
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Drug Discov., 08 July 2022
Sec. In silico Methods and Artificial Intelligence for Drug Discovery
Volume 2 - 2022 | https://doi.org/10.3389/fddsv.2022.908870
 Jonathan Allcock1*
Jonathan Allcock1* Anna Vangone2*
Anna Vangone2* Agnes Meyder3
Agnes Meyder3 Stanislaw Adaszewski3*Martin Strahm3
Stanislaw Adaszewski3*Martin Strahm3 Chang-Yu Hsieh1Shengyu Zhang1*
Chang-Yu Hsieh1Shengyu Zhang1*Quantum computing for the biological sciences is an area of rapidly growing interest, but specific industrial applications remain elusive. Quantum Markov chain Monte Carlo has been proposed as a method for accelerating a broad class of computational problems, including problems of pharmaceutical interest. Here we investigate the prospects of quantum advantage via this approach, by applying it to the problem of modelling antibody structure, a crucial task in drug development. To minimize the resources required while maintaining pharmaceutical-level accuracy, we propose a specific encoding of molecular dihedral angles into registers of qubits and a method for implementing, in quantum superposition, a Markov chain Monte Carlo update step based on a classical all-atom force field. We give the first detailed analysis of the resources required to solve a problem of industrial size and relevance and find that, though the time and space requirements of using a quantum computer in this way are considerable, continued technological improvements could bring the required resources within reach in the future.
The last few years have seen a dramatic increase in global investment in quantum computing, accompanied by a concerted effort to find industrial applications that can outperform existing computational methods. In the biological sciences, a number of initial studies on quantum computing for protein folding have been made. In particular, the works of (Perdomo et al., 2008; Babbush et al., 2012; Perdomo-Ortiz et al., 2012; Babej et al., 2018; Outeiral et al., 2021a) investigate simulating lattice-based models of proteins using analog quantum computers such as quantum annealers; For gate-based quantum computing, lattice-models of proteins have also been considered in (Fingerhuth et al., 2018; Robert et al., 2021) using variations of the quantum approximate optimization algorithm (QAOA) (Farhi et al., 2014) and the variational quantum eigensolver (VQE) (Peruzzo et al., 2014). More realistic, non-lattice based models have also been studied, with (Mulligan et al., 2020) using the D-Wave quantum annealer in conjunction with a Rosetta energy function and side-chain rotamer library, and (Casares et al., 2022) proposing a method which combines the AlphaFold (Senior et al., 2020) algorithm with quantum walks1. While these works have made important contributions to our understanding of how quantum computers may be applied to this domain, no methods have yet been proposed that might solve specific problems to a speed and accuracy that would make them attractive to industry.
Motivated by the growing need to understand the true potential of quantum computing for solving real-world problems of industrial size and commercial relevance, here we show how classical Markov chain Monte Carlo (MCMC) methods based on torsion space conformation updates and all-atom force fields, such as those used in the Rosetta software package, can be adapted into a quantum computing procedure to predict the 3D structure of protein loops starting from their amino acid sequence. As a potential application, we have in mind the modelling of antibody loops—in particular, the H3 loop—a crucial task in the development of therapeutic antibodies. This problem lies in the sweet spot of 1) being of practical importance to the pharmaceutical industry, as existing computational methods cannot predict H3 loop structures to the required near-atomic-level accuracy quickly enough to be part of an industrial workflow2; and 2) involving molecules of a size (typically 3 to 30 amino acid residues long) that, as we show, the problem can be tackled on a quantum computer with resources that are plausibly within reach in the future.
Structurally, antibodies consist of two identical pairs of polypeptides chains, with each pair comprising a heavy chain (containing approx. 500 amino acid residues) and a light chain (approx. 200 amino acids) Their ability to bind to a large variety of molecular targets with high affinity and specificity has led to antibodies becoming the predominant class of new therapeutics and diagnostics tools in recent years. The function of antibodies, together with their desired drug profile (e.g. affinity, stability, half-life, tissue penetration (Kim et al., 2005)), is a direct consequence of their structure. As experimental structure determination of antibodies is time-consuming and costly, computational structure prediction plays a crucial role in accelerating and facilitating the development of antibody-therapeutics.
Antibody binding occurs via a specific antigen-binding region, characterized by 6 hypervariable loops—called complementarity-determining regions (CDRs)—located on the variable domains of the light (L1, L2, L3) and heavy (H1, H2, H3) chains (Figure 1). While antibodies are typically more rigid and stable than other proteins, they are known to retain a certain amount of plasticity to accommodate for different antigens, with a degree of flexibility inherent to many CDRs (Fernández-Quintero et al., 2019; Fernández-Quintero et al., 2020; Fernández-Quintero et al., 2021). For 5 of the CDRs loops (L1, L2, L3, H1 and H2) though, limited shapes have been observed, leading to the identification of definite canonical structures based on their sequences. In contrast, H3 loops exhibit high diversity—with longer loops typically displaying more conformal variety—and cannot be classified into canonical groups. Consequently, the main antibody modelling problem of interest is the accurate prediction of the H3 loop. While atomic resolutions (i.e. 1 Å = 10−10 m) can be reached for canonical CDRs, accuracy ranging from 1.5–3 Å or worse can be expected for H3. Furthermore, such loops do not exist in a unique structural conformation, but rather as an ensemble of different states that can occur on different timescales and with different probabilities. As experimental structures for antibodies are, in most cases, derived by X-ray crystallography at low temperature (
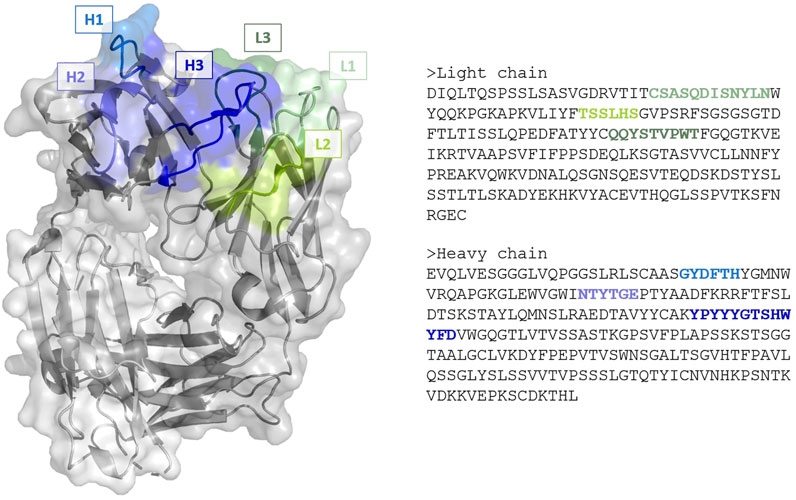
FIGURE 1. (Left) Cartoon and surface representation of the Fab region of the anti-VEGF antibody with PDB code 1CZ8. Framework is represented in gray, complementarity-determining regions in green (light chain loops L1, L2 and L3) and blue (heavy chains loops, H1, H2 and H3). (Right) The corresponding amino acid sequences of the chains. Image created with PyMOL (Schrödinger, 2021).
Classical MCMC methods for loop modelling sample from the Boltzmann distribution based on a chosen state space Ω of possible configurations of the molecule. Starting from an initial configuration x ∈ Ω of the molecule, the following steps are then iterated:
1. Propose a random update x → x′ ∈ Ω.
2. Accept the update with probability
Here, E(x) is the energy of a configuration and T is the temperature. This procedure—if run for a sufficiently long time—is guaranteed to converge to a configuration drawn from the distribution p(x) ∼ e−E(x)/T, and has effectiveness that depends on defining a state space that can capture the biologically relevant structures at appropriate levels of granularity, and update rules that can efficiently explore this space, avoiding spending too long stuck in energetically unfavorable configurations. In practice, the accurate MCMC modelling of pharmaceutically relevant loops can take days to weeks to complete—too long to be part of a commercially feasible workflow.
Here we are interested in using quantum computing to accelerate the MCMC process, and give the first detailed analysis of the resources required to solve a protein structure problem of industrial size and relevance. Our focus is specifically on fault-tolerant gate-based quantum computing (see Section 2.1). While it may be several decades before large-scale fault-tolerant devices are available (Sevilla and Riedel, 2020), they are widely believed to offer the best long term prospects for practical advantage of classically-intractable problems, with mathematically provable efficiency of algorithms possible in some cases. In contrast, other models of quantum computing may be available sooner, but may not guarantee the same long-term advantages. Analog approaches to quantum computing such as adiabatic quantum computing (Farhi et al., 2000), quantum annealing or continuous time quantum walks (Farhi and Gutmann, 1998) lack practical means of error-correction, which may limit the size of computations that can be performed; and the embedding of computational problems in a form amenable to annealing makes the application of realistic energy models challenging (e.g. (Marchand et al., 2019; Mulligan et al., 2020) require non-trivial procedures interleaving classical and quantum computation). Hybrid quantum-classical approaches such as VQE and QAOA, which are gate-based but primarily targeted at noisy, non-error-corrected quantum computers, similarly lack strong evidence for practical advantage or scalability. For an overview on the prospects of various quantum computing technologies see (National Academies of Sciences, Engineering and Medicine, 2019).
In the standard circuit model of quantum computing, computational tasks are carried out by applying operations, known as gates, to registers (i.e., groups) of qubits. At the end of a sequence of gates, one or more of the qubits are measured and the results recorded. The size and complexity of the quantum circuit required to solve a particular task determines the overall algorithmic running time and, in particular, whether or not an advantage can be gained over existing classical computational methods. As individual qubits and gates are invariably error prone, quantum error correction procedures must be applied for long circuits to be computed. The aim of error correction is to use multiple noisy physical qubits to encode a single error-free logical qubit, which comes at the cost of additional qubits and computational time. As the error correction operations themselves may be faulty, one must take care to ensure that the net effect is an overall reduction in error. This is referred to as fault-tolerance and, if achieved, can be used to drive errors arbitrarily low, enabling large scale computations to be implemented. For further background, we refer readers to (Outeiral et al., 2021b) for a good introduction to quantum computing from a biological sciences perspective. In the supplementary material we give additional details on topics specific to this work, including accelerating MCMC via quantum walks, resource overheads required for error correction, and evaluating complicated functions in a quantum circuit.
Quantum Markov chain Monte Carlo (Szegedy, 2004) is an approach to speeding up classical MCMC methods on a fault-tolerant quantum computer (FTQC). By encoding the state of the system of interest in a number of qubits and translating the update and acceptance rules into a sequence of quantum gates, the number of update steps required can be reduced to roughly the square root of the number of steps required classically (see Supplementary Material S1). While this quadratic reduction provides an opportunity for quantum advantage, the time required for each step may be longer in the quantum case, and care is needed in analyzing whether a speedup can be obtained. Furthermore, the success of the approach depends, among other things, on finding an efficient quantum encoding of the 3D structure of the antibody loop. That is, a way of representing the antibody structure in the state of multiple qubits.
To evaluate the feasibility of quantum MCMC for antibody loop modelling, we propose a specific encoding of molecular dihedral angles into registers of qubits and a method for implementing the MCMC update step coherently in quantum superposition. To enable the latter, we propose a quantum subroutine (Quantum SN-NeRF) based on the classical Self-Normalizing Natural extension Reference Frame (Parsons et al., 2005) method for coherently converting from dihedral angles to Cartesian coordinates. We estimate the number of qubits and time required to implement such an approach on an FTQC and find that, while there are limited prospects for an advantage on a first generation FTQC, continued technological improvements could bring the required resources within reach on future quantum devices.
We consider polypeptides consisting of L amino acid residues, containing N heavy (non-hydrogen) atoms. Atomic positions can be described by Cartesian coordinates in 3D space, or relative to one another using dihedral (or torsion) angles. While the Cartesian representation is convenient for computing atomic forces and determining deviations of predicted atomic positions from experimentally determined positions, the dihedral representation can be preferable for generating perturbations to the molecular structure.
In the dihedral formalism, any four consecutive backbone atoms A-B-C-D define two planes, containing A-B-C, and B-C-D respectively. The angle between these two planes is the associated dihedral angle and, in a polypeptide backbone, each residue has three associated dihedral angles labelled φ, ψ and ω. A complete internal representation of the backbone is given by specifying each of the dihedral angles and bond lengths between consecutive backbone atoms, as well as the bond angles between any three consecutive atoms. The side chains of a polypeptide can similarly be described by dihedral angles χi, where i = 1, 2, 3, … depending on the length of the side chain (see Figure 2).
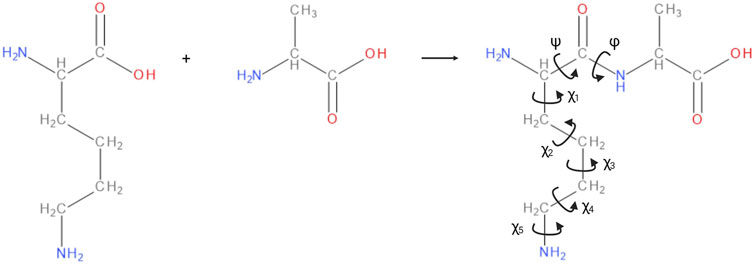
FIGURE 2. The amino acids leucine and alanine forming a dipeptide. The backbone angles φ, ψ, and side chain angles χi of alanine are annotated. In any polypeptide, φi is defined by atoms Ci−1 - Ni - Cαi - Ci. ψi is defined by atoms Ni - Cαi - Ci - Ni+1, and ωi is defined by atoms Cαi−1 - Ci−1 - Ni - Cαi. The ωi angle is not shown in the figure as it is nearly always close to 180°. Image created with BIOVIA Draw and Biorender.com.
Given a classical Markov chain with state space Ω, let
In our approach, we follow the LHPST (Lemieux et al., 2020) quantum MCMC framework, where a quantum walk operator W is implemented by four other quantum operators (V, B, F, R), acting on System (S), Move (M) and Coin (C) quantum registers. Mathematical definitions of these operators are given in Supplementary Material S4, but their functions, and that of the various registers, can be understood by analogy with the classical MCMC process. In what follows we use Dirac notation
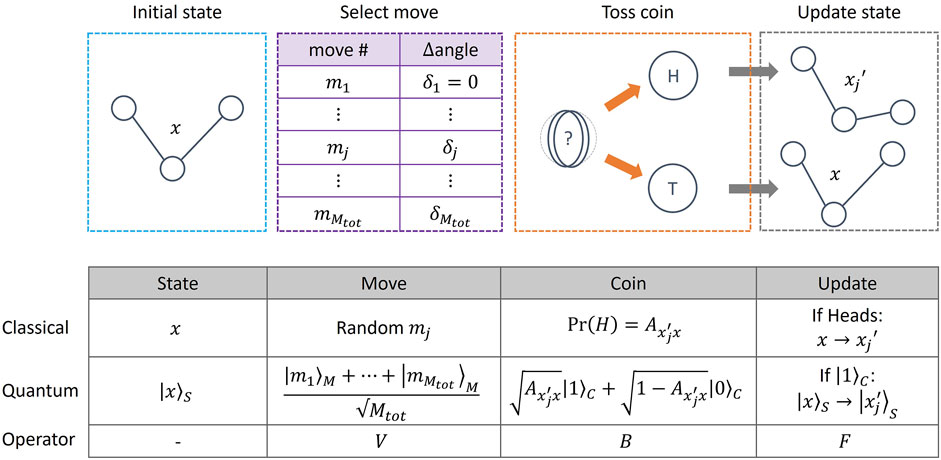
FIGURE 3. (Top) Schematic of the (classical and quantum) MCMC process, illustrated with a toy model consisting of a 3 atom molecule defined by a single angle x. Given an initial state of the system, an update move is proposed from a list of possible moves. In this example, a move mj corresponds to increasing the angle x by an amount δj. A coin is then tossed and, if a heads is obtained, the state is updated to
The Coin register consists of a single qubit, while the number of qubits in the System and Move registers is problem-dependent. The quantum walk operator W is then defined as
To apply this framework to the problem of antibody loop modelling, we make the following design choices:
Dihedral angle encoding. We represent each amino acid residue in the loop by its backbone and side chain dihedral angles, assuming ideal bond lengths and angles. Lookup tables of these angles can be constructed by sampling data from Ramachandran plots (Ramachandran et al., 1963) and backbone-dependent rotamer libraries, e.g. (Shapovalov and Dunbrack, 2011). By specifying an index into the tables for each residue, the biologically relevant structures of the loop can be given in compact form for encoding in a register of qubits.
Update rules. The dihedral angle encoding allows for an efficiently implementable Monte Carlo update step corresponding to replacing a randomly chosen backbone or side chain dihedral angle with another randomly chosen value from the corresponding lookup table3.
Energy function. We are interested in high-accuracy modelling and thus take E(x) to be specified by a classical all-atom force field such as CHARMM36m (Huang et al., 2017).
Conversion to 3D coordinates. Evaluation of E(x) necessitates conversion of the dihedral angle representation of the loop into 3D coordinates. To do so, we first use a Quantum Read-Only Memory (QROM) (Babbush et al., 2018) approach to convert the dihedral lookup table indices into their corresponding angles. Then, we adapt a variant (Parsons et al., 2005) of the classical Natural-extension Reference Frame (NerF) algorithm used by the Rosetta software package to give a quantum procedure (QSN-NeRF) for coherently converting from dihedral angles to 3D coordinates in quantum superposition.
Energy calculation and Metropolis update. After conversion to 3D coordinates, the energy of the existing and proposed configurations can be computed. Implementing the Metropolis update in the LHPST framework requires evaluating
With these design choices we obtain estimates of the resources required to implement the LPHST quantum walk given in Table 1 (see Supplementary Material S4 for details). In leading candidate proposals for fault-tolerant quantum computing such as via the surface code, the Toffoli (controlled-controlled-NOT) gate is expected to take orders of magnitude longer to implement than other gates, as each Toffoli gate first requires the production of an associated magic state via an expensive process known as distillation. We therefore estimate the computational time and number of qubits required to implement the various quantum operators required by our approach by the number of Toffoli gates needed.
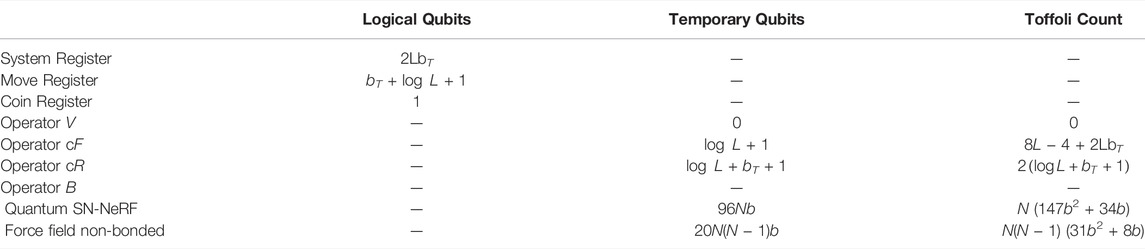
TABLE 1. Resources Summary. L = number of residues, N = total number of heavy atoms in loop, bT = number of bits in lookup table keys, b = number of bits of precision used to store Cartesian coordinates. B operator resources are estimated from contributions from quantum SN-NeRF and non-bonded energy terms. Values for quantum SN-NeRF and non-bonded force field terms are double the numbers given in Supplementary Material 4, as the computations are performed for the current and proposed update states.
To benchmark our approach and understand the technological requirements necessary for a quantum advantage, we consider the MKHMAGAAAAGAVV H1 loop from the Syrian hamster prion protein, which was modelled to high accuracy via classical Monte Carlo methods in (Ulmschneider et al., 2006) using the OPLS-AA all-atom force field, GBSA implicit solvent model and concerted rotation updates. While some of these details differ from our quantum procedure, and the loop considered is an H1 rather than H3 loop, the loop size (L = 14, N = 88) and the number of classical MCMC steps (106 per structural sample) serve as a useful baseline for comparison. Estimates of the resources required to solve the same problem using our quantum approach are given in Table 2 (see Supplementary Material S5 for more details). The number of qubits needed and the quantum running time per step are based on a superconducting quantum processor running the surface code (Bravyi and Kitaev, 1998; Dennis et al., 2002), a leading candidate for error-corrected quantum computing. As mentioned, for such a system, the resource bottleneck is the time and qubits required for the magic state distillation used to implement non-Clifford operations such as the Toffoli gate.

TABLE 2. Resource estimates for the 14 residue MKHMAGAAAAGAVV antibody loop, giving the number nsteps of MCMC steps required per structural sample, the time tsample required per sample, and the total time required to obtain 103 samples. The first row corresponds to the classical MCMC result from (Ulmschneider et al., 2006). Quantum estimates correspond to different assumptions on physical error rates pe and surface code cycle times tS. For first generation (Gen.1) large scale FTQC, we assume pe = 10–4, tS = 1μs. For future quantum devices, pe = 10–5, tS = 200ns are plausibly achievable (Fowler et al., 2012; Sevilla and Riedel, 2020). The TF column indicates the number of parallel Toffoli distillation factories that are assumed to be available.
Using the state-of-the-art
In terms of qubit numbers, we assume that the first generations of large-scale fault-tolerant devices will be limited to O (106) to O (107) physical qubits, but that these numbers may increase by one or two orders of magnitude in the longer term. Table 2 gives estimates based on these first generation and future FTQC computing parameter regimes. As the number of Toffoli gates required is significant, a single distillation factory may not suffice, and we thus give estimates assuming parallel access to varying numbers of factories. The single step time estimates are based on the Toffoli gate count required to implement the quantum SN-NeRF conversion from dihedral to 3D coordinates, and the quantum circuit computation of the non-bonded energy terms in the force field (assuming no cut-off radius). These steps dominate the classical computation time and, as the form of the non-bonded terms is similar to those in other popular force fields, do not tie our results to a specific force field. The number of qubits includes additional ancillary registers required to store temporary arithmetic values prior to uncomputation, but ignores ancilla used in various quantum arithmetic primitives which are highly implementation-specific and depend on specific choices of quantum arithmetic circuits. These simplifications are sufficient for our goal of understanding the order of magnitude of technological performance required to obtain a quantum advantage. We find that, in spite of the careful design decisions made to minimize the resources required, first generation FTQC are unlikely to provide an advantage over classical MCMC techniques, with estimated computational times that greatly exceed those required classically. However, with plausible improvements to physical qubit error rates and error correction speed, future quantum devices, with sufficient qubit numbers, may close the gap with existing MCMC approaches to the point where quantum walk methods may be competitive. Further improvements to both hardware and algorithm design could eventually yield an overall quantum advantage.
In this work, we set out to understand the feasibility of quantum computing to accelerate MCMC for antibody loop modelling, and proposed a suitable state space and update rule for such a computation. Our method is based on a dihedral angle encoding of the atoms involved, and a procedure (Quantum SN-NeRF) for coherently converting from dihedrals to Cartesian coordinates so that the classical force field potential energy function of each configuration can be evaluated in quantum superposition.
While the encoding and conversions can be carried out efficiently, the energy function evaluation is costly and imposes a trade-off between the quadratic reduction in number of MCMC steps needed for the quantum approach, and a significant constant factor increases in the time required for each step. These long step times are in large part due to the time needed to implement fault-tolerant Toffoli gates used to carry out basic arithmetic operations. We find that for system sizes of practical interest, this trade-off is not yet in favor of quantum computers, and that further developments in both algorithms and quantum hardware are likely needed in order for quantum computing to be practical in this domain.
Our analysis indicates limitations of directly applying the quantum walk approach to classical MCMC methods and suggests that, without several orders of magnitude increases in fault-tolerant hardware efficiency, new quantum algorithms or design improvements to our scheme (e.g., alternative encodings of molecular states in qubits) will be needed to make protein folding practical on quantum computers. These findings are in line with those in (Babbush et al., 2020), which show significant challenges for constructing efficient quantum solutions to a number of non-toy-model optimization problems. An interesting open problem is to investigate whether new force fields can be designed (or indeed, machine-learned (Botu et al., 2017; Unke et al., 2021)) to be efficiently implementable on quantum computers while still delivering sufficiently accurate results.
As investment and hype continue to grow in quantum computing, for meaningful and informed progress to be made, detailed analyses of specific problems facing industry must continue to be carried out and disseminated even (and indeed especially) if they show limitations or challenges with quantum computing. It is our hope that the results presented here shed some light on the future prospects of quantum computing as a tool for modelling antibody loops, and serve as a useful starting point for further improvements. As a first step in this direction, our approach can be further refined, for instance by accounting for the presence of solvents, or constraining the ends of the loop at fixed anchor points (e.g. by kinematic closure). In addition, our proposed method can be applied to more general protein folding problems beyond antibody loop modelling, which may have different time and accuracy requirements for demonstrating quantum advantage. As quantum technology continues to improve, its viability as a competitive resource for the pharmaceutical industry will need to be continually reassessed.
AV, AM, SA and MS guided the project from the pharmaceutical side. JA, C-YH and SZ guided the project from the quantum computing side. All authors contributed to the analysis of the results and the writing and review of the manuscript.
JA, C-YH and SZ are employed by Tencent, AV is employed by Roche, Germany. AM, SA and MS are employed by Roche, Switzerland.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to express our gratitude to Guy Georges, Alexander Bujotzek, Hubert Kettenberger, Detlef Wolf, Yvonna Li, Xavier Lucas, Bryn Roberts and Mariëlle van de Pol for their expertise, support, and encouragement throughout the course of this project. JA is grateful to Yicong Zheng for many helpful discussions and feedback during the preparation of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2022.908870/full#supplementary-material
1Of the previous quantum studies on protein folding, the paper of (Casares et al., 2022)—the preprint of which appeared while our manuscript was in preparation—is closest to ours. Like this manuscript, their method is also based on quantum Markov Chain Monte Carlo, but they do not consider the implementation of the key quantum operations, viewing these as being performed by a “black box”.
2While advances continue to be made in machine learning for protein folding—most notably with the announcement of AlphaFold 2 (DeepMind, 2020) in the CASP 14 protein structure prediction competition—not all protein folding problems can be solved with these new methods, and there is the need to continue to explore the potential of quantum computing for this domain.
3The lookup table approach to encoding and updating can be extended to other schemes based on dihedral angles, e.g. the backbone fragment insertion scheme of Rosetta where a multiple-residue fragment is randomly selected and the associated torsion angles replaced with the torsion angles from another fragment from a precomputed list.
4Cycle times of this order of magnitude have essentially already been demonstrated experimentally (Chen et al., 2021; Zhao et al., 2021).
Babbush, R., Perdomo-Ortiz, A., O'Gorman, B., Macready, W., and Aspuru-Guzik, A. (2012). Construction of Energy Functions for Lattice Heteropolymer Models: Efficient Encodings for Constraint Satisfaction Programming and Quantum Annealing. Adv. Chem. Phys. 155, 201–244. arXiv:1211.3422 [quant-ph]. doi:10.1002/9781118755815.ch05
Babbush, R., Gidney, C., Berry, D. W., Wiebe, N., McClean, J., Paler, A., et al. (2018). Encoding Electronic Spectra in Quantum Circuits with Linear T Complexity. Phys. Rev. X 8, 041015. doi:10.1103/physrevx.8.041015
Babbush, R., McClean, J., Newman, M., Gidney, C., Boixo, S., and Neven, H. (2020). Focus beyond Quadratic Speedups for Error-Corrected Quantum Advantage. PRX Quantum 2, 010103. arXiv:2011.04149 [quant-ph]. doi:10.1103/prxquantum.2.010103
Babej, T., Ing, C., and Fingerhuth, M. (2018). Coarse-grained Lattice Protein Folding on a Quantum Annealer. arXiv:1811.00713 [quant-ph].
Botu, V., Batra, R., Chapman, J., and Ramprasad, R. (2017). Machine Learning Force Fields: Construction, Validation, and Outlook. J. Phys. Chem. C 121, 511–522. doi:10.1021/acs.jpcc.6b10908
Bravyi, S. B., and Kitaev, A. Y. (1998). Quantum Codes on a Lattice with Boundary. arXiv:quant-ph/9811052.
Casares, P. A. M., Campos, R., and Martin-Delgado, M. A. (2022). Qfold: Quantum Walks and Deep Learning to Solve Protein Folding. Quantum Sci. Technol. 7, 025013. doi:10.1088/2058-9565/ac4f2f
Chen, Z., Satzinger, K. J., Atalaya, J., Korotkov, A. N., Dunsworth, A., Sank, D., et al. (2021). Exponential Suppression of Bit or Phase Flip Errors with Repetitive Error Correction. arXiv:2102.06132 [quant-ph].
DeepMind (2020). AlphaFold: A Solution to a 50-Year-Old Grand Challenge in Biology. Available at: https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology.
Dennis, E., Kitaev, A., Landahl, A., and Preskill, J. (2002). Topological Quantum Memory. J. Math. Phys. 43, 4452–4505. doi:10.1063/1.1499754
Farhi, E., Goldstone, J., and Gutmann, S. (2014). A Quantum Approximate Optimization Algorithm. arXiv preprint arXiv:1411.4028.
Farhi, E., Goldstone, J., Gutmann, S., and Sipser, M. (2000). Quantum Computation by Adiabatic Evolution. arXiv preprint quant-ph/0001106.
Farhi, E., and Gutmann, S. (1998). Quantum Computation and Decision Trees. Phys. Rev. A 58, 915–928. doi:10.1103/physreva.58.915
Fernández-Quintero, M. L., Kraml, J., Georges, G., and Liedl, K. R. (2019). CDR-H3 Loop Ensemble in Solution - Conformational Selection upon Antibody Binding. MAbs 11 (6), 1077–1088. Taylor & Francis. doi:10.1080/19420862.2019.1618676
Fernández-Quintero, M. L., Pomarici, N. D., Math, B. A., Kroell, K. B., Waibl, F., and Bujotzek, A. (2020). Antibodies Exhibit Multiple Paratope States Influencing VH–VL Domain Orientations. Commun. Biol. 3, 1–14. doi:10.1038/s42003-020-01319-z
Fernández-Quintero, M. L., Georges, G., Varga, J. M., and Liedl, K. R. (2021). Ensembles in Solution as a New Paradigm for Antibody Structure Prediction and Design. Mabs 13, 1923122. Taylor & Francis. doi:10.1080/19420862.2021.1923122
Fingerhuth, M., Babej, T., and Ing, C. (2018). A Quantum Alternating Operator Ansatz with Hard and Soft Constraints for Lattice Protein Folding. arXiv:1810.13411 [quant-ph].
Fowler, A. G., Mariantoni, M., Martinis, J. M., and Cleland, A. N. (2012). Surface Codes: Towards Practical Large-Scale Quantum Computation. Phys. Rev. A 86, 032324. doi:10.1103/physreva.86.032324
Gidney, C., and Fowler, A. G. (2019). Efficient Magic State Factories with a Catalyzed |CCZ⟩to2|T⟩ Transformation. Quantum 3, 135. doi:10.22331/q-2019-04-30-135
Huang, J., Rauscher, S., Nawrocki, G., Ran, T., Feig, M., De Groot, B. L., et al. (2017). CHARMM36m: an Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. methods 14, 71–73. doi:10.1038/nmeth.4067
Kim, S. J., Park, Y., and Hong, H. J. (2005). Antibody Engineering for the Development of Therapeutic Antibodies. Mol. Cells 20, 17–29.
Lemieux, J., Heim, B., Poulin, D., Svore, K., and Troyer, M. (2020). Efficient Quantum Walk Circuits for Metropolis-Hastings Algorithm. Quantum 4, 287. doi:10.22331/q-2020-06-29-287
Marchand, D., Noori, M., Roberts, A., Rosenberg, G., Woods, B., Yildiz, U., et al. (2019). A Variable Neighbourhood Descent Heuristic for Conformational Search Using a Quantum Annealer. Sci. Rep. 9, 1–13. doi:10.1038/s41598-019-47298-y
Mulligan, V. K., Melo, H., Merritt, H. I., Slocum, S., Weitzner, B. D., Watkins, A. M., et al. (2020). Designing Peptides on a Quantum Computer. bioRxiv [Preprint]. doi:10.1101/752485
National Academies of Sciences, Engineering and Medicine (2019). Quantum Computing: Progress and Prospects. Washington, DC: National Academies Press.
Outeiral, C., Morris, G. M., Shi, J., Strahm, M., Benjamin, S. C., and Deane, C. M. (2021a). Investigating the Potential for a Limited Quantum Speedup on Protein Lattice Problems. New J. Phys. 23, 103030. doi:10.1088/1367-2630/ac29ff
Outeiral, C., Strahm, M., Shi, J., Morris, G. M., Benjamin, S. C., and Deane, C. M. (2021b). The Prospects of Quantum Computing in Computational Molecular Biology. Wiley Interdiscip. Rev. Comput. Mol. Sci. 11, e1481. doi:10.1002/wcms.1481
Parsons, J., Holmes, J. B., Rojas, J. M., Tsai, J., and Strauss, C. E. (2005). Practical Conversion from Torsion Space to Cartesian Space for In Silico Protein Synthesis. J. Comput. Chem. 26, 1063–1068. doi:10.1002/jcc.20237
Perdomo, A., Truncik, C., Tubert-Brohman, I., Rose, G., and Aspuru-Guzik, A. (2008). Construction of Model Hamiltonians for Adiabatic Quantum Computation and its Application to Finding Low-Energy Conformations of Lattice Protein Models. Phys. Rev. A 78, 012320. doi:10.1103/physreva.78.012320
Perdomo-Ortiz, A., Dickson, N., Drew-Brook, M., Rose, G., and Aspuru-Guzik, A. (2012). Finding Low-Energy Conformations of Lattice Protein Models by Quantum Annealing. Sci. Rep. 2, 571. doi:10.1038/srep00571
Peruzzo, A., McClean, J., Shadbolt, P., Yung, M.-H., Zhou, X.-Q., Love, P. J., et al. (2014). A Variational Eigenvalue Solver on a Photonic Quantum Processor. Nat. Commun. 5, 1–7. doi:10.1038/ncomms5213
Ramachandran, G., Ramakrishnan, C., and Sasisekhran, V. (1963). Stereochemistry of Polypeptide Chain Configurations. J. Mol. Biol. 7, 95–99. doi:10.1016/s0022-2836(63)80023-6
Robert, A., Barkoutsos, P. K., Woerner, S., and Tavernelli, I. (2021). Resource-efficient Quantum Algorithm for Protein Folding. npj Quantum Inf. 7, 1–5. doi:10.1038/s41534-021-00368-4
Sanders, Y. R., Berry, D. W., Costa, P. C., Tessler, L. W., Wiebe, N., Gidney, C., et al. (2020). Compilation of Fault-Tolerant Quantum Heuristics for Combinatorial Optimization. PRX Quantum 1, 020312. doi:10.1103/prxquantum.1.020312
Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., et al. (2020). Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 577, 706–710. doi:10.1038/s41586-019-1923-7
Sevilla, J., and Riedel, C. J. (2020). Forecasting Timelines of Quantum Computing. arXiv:2009.05045 [quant-ph].
Shapovalov, M. V., and Dunbrack, R. L. (2011). A Smoothed Backbone-dependent Rotamer Library for Proteins Derived from Adaptive Kernel Density Estimates and Regressions. Structure 19, 844–858. doi:10.1016/j.str.2011.03.019
Szegedy, M. (2004). “Quantum Speed-Up of Markov Chain Based Algorithms,” in Proceeding of the 45th Annual IEEE symposium on foundations of computer science, Rome, Italy, October 2004 (IEEE), 32–41.
Ulmschneider, J. P., Ulmschneider, M. B., and Di Nola, A. (2006). Monte Carlo vs Molecular Dynamics for All-Atom Polypeptide Folding Simulations. J. Phys. Chem. B 110, 16733–16742. doi:10.1021/jp061619b
Unke, O. T., Chmiela, S., Sauceda, H. E., Gastegger, M., Poltavsky, I., Schuütt, K. T., et al. (2021). Machine Learning Force Fields. Chem. Rev. 121, 10142–10186. doi:10.1021/acs.chemrev.0c01111
Keywords: quantum computing, protein folding, Monte Carlo methods, in silco studies, antibody loop
Citation: Allcock J, Vangone A, Meyder A, Adaszewski S, Strahm M, Hsieh C-Y and Zhang S (2022) The Prospects of Monte Carlo Antibody Loop Modelling on a Fault-Tolerant Quantum Computer. Front. Drug. Discov. 2:908870. doi: 10.3389/fddsv.2022.908870
Received: 31 March 2022; Accepted: 09 June 2022;
Published: 08 July 2022.
Edited by:
Ho Leung Ng, Atomwise Inc., United StatesReviewed by:
U Deva Priyakumar, International Institute of Information Technology, IndiaCopyright © 2022 Allcock, Vangone, Meyder, Adaszewski, Strahm, Hsieh and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Allcock, am9uYWxsY29ja0B0ZW5jZW50LmNvbQ==; Anna Vangone, YW5uYS52YW5nb25lQHJvY2hlLmNvbQ==; Stanislaw Adaszewski, c3RhbmlzbGF3LmFkYXN6ZXdza2lAcm9jaGUuY29t; Shengyu Zhang, c2hlbmd5emhhbmdAdGVuY2VudC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.