
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Drug Discov., 24 May 2023
Sec. Technologies and Strategies to Enable Drug Discovery
Volume 3 - 2023 | https://doi.org/10.3389/fddsv.2023.1201419
This article is part of the Research TopicDrug Discovery and Development Explained: Introductory Notes for the General PublicView all 11 articles
 Natesh Singh1,2†Philippe Vayer2Shivalika Tanwar2
Natesh Singh1,2†Philippe Vayer2Shivalika Tanwar2 Jean-Luc Poyet3Katya Tsaioun4,5
Jean-Luc Poyet3Katya Tsaioun4,5 Bruno O. Villoutreix2,5*
Bruno O. Villoutreix2,5*Finding new drugs usually consists of five main stages: 1) a pre-discovery stage in which basic research is performed to try to understand the mechanisms leading to diseases and propose possible targets (e.g., proteins); 2) the drug discovery stage, during which scientists search for molecules (two main large families, small molecules and biologics) or other therapeutic strategies that interfere or cure the investigated disease or at least alleviate the symptoms; 3) the preclinical development stage that focuses on clarifying the mode of action of the drug candidates, investigates potential toxicity, validates efficacy on various in vitro and in vivo models, and starts evaluate formulation; 4) the clinical stage that investigates the drug candidate in humans; 5) the reviewing, approval and post-market monitoring stage during which the drug is approved or not. In practice, finding new treatments is very challenging. Despite advances in the understanding of biological systems and the development of cutting-edge technologies, the process is still long, costly with a high attrition rate. New approaches, such as artificial intelligence and novel in vitro technologies, are being used in an attempt to rationalize R&D and bring new drugs to patients faster, but several obstacles remain. Our hope is that one day, it becomes possible to rapidly design inexpensive, more specific, more effective, non-toxic, and personalized drugs. This is a goal towards which all authors of this article have devoted most of their careers.
Drug discovery has a long history and dates back to the early days of human civilization. In those ancient times, treatments were often discovered by chance or resulted from observation of nature, typically but not exclusively, using ingredients extracted from plants/animals, and not just used for physical remedy but also for spiritual healing. Modern drug discovery research started to being performed around the early 1900s. Nowadays, the development of a new medicine usually starts when basic research, often performed in academia, identifies a macromolecule (i.e., a molecule with a large molecular weight like genes/proteins), or a dysfunctional signaling pathway or a molecular mechanism apparently linked to a disease condition (pre-discovery stage) (Figure 1; Table 1) (Hefti, 2008; Hughes et al., 2011; Mohs and Greig, 2017; Villoutreix, 2021). In general, at this stage, research teams attempt to identify the so-called therapeutic targets (often a protein) that are linked to the disease state (Gashaw et al., 2012). To be nominated therapeutic target, scientists will also have to find therapeutic agents that modify the function of the perturbed target and restore health or alleviate symptoms. Finding the right target is however extremely challenging. Further, drugs are efficient in humans because of specific actions on the intended therapeutic target but also due to interactions with other, unintended (often unknown) targets! The process continues with the search of therapeutic agents followed by a preclinical phase, during which potential drugs are tested in a battery of animal models, to demonstrate safety and select drug candidates (novel strategies to avoid animal testing are being developed, see below). Clinical studies in humans can then get started to establish safety and efficacy of the drugs in patients with the highest benefit-to-risk ratio (Kandi and Vadakedath, 2023). The studies are then submitted to regulatory agencies, which review the documents and decide about market approval. If the review is positive, the drug can then be released to the market and be administrated to patients. Once a drug has been approved, investigations continue to monitor putative side effects that could be caused, over time, by the new treatment. This last step is often referred to as pharmacovigilance studies (or real-world evidence), generally dubbed “phase 4” clinical trial. The entire drug discovery and development process involves many disciplines, years of efforts and is very expensive. It also implies the generation and use of vast amount of data usually obtained via different types of high-throughput technologies. Many of these experiments and the analysis of the results can be automated via computer-assisted methods to speed-up some steps of the process, gain knowledge and reduce mistakes.
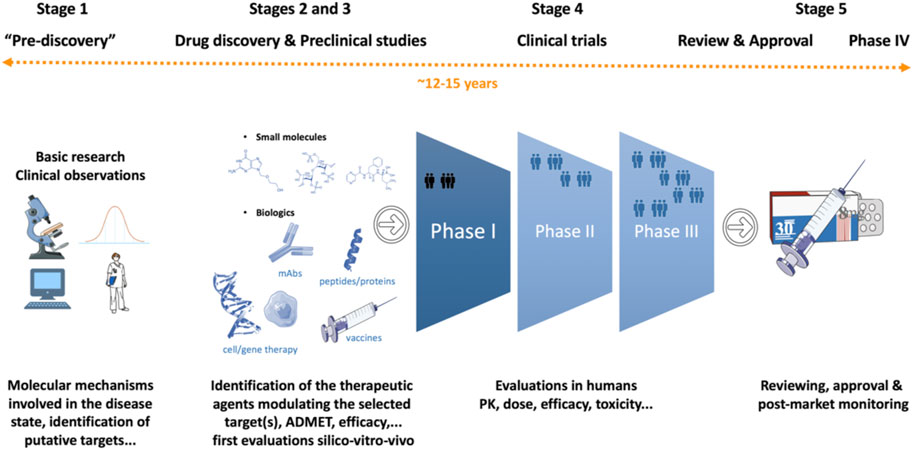
FIGURE 1. Drug discovery and development. The main stages are represented in a highly simplified manner. The process varies depending on the molecular mechanisms expected to be linked to the disease and the type of therapeutic agents that needs to be developed. The approximate cost is around US $2.8 billion and the time needed to complete the entire process is around 12–15 years.
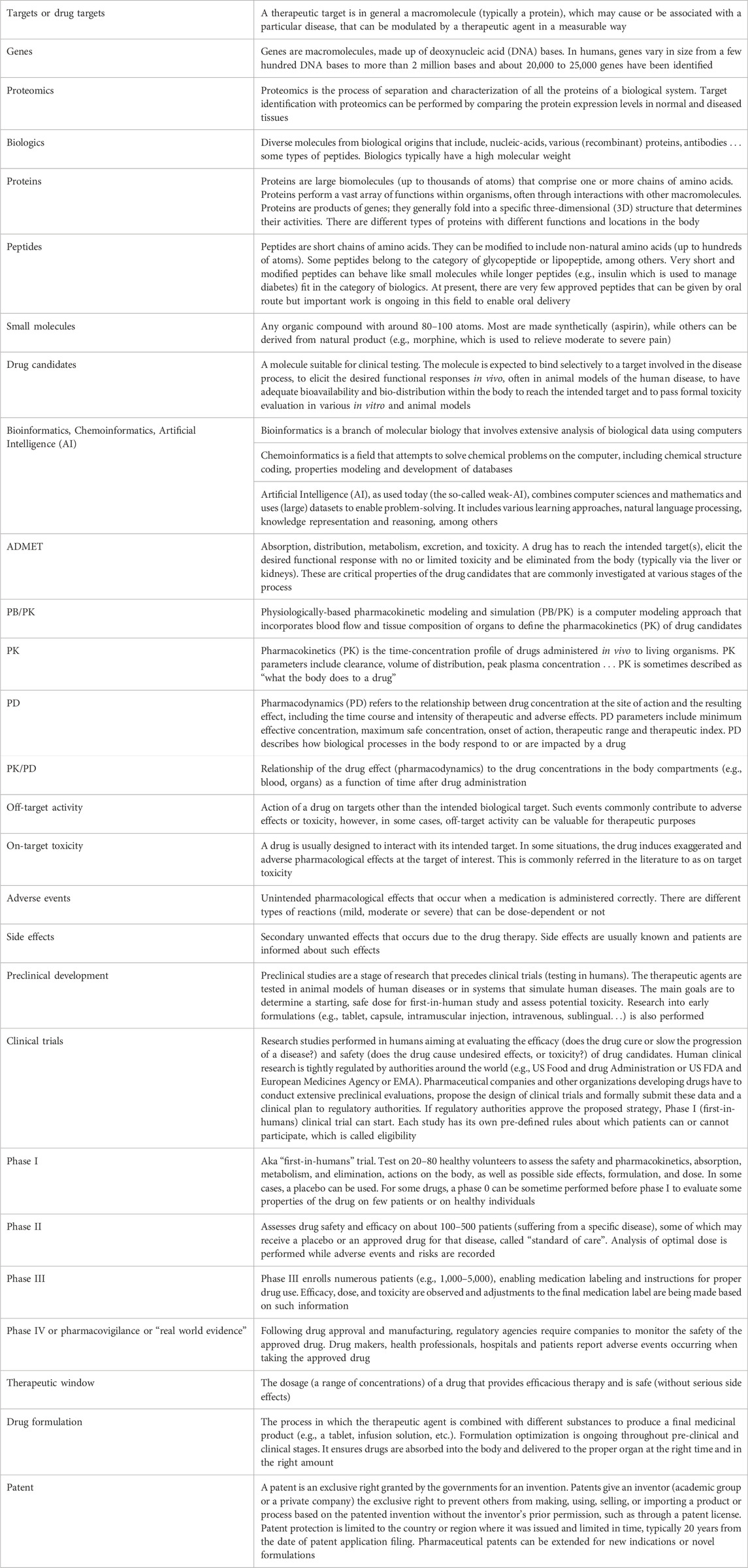
TABLE 1. Glossary.
As mentioned above, to act on a disease, the problematic target(s) have to be modulated by a therapeutic agent (or several). There is a wide variety of agents that traditionally fits into two major classes, the so-called “small molecules” (small chemical compounds, some modified short peptides…) and the “biologics” (typically macromolecules such as recombinant proteins, antibodies, siRNAs, long peptides, cells, genes … and vaccines). There are major differences between biologics and small molecules (Figure 2; Table 2) and we will essentially focus here on small molecules. It is also important to note that gene therapy is different from the other types of therapeutic agents because it is a technique that modifies a person’s genes to treat or cure a disease. In this case, the target is a disease-causing gene which has to be modified with a healthy copy of the gene, or the disease causing gene could be inactivated. Thus, beside technical issues, there are a number of ethical questions surrounding gene therapy and genome editing strategies that are not easy to answer. Further, some therapeutic agents are not acceptable to some parts of the population, as seen during the COVID-19 crisis and vaccine hesitancy. This is often due to misunderstanding of the biological processes and misinformation, resulting in fears, but yet this has to be considered. Also, about 5%–10% of the population are non-responders and have to receive other medications than vaccines. The division into small molecules and biologics is far from being perfect as some therapeutic agents combine a small molecule grafted onto a biologic (e.g., tisotumab vedotin is an antibody-drug conjugate used to treat cervical cancer). Therapeutic agents can be administrated to patients via different routes, called “routes of administration”. Small molecules can in general be administrated orally (the most convenient route for patients), while biologics usually need to be injected. The choice of a route of administration is also governed by the patient’s condition, for instance, in acute situations in hospitals, drugs are most often given intravenously. Other critical medical interventions that will not be discussed here are surgery, radiotherapy and psychological support.
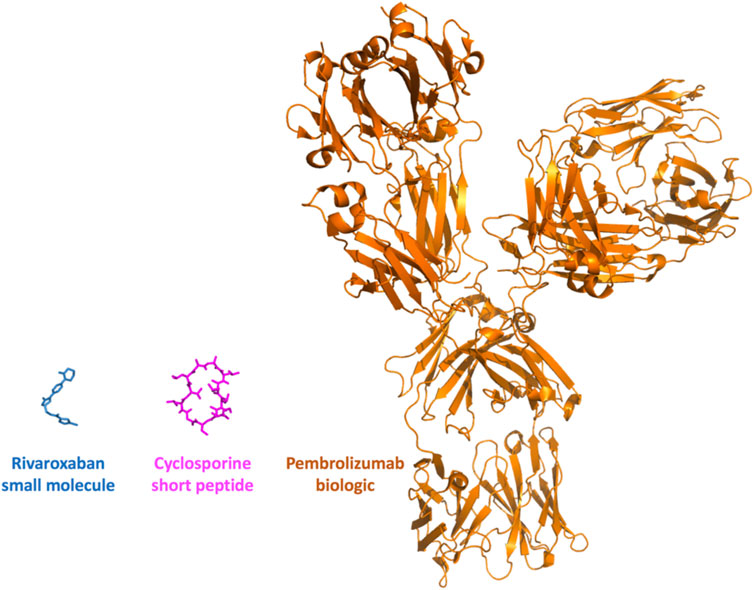
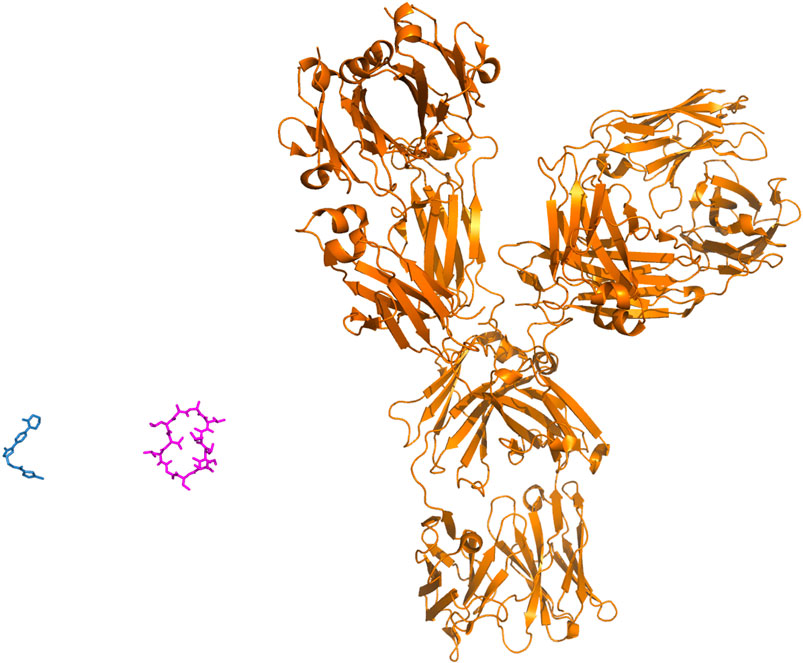
FIGURE 2. Small molecules, peptides and biologics. The properties and sizes of the therapeutic agents vary greatly. Three molecules are presented at the same scale, these involve rivaroxaban, a small chemical molecule used to treat thrombosis and pulmonary embolism, cyclosporine, a short immunosuppressive cyclic peptide (11 amino-acids, a biologic that still resembles to a certain extent to a small molecule) used to treat post-transplant organ rejection and a biologic, pembrolizumab (antibody, over 1300 amino-acids), used to treat various types of cancer.
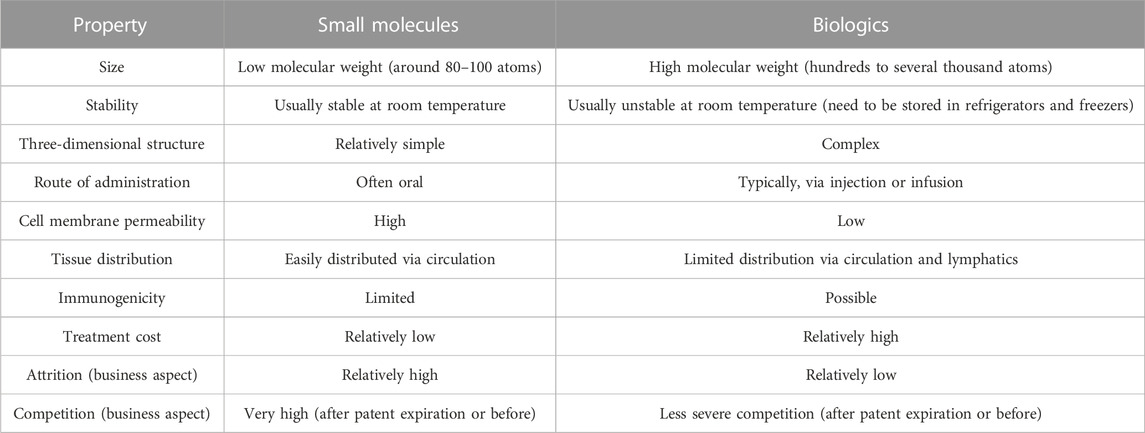
TABLE 2. General characteristics of small molecule drugs and biologics.
There are several stages in the drug discovery process that require numerous skills and the use of various advanced technological platforms (often a combination of computational and experimental approaches) to validate targets and search for therapeutic agents. When initial experimental compounds have been sufficiently optimized to be selective, potent and safe in preliminary in vitro experiments and animal models, they can be nominated as drug candidates. At this stage, the project focus shifts from drug discovery to drug development to enable human clinical trials. If the therapeutic agent is successful in all three phases of the clinical trials, it goes through regulatory registration and the drug can be marketed (Hefti, 2008; Hughes et al., 2011; Mohs and Greig, 2017).
Now, we will take a closer look at the process with the discovery of small molecules as an example. The process usually begins by focusing on a disease and the search of possible targets, often proteins, that can be modulated by small compounds (Hughes et al., 2011) (Figure 1). These compounds are expected to interfere or prevent the disease or at least limit the development of symptoms. These targets can be identified using cellular assays, genomic studies, proteomic studies, among many others. Then, thousands (to millions or even billions when using computer-aided drug design approaches prior to vitro assays) of small molecules have to be tested in various types of assays and a few promising molecules are then evaluated in animal models (and in alternative in vitro models) of human diseases. It is worth mentioning here that animal models can be misleading (e.g., a drug found toxic in animal models may not be toxic to humans or the opposite) (Pognan et al., 2023). At the same time, absorption, distribution and elimination studies (ADME) are conducted. After years of research, a few compounds will hopefully be safe and effective enough to take forward to trials in patients. The different stages can have different names in the scientific literature, often they are referred to as: the pre-discovery and basic research stage (around 5–6 years) in which targets and modifying small molecules are searched in silico (i.e., using a computer), in vitro (i.e., in the test tube), ex vivo (e.g., on tissues or organs) and in vivo using simple animal models (i.e., in a living organism, typically rats or mice) and a preclinical stage (2–3 years) during which the best small molecules are selected using various in silico, in vitro and in vivo experiments. In general, after all these steps, only a few compounds progress to the next stage. Toxicity is investigated further on at least two animal models [one rodent (e.g., rat) and one non-rodent (e.g., dogs, mini-pigs)] often using different administration routes before they become nominated clinical candidates and get a regulatory permission to proceed to human clinical trials. Prior to starting clinical trials, a so-called Investigational New Drug (IND) application is submitted to regulatory agencies (e.g., the Food and Drug Administration in the United States). Such documents, at least up to now (see below), usually include animal efficacy data and toxicity (Good Laboratory Practice (GLP)-compliant animal toxicology data are performed supporting the dose, dosing schedule, administration), manufacturing information, clinical protocols (e.g., patient population, number of patients, duration of the study) proposed for the clinical trials and information about the investigators of the study.
If the IND is approved, then clinical trials start (4–7 years) (Kandi and Vadakedath, 2023). In some specific cases such as cancer, a so-called phase 0 may get started, which involves the use of very small doses of the new drug in a limited number of people and sometimes in patients. This is an exploratory study with the goal of quickly exploring if and how the drug may work. In Phase I, the safety, and tolerability of the therapeutic agent (usually a single dose at first and then short-term multi-dose studies) is tested in a small number of healthy individuals (e.g., 20–80 people). Other parameters are investigated including the dose. Phase II typically involves 100–500 patients and the study can take place in several hospitals located in different countries. The study is designed to determine whether or not the therapeutic agent provides the desired therapeutic effect. Safety studies continue through the phase II trials. In the first part of phase II, referred to as phase IIa, the goal is to further refine the dose required to provide the desired therapeutic impact or monitored endpoints for the clinical candidate. Once the proper dose levels are determined, phase IIb studies can be initiated. The goal of the phase IIb is to determine the overall efficacy of the candidate drugs in a limited population of subjects. Numerous drug candidates fail in phase II due to safety issues or lack of efficacy. In phase III, the efficacy of the drug candidate is evaluated in a larger patient population. These studies are typically randomized and involve 1,000–5,000 patients at multiple clinical trial centers and are designed to determine the efficacy of the candidate compound relative to the current standard of care or a placebo, possible interactions with other medications and re-assess different doses (optimal dose is important for medication effectiveness). When neither the clinicians nor the patients know which of the treatments the patient is getting, the study is said to be double-blind. The cost and time associated with this phase can vary dramatically depending on the disease and the clinical endpoint under investigation. Phase III clinical trials are the most expensive part of drug discovery and development as it has a complex design and requires a large number of patients. Last but not least, formulation and stability studies are performed during the development stage to characterize the impurities present (either in batches or during storage conditions worldwide), and to determine the best formulation. Upon completion of the phase III trial, a New Drug Application (NDA) is submitted to the regulatory agencies to demonstrate drug safety and efficacy. Regulatory reviews can lead to requests for additional information, or even additional clinical trials to further establish either safety or efficacy. Ideally, these reviews lead to regulatory approval, including labelling requirements, and approval to market (review and approval ∼1–2 years). For approval, the drug must have adequate pharmaceutical quality, therapeutic effectiveness, and safety. It has to have a favorable “risk-benefit ratio”. Drugs offering important advances in treatment of a condition are given priority. Approval of regulatory bodies does not, however, signal the end of clinical trials. In many cases, regulatory agencies will require additional follow-up studies, often referred to as phase IV or post-marketing surveillance (“real-world evidence” trials) with infinite duration. In general, these studies are designed to detect rare adverse effects across a much larger population of patients or long-term adverse effects. The impact of phase IV studies can include alterations to labelling based on safety observations, contraindications for use of the new drug in combination with other medications, or even the withdrawal of marketing approval if the findings are severe enough.
Drug repurposing or repositioning aims to take a drug (approved or in advanced clinical stages or even a drug that has been withdrawn from the market, most of the time it involves small molecules but biologics like antibodies are also explored), thus a molecule that has undergone extensive safety and efficacy testing, and use it for an additional or unrelated indication (van den Berg et al., 2021; Roessler et al., 2021; Schipper et al., 2022). In some situations, even a withdrawn drug can be repurposed like thalidomide, originally intended as a sedative and then used for treating a wide range of other conditions, including morning sickness in pregnant women. Thalidomide was then withdrawn due to causing birth defects but then was approved to treat leprosy (in 1998) and multiple myeloma (in 2006) (Begley et al., 2021). Drug repurposing approach can be very valuable in most cases including emergency situation like a pandemic, for rare and neglected diseases [for which specific drug developments are in general missing in pharmaceutical companies (Scherman and Fetro, 2020; Roessler et al., 2021)]. This strategy is promoted as a cost- and time-effective approach for providing novel medicines. It is often claimed that repurposing drugs can be faster, more economical, less risky, and carry higher success rates as compared to traditional approaches, primarily because it is in theory possible to bypass early stages of development such as establishing drug safety. Other benefits that come with this approach include readily available products and manufacturing supply chains. Drug repurposing can be very profitable as in the case of fenfluramine (in 2022, acquisition of Zogenix by UCB Pharma for about US$ 1.9 billion, https://www.ucb.com/stories-media/Press-Releases/article/UCB-Completes-Acquisition-of-Zogenix-Inc), a drug initially developed for weight loss, withdrawn and now used in several countries for the treatment of some forms of epilepsy (Odi et al., 2021). Yet, despite advantages, drug repurposing suffers from several issues. One problem is that there are no possibilities for optimization of the therapeutic molecule without losing the repurposing potential because any small change in the structure of the therapeutic agent means a new full manufacture process validation and preclinical safety development. Identifying an optimal dosage and formulation for the new disease indication can also be time consuming and requires novel investigations while side effects can indeed arise due to the new indication or in cases doses need to be changed. Also, assessing the patent status of the drug to repurpose requires very specific skills. The molecules that are investigated for repurposing are either patented or off-patent, and in some cases the intellectual property protection for the new indication may not be strong enough to engage in such project. Overall, while drug repurposing is intuitively attractive as it offers shorter routes to the clinic, challenges throughout the entire process are usually substantial. Investigating molecular mechanisms behind repurposing can however be very valuable as it can help identifying novel targets and as the repurposed drugs could be considered as starting point for the development of novel compounds (e.g., lenalidomide and pomalidomide are superior molecules derived from thalidomide) and as such emerge as breakthrough innovation in a reduced amount of time and still reduced cost compared to starting from scratch. It could also be of interest to combine several approved drugs (in some cases with a newer drug) to increase effectiveness.
Providing efficient and safe drug to patients is a long and complex process. The amount of data generated during this process or that can be collected from various sources is massive. It is thus necessary to integrate as much as possible quality data so as to be able to make decision in real time. Artificial Intelligence (AI or indeed, most of the time, machine learning) can definitely contribute here as it involves the use of powerful computers and efficient program algorithms to integrate large volume of data to train expert systems to perform a complex task (Brogi and Calderone, 2021; Ruffolo et al., 2021; Jayatunga et al., 2022; Sadybekov and Katritch, 2023). During the early discovery phases, AI is used to rationalize processes, and to assist in project management (e.g., definition of a target product profile that allows to locate each compound with regard to the expected final drug specifications in a complex multi-dimensional space), to summarize information, to understand better complex biological systems (e.g., using for instance system biology and chemogenomics approaches), or to propose original compounds or biologics (e.g., small molecules, peptides) generated by the machine under various types of constraints (e.g., ADMET constraints or affinity to the target) (Lambert, 2010; Gupta et al., 2021; Paul et al., 2021; Kontoyianni, 2022; Vijayan, et al., 2022). Most of the well-known success stories of AI have been in image recognition (e.g., in the early days, the approach was trained to for instance recognize cat and dog images, but today the method can be used to analyze biopsies or guide surgery) while also advertised in reducing time to reach phase I clinical trial. In the latter case, one can site the story of compound DSP-1181, developed by Exscientia and Sumitomo Dainippon Pharma, intended to treat obsessive compulsive disorder where time from first screening to the development stage was 4 time faster than using a conventional approach (although, unfortunately, the molecule failed in phase I, for numerous reasons including a difficult target while it was also observed that the molecules generated by AI were not novel) (Santa Maria Jr et al., 2023) (https://www.science.org/content/blog-post/another-ai-generated-drug; https://www.cas.org/resources/cas-insights/drug-discovery/ai-designed-drug-candidates). Similar observations have been posted by hundreds of financial analysts and research scientists about results obtained by other AI companies. In other words, the AI predictions are not perfect and indeed cannot be perfect at present (Bajorath, 2021; Bender and Cortés-Ciriano, 2021). This situation reflects the dependency of AI/machine learning to quality, size and diversity of the data used to train the mathematical models. There are millions of compounds (most will never be a drug) tested via standard experiments available in various databases, but there are only a few thousand approved in humans that are annotated on which to learn from, highlighting the so-called data gap (i.e., there are billions of pictures of dogs and cats to learn from, but a limited amount of quality data is available in the field of drug discovery despite the use of numerous the high-throughput approaches). The predictions can thus be misleading, because we do not have enough quality data as input and/or because we do not understand enough the complexity of the biological systems (Moingeon et al., 2022). During the drug development phases, in human, AI is associated to data-mining to for instance model some properties (e.g., PB/PK, PK/PD or population-based simulations and analysis, prediction of drug-drug interactions …). At this stage, these computer approaches can also be used to select the most informative population profile to be included in clinical trials or to explain the variability of effects, or provide « virtual » patients or populations, and applied to, for example, pediatric formulation using as input data collected on adults (Lang et al., 2021). Related to these, the concept of digital twins (which has been around for a while in other areas of research), now starts to be explored in the context of drug discovery and development. The overall idea would be to collect data about a particular disease, how it progresses, about the current treatments, about specific patients, and about a whole population, encapsulate all these data into a computer model so as to create a digital representation of a biological system or of a person and be able to simulate, for example, what might happen if one were to take a novel drug. While the concept is attractive, there are still major challenges and obstacles ahead but progresses are being made (An and Cockrell, 2022). Overall, AI, in the field of drug discovery and development, is still in the infancy stage and it will take time to fully integrate the technology into the R&D process (Hillisch et al., 2015). AI-discovered drugs do not guarantee success in clinical trials. The understanding of the data used as well as the critical mind of the scientists are key points that lead to the success or failure of AI-assisted drug research and development processes. The technology, in some circumstances, can make the process faster and more cost-effective, however, AI needs quality data to produce meaningful results and still today requires significant experimental validation. As such, it is important to trust AI, but verify the predictions (Schneider et al., 2020; Bajorath, 2021).
Analyses across all therapeutic areas indicate that the development of a new medicine, from target identification through approval for marketing, takes around 12–15 years and often longer. The cost to develop a new drug is very high, in part because failure is endemic in drug discovery, and success is rare. While various numbers have been reported, the latest formal assessment is around US $2.8 billion (DiMasi, 2020). There are many factors that contribute to this situation: the lack of understanding of what causes the disease can lead to the selection of the wrong therapeutic target; the impossibility of reaching the target with a sufficient concentration of drug in vivo without leading to adverse effects; no formulation compatible with the use of the drug in human; the therapeutic agent developed during years is found in phase III to have very low efficacy; the therapeutic agents or a metabolite (e.g., case of a small molecule) can interacts, specifically or not, with other drugs or with hundreds of molecules in the body, these interactions are usually not known in details and can lead to numerous adverse effects; animal experiments that are used to evaluate potency, selectivity, and toxicity during the different stages of the process can be highly misleading; stricter regulatory guidelines; duration of patents; the identified therapeutic molecule can be toxic in some patients but this could not be anticipated during the clinical trials due to the relatively small number of patients treated. Next and related to the cost of R&D, comes the cost of the treatments. Although there is a very complex protocol to determine the price tag of a drug (it varies from country to country, it can consider the insurance system, whether the drug is curative and represents a major advance to both patients and the health system or it has a minor effect on the disease), but in the end, biologics are generally much more expensive than small molecules, in part due to the complex manufacturing process. Studies suggest that on average, the daily dose of biologics costs 22 times more than a small molecule (Makurvet, 2021). It is important to keep in mind that the healthcare systems, in many countries, are about to collapse and that about half of the world population cannot get access to basic treatments (Ozawa et al., 2019). Biologics have been here for several decades already and are becoming increasingly important in several therapeutic areas. For example, cancer checkpoint inhibitors (e.g., the antibody ipilimumab and about 4–5 others at the time of writing) have received considerable and broad interest because of their ability to generate responses in many hitherto intractable malignant tumors. Yet, many recent studies suggest that such molecules lead to responses in less than 10%–15% of patients with cancer. Clearly, such molecules offer hope but also rise many questions (Fojo et al., 2014; Kantarjian and Rajkumar, 2015). That is, in some cases, biologics are real innovative breakthroughs, but in other situations, the strategy is pursued only for commercial reasons and alternative molecules such as small molecules are not even considered. These questions are, in theory, investigated by regulatory agencies [The United States Food and Drug Administration (FDA), European Medicines Agency (EMA), Pharmaceuticals and Medical Devices Agency (PMDA)] so as to try to avoid speculative drugs but more transparent processes would certainly be beneficial to patients and the general population. Although finding new treatments is very difficult, it is a profitable market, with global drug sales expected to grow to US$ 1.9 trillion by 2027 (Mullard, 2023).
It is important to note that, in step with the scientific progress in human tissue models research in the past decades, in the US, new medicines may not have to be tested in animals, according to legislation signed by the President Joe Biden in late December 2022 (“Text–S.5002–117th Congress (2021–2022): FDA Modernization Act 2.0.” 29 September 2022. https://www.congress.gov/bill/117th-congress/senate-bill/5002/text). Accordingly, US FDA is already accepting data from in vitro studies as part of the formal submission to the Agency (Wadman, 2023). Additionally, at the same time, following the leadership of some academic researchers (e.g., Guzelian et al., 2005; Hoffmann and Hartung, 2006), major European and US agencies started using evidence-based methodologies, such as systematic reviews and systematic maps, in toxicological assessment. These methodologies were developed and tested over the last 40 years in clinical research, spearheaded by Cochrane Collaboration (www.Cochrane.org) to compare the effectiveness of treatments, and have been applied to toxicological assessment of data-rich substances by the European Food Safety Authority (EFSA, 2017) and US Environmental Protection Agency’s (US EPA, https://cfpub.epa.gov/ncea/iris_drafts/recordisplay.cfm?deid=356370). While some of the aspects of these methodologies are not entirely applicable to drug-discovery because of the proprietary nature of the work, the main principles of evidence-based approaches, which encourage pre-publishing the methodologies before the research is conducted, comprehensiveness and transparency in data selection, minimization of bias (or systematic error), are in line with basic principles of the scientific method, and are applicable to drug discovery. Programs and drug candidates are all too frequently selected based on a biased opinion of a few scientists who are bound by similar training, scientific methodologies and beliefs. Opening-up drug discovery to scrutiny by other scientists with different training and opinions may lead to more failures in the earlier discovery stage, but less failures in the clinic, resulting in enhanced efficiency and more successes, benefiting the patients who need new treatments, first and foremost.
Drug discovery and development is a long and difficult endeavor; all novel ideas and strategies that can improve the process are valuable to explore. It is interesting to note that despite the steady increase in research and development expenditure, and major scientific advances in proteomics and genomics, the discovery of new drugs either seems to be drying-up some years or to remain essentially stable (Laermann-Nguyen and Backfisch, 2021). This situation has various origins (e.g., many diseases with no treatment are extremely difficult to study), while, certainly, industry scientists would benefit from greater exposure to new ideas from public research and public researchers would benefit from the private sector to move beyond exploration of molecular mechanisms towards the end goal of efficient development of candidate therapeutic agents. Along these lines, some countries like the United States and United Kingdom have been working extensively at improving academic drug discovery (e.g., all the skills and platforms connected via open research networks with rational protocols) but in the others, the process is fragmented (no coordination, no intent, duplication of efforts and inefficient investments …) and, thus, not capable of producing desired results compared to the time, energy and money spent. A first step could be to develop strong academic drug discovery networks in countries where this type of activity is not coordinated or not considered. Strong collaborations between the private sector, academia and not-for-profit institutions are clearly of major importance and have led to some successes in the past but such partnerships can be difficult to maintain over a long period of time (Yildirim et al., 2016; Takebe et al., 2018). The rationale being that open interconnections between the different scientific disciplines involved in drug research allow a “cross-fertilization”, each of them benefiting from the advances of the other fields. Obviously, such collaborations tend to be easier when academic and private research teams are located on the same campus, with possibilities of sharing ideas or technologies. Other types of collaboration imply building consortia, often for around 4–5 years, with research teams located in different cities or countries (unfortunately, most of the time, when the consortia have been built, they function as closed systems not allowing new scientists or novel research teams to join). Therefore, novel strategies need to be pursued, and among the novel public-private models that are being investigated, open science partnerships, could be of interest, if correctly implemented (e.g., the system must be open to all interested scientists, teams and relevant disciplines) (Gold and Edwards, 2022). Open science projects (Chodera et al., 2020), like the consortia models discussed above, are built on the differential expertise of the various partners, with generally academic and governmental partners taking on a larger role in the earlier stages and big pharmas leading in the later stages (e.g., advanced preclinical investigations, product development, manufacturing, and distribution). But in open science projects, results, publications, data, tools, and materials are open without regard for intellectual property. At some points, the various partners are free to use the results and develop their own proprietary products if deemed appropriate.
Next, novel technologies including AI could be a game changer in the years to come, even more so once we get past the hype stage. Novel approaches to replace animal models by more efficient, ethical, human-biology-based in vitro approaches could also play a significant role this next decade. Indeed, new tools and understanding, in, for instance, the area of investigative toxicology, are continually being implemented to reduce safety-related attrition in drug development (Aleo et al., 2020). Combining all these strategies, methods and know-how should definitively facilitate the design of more specific, effective, non-toxic, and patient-tailored drugs, thereby, providing a more optimistic outlook to the field. As a last note, we encourage the general public and patients to become more curious about the process of finding novel therapies, from the pre-discovery to the post-marketing stages. Further, crowd-funded citizen science initiatives are emerging in various areas of drug discovery and development (e.g.,https://www.clinicaltrialsarena.com/news/citizen-science-as-an-open-trials-tool-for-post-marketing-and-drug-repurposing-5909331-2/; see also the CTSA program at NIH), these projects are definitively valuable to the field.
NS, PV, and BV conceptualized the topic and drafted the first version of the manuscript. All authors contributed to the article and approved the submitted version.
Support from the INSERM institute is appreciated. This article is based upon work supported by the National Science Foundation under Grant No. 2136307.
We thank Stephane Auvin, Epileptologist and Child Neurologist, Head of the Pediatric Neurology Department, Robert Debré Hospital, for interesting discussions about the use of fenfluramine for the treatment of some forms of epilepsy.
Authors KT and BV were employed by the company Akttyva Therapeutics, Inc. Author NS is employed by Evotech Se.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aleo, M. D., Shah, F., Allen, S., Barton, H. A., Costales, C., Lazzaro, S., et al. (2020). Moving beyond binary predictions of human drug-induced liver injury (DILI) toward contrasting relative risk potential. Chem. Res. Toxicol. 33, 223–238. doi:10.1021/acs.chemrestox.9b00262
An, G., and Cockrell, C. (2022). Drug development digital twins for drug discovery, testing and repurposing: A schema for requirements and development. Front. Syst. Biol. 2, 928387. doi:10.3389/fsysb.2022.928387
Bajorath, J. (2021). State-of-the-art of artificial intelligence in medicinal chemistry. Future Sci. oa. 7, FSO702. doi:10.2144/fsoa-2021-0030
Begley, C. G., Ashton, M., Baell, J., Bettess, M., Brown, M. P., Carter, B., et al. (2021). Drug repurposing: Misconceptions, challenges, and opportunities for academic researchers. Sci. Transl. Med. 13, eabd5524. doi:10.1126/scitranslmed.abd5524
Bender, A., and Cortés-Ciriano, I. (2021). Artificial intelligence in drug discovery: What is realistic, what are illusions? Drug Discov. Today. 26, 511–524. doi:10.1016/j.drudis.2020.12.009
Brogi, S., and Calderone, V. (2021). Artificial intelligence in translational medicine. Int. J. Transl. Med. 1, 223–285. doi:10.3390/ijtm1030016
Chodera, J., Lee, A. A., London, N., and von Delft, F. (2020). Crowdsourcing drug discovery for pandemics. Nat. Chem. 12, 581. doi:10.1038/s41557-020-0496-2
DiMasi, J. A. (2020). Research and development costs of new drugs. JAMA 324, 517. doi:10.1001/jama.2020.8648
EFSA (2017). Protocol for a systematic review on health outcomes related to the age of introduction of complementary food for the scientific assessment of the appropriate age of introduction of complementary feeding into an infant's diet. EFSA J. 15, e04969. doi:10.2903/j.efsa.2017.4969
Fojo, T., Mailankody, S., and Lo, A. (2014). Unintended consequences of expensive cancer therapeutics—the pursuit of marginal indications and a me-too mentality that stifles innovation and creativity: The john conley lecture. JAMA Otolaryngol. Head. Neck Surg. 140, 1225–1236. doi:10.1001/jamaoto.2014.1570
Gashaw, I., Ellinghaus, P., Sommer, A., and Asadullah, K. (2012). What makes a good drug target? Drug Discov. Today Suppl, S24–S30. doi:10.1016/j.drudis.2011.12.008
Gold, E. R., and Edwards, A. M. (2022). Overcoming market failures in pandemic drug discovery through open science: A Canadian solution. Front. Drug. Discov. 2. doi:10.3389/fddsv.2022.898654
Gupta, R., Srivastava, D., Sahu, M., Tiwari, S., Ambasta, R., and Kumar, P. (2021). Artificial intelligence to deep learning: Machine intelligence approach for drug discovery. Mol. Divers. 25, 1315–1360. doi:10.1007/s11030-021-10217-3
Guzelian, P. S., Victoroff, M. S., Halmes, N. C., James, R. C., and Guzelian, C. P. (2005). Evidence-based toxicology: A comprehensive framework for causation. Hum. Exp. Toxicol. 24, 161–201. doi:10.1191/0960327105ht517oa
Hefti, F. F. (2008). Requirements for a lead compound to become a clinical candidate. BMC Neurosci. 9, S7. doi:10.1186/1471-2202-9-S3-S7
Hillisch, A., Heinrich, N., and Wild, H. (2015). Computational chemistry in the pharmaceutical industry: From childhood to adolescence. ChemMedChem 10, 1958–1962. doi:10.1002/cmdc.201500346
Hoffmann, S., and Hartung, T. (2006). Toward an evidence-based toxicology. Hum. Exp. Toxicol. 25, 497–513. doi:10.1191/0960327106het648oa
Hughes, J. P., Rees, S., Kalindjian, S. B., and Philpott, K. L. (2011). Principles of early drug discovery. Br. J. Pharmacol. 162, 1239–1249. doi:10.1111/j.1476-5381.2010.01127.x
Jayatunga, M. K. P., Xie, W., Ruder, L., Schulze, U., and Meier, C. (2022). AI in small-molecule drug discovery: A coming wave? Nat. Rev. Drug Discov. 21, 175–176. doi:10.1038/d41573-022-00025-1
Kandi, V., and Vadakedath, S. (2023). Clinical trials and clinical research: A comprehensive review. Cureus 15, e35077. doi:10.7759/cureus.35077
Kantarjian, H., and Rajkumar, S. V. (2015). Why are cancer drugs so expensive in the United States, and what are the solutions? Mayo Clin. Proc. 90, 500–504. doi:10.1016/j.mayocp.2015.01.014
Kontoyianni, M. (2022). Library size in virtual screening: Is it truly a number's game? Expert Opin. Drug Discov. 17, 1177–1179. doi:10.1080/17460441.2022.2130244
Laermann-Nguyen, U., and Backfisch, M. (2021). Innovation crisis in the pharmaceutical industry? A survey. SN Bus. Econ. 1, 164. doi:10.1007/s43546-021-00163-5
Lambert, W. (2010). Considerations in developing a target product profile for parenteral pharmaceutical products. AAPS Pharm. Sci. Tech. 11, 1476–1481. doi:10.1208/s12249-010-9521-x
Lang, J., Vincent, L., Chenel, M., Ogungbenro, K., and Galetin, A. (2021). Impact of hepatic CYP3A4 ontogeny functions on drug-drug interaction risk in pediatric physiologically-based pharmacokinetic/pharmacodynamic modeling: Critical literature review and ivabradine case study. Clin. Pharmacol. Ther. 109, 1618–1630. doi:10.1002/cpt.2134
Makurvet, F. D. (2021). Biologics vs. small molecules: Drug costs and patient access. Med. Drug Discov. 9, 100075. doi:10.1016/j.medidd.2020.100075
Mohs, R. C., and Greig, N. H. (2017). Drug discovery and development: Role of basic biological research. Alzheimers Dement. (NY) 3, 651–657. doi:10.1016/j.trci.2017.10.005
Moingeon, P., Kuenemann, M., and Guedj, M. (2022). Artificial intelligence-enhanced drug design and development: Toward a computational precision medicine. Drug Discov. Today. 27, 215–222. doi:10.1016/j.drudis.2021.09.006
Mullard, A. (2023). Drug sales to reach $1.9 trillion within 5 years? Nat. Rev. Drug. Discov. 22, 172. doi:10.1038/d41573-023-00026-8
Odi, R., Invernizzi, R. W., Gallily, T., Bialer, M., and Perucca, E. (2021). Fenfluramine repurposing from weight loss to epilepsy: What we do and do not know. Pharmacol. Ther. 226, 107866. doi:10.1016/j.pharmthera.2021.107866
Ozawa, S., Shankar, R., Leopold, C., and Orubu, S. (2019). Access to medicines through health systems in low- and middle-income countries. Health Policy Plan. 34, iii1–iii3. doi:10.1093/heapol/czz119
Paul, D., Sanap, G., Shenoy, S., Kalyane, D., Kalia, K., and Tekade, R. (2021). Artificial intelligence in drug discovery and development. Drug Discov. Today. 26, 80–93. doi:10.1016/j.drudis.2020.10.010
Pognan, F., Beilmann, M., Boonen, H. C. M., Czich, A., Dear, G., Hewitt, P., et al. (2023). The evolving role of investigative toxicology in the pharmaceutical industry. Nat. Rev. Drug. Discov. 1, 317–335. doi:10.1038/s41573-022-00633-x
Roessler, H. I., Knoers, N. V. A. M., van Haelst, M. M., and van Haaften, G. (2021). Drug repurposing for rare diseases. Trends Pharmacol. Sci. 42, 255–267. doi:10.1016/j.tips.2021.01.003
Ruffolo, J. A., Sulam, J., and Gray, J. J. (2021). Antibody structure prediction using interpretable deep learning. Patterns (N Y). 3, 100406. doi:10.1016/j.patter.2021.100406
Sadybekov, A. V., and Katritch, V. (2023). Computational approaches streamlining drug discovery. Nature 616, 673–685. doi:10.1038/s41586-023-05905-z
Santa Maria, J. P., Wang, Y., and Camargo, L. M. (2023). Perspective on the challenges and opportunities of accelerating drug discovery with artificial intelligence. Front. Bioinform. 3, 1121591. doi:10.3389/fbinf.2023.1121591
Scherman, D., and Fetro, C. (2020). Drug repositioning for rare diseases: Knowledge-based success stories. Therapie 75, 161–167. doi:10.1016/j.therap.2020.02.007
Schipper, L. J., Zeverijn, L. J., Garnett, M. J., and Voest, E. E. (2022). Can drug repurposing accelerate precision oncology? Cancer Discov. 12, 1634–1641. doi:10.1158/2159-8290.CD-21-0612
Schneider, P., Walters, W. P., Plowright, A. T., Sieroka, N., Listgarten, J., Goodnow, R. A., et al. (2020). Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 19, 353–364. doi:10.1038/s41573-019-0050-3
Takebe, T., Imai, R., and Ono, S. (2018). The current status of drug discovery and development as originated in United States academia: The influence of industrial and academic collaboration on drug discovery and development. Clin. Transl. Sci. 11, 597–606. doi:10.1111/cts.12577
van den Berg, S., de Visser, S., Leufkens, H. G. M., and Hollak, C. E. M. (2021). Drug repurposing for rare diseases: A role for academia. Front. Pharmacol. 12, 746987. doi:10.3389/fphar.2021.746987
Vijayan, R. S. K., Kihlberg, J., Cross, J. B., and Poongavanam, V. (2022). Enhancing preclinical drug discovery with artificial intelligence. Drug Discov. Today. 27, 967–984. doi:10.1016/j.drudis.2021.11.023
Villoutreix, B., O. (2021). Post-pandemic drug discovery and development: Facing present and future challenges. Front. Drug. Discov. 1. doi:10.3389/fddsv.2021.728469
Wadman, M. (2023). FDA no longer has to require animal testing for new drugs. Science 379, 127–128. doi:10.1126/science.adg6276
Keywords: drug discovery, drug development, therapeutic agent, biologics, small molecules, artificial intelligence (AI)
Citation: Singh N, Vayer P, Tanwar S, Poyet J-L, Tsaioun K and Villoutreix BO (2023) Drug discovery and development: introduction to the general public and patient groups. Front. Drug Discov. 3:1201419. doi: 10.3389/fddsv.2023.1201419
Received: 06 April 2023; Accepted: 08 May 2023;
Published: 24 May 2023.
Edited by:
Vasanthanathan Poongavanam, Uppsala University, SwedenReviewed by:
Duc Duy Vo, Uppsala University, SwedenCopyright © 2023 Singh, Vayer, Tanwar, Poyet, Tsaioun and Villoutreix. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno O. Villoutreix, QnJ1bm8udmlsbG91dHJlaXhAaW5zZXJtLmZy
†Present address: Natesh Singh, Evotec SE, Molecular Architects, Integrated Drug Discovery, Campus Curie, 195 Rte d’Espagne, 31100 Toulouse, France
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.