 Keerthy Reghunandanan
Keerthy Reghunandanan Rajesh Chandramohanadas
Rajesh Chandramohanadas
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Drug Discov. , 12 October 2022
Sec. Anti-Infective Agents
Volume 2 - 2022 | https://doi.org/10.3389/fddsv.2022.920850
This article is part of the Research Topic Advances in Anti-Malarial Drug Discovery View all 5 articles
Malaria remains a health and economic burden, particularly in marginalized populations worldwide. The current strategies for combating malaria rely on eliminating the mosquito vector, using insecticide-treated nets, and other management policies or through the administration of small molecule drugs to perturb the intra-erythrocytic development of the parasite. However, resistance against commonly used drugs such as artemisinin has recently become a concern necessitating the identification of novel pharmacophores with unique mechanisms of action. This review summarizes the various life-stage events of the malaria parasite, Plasmodium falciparum, during the in vitro development, which can be targeted by different classes of small molecules. We also describe various chemically induced phenotypes and methods to ascertain and validate drug-induced changes to derive early insights into which cellular mechanisms are affected.
Malaria remains a global concern, with an estimated 1.7 billion cases and 10.6 million deaths between 2000 and 2020 (“World Malaria Report 2021”, 2021). Plasmodium falciparum is responsible for a significant proportion of malaria-related morbidity and mortality. Increasing resistance to artemisinin and other drugs is of paramount concern, warranting new research and innovations toward improved therapies (Shibeshi et al., 2020). These efforts rely on identifying alternate drug targets and new partner drugs with alternate mechanisms of action.
P. falciparum has a complex lifecycle involving developmental and transmissive stages in humans and mosquitoes, with the blood-stage cycle responsible for most of the disease manifestations. Infection begins when female Anopheles mosquitoes harboring Plasmodium sporozoites inject them into the human skin during a blood meal. These sporozoites traverse the bloodstream and invade and multiply within human hepatocytes to eventually release thousands of merozoites into circulation. The subsequent invasion of merozoites into human red blood cells (RBCs) marks the beginning of the symptomatic asexual lifecycle, with each round occurring in ∼48 h (Tilley et al., 2011).
Upon invasion, merozoites develop within the infected RBCs (iRBCs), which initiates with ring and trophozoite stages followed by parasite multiplication during schizogony to form invasive merozoites that can continue the cycle of infection. The asexual lifecycle ends when the iRBCs burst to release ∼20–30 daughter merozoites (Bannister et al., 2000). Blood stage development and transitions have been a significant focus of antimalarial drug development owing to its significance in the overall infectious cycle and the availability of a robust in vitro culture system to maintain and screen bioactive molecules. Furthermore, erythrocytic development is amenable to robust genetic manipulations, making it the most viable stage for drug screening and mechanism/target validation to advance therapeutic interventions (Burns et al., 2019).
Like other parasitic members of the Apicomplexan family, P. falciparum contains unique organelles that aid parasite survival and growth within host RBCs. For instance, invading merozoites contain secretory organelles such as micronemes and rhoptries essential for releasing proteins/ligands critical for host invasion (Maier et al., 2009). Rhoptries possess about 45 proteins that help to remodel host RBC architecture (Liffner et al., 2021). During the transition from rings to trophozoites, the food/digestive vacuole (DV), where most hemoglobin degradation occurs, becomes evident (Bakar et al., 2010). Parasites use endocytic structures (cytostomes) to consume tiny packets of the host cell cytoplasm containing hemoglobin and transfer it to the DV (Bakar et al., 2010). Next, proteases in the DV break down hemoglobin; some of the freed amino acids are used for protein synthesis, while the rest is transferred to the extracellular media (Goldberg, 2005). The by-product of hemoglobin digestion, hematin, is sequestered into a crystalline form known as hemozoin (or malaria pigment), which is stored within the digestive vacuole (Pagola et al., 2000).
Plasmodium parasites also synthesize and export a variety of proteins to the iRBC cytoplasm and membrane to tackle various aspects related to immune evasion and cytoadherence and to establish antigenic variation. One interesting parasite-driven RBC modification is sequestration, wherein the cytoadherence to the blood capillaries during the intra-erythrocytic development of trophozoites and schizonts prevents the splenic clearance of parasitized RBCs (Maier et al., 2009). iRBCs can also adhere to non-infected erythrocytes, forming rosettes, and can form clumps through platelet-mediated binding to other parasitized RBCs (Lee et al., 2019). Many of these life stage events can be targeted using small molecules, leading to growth arrest or inhibition of a process, often resulting in a phenotypic outcome.
A typical drug development pipeline follows target- and/or phenotype-based approaches. The ability to utilize knowledge about the target and/or its active site, which can guide a logical structure-based drug design strategy, offers a clear advantage to target-based screens (Swinney and Anthony 2011; Love and McNamara 2020). In contrast, phenotypic screening is an approach to discovering molecules in a complex biological environment that exerts a measurable effect on one or more aspects of the biological system (Lansdowne, 2017). As such, phenotype-based screening alone is insufficient to determine the biological activities of novel molecular entities or their actions and must be coupled with appropriate mode of action (MOA) studies specific to the process under investigation (Wagner and Schreiber 2016). Notably, phenotypic approaches have shown more translational success for the discovery of first-in-class drugs, whereas follow-on drugs are much more likely to emerge from target-based screening (Swinney and Anthony 2011). In the case of Plasmodium, its intra-erythrocytic development is characterized by distinct morphological and phenotypic features (Bannister et al., 2000; Rudlaff et al., 2020), which allows for both activity and phenotype-based screening in various degrees of throughputs.
Most studies of pharmacological or biological (i.e., antibodies) growth inhibitors of Plasmodium include adding the inhibitor to in vitro parasite cultures and measuring the parasitemia after a specified time by counting Giemsa-stained blood smears. Radiometric measurements that calculated the parasite’s incorporation of labeled metabolites, such as [3H]-hypoxanthine (Desjardins et al., 1979), were previously widely used. More efficient alternative techniques have arisen since, including robust flow cytometric (YOYO-1 dye) (Li et al., 2007) and fluorescence-based assays utilizing stains such as Hoechst, PicoGreen (Quashie et al., 2006), SYBR green (Johnson et al., 2007), and 4,6-diamidino-2-phenylindole (DAPI) (Baniecki et al., 2007), which leverage the progressive changes in parasite DNA content across developmental stages. Another high-throughput screening (HTS)-compatible technique utilizes parasites expressing the firefly luciferase reporter gene (Che et al., 2012). Various assays based on fluorescent DNA dyes are readily adaptable to high throughput (Baniecki et al., 2007; Plouffe et al., 2008); the advent of high-throughput image-based screening technologies has led to the development of more data-rich and informative image-based assays (Sykes and Avery, 2013). However, these approaches often provide limited data on whether the inhibitor blocks a specific stage of growth or stage transitions (for example, egress or invasion) and how quickly these effects occur. Moreover, some small molecules may be fast-acting, making it critical to examine blood smears at shorter intervals for growth/maturation arrest and associated phenotypes (Subramanian et al., 2018).
In addition to general parasite death, the phenotypic characteristics of parasitized RBCs can significantly change upon inhibitor treatment, which can provide early indications of the stage (and potentially pathways) perturbed by small molecule inhibitors. This review summarizes the phenotypic consequences in intraerythrocytic Plasmodium parasites treated with distinct classes of small molecules/biologically active compounds. Hence, we have carefully chosen inhibitors that induce a characteristic phenotype and attempted to link phenotype-mechanism-chemical classes in the context of parasite invasion, egress, digestive vacuole function, cyto-adhesion, and rosetting. These compounds, along with their chemical structures and possible targets, are presented in Supplementary Table S1.
Blood-stage development begins when plasmodial merozoites invade erythrocytes. While invaded, the host erythrocytes are made to undergo extensive membrane remodeling events through distinct stages including 1) initial attachment, 2) apical tip reorientation, 3) tight junction formation, 4) internalization following PV (parasitophorous vacuole) formation, and 5) echinocytosis (Figure 1). These steps are facilitated by specific protein-ligand interactions between the merozoite surface ligands and the RBC membrane (Kumar and Tolia, 2019), proteolytic events, and cytoskeletal deformation. Merozoite invasion ligands (MILs) secreted by rhoptries and micronemes are described as merozoite surface proteins and alternative pathway ligands, in which the individual ligands of the latter group are functionally redundant but collectively aid in the invasion process (Tham et al., 2015). Blocking/halting invasion itself can prevent a cycle from occurring and, hence, eliminate the possibility of developing drug resistance and the formation of transmissive parasite forms.
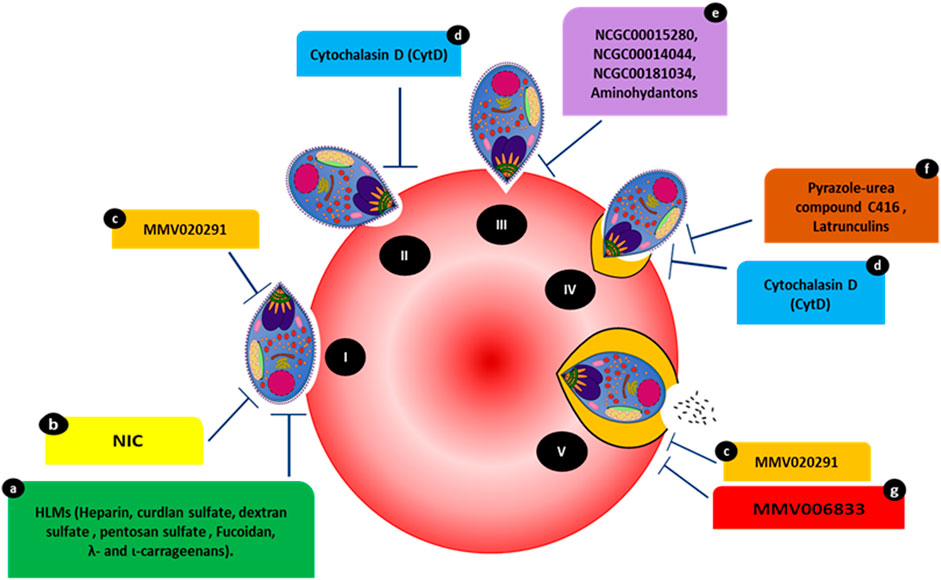
FIGURE 1. Compounds targeting various events occurring during the steps of host invasion. (I) Initial attachment. (II) Apical tip reorientation. (III) Tight junction formation. (IV) Internalization complemented with PVM formation. (V) Complete invasion. Compounds HLMs (a), NIC (b), and MMV020291 (c) reportedly inhibit membrane deformation. MMV02091 (c) also reportedly prolongs the echinocytosis duration of the RBC membrane. Cytochalasin D (cytD) (d) blocks the merozoite’s actin-myosin invasion motor and inhibits deformation. Three compounds, NCGC00015280 (e), NCGC00014044 (dimetacrine) (e), NCGC00181034 (liarozole hydrochloride) (e) and aminohydantons (e) inhibit junction formation. Pyrazole-urea compound C416 (f) reportedly targets the MyoA/MTIP interaction, while latrunculins (f) act against F-actin polymerization. MMV006833 (g) slows the invasion process and prevents echinocytosis recovery in RBCs.
The kinetics of merozoite invasion and ligand-receptor interactions aiding this activity have been widely studied for more than a decade (Gilson and Crabb n.d.; Geoghegan et al., 2021; Boyle, Wilson, and Beeson, 2013). The initial attachment is a pre-invasion event in which the merozoite surface proteins mediate a low-affinity attachment with the RBC plasma membrane. The most abundant component on the merozoite surface, MSP-1 (a leading candidate in multiple subunit vaccine development efforts), is associated with a localized weak deformation of the RBC surface followed by a stronger deformation (Weiss et al., 2015). MSP-1 binds to glycophorin A (GPA) and erythrocyte membrane band 3, which are among the most abundant membrane proteins in RBCs and form a tight stoichiometric complex with GPA. The MSP-GPA/Band3 interactions also lead to changes in RBC membrane properties, which facilitate parasite reorientation and erythrocyte penetration (Baldwin et al., 2015).
Strong deformation is associated with successful invasion, which can be a target of inhibitors. Compounds such as cytochalasin D (cytD) (Figure 1) not only block the merozoite’s actin-myosin invasion motor but also inhibit deformation, with 88.4% of treated merozoites weakly or unable to deform erythrocytes (Weiss et al., 2015). Molecules affecting invasion can lead to visible phenotypes and are often characterized by merozoites making an initial attempt to recognize and attach to RBCs. This is accompanied by merozoite inability to form ring-stage infections compared to controls (treated with DMSO or PBS). For instance, heparin-like-molecules (HLMs) and other small molecules such as 2-butyl-5-chloro-3-(4-nitro-benzyl)-3H-imidazole-4-carbaldehyde (NIC) prevent merozoite invasion based on microscopic phenotypes (Figure 1) (Boyle et al., 2010; Chandramohanadas et al., 2014). The effects of heparin were quantified via live video microscopy, in which treatment significantly reduced the weak deformation of the RBC surface. Although heparin binds to other invasion ligands, MSP-1 (MSP142 fragment of MSP1) could be the primary target of heparin since it blocks the early phase of the invasion; i.e., the weak deformation (Kobayashi et al., 2013; Weiss et al., 2015).
Heparin contains abundant sulfate groups that contribute to inhibitory activity. Various other HLMs (Figure 1), sulfated polysaccharides such as curdlan sulfate, dextran sulfate, pentosan sulfate and fucoidan, λ- and ι-carrageenans, as well as heparan sulfate (HS), inhibit the growth of blood-stage parasites in vitro, forming invasion-arrested phenotypes (Butcher et al., 1988; Clark et al. 1997; Evans et al., 1998; Adams et al., 2005). Moreover, the small molecule NIC (Figure 1) was designed to bind to the EGF-like domain of PfMSP-119 through its ionic nitro-group on the benzene ring. Inhibition by NIC was confirmed by visualizing the invasion process through live video microscopy. In the presence of NIC, the schizonts appeared to burst slowly and, despite making initial contact with RBCs, the merozoites failed to invade even after several minutes (Chandramohanadas et al., 2014). This was confirmed by microscopic examination of Giemsa-stained smears made post-NIC treatment, in which the merozoites were stuck on the RBC surface for several hours.
Strong attachment to the RBC surface is mediated by reticulocyte-binding-like homologs (RHs) and erythrocyte-binding-like proteins (EBA-175, EBA-140, EBA-181, and EBL1-alternative pathway ligands members) (Harvey et al., 2012; Groomes et al., 2022). Reorientation of the merozoite’s apical end is enabled by rapid and localized membrane deformations with the help of EBAs and RHs (Tham et al., 2015; Koch et al., 2017; Hillringhaus et al., 2019).
The interactions of P. falciparum RH5 (reticulocyte-binding protein homolog) and basigin simultaneously evoke a cross-talk (Ca2+ signaling cascade) to the erythrocyte membrane, causing biophysical changes in the RBC cytoskeleton, a pre-requisite for merozoite invasion (Aniweh et al., 2017). The PfRH5–basigin interaction and, hence, invasion can be blocked by antibodies to either protein; however, the precise role of this interaction in the context of host invasion requires further study (Bustamante et al., 2013; Douglas et al., 2014).
The tight junction between the invading merozoite and RBC occurs through PfAMA1 (apical membrane antigen-1, a micronemal protein) and the RON (rhoptry neck proteins) complex interaction (Srinivasan et al., 2011). The junction provides a firm anchor for the parasite to invaginate into the RBC using its actin-myosin motor (the interaction between the carboxy-terminal tail of myosin A [MyoA] and the myosin tail interacting protein [MTIP]), which ultimately leads to the formation of the parasitophorous vacuole (PV) membrane (Geoghegan et al., 2021). At the same time, the secretion of lipid-rich rhoptry content also happens, with a predicted role in PV membrane establishment, which then fuses to surround the invading parasite, completing the invasion (Lingelbach and Joiner, 1998).
The PfAMA-RON complex plays a critical part in merozoite invasion as inhibiting the PfAMA-RON interaction prevents erythrocyte entry (Collins et al., 2009). The compounds NCGC00015280 (7-cyclopentyl-5-(4phenoxy)phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamine), NCGC00014044 (dimetacrine) and NCGC00181034 (liarozole hydrochloride) block AMA1 binding to RON2, preventing junction formation (Figure 1) and, therefore, merozoite invasion at micromolar concentrations. Alpha screen assays (Amplified Luminescent Proximity Homogeneous Assay) were used to screen small-molecule inhibitors of AMA1-RON2 interaction. The phenotypic change due to the inhibitor was characterized using Alpha screen assay-purified merozoites, which were allowed to invade fresh RBCs in the presence of varying compound concentrations. The efficiency of the compounds to inhibit invasion was measured by counting newly invaded rings and confirmed by transmission electron microscopy (Srinivasan et al., 2013). In addition, in-silico structure based-screening identified the pyrazole-urea compound C416 (Figure 1) to target MyoA/MTIP interaction, which inhibited P. falciparum growth, with an EC50 value of 145 nM (Kortagere et al., 2010; Burns et al., 2019). Compounds belonging to the class of latrunculins (derived from the marine sponge Negombata magnifica) are potent actin-disrupting agents that disrupt actin polymerization dynamics and ultimately arrest RBC invasion (Figure 1) (Johnson et al., 2016). Latrunculin B analog 15 exhibited selective activity against P. falciparum with reduced toxicity against mammalian cells. Pyrene fluorescence assays were used to determine the actin-myosin disruption mechanism of these analogs. Pyrene fluorescence is a powerful approach used to study actin filament formation based on the enhancement of fluorescence when pyrene-labeled G-actin (monomers) assemble to form prenylated actin filaments (Johnson et al., 2016).
Treatment with the Medicines for Malaria Venture (MMV) pathogen box compound (Figure 1) MMV02091 (sulfonylpiperazine), a multi-step invasion inhibitor, specifically blocked P. falciparum merozoite invasion to a similar degree as that observed for heparin. MMV02091 treatment caused weaker RBC deformation and prevented the merozoite from pushing a furrow into the RBC surface. This effect was confirmed through merozoite invasion assays and live-cell imaging, which highlighted reduced gliding motility of merozoites across the erythrocyte surface, indicative of the likely weaker engagement by EBA and PfRH interactions and AMA1-RON2 interactions (Tham et al., 2015; Koch et al., 2017; Dans et al., 2020).
Echinocytosis occurs upon the accomplishment of invasion and is speculated to be driven by the entry of Ca2+, ending in iRBC shrinkage (Gilson and Crabb, 2009). MMV02091 greatly prolonged echinocytosis duration in the RBC membrane. In the same study, MMV006833 (Figure 1) slowed the invasion process, blocked ring formation, and prevented RBC recovery of echinocytosis (Dans et al., 2020). All compounds targeting various invasion events with visible phenotypic changes are shown in Figure 1.
Post invasion, the parasites undergo distinct stages as rings and trophozoites before they enter schizogony. Ultrastructural analysis shows that the ring stages are biconcave cup or disc-shaped, although they can occasionally present amoeboid forms and constitute the major asexual forms circulating in the peripheral blood, in which trophozoites adhere to the endothelium (Aikawa et al., 1967; Langreth et al., 1978). A major developmental characteristic during the ring-trophozoite transition is the export of proteins to the host cell to express antigenic determinants, increase membrane permeability, and modulate cyto-adhesion to the human endothelium (Grüring et al., 2011).
During the trophozoite stage, rapid growth and metabolism occur, which leads to the formation of hemozoin (a remnant of digested hemoglobin). Hemozoin appears as brown crystals in the parasite’s acidic digestive vacuole. The internalization of hemoglobin remains debated and probably includes multiple ingestion pathways (Elliott et al., 2008; Lazarus et al., 2008; Bakar et al., 2010). Several enzymes involved in hemoglobin breakdown are most active around pH 4.5–5.0; thus, the DV may have to retain a comparable pH to allow effective proteolysis (Francis et al., 1997). The inhibition of hemoglobin degradation and, thereby, DV function has long been an interesting premise for chemotherapy development.
Digestive vacuole compromise is primarily attributed to the disruption of the parasite’s calcium dynamics, altered pH, disruption of enzymes involved in Hb metabolism, or failure to detoxify heme, resulting in toxic reactive oxygen species (ROS) release. Phenotypes with swollen DV occur when the degradation of oligopeptides derived from hemoglobin is prevented, causing undigested oligopeptides to accumulate in the DV. Aminopeptidase inhibitors based on bestatin target PfA-M1 (metalloaminopeptidase-M1 alanyl-aminopeptidase), which is localized primarily to the digestive vacuole, disrupts Hb digestion, and causes swelling of parasite’s DV (Gavigan et al., 2001; Harbut et al., 2011).
Permeabilization of the DV membrane at the trophozoite stage by lysosomotropic compounds may result in the release of calcium from the vacuolar compartment into the cytosol (Ch’ng et al., 2011). The classical action of chloroquine (CQ), a well-studied antimalarial is the inhibition of heme crystallization (Slater, 1993), which caused a dose-dependent decrease in hemozoin and concomitant increase in toxic-free heme in cultured P. falciparum. Transmission electron microscopy further highlighted the cellular phenotypes, in which heme is redistributed from the parasite digestive vacuole to the cytoplasm and CQ disrupts hemozoin crystal growth, resulting in mosaic boundaries in the formed crystals (Combrinck et al., 2013). The apoptosis-like phenotypes arising from CQ (Figure 2) treatment and digestive vacuole compromise are associated with uncontrolled and rapid calcium leakage, loss of mitochondrial membrane potential, DNA degradation, and protease release, leading to the parasite’s eventual death (Ch’ng et al., 2010). While these are interesting observations, the source and dynamics of calcium underpinning this phenotype require additional careful dissection.

FIGURE 2. Phenotypes indicative of digestive vacuole compromise. Chloroquine, E64d, MMV006087, MMV665879, MMV085071, ferroquine, ferrocenyl phosphine compounds, TCMDC-136230, TCMDC-125431, and TCMDC-125457 reportedly affect DV integrity and induce programmed cell death-like features. The adjacent organelles (mitochondria, Golgi apparatus, endoplasmic reticulum, and nucleus) in the cytosol are assumed to be affected due to the leakage of DV contents.
Cysteine protease inhibitors such as E64d (Figure 2) can also cause the accumulation of undegraded Hb in the parasite DV. Trophozoite-stage parasites treated with E64d for up to 3 h developed a characteristic swollen digestive vacuole (Giannangelo et al., 2020). Recent work demonstrates that several compounds from the MMV collection destabilize the DV, resulting in eventual parasite death. These studies benefitted from an imaging cytometer-based assay using the calcium fluorophore (Fluo-4 AM) as a surrogate marker for DV integrity. The compounds MMV006087 and MMV665879 (Figure 2) from the malaria box library affected DV integrity (Subramanian et al., 2018). In addition, MMV Box compound MMV085071 (Figure 2) was a potent, non-CQ and non-artemisinin-cross-resistant, rapidly parasiticidal, and DV-permeabilizing agent that demonstrated programmed cell death-like features due to DV membrane perturbation (Tong et al., 2018).
Ferrocene and its derivatives have been widely explored as antimicrobial agents due to their chemical stability and lipophilic nature, which helps them to cross the lipophilic membranes of intracellular parasites. Ferroquine (FQ) (Figure 2), a prime example of a ferrocene derivative (an analog of chloroquine created by covalently bonding ferrocene to 4-aminoquinoline), demonstrated superior efficacy against multi-drug-resistant malarial parasites (Held et al., 2015; Wells and van Huijsduijnen 2015) FQ, like CQ by nature, is a weak base that can accumulate in the parasite DV. FQ also produced significant amounts of hydroxyl radicals in the DV (acidic pH and H2O2), compromising its membrane integrity (Wani et al., 2015). In addition, several ferrocenyl phosphine-class (Figure 2) compounds showed phenotypes in which the DV was swollen and compromised, possibly due to the leakage of its contents into the cytosol. Among them, G3, a gold complex generated from the hydrophosphination of the terminal furan bearing an enone substrate, had the strongest inhibitory potential. G3 dramatically altered heme de-toxification, causing apparent oxidative stress and eventual parasite death (Subramanian et al., 2019).
More recently, molecules belonging to the TCAMS (Tres Cantos Anti-Malarial Set) drug library (Gamo et al., 2010) were shown to induce DV phenotype against P. falciparum. Treatment with TCMDC-136230, TCMD-125431, and TCMD-125457 (Figure 2) resulted in calcium redistribution. Among them, TCMDC-125457 killed 44% of artemisinin-resistant ring-stage P. falciparum (MRA-1240 strain) as a single drug and close to 100% in combination with artesunate. The phenotypic changes (apoptotic-like features) were characterized using conventional fluorescence microscopy (Chia et al., 2021). The increase in cytosolic calcium was tracked using Fluo-4 and the increase in fluorescence area was used as an indicator of the overall disruption of parasite calcium dynamics (Chia et al., 2021).
Most of the commonly used drugs are inhibitory to the ring stage to trophozoite transition or inhibit trophozoite development. For example, Wilson et al. (2013) reported a panel of drugs (atovaquone, trichostatin A, cycloheximide, artemisinin, and artesunate that disrupted ring-trophozoite transitions (Figure 3). All aminoquinoline-related compounds, including chloroquine, amodiaquine, mefloquine, halofantrine, quinine, lumefantrine, and piperaquine, caused parasite death during the trophozoite stage with no effect on schizont rupture (Wilson et al., 2013). Phenotype characterization was carried out via flow cytometry and light microscopy, the latter highlighting death indicated by dark blue staining of parasitic material within the iRBCs (Wilson et al., 2013).
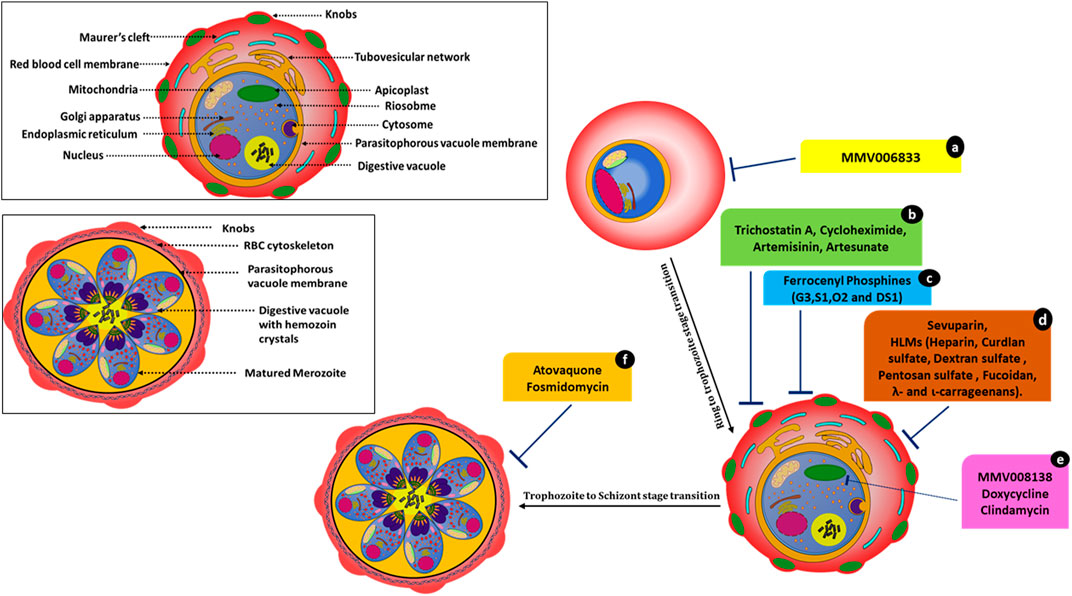
FIGURE 3. Molecules affecting parasite development and associated events. MMV006833 (a) prevents the development of ring-stage parasites. Trichostatin A (b), cycloheximide (b), artemisinin (b), and artesunate (b) actively disrupt ring-to-trophozoite transitions and arrest the parasites at the ring stage. Ferrocenyl phosphine compounds (c) G3, S1, O2, and DS1 inhibit trophozoite development and schizont maturation. Sevuparin (d) and HLMs (d) disrupt the rosetting and sequestration of infected RBCs. Compounds MMV008138 (e), doxycycline (e), and clindamycin (e) cause apicoplast developmental arrest. Atovaquone (f) and fosmidomycin (f) cause incomplete schizont development.
Atovaquone disrupts electron transport in mitochondria but kills Plasmodium through its effects on pyrimidine synthesis (Figure 3). Late-stage parasite accumulation was observed upon treating parasites at the ring stage; i.e., there was the development of parasites with size and staining patterns similar to those of schizonts but without the formation of daughter merozoites. Fosmidomycin-treated parasites showed sizes nearly equivalent to those of late-stage parasites but lacked merozoites (Figure 3) (Nair et al., 2011a; Wilson et al., 2013). Artesunate-treated iRBCs did not develop at all and were transformed into small, shrunken, and pyknotic cells. Furthermore, they showed no expansion of the pigment granule or other cellular features within 1–2 h of drug exposure. Artesunate-retarded parasite development caused a reduction in the cytoplasm and nucleus size, evident by light microscopy. Under electron microscopy, these parasites showed changes in their limiting membranes and alterations in the ribosomal organization (Wilson et al., 2013).
Artesunate/artemisinin treatment at the ring stages resulted in a loss of entire developmental stages. However, the effects of artemisinin and artesunate varied significantly in the schizont rupture assay. Artemisinin treatment led to a pronounced reduction in schizont rupture and the formation of new ring-stage parasites, whereas treatment with artesunate showed no effect at this stage. In contrast, treating parasites with trichostatin A and cycloheximide resulted in novel inhibitory phenotypes with instant effects on ring stages. These compounds also showed a substantial arrest of late-stage parasite development when schizonts were treated. In this case, neither free merozoites nor rings were established (Wilson et al., 2013). After trichostatin A treatment, the remnant ring-stage parasites appeared to mature beyond early rings before growth ceased. Cycloheximide-treated rings progressed little and did not mature while appearing healthy. Cycloheximide affected protein expression by blocking cytosolic protein translation (Geary, 1983), and, hence, immediate effects on parasite development were expected. The use of histone deacetylase inhibitor trichostatin A on late-stage parasites caused gene expression changes within 2 h of exposure (Andrews et al., 2012).
Ferrocenyl phosphines; G3, S1, O2, and DS1 also inhibited trophozoite development (disrupting the ring to trophozoite transition) and schizont maturation. Compound G3 (Gold(I) ferrocenyl phosphines) also showed a significantly greater inhibitory effect in the early erythrocytic stage (ring to trophozoite) compared to the other ferrocenyl phosphine derivatives. G3 also showed hematin inhibition in the β-hematin assay (Subramanian et al., 2019).
The apicoplast, an essential plastid organelle conserved across the apicomplexan family, is essential for both blood-stage and intra-hepatic development of Plasmodium. It contains several clinically validated antimalarial drug targets (Wu et al., 2015). Antibiotics such as doxycycline and clindamycin (tetracyclines that inhibit prokaryotic transcription and translation) blocked expression of the apicoplast genome and are active against Plasmodium (Dahl et al., 2006), leading to a “delayed death” phenotype with growth inhibition after only two replication cycles (96 h). Recently, MMV compound MMV008138 (Figure 3) was shown to impair apicoplast development and arrest of apicoplast elongation and its division, in addition to disturbing the mitochondrial membrane potential. The same study compared the efficacy of apicoplast targeting by fosmidomycin with that by MMV008138. Phenotypes were captured via fluorescence microscopy (Nair et al., 2011b; Bowman et al., 2014). The molecular target of MMV008138 was later identified as PfIspD, an enzyme in the key MEP isoprenoid precursor biosynthesis pathway in the apicoplast (Wu et al., 2015).
In addition to the above-mentioned phenotypes, HLMs (heparin-like molecules) such as sevuparin block rosetting and sequestration of infected RBCs (Figure 3) (Vogt et al., 2006; Kyriacou et al., 2007; Skidmore et al., 2008; Bastos et al., 2014; Saiwaew et al., 2017). Thus, HLMs provide dual protective modes of action (invasion arrest and hindering parasite maturation) against the disease.
Toward the end of the intra-erythrocytic cycle, the parasites undergo segmentation to generate merozoites that are released from the host cell in a stepwise lytic process known as egress, which is amenable to chemical manipulation. Figure 4 summarizes the compounds targeting various stages of the egress pathway that induce visible phenotypes.
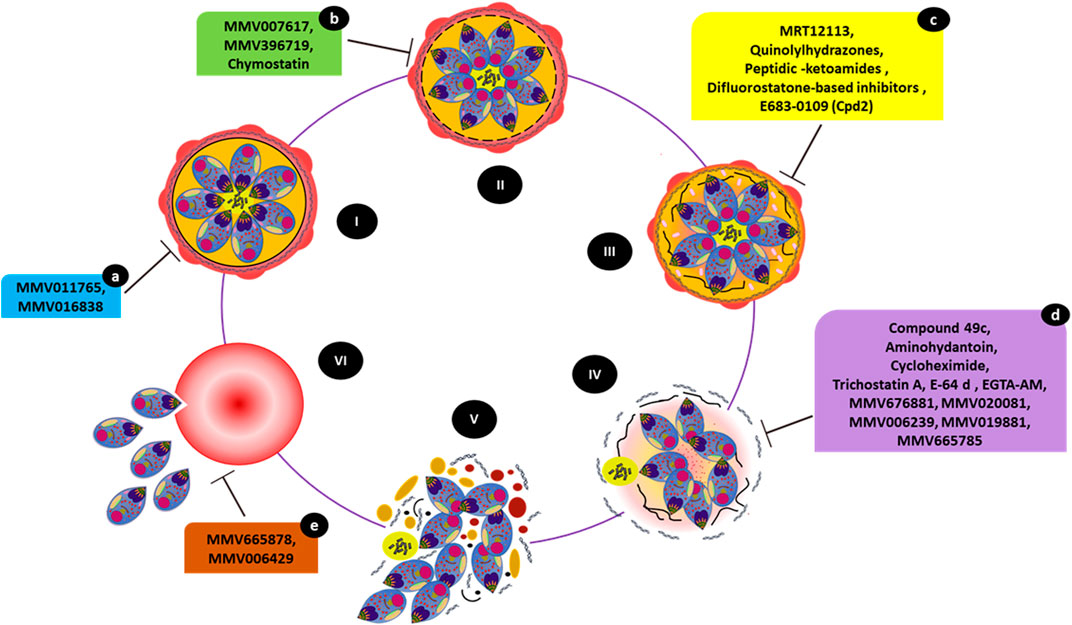
FIGURE 4. Small molecules causing egress-associated phenotypes. The figure illustrates the steps involved in parasite egress starting with a mature schizont (I), (II), and (III) progressive disintegration of the red cell cytoskeleton and PV inside iRBCs, (IV) destabilization of the iRBC membrane followed by (V) host cell cytolysis. Eventually, released merozoites invade a new RBC (VI). Compounds MMV011765 and MMV016838 (a) block late schizont maturation. MMV007617, MMV396719, and chymostatin (b) induce a phenotype in which merozoites appear to have arrested within the PV inside intact iRBCs. MRT12113, quinolylhydrazones, peptidic-ketoamides, difluorostatone-based inhibitors, and E683-0109 (Cpd2) (c) prevent PVM rupture. Compounds 49c, aminohydantoin, cycloheximide, trichostatin A, E-64d, EGTA-AM, purine nitrile MMV676881, MMV020081, MMV006239, MMV019881, and MV665785 (d) prevent RBCM rupture. While treatment with MMV665878 or MMV006429 (e) does not interfere with egress, the merozoites remain attached to the RBC surface, preventing subsequent invasion.
Egress involves breaking open the two membrane compartments (PV and iRBC membrane) surrounding the parasites, simultaneously destabilizing the erythrocyte cytoskeleton. The structural changes of infected erythrocytes before parasite egress are complex, involving the loss of membrane tension and eventual collapse onto the schizont (Glushakova et al., 2005; Glushakova et al., 2009; Hale et al., 2017). Minutes before egress, the PVM alters in shape, rounding up without apparent swelling. PVM rounding and likely all downstream steps are dependent on the mobilization of intracellular Ca2+ (Glushakova et al., 2013; Brochet et al., 2014), itself regulated by PKG activation. PKG activity and Ca2+ are also required for discharge and SUB1 (subtilisin-like protease) activity from exonemes into the PV lumen (Yeoh et al., 2007; Agarwal et al., 2013; Withers-Martinez et al., 2014). SUB1 initiates the coordinated maturation of both cysteine-like protease SERA6 (Serine Repeat Antigen 6) and MSA180 (co-factor of SERA6) by proteolysis, completing the formation of an active SERA6-MSA180 complex. PVM rupture allows the active SERA6-MSA180 complex to access the RBC cytoskeleton, where it cleaves β-spectrin, detaching the spectrin network from the RBC membrane. This rapidly leads to RBCM rupture and merozoite egress (Thomas et al., 2018).
Plasmepsin IX (PMIX) and Plasmepsin X (PMX) are two homologs from the Plasmepsin family, among which Plasmepsin X, the most upstream protease known in the egress cascade, is located in merozoite exonemes (secreted from the merozoite before rupture). This protein is involved in activating the SUB1 protease essential for invasion and schizont rupture, as well as directly processing ligands released from the microneme (Nasamu et al., 2017; Pino et al., 2017). Cleavage of SUB1 by Plasmepsin X leads to the activation of the egress-regulating proteases SERA5 and SERA6. SERA6 is particularly essential for the final stages of egress; i.e., rupture of the RBC membrane, where it regulates β-spectrin proteolytic cleavage, leading to a loss of structural integrity and weakening of the RBC cytoskeleton (Thomas et al., 2018).
As the inhibition of PfSUB1 prevents SERAs maturation and, therefore, egress, PfSUB1 may be a potential target for the development of antimalarial chemotherapeutics (Withers-Martinez et al., 2012; Giovani et al., 2014). The invasion ligand MSPs (MSP1, MSP6, and MSP7), as well as AMA1 and RAP1, are cleaved by PfSUB1 (Yeoh et al., 2007; Koussis et al., 2009; MonerriSilmon de et al., 2011) are involved in initiating the proteolytic cascade leading to merozoite egress (Yeoh et al., 2007). As PfSUB1 is a dual-acting (invasion and egress) target, chemical classes such as MRT12113 (Yeoh et al., 2007), auinolylhydrazones (Gemma et al., 2012), peptidic-ketoamides (Kher et al., 2014), difluorostatone-based inhibitors (Giovani et al., 2014), and E683-0109 (Cpd2) (Bouillon et al., 2013) (Figure 4) were explored as inhibitors of invasion and egress, generating typical phenotypes. Compound Cpd2, a “pan-species” (Figure 4) inhibitor, inhibited the activity of recombinant PvSUB1 and PfSUB1 (Bouillon et al., 2013). A follow-up study showed that even the partial inhibition of MSP1 processing by MRT12113 tended to halt merozoite entry into RBCs. This finding established the sensitivity of merozoite ingress to chemical inhibition (Koussis et al., 2009).
As shown in Figure 4, the peptidomimetic compound 49c inhibits the protease activities of both PMX (preventing the commencement of the egress/invasion via PfSUB1 and processing of invasion ligands) and PMIX (halting rhoptry biogenesis and invasion ligand processing) where it prevents RBCM breakage and merozoites remained trapped and enveloped by the iRBC membrane (Ciana et al., 2013; Nasamu et al., 2017; Pino et al., 2017). Aminohydantoin is another class of compounds specific to PMX that inhibit merozoite egress and invasion (Meyers et al., 2014).
The compounds cycloheximide and trichostatin A inhibit schizont rupture and free release of merozoites (Figure 4). This was inferred from the presence or absence of early-stage, late-stage, and pyknotic parasites in flow cytometry and microscopy analyses (Boyle et al., 2013).
The broadly active protease inhibitor E-64 and its analogs block RBCM rupture (Figure 4), but not RBCM poration or PVM rupture. E64d, a membrane-permeable analog of E-64, acts on SERA6 maturation during the final step of egress (Wickham et al., 2003; Glushakova et al., 2010; Millholland et al., 2011; Dans et al., 2020). The egress inhibitory potential of molecules derived from the E64 scaffold was also associated with the targeting of host calpains, an abundant calcium-dependent thiol protease from the iRBCs, which are activated due to calcium redistribution during parasite release (Chandramohanadas et al., 2009). Egress-arrested phenotypes caused by E64d treatment were further characterized in a 3D-refractive index (3D-RI) mapping study (Chandramohanadas et al., 2011) together with other inhibitors EGTA-AM (a general calcium chelator) and chymostatin. Giemsa-stained smears showed remarkable rupture-arrested phenotypes. Merozoites from inhibitor-treated iRBCs were unable to break open their host RBCs as late as 52 hpi, forming membrane-enclosed structures. The rupture-arrested phenotypes observed at ∼52 hpi caused by E64d and EGTA-AM appeared to be unique flattened structures that differed significantly from the classical spherical schizonts, but without an intact PV. EGTA-AM supported PV breakdown but inhibited iRBC membrane rupture, generating flattened structures that had lost the characteristic schizont shape. In contrast, chymostatin treatment showed no 3D-RI changes and caused elevated fluctuations solely within the PV, where merozoites reside, indicating that both membranes were intact after inhibitor treatment (Chandramohanadas et al., 2011).
The MMV compound MMV676881 showed a similar egress inhibitory phenotype (RBCM rupture prevention). This effect was examined by luminescence-based assays using a bioluminescent reporter, Nluc, combined with live-cell microscopy (Dans et al., 2020). The study also identified several other MMV compounds with unique phenotypic consequences, including MMV011765 and MMV016838, which blocked the maturation of late schizonts (Figure 4).
Another phenotypic screening attempt using the MMV Malaria Box identified 26 molecules that inhibited P. falciparum merozoite release from iRBCs with micromolar to nanomolar potency. This assay combined flow-cytometry and microscopic techniques (Subramanian et al., 2018) to eventually identify several egress inhibitors (including MMV000653, MMV000642, MMV007617, MMV396715, MMV396719, MMV006429, MMV396749, MMV006427, MMV665878, and MMV666025). Several of these molecules affect parasite Na+ and pH homeostasis in a model similar to that of the PfATP4 inhibitor spiroindolone KAE609 (NITD609/Cipargamin) (Spillman et al., 2013; Lehane et al., 2014). In vitro sensitivity assays using P. falciparum at the ring, trophozoite, and schizont phases of the human blood stages determined that NITD609 was most effective against schizonts (Rottmann et al., 2010). Among the MMV molecules, MMV019881 and MMV665785 induced phenotypes in which the merozoites were trapped within iRBCs, even as the PV appeared to have disintegrated (Figure 4). In contrast, treatment with MMV007617 and MMV396719 resulted in phenotypes in which merozoites were locked within the PV (Figure 4). Interestingly, treatment of schizonts with MMV665878 or MMV006429 did not affect merozoite release and they remained attached to the RBC surface (Subramanian et al., 2018), failing to invade (Figure 4). This phenotype is typical of invasion inhibitors such as heparin (Kulane et al., 1992; Boyle et al., 2010).
The screening of compounds from MMV Pathogen Box identified nearly 20 molecules active against late-stage development and egress. Specific inhibitors including MMV020520, MMV020710, MMV020391, MMV006239, MMV020623, MMV675968, MMV010576, MMV020670, MMV026356, MMV085071, MMV024443, and MMV020081 showed inhibition of the schizont to ring transition (Patra et al., 2020). Microscopic examinations following treatment with MMV020081 and MMV006239 showed similar cellular phenotypes as those exhibited with E-64 treatment (Patra et al., 2020), highlighted in Figure 4.
Phenotype-based screening offers an attractive strategy for prioritizing molecules active against Plasmodium parasites. Thorough documentation of the morphological and physiological changes across blood-stage development provides a feasible method to apply a variety of cell biological and imaging tools to systematically capture the cellular consequences of small-molecule treatment. This approach can facilitate the identification of molecules with unique inhibitory potential and provide information on phenotypic outcomes to better understand the specific modes of action and cellular targets and, eventually, inform the development of future antimalarials.
KR and RC designed the manuscript, collected the information, and wrote the paper.
KR and RC acknowledge the funding and infrastructure support provided by the DBT-Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2022.920850/full#supplementary-material
Adams, Yvonne, Smith, Simone L., Schwartz-Albiez, Reinhard, and KatherineAndrews, T. (2005). Carrageenans inhibit the in vitro growth of Plasmodium falciparum and cytoadhesion to CD36. Parasitol. Res. 97 (4), 290–294. doi:10.1007/s00436-005-1426-3
Agarwal, Shalini, Singh, Maneesh Kumar, Garg, Swati, Chitnis, Chetan E., and Singh, Shailja (2013). Ca2+-Mediated exocytosis of subtilisin-like protease 1: A key step in egress of Plasmodium falciparum merozoites. Cell. Microbiol. 15 (6), 910–921. doi:10.1111/CMI.12086
Aikawa, M., Huff, C. G., and Sprinz, H. (1967). Fine structure of the asexual stages of plasmodium elongatum. J. Cell Biol. 34 (1), 229–249. doi:10.1083/JCB.34.1.229
Andrews, Katherine T., Tran, Thanh N., and Fairlie, David P. (2012). Towards Histone Deacetylase Inhibitors as New Antimalarial DrugsCurrent Pharmaceutical Design, 18. Available at: www.PlasmoDB.org.
Aniweh, Yaw, Gao, Xiaohong, Hao, Piliang, Meng, Wei, Lai, Soak Kuan, Gunalan, Karthigayan, et al. (2017). P. Falciparum RH5-basigin interaction induces changes in the cytoskeleton of the host RBC. Cell. Microbiol. 19 (9), e12747. doi:10.1111/CMI.12747
Bakar, Nurhidanatasha Abu, Klonis, Nectarios, Hanssen, Eric, Chan, Cherrine, and Tilley, Leann (2010). Digestive-vacuole genesis and endocytic processes in the early intraerythrocytic stages of Plasmodium falciparum. J. Cell Sci. 123 (3), 441–450. doi:10.1242/JCS.061499
Baldwin, Michael R., Li, Xuerong, Hanada, Toshihiko, Liu, Shih Chun, and Chishti, Athar H. (2015). Merozoite surface protein 1 recognition of host Glycophorin A mediates malaria parasite invasion of red blood cells. Blood 125 (17), 2704–2711. doi:10.1182/BLOOD-2014-11-611707
Baniecki, Mary Lynn, Wirth, Dyann F., and Clardy, Jon (2007). High-throughput Plasmodium falciparum growth assay for malaria drug discovery. Antimicrob. Agents Chemother. 51 (2), 716–723. doi:10.1128/AAC.01144-06
Bannister, L. H., Hopkins, J. M., Fowler, R. E., Krishna, S., and Mitchell, G. H. (2000). A brief illustrated guide to the ultrastructure of Plasmodium falciparum asexual blood stages. Parasitol. Today 16 (10), 427–433. doi:10.1016/S0169-4758(00)01755-5
Bastos, M. F., Albrecht, L., Kozlowski, E. O., Lopes, S. C., Blanco, Y. C., Carlos, B. C, et al. (2014).KozlowskiFucosylated chondroitin sulfate inhibits Plasmodium falciparum cytoadhesion and merozoite invasion, Antimicrob. Agents Chemother., 58, 1862–1871. doi:10.1128/AAC.00686-13/SUPPL_FILE/ZAC004142700SO1
Bouillon, Anthony, Giganti, David, Benedet, Christophe, Gorgette, Olivier, Petres, Stephane, Crublet, Elodie, et al. (2013).In silico screening on the three-dimensional model of the Plasmodium vivax Sub1 protease leads to the validation of a novel anti-parasite compound, J. Biol. Chem., 288, 18561–18573. doi:10.1074/JBC.M113.456764/ATTACHMENT/11538B9B-C096-4697-867A-AF7A507C77DE/MMC1
Bowman, Jessica D., Merino, Emilio F., Brooks, Carrie F., Striepen, Boris, Carlier, Paul R., and Cassera, Maria B. (2014).Antiapicoplast and gametocytocidal screening to identify the mechanisms of action of compounds within the malaria box, Antimicrob. Agents Chemother., 58, 811–819. doi:10.1128/AAC.01500-13/SUPPL_FILE/ZAC002142549SO1
Boyle, M. J., Wilson, D. W., Richards, J. S., Riglar, D. T., Tetteh, K. K., Conway, D. J., et al. (2010). Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc. Natl. Acad. Sci. U. S. A. 107 (32), 14378–14383. doi:10.1073/pnas.1009198107
Boyle, Michelle J., Wilson, Danny W., and Beeson, James G. (2013). New approaches to studying Plasmodium falciparum merozoite invasion and insights into invasion biology. Int. J. Parasitol. 43, 1–10. doi:10.1016/j.ijpara.2012.11.002
Brochet, Mathieu, Collins, Mark O., Smith, Terry K., Thompson, Eloise, Sebastian, Sarah, Volkmann, Katrin, et al. (2014). Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca²⁺ signals at key decision points in the life cycle of malaria parasites. PLoS Biol. 12 (3), e1001806. doi:10.1371/JOURNAL.PBIO.1001806
Burns, Amy L., MadelineDans, G. Juan M. Balbin, Tania F., Gilson, Paul R., Beeson, James G., Boyle, Michelle J., and Wilson, Danny W. (2019). de Koning-WardTargeting malaria parasite invasion of red blood cells as an antimalarial strategy. Oxford University Press. Oxford, doi:10.1093/femsre/fuz005
Bustamante, Leyla Y., Josefin Bartholdson, S., Crosnier, Cecile, Campos, Marta G., Wanaguru, Madushi, Nguon, Chea, et al. (2013). A full-length recombinant Plasmodium falciparum PfRH5 protein induces inhibitory antibodies that are effective across common PfRH5 genetic variants. Vaccine 31 (2), 373–379. doi:10.1016/J.VACCINE.2012.10.106
Butcher, G. A., Parish, C. R., and Cowden, W. B. (1988).Inhibition of growth in vitro of Plasmodium falciparum by complex polysaccharides, Trans. R. Soc. Trop. Med. Hyg., 82, 558–559. doi:10.1016/0035-9203(88)90504-4/2/82-4-558
Chandramohanadas, Rajesh, Park, Yong Keun, Lui, Lena, Li, Ang, Quinn, David, Liew, Kingsley, et al. (2011). Biophysics of malarial parasite exit from infected erythrocytes. PLOS ONE 6 (6), e20869. doi:10.1371/JOURNAL.PONE.0020869
Chandramohanadas, Rajesh, Russell, Bruce, Liew, Kingsley, Yin, Hoe Yau, Chong, Alvin, Liu, Min, et al. (2014). Small molecule targeting malaria merozoite surface protein-1 (MSP-1) prevent host invasion of divergent plasmodial species, Journal of infectious diseases advance access. Available at: http://jid.oxfordjournals.org/http://jid.oxfordjournals.org/.
Che, Pulin, Cui, Long, Kutsch, Olaf, Cui, Liwang, and Li, Qianjun (2012). Validating a firefly luciferase-based high-throughput screening assay for antimalarial drug discovery. Assay. Drug Dev. Technol. 10 (1), 61–68. doi:10.1089/ADT.2011.0378
Chia, Wanni, Maria Gomez-Lorenzo, G., Castellote, I., Tong, J. X., Chandramohanadas, R., Thu Chu, T. T., et al. (2021). High-Content Phenotypic Screen of a Focused TCAMS Drug Library Identifies Novel Disruptors of the Malaria Parasite Calcium Dynamics. ACS Chem. Biol. 16 (11), 2348–2372. doi:10.1021/acschembio.1c00512
Ch’ng, J. H., Kotturi, S. R., Chong, A. G. L., Lear, M. J., and Tan, K. S. W. (2010). A programmed cell death pathway in the malaria parasite Plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis. , 2010, e26. doi:10.1038/cddis.2010.2
Ch’ng, J. H., Liew, K., Goh, A. S. P., Sidhartha, E., and Tan, K. S. W. 2011. “Drug-induced permeabilization of parasite’s digestive vacuole is a key trigger of programmed cell death in Plasmodium falciparum.” Cell Death Dis.:10 2 (10): e216. doi:10.1038/cddis.2011.97
Ciana, Claire Lise, Siegrist, Romain, Aissaoui, Hamed, Marx, Léo, Racine, Sophie, Meyer, Solange, et al. (2013). Novel in vivo active anti-malarials based on a hydroxy-ethyl-amine scaffold. Bioorg. Med. Chem. Lett. 23 (3), 658–662. doi:10.1016/J.BMCL.2012.11.118
Clark, Diana L., Su, Shidong, and Davidson, Eugene A. (1997). Saccharide anions as inhibitors of the malaria parasite. Glycoconj. J. 144 14 (4), 473–479. doi:10.1023/A:1018551518610
Collins, Christine R., Withers-Martinez, Chrislaine, Hackett, Fiona, and MichaelBlackman, J. (2009). An inhibitory antibody blocks interactions between components of the malarial invasion machinery. PLoS Pathog. 5 (1), e1000273. doi:10.1371/JOURNAL.PPAT.1000273
Combrinck, Jill M., Mabotha, Tebogo E., Ncokazi, Melvin A. Ambele, Dale Taylor, Peter J. Smith, Heinrich, Kanyile K., Smith, P. J., Hoppe, C., Egan, Timothy J., et al. (2013). Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem. Biol. 8 (1), 133–137. doi:10.1021/CB300454T
Dahl, Erica L., Shock, Jennifer L., Shenai, Bhaskar R., DeRisi, Joseph L., and Rosenthal, Philip J. (2006). Jiri GutTetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob. Agents Chemother. 50 (9), 3124–3131. doi:10.1128/AAC.00394-06
Dans, Madeline G., Weiss, Greta E., Wilson, Danny W., Sleebs, Brad E., Crabb, Brendan S., de Koning-Ward, Tania F., et al. (2020). Screening the Medicines for malaria venture pathogen box for invasion and egress inhibitors of the blood stage of Plasmodium falciparum reveals several inhibitory compounds. Int. J. Parasitol. 50 (3), 235–252. doi:10.1016/J.IJPARA.2020.01.002
Desjardins, R. E., Canfield, C. J., Haynes, J. D., and Chulay, J. D. (1979). Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 16 (6), 710–718. doi:10.1128/AAC.16.6.710
Douglas, Alexander D., Williams, Andrew R., Knuepfer, Ellen, Illingworth, Joseph J., Furze, Julie M., Crosnier, Cécile, et al. (2014). Neutralization of Plasmodium falciparum merozoites by antibodies against PfRH5. J. Immunol. 192 (1), 245–258. doi:10.4049/JIMMUNOL.1302045
Elliott, David A., McIntosh, Michael T., Dean, H. Hosgood, Chen, Shuo, Zhang, Gina, Baevova, Pavlina, et al. (2008). Four distinct pathways of hemoglobin uptake in the malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 105 (7), 2463–2468. doi:10.1073/PNAS.0711067105
Evans, S. G., Morrison, D., Kaneko, Y., and Havlik, I. (1998). The effect of curdlan sulphate on development in vitro of Plasmodium falciparum. Trans. R. Soc. Trop. Med. Hyg. 92 (1), 87–89. doi:10.1016/S0035-9203(98)90969-5
Francis, S. E., SullivanGoldberg, D. E., and Sullivan, D. (1997). Hemoglobin metabolism in the malaria parasite plasmodium falciparum. Annu. Rev. Microbiol. 51, 97–123. doi:10.1146/annurev.micro.51.1.97
Gamo, Francisco Javier, Sanz, Laura M., Vidal, Jaume, de Cozar, Cristina, Alvarez, Emilio, Lavandera, Jose Luis, et al. (2010). Thousands of chemical starting points for antimalarial lead identification. Nature 465, 7296–465310. doi:10.1038/nature09107
Gavigan, Clare S., Dalton, John P., Bell., Angus, and Bell, A. (2001). The role of aminopeptidases in haemoglobin degradation in Plasmodium falciparum-infected erythrocytes. Mol. Biochem. Parasitol. 117 (1), 37–48. doi:10.1016/S0166-6851(01)00327-9
Geary, Timothy (1983). Effects of antibiotics on Plasmodium falciparum in vitro * characterization of schistosoma mansoni-derived extracellular vesicles view project. doi:10.4269/ajtmh.1983.32.221
Gemma, Sandra, Giovani, Simone, Brindisi, Margherita, Tripaldi, Pierangela, Brogi, Simone, Savini, Luisa, et al. (2012). Quinolylhydrazones as novel inhibitors of Plasmodium falciparum serine protease PfSUB1. Bioorg. Med. Chem. Lett. 22 (16), 5317–5321. doi:10.1016/J.BMCL.2012.06.023
Geoghegan, N. D., Evelyn, C., Whitehead, L. W., Pasternak, M., McDonald, P., Triglia, T., et al. (2021). 4D analysis of malaria parasite invasion offers insights into erythrocyte membrane remodeling and parasitophorous vacuole formation. Nat. Commun., 3620–3716. 2021, doi:10.1038/s41467-021-23626-7
Giannangelo, Carlo, Siddiqui, Ghizal, de Paoli, Amanda, Anderson, Bethany M., Edgington-Mitchell, Laura E., Charman, Susan A., et al. (2020). System-wide biochemical analysis reveals ozonide antimalarials initially act by disrupting Plasmodium falciparum haemoglobin digestion. PLoS Pathog. 16 (6), e1008485. doi:10.1371/JOURNAL.PPAT.1008485
Gilson, Paul R., and BrendanCrabb, S. (2009). Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int. J. Parasitol. 39, 91–96. doi:10.1016/j.ijpara.2008.09.007
Giovani, Simone, Penzo, Maria, Brogi, Simone, Brindisi, Margherita, Gemma, Sandra, Novellino, Ettore, et al. (2014). Rational design of the first difluorostatone-based PfSUB1 inhibitors. Bioorg. Med. Chem. Lett. 24 (15), 3582–3586. doi:10.1016/J.BMCL.2014.05.044
Glushakova, Svetlana, Humphrey, Glen, Leikina, Evgenia, Balaban, Amanda, Miller, Jeffrey, and Zimmerberg, Joshua (2010). New stages in the program of malaria parasite egress imaged in normal and sickle erythrocytes. Curr. Biol. 20 (12), 1117–1121. doi:10.1016/J.CUB.2010.04.051
Glushakova, Svetlana, Lizunov, Vladimir, Paul, S., Melikov, K., Humphrey, G., and Zimmerberg, J. (2013). Blank, kamran melikov, glen humphrey, and joshua ZimmerbergCytoplasmic free Ca2+ is essential for multiple steps in malaria parasite egress from infected erythrocytes. Malar. J. 12 (1), 41–12. doi:10.1186/1475-2875-12-41
Glushakova, Svetlana, Mazar, Julia, MartinHohmann-marriott, F., Hama, Erinn, and Zimmerberg, Joshua (2009). Irreversible effect of cysteine protease inhibitors on the release of malaria parasites from infected erythrocytes. Cell. Microbiol. 11 (1), 95–105. doi:10.1111/J.1462-5822.2008.01242.X
Glushakova, Svetlana, Yin, Dan, Li, Tao, and Zimmerberg, Joshua (2005). Membrane transformation during malaria parasite release from human red blood cells. Curr. Biol. 15 (18), 1645–1650. doi:10.1016/J.CUB.2005.07.067
Goldberg, D. E. (2005). Hemoglobin degradation. Curr. Top. Microbiol. Immunol. 295, 275–291. doi:10.1007/3-540-29088-5_11
Groomes, Patrice v., Kanjee, Usheer, and Duraisingh, Manoj T. (2022). RBC membrane biomechanics and Plasmodium falciparum invasion: Probing beyond ligand–receptor interactions. Trends Parasitol. 38 (4), 302–315. doi:10.1016/J.PT.2021.12.005
Grüring, C., Heiber, A., Kruse, F., Ungefehr, J., Tim, W. G., and Tobias, S. (2011). Development and host cell modifications of Plasmodium falciparum blood stages in four dimensions. Nat. Commun., 2011, (1 21), 165–211. doi:10.1038/ncomms1169
Hale, Victoria L., Watermeyer, Jean M., Hackett, Fiona, Vizcay-Barrena, Gema, Christiaan van Ooij, , Thomas, James A., et al. (2017). Parasitophorous vacuole poration precedes its rupture and rapid host erythrocyte cytoskeleton collapse in Plasmodium falciparum egress. Proc. Natl. Acad. Sci. U. S. A. 114 (13), 3439–3444. doi:10.1073/PNAS.1619441114
Harbut, Michael B., Velmourougane, Geetha, Dalal, Seema, Reiss, Gilana, Whisstock, James C., Onder, Ozlem, et al. (2011).Bestatin-based chemical biology strategy reveals distinct roles for malaria M1- and M17-family aminopeptidases, Proc. Natl. Acad. Sci. U. S., 108. doi:10.1073/PNAS.1105601108/ASSET/A9FFA89F-F025-472E-A84A-95558144EC8A/
Harvey, Katherine L., Gilson, Paul R., and Crabb, Brendan S. (2012). A model for the progression of receptor–ligand interactions during erythrocyte invasion by Plasmodium falciparum. Int. J. Parasitol. 42 (6), 567–573. doi:10.1016/J.IJPARA.2012.02.011
Held, Jana, Supan, Christian, Carmen, L., Salazar, O., Tinto, Halidou, Bonkian, Léa N., et al. (2015). Ferroquine and artesunate in african adults and children with Plasmodium falciparum malaria: A phase 2, multicentre, randomised, double-blind, dose-ranging, non-inferiority study. Lancet. Infect. Dis. 15 (12), 1409–1419. doi:10.1016/S1473-3099(15)00079-1
Hillringhaus, Sebastian, Dasanna, Anil K., Gompper, Gerhard, and Fedosov, Dmitry A. (2019). Importance of erythrocyte deformability for the alignment of malaria parasite upon invasion. Biophys. J. 117 (7), 1202–1214. doi:10.1016/J.BPJ.2019.08.027
Johnson, Jacob D., Dennull, Richard A., Gerena, Lucia, Lopez-Sanchez, Miriam, Roncal, Norma E., and Waters, Norman C. (2007). Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrob. Agents Chemother. 51 (6), 1926–1933. doi:10.1128/AAC.01607-06
Johnson, Swapna, Rahmani, Raphaël, Damien, R. DrewMelanie J. Williams, JohnnyWilkinson, X. HuangMark Dale, Tan, Yan Hong, Tan, Y. H., et al. (2016). Truncated latrunculins as actin inhibitors targeting Plasmodium falciparum motility and host-cell invasion. J. Med. Chem. 59, 10994–11005. doi:10.1021/acs.jmedchem.6b01109
Kher, Samir S., Penzo, Maria, Fulle, Simone, Paul, W., Finn, Michael J. Blackman, and Jirgensons, Aigars (2014). Substrate derived peptidic α-ketoamides as inhibitors of the malarial protease PfSUB1. Bioorg. Med. Chem. Lett. 24 (18), 4486–4489. doi:10.1016/J.BMCL.2014.07.086
Kobayashi, Kyousuke, Takano, Ryo, Takemae, Hitoshi, Sugi, Tatsuki, Ishiwa, Akiko, Gong, Haiyan, et al. (2013). Analyses of interactions between heparin and the apical surface proteins of Plasmodium falciparum. Sci. Rep. , 2013, 3178–3211. doi:10.1038/srep03178
Koch, Marion, Wright, Katherine E., Otto, Oliver, Herbig, Maik, D Salinas, Nichole, H Tolia, Niraj., et al. (2017). Plasmodium falciparum erythrocyte-binding antigen 175 triggers a biophysical change in the red blood cell that facilitates invasion. Proc. Natl. Acad. Sci. U. S. A. 114, 4225–4230. doi:10.1073/PNAS.1620843114
Kortagere, Sandhya, Welsh, William J., Morrisey, Joanne M., Daly, Thomas, Ejigiri, Ijeoma, Sinnis, Photini, et al. (2010). Structure-based design of novel small-molecule inhibitors of Plasmodium falciparum. J. Chem. Inf. Model. 50 (5), 840–849. doi:10.1021/ci100039k
Koussis, Konstantinos, Withers-Martinez, Chrislaine, Yeoh, Sharon, Child, Matthew, Hackett, Fiona, Knuepfer, Ellen, et al. (2009). A multifunctional serine protease primes the malaria parasite for red blood cell invasion. EMBO J. 28 (6), 725–735. doi:10.1038/EMBOJ.2009.22
Kulane, A., Ekre, H. P., Perlmann, P., Rombo, L., Wahlgren, M., and Wahlin, B. (1992). Effect of different fractions of heparin on Plasmodium falciparum merozoite invasion of red blood cells in vitro. Am. J. Trop. Med. Hyg. 46 (5), 589–594. doi:10.4269/AJTMH.1992.46.589
Kumar, Hirdesh, and Tolia, Niraj H. (2019). Getting in: The structural biology of malaria invasion. PLoS Pathog. 15 (9), e1007943. doi:10.1371/JOURNAL.PPAT.1007943
Kyriacou, Helen M., Steen, Katie E., Ahmed, Raza, Arman, Monica, George, Warimwe, Bull, Peter C., et al. (2007).In vitro inhibition of Plasmodium falciparum rosette formation by curdlan sulfate, Antimicrob. Agents Chemother., 51, 1321–1326. doi:10.1128/AAC.01216-06/ASSET/1F4E0DBF-A8C5-49D4-8E49-BB9CC3EFD4FC/ASSETS/GRAPHIC/ZAC0040764670002
Lazarus, Michelle D., Schneider, Timothy G., and Taraschi, Theodore F. (2008). A new model for hemoglobin ingestion and transport by the human malaria parasite Plasmodium falciparum. J. Cell Sci. 121 (11), 1937–1949. doi:10.1242/JCS.023150
Langreth, S. U. S. A. N. G., Jamesjensen, B., Robertreese, T., and William, T. R. A. G. E. R. (1978). Fine structure of human malaria in vitro. J. Protozool. 25 (4), 443–452. doi:10.1111/J.1550-7408.1978.TB04167.X
Lee, Wenn Chyau, Russell, Bruce, and Rénia, Laurent (2019). Sticking for a cause: The falciparum malaria parasites cytoadherence paradigm. Front. Immunol. 10, 1444. doi:10.3389/fimmu.2019.01444
Lehane, Adele M., Ridgway, Melanie C., Baker, Eileen, and Kirk, Kiaran (2014). Diverse chemotypes disrupt ion homeostasis in the malaria parasite. Mol. Microbiol. 94 (2), 327–339. doi:10.1111/MMI.12765
Li, Q., Gerena, L., Xie, L., Zhang, J., and Kyle, D. (2007)., 71. Development and validation of flow cytometric measurement for parasitemia in cultures of P falciparum vitally stained with YOYO, Cytom. Part A undefined, 297–307. doi:10.1002/cyto.a.20380
Liffner, Benjamin, Juan, M. B., Stephan Wichers, J., Tim, W. G., and Wilson, Danny W. (2021). The ins and outs of Plasmodium rhoptries, focusing on the cytosolic side. Trends Parasitol. 37 (7), 638–650. doi:10.1016/J.PT.2021.03.006
Lingelbach, Klaus, and Joiner, Keith A. (1998). The parasitophorous vacuole membrane surrounding Plasmodium and toxoplasma: An unusual compartment in infected cells. J. Cell Sci. 111 (11), 1467–1475. doi:10.1242/JCS.111.11.1467
Love, M. S., and McNamara, C. W. 2020. “Phenotypic screening techniques for cryptosporidium drug discovery.”: 59–74. doi:10.1080/17460441.2020.1812577
Maier, A. G., Cooke, B. M., Cowman, A. F., and Leann, T. (2009). Malaria parasite proteins that remodel the host erythrocyte. Nat. Rev. Microbiol. 2009, 341–354. doi:10.1038/nrmicro2110
Meyers, Marvin J., Tortorella, Micky D., Xu, Jing, Qin, Limei, He, Zhengxiang, Lang, Xingfen, et al. (2014). Evaluation of aminohydantoins as a novel class of antimalarial agents. ACS Med. Chem. Lett. 5 (1), 89–93. doi:10.1021/ml400412x
Millholland, M. G., Chandramohanadas, R., Pizzarro, A., Wehr, A., Shi, H., Darling, C., Chwee, T. L., Doron, C., and Greenbaum, M. (2011).The malaria parasite progressively dismantles the host erythrocyte cytoskeleton for efficient egress, Mol. Cell. Proteomics, 10. doi:10.1074/MCP.M111.010678/ATTACHMENT/D3E5B08A-09BD-442D-A6CD-07F2F9E1FF71/MMC1
MonerriSilmon de, Natalie C., Flynn, Helen R., Campos, Marta G., Hackett, Fiona, Koussis, Konstantinos, Withers-Martinez, Chrislaine, et al. (2011).Global identification of multiple substrates for Plasmodium falciparum SUB1, an essential malarial processing protease, Infect. Immun., 79, 1086–1097. doi:10.1128/IAI.00902-10/SUPPL_FILE/SUPPLEMENTAL_DATA_FINAL_SLMON_DE_MONERRI_REVISED
Nair, S. C., Brooks, C. F., and Goodman, C. D. (2011a). apicoplast isoprenoid precursor synthesis and the molecular basis of Fosmidomycin resistance in toxoplasma gondii, Rupress. Org. Available at: https://rupress.org/jem/article-abstract/208/7/1547/41139 (Accessed May 29, 2022).
Nair, Sethu C., Brooks, Carrie F., Angelika, S., Sandeep, S., Anglin, Justin L., Song, Yongcheng, Moreno, Silvia N. J., Striepen, Boris, et al. (2011b). Apicoplast isoprenoid precursor synthesis and the molecular basis of Fosmidomycin resistance in toxoplasma gondii. J. Exp. Med. 208 (7), 1547–1559. doi:10.1084/JEM.20110039
Nasamu, Armiyaw S., Glushakova, Svetlana, Russo, Ilaria, Vaupel, Barbara, Anna, Oksman, Kim, Arthur S., et al. (2017).Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion, Science, 358, 518–522. doi:10.1126/SCIENCE.AAN1478/SUPPL_FILE/
Pagola, Silvina, Stephens, Peter W., Scott, D. Bohle, Kosar, Andrew D., and Madsen, Sara K. (2000). The structure of malaria pigment β-haematin. Nature 404, 6775–404310. doi:10.1038/35005132
Patra, Alok Tanala, Hingamire, Tejashri, Belekar, Meenakshi A., Xiong, Aoli, Subramanian, G., Bozdech, Z., et al. (2020). Gowtham subramanian, zbynek bozdech, peter preiser, dhanasekaran shanmugam, and rajesh ChandramohanadasWhole-cell phenotypic screening of Medicines for malaria venture pathogen box identifies specific inhibitors of Plasmodium falciparum late-stage development and egress. Antimicrob. Agents Chemother. 64 (5), e01802. doi:10.1128/AAC.01802-19
Pino, Paco, Caldelari, Reto, Mukherjee, Budhaditya, Vahokoski, Juha, Klages, Natacha, Maco, Bohumil, et al. (2017). A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science 358 (6362), 522–528. doi:10.1126/SCIENCE.AAF8675
Plouffe, D., Brinker, A., McNamara, C., Henson, K., Kato, N., Kuhen, K., Nagle, A., et al. (2008). “Silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen,” in Proceedings of the National Academy of Sciences of the United States of America 105 (26), 9059–9064. Available at: https://doi.org/10.1073/PNAS.0802982105/SUPPL_FILE/0802982105SI.PDF
Quashie, Neils B., de Koning, Harry P., and Ranford-Cartwright, Lisa C. (2006). An improved and highly sensitive microfluorimetric method for assessing susceptibility of Plasmodium falciparum to antimalarial drugs in vitro. Malar. J. 5, 95. doi:10.1186/1475-2875-5-95
Rottmann, Matthias, McNamara, Case, Bryan, K., Yeung, S., Lee, Marcus C. S., Zou, Bin, et al. (2010). Spiroindolones, a potent compound class for the treatment of malaria. Science 329 (5996), 1175–1180. doi:10.1126/SCIENCE.1193225
Rudlaff, Rachel M., Kraemer, Stephan, Marshman, Jeffrey, and Dvorin, Jeffrey D. (2020). Three-dimensional ultrastructure of Plasmodium falciparum throughout cytokinesis. PLoS Pathog. 16 (6), e1008587. doi:10.1371/JOURNAL.PPAT.1008587
Saiwaew, S., Sritabal, J., Piaraksa, N., Keayarsa, S., Ruengweerayut, R., Utaisin, C., et al. (2017). Effects of Sevuparin on rosette formation and cytoadherence of Plasmodium falciparum infected erythrocytes. PLOS ONE 12 (3), e0172718. doi:10.1371/JOURNAL.PONE.0172718
Shibeshi, M. A., Zemene, D. K., and Seyfe, A. A. (2020). Antimalarial drug resistance and novel targets for antimalarial drug discovery. Infect. Drug Resist. 13, 4047–4060. doi:10.2147/IDR.S279433
Skidmore, Mark A., Dumax-Vorzet, Audrey F., ScottGuimondEdwards, E. Timothy R. Rudd, Elizabeth A., Turnbull, Jeremy E., Craig, Alister G., and Yates, Edwin A. (2008).Disruption of rosetting in Plasmodium falciparum malaria with chemically modified heparin and low molecular weight derivatives possessing reduced anticoagulant and other serine protease inhibition activities, J. Med. Chem., 51, 1453–1458. doi:10.1021/JM701337T/SUPPL_FILE/JM701337T-FILE003
Slater, Andrew F. G. (1993). Chloroquine: Mechanism of drug action and resistance in Plasmodium falciparum. Pharmacol. Ther. 57 (2–3), 203–235. doi:10.1016/0163-7258(93)90056-J
Spillman, N. J., Allen, R. J. W., McNamara, C. W., Yeung, B. K. S., Winzeler, E. A., Diagana, T. T., et al. (2013). Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the Spiroindolone antimalarials. Cell Host Microbe 13 (2), 227–237. doi:10.1016/J.CHOM.2012.12.006
Srinivasan, Prakash, Adam, Yasgar, Luci, Diane K., Beatty, Wandy L., Hu, Xin, Andersen, John, et al. (2013). Disrupting malaria parasite AMA1-RON2 interaction with a small molecule prevents erythrocyte invasion. Nat. Commun. 4, 2261. doi:10.1038/ncomms3261
Srinivasan, Prakash, Beatty, Wandy L., Diouf, Ababacar, Herrera, Raul, Ambroggio, Xavier, Kathleen Moch, J., et al. (2011). Binding of Plasmodium merozoite proteins RON2 and AMA1 triggers commitment to invasion. Proc. Natl. Acad. Sci. U. S. A. 108 (32), 13275–13280. doi:10.1073/PNAS.1110303108
Subramanian, G., Sadeer, A., Mukherjee, K., Kojima, T., Tripathi, P., Naidu, R., et al. (2019). Evaluation of ferrocenyl phosphines as potent antimalarials targeting the digestive vacuole function of Plasmodium falciparum. Dalton Trans. 48 (3), 1108–1117. doi:10.1039/c8dt04263b
Subramanian, Gowtham, Belekar, Meenakshi A., Shukla, Anurag, Tong, Jie Xin, Sinha, Ameya, Trang, T., et al. (2018).Targeted phenotypic screening in Plasmodium falciparum and toxoplasma gondii reveals novel modes of action of Medicines for malaria venture malaria box molecules, MSphere, 3. doi:10.1128/MSPHERE.00534-17/ASSET/D26FC6C6-5B58-4EBA-8847-00A46B0E47A6/ASSETS/GRAPHIC/SPH0011824570007
Swinney, David C., and Anthony., Jason (2011). How were new Medicines discovered? Nat. Rev. Drug Discov. 7 10 (7), 507–519. doi:10.1038/nrd3480
Sykes, Melissa L., and Avery., Vicky M. (2013). Approaches to Protozoan drug discovery: Phenotypic screening. J. Med. Chem. 56 (20), 7727–7740. doi:10.1021/jm4004279
Tham, Wai Hong, Nicholas, T., Lim, Y., Weiss, Greta E., Lopaticki, Sash, BrendanAnsell, R. E., et al. (2015). Plasmodium falciparum adhesins play an essential role in signalling and activation of invasion into human erythrocytes. PLoS Pathog. 11 (12), e1005343. doi:10.1371/JOURNAL.PPAT.1005343
Thomas, James A., Tan, Michele S. Y., Bisson, Claudine, Borg, Aaron, Umrekar, Trishant R., Hackett, Fiona, et al. (2018). A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells. Nat. Microbiol. 4 3 (4), 447–455. doi:10.1038/s41564-018-0111-0
Tilley, Leann, Dixon, Matthew W. A., and Kirk., Kiaran (2011). The Plasmodium falciparum-infected red blood cell, International journal of biochemistry and cell biology. Elsevier. doi:10.1016/j.biocel.2011.03.012
Tong, Jie Xin, Chandramohanadas, Rajesh, and Tan, Kevin Shyong-Wei (2018). High-content screening of the Medicines for malaria venture pathogen box for Plasmodium falciparum digestive vacuole-disrupting molecules reveals valuable starting points for drug discovery. Antimicrob. Agents Chemother. 62 (3), 020311. doi:10.1128/AAC.02031-17
Vogt, Anna M., Moll, K., Jonsson, C., Normark, J., Ribacke, U., et al. (2006). Fredrik pettersson, kirsten moll, cathrine jonsson, johan normark, ulf ribacke, thomas G. Egwang, etRelease of sequestered malaria parasites upon injection of a glycosaminoglycan. PLoS Pathog. 2 (9), e100. doi:10.1371/JOURNAL.PPAT.0020100
Wagner, Bridget K., and Schreiber., Stuart L. (2016). The power of sophisticated phenotypic screening and modern mechanism-of-action methods. Cell Chem. Biol. 23 (1), 3–9. doi:10.1016/J.CHEMBIOL.2015.11.008
Wani, W. A., Jameel, E., Baig, U., Mumtazuddin, S., and Hun, L. T. (2015). Ehtesham jameel, umair baig, syed mumtazuddin, and lee ting HunFerroquine and its derivatives: New generation of antimalarial agents. Eur. J. Med. Chem. 101 (August), 534–551. doi:10.1016/J.EJMECH.2015.07.009
Weiss, Greta E., Gilson, Paul R., Taechalertpaisarn, T., Tham, Wai Hong., de Jong, N. W., Harvey, K. L., et al. (2015). Revealing the sequence and resulting cellular morphology of receptor-ligand interactions during Plasmodium falciparum invasion of erythrocytes. PLoS Pathog. 11 (2), e1004670. doi:10.1371/JOURNAL.PPAT.1004670
Wells, Timothy N. C., and Robvan Huijsduijnen, Hooft (2015). Ferroquine: Welcome to the next generation of antimalarials. Lancet. Infect. Dis. 15 (12), 1365–1366. doi:10.1016/S1473-3099(15)00148-6
Wickham, Mark E., Culvenor, Janetta G., and Cowman, Alan F. (2003). Selective inhibition of a two-step egress of malaria parasites from the host erythrocyte. J. Biol. Chem. 278 (39), 37658–37663. doi:10.1074/JBC.M305252200
Wilson, Danny W., Christine Langer, Christopher D. Goodman, Geoffrey I. McFadden, and Beeson, James G. (2013).Defining the timing of action of antimalarial drugs against Plasmodium falciparum, Antimicrob. Agents Chemother., 57, 1455–1467. doi:10.1128/AAC.01881-12/SUPPL_FILE/ZAC999101679SO1
Withers-Martinez, Chrislaine, Suarez, Catherine, Fulle, Simone, Kher, Samir, Penzo, Maria, Paul Ebejer, Jean, et al. (2012). Plasmodium subtilisin-like protease 1 (SUB1): Insights into the active-site structure, specificity and function of a pan-malaria drug target. Int. J. Parasitol. 42 (6), 597–612. doi:10.1016/J.IJPARA.2012.04.005
Withers-Martinez, Chrislaine, Strath, Malcolm, Hackett, Fiona, Haire, Lesley F., Howell, Steven A. Philip A. Walker, Evangelos, Christodoulou, et al. (2014). The malaria parasite egress protease SUB1 is a calcium-dependent redox switch subtilisin. Nat. Commun. 2014, 3726–3811. doi:10.1038/ncomms4726
Wu, Wesley, Herrera, Zachary, Ebert, Danny, Baska, Katie, Cho, Seok H., DeRisi, Joseph L., et al. (2015).A chemical rescue screen identifies a Plasmodium falciparum apicoplast inhibitor targeting MEP isoprenoid precursor biosynthesis, Antimicrob. Agents Chemother., 59, 356–364. doi:10.1128/AAC.03342-14/SUPPL_FILE/ZAC001153600SO1
Yeoh, Sharon, O’Donnell, Rebecca A., Koussis, Konstantinos, Dluzewski, Anton R., Ansell, Keith H., Osborne, Simon A., et al. (2007). Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 131 (6), 1072–1083. doi:10.1016/J.CELL.2007.10.049
Keywords: Plasmodium falciparum, phenotypes, egress, invasion, microscopy, therapeutic development
Citation: Reghunandanan K and Chandramohanadas R (2022) Chemically induced phenotypes during the blood stage development of Plasmodium falciparum as indicators of the drug mode of action. Front. Drug. Discov. 2:920850. doi: 10.3389/fddsv.2022.920850
Received: 15 April 2022; Accepted: 20 September 2022;
Published: 12 October 2022.
Edited by:
Cátia Teixeira, LAQV Network of Chemistry and Technology, PortugalReviewed by:
Carlo Giannangelo, Monash Institute of Pharmaceutical Sciences, Monash University, AustraliaCopyright © 2022 Reghunandanan and Chandramohanadas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajesh Chandramohanadas, cmFqZXNoY2Rhc0ByZ2NiLnJlcy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.