
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Drug Discov., 25 July 2023
Sec. In silico Methods and Artificial Intelligence for Drug Discovery
Volume 3 - 2023 | https://doi.org/10.3389/fddsv.2023.1237655
 Mateus Sá Magalhães Serafim1,2†
Mateus Sá Magalhães Serafim1,2† Simone Queiroz Pantaleão3†
Simone Queiroz Pantaleão3† Elany Barbosa da Silva2James H. McKerrow2
Elany Barbosa da Silva2James H. McKerrow2 Anthony J. O’Donoghue2
Anthony J. O’Donoghue2 Bruno Eduardo Fernandes Mota4Kathia Maria Honorio5,6
Bruno Eduardo Fernandes Mota4Kathia Maria Honorio5,6 Vinícius Gonçalves Maltarollo7*
Vinícius Gonçalves Maltarollo7*Computer-Aided Drug Design (CADD) approaches, such as those employing quantitative structure-activity relationship (QSAR) methods, are known for their ability to uncover novel data from large databases. These approaches can help alleviate the lack of biological and chemical data, but some predictions do not generate sufficient positive information to be useful for biological screenings. QSAR models are often employed to explain biological data of chemicals and to design new chemicals based on their predictions. In this review, we discuss the importance of data set size with a focus on false hits for QSAR approaches. We assess the challenges and reliability of an initial in silico strategy for the virtual screening of bioactive molecules. Lastly, we present a case study reporting a combination approach of hologram-based quantitative structure-activity relationship (HQSAR) models and random forest-based QSAR (RF-QSAR), based on the 3D structures of 25 synthetic SARS-CoV-2 Mpro inhibitors, to virtually screen new compounds for potential inhibitors of enzyme activity. In this study, optimal models were selected and employed to predict Mpro inhibitors from the database Brazilian Compound Library (BraCoLi). Twenty-four compounds were then assessed against SARS-CoV-2 Mpro at 10 µM. At the time of this study (March 2021), the availability of varied and different Mpro inhibitors that were reported definitely affected the reliability of our work. Since no hits were obtained, the data set size, parameters employed, external validations, as well as the applicability domain (AD) could be considered regarding false hits data contribution, aiming to enhance the design and discovery of new bioactive molecules.
Computational approaches, such as machine learning (ML) techniques (Rodríguez-Pérez and Bajorath, 2021), have helped strategies into the drug design and discovery scenario (Lima et al., 2016), such as obtaining new compounds with antibacterial (Serafim et al., 2020), antiparasitic (Veríssimo et al., 2019), and antiviral (Serafim et al., 2021a) activity. Quantitative structure–activity relationship (QSAR) methods have the potential to predict physicochemical properties as quantitative structure-property relationship (QSPR) (Lu et al., 2019) and various biological activities (i.e., different end-points), such as protein/enzyme inhibitors, toxicity, and mutagenicity (Gramatica, 2020). In addition, to ensure higher predictive ability and reliability of different models, rigorous (external) validations, and applicability domain (AD) calculations (i.e., the chemical space defined by molecules in a training set) are required (Mathea et al., 2016). Developed in the early 1960s (Hansch and Fujita, 1964), QSAR is a computational tool that uses data collected from one or various databases and literature (Neves et al., 2018) to establish a statistically significant correlation between a given chemical structure and a particular biological activity, property, or category (e.g., active, or inactive) (Cherkasov et al., 2014).
QSAR models have been applied in combination with different methods that either enhance their predictive accuracy or complement the predicted data. These methods include: i) molecular docking analysis, as demonstrated for the design of tyrosinase inhibitors (Dong et al., 2018); ii) ML techniques, such as random forest (RF), as demonstrated for the prediction of synergism between anti-cancer drugs (Sidorov et al., 2019); iii) molecular dynamics (MD) simulations (Rafi et al., 2022) as described for the discovery of SARS-CoV-2 drug candidates; iv) virtual screening (VS.) campaigns for repurposing drugs against COVID-19 (Alves et al., 2021); and even v) principal component analysis (PCA), for prediction of pollutants and hazardous chemicals (Gramatica et al., 2018). Furthermore, QSAR has also been developed in combination with other ML and artificial intelligence (AI) methods (Mao et al., 2021), such as neural networks (NN) with diverse architectures (Chakravarti and Alla, 2019), expanding its applicability to multiple targets, various biological activities, or different property predictions.
Multi-target QSAR models can simultaneously predict compounds’ activity or affinity for multiple targets (e.g., protein), and can be performed using independent target based QSAR models to an integrated approach, such as multitask or multi-target deep neural networks (DNN). Nevertheless, the performance of multitask approaches can be affected by the availability of biological data for multiple targets, suggesting an influence of ML synergies when compared to single target approaches (Rodríguez-Pérez and Bajorath, 2021). In addition, multi-target models may show lower overall performances than single approaches when assessing larger data sets (e.g., 143,310 compounds (Rodríguez-Pérez and Bajorath, 2018)).
One could argue that differences in data sets, such as small data sets (e.g., <1,000 compounds), could be regarded as a limitation to predictive accuracy and performance of computational approaches, as drug discovery is usually favored by large databases (e.g., >10,000) or availability of diverse biological data from experimental determination (Veríssimo et al., 2022). For instance, one-shot learning approaches and techniques could solve these different issues when facing data scarcity, considering approaches to support generating or obtaining enough data to improve existing biological and/or computational methods. Herein, other methods could be employed, such as MD simulations, scoring function space (SFS), and quantum mechanics (QM) (Veríssimo et al., 2022), which do not require learning from external data. However, this is not the reality of QSAR or ML predictors, which require a high amount of data to improve their predictive ability and accuracy.
In this sense, the rate of drug discovery derived from research with scarce available data could be enhanced by a combination of QSAR models with additional methods, such as MD simulations (Rafi et al., 2022). For instance, before in vitro or in vivo experiments are performed (Tolah et al., 2021), computational methods can quickly provide enough data in a cost-effective manner (Sadybekov and Katritch, 2023). To verify accuracy, it is important to then perform experimental validation of the computational hits (Azevedo et al., 2022), such as those obtained from QSAR-based methods (Neves et al., 2018; Tolah et al., 2021), including QSAR-based VS. (Kar and Roy, 2013), a consensus approach used to identify few compounds against a given target (e.g., dopamine receptors) (Cherkasov et al., 2014). This is especially important as we can expect about 12% of predicted compounds from different VS. approaches, against different protein targets, presenting a biological activity (Irwin and Shoichet, 2016), which would contrast to almost 90% of results as false hits.
QSAR models have been used in a consensus VS. strategy to obtain inhibitors against various targets from small, curated data sets. For instance, 2D- and 3D-QSAR models were combined to perform a VS. aiming to select 2′-deoxyuridine 5′-triphosphate nucleotide hydrolase (dUTPase) inhibitors that specifically target the enzyme in chloroquine-sensitive and resistant strains of Plasmodium falciparum, the causative agent of malaria. Here, 127 compounds extracted from the literature were screened to identify hits (Lima et al., 2018). A hologram-based quantitative structure-activity relationship (HQSAR) was used to predict the chemical contributions of compounds, and 3D-QSAR methods were used to assess regions in molecules with favorable and unfavorable interactions. These predictions identified a positive contribution of a trityl ring against P. falciparum dUTPase, as well as regions where the trityl groups are favorable for both inhibition and selectivity. These studies corroborated previous data (Ojha and Roy, 2013). Three of the five hits showed inhibitory activity against different P. falciparum strains, with IC50 values ranging from 6.1 ± 1.95 to 17.1 ± 16.2 µM and with selectivity indexes (SI) over COS7 (monkey kidney fibroblast-like) cells ranging from 2.7 to 11.7 (Lima et al., 2018).
Asse Junior et al. (2020) also employed a fragment-based HQSAR method (Kronenberger et al., 2017) after a VS. of six different chemical libraries (AfroDbNatural Products, BraCoLi, Clean drug-like database from ZINC, FDA approved drugs from ZINC, NuBBE, and Traditional Chinese Medicine database) to select potential inhibitors against enoyl-ACP reductase (FabI) (Asse Junior et al., 2020). Authors included 166 known Staphylococcus aureus FabI compounds classified as active, (IC50 < 1 μM) and inactive (IC50 > 1 μM). Compounds were used to generate models, along with fifty additional decoys generated using the Database of Useful Decoys (Mysinger et al., 2012), as a validation approach. The inhibitory activity against S. aureus FabI was predicted with HQSAR and compounds with desirable prediction values were selected for experimental testing. Among the 14 hits selected from the VS., four showed activity as minimal inhibitory concentration (MIC) against S. aureus clinical isolates ranging from 15.62 to 250 μM, with selectivity indexes (SI) ranging from 0.02 to 641.03 (Asse Junior et al., 2020) over Vero cells.
In another example, HQSAR and random forest-based QSAR (RF-QSAR) models were used to predict the biological activity of nitroimidazole derivatives against Trichomonas vaginalis, the causative agent of trichomoniasis. The model predicted compounds with 5-nitroimidazole with better inhibitory activity than those with 4-nitroimidazole. Additionally, a consensus of both QSAR methods’ predictions resulted in 16 selected compounds, three of which were newly planned nitroimidazole derivatives with confirmed activity against T. vaginalis strains. This is an example of a successful approach gathering information from molecular fragments to explain the importance of different chemical structures and their corresponding anti-infective activity (Veríssimo et al., 2019).
Still, despite successful approaches, false hit predictions are expected from any given predictive model, usually resulting from different methods assessed individually or combined, including classification ML (i.e., probability of a prediction classified as positive or negative using a cutoff) (Handler et al., 2022), QSAR (i.e., calculation of each parameter’s and descriptor’s influence in a model) (Gramatica, 2020), and also VS. (e.g., consensus approach) (Adeshina et al., 2020), which will be further discussed in the case study presented in this work. Notwithstanding, true- and false-positive, as well as negative predictions, can be used to statistically evaluate the robustness of a specific predictor (Tharwat, 2020; Handler et al., 2022). In this sense, one could argue the importance of considering false hits as input for a given predictive model, thus training from true negatives and potentially removing false negatives (Tharwat, 2020), and also of considering the potential induced bias of negative sampling (Sidorczuk et al., 2022). Moreover, there may also be a limited number of activity data available, and an imbalance between the activity classes assessed in QSAR models (Bosc et al., 2019), usually with a small number of active compounds and a large number of inactive compounds (Zakharov et al., 2014), and improving balance should be considered.
For instance, Sidorczuk et al. (2022) generated 660 predictive models with 12 ML architectures (one positive and 11 “negative data sets”), to assess the impact of false hits for antimicrobial peptide predictions. Authors observed that similar training data sets (e.g., peptide sequences from 5 to 100 amino acid residues), generated by the same or by a similar data sampling method (e.g., removing sequences with identity >40%, 70%, or 90%), influenced the predictive model’s performance, thus biasing the analysis. Thus, not only can a model be biased, but it is not possible to know which model would be the most accurate (Sidorczuk et al., 2022). Moreover, Cortes-Ciriano et al. (2015) discussed the effect of random experimental errors (i.e., noise) in the predictive ability of QSAR models, as it is related to the choice and performance of the test set, especially when using algorithms that simulate the bioactivity values of a set. Authors evaluated 12 learning algorithms on 12 data sets, and regardless of the algorithm used, there was a margin of noise involved, highlighting the need to also apply the use of replicates in the generation of QSAR models and use other techniques to jointly decide between the molecules that will be subsequently tested in vitro (Cortes-Ciriano et al., 2015).
To avoid such issues or potentially biased analysis, including obtaining false hits, some practices (Figure 1) for QSAR models’ development, validation, and exploitation can be employed (Tropsha, 2010), aiming to properly curate a given data set, assess a model’s predictive ability, and rank, select or virtually screen compounds in a consensus VS. approach towards experimental validation. Those will be discussed in the topics as follows.
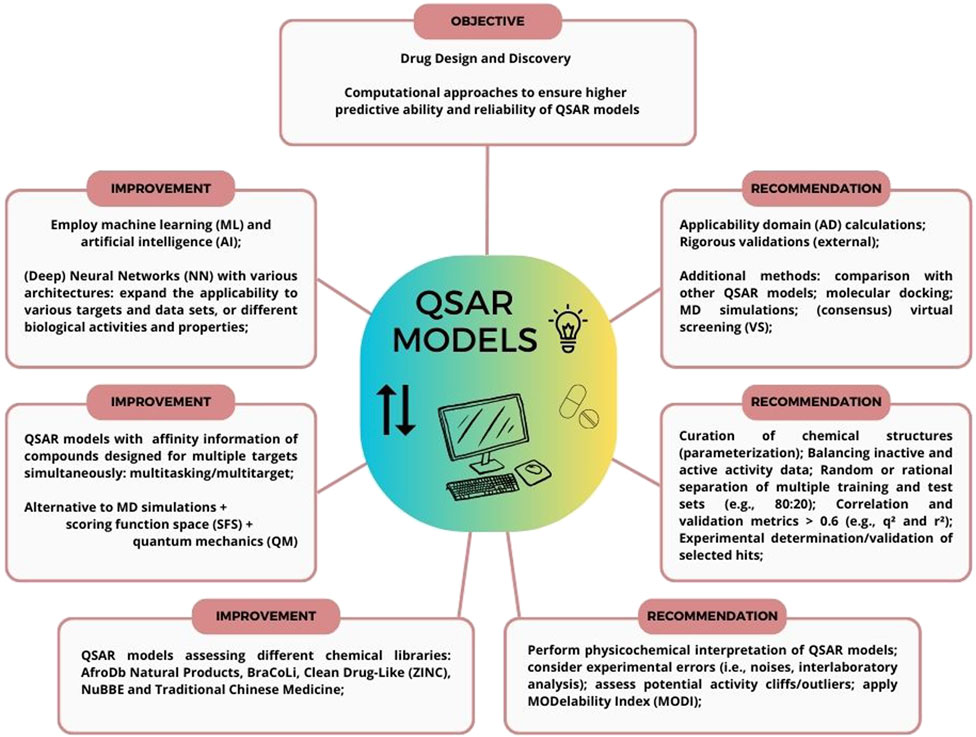
FIGURE 1. Practices for QSAR models’ development, validation, and exploitation. Aiming for drug design and discovery of compounds, QSAR models can be employed with recommendations (e.g., external validations and AD calculation), combined with other computational methods (e.g., MD simulations and VS.), and improved with additional strategies (e.g., neural networks and multitasking).
After approximately 60 years since the report of the first QSAR study (Hansch and Fujita, 1964), arguably one of the major computational and molecular modeling methods available, are characterized by well-defined protocols and procedures aiming to explore and potentially drive any given biological activity prediction from a chemical structure or chemical compound (Tropsha, 2010). To explore and exploit a relationship between a chemical structure and a biological activity, a modeling workflow must be considered to first validate a model and ultimately consider computational hits to experimental validation, such as lead compound optimization (Muchmore et al., 2010). For instance, some points should be addressed to ensure quality and performance of a model, aiming to avoid negative outcomes from predictions, such as i) curating chemical structures and their biological activities; ii) randomly or rationally separating one or multiple training and test sets (e.g., 80:20% ratio); iii) establishing and calculating correlation and validation metrics (e.g., q2 and r2 > 0.6); iv) calculation and use of an AD; v) VS. (e.g., consensus approach); or selection of hits; and vi) experimental validation (Tropsha, 2010; Gramatica, 2020).
Some principles for validation and regulatory purposes of (Q)SAR models were agreed by the Organisation for Economic Co-operation and Development (OECD) in 2004 (OECD, 2004), as recommendations to be applied in the field (Gramatica, 2007), and discussed even for future works with different QSAR models (Lowe et al., 2023). These principles are associated with the following information: a defined endpoint, an unambiguous algorithm, a defined AD, measurements of robustness and predictive ability, and, when possible, a mechanistic interpretation. First, when properly defined, a predicted endpoint would ensure clarity for a given QSAR model, so it can be determined by different experimental validation protocols or conditions. Secondly, it would be essential to ensure the transparency of the model algorithm, allowing for reproducibility. In addition, as QSAR models are directly dependent on assessed chemical structures and their mechanisms of action to generate predictions, defining an AD would also be required. Then, measurements of robustness (internal) and predictive ability (external) should be properly conducted with appropriate methods to determine models’ validations. Lastly, when possible, ensuring the possibility of a mechanistic association between descriptors and the endpoint being predicted would be of interest, thus determining a directed causality between a given structure and its given activity (OECD, 2004).
As the predictive ability of QSAR models is influenced by the size of the data sets, chemical diversity, availability of biological activity, nature of the biological experiment, as well as influences from the workflow conditions (e.g., variables’ selection, external validation, and the use of AD), one could suggest the employment of combined approaches, such as the use of ML (Golbraikh et al., 2014). However, not all approaches can be built with significant predictive abilities even with the combination of different algorithms and rigorous workflow designs (Thomas et al., 2012), thus potentially resulting in false hits. In this sense, data set curation and modelability are decisive starting points for any given QSAR, making it a priority to estimate the feasibility of obtaining reliable predictive QSAR models for a given data set of bioactive molecules (Golbraikh et al., 2014). Further, conformers with experimental properties (i.e., bioactive conformation) could be of interest, but would require a data set with both the conformer and the experimental data available (Axelrod and Gómez-Bombarelli, 2022). As the bioactive conformation in data sets is usually unknown, “stable minima” conformers or 2D structures could be employed to approximate or coincide with the bioactive conformation (Guimarães et al., 2016).
To this end, some tools could be applied, such as the MODelability Index (MODI) proposed by Golbraikh et al. (2014), which allows the prediction of a bioactive molecule being in the same or in a different activity class of its nearest neighbor (i.e., Euclidean distance), improving data curation (Golbraikh et al., 2014). This approach could predict the so-called activity cliffs, where compounds with similar structure have very different activities (Maggiora, 2006), which could result in many issues in computational analysis (Stumpfe et al., 2014). Luque Ruiz and Gómez-Nieto. (2018a) also proposed a reformulated calculation of MODI based on the distance between the first nearest neighbors in a data set (Luque Ruiz and Gómez-Nieto, 2018b), which contributes to measure the ability of molecules to be properly classifiable by rivality index values (Luque Ruiz and Gómez-Nieto, 2019), that is, correctly predicting a biological activity by a statistic algorithm. Additionally, the formulation of a regression modelability index (Luque Ruiz and Gómez-Nieto, 2018a), specifically designed for QSAR regression models, would also help regression algorithms correctly predict each molecule’s applicability in a data set.
Defining the applicability of a data set is also important to ensure the quality of different data sources into different experimental protocols. Therefore, evaluating any experimental errors observed in modeling sets that may lead to lower predictive ability of given QSAR models is also important to minimize erroneous selection of new compounds of interest (Zhao et al., 2017). Moreover, the usefulness of a chemical library can be questionable due to the potential lack of necessary quality control (Williams and Ekins, 2011), such as incorrect representation of chemical structures and inaccurate information regarding its biological activities, which would ultimately reduce a model’s accuracy. Lastly, in addition to the importance of selecting a good data set and subsequently performing its data curation, one should also consider its training and test sets’ ratio and size for the QSAR model (Andrada et al., 2017).
Roy et al. (2008) discussed this matter when assessing three different data sets containing compounds that inhibit the human immunodeficiency virus (HIV) multiplication (62 thiocarbamates, 107 HEPT derivatives, and 122 diverse nonionic organic functional compounds), to evaluate the importance and/or dependency of sets’ sizes and differences to QSAR models. Data sets were divided into different combinations of training sets (85%, 75%, 60%, 50%, or 40% to the first data set, and 75%, 60%, 50%, 40%, or 25% to the second and third) in several iterations (i.e., repetitions). The first data set showed a decrease in prediction r2 (external) values, including negative predictive values, as the number of available compounds in the training set decreased. The second set, more than 70% larger than the first, also showed a decreasing predictive ability, but less pronounced. Lastly, the third set showed good predictive r2 values, close to or higher than 0.9 in most of the cases, regardless of the training sets’ size. These would suggest not only the impact of smaller-larger data sets and training-test ratios, but also infer that diverse chemical structures could help with predictive ability (Roy et al., 2008).
To this end Andrada et al. (2017) further discussed the rational selection of training and test sets in a more molecular homogeneous data set. Here, authors assessed three different data sets of influenza virus (H1N1) neuraminidase inhibitors (n = 40, n = 26 and n = 29), with chemical structural similarities within each set. Training and test sets were divided based on k-means (i.e., Euclidean distance), the Kennard-Stone algorithm, and based on activity (4:1 selection ratio), which could leverage more reliable results instead of the mean of three random selections. A total of 31,490 linear regression models (r2) were used for analysis, suggesting a slight influence on the quality of the models depending on the selection method used. Interestingly, models with higher predictive ability were developed using the k-means algorithm, while the use of the mean of three random selections led to erroneous outcomes, especially when assessing a data set with more similar chemical structures (Andrada et al., 2017).
Apart from diversified or homogeneous chemical structures, Rácz, Bajusz and Héberger (2021) also corroborated findings showing differences among applied machine learning algorithms to select training and test sets, as well as between data sets’ sizes and training and test ratios (Rácz et al., 2021). Measures combining different numbers of compounds and ratios were assessed in five iterations each, with 100, 500, or 1,000 compounds randomly selected five times, or simply the total number of molecules being kept. Next, training sets of 80%, 70%, 60%, or 50% were also repeated five times for each ratio. Results suggested that training and test ratios exert a significant effect on classification performance (r2), corroborating previous data shown in the literature (Roy et al., 2008; Andrada et al., 2017), and that outcomes are also influenced by specific chemical structures within the data sets, once again suggesting the importance of selecting chemically different structures containing data sets.
Furthermore, one should consider that the validations performed for each model, including size, diversity, and training-test ratio, may also influence QSAR predictive abilities. For example, r2 value is a simple parameter to evaluate the correlation between predictions and experimentation (Shayanfar and Shayanfar, 2022). However, a high r2 value in QSAR does not necessarily have an acceptable validity (Kaneko, 2019), and can ultimately overpredict or underpredict results’ values. For instance, Shayanfar and Shayanfar (2022) discussed that r2 alone could not validate a QSAR model, after calculating various statistical parameters for external validation of 44 different QSAR models (Shayanfar and Shayanfar, 2022), showing that different criteria and metrics for external validation should be used, and that some have specific advantages and disadvantages to be considered (Shayanfar and Ershadi, 2019).
In addition to an external r2 and other validation metrics (Gramatica and Sangion, 2016), validations such as Y-randomization (Rücker et al., 2007), different cross-validation approaches (Konovalov et al., 2008), such as leave-many-out (LMO) (Kiralj and Ferreira, 2009), can be employed to select the optimal QSAR models, supporting their robustness. For instance, Y-randomization calculates the predictability of QSAR models generated with scrambled biological activities (which are expected to be highly non-predictive) in comparison to a given QSAR model, that is, not randomly generated (Rücker et al., 2007). Cross-validation coefficients, on the other hand, assesses the impact of excluding of compounds from the test set, and the effect of a single sample (i.e., one compound) removal compared to the original model assessed, for example, not being sensitive to variations in the training set (Konovalov et al., 2008). LMO, for example, statistically correlates a compound removal randomness in a training set by different-sized groups (e.g., multiples of two, five, or ten) being validated in replicates (e.g., triplicate) with standard deviation (Kiralj and Ferreira, 2009). Additionally, classification models should be evaluated according to the predictive ability by calculating metrics such as true negative and true positive rates, specificity and sensitivity, accuracy, Matthews’ correlation coefficient, and others (Matveieva and Polishchuk, 2021; Pradeep et al., 2021).
Moreover, in the absence of a true external data set, showing the importance of performing a statistical external validation, and especially because a data set division in training and test sets usually relies on a random division (Martin et al., 2012). However, one could argue what the influence of a random division could be in comparison to a rational division regarding an external validation. Martin et al. (2012) assessed random and rational division of a training set in an 80:20% ratio, and despite higher statistical external values obtained for rational division, no significant differences in predictive ability from QSAR models were observed (Martin et al., 2012). In addition, even if rationally selected training and test sets may establish a reliable QSAR model, external validation is required to assess the correlation between training set validation values (e.g., q2) and the overall external accuracy of prediction for the test set (r2), being a general property of any QSAR model developed (Golbraikh et al., 2003).
Additionally, internal and external validations that are often used in the absence of a true external data set may not comprise all features related to a particular SAR analysis due to omission of some compounds in each set, especially for small data sets. In this sense, Masand et al. (2015) discussed that rational splitting could favor small sets, as the predictive ability of a given QSAR model is influenced by the method of splitting (i.e., random or rational) and the distribution of training and test sets. Authors observed that external validation based on a single split is insufficient to guarantee the true predictive ability of a QSAR model (Masand et al., 2015), which would potentially result in predictive failure or false hits.
Finally, errors in predictions may also be dependent on the lack of an AD, as well as being negatively influenced by using a normal correlation coefficient to describe a given QSAR model predictive ability (Roy et al., 2017). Even with recent technological advances in QSAR modeling, such as the combination of improved algorithms and validation practices to different areas, such as biomaterials, clinicals, nanotechnology, and synthesis planning (Muratov et al., 2020). In this sense, even if all steps or conditions in a workflow are followed thoroughly, and different parameters, metrics, validations, and approaches are used, lower predictive ability (Thomas et al., 2012) and false hits could still be expected. Additionally, the inactive data may not be available, which would also directly impact the accuracy of QSAR models (López-López et al., 2022). Here, an inactivity data gap in the literature could limit the employment of additional in silico approaches, such as consensus VS. approaches (e.g., SARS-CoV-2 Mpro (Alves et al., 2021)), resulting in potential false hits that could be removed according to previous inactive reports or predictions of active and inactive compounds in cellular or target-based studies (Rodríguez-Pérez et al., 2018).
Nevertheless, knowledge and data obtained from these robust data-driven models are important and can become essential for scientists in future research (Muratov et al., 2020), including those from false hits predictions. For instance, publishing negative findings or results from experimental determination can help others avoid investing in specific approaches, but also encourage looking for alternatives (Taragin, 2019). Negative outcomes are important for the broader field where they could be relevant, not only to interpret selective information (i.e., positive results) that might have been obtained in related studies, but also to improve further analysis and potential modifications (Weintraub, 2016). Notwithstanding, single tested values or weak inhibitors (e.g., IC50 > 100 µM (Zhang et al., 2021)) should also be considered, leading further optimization studies (Deshmukh et al., 2021). Thus, considering a timely disclosure of these results (Nimpf and Keays, 2020) is also important to understand the strength of a given initial hypothesis (negative, neutral, or null results), which may not have yet produced desired outcomes, but should and must be turn public to the scientific community (Bespalov et al., 2019), either as publications in journals or available in public databases.
Taken together, we bring the discussion of sharing false hits towards QSAR approaches, considering the importance of supporting biological conclusions of any positive, negative, conflicting, or inconclusive QSAR predictions (Honma et al., 2019), such as those of inactive compounds. Thus, the design and applicability of a QSAR model for classification, biological activity prediction and/or selection of compounds for experimental validation, could include unexpected results and still be relevant to the scientific community. Herein, considering similar potential cases, we briefly present a case of false hits resulting from a combined QSAR approach to select potential SARS-CoV-2 main protease (Mpro) inhibitors, discussing employed parameters, potential flaws, and future improvements, aiming to maximize sensitivity for identifying potential bioactive molecules in future studies.
As of July 5, 2023, over 767.726 million cases of the severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) infections were reported worldwide, with over 6.948 million deaths reported for the coronavirus disease 2019 (COVID-19) (WHO, 2023). Vaccines’ emergency approval happened quickly, such as the mRNA-based vaccine by Pfizer-BioNTech (Polack et al., 2020) in less than a year. However, the existence of different SARS-CoV-2 strains (Rubin, 2021), an unequal distribution and acquisition of vaccines worldwide (Duan et al., 2021), as well as individual hesitance to vaccination (Gorman et al., 2021; Ullah et al., 2021), have hampered vaccination coverage globally, worsening the disease dissemination (Olivera Mesa et al., 2022).
In this sense, drug design and development were essential to tackle the COVID-19 pandemic scenario. Few therapeutic agents reached approval, such as the combined oral therapy of nirmatrelvir, a SARS-CoV-2 Mpro inhibitor, and ritonavir (Owen et al., 2021) (commercially as Paxlovid™), which showed up to 89% of hospitalization relative risk reduction in clinical trials (II to III) (Hammond et al., 2022; Hung et al., 2022). Yet, selection of SARS-CoV-2 resistant strains were reported in vitro (Zhou et al., 2022) and in vivo (Abdelnabi et al., 2023), and the already known limitations and risk of drug interactions of Paxlovid™ (Girardin et al., 2022) (e.g., ritonavir inhibition of CYP450) (Stader et al., 2021; Lange et al., 2022), have threatened the continuity of this therapeutic option, similar to what was observed for HIV treatment throughout the last decades (Tseng et al., 2015; Forsythe et al., 2019).
Considering this scenario, therapeutic drugs against SARS-CoV-2 are greatly needed, and the employment of computer-aided drug design (CADD) techniques, such as ligand-based drug design (LBDD) (Lima et al., 2016), may enhance the discovery of potential bioactive compounds, especially new antivirals (Serafim et al., 2021b; 2021a). For instance, oseltamivir (Talele et al., 2010), boceprevir (Njoroge et al., 2008), a hepatitis C virus (HCV) protease inhibitor, as well as lopinavir and ritonavir (Wlodawer, 2002), both protease inhibitors of HIV, were developed from initial computational approaches. Additionally, facing future outbreaks, epidemic, and pandemic scenarios (Morens and Fauci, 2020), new broad-spectrum antivirals would be of interest to treat COVID-19 (Santos et al., 2020; Serafim et al., 2021a), when considering the potential of mutated SARS-CoV-2 strains (Young et al., 2020), as well as the emergence of new coronaviruses and their associated diseases (Deng et al., 2014; Santos et al., 2020).
The main protease (Mpro) of SARS-CoV-2 is an essential enzyme in the viral replication cycle, responsible for cleaving translated polyproteins into individual nonstructural, structural (V’kovski et al., 2021), and accessory (Redondo et al., 2021) proteins. This enzyme is highly conserved among coronaviruses species (Ramajayam et al., 2011; Zhang et al., 2020), thereby it can be considered as a potential pancoronavirus target (Dong et al., 2020; Serafim et al., 2021b). This target has been considered for the design and development of inhibitors (Ferreira et al., 2021; Pillaiyar et al., 2022) by computational approaches (Serafim et al., 2021b), including of many different consensus VS. approaches that employed QSAR models (Table 1), that is, VS. approaches without QSAR were not taken into consideration.
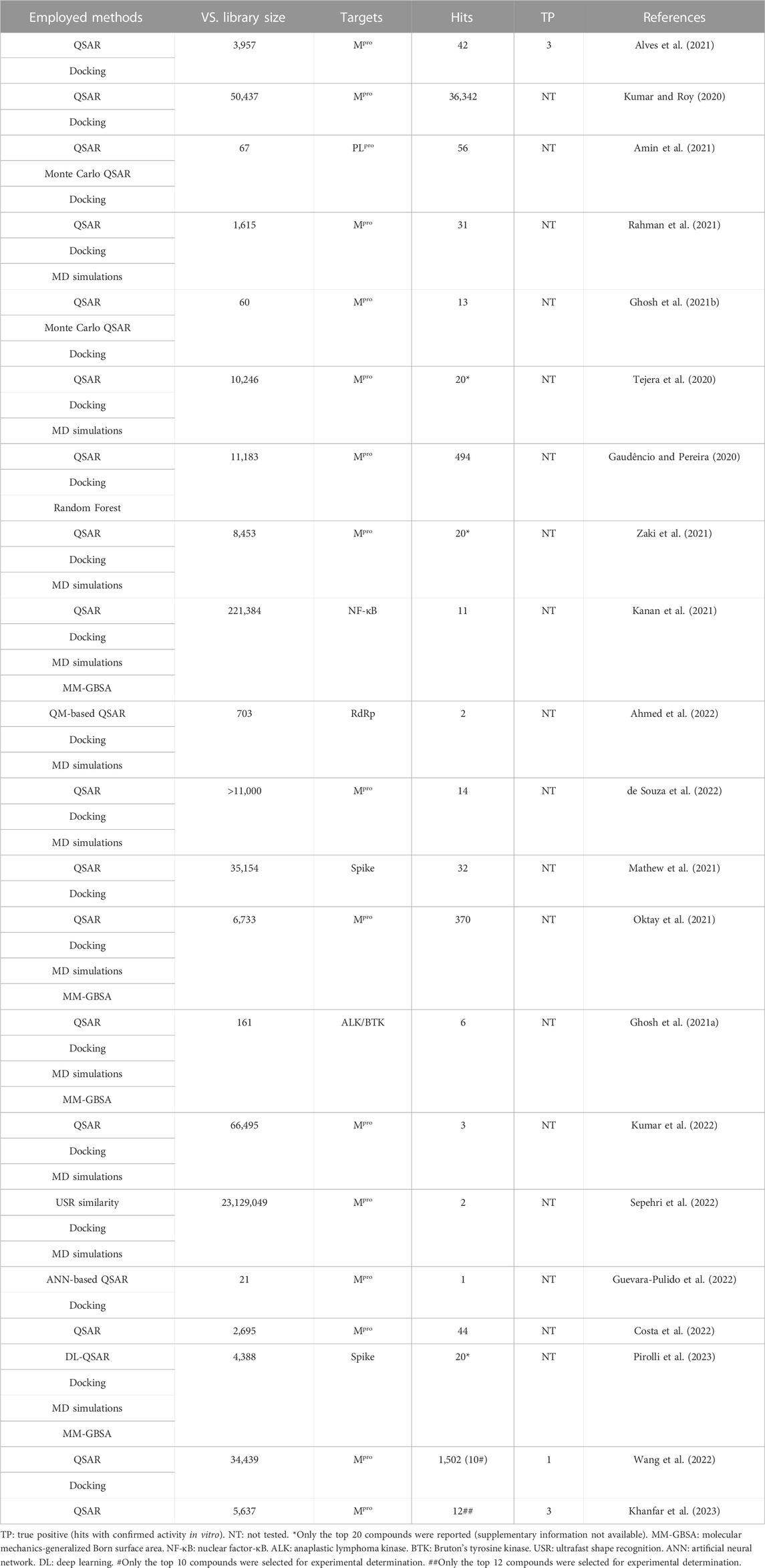
TABLE 1. Consensus QSAR/VS. approaches employed against SARS-CoV-2.
Most of these studies did not experimentally evaluate VS.’s hits, and only three presented proper validation against the same target assessed in our study (SARS-CoV-2 Mpro). Additionally, among these studies, it is notable that false hits would still comprise most of the outcomes from those consensus VS. approaches, such as >75% (Khanfar et al., 2023), >90% (Wang et al., 2022), and 92.86% (Alves et al., 2021) of false hits, which would be consistent with almost 90% of false hits expected from different VS. against different protein targets (Irwin and Shoichet, 2016). Taking these into consideration, we aimed to obtain novel bioactive molecules from known inhibitors of SARS-CoV-2 Mpro, performing a VS. approach based on a HQSAR model. In addition, we employed enzymatic inhibition assays to confirm whether the predictions were correct, thus identifying potential protease inhibitors. As of March 2021, a series containing a total of 25 inhibitors from the same chemical class synthesized and tested by the same research group, which presented IC50 values determined against the SARS-CoV-2 Mpro (Zhang et al., 2021) were selected (Table 2). At the time, some studies discussing a complete class of SARS-CoV-2 Mpro inhibitors were available, with few compounds (n < 10) per data set (Rathnayake et al., 2020; Sacco et al., 2020) and with different structures’ scaffold (Ghahremanpour et al., 2020; Jin et al., 2020), which could limit this HQSAR study.
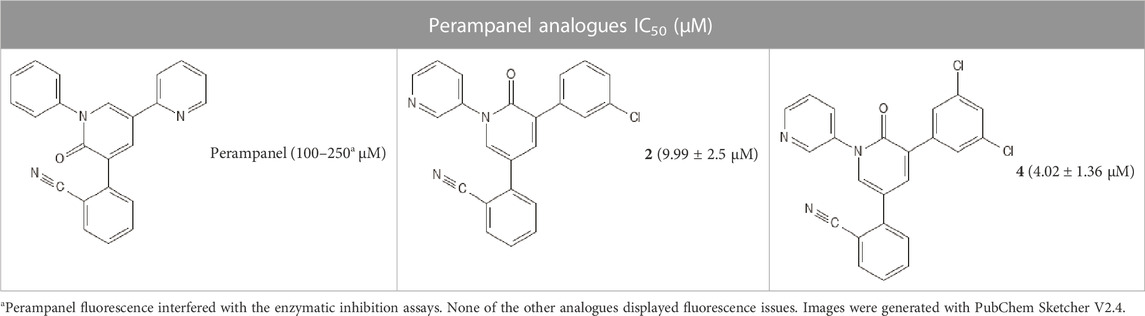
TABLE 2. Perampanel analogues with determined IC50 values (μM) against SARS-CoV-2 Mpro.
A review study at the end of 2021 (Macip et al., 2022) presented a total of 758 compounds extracted from peer-reviewed articles (January 2020 to August 2021) that were tested against SARS-CoV-2 Mpro. Still in March 2021, 32 compounds were synthesized and experimentally validated as inhibitors assessed against the proposed target (Qiao et al., 2021), and another approach with 116 compounds with confirmed enzyme inhibition would become available (Kuzikov et al., 2021). Here, we intended to carry out a fast and reliable drug discovery approach due to the availability of physical samples and partnerships that could promptly evaluate experimentally our hits (March 2021) in the urge of the pandemic scenario. Finally, we tried to perform a QSAR approach to compare with other computational methods that were being developed in parallel (e.g., docking), aiming to retrieve different hits (Maltarollo et al. in press).
Notwithstanding, HQSAR as other QSAR models can be easily and quickly generated and validated for their predictive ability in correlating a given chemical structure towards one biological activity (Cherkasov et al., 2014). Results from HQSAR can also improve other QSAR models predictive ability (Chavda and Bhatt, 2019; Veríssimo et al., 2019), and may glimpse a biological mechanism of studied chemicals and enlighten molecular descriptors related to the proposed end-point of interest (Gramatica, 2020). Additionally, a QSAR model can also be combined with other models to be applied in a consensus VS. approach against SARS-CoV-2 Mpro (Alves et al., 2021).
The 25 inhibitors (analogues 2–8 and 10–27 of the antiepileptic drug perampanel) had IC50 values between 0.018 and 9.99 µM (Zhang et al., 2021). Selected compounds presented some properties that favor HQSAR analysis, such as a conserved chemical moiety, a relatively small size of the structures, and the variety of substituents available. However, an ideal model should also consider a larger set of structurally diverse compounds (Sadeghi et al., 2022) with different moieties (e.g., retrieved from different studies), and also comprising natural and synthetic compounds, which would comprise different physicochemical properties (e.g., size and polar surface area (Radhakrishnan et al., 2022)) and synthesis accessibility (Atanasov et al., 2021). Considering the small data set assessed here, a second training set was generated considering the inclusion of the standard deviation (SD) for each IC50 values in the HQSAR models, herein termed a triplicate model (m: subtracting the SD value; p: adding the SD value). This model is used to enhance accuracy and lower errors in predictions, thus potentially increasing robustness towards the training set (Kronenberger et al., 2018). The original training and test sets were also used as a control to compare both models’ predictive ability, which indeed helped to minimize prediction errors but did not eliminate them completely. Thus, it would be of interest to work with larger sets of molecules that could also improve statistical control of the models.
Training and test sets were randomly divided with an 80:20% ratio, that is, 20 or 60 (triplicate) compounds for the training set and five compounds for the test set. HQSAR models were then generated by fixing fragments size (Supplementary Table SA), selecting the highest q2 value, if they presented q2 values >0.5 (Golbraikh and Tropsha, 2002), and r2 values >0.6 (Golbraikh et al., 2003; Gramatica and Sangion, 2016). Herein, the selected model was built using fragment distinction, containing atoms A), bonds B), connectivity C), hydrogen atoms H), chirality (Ch), and donor or acceptor hydrogen atoms (DA) presented acceptable values of q2 (0.885) and r2 (0.977) was then outperformed by its triplicate model, q2 (0.964) and r2 (0.975). Aiming to increase the selected model predictive abilities, atoms’ fragment size variation was employed (1–4 up to 7–10), maintaining the HQSAR descriptors, but none outperformed the original model (Supplementary Table SB).
The selected triplicate model was submitted to external validation, resulting in all tested metrics above 0.6 (Supplementary Table SC). Chemical contribution maps were generated (Figure 2), highlighting positive (green and yellow), neutral (white), and negative (orange and red) contributions of different regions of the molecules to the biological activity. Specifically, predictions showed phenyl and pyridinone rings, as well as chlorine and fluorine substituents as important substructures (compounds 23 and 21 m) for Mpro inhibition, based on their corresponding negative log of IC50 (pIC50), as summarized in (Supplementary Table SD). These positive contributions are also consistent with crystallographic results observed from the chosen 25 inhibitors, in which the pyridinone oxygen, the pyridine nitrogen, and the chlorophenyl edge are highlighted with hydrogen bonds to important residues (Glu166, His163, and His41, respectively) (Zhang et al., 2021).
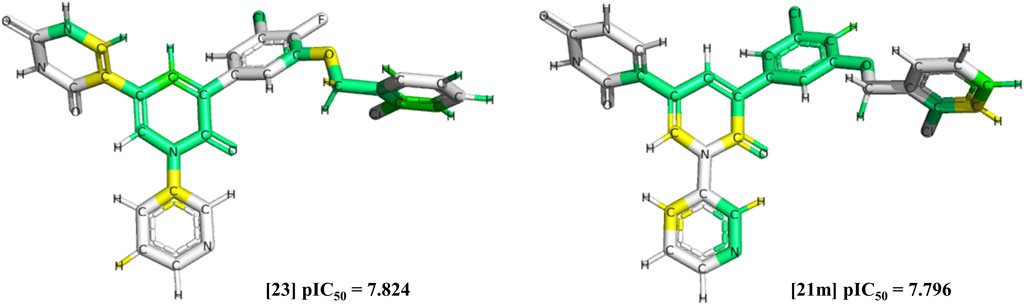
FIGURE 2. Chemical contribution maps of compounds with highest pIC50 values. Positive (green and yellow), neutral (white) and negative (orange and red) contribution regions of compounds 23 and 21 m (21 IC50 minus SD) were colored accordingly. Carbon, chlorine, fluoride, hydrogen, nitrogen, and oxygen atoms were highlighted in black. Images were generated with SYBYL-X 8.1.
The optimal HQSAR model was then used to predict biological activity of the 60 compounds in the triplicate training set and the five compounds in the test set (Supplementary Table SE), resulting in corresponding values, thus being employed for biological prediction against BraCoLi compounds. Additionally, PaDEL (Yap, 2011) software was used to calculate molecular descriptors (e.g., Fingerprinter, Extended Fingerprinter, MACCS, PubChem, Substructure, Substructure Count, Klekota Roth, Klekota Roth Count, AtomPairs2D, and AtomPairs2Dcount) for the training and test sets. Then, we generated RF-QSAR models for each descriptor set, considering MaxLevels from 10 to 50 levels, and Nmodels from 10,000 to 50,000 models for selection of potentially bioactive compounds, aiming to select a more adequate descriptor able to predict other compounds as potential SARS-CoV-2 Mpro inhibitors within BraCoLi.
Lastly, RF-QSAR models were also validated in KNIME® for their predictive ability, classified according to their concordance correlation coefficient (CCC), and for their robustness, according to higher q2 values (internal correlation). Here, PubChem and AtomPairs2Dcount showed higher regression values among the assessed metrics, over 0.85 for CCC and over 0.75 for q2, overcoming the other descriptors assessed. AtomPairs2D is a topological fingerprint, which is defined in terms of the so-called atomic environment of a chemical structure, and also the shortest path separations between its pairs of atoms (Carhart et al., 1985), while PubChem is a substructure fingerprint, which generates structural types that correspond to fragments (i.e., substructures) of all compounds in the PubChem database (Wang et al., 2017). Thus, these two molecular descriptors were considered for the RF-QSAR VS. approach, after being comparatively assessed considering their predictive ability (Figure 3).
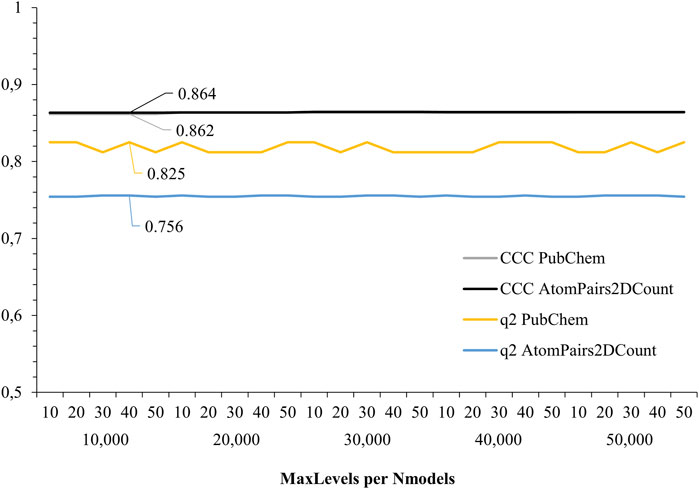
FIGURE 3. Regression analysis results for selected RF-QSAR models from PaDEL descriptors. Molecular descriptors PubChem and AtomPairs2DCount showed higher CCC (0.825 and 0.756) and q2 values (0.862 and 0.864), respectively.
Finally, the VS. of BraCoLi compounds was conducted with a consensus of both RF-QSAR and HQSAR models, resulting in 24 compounds, 20 from PubChem and four from AtomPairs2DCount, considering a pIC50 cutoff of 6.5 (corresponding to 0.3 µM). For instance, PubChem showed an overall higher specificity for synthetic compounds’ classification in contrast to natural products’ classification, with the lowest average for false positives and highest for true negatives (Seo et al., 2020), which would also befit this study accessing the BraCoLi database. At the time (March 2021), with the urgency of the COVID-19 pandemic, the selected compounds were evaluated in enzymatic inhibition assays against SARS-CoV-2 Mpro, without additional analysis, such as the definition of an AD, or employing other methods, such as molecular docking or MD simulations.
Briefly, enzymatic inhibition assays involve monitoring the cleavage of a fluorogenic substrate over time. When assessing the inhibition of enzyme activity, the difference between readings of the enzyme in the absence of test compound (i.e., 0% inhibition) is compared with the activity in the presence of compound (i.e., up to 100% inhibition) (Mellott et al., 2021). Among the 24 compounds selected from the VS. campaign, none presented inhibition of enzymatic activity by more than 50%, when assessed at 10 µM (Table 3).
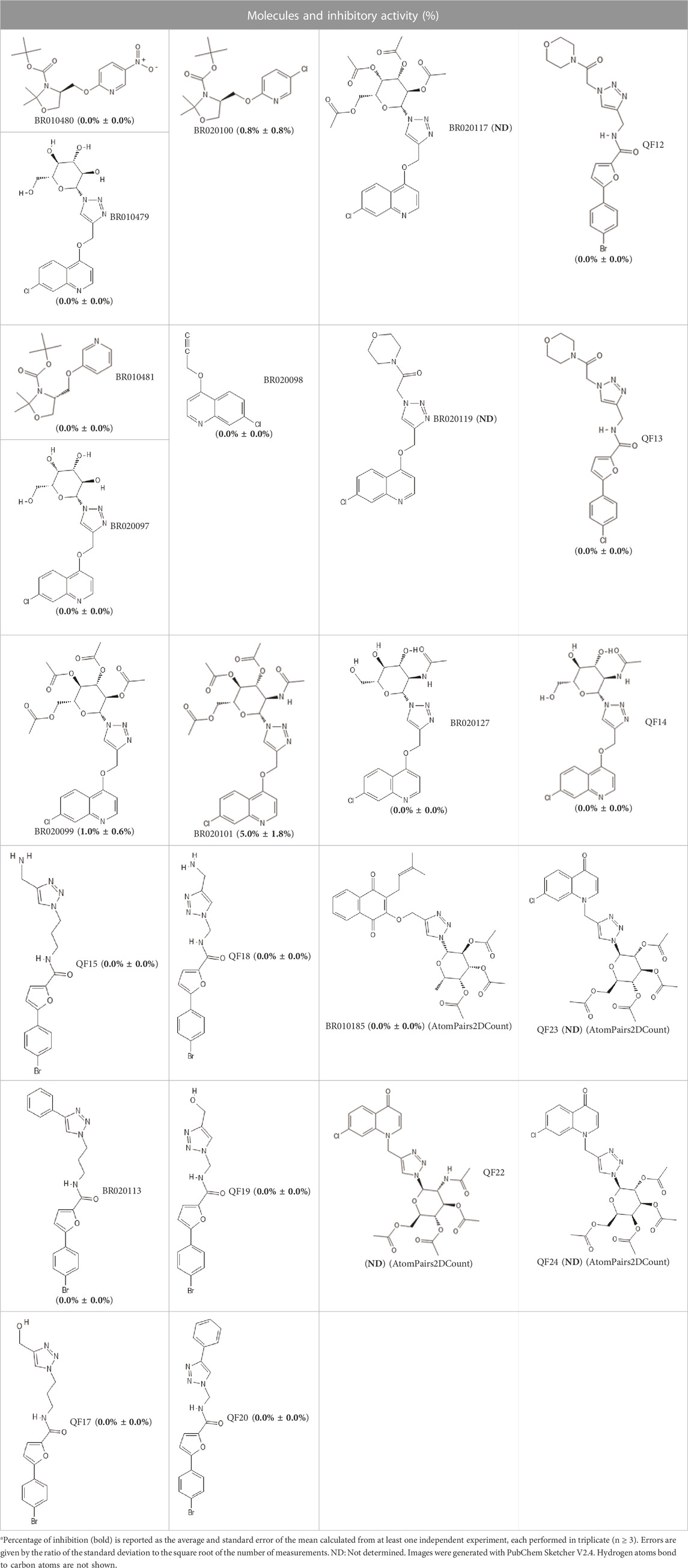
TABLE 3. Inhibitory activity (%) at 10 µM of selected molecules against SARS-CoV-2 Mpro.
Interestingly, no inhibitory activity (>50%) against SARS-CoV-2 Mpro was observed from the 24 screened compounds when assessed at a concentration of 10 µM. As discussed in this review, some parameters and conditions could explain this low accuracy and the prediction of false hits. Firstly, the definition of an AD is important for QSAR models, as predictions are considered reliable when molecules assessed are inserted in a specific domain (Mathea et al., 2016). Yet, AD is also restricted to the size and diversity of the training set, and once a given chemical structure is out of a given domain, predictions may be erroneous, corroborating the importance of calculating AD to the applicability of a QSAR model to the prediction of compounds from chemical libraries (Tropsha, 2010).
Likewise, as mentioned, only r2 values alone would not be able to validate a given QSAR model (Shayanfar and Shayanfar, 2022), thus requiring additional approaches, and considering their specific advantages and disadvantages (Shayanfar and Ershadi, 2019). These would potentially improve predictions able of identifying favorable inhibitors and a given target selectivity when employing independent validation tests to evaluate the robustness of the model, such as a cross-validation, y-scrambling, and LMO (Kiralj and Ferreira, 2009). In addition, considering the two major HQSAR parameters together (q2 and r2) is important to corroborate different regression metrics, such as q2 validation coefficients and r2 averages, which could testify to the robustness of the assessed model (Chirico and Gramatica, 2011; Chai and Draxler, 2014). This is important as q2 is considered a metric that can underestimate the predictive quality of a model when assessing the compounds in a data set (Golbraikh and Tropsha, 2002), which would require more external and cross-validation metrics to assess the predictive ability of different QSAR models (Gramatica and Sangion, 2016).
Moreover, changes in variables employed in the HQSAR model could improve its robustness towards predictions, such as selecting the top three or five models with higher q2 values in the first model selection, in addition to assessing fragments’ size variations (e.g., intervals of 2, 3 and 4 atoms) (Tropsha et al., 2003; Gramatica, 2020). More than one fragment’s size variations have shown better predictive ability when employed, such as six distinct fragment sizes (2–5, 3 to 6, 4 to 7, 5 to 8, 6 to 9, and 7 to 10 atoms), as well as a larger series of hologram lengths (53–997), highlighting important fragments from chemical contribution maps, as shown by (Lima et al., 2018). In this work, chemical contribution maps from the HQSAR predicted phenyl and pyridinone rings, and chlorine and fluorine substituents as important substructures (Figure 2) for Mpro inhibition. Similarly, aromatic rings have been previously predicted to bind in hydrophobic pockets within SARS-CoV-2 Mpro, such as bonds with Leu167 and Pro168, by molecular docking analysis (Zhang et al., 2021). Further, one could suggest that the presence of nitrogen atoms (pyridinone) and chlorine and fluorine substituents (chlorophenyl and fluorophenyl) would also favor hydrogen bonds. These substituents were also predicted to form hydrogen bonds with His163 and Glu166, as well as with the imidazole from His41 in the active site (ZHANG et al., 2021).
Furthermore, Aljuhani et al. (2022) also assessed derivatives of pyridine analogues (e.g., chlorophenyl) aiming to inhibit the enzymatic activity of SARS-CoV-2 Mpro, as well as to inhibit SARS-CoV and SARS-CoV-2 multiplication in Vero cells. Here, IC50 values up to 0.67 µM were obtained against SARS-CoV-2 Mpro, in addition to effective concentration of 50% (EC50) values of up to 0.021 µM against SARS-CoV-2, and 0.03 µM against SARS-CoV (Aljuhani et al., 2022). Additional docking analysis also showed binding predictions with His41, Cys145, His163, and Glu166, similar to those discussed by (Zhang et al., 2021). Luo et al. (2022) also predicted positive contributions from pyridinone rings, as well as chlorine and fluorine substituents from HQSAR analysis assessing SARS-CoV-2 Mpro inhibitors, as well as predicting rings’ positions where substituents would contribute negatively to the biological activity (Luo et al., 2022).
It is also of interest to perform a combination of QSAR and other LBDD methods aiming to improve the accuracy of different approaches (Lima et al., 2016), such as molecular docking, potentially helping to discover bioactive compounds against SARS-CoV-2 and even other coronaviruses (Wu et al., 2020; Serafim et al., 2021b; Pant et al., 2021). In addition, such approaches may also benefit from calculating decoys (putative inactive compounds) to increase predictive accuracy of true negatives and validate, for example, consensus VS. approaches (Réau et al., 2018), as shown by (Asse Junior et al., 2020). Furthermore, obtaining these true negative compounds or even identifying false hits is important for subsequent predictive models (Réau et al., 2018), thus improving screening protocols and potentially obtaining designed bioactive compounds (Gimeno et al., 2019).
Additionally, the design of pharmacophore models (Lu et al., 2018), which consists of determining specific properties related to ligands interactions in a given target (e.g., SARS-CoV-2 Mpro (Hayek-Orduz et al., 2022)), could improve comparisons to different compounds of interest (Hayek-Orduz et al., 2022), in such VS. campaigns (Arun et al., 2021; Bouback et al., 2021), thus potentially improving the reliability of selected compounds. Lastly, combining LBDD with structure-based drug design (SBDD) strategies (Lima et al., 2016) can potentially increase the accuracy, robustness, and predictive ability of computational methods (Azevedo et al., 2022), including QSAR models (Vázquez et al., 2020), ultimately filtering and selecting potential inhibitors specifically against a given molecular target structure, such as SARS-CoV-2 Mpro. Furthermore, increasing the number of different validation applications for the HQSAR models and selecting larger and more chemically diverse set of the compounds may also favor more reliable hit rates. The importance of building models considering key strategies was emphasized (Figure 1), and by evaluating their sensitivities, structural hints could be provided for the potential of more robust models with higher predictive ability in the future.
Finally, some thoughts could be raised from this case report. For instance, the amount of data is proportional to the quality of predictions, not in terms of accuracy, but in the sense of ability to predict biological activity from a broad spectrum of structural diversity. In other words, models with few data set samples should be carefully employed in further drug design and discovery, taking into consideration the impact of data set size and diversity on the conclusions. Considering the quality of biological data, unfortunately little can be discussed about direct evidence of inappropriate experimental practices, but different laboratories and methods can obtain different IC50 values (e.g., GC376 against SARS-CoV-2 Mpro (Macip et al., 2022)). However, we can suggest that mixing biological data from different sources could introduce noise and bias in the data set (which was not done in present work), but can drastically improve the structural diversity in the data set, and it can be avoided by using some ML models. Furthermore, the influence of descriptors is an essential topic that must be considered during QSAR modeling aiming to find the best set of variables that describe the physicochemical properties of the modeled biological phenomena. Therefore, multiple QSAR approaches could also be employed (e.g., 3D-QSAR, descriptor-based QSAR) combined or not as additional methods.
QSAR approaches are well known for their reliability and ability to predict a given biological activity from distinct chemical structures and their contribution to select potential bioactive compounds from various data sets (i.e., databases). Either single-handling predictions or in combination with different methods to improve predictive ability QSAR models can be influenced by many different conditions (e.g., training and test set sizes and ratios, and their chemical diversity), and may result in varied predictive values depending on which validation metrics were employed. Ultimately, these may reflect a model’s overall accuracy and its predicted outcomes. Herein, these predictions can be used for VS. campaigns, aiming to select designed bioactive compounds for a specific target. However, one should bear in mind that even the combination of good practices in a QSAR-driven workflow could provide false hits in VS. protocols. Altogether, although false hits may occur, these should be disclosed to the scientific community and must be considered to improve computational approaches in future studies, either by feeding true negatives or removing inactive data.
BM, KH, and VM contributed to conception and design of the study. MS and SP performed the HQSAR analysis. ES and MS performed the enzymatic assays. MS and SP performed the statistical analysis. MS and SP wrote the first draft of the manuscript. ES and VM wrote sections of the manuscript. AO, BM, JM, KH, and VM supervision and funding acquisition. All authors contributed to the article and approved the submitted version.
This research was funded by CAPES foundation, grant numbers 88887.595578/2020-00 and 88887.684031/2022-00; CNPq, grant numbers 302081/2018-6; FAPEMIG, grant numbers APQ-03116-18.
Authors would like to thank OpenEye Scientific for the academic licenses, UCSD and UFMG for intramural funds, and Professors Renata Barbosa de Oliveira and Ricardo José Alves for providing chemicals for the VS. approach.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2023.1237655/full#supplementary-material
Abdelnabi, R., Jochmans, D., Donckers, K., Trüeb, B., Ebert, N., Weynand, B., et al. (2023). Nirmatrelvir-resistant SARS-CoV-2 is efficiently transmitted in female Syrian hamsters and retains partial susceptibility to treatment. Nat. Commun. 14, 2124. doi:10.1038/s41467-023-37773-6
Adeshina, Y. O., Deeds, E. J., and Karanicolas, J. (2020). Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl. Acad. Sci. 117, 18477–18488. doi:10.1073/pnas.2000585117
Ahmed, S., Mahtarin, R., Islam, Md. S., Das, S., Al Mamun, A., Ahmed, S. S., et al. (2022). Remdesivir analogs against SARS-CoV-2 RNA-dependent RNA polymerase. J. Biomol. Struct. Dyn. 40, 11111–11124. doi:10.1080/07391102.2021.1955743
Aljuhani, A., Ahmed, A. H. E., Ihmaid, K., Omar, S. M., Althagfan, A. S., Alahmadi, Y. M., et al. (2022). In vitro and computational investigations of novel synthetic carboxamide-linked pyridopyrrolopyrimidines with potent activity as SARS-CoV-2-M Pro inhibitors. RSC Adv. 12, 26895–26907. doi:10.1039/D2RA04015H
Alves, V. M., Bobrowski, T., Melo-Filho, C. C., Korn, D., Auerbach, S., Schmitt, C., et al. (2021). QSAR modeling of SARS-CoV Mpro inhibitors identifies sufugolix, cenicriviroc, proglumetacin, and other drugs as candidates for repurposing against SARS-CoV-2. Mol. Inf. 40, 2000113. doi:10.1002/minf.202000113
Amin, S. A., Ghosh, K., Gayen, S., and Jha, T. (2021). Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J. Biomol. Struct. Dyn. 39, 4764–4773. doi:10.1080/07391102.2020.1780946
Andrada, M. F., Vega-Hissi, E. G., Estrada, M. R., and Garro Martinez, J. C. (2017). Impact assessment of the rational selection of training and test sets on the predictive ability of QSAR models. Sar. QSAR Environ. Res. 28, 1011–1023. doi:10.1080/1062936X.2017.1397056
Arun, K. G., Sharanya, C. S., Abhithaj, J., Francis, D., and Sadasivan, C. (2021). Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J. Biomol. Struct. Dyn. 39, 4647–4658. doi:10.1080/07391102.2020.1779819
Asse Junior, L. R., Kronenberger, T., Magalhães Serafim, M. S., Sousa, Y. V., Franco, I. D., Valli, M., et al. (2020). Virtual screening of antibacterial compounds by similarity search of Enoyl-ACP reductase (FabI) inhibitors. Future Med. Chem. 12, 51–68. doi:10.4155/fmc-2019-0158
Atanasov, A. G., Zotchev, S. B., Dirsch, V. M., and Supuran, C. T. (2021). Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 20, 200–216. doi:10.1038/s41573-020-00114-z
Axelrod, S., and Gómez-Bombarelli, R. (2022). GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Sci. Data 9, 185. doi:10.1038/s41597-022-01288-4
Azevedo, L., Serafim, M. S. M., Maltarollo, V. G., Grabrucker, A. M., and Granato, D. (2022). Atherosclerosis fate in the era of tailored functional foods: Evidence-based guidelines elicited from structure- and ligand-based approaches. Trends Food Sci. Technol. 128, 75–89. doi:10.1016/j.tifs.2022.07.010
Bespalov, A., Steckler, T., and Skolnick, P. (2019). Be positive about negatives-recommendations for the publication of negative (or null) results. Eur. Neuropsychopharmacol. 29, 1312–1320. doi:10.1016/j.euroneuro.2019.10.007
Bosc, N., Atkinson, F., Felix, E., Gaulton, A., Hersey, A., and Leach, A. R. (2019). Large scale comparison of QSAR and conformal prediction methods and their applications in drug discovery. J. Cheminformatics 11, 4. doi:10.1186/s13321-018-0325-4
Bouback, T. A., Pokhrel, S., Albeshri, A., Aljohani, A. M., Samad, A., Alam, R., et al. (2021). Pharmacophore-based virtual screening, quantum mechanics calculations, and molecular dynamics simulation approaches identified potential natural antiviral drug candidates against MERS-CoV S1-NTD. Molecules 26, 4961. doi:10.3390/molecules26164961
Carhart, R. E., Smith, D. H., and Venkataraghavan, R. (1985). Atom pairs as molecular features in structure-activity studies: definition and applications. J. Chem. Inf. Comput. Sci. 25, 64–73. doi:10.1021/ci00046a002
Chai, T., and Draxler, R. R. (2014). Root mean square error (RMSE) or mean absolute error (MAE)? – Arguments against avoiding RMSE in the literature. Geosci. Model. Dev. 7, 1247–1250. doi:10.5194/gmd-7-1247-2014
Chakravarti, S. K., and Alla, S. R. M. (2019). Descriptor free QSAR modeling using deep learning with long short-term memory neural networks. Front. Artif. Intell. 2, 17. doi:10.3389/frai.2019.00017
Chavda, J., and Bhatt, H. (2019). 3D-QSAR (CoMFA, CoMSIA, HQSAR and topomer CoMFA), MD simulations and molecular docking studies on purinylpyridine derivatives as B-Raf inhibitors for the treatment of melanoma cancer. Struct. Chem. 30, 2093–2107. doi:10.1007/s11224-019-01334-9
Cherkasov, A., Muratov, E. N., Fourches, D., Varnek, A., Baskin, I. I., Cronin, M., et al. (2014). QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 57, 4977–5010. doi:10.1021/jm4004285
Chirico, N., and Gramatica, P. (2011). Real external predictivity of QSAR models: How to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 51, 2320–2335. doi:10.1021/ci200211n
Cortes-Ciriano, I., Bender, A., and Malliavin, T. E. (2015). Comparing the influence of simulated experimental errors on 12 machine learning algorithms in bioactivity modeling using 12 diverse data sets. J. Chem. Inf. Model. 55, 1413–1425. doi:10.1021/acs.jcim.5b00101
Costa, A. S., Martins, J. P. A., and de Melo, E. B. (2022). SMILES-based 2D-QSAR and similarity search for identification of potential new scaffolds for development of SARS-CoV-2 MPRO inhibitors. Struct. Chem. 33, 1691–1706. doi:10.1007/s11224-022-02008-9
de Souza, A. S., de Souza, R. F., and Guzzo, C. R. (2022). Quantitative structure-activity relationships, molecular docking and molecular dynamics simulations reveal drug repurposing candidates as potent SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 40, 11339–11356. doi:10.1080/07391102.2021.1958700
Deng, X., StJohn, S. E., Osswald, H. L., O’Brien, A., Banach, B. S., Sleeman, K., et al. (2014). Coronaviruses resistant to a 3C-like protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance. J. Virol. 88, 11886–11898. doi:10.1128/JVI.01528-14
Deshmukh, M. G., Ippolito, J. A., Zhang, C.-H., Stone, E. A., Reilly, R. A., Miller, S. J., et al. (2021). Structure-guided design of a perampanel-derived pharmacophore targeting the SARS-CoV-2 main protease. Structure 29, 823–833.e5. doi:10.1016/j.str.2021.06.002
Dong, H., Liu, J., Liu, X., Yu, Y., and Cao, S. (2018). Combining molecular docking and QSAR studies for modeling the anti-tyrosinase activity of aromatic heterocycle thiosemicarbazone analogues. J. Mol. Struct. 1151, 353–365. doi:10.1016/j.molstruc.2017.08.034
Dong, S., Sun, J., Mao, Z., Wang, L., Lu, Y.-L., and Li, J. (2020). A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J. Med. Virol. 92, 1542–1548. doi:10.1002/jmv.25768
Duan, Y., Shi, J., Wang, Z., Zhou, S., Jin, Y., and Zheng, Z.-J. (2021). Disparities in COVID-19 vaccination among low-middle-and high-income countries: The mediating role of vaccination policy. Vaccines (Basel) 9, 905. doi:10.3390/vaccines9080905
Ferreira, G. M., Kronenberger, T., Tonduru, A. K., Hirata, R. D. C., Hirata, M. H., and Poso, A. (2021). SARS-COV-2 Mpro conformational changes induced by covalently bound ligands. J. Biomol. Struct. Dyn. 40, 12347–12357. doi:10.1080/07391102.2021.1970626
Forsythe, S. S., McGreevey, W., Whiteside, A., Shah, M., Cohen, J., Hecht, R., et al. (2019). Twenty years of antiretroviral therapy for people living with HIV: Global costs, health achievements, economic benefits. Health Aff. 38, 1163–1172. doi:10.1377/hlthaff.2018.05391
Gaudêncio, S. P., and Pereira, F. (2020). A computer-aided drug design approach to predict marine drug-like leads for SARS-CoV-2 main protease inhibition. Mar. Drugs 18, 633. doi:10.3390/md18120633
Ghahremanpour, M. M., Tirado-Rives, J., Deshmukh, M., Ippolito, J. A., Zhang, C.-H., Cabeza de Vaca, I., et al. (2020). Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med. Chem. Lett. 11, 2526–2533. doi:10.1021/acsmedchemlett.0c00521
Ghosh, A., Mukerjee, N., Sharma, B., Pant, A., Kishore Mohanta, Y., Jawarkar, R. D., et al. (2021a). Target specific inhibition of protein tyrosine kinase in conjunction with cancer and SARS-COV-2 by olive nutraceuticals. Front. Pharmacol. 12, 812565. doi:10.3389/fphar.2021.812565
Ghosh, K., Amin, S. A., Gayen, S., and Jha, T. (2021b). Chemical-informatics approach to COVID-19 drug discovery: Exploration of important fragments and data mining based prediction of some hits from natural origins as main protease (Mpro) inhibitors. J. Mol. Struct. 1224, 129026. doi:10.1016/j.molstruc.2020.129026
Gimeno, A., Ojeda-Montes, M. J., Tomás-Hernández, S., Cereto-Massagué, A., Beltrán-Debón, R., Mulero, M., et al. (2019). The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 20, 1375. doi:10.3390/ijms20061375
Girardin, F., Manuel, O., Marzolini, C., and Buclin, T. (2022). Evaluating the risk of drug-drug interactions with pharmacokinetic boosters: The case of ritonavir-enhanced nirmatrelvir to prevent severe COVID-19. Clin. Microbiol. Infect. 28, 1044–1046. doi:10.1016/j.cmi.2022.03.030
Golbraikh, A., Muratov, E., Fourches, D., and Tropsha, A. (2014). Data set modelability by QSAR. J. Chem. Inf. Model. 54, 1–4. doi:10.1021/ci400572x
Golbraikh, A., Shen, M., Xiao, Z., Xiao, Y.-D., Lee, K.-H., and Tropsha, A. (2003). Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 17, 241–253. doi:10.1023/A:1025386326946
Golbraikh, A., and Tropsha, A. (2002). Beware of q2! J. Mol. Graph. Model. 20, 269–276. doi:10.1016/S1093-3263(01)00123-1
Gorman, J. M., Gorman, S. E., Sandy, W., Gregorian, N., and Scales, D. A. (2021). Implications of COVID-19 vaccine hesitancy: Results of online bulletin board interviews. Front. Public Health 9, 757283. doi:10.3389/fpubh.2021.757283
Gramatica, P., Papa, E., and Sangion, A. (2018). QSAR modeling of cumulative environmental end-points for the prioritization of hazardous chemicals. Environ. Sci. Process. Impacts 20, 38–47. doi:10.1039/C7EM00519A
Gramatica, P. (2020). Principles of QSAR modeling: Comments and suggestions from personal experience. Int. J. Quantitative Structure-Property Relat. 5, 61–97. doi:10.4018/IJQSPR.20200701.oa1
Gramatica, P. (2007). Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 26, 694–701. doi:10.1002/qsar.200610151
Gramatica, P., and Sangion, A. (2016). A historical Excursus on the statistical validation parameters for QSAR models: A clarification concerning metrics and terminology. J. Chem. Inf. Model. 56, 1127–1131. doi:10.1021/acs.jcim.6b00088
Guevara-Pulido, J., Jiménez, R. A., Morantes, S. J., Jaramillo, D. N., and Acosta-Guzmán, P. (2022). Design, synthesis, and development of 4-[(7-Chloroquinoline-4-yl)amino]phenol as a potential SARS-CoV-2 Mpro inhibitor. ChemistrySelect 7, e202200125. doi:10.1002/slct.202200125
Guimarães, M. C., Duarte, M. H., Silla, J. M., and Freitas, M. P. (2016). Is conformation a fundamental descriptor in QSAR? A case for halogenated anesthetics. Beilstein J. Org. Chem. 12, 760–768. doi:10.3762/bjoc.12.76
Hammond, J., Leister-Tebbe, H., Gardner, A., Abreu, P., Bao, W., Wisemandle, W., et al. (2022). Oral nirmatrelvir for high-risk, nonhospitalized adults with covid-19. N. Engl. J. Med. 386, 1397–1408. doi:10.1056/NEJMoa2118542
Handler, J. A., Feied, C. F., and Gillam, M. T. (2022). Novel techniques to assess predictive systems and reduce their alarm burden. IEEE J. Biomed. Health Inf. 26, 5267–5278. doi:10.1109/JBHI.2022.3189312
Hansch, C., and Fujita, T. (1964). p-σ-π analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 86, 1616–1626. doi:10.1021/ja01062a035
Hayek-Orduz, Y., Vásquez, A. F., Villegas-Torres, M. F., Caicedo, P. A., Achenie, L. E. K., and González Barrios, A. F. (2022). Novel covalent and non-covalent complex-based pharmacophore models of SARS-CoV-2 main protease (Mpro) elucidated by microsecond MD simulations. Sci. Rep. 12, 14030. doi:10.1038/s41598-022-17204-0
Honma, M., Kitazawa, A., Cayley, A., Williams, R. V., Barber, C., Hanser, T., et al. (2019). Improvement of quantitative structure-activity relationship (QSAR) tools for predicting ames mutagenicity: Outcomes of the ames/QSAR international challenge project. Mutagenesis 34, 3–16. doi:10.1093/mutage/gey031
Hung, Y.-P., Lee, J.-C., Chiu, C.-W., Lee, C.-C., Tsai, P.-J., Hsu, I.-L., et al. (2022). Oral nirmatrelvir/ritonavir therapy for COVID-19: The dawn in the dark? Antibiot. (Basel) 11, 220. doi:10.3390/antibiotics11020220
Irwin, J. J., and Shoichet, B. K. (2016). Docking screens for novel ligands conferring new biology. J. Med. Chem. 59, 4103–4120. doi:10.1021/acs.jmedchem.5b02008
Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., et al. (2020). Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293. doi:10.1038/s41586-020-2223-y
Kanan, T., Kanan, D., Al Shardoub, E. J., and Durdagi, S. (2021). Transcription factor NF-κB as target for SARS-CoV-2 drug discovery efforts using inflammation-based QSAR screening model. J. Mol. Graph Model. 108, 107968. doi:10.1016/j.jmgm.2021.107968
Kaneko, H. (2019). Beware of r2 even for test datasets: Using the latest measured y-values (r2LM) in time series data analysis. J. Chemom. 33, e3093. doi:10.1002/cem.3093
Kar, S., and Roy, K. (2013). How far can virtual screening take us in drug discovery? Expert Opin. Drug Discov. 8, 245–261. doi:10.1517/17460441.2013.761204
Khanfar, M. A., Salaas, N., and Abumostafa, R. (2023). Discovery of natural-derived Mpro inhibitors as therapeutic candidates for COVID-19: Structure-based pharmacophore screening combined with QSAR analysis. Mol. Inf. 42, 2200198. doi:10.1002/minf.202200198
Kiralj, R., and Ferreira, M. M. C. (2009). Basic validation procedures for regression models in QSAR and QSPR studies: Theory and application. J. Braz. Chem. Soc. 20, 770–787. doi:10.1590/S0103-50532009000400021
Konovalov, D. A., Llewellyn, L. E., Vander Heyden, Y., and Coomans, D. (2008). Robust cross-validation of linear regression QSAR models. J. Chem. Inf. Model. 48, 2081–2094. doi:10.1021/ci800209k
Kronenberger, T., Asse, L. R., Wrenger, C., Trossini, G. H. G., Honorio, K. M., and Maltarollo, V. G. (2017). Studies of Staphylococcus aureus FabI inhibitors: Fragment-based approach based on holographic structure-activity relationship analyses. Future Med. Chem. 9, 135–151. doi:10.4155/fmc-2016-0179
Kronenberger, T., Windshügel, B., Wrenger, C., Honorio, K. M., and Maltarollo, V. G. (2018). On the relationship of anthranilic derivatives structure and the FXR (Farnesoid X receptor) agonist activity. J. Biomol. Struct. Dyn. 36, 4378–4391. doi:10.1080/07391102.2017.1417161
Kumar, V., Kar, S., De, P., Roy, K., and Leszczynski, J. (2022). Identification of potential antivirals against 3CLpro enzyme for the treatment of SARS-CoV-2: A multi-step virtual screening study. Sar. QSAR Environ. Res. 33, 357–386. doi:10.1080/1062936X.2022.2055140
Kumar, V., and Roy, K. (2020). Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3C-like protease (3CLpro) enzyme inhibitors against SARS-CoV diseases. Sar. QSAR Environ. Res. 31, 511–526. doi:10.1080/1062936X.2020.1776388
Kuzikov, M., Costanzi, E., Reinshagen, J., Esposito, F., Vangeel, L., Wolf, M., et al. (2021). Identification of inhibitors of SARS-CoV-2 3CL-pro enzymatic activity using a small molecule in vitro repurposing screen. ACS Pharmacol. Transl. Sci. 4, 1096–1110. doi:10.1021/acsptsci.0c00216
Lange, N. W., Salerno, D. M., Jennings, D. L., Choe, J., Hedvat, J., Kovac, D., et al. (2022). Nirmatrelvir/ritonavir use: Managing clinically significant drug-drug interactions with transplant immunosuppressants. Am. J. Transplant. 22, 1925–1926. doi:10.1111/ajt.16955
Lima, A. N., Philot, E. A., Trossini, G. H. G., Scott, L. P. B., Maltarollo, V. G., and Honorio, K. M. (2016). Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 11, 225–239. doi:10.1517/17460441.2016.1146250
Lima, M. N. N., Melo-Filho, C. C., Cassiano, G. C., Neves, B. J., Alves, V. M., Braga, R. C., et al. (2018). QSAR-driven design and discovery of novel compounds with antiplasmodial and transmission blocking activities. Front. Pharmacol. 9, 146. doi:10.3389/fphar.2018.00146
López-López, E., Fernández-de Gortari, E., and Medina-Franco, J. L. (2022). Yes SIR! On the structure–inactivity relationships in drug discovery. Drug Discov. Today 27, 2353–2362. doi:10.1016/j.drudis.2022.05.005
Lowe, C. N., Charest, N., Ramsland, C., Chang, D. T., Martin, T. M., and Williams, A. J. (2023). Transparency in modeling through careful application of OECD’s QSAR/QSPR principles via a curated water solubility data set. Chem. Res. Toxicol. 36, 465–478. doi:10.1021/acs.chemrestox.2c00379
Lu, X., Deng, L., Gin, S., and Du, J. (2019). Quantitative structure-property relationship (QSPR) analysis of ZrO2-containing soda-lime borosilicate glasses. J. Phys. Chem. B 123, 1412–1422. doi:10.1021/acs.jpcb.8b11108
Lu, X., Yang, H., Chen, Y., Li, Q., He, S.-Y., Jiang, X., et al. (2018). The development of pharmacophore modeling: Generation and recent applications in drug discovery. Curr. Pharm. Des. 24, 3424–3439. doi:10.2174/1381612824666180810162944
Luo, D., Tong, J.-B., Zhang, X., Xiao, X.-C., and Bian, S. (2022). Computational strategies towards developing novel SARS-CoV-2 Mpro inhibitors against COVID-19. J. Mol. Struct. 1247, 131378. doi:10.1016/j.molstruc.2021.131378
Luque Ruiz, I., and Gómez-Nieto, M. Á. (2019). Prediction of the datasets modelability for the building of QSAR classification models by means of the centroid based rivality index. J. Math. Chem. 57, 1374–1393. doi:10.1007/s10910-018-0972-8
Luque Ruiz, I., and Gómez-Nieto, M. Á. (2018a). Regression modelability index: A new index for prediction of the modelability of data sets in the development of QSAR regression models. J. Chem. Inf. Model. 58, 2069–2084. doi:10.1021/acs.jcim.8b00313
Luque Ruiz, I., and Gómez-Nieto, M. Á. (2018b). Study of data set modelability: Modelability, rivality, and weighted modelability indexes. J. Chem. Inf. Model. 58, 1798–1814. doi:10.1021/acs.jcim.8b00188
Macip, G., Garcia-Segura, P., Mestres-Truyol, J., Saldivar-Espinoza, B., Pujadas, G., and Garcia-Vallvé, S. (2022). A review of the current landscape of SARS-CoV-2 main protease inhibitors: Have we hit the bullseye yet? Int. J. Mol. Sci. 23, 259. doi:10.3390/ijms23010259
Maggiora, G. M. (2006). On outliers and activity cliffs-why QSAR often disappoints. J. Chem. Inf. Model. 46, 1535. doi:10.1021/ci060117s
Mao, J., Akhtar, J., Zhang, X., Sun, L., Guan, S., Li, X., et al. (2021). Comprehensive strategies of machine-learning-based quantitative structure-activity relationship models. iScience 24, 103052. doi:10.1016/j.isci.2021.103052
Martin, T. M., Harten, P., Young, D. M., Muratov, E. N., Golbraikh, A., Zhu, H., et al. (2012). Does rational selection of training and test sets improve the outcome of QSAR modeling? J. Chem. Inf. Model. 52, 2570–2578. doi:10.1021/ci300338w
Masand, V. H., Mahajan, D. T., Nazeruddin, G. M., Hadda, T. B., Rastija, V., and Alfeefy, A. M. (2015). Effect of information leakage and method of splitting (rational and random) on external predictive ability and behavior of different statistical parameters of QSAR model. Med. Chem. Res. 24, 1241–1264. doi:10.1007/s00044-014-1193-8
Mathea, M., Klingspohn, W., and Baumann, K. (2016). Chemoinformatic classification methods and their applicability domain. Mol. Inf. 35, 160–180. doi:10.1002/minf.201501019
Mathew, S. M., Benslimane, F., Althani, A. A., and Yassine, H. M. (2021). Identification of potential natural inhibitors of the receptor-binding domain of the SARS-CoV-2 spike protein using a computational docking approach. Qatar Med. J. 2021, 12. doi:10.5339/qmj.2021.12
Matveieva, M., and Polishchuk, P. (2021). Benchmarks for interpretation of QSAR models. J. Cheminformatics 13, 41. doi:10.1186/s13321-021-00519-x
Mellott, D. M., Tseng, C.-T., Drelich, A., Fajtová, P., Chenna, B. C., Kostomiris, D. H., et al. (2021). A clinical-stage cysteine protease inhibitor blocks SARS-CoV-2 infection of human and monkey cells. ACS Chem. Biol. 16, 642–650. doi:10.1021/acschembio.0c00875
Morens, D. M., and Fauci, A. S. (2020). Emerging pandemic diseases: How we got to COVID-19. Cell. 182, 1077–1092. doi:10.1016/j.cell.2020.08.021
Muchmore, S. W., Edmunds, J. J., Stewart, K. D., and Hajduk, P. J. (2010). Cheminformatic tools for medicinal chemists. J. Med. Chem. 53, 4830–4841. doi:10.1021/jm100164z
Muratov, E. N., Bajorath, J., Sheridan, R. P., Tetko, I. V., Filimonov, D., Poroikov, V., et al. (2020). QSAR without borders. Chem. Soc. Rev. 49, 3525–3564. doi:10.1039/d0cs00098a
Mysinger, M. M., Carchia, M., Irwin, J. J., and Shoichet, B. K. (2012). Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582–6594. doi:10.1021/jm300687e
Neves, B. J., Braga, R. C., Melo-Filho, C. C., Moreira-Filho, J. T., Muratov, E. N., and Andrade, C. H. (2018). QSAR-based virtual screening: Advances and applications in drug discovery. Front. Pharmacol. 9, 1275. doi:10.3389/fphar.2018.01275
Nimpf, S., and Keays, D. A. (2020). Why (and how) we should publish negative data. EMBO Rep. 21, e49775. doi:10.15252/embr.201949775
Njoroge, F. G., Chen, K. X., Shih, N.-Y., and Piwinski, J. J. (2008). Challenges in modern drug discovery: A case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection. Acc. Chem. Res. 41, 50–59. doi:10.1021/ar700109k
OECD (2004). Validation of (Q)SAR models - oecd. Available at: https://www.oecd.org/chemicalsafety/risk-assessment/validationofqsarmodels.htm (Accessed July 6, 2023).
Ojha, P. K., and Roy, K. (2013). Exploring structural requirements for a class of nucleoside inhibitors (PfdUTPase) as antimalarials: First report on QSAR, pharmacophore mapping and multiple docking studies. Comb. Chem. High. Throughput Screen 16, 739–757. doi:10.2174/13862073113169990002
Oktay, L., Erdemoğlu, E., Tolu, İ., Yumak, Y., Özcan, A., Acar, E., et al. (2021). Binary-QSAR guided virtual screening of FDA approved drugs and compounds in clinical investigation against SARS-CoV-2 main protease. Turkish J. Biol. 45, 459–468. doi:10.3906/biy-2106-61
Olivera Mesa, D., Hogan, A. B., Watson, O. J., Charles, G. D., Hauck, K., Ghani, A. C., et al. (2022). Modelling the impact of vaccine hesitancy in prolonging the need for Non-Pharmaceutical Interventions to control the COVID-19 pandemic. Commun. Med. 2, 14–18. doi:10.1038/s43856-022-00075-x
Owen, D. R., Allerton, C. M. N., Anderson, A. S., Aschenbrenner, L., Avery, M., Berritt, S., et al. (2021). An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593. doi:10.1126/science.abl4784
Pant, S., Singh, M., Ravichandiran, V., Murty, U. S. N., and Srivastava, H. K. (2021). Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 39, 2904–2913. doi:10.1080/07391102.2020.1757510
Pillaiyar, T., Flury, P., Krüger, N., Su, H., Schäkel, L., Barbosa Da Silva, E., et al. (2022). Small-molecule thioesters as SARS-CoV-2 main protease inhibitors: Enzyme inhibition, structure-activity relationships, antiviral activity, and X-ray structure determination. J. Med. Chem. 65, 9376–9395. doi:10.1021/acs.jmedchem.2c00636
Pirolli, D., Righino, B., Camponeschi, C., Ria, F., Di Sante, G., and De Rosa, M. C. (2023). Virtual screening and molecular dynamics simulations provide insight into repurposing drugs against SARS-CoV-2 variants Spike protein/ACE2 interface. Sci. Rep. 13, 1494. doi:10.1038/s41598-023-28716-8
Polack, F. P., Thomas, S. J., Kitchin, N., Absalon, J., Gurtman, A., Lockhart, S., et al. (2020). Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615. doi:10.1056/NEJMoa2034577
Pradeep, P., Judson, R., DeMarini, D. M., Keshava, N., Martin, T. M., Dean, J., et al. (2021). An evaluation of existing QSAR models and structural alerts and development of new ensemble models for genotoxicity using a newly compiled experimental dataset. Comput. Toxicol. 18, 100167. doi:10.1016/j.comtox.2021.100167
Qiao, J., Li, Y.-S., Zeng, R., Liu, F.-L., Luo, R.-H., Huang, C., et al. (2021). SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 371, 1374–1378. doi:10.1126/science.abf1611
Rácz, A., Bajusz, D., and Héberger, K. (2021). Effect of dataset size and train/test split ratios in QSAR/QSPR multiclass classification. Molecules 26, 1111. doi:10.3390/molecules26041111
Radhakrishnan, S., Hoff, O., and Muellner, M. K. (2022). Current challenges in small molecule proximity-inducing compound development for targeted protein degradation using the ubiquitin proteasomal system. Molecules 27, 8119. doi:10.3390/molecules27238119
Rafi, Md. O., Bhattacharje, G., Al-Khafaji, K., Taskin-Tok, T., Alfasane, Md. A., Das, A. K., et al. (2022). Combination of QSAR, molecular docking, molecular dynamic simulation and MM-PBSA: Analogues of lopinavir and favipiravir as potential drug candidates against COVID-19. J. Biomol. Struct. Dyn. 40, 3711–3730. doi:10.1080/07391102.2020.1850355
Rahman, M. M., Saha, T., Islam, K. J., Suman, R. H., Biswas, S., Rahat, E. U., et al. (2021). Virtual screening, molecular dynamics and structure-activity relationship studies to identify potent approved drugs for Covid-19 treatment. J. Biomol. Struct. Dyn. 39, 6231–6241. doi:10.1080/07391102.2020.1794974
Ramajayam, R., Tan, K. P., and Liang, P. H. (2011). Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 39, 1371–1375. doi:10.1042/BST0391371
Rathnayake, A. D., Zheng, J., Kim, Y., Perera, K. D., Mackin, S., Meyerholz, D. K., et al. (2020). 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV–infected mice. Sci. Transl. Med. 12, eabc5332. doi:10.1126/scitranslmed.abc5332
Réau, M., Langenfeld, F., Zagury, J.-F., Lagarde, N., and Montes, M. (2018). Decoys selection in benchmarking datasets: Overview and perspectives. Front. Pharmacol. 9, 11. doi:10.3389/fphar.2018.00011
Redondo, N., Zaldívar-López, S., Garrido, J. J., and Montoya, M. (2021). SARS-CoV-2 accessory proteins in viral pathogenesis: Knowns and unknowns. Front. Immunol. 12, 708264. doi:10.3389/fimmu.2021.708264
Rodríguez-Pérez, R., and Bajorath, J. (2021). Evaluation of multi-target deep neural network models for compound potency prediction under increasingly challenging test conditions. J. Comput. Aided Mol. Des. 35, 285–295. doi:10.1007/s10822-021-00376-8
Rodríguez-Pérez, R., and Bajorath, J. (2018). Prediction of compound profiling matrices, Part II: Relative performance of multitask deep learning and random forest classification on the basis of varying amounts of training data. ACS Omega 3, 12033–12040. doi:10.1021/acsomega.8b01682
Rodríguez-Pérez, R., Miyao, T., Jasial, S., Vogt, M., and Bajorath, J. (2018). Prediction of compound profiling matrices using machine learning. ACS Omega 3, 4713–4723. doi:10.1021/acsomega.8b00462
Roy, K., Ambure, P., and Aher, R. B. (2017). How important is to detect systematic error in predictions and understand statistical applicability domain of QSAR models? Chemom. Intelligent Laboratory Syst. 162, 44–54. doi:10.1016/j.chemolab.2017.01.010
Roy, P. P., Leonard, J. T., and Roy, K. (2008). Exploring the impact of size of training sets for the development of predictive QSAR models. Chemom. Intelligent Laboratory Syst. 90, 31–42. doi:10.1016/j.chemolab.2007.07.004
Rubin, R. (2021). COVID-19 vaccines vs variants—determining how much immunity is enough. JAMA 325, 1241–1243. doi:10.1001/jama.2021.3370
Rücker, C., Rücker, G., and Meringer, M. (2007). y-Randomization and its Variants in QSPR/QSAR. J. Chem. Inf. Model. 47, 2345–2357. doi:10.1021/ci700157b
Sacco, M. D., Ma, C., Lagarias, P., Gao, A., Townsend, J. A., Meng, X., et al. (2020). Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 6, eabe0751. doi:10.1126/sciadv.abe0751
Sadeghi, F., Afkhami, A., Madrakian, T., and Ghavami, R. (2022). QSAR analysis on a large and diverse set of potent phosphoinositide 3-kinase gamma (PI3Kγ) inhibitors using MLR and ANN methods. Sci. Rep. 12, 6090. doi:10.1038/s41598-022-09843-0
Sadybekov, A. V., and Katritch, V. (2023). Computational approaches streamlining drug discovery. Nature 616, 673–685. doi:10.1038/s41586-023-05905-z
Santos, I. de A., Grosche, V. R., Bergamini, F. R. G., Sabino-Silva, R., and Jardim, A. C. G. (2020). Antivirals against coronaviruses: Candidate drugs for SARS-CoV-2 treatment? Front. Microbiol. 11, 1818. doi:10.3389/fmicb.2020.01818
Seo, M., Shin, H. K., Myung, Y., Hwang, S., and No, K. T. (2020). Development of natural compound molecular fingerprint (NC-mfp) with the dictionary of natural products (DNP) for natural product-based drug development. J. Cheminform 12, 6. doi:10.1186/s13321-020-0410-3
Sepehri, B., Ghavami, R., Mahmoudi, F., Irani, M., Ahmadi, R., and Moradi, D. (2022). Identifying SARS-CoV-2 main protease inhibitors by applying the computer screening of a large database of molecules. SAR QSAR Environ. Res. 33, 341–356. doi:10.1080/1062936X.2022.2050424
Serafim, M. S. M., Dos Santos Júnior, V. S., Gertrudes, J. C., Maltarollo, V. G., and Honorio, K. M. (2021a). Machine learning techniques applied to the drug design and discovery of new antivirals: A brief look over the past decade. Expert Opin. Drug Discov. 16, 961–975. doi:10.1080/17460441.2021.1918098
Serafim, M. S. M., Gertrudes, J. C., Costa, D. M. A., Oliveira, P. R., Maltarollo, V. G., and Honorio, K. M. (2021b). Knowing and combating the enemy: A brief review on SARS-CoV-2 and computational approaches applied to the discovery of drug candidates. Biosci. Rep. 41, BSR20202616. doi:10.1042/BSR20202616
Serafim, M. S. M., Kronenberger, T., Oliveira, P. R., Poso, A., Honório, K. M., Mota, B. E. F., et al. (2020). The application of machine learning techniques to innovative antibacterial discovery and development. Expert Opin. Drug Discov. 15, 1165–1180. doi:10.1080/17460441.2020.1776696
Shayanfar, A., and Ershadi, S. (2019). Developing new criteria for validity evaluation of analytical methods. J. AOAC Int. 102, 1908–1916. doi:10.5740/jaoacint.19-0007
Shayanfar, S., and Shayanfar, A. (2022). Comparison of various methods for validity evaluation of QSAR models. BMC Chem. 16, 63. doi:10.1186/s13065-022-00856-4
Sidorczuk, K., Gagat, P., Pietluch, F., Kała, J., Rafacz, D., Bąkała, L., et al. (2022). Benchmarks in antimicrobial peptide prediction are biased due to the selection of negative data. Brief. Bioinform 23, bbac343. doi:10.1093/bib/bbac343
Sidorov, P., Naulaerts, S., Ariey-Bonnet, J., Pasquier, E., and Ballester, P. J. (2019). Predicting synergism of cancer drug combinations using NCI-almanac data. Front. Chem. 7, 509. doi:10.3389/fchem.2019.00509
Stader, F., Battegay, M., and Marzolini, C. (2021). Physiologically-based pharmacokinetic modeling to support the clinical management of drug-drug interactions with bictegravir. Clin. Pharmacol. Ther. 110, 1231–1239. doi:10.1002/cpt.2221
Stumpfe, D., Hu, Y., Dimova, D., and Bajorath, J. (2014). Recent progress in understanding activity cliffs and their utility in medicinal chemistry. J. Med. Chem. 57, 18–28. doi:10.1021/jm401120g
Talele, T. T., Khedkar, S. A., and Rigby, A. C. (2010). Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 10, 127–141. doi:10.2174/156802610790232251
Taragin, M. I. (2019). Learning from negative findings. Israel J. Health Policy Res. 8, 38. doi:10.1186/s13584-019-0309-5
Tejera, E., Munteanu, C. R., López-Cortés, A., Cabrera-Andrade, A., and Pérez-Castillo, Y. (2020). Drugs repurposing using QSAR, docking and molecular dynamics for possible inhibitors of the SARS-CoV-2 Mpro protease. Molecules 25, 5172. doi:10.3390/molecules25215172
Tharwat, A. (2020). Classification assessment methods. Appl. Comput. Inf. 17, 168–192. doi:10.1016/j.aci.2018.08.003
Thomas, R. S., Black, M. B., Li, L., Healy, E., Chu, T.-M., Bao, W., et al. (2012). A comprehensive statistical analysis of predicting in vivo hazard using high-throughput in vitro screening. Toxicol. Sci. 128, 398–417. doi:10.1093/toxsci/kfs159
Tolah, A. M., Altayeb, L. M., Alandijany, T. A., Dwivedi, V. D., El-Kafrawy, S. A., and Azhar, E. I. (2021). Computational and in vitro experimental investigations reveal anti-viral activity of licorice and glycyrrhizin against severe acute respiratory syndrome coronavirus 2. Pharm. (Basel) 14, 1216. doi:10.3390/ph14121216
Tropsha, A. (2010). Best practices for QSAR model development, validation, and exploitation. Mol. Inf. 29, 476–488. doi:10.1002/minf.201000061
Tropsha, A., Gramatica, P., and Gombar, V. (2003). The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 22, 69–77. doi:10.1002/qsar.200390007
Tseng, A., Seet, J., and Phillips, E. J. (2015). The evolution of three decades of antiretroviral therapy: Challenges, triumphs and the promise of the future. Br. J. Clin. Pharmacol. 79, 182–194. doi:10.1111/bcp.12403
Ullah, I., Khan, K. S., Tahir, M. J., Ahmed, A., and Harapan, H. (2021). Myths and conspiracy theories on vaccines and COVID-19: Potential effect on global vaccine refusals. Vacunas Engl. Ed. 22, 93–97. doi:10.1016/j.vacun.2021.01.001
Vázquez, J., López, M., Gibert, E., Herrero, E., and Luque, F. J. (2020). Merging ligand-based and structure-based methods in drug discovery: An overview of combined virtual screening approaches. Molecules 25, 4723. doi:10.3390/molecules25204723
Veríssimo, G. C., Menezes Dutra, E. F., Teotonio Dias, A. L., de Oliveira Fernandes, P., Kronenberger, T., Gomes, M. A., et al. (2019). HQSAR and random forest-based QSAR models for anti-T. vaginalis activities of nitroimidazoles derivatives. J. Mol. Graph Model. 90, 180–191. doi:10.1016/j.jmgm.2019.04.007
Veríssimo, G. C., Serafim, M. S. M., Kronenberger, T., Ferreira, R. S., Honorio, K. M., and Maltarollo, V. G. (2022). Designing drugs when there is low data availability: One-shot learning and other approaches to face the issues of a long-term concern. Expert Opin. Drug Discov. 17, 929–947. doi:10.1080/17460441.2022.2114451
V’kovski, P., Kratzel, A., Steiner, S., Stalder, H., and Thiel, V. (2021). Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170. doi:10.1038/s41579-020-00468-6
Wang, J., Jiang, Y., Wu, Y., Yu, H., Wang, Z., and Ma, Y. (2022). Pharmacophore-based virtual screening of potential SARS-CoV-2 main protease inhibitors from library of natural products. Nat. Product. Commun. 17, 1934578X2211436. doi:10.1177/1934578X221143635
Wang, Y., Bryant, S. H., Cheng, T., Wang, J., Gindulyte, A., Shoemaker, B. A., et al. (2017). PubChem BioAssay: 2017 update. Nucleic Acids Res. 45, D955–D963. doi:10.1093/nar/gkw1118
Weintraub, P. G. (2016). The importance of publishing negative results. J. Insect Sci. 16, 109. doi:10.1093/jisesa/iew092
WHO (2023). WHO coronavirus (COVID-19) dashboard. Available at: https://covid19.who.int (Accessed July 10, 2023).
Williams, A. J., and Ekins, S. (2011). A quality alert and call for improved curation of public chemistry databases. Drug Discov. Today 16, 747–750. doi:10.1016/j.drudis.2011.07.007
Wlodawer, A. (2002). Rational approach to AIDS drug design through structural biology. Annu. Rev. Med. 53, 595–614. doi:10.1146/annurev.med.53.052901.131947
Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., Wang, Y., et al. (2020). Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 10, 766–788. doi:10.1016/j.apsb.2020.02.008
Yap, C. W. (2011). PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 32, 1466–1474. doi:10.1002/jcc.21707
Young, B. E., Fong, S.-W., Chan, Y.-H., Mak, T.-M., Ang, L. W., Anderson, D. E., et al. (2020). Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 396, 603–611. doi:10.1016/S0140-6736(20)31757-8
Zakharov, A. V., Peach, M. L., Sitzmann, M., and Nicklaus, M. C. (2014). QSAR modeling of imbalanced high-throughput screening data in PubChem. J. Chem. Inf. Model. 54, 705–712. doi:10.1021/ci400737s
Zaki, M. E. A., Al-Hussain, S. A., Masand, V. H., Akasapu, S., Bajaj, S. O., El-Sayed, N. N. E., et al. (2021). Identification of anti-SARS-CoV-2 compounds from food using QSAR-based virtual screening, molecular docking, and molecular dynamics simulation analysis. Pharmaceuticals 14, 357. doi:10.3390/ph14040357
Zhang, C.-H., Stone, E. A., Deshmukh, M., Ippolito, J. A., Ghahremanpour, M. M., Tirado-Rives, J., et al. (2021). Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 7, 467–475. doi:10.1021/acscentsci.1c00039
Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., et al. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 368, 409–412. doi:10.1126/science.abb3405
Zhao, L., Wang, W., Sedykh, A., and Zhu, H. (2017). Experimental errors in QSAR modeling sets: What we can do and what we cannot do. ACS Omega 2, 2805–2812. doi:10.1021/acsomega.7b00274
Keywords: enzymatic inhibition, HQSAR, Mpro, QSAR, SARS-CoV-2
Citation: Serafim MSM, Pantaleão SQ, da Silva EB, McKerrow JH, O’Donoghue AJ, Mota BEF, Honorio KM and Maltarollo VG (2023) The importance of good practices and false hits for QSAR-driven virtual screening real application: a SARS-CoV-2 main protease (Mpro) case study. Front. Drug Discov. 3:1237655. doi: 10.3389/fddsv.2023.1237655
Received: 09 June 2023; Accepted: 13 July 2023;
Published: 25 July 2023.
Edited by:
José L. Medina-Franco, National Autonomous University of Mexico, MexicoReviewed by:
Edgar López-López, National Polytechnic Institute of Mexico (CINVESTAV), MexicoCopyright © 2023 Serafim, Pantaleão, da Silva, McKerrow, O’Donoghue, Mota, Honorio and Maltarollo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinícius Gonçalves Maltarollo, dmluaWNpdXNtYWx0YXJvbGxvQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.