et al., 2018). This is a critical area of study, per se, where chemical fingerprints are related to a functional endpoint. The latter is where ML comes into play and is essentially a more complex application of structure-activity relationships. Classical 2D linear regression analysis would utilize indices such as logP, Hammett constants, and acid ionization constants pKa (to name a few) and correlate these to the EC50 (LC50, IC50) of a desired effect. This type of analysis is named after Corwin Hansch from Pomona College, co-founded by Fujita, who founded this area in the 1960s (Hansch and Fujita, 1964). Free-Wilson analysis emerged around the same time (Free and Wilson, 1964; Martin, 2012). Such relationships are essentially linear free energy relationships and are fundamentally modelled by the earliest equations relating function with structure (Kubinyi, 2002)
where the change in physiological activity (ΔΦ) is a function of the chemical constitution (C). Analogous to anatomy, structure is related to function. Note that the “C” for chemical constitution can easily be replaced with a “B” for Bioinformatics. These linear free energy relationships are still useful today, perhaps due to understanding the basis for computational process and the accessibility of analysis tools. A recent study used such approaches to investigate the potency of monoamine oxidase inhibition (Pisani et al., 2023). A model in ML is typically a mathematical function that maps input data to desired output:
where that function could be, for example, in linear regression,
where
Linear modeling approaches are still useful, but sometimes they encounter challenges with large datasets and the need to predict toxicity in both existing and new chemicals, some of which have unknown properties. The California Department of Toxic Substances Control reports that the US has more than 80,000 commercially available chemicals, of which 2,500 are produced at >1 million pounds/year, almost half of which lack adequate toxicology testing (https://dtsc.ca.gov/emerging-chemicals-of-concern/). As such, a workflow is needed to reliably address the information gap on the numerous chemicals found in the environment and to which humans are exposed. The term molecular informatics combines both chemoinformatics and bioinformatics. The latter can involve data from various omics platforms as the biological readout. As one can imagine, this vast amount of data demands powerful computational algorithms to produce valuable conclusions from the data (Figure 7). Furthermore, regulatory agencies face the daunting task of risk management for large numbers of anthropogenic compounds (discussed further in Section 1.4.5). Regulatory agencies rely on experimental data readouts to carry out risk assessment strategies, which becomes challenging with producing new and dealing with existing chemicals concurrently (Yauk et al., 2019).
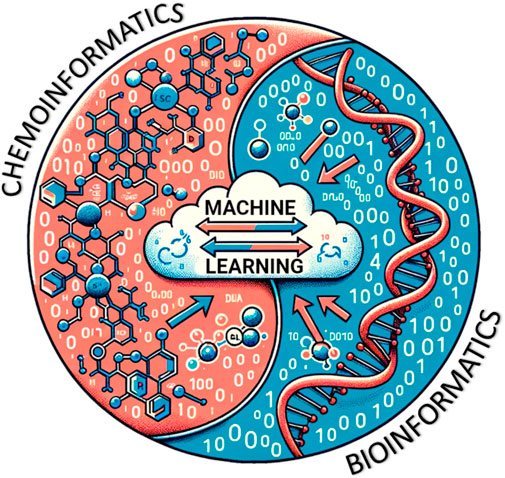
FIGURE 7. Conceptual interplay between chemoinformatics and bioinformatics and the synthesis of ML models using relevant databases (Generated by DALL-E and modified by author).
1.4.2 Toxicity prediction
The combination of data sets described above using appropriate ML models can contribute to the accurate prediction of toxicity, enabling early identification of potential hazards associated with chemical compounds. The type of toxicity will depend on the particular study and is quite diverse. There appear to be ML applications in almost every area of toxicology, including (but not limited to) drug discovery and development (Gupta et al., 2021), oncologic drug efficacy and toxicity (Badwan et al., 2023), human health hazard assessment of industrial azo dyes (Keshava et al., 2023), and histopathology from toxicological findings to name a few (Mehrvar et al., 2021). Other studies include small molecule-induced mitochondrial toxicity (Zhao et al., 2021; Garcia de Lomana et al., 2023). More broad endpoints for toxicology include hepatotoxicity, cardiotoxicity, respiratory toxicity, nuclear receptor binding, mutagenicity, carcinogenicity, and acute oral toxicity (reviewed in (Cavasotto and Scardino, 2022; Yang et al., 2018)). What has made such approaches to toxicity prediction feasible is the availability of big data, i.e., publicly available toxicology databases (Yang et al., 2018). In silico drug toxicity prediction offers clear advantages, especially considering that drug attrition rates are always a concern. Analogous to organ-on-a-chip technologies, ML prediction models for multiple organs have been reported (Hu et al., 2023). This study applied six algorithms to model carcinogenicity, cardiotoxicity, developmental toxicity, hepatotoxicity, nephrotoxicity, neurotoxicity, reproductive toxicity, and skin sensitization. This systems toxicology approach is important and has great potential to provide a comprehensive prediction assessment.
1.4.3 Adverse drug reactions
Adverse drug reactions (ADRs) are unwanted, undesirable, unexpected, or harmful effects that result from normal pharmacotherapy. Predicting ADRs through machine learning may enhance drug safety assessments during development and post-market surveillance (i.e., approved drugs on the market). ML applications during drug development can aid in minimizing or preventing ADRs once a drug reaches the market. However, ADRs happen in various settings, and many jurisdictions have significant pharmacovigilance strategies in place, such as the Canada Vigilance Adverse Reaction Online Database, the FDA’s Adverse Event Reporting System (FAERS), the European Database of Suspected Adverse Drug Reaction Reports, the Database of Adverse Event Notifications (Australia), and the Japanese Adverse Drug Event Report (JADER) Database. One specific study used VigiBase® (described below) to determine individual case reports of ADRs occurring in African countries (Ampadu et al., 2016). To capture the global implications of the side effects of medications, the World Health Organization launched VigiAccess in 2015 (vigiaccess.org), which is reportedly the largest database of its kind; in collaboration with the WHO, the Uppsala Monitoring Centre (UMC) operates and maintains VigiBase® on a cost-recovery basis (who-umc.org/vigiBase/). Another resource called “SIDER” (http://sideeffects.embl.de/) also contains a searchable database for the end user. The DrugBank (https://go.drugbank.com/) contains diverse information that also includes drug side effects. Other useful datasets include ClinTox, Tox21, and RTECS (discussed later). A challenge with these approaches is their reliance on user input to populate these databases and data curation.
On a population scale, ADRs can affect a significant proportion of individuals. ADRs account for 6%–12% of hospital admissions among older patients, costing almost $36 million annually (Parameswaran Nair et al., 2016). About 1/10th of elderly individuals discharged from a hospital have been shown to experience drug-related readmission, and half of those were considered preventable (Prasad et al., 2023). The American Society of Health-System Pharmacists defines an ADR as an occurrence where a person is adversely affected by a drug, encompassing both medication errors and ADRs (Demler and Chehovich, 2021). The demand for ML, in this instance, is particularly unique because not all ADRs are predictable. This has been of particular significance and detriment to patients and the pharmaceutical companies. There are different classifications of ADRs (Table 1), which have remained the same for the past two decades (Edwards and Aronson, 2000; Besco et al., 2022).
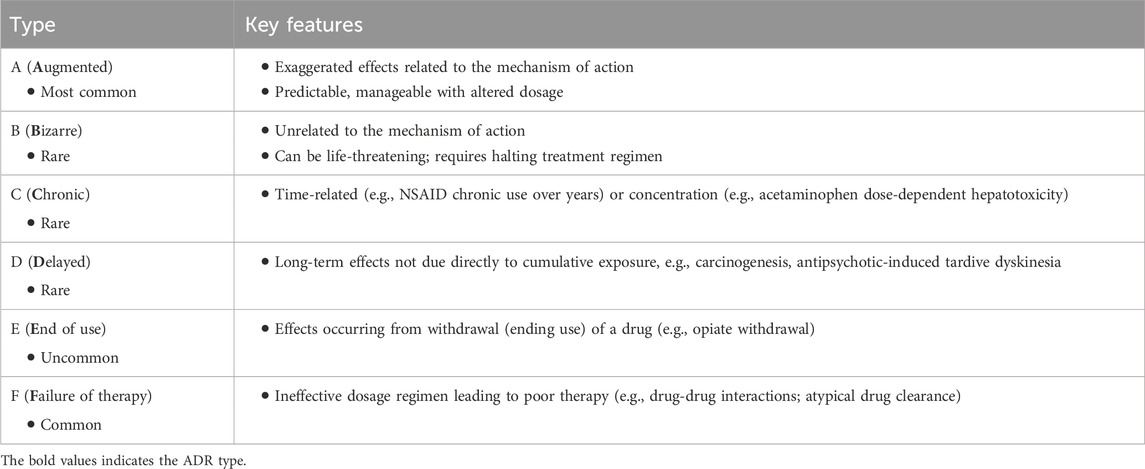
TABLE 1. ADRs classified by type.
Although this classification of ADRs by type is functional, it only sometimes captures the severity of the reactions. Furthermore, the specific organ or organ systems are also an important classification parameter. Another method of classification could involve severity (Table 2).
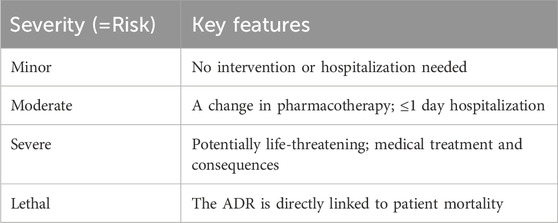
TABLE 2. ADRs classified by severity.
Based on the classifications described in Tables 1 and 2, there are numerous possibilities to use ML in pharmacovigilance and ADRs. One critical review proposed that deep learning strategies were more powerful than earlier approaches using supervised learning; this is considered feasible due to the multiple layers used in deep learning (Lee and Chen, 2019). As described, the presence of the ADR is not associated with the severity of the ADR, e.g., drug-induced liver injury (DILI) can range from a marginal increase in liver enzyme levels to fulminant hepatic failure. However, if the readout for an ADR is “yes” or “no,” a binary classifier, it is worthwhile considering decision trees or random forests. The SIDER database and DrugBank were used to create a knowledge graph that could be used to vectorize drugs and ADRs and utilize a binary classifier based on logistic regression for the causality of the ADRs. This model was also applied to discriminate for DILI (using the DILIrank dataset), which was able to discriminate liver injury but not predict severity (Zhang et al., 2021). The DILIrank dataset contains 1,036 drugs that rank drugs by their risk for developing DILI (Chen et al., 2016).
DILI is one of the most frequent causes of drug attrition, black-box warnings, or withdrawal from the market. As indicated above, there is a broad spectrum of harm that drugs can cause to the liver, with varying degrees of severity. The liver, responsible for the majority of drug metabolism, may produce potentially harmful by-products such as reactive metabolites and free radicals during this process (Chalasani et al., 2008). Some cases of DILI are dose-dependent (e.g., acetaminophen overdose, a type C ADR), while others could occur unpredictably at therapeutic doses (Type B ADR). The exact mechanisms underlying the latter are still being studied, but it is thought that both drug-related and host-related factors play roles. In one study, a model was trained on a database called TG-GATEs (a toxicogenomics database from Japan) (Igarashi et al., 2014) and used two external databases (JNJ-I, JNJ-II) for test data sets (Moein et al., 2023); this challenging yet crucial area employs several different ML algorithms (support vector machine, linear regression, and random forest). The authors faced challenges related to the difference between the compounds (and, thus, the chemical space) in each data set. The TG-GATE system, which contains liver imaging for the 170 drugs tested, was analyzed with PathologAI (referred to by the authors as a weakly supervised deep learning network) to predict liver necrosis for over 800 compounds from whole slide images, where the classification accuracy was over 80% (Bussola et al., 2023).
A study utilizing the SIDER database focused on developing ML models to link cheminformatics with drug-induced autoimmune disease (Guo et al., 2022). This study used approximately 600 drugs (combining training and validation sets) and tested 35 classification-based models; the selected models were based on support vector machines and MACCS keys methods that associated physicochemical properties with autoimmune reactions and developed a website based on the findings (http://diad.sapredictor.cn/). Off-target toxicity, which can become apparent in a Type A ADR, is an important consideration both during drug development and post-market release. One study used a feed-forward neural network (with TensorFlow and Keras) to identify bias in public data compared to proprietary data. In addition, using both datasets revealed with certainty specific enzyme inhibition by two compounds, suggesting that consensus scores from models using different datasets can be an advantage (Smajić et al., 2023). An important aspect to consider is that enzyme inhibition can lead to drug-drug interactions. One approach on cytochrome P450 2D6, an important drug-metabolizing enzyme, compared some ML algorithms. The MACCS fingerprint models performed best (based on accuracy and area under the ROC curve) (Li et al., 2023). Another study focused more on the dominant drug-metabolizing isoform, cytochrome P450 3A4, found that regression-based ML (using a support vector regressor) was most effective for drugs from 120 studies (Gill et al., 2023).
In certain instances, a drug-drug interaction is desirable. For example, nirmatrelvir and ritonavir are sold under the brand name Paxlovid™. Ritonavir inhibits cytochrome P450 3A4, consequently inhibiting the metabolism of nirmatrelvir and is considered a pharmacokinetic enhancer. A specific application to predict synergistic drug-drug interactions in cancer patients used a deep-learning model (combining convolutional neural networks, recurrent neural networks, and mixture density networks). The latter was concluded to outperform existing ML models based on a smaller root MSE (Kumar Shukla et al., 2020).
1.4.4 Drug discovery toxicology
ML can enhance drug discovery by optimizing candidate selection and predicting the likelihood of success in the development pipeline by mitigating toxicity. Toxicity has traditionally been a significant cause of drug attrition, which is why significant efforts are made to root out potentially harmful compounds during the hit or hit-to-lead phase of active pharmaceutical ingredient identification. ADMET (absorption, distribution, metabolism, excretion, toxicity) prediction is of significant importance in this area. Tools for the end users are becoming increasingly accessible. This includes ADMET Predictor®, which is becoming increasingly important concerning time and cost saving for researchers and industry. It appears that supervised learning algorithms for ADMET are more frequently applied, although this is continually evolving (Maltarollo et al., 2015). Fast supervised learning methods (random forest, deep and graphical neural networks) are used for toxicity (and efficacy). In contrast, unsupervised learning methods (including clustering, biomarker extraction, and generative autoencoders) are used for drug design (Badwan et al., 2023).
1.4.5 Risk assessment
ML contributes to risk assessment models, improving our ability to evaluate and manage potential risks associated with chemical exposures. A combination of large data sets, such as toxicogenomics and high-throughput screening (in vitro) with ML, aids in unravelling toxicity pathways, analyzing genomic data, and screening large datasets, advancing our understanding of toxicological mechanisms but also risk. Risk assessment or chemical safety assessment relies heavily on large datasets, but molecular informatics (chemoinformatics) can be used to draw reliable conclusions for policymakers. The use of computational approaches appears to be the path forward in this respect. Considerable effort is being placed on the next-generation of risk assessment. The ChemTunes•ToxGPS® platform is a commercial product, which draws on the combination of integrating numerous databases, including physicochemical parameters, xenobiotic metabolism, toxicokinetics, ToxCast/Tox21 database (EPA, 2023), and others. This study used a hybrid platform to combine ML methods with quantitative structure-toxicity relationships for a final assessment of a given compound (Yang et al., 2023). The study focused on cosmetic products, for which testing has evolved from animal (in vivo) to in vitro to in silico approaches over the last few decades. The term read-across is usually found in large-scale risk assessment strategies to provide a method for hazard identification in instances, where all the data for a specific compound is unknown or unavailable (e.g., unknown physicochemical parameters, toxicity, etc.). The read-across approach is based on analogs to fill the data gap in these instances, which can be significant when considering thousands of chemicals.
An important consideration for risk assessment involves the in silico adverse outcome pathways (AOPs) and integrated approaches to testing and assessment (IATA). In silico modelling can aid in moving from structure-activity relationships to more advanced methods of deep learning within the context of AOPs. Machine learning models have evolved from structure-activity approaches, but are critiqued if using black box algorithms. The approach to utilize AOPs can relate an exposure to a molecular initiating event, which links to a key event that can form an AOP network (Hemmerich and Ecker, 2020). Some examples have been successfully applied to neurotoxicity studies in producing models that predict xenobiotics agents that can cause deleterious effects, such as Parkinsonian motor deficits (Kan et al., 2022; Dong et al., 2023). While AOPs tend to focus mainly on mechanistic understandings for prediction, the IATA represent a broader framework that integrates data and methods from various sources, including AOPs. As such, an IATA represents a more holistic assessment of toxicity that is useful for regulatory decision making (Willett, 2019). The approach of IATA or next-generation risk assessment should utilize animal data (keeping the 3 R’s in mind) in combination with in vitro data, in silico data, non-mammalian data, biomarkers and epidemiology studies for a comprehensive assessment of chemical hazards (Tollefsen et al., 2014; Halappanavar et al., 2020; Bajard et al., 2023).
1.5 Some applications of supervised and unsupervised learning for toxicological problems
1.5.1 The iris classification using decision trees
The primary goal of a decision tree is to create a model that predicts the value of a target variable based on several input features. This approach comprises training, model building, prediction, and evaluation. The decision tree approach can be used for classification (e.g., toxic vs. non-toxic) or regression (e.g., structure-activity relationships). The key difference between these approaches is that classification outputs a label, whereas regression produces a continuum of values (e.g., IC50 concentrations to halt cancer cell growth). A combination of these methods is called CARTs. Just like any approach, the decision tree algorithm has advantages and disadvantages.
The classic example of a decision tree that is frequently encountered is the iris flower classification. This illustrative example is based on work relating the flower petal characteristics (inputs) to its species (output). The original study was presented by the statistician Ronald Fisher, from the iris flower derived from the dataset from Edgar Anderson in the Gaspe peninsula, Quebec, Canada (Fisher, 1936). The decision tree still appears most useful for this application, although others have been proposed (Poojithaa and Malathib, 2022).
1.5.2 Classification of cannabis strains and their effects
Data availability is a critical aspect of any successful ML application. In the case of cannabis, the website Leafly.com contains products and consumer-reported behavioural profiles of cannabis products. Though the abundance of data is essential, it is just as important to have good-quality data. Relatable examples of toxicological questions today can utilize cannabis classification. It is assumed that consumer cannabis consumption is relatively innocuous, and the consumer can achieve their desired effect based on the selected type (i.e., indica, sativa, or hybrid). However, there are a wide variety of strains that have diverse, potentially adverse effects in some situations. A GitHub repository used logistic regression to analyze the dataset containing 2,350 unique strains of cannabis. Based on the provided input, this repository reported a 63.4% accuracy in predicting new strain types (Kowel, 2019). Part of the challenge is that the data relies on user responses. For example, the indica type “9-pound hammer” strain produces “Relaxed, Sleepy, Euphoric, Happy, Hungry” effects, whereas the indica type “Athabasca” strain yields “Talkative, Uplifted, Happy, Relaxed, Giggly” effects. Though both strains have relaxed as a common characteristic, the former also produces sleepiness, which may be more of a concern for impairment of activities. A comprehensive study that associated subject response with flavour and chemical composition used methods including the random forest to discriminate between effect and flavour vs. strain type (de la Fuente et al., 2020). The findings suggested that flavour perception could be a reliable marker for the behavioural (e.g., psychoactive) effects of specific strains of cannabis.
1.5.3 Predicting carcinogenicity and mutagenicity
Another important application of decision trees involves the classification of carcinogens. According to the International Agency for Research on Cancer classifications, chemicals can be classified based on evidence for causing human cancer: Group 1 (carcinogenic to humans), Group 2A (probably carcinogenic to humans), Group 2B (possibly carcinogenic to humans), and Group 3 (not classifiable as to its carcinogenicity to humans) (https://monographs.iarc.who.int/agents-classified-by-the-iarc/). About five decades ago, a publication on estimating a toxic hazard proposed a decision tree of 33 levels (questions) to categorize a compound as low, moderate, or seriously toxic (Cramer et al., 1976). More recently, a combined model used a hybrid neural network compared with other machine learning algorithms, including a modified decision tree, random forest, and others (Limbu and Dakshanamurthy, 2022). Although the “bagged” decision tree (which reduces variance and overfitting) was found to provide statistically indistinguishable accuracy, AUC, sensitivity, and specificity for a dataset of carcinogens, the authors suggested their hybrid neural network was superior (Limbu and Dakshanamurthy, 2022). The employed algorithms appear critically important for the predictive outcome. A random forest method that used physicochemical and structural parameters could predict over 70% mutagens in a test set of 1,400 carcinogenic compounds (Moorthy et al., 2017). The gold standard for mutagenicity testing is the Ames test, which is based on the ability of a xenobiotic to cause mutations in the bacterium, S. typhimurium. The bacteria that are used have a defect preventing them from growing without histidine. If the xenobiotic corrects this defect (histidine reversion), the bacteria can grow, suggesting that the chemical is likely mutagenic (and thus, potentially carcinogenic). Xenobiotic metabolites can be assessed via an activating system, such as liver enzyme fractions. There has been a sufficient buildup of data to facilitate predictive techniques for mutagenicity. A study sought to enhance mutagen prediction by employing “multiple instance learning,” aiming to augment traditional “single instance” learning methods. This approach aimed to capture various aspects of xenobiotics, including distinct metabolite activities arising from enzymatic bioactivation, which often differ from the mutagenic activity of the parent compound (Feeney et al., 2023). Another important consideration relates to the input of data from the bacteria themselves. A study using neural network ML models employed splitting of strain tasks. Multitask neural networks were more accurate than single-task neural networks, and grouped multitask neural networks were superior, most likely because the latter was grouped rationally by mutagenic and metabolic mechanisms (Lui et al., 2023). However, complete automation and reliance on the ML models may not produce the most accurate results.
1.5.4 Using Hidden Markov Models
Hidden Markov Models (HMMs) are characterized by a stochastic chain of events or states that adhere to the Markov property. The Markov property states that the probability of transitioning from one state to another depends only on the current state and not on the events that preceded it. This characteristic has been described as being “memoryless” (Kouemou and Dymarski, 2011).
HMMs represent a specific type of probabilistic model, wherein the system behaves as a Markov process with “hidden” states being not directly observable. These hidden states influence the observed events, introducing a level of abstraction to the modelling process. HMMs assume a hidden process, A, that can be inferred upon observable states, B. The goal is to learn about A (hidden) by observing B (apparent). As HMMs are built on a time course, the effect of A can be inferred by observing B during each time interval, for which there is an associated probability distribution. These hidden states form a Markov chain, where each state represents a particular situation at a given time.
The Central Limit Theorem can apply to Markov chains under conditions like irreducibility and aperiodicity. With enough steps, the distribution of state values converges to a normal distribution.
In the context of HMMs, the transition between hidden states is governed by transition probabilities. These probabilities determine the likelihood of moving from one hidden state to another, providing a framework for understanding the dynamic nature of the system (Gámiz et al., 2023). The utilization of hidden states in HMMs allows for modelling scenarios where certain aspects of the system are not directly measurable, making it a valuable tool in various fields, including finance, speech recognition, and bioinformatics.
1.5.5 Application of HMMs in toxicology
A significant advance in the application of HMMs evolved due to ample sequenced bacterial genomes in the late 1990s. The HMMs are used in bioinformatics, for example, identifying open reading frames (ORFs) in nucleotide sequences. An open reading frame is a part of a reading frame in a DNA sequence that has the potential to be translated into a protein. In prokaryotic nucleotide sequences, HMMs can be used to identify ORFs by modeling various hidden states. These hidden states include start and stop codons (which signal the beginning and end of a protein-coding region), non-coding regions, and coding regions in both the forward and reverse directions (Lukashin and Borodovsky, 1998). Similarly, HMMs have also been constructed for use in modelling eukaryotic genes (Yoon, 2009). Furthermore, an interesting application of gene findings for toxins (e.g., Shiga toxin) was reported to use a modified HMM to identify potential bacterial toxins, including sequences related to virulence factor and antimicrobial resistance. This HMM model was reported to be faster and more specific than other approaches that depend on BLAST searches (Xie and Fair, 2021).
Key advances should be discussed regarding the application of HMMs to toxicology, which appear to be less commonly used than other algorithms. The potential for biomarker discovery, toxicokinetics, xenobiotic exposure, and toxicological responses are all attainable objectives in using HMMs. The models can be used to analyze genes (toxicogenomics), protein expression (toxicoproteomics), and metabolite analysis (toxicometabolomics) patterns in response to a toxin or toxicant, where the hidden states can be indicative of stage or pathobiology severity. The power of this approach is that the algorithms are data-driven, not rule-based, which can provide findings that could be overlooked (Martinelli, 2023).
There are opportunities to apply HMMs to toxicological problems, although there are few applications in this area. Employing an HMM model for ADMET parameters of toxicant exposure in an organism or model system, where the hidden states in this scenario could reveal specific transporters, drug-metabolizing enzymes, and physicochemical properties of the toxicant itself. The toxicity (hidden states) occurring from toxicant exposure could be revealed based on the toxidrome (symptoms) in a clinical setting. For example, an HMM was recently used to predict schizophrenia (the hidden state) based on motor activity (the observed state) (Boeker et al., 2023). Also, a study evaluating different states of depression in youth over 90 weeks (time-series data is well-suited for this algorithm) used HMMs to report that some were more likely to transition from a low to a highly depressed state and the need for intervention (Liu et al., 2023). In evaluating opioid use disorder patients over 12 months, an HMM model could predict the effect of addiction consult services on the disposition of those individuals (King et al., 2021).
2 Looking forward—unlocking the future of ML in toxicological sciences
This foundational exploration of ML applications in toxicological sciences presented will continue to grow [the term ToxAIcology was coined recently (Hartung, 2023)]. This short review has covered fundamental concepts, highlighted key algorithms, and introduced the workflow that guides effective ML model implementation in specific areas of toxicology. The algorithms used in toxicological studies range from neural networks and decision trees as the most popular, followed by deep learning, support vector machines, random forests, and HMMs. However, there are essential considerations from an overall vantage point that should be emphasized when undertaking an ML-based approach to scientific inquiry. A growing number of studies are evolving beyond the single-instance ML approach and are using multi-instance ML techniques across platforms. Findings from clinical studies can positively interplay with risk assessment in the sense that there are large numbers of patients or individuals for which different sources of risk must be mitigated (i.e., drugs vs. industrial chemicals). Combining in vitro, in vivo, and clinical data with ML algorithms using multi-task deep neural networks is a beneficial and powerful approach. However, in the study of Sharma et al. (Figure 8), clinical toxicity predictability was more accurately modelled (based on AUC) from in vitro data rather than mouse in vivo data (Sharma et al., 2023). Despite its seemingly contradictory nature, this outcome highlights the critical need for vigilance against bias, as it arises from the chemical parallels in the in vitro (Tox21) and clinical (ClinTox) databases.
FIGURE 8. A representation of learning transfer from in vitro and in vivo platforms to clinical toxicity. It will be important to determine which models more accurately predict clinical toxicity, discussed in Sharma et al. (2023).
2.1 Evaluating prospects: navigating challenges and embracing opportunities
A foundational requirement for any ML model is high-quality and available data. Toxicological datasets often come from varied sources, potentially leading to inconsistencies. While there are techniques to delineate the distinctions, particularly in database comparison, these can nonetheless impede the pace of advancement. The multifaceted challenges of big data are often represented by a series of “V”s: volume, velocity, variety, veracity, variability, validity, visibility, visualization, volatility, and value, each reflecting a critical dimension of this complex field (Richarz et al., 2019). Furthermore, issues can arise in terms of the availability of data within a particular chemical space. Lastly, if datasets are biased towards certain compounds or outcomes, models can perpetuate and amplify those biases, leading to misclassifications or missing potential toxicants.
• Opportunity: Rigorous data curation and addressing biases can enhance model accuracy, which is achievable. Enriched analyses can realize personalized toxicology to assess individual risk, and the intercommunications of large omics data sets can bring significant progress. Prediction of unknown toxicological entities with accuracy is an ideal outcome.
• Challenge: Ensuring representativeness and fairness (i.e., no bias) in datasets remains challenging. General rules may miss rare ADRs or rare toxicological events.
The interpretation of a given model can also become a challenge for some ML models. The “black box” models refer to a level of complexity in how the ML methods work that is unclear to the individual involved. For example, clinicians who have relied on linear or logistic regression find barriers if the ML algorithm is not transparent (Vellido, 2020). This has been a concern for clinicians for some time, which is quite understandable if it can affect patients’ outcomes (i.e., whether they live or die) (Stiglic et al., 2020; Petch et al., 2022). This kind of “black box” (as with the black box warning) can be problematic in a field like toxicology, where understanding the underlying mechanisms is vital.
• Opportunity: understanding how models arrive at specific predictions will provide transparency, enhance trust and facilitate broader acceptance and uptake.
• Challenge: the trade-off between model complexity and interpretability can limit the predictive power of simpler models. Complex models may provide superior accuracy but lack interpretability. Bridging the gap between traditional toxicological methods and the reliability of ML algorithms remains an ongoing challenge.
There are numerous ethical considerations for using ML in toxicology, which will depend on the specific setting. Key considerations across toxicology disciplines include accuracy, reliability, and reproducibility. Experimentalists need to have a clear understanding of the algorithms for the sake of transparency and mechanistic understanding. Such issues also affect the uptake in regulatory affairs and risk management. The accountability of such systems currently rests with the individuals creating the ML toxicology applications. Some of the issues described above regarding data sets’ accuracy and reliability tie into the ethical considerations here, especially if they are used to make a decision that affects populations and society as a whole (Guan et al., 2022).
• Opportunity: Enhance the 3 R’s (replacement, reduction, refinement) for animal welfare (Hubrecht and Carter, 2019), guide alternative testing for in vitro—in silico models, and engage automated systems for patient health management and protect populations efficiently.
• Challenges: Reliance on ML algorithms for direction and guidance that brings harm, who (or what) is ultimately accountable in decision-making, and transparency for the widespread use of complex “black box” ML models in regulatory and public policy settings. Striking the right balance and ensuring collaboration between ML and human experts is critical.
Machine learning is a powerful tool in toxicology research that has the potential to revolutionize the field. By enabling the rapid identification of potential toxicants and new drug targets, machine learning can help develop safe and effective drugs and reduce the impact of environmental pollution on human health. However, the use of machine learning in toxicology also raises ethical and regulatory concerns that must be addressed to ensure this technology is safe and responsible use. Just as these techniques can be used to optimize drug efficacy, the same principles can be used for developing poisons. The calls for regulatory oversight must be addressed (Meskó and Topol, 2023).
Author contributions
LT: Conceptualization, Visualization, Writing–original draft, Writing–review and editing. AS: Writing–review and editing.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ampadu, H. H., Hoekman, J., de Bruin, M. L., Pal, S. N., Olsson, S., Sartori, D., et al. (2016). Adverse drug reaction reporting in africa and a comparison of individual case safety report characteristics between africa and the rest of the world: analyses of spontaneous reports in VigiBase. Drug Saf. 39(4), 335–345. doi:10.1007/s40264-015-0387-4
Badwan, B. A., Liaropoulos, G., Kyrodimos, E., Skaltsas, D., Tsirigos, A., and Gorgoulis, V. G. (2023). Machine learning approaches to predict drug efficacy and toxicity in oncology. Cell. Rep. Methods 3 (2), 100413. doi:10.1016/j.crmeth.2023.100413
Bajard, L., Adamovsky, O., Audouze, K., Baken, K., Barouki, R., Beltman, J. B., et al. (2023). Application of AOPs to assist regulatory assessment of chemical risks - case studies, needs and recommendations. Environ. Res. 217, 114650. doi:10.1016/j.envres.2022.114650
Besco, K., and Keller, M. E. (2022). “Medication safety I: adverse drug reactions,” in Drug Information: a Guide for Pharmacists, 7e. Editors P. M. Malone, B. A. Witt, M. J. Malone, and D. M. Peterson (New York, NY: McGraw Hill).
Boeker, M., Hammer, H. L., Riegler, M. A., Halvorsen, P., and Jakobsen, P. (2023). Prediction of schizophrenia from activity data using hidden Markov model parameters. Neural Comput. Appl. 35 (8), 5619–5630. doi:10.1007/s00521-022-07845-7
Bussola, N., Xu, J., Wu, L., Gorini, L., Zhang, Y., Furlanello, C., et al. (2023). A weakly supervised deep learning framework for whole slide classification to facilitate digital pathology in animal study. Chem. Res. Toxicol. 36 (8), 1321–1331. doi:10.1021/acs.chemrestox.3c00058
Cavasotto, C. N., and Scardino, V. (2022). Machine learning toxicity prediction: latest advances by toxicity end point. ACS Omega 7 (51), 47536–47546. doi:10.1021/acsomega.2c05693
Chalasani, N., Fontana, R. J., Bonkovsky, H. L., Watkins, P. B., Davern, T., Serrano, J., et al. (2008). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 135 (6), 1924–1934. doi:10.1053/j.gastro.2008.09.011
Chen, M., Suzuki, A., Thakkar, S., Yu, K., Hu, C., and Tong, W. (2016). DILIrank: the largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov. Today 21 (4), 648–653. doi:10.1016/j.drudis.2016.02.015
Cramer, G. M., Ford, R. A., and Hall, R. L. (1976). Estimation of toxic hazard—a decision tree approach. Food Cosmet. Toxicol. 16 (3), 255–276. doi:10.1016/s0015-6264(76)80522-6
de la Fuente, A., Zamberlan, F., Sánchez Ferrán, A., Carrillo, F., Tagliazucchi, E., and Pallavicini, C. (2020). Relationship among subjective responses, flavor, and chemical composition across more than 800 commercial cannabis varieties. J. Cannabis Res. 2 (1), 21. doi:10.1186/s42238-020-00028-y
Demler, T. L., and Chehovich, C. (2021). Trends of adverse drug reaction reports in a hospitalized psychiatric population: exploring prescriber discontinuations as potential unreported adverse drug events. Innov. Clin. Neurosci. 18 (7-9), 29–38.
Dong, L., Wang, S., Wang, X., Wang, Z., Liu, D., and You, H. (2023). Investigating the adverse outcome pathways (AOP) of neurotoxicity induced by DBDPE with a combination of in vitro and in silico approaches. J. Hazard Mater 449, 131021. doi:10.1016/j.jhazmat.2023.131021
Duffull, S., and Isbister, G. (2022). Challenges faced when modeling clinical toxicology and toxinology events. CPT Pharmacometrics Syst. Pharmacol. 11 (5), 532–534. doi:10.1002/psp4.12792
Edwards, I. R., and Aronson, J. K. (2000). Adverse drug reactions: definitions, diagnosis, and management. Lancet 356 (9237), 1255–1259. doi:10.1016/S0140-6736(00)02799-9
Epa, U. (2023). Exploring ToxCast Data Available at: https://www.epa.gov/chemical-research/exploring-toxcast-data.
Feeney, S. V., Lui, R., Guan, D., and Matthews, S. (2023). Multiple instance learning improves ames mutagenicity prediction for problematic molecular species. Chem. Res. Toxicol. 36 (8), 1227–1237. doi:10.1021/acs.chemrestox.2c00372
Fisher, R. A. (1936). The use of multiple measurements in taxonomic problems. Ann. Eugen. 7, 179–188. doi:10.1111/j.1469-1809.1936.tb02137.x
Free, S. M., and Wilson, J. W. (1964). A mathematical contribution to structure-activity studies. J. Med. Chem. 7 (4), 395–399. doi:10.1021/jm00334a001
Gámiz, M. L., Limnios, N., and Segovia-García, M. C. (2023). Hidden markov models in reliability and maintenance. Eur. J. Operational Res. 304 (3), 1242–1255. doi:10.1016/j.ejor.2022.05.006
Garcia de Lomana, M., Marin Zapata, P. A., and Montanari, F. (2023). Predicting the mitochondrial toxicity of small molecules: insights from mechanistic assays and cell painting data. Chem. Res. Toxicol. 36 (7), 1107–1120. doi:10.1021/acs.chemrestox.3c00086
Gill, J., Moullet, M., Martinsson, A., Miljković, F., Williamson, B., Arends, R. H., et al. (2023). Evaluating the performance of machine-learning regression models for pharmacokinetic drug–drug interactions. CPT Pharmacometrics Syst. Pharmacol. 12 (1), 122–134. doi:10.1002/psp4.12884
Guan, H., Dong, L., and Zhao, A. (2022). Ethical risk factors and mechanisms in artificial intelligence decision making. Behav. Sci. (Basel). 12 (9), 343. doi:10.3390/bs12090343
Guo, H., Zhang, P., Zhang, R., Hua, Y., Zhang, P., Cui, X., et al. (2022). The use of Kumpfer's resilience framework in understanding the breastfeeding experience of employed mothers after returning to work: a qualitative study in China. Front. Immunol. 17, 13. doi:10.1186/s13006-022-00459-8
Gupta, R., Srivastava, D., Sahu, M., Tiwari, S., Ambasta, R. K., and Kumar, P. (2021). Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol. Divers. 25 (3), 1315–1360. doi:10.1007/s11030-021-10217-3
Halappanavar, S., van den Brule, S., Nymark, P., Gaté, L., Seidel, C., Valentino, S., et al. (2020). Adverse outcome pathways as a tool for the design of testing strategies to support the safety assessment of emerging advanced materials at the nanoscale. Part Fibre Toxicol. 17 (1), 16. doi:10.1186/s12989-020-00344-4
Hansch, C., and Fujita, T. (1964). p-σ-π analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 86 (8), 1616–1626. doi:10.1021/ja01062a035
Hartung, T. (2023). ToxAIcology - the evolving role of artificial intelligence in advancing toxicology and modernizing regulatory science. Altex 40 (4), 559–570. doi:10.14573/altex.2309191
Hemmerich, J., and Ecker, G. F. (2020). In silico toxicology: from structure-activity relationships towards deep learning and adverse outcome pathways. Wiley Interdiscip. Rev. Comput. Mol. Sci. 10 (4), e1475. doi:10.1002/wcms.1475
Hu, Y., Ren, Q., Liu, X., Gao, L., Xiao, L., and Yu, W. (2023). In silico prediction of human organ toxicity via artificial intelligence methods. Chem. Res. Toxicol. 36 (7), 1044–1054. doi:10.1021/acs.chemrestox.2c00411
Hubrecht, R. C., and Carter, E. (2019). The 3Rs and humane experimental technique: implementing change. Anim. (Basel) 9 (10), 754. doi:10.3390/ani9100754
Igarashi, Y., Nakatsu, N., Yamashita, T., Ono, A., Ohno, Y., Urushidani, T., et al. (2014). Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Res. 43 (D1), D921–D927. doi:10.1093/nar/gku955
Kan, H. L., Tung, C. W., Chang, S. E., and Lin, Y. C. (2022). In silico prediction of parkinsonian motor deficits-related neurotoxicants based on the adverse outcome pathway concept. Arch. Toxicol. 96 (12), 3305–3314. doi:10.1007/s00204-022-03376-1
Keshava, C., Nicolai, S., Vulimiri, S. V., Cruz, F. A., Ghoreishi, N., Knueppel, S., et al. (2023). Application of systematic evidence mapping to identify available data on the potential human health hazards of selected market-relevant azo dyes. Environ. Int. 176, 107952. doi:10.1016/j.envint.2023.107952
King, C. A., Englander, H., Korthuis, P. T., Barocas, J. A., McConnell, K. J., Morris, C. D., et al. (2021). Designing and validating a Markov model for hospital-based addiction consult service impact on 12-month drug and non-drug related mortality. PLoS One 16 (9), e0256793. doi:10.1371/journal.pone.0256793
Kouemou, G. L., and Dymarski, D. P. (2011). History and theoretical basics of hidden Markov models. in Hidden Markov models, theory and applications, 1.
Kubinyi, H. (2002). From narcosis to hyperspace: the history of QSAR. Quant. Structure-Activity Relat. 21 (4), 348–356. doi:10.1002/1521-3838(200210)21:4<348::aid-qsar348>3.0.co;2-d
Kumar Shukla, P., Kumar Shukla, P., Sharma, P., Rawat, P., Samar, J., Moriwal, R., et al. (2020). Efficient prediction of drug-drug interaction using deep learning models. IET Syst. Biol. 14 (4), 211–216. doi:10.1049/iet-syb.2019.0116
Lee, C. Y., and Chen, Y.-P. P. (2019). Machine learning on adverse drug reactions for pharmacovigilance. Drug Discov. Today 24 (7), 1332–1343. doi:10.1016/j.drudis.2019.03.003
Li, L., Lu, Z., Liu, G., Tang, Y., and Li, W. (2023). Machine learning models to predict cytochrome P450 2B6 inhibitors and substrates. Chem. Res. Toxicol. 36 (8), 1332–1344. doi:10.1021/acs.chemrestox.3c00065
Limbu, S., and Dakshanamurthy, S. (2022). Predicting chemical carcinogens using a hybrid neural network deep learning method. Sensors 22 (21), 8185. doi:10.3390/s22218185
Liu, Q., Cole, D., Tran, T., Quinn, M., McCauley, E., Diamond, G., et al. (2023). Intraindividual phenotyping of depression in high-risk youth: an application of a multilevel hidden Markov model. Dev. Psychopathol., 1–10. doi:10.1017/S0954579423000500
Lo, Y. C., Rensi, S. E., Torng, W., and Altman, R. B. (2018). Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 23 (8), 1538–1546. doi:10.1016/j.drudis.2018.05.010
Lui, R., Guan, D., and Matthews, S. (2023). Mechanistic task groupings enhance multitask deep learning of strain-specific ames mutagenicity. Chem. Res. Toxicol. 36 (8), 1248–1254. doi:10.1021/acs.chemrestox.2c00385
Lukashin, A. V., and Borodovsky, M. (1998). GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26 (4), 1107–1115. doi:10.1093/nar/26.4.1107
Maltarollo, V. G., Gertrudes, J. C., Oliveira, P. R., and Honorio, K. M. (2015). Applying machine learning techniques for ADME-Tox prediction: a review. Expert Opin. Drug Metabolism Toxicol. 11 (2), 259–271. doi:10.1517/17425255.2015.980814
Martin, Y. C. (2012). Hansch analysis 50 years on. WIREs Comput. Mol. Sci. 2 (3), 435–442. doi:10.1002/wcms.1096
Martinelli, D. D. (2023). Machine learning for metabolomics research in drug discovery. Intelligence-Based Med. 8, 100101. doi:10.1016/j.ibmed.2023.100101
Mehrvar, S., Himmel, L. E., Babburi, P., Goldberg, A. L., Guffroy, M., Janardhan, K., et al. (2021). Deep learning approaches and applications in toxicologic histopathology: current status and future perspectives. J. Pathology Inf. 12 (1), 42. doi:10.4103/jpi.jpi_36_21
Meskó, B., and Topol, E. J. (2023). The imperative for regulatory oversight of large language models (or generative AI) in healthcare. npj Digit. Med. 6 (1), 120. doi:10.1038/s41746-023-00873-0
Moein, M., Heinonen, M., Mesens, N., Chamanza, R., Amuzie, C., Will, Y., et al. (2023). Chemistry-based modeling on phenotype-based drug-induced liver injury annotation: from public to proprietary data. Chem. Res. Toxicol. 36 (8), 1238–1247. doi:10.1021/acs.chemrestox.2c00378
Moorthy, NSHN, Kumar, S., and Poongavanam, V. (2017). Classification of carcinogenic and mutagenic properties using machine learning method. Comput. Toxicol. 3, 33–43. doi:10.1016/j.comtox.2017.07.002
M. Muntean, and F.-D. Militaru (2023). Metrics for evaluating classification Algorithms2023 (Singapore: Springer Nature Singapore).
Parameswaran Nair, N., Chalmers, L., Peterson, G. M., Bereznicki, B. J., Castelino, R. L., and Bereznicki, L. R. (2016). Hospitalization in older patients due to adverse drug reactions -the need for a prediction tool. Clin. Interv. Aging 11, 497–505. doi:10.2147/CIA.S99097
Petch, J., Di, S., and Nelson, W. (2022). Opening the black box: the promise and limitations of explainable machine learning in cardiology. Can. J. Cardiol. 38 (2), 204–213. doi:10.1016/j.cjca.2021.09.004
Pisani, L., de Candia, M., Rullo, M., and Altomare, C. D. (2023). Hansch-type QSAR models for the rational design of MAO inhibitors: basic principles and methodology. Methods Mol. Biol. 2558, 207–220. doi:10.1007/978-1-0716-2643-6_16
Poojithaa, M., and Malathib, K. (2022). Decision tree over support vector machine for better accuracy in identifying the problem based on the Iris flower. Adv. Parallel Comput. Algorithms, Tools Paradigms 41, 209. doi:10.3233/apc220028
Prasad, N., Lau, E. C. Y., Wojt, I., Penm, J., Dai, Z., and Tan, E. C. K. (2023). Prevalence of and risk factors for drug-related readmissions in older adults: a systematic review and meta-analysis. Drugs Aging 41, 1–11. doi:10.1007/s40266-023-01076-8
Rashidi, H. H., Albahra, S., Robertson, S., Tran, N. K., and Hu, B. (2023). Common statistical concepts in the supervised Machine Learning arena. Front. Oncol. 13, 1130229. doi:10.3389/fonc.2023.1130229
Ribba, B., Dudal, S., Lavé, T., and Peck, R. W. (2020). Model-informed artificial intelligence: reinforcement learning for precision dosing. Clin. Pharmacol. Ther. 107, 853–857. doi:10.1002/cpt.1777
Richarz, A.-N. (2019). “Big data in predictive toxicology: challenges, opportunities and perspectives,” in Big data in predictive toxicology. Editors D. Neagu, and A.-N. Richarz (Croydon, England: CPI Group (UK) Ltd).
Sharma, B., Chenthamarakshan, V., Dhurandhar, A., Pereira, S., Hendler, J. A., Dordick, J. S., et al. (2023). Accurate clinical toxicity prediction using multi-task deep neural nets and contrastive molecular explanations. Sci. Rep. 13 (1), 4908. doi:10.1038/s41598-023-31169-8
Sinha, K., Ghosh, N., and Sil, P. C. (2023). A review on the recent applications of deep learning in predictive drug toxicological studies. Chem. Res. Toxicol. 36 (8), 1174–1205. doi:10.1021/acs.chemrestox.2c00375
Smajić, A., Rami, I., Sosnin, S., and Ecker, G. F. (2023). Identifying differences in the performance of machine learning models for off-targets trained on publicly available and proprietary data sets. Chem. Res. Toxicol. 36 (8), 1300–1312. doi:10.1021/acs.chemrestox.3c00042
Stiglic, G., Kocbek, P., Fijacko, N., Zitnik, M., Verbert, K., and Cilar, L. (2020). Interpretability of machine learning-based prediction models in healthcare. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 10 (5), e1379. doi:10.1002/widm.1379
Tollefsen, K. E., Scholz, S., Cronin, M. T., Edwards, S. W., de Knecht, J., Crofton, K., et al. (2014). Applying adverse outcome pathways (AOPs) to support integrated approaches to testing and assessment (IATA). Regul. Toxicol. Pharmacol. 70 (3), 629–640. doi:10.1016/j.yrtph.2014.09.009
Valero-Carreras, D., Alcaraz, J., and Landete, M. (2023). Comparing two SVM models through different metrics based on the confusion matrix. Comput. Operations Res. 152, 106131. doi:10.1016/j.cor.2022.106131
Vellido, A. (2020). The importance of interpretability and visualization in machine learning for applications in medicine and health care. Neural Comput. Appl. 32 (24), 18069–18083. doi:10.1007/s00521-019-04051-w
C. Willett (2019). The use of adverse outcome pathways (AOPs) to support chemical safety decisions within the context of integrated approaches to testing and assessment (IATA) (Singapore: Springer Singapore).
Xie, G., and Fair, J. M. (2021). Hidden Markov Model: a shortest unique representative approach to detect the protein toxins, virulence factors and antibiotic resistance genes. BMC Res. Notes 14 (1), 122. doi:10.1186/s13104-021-05531-w
Yang, C., Rathman, J. F., Bienfait, B., Burbank, M., Detroyer, A., Enoch, S. J., et al. (2023). The role of a molecular informatics platform to support next generation risk assessment. Comput. Toxicol. 26, 100272. doi:10.1016/j.comtox.2023.100272
Yang, H., Sun, L., Li, W., Liu, G., and Tang, Y. (2018). Corrigendum: in silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front. Chem. 6, 129. doi:10.3389/fchem.2018.00129
Yauk, C. L., Cheung, C., Barton-Maclaren, T. S., Boucher, S., Bourdon-Lacombe, J., Chauhan, V., et al. (2019). Toxicogenomic applications in risk assessment at Health Canada. Curr. Opin. Toxicol. 18, 34–45. doi:10.1016/j.cotox.2019.02.005
Yoon, B. J. (2009). Hidden markov models and their applications in biological sequence analysis. Curr. Genomics 10 (6), 402–415. doi:10.2174/138920209789177575
Zhang, F., Sun, B., Diao, X., Zhao, W., and Shu, T. (2021). Prediction of adverse drug reactions based on knowledge graph embedding. BMC Med. Inf. Decis. Mak. 21 (1), 38. doi:10.1186/s12911-021-01402-3
Keywords: artificial intelligence (AI), machine learning (ML), mean squared error (MSE), adverse drug reaction (ADR), drug-induced liver injury (DILI), toxicology, drug toxicity, metabolism
Citation: Tonoyan L and Siraki AG (2024) Machine learning in toxicological sciences: opportunities for assessing drug toxicity. Front. Drug Discov. 4:1336025. doi: 10.3389/fddsv.2024.1336025
Received: 09 November 2023; Accepted: 18 January 2024;
Published: 08 February 2024.
Edited by:
Md Saifur Rahman Khan, University of Toronto, CanadaReviewed by:
Rodolpho C. Braga, InsilicAll, BrazilFernando Prieto-Martínez, National Autonomous University of Mexico, Mexico
Copyright © 2024 Tonoyan and Siraki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arno G. Siraki, c2lyYWtpQHVhbGJlcnRhLmNh