 Jürgen Bajorath
Jürgen Bajorath- Department of Life Science Informatics and Data Science, B-IT, Rheinische Friedrich-Wilhelms-Universität, Bonn, Germany
In recent years, deep learning (DL) has led to new scientific developments with immediate implications for computer-aided drug design (CADD). These include advances in both small molecular and macromolecular modeling, as highlighted herein. Going forward, these developments also challenge CADD in different ways and require further progress to fully realize their potential for drug discovery. For CADD, these are exciting times and at the very least, the dynamics of the discipline will further increase.
Introduction
The computer-aided drug design (CADD) field encompasses a wide range of computational approaches for small molecular and macromolecular modeling as well as for the analysis and prediction of protein-ligand interactions (Jorgensen, 2004; Bajorath, 2015). In addition, a variety of molecular property calculations are a part of this methodological spectrum. CADD can roughly be divided into structure- and ligand-based approaches, augmented by bio- and chemoinformatics, respectively, where the scientific boundaries are rather fluid (Bajorath, 2015). While mostly dominated by quantitative structure-activity relationship (QSAR) methods since the 1960s and pharmacophore modeling since the 1970s, the field substantially expanded through increasing focus on structure-based drug design beginning in the mid-1980s and the advent of machine learning (ML), which experienced increasing attention during the 1990s. Structure-based design was strongly supported by advances in X-ray crystallography, high-resolution computer graphics, and the development of docking algorithms. It was further extended though fragment-based design approaches from the mid-1990s on. Concomitantly increasing computational power also triggered the application of molecular dynamics (MD) and other simulation techniques to large biomolecular systems. Docking methods opened the door to structure-based virtual compound screening while similarity searching and ML were applied to ligand-based virtual screening. Combined quantum mechanics/molecular mechanics (QM/MM) simulations originated in the 1970s and were increasingly applied to biomolecular systems over the next 2 decades. Taken together, these and other developments shaped the CADD field for years to come. In the 2 decades since the turn of the century, many incremental improvements have been made to existing approaches for ligand- and structure-based design, but truly novel methodological concepts have been rarely introduced. During this time, methodologies receiving high levels of attention in the field were essentially extensions of approaches originally introduced much earlier. Prominent examples include free energy perturbation for the calculation of relative ligand binding energies (Williams-Noonan et al., 2018) or deep neural networks (DNNs) (Chen et al., 2018) enabling deep (machine) learning (DL). DNNs have extended the traditional use of shallow neural networks (NNs) in chemoinformatics (vide infra) and put NN modeling onto a new level.
In this Perspective, exemplary applications of DL are highlighted that currently generate excitement and promise for CADD going forward. By its very nature, this Perspective is selective and far from being comprehensive and conclusions drawn are partly subjective. It is also attempted to put these recent developments into scientific context by providing an overview of the development and foundations of the CADD field. While scientific views might certainly differ, the discussion presented herein is intended to emphasize selected applications of DL having the potential to substantially influence and shape CADD as it further evolves.
Foundations
One of the characteristics of the CADD field as it has evolved is that despite many methodological developments reported over the past 20+ years, some of the foundations of CADD and underlying approximations have remained essentially unchanged. For example, the way in which the physical basis of biomolecular interactions was approximated by MM force fields and force field-based scoring functions for protein-ligand interactions has not fundamentally changed since the early days, although many refinements have been introduced over the years such as the inclusion of various solvation models in force field calculations. During the past decade, a hot topic in CADD has been the use of FEP calculations to estimate differences in the affinities of congeneric compounds (analogs) binding to pharmaceutical targets. FEP has its roots in the 1980s and employs MD and techniques such as umbrella sampling for simulating ligand-target systems and compound modifications (Williams-Noonan et al., 2018). Successful FEP case studies reported over the past decade essentially employed the same force field framework as in the early days of biomolecular simulations (Wang et al., 2015). Advances in FEP calculations were largely attributed to increasing computational resources available for prolonged simulations and more refined sampling techniques, rather than fundamental changes in the underlying theory or the way in which physical reality was approximated. Recently, the interest in FEP applications appeared to decrease again. However, going beyond FEP analysis, no conceptually new methodology has become available to this date to consistently and reliably predict free energies of small molecule binding across pharmaceutical targets with known three-dimensional (3D) structures, which would represent a true milestone event for CADD.
As another example for long lasting foundations and approximations, molecular similarity analysis and similarity searching in virtual screening still employ the same compound representations for the past 20 or more years (Willett, 2009; Maggiora et al., 2014). Moreover, while descriptors for known bioactivities have been introduced, in similarity searching and ML-based virtual screening, one continues to extrapolate from calculated molecular similarity to property similarity, for example, in predicting new active compounds, without taking bioactivity measures directly into account (Maggiora et al., 2014).
Deep Neural Networks
Over the past few years, DL using DNNs has become increasingly popular in chemistry, chemoinformatics, and CADD (Baskin et al., 2016; Chen et al., 2018), just as in many other scientific fields, spurred on by high predictive performance of DL achieved in computer vision or natural language processing (LeCun et al., 2015). Shallow NNs were popular in chemoinformatics during the early days of ML, but have largely been replaced over the years with other ML methods, mostly due to the tendency of NNs to overfit models for small data sets and their notorious black box character (Castelvecchi, 2016). Table 1 presents a summary of widely used ML methods. In recent years, the situation has fundamentally changed again. Currently, DNNs are widely applied for predictions at the small molecular as well as the macromolecular level and the popularity of DL is further increasing. Table 1 also includes examples of specialized DNN architectures that have become popular in chemistry.

TABLE 1. Popular machine learning methods for drug discovery and design.
A variety of DNN architectures have been introduced for molecular applications (Baskin et al., 2016). Figure 1 shows examples of specialized DNN architectures that are frequently applied in chemoinformatics and CADD. In Figure 1A, a multi-task DNN is shown where each output node represents a different prediction task. While single-task DNNs have a single output node reporting the prediction, multi-task DNNs are designed to address several related predictions tasks simultaneously, attempting to exploit synergies between these tasks. Figure 1B shows a convolutional DNN that is typically used for learning from image data or molecular graphs. Figure 1C depicts one of the preferred DNN architectures for generative de novo compound design, as further discussed below.
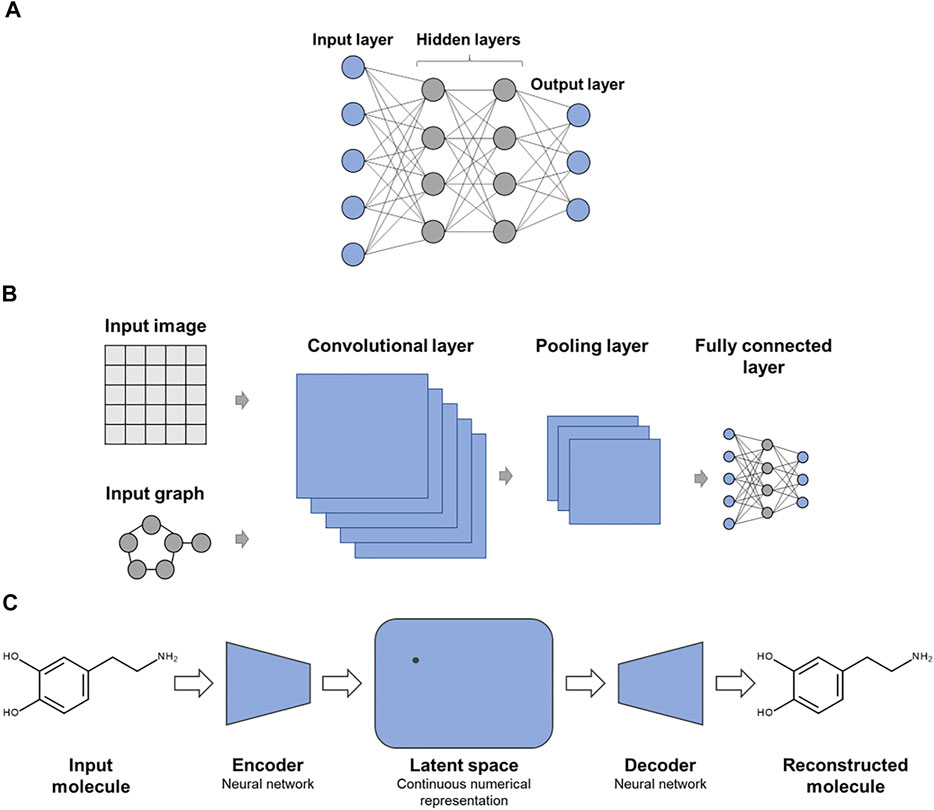
FIGURE 1. Exemplary advanced deep neural networks. Shown are specialized DNN architectures including (A) multi-task DNN, (B) convolutional DNN for learning from molecular images or graphs, and (C) autoencoder as an exemplary DNN architecture for generative de novo design.
The great variety of available DNN architectures, fittingly termed the “neural network zoo” (van Venn, 2016), makes it often difficult to scientifically judge choices of DNNs for specific applications and their performance compared to simpler computational approaches. DNNs can be rendered increasingly complex through addition of multiple layers with different functions. With the popularity of DL on the rise, there is a tendency to apply complex DNN architectures for standard prediction tasks and omit performance controls using simpler ML methods. In chemoinformatics, this especially applies to compound property predictions where relatively small data sets are available for learning and well-defined molecular representations are typically used. In benchmark settings using compound activity classes of limited size as test cases, reported prediction accuracies using other ML methods (Table 1) are typically high (often artificially high when compared to prospective applications) and in these cases, it is difficult to demonstrate significant advantages of DNNs over other widely used ML approaches (Bajorath, 2021). In property prediction, these ML methods often perform better than DNNs. While representation learning from large volumes of low-resolution data (such as pixels in image analysis) is a noted strength of DL, this situation usually does not apply to chemoinformatics/CADD applications. Accordingly, graph-based DNNs including message passing networks have increasingly been investigated for learning model-internal representations from molecular structure (Chuang et al., 2020). In addition to graph-based representation learning, DL has recently also been applied to predict biological signatures of test compounds (Bertoni et al., 2021), which might be combined with standard structural descriptors in virtual screening (vide supra). However, on the basis of currently available data, it remains to be determined whether alternative molecular representations–be they learned from graphs or predicted–might yield higher performance in ML and other applications than long-used standards such as molecular fingerprints or numerical descriptors. Although DNNs are unlikely to become a general performance booster in molecular ML, the versatility and adaptability of DNN architectures referred to above opens the door to new DL applications that were difficult, if not impossible to tackle before. These include, for example, new electron density calculations in quantum chemistry, molecular image-based learning, computer-assisted synthesis planning, generative de novo compound design, or de novo protein structure prediction in bioinformatics. Enabling access to previously infeasible applications represents a major attraction of DNNs, which sets DL indeed apart from other methodological developments in CADD over the past 2 decades. In the following, two selected applications are highlighted, in macromolecular and small molecular design, which represent particular growth areas for DL.
Focus Areas
The discussion of these two focal points begins with protein structure prediction, which has thus far served as a supporting approach for structure-based drug design. Here, an unprecedented breakthrough of DL with immediate relevance for CADD has been achieved in de novo protein structure prediction from sequence. Over the past decade, the accuracy of de novo protein modeling significantly increased through inclusion of residue coevolution analysis in computational design protocols (Marks et al., 2011). The residue coevolution concept accounts for the fact that amino acids distant in sequence but proximal in 3D structure undergo compensatory mutations during evolution. The identification of coevolving residue positions then defines spatial constraints for model building. By combining residue coevolution with sequence-structure correlation and conformational analysis, encouraging results have been reported in protein structure prediction and design, as perhaps best exemplified by the Rosetta approach (Leman et al., 2020). Recently, however, de novo protein structure prediction has reached a new level through DL. Building upon the coevolution and structural fragment/template matching framework, AlphaFold2 was able to predict single-domain protein structures consistently with an accuracy of ∼2 Å compared to experimental structures, as revealed by the 14th critical assessment of protein structure prediction (CASP14) blind test competition (Jumper et al., 2021). These findings were unprecedented, especially in their consistency. An accuracy of ∼2 Å is often within the limits of crystallographic resolution and protein models at this accuracy level rival experimental structures as templates for CADD. The AlphaFold2 approach uses multiple sequence alignments or template structures as input for an attention-based DNN to identify important sequence segments and structural patterns for model building. Beyond CASP14, the AlphaFold DL methodology was also applied to generate protein domain models for nearly the entire human proteome at an overall high level of confidence (Tunyasuvunakool et al., 2021) and further extended to systematically predict protein-protein complexes (Humphreys et al., 2021). Taken together, these results are regarded as a milestone event in solving the protein folding problem and one of the premier global achievements of artificial intelligence (AlQuraishi, 2021; Eisenberg, 2021).
In chemoinformatics and CADD, DL has in recent years substantially impacted chemical reaction prediction and automation of synthesis (e.g., Segler et al., 2018; Coley et al., 2019) as well as de novo compound design (e.g., Blaschke et al., 2020; Kotsias et al., 2020). These two areas have essentially dominated the use of DNNs in small molecule modeling and design and methodological aspects have been comprehensively reviewed (Struble et al., 2020; Tong et al., 2021). A flurry of recent publications reports different DNN architectures and protocols for generative de novo compound design. The principal goal of the approach is the generation of novel chemical entities with desired properties such as a specific biological activity. Generative modeling is expected to further extend the coverage of biologically relevant chemical space with novel chemical structures. A characteristic feature of this field and other applications of DL in early-phase drug discovery is that these efforts are currently dominated by methodological developments, whereas practical proof-of-concept applications are still rare (Bajorath et al., 2020). This characteristic and other aspects have implications for the future of DL in CADD.
Implications and Challenges for Deep Learning
Intense scientific efforts investigating DL in different CADD-relevant areas, as discussed above, provide an opportunity for CADD to enter its next phase and further expand. However, realizing this potential will critically depend on a number of adjustments requiring substantial scientific efforts. First and foremost, early-phase drug discovery is not a data-rich discipline. Rather, it is characterized by data heterogeneity and the availability of only limited chemical and biological data for many discovery projects, which poses a problem for data-hungry DL approaches. Accordingly, attempts are being made to investigate the ability of generative design to operate on the basis of confined data sets (Skinnider et al., 2021). Limited data availability in drug discovery also implies that more attention should be paid to learning strategies for low-data regimes such as transfer, multi-class, one-shot, few-shot, meta learning, or active learning (Baskin, 2019; Ding et al., 2021; Stanley et al., 2021). Furthermore, ML/DL in the life sciences and drug design is still lacking generally applicable criteria and standards for performance evaluation, assessment of design validity, and ensuring reproducibility, which hinders methodological progress and limits the impact on drug discovery. These issues are now beginning to be addressed (Walters and Murcko, 2020; Heil et al., 2021; Walsh et al., 2021), which is a positive trend in the field. Moreover, it will be of critical importance for the future of DL in CADD to concentrate on prospective applications with measurable impact on experimental programs. To these ends, it will also be crucial to reduce the black box character of DL by integrating methods for explaining predictions (Fisher et al., 2019; Murdoch et al., 2019; Lundberg et al., 2020; Matveieva and Polishchuk, 2021; Rodríguez-Pérez and Bajorath, 2021). The ability to rationalize DNN predictions will further increase the acceptance of DL for experimental design.
Milestone achievements in protein structure prediction discussed above provide new opportunities for structure-based drug design. This is perhaps the largest growth area for CADD as a whole. The availability of high-quality models with near experimental accuracy for the human proteome puts structure-based virtual screening onto a new level, but also challenges it more than ever before. To positively impact drug discovery, the computational efficiency of high-throughput flexible ligand docking will need to further increase through methodological adjustments or increasing hardware resources, as exemplified by docking complemented with DL models derived from scores (Gentile et al., 2020) or docking of ultra-large virtual libraries (Alon et al., 2021). Furthermore, for 3D molecular representations, methods for geometric DL incorporating symmetry relationships such as 3D CNNs, which are entering the molecular modeling arena (Atz et al., 2021), also have the potential to advance structure-based design including QM/MM approaches. Most importantly, however, substantial improvements of the accuracy of scoring functions will be essential, as volumes of computational screening data grow. Without such improvements, many large-scale virtual screening campaigns will be doomed to fail. This should encourage CADD investigators to concentrate on the development of approaches for more reliable estimation of ligand binding energies, which represents a principal shortcoming of current scoring schemes and one of the grand challenges ahead.
There also are important consequences for the interplay between computation and experiment. AlphaFold2 models will globally reduce the burden of experimental structure determination in an unexpected manner. Single domain-proteins might already be covered with models that are sufficiently accurate for CADD applications across the proteome (Tunyasuvunakool et al., 2021). However, this will not alleviate the need for experimental structure elucidation of complex protein targets such as multi-domain membrane receptors using X-ray crystallography or, more likely, cryo electron microscopy. Although first attempts have been made to systematically predict protein-protein complexes using AlphaFold2 (vide supra), coverage is lower than for single-domain proteins. The prediction of protein-protein complexes is closely related to the prediction of domain assemblies in multi-domain proteins, which is still in its infancy at present. Furthermore, additional experimental efforts of considerable magnitude will be required to make meaningful use of structural models for CADD. For novel targets, putative small molecule binding sites will need to be mapped and experimentally confirmed. In addition, structures of target complexes with interesting active compounds might still need to be determined experimentally at high resolution to guide chemical optimization efforts in a meaningful way. Hence, anticipated growth of structure-based design will not only depend on computational advances, but require interdisciplinary research efforts. This also applies to the analysis of increasing numbers of available models of protein-protein complexes, which are expected to aid in developing new protein-protein interaction inhibitors.
Conclusion
Herein, a brief overview of the CADD field has been presented as it has evolved over time and recent scientific developments have been highlighted that are likely to strongly impact CADD and contribute to its further growth. Most of these developments depend on DL applied to macromolecular or small molecular design. While CADD may have experienced a relatively dormant phase over the past years, with only few methodological breakthroughs, these recent developments will likely increase the dynamics of the field. Hence, CADD might well enter its next phase, provided key challenges outlined herein will be met. These include further improved validation of DL at different levels, with particular emphasis on prospective applications, but also improvements of core methodologies such as molecular docking. To these ends, off-the-beaten path scientific concepts might need to be explored, for example, to arrive at reliable estimates of ligand binding energies. Last but not least, the integration of novel computational approaches, as they are shaping up, into the practical workflows of drug discovery programs will be of central relevance for CADD going forward.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alon, A., Lyu, J., Braz, J. M., Tummino, T. A., Craik, V., O’Meara, M. J., et al. (2021). Structures of the σ2 Receptor Enable Docking for Bioactive Ligand Discovery. Nature 600 (7890), 759–764. doi:10.1038/s41586-021-04175-x
AlQuraishi, M. (2021). Machine Learning in Protein Structure Prediction. Curr. Opin. Chem. Biol. 65 (1), 1–8. doi:10.1016/j.cbpa.2021.04.005
Atz, K., Grisoni, F., and Schneider, G. (2021). Geometric Deep Learning on Molecular Representations. Nat. Mach. Intell. 3 (12), 1023–1032. doi:10.1038/s42256-021-00418-8
Bajorath, J., Kearnes, S., Walters, W. P., Meanwell, N. A., Georg, G. I., and Wang, S. (2020). Artificial Intelligence in Drug Discovery: Into the Great Wide Open. J. Med. Chem. 63 (16), 8651–8652. doi:10.1021/acs.jmedchem.0c01077
Bajorath, J. (2015). Computer-aided Drug Discovery. F1000Res 4 (1), 630. Faculty Rev-630. doi:10.12688/f1000research.6653.1
Bajorath, J. (2021). State-of-the-art of Artificial Intelligence in Medicinal Chemistry. Future Sci. OA 7 (1), FSO702. doi:10.2144/fsoa-2021-0030
Baskin, I. I., Winkler, D., and Tetko, I. V. (2016). A Renaissance of Neural Networks in Drug Discovery. Expert Opin. Drug Discov. 11 (8), 785–795. doi:10.1080/17460441.2016.1201262
Baskin, I. I. (2019). Is One-Shot Learning a Viable Option in Drug Discovery. Expert Opin. Drug Discov. 14 (7), 601–603. doi:10.1080/17460441.2019.1593368
Bertoni, M., Duran-Frigola, M., Badia-i-Mompel, P., Pauls, E., Orozco-Ruiz, M., Guitart-Pla, O., et al. (2021). Bioactivity Descriptors for Uncharacterized Chemical Compounds. Nat. Commun. 12 (1), 3932. doi:10.1038/s41467-021-24150-4
Blaschke, T., Arús-Pous, J., Chen, H., Margreitter, C., Tyrchan, C., Engkvist, O., et al. (2020). REINVENT 2.0: An AI Tool for De Novo Drug Design. J. Chem. Inf. Model. 60 (12), 5918–5922. doi:10.1021/acs.jcim.0c00915
Castelvecchi, D. (2016). Can We Open the Black Box of AI. Nature 538 (7623), 20–23. doi:10.1038/538020a
Chen, H., Engkvist, O., Wang, Y., Olivecrona, M., and Blaschke, T. (2018). The Rise of Deep Learning in Drug Discovery. Drug Discov. Today 23 (6), 1241–1250. doi:10.1016/j.drudis.2018.01.039
Chuang, K. V., Gunsalus, L. M., and Keiser, M. J. (2020). Learning Molecular Representations for Medicinal Chemistry. J. Med. Chem. 63 (16), 8705–8722. doi:10.1021/acs.jmedchem.0c00385
Coley, C. W., Thomas, D. A., Lummiss, J. A. M., Jaworski, J. N., Breen, C. P., and Schultz, V. (2019). A Robotic Platform for Flow Synthesis of Organic Compounds Informed by AI Planning. Science 365 (6453), eaax1566. doi:10.1126/science.aax1566
Ding, X., Cui, R., Yu, J., Liu, T., Zhu, T., Wang, D., et al. (2021). Active Learning for Drug Design: A Case Study on the Plasma Exposure of Orally Administered Drugs. J. Med. Chem. 64 (22), 16838–16853. doi:10.1021/acs.jmedchem.1c01683
Eisenberg, M. (2021). Artificial Intelligence Proves its Protein-Folding Power. Nature 599 (7886), 706–708. https://www.nature.com/articles/d41586-021-03499-y.
Fisher, A., Rudin, C., and Dominici, F. (2019). All Models Are Wrong, but Many Are Useful: Learning a Variable’s Importance by Studying an Entire Class of Prediction Models Simultaneously. J. Mach. Learn. Res. 20, 1–81. http://jmlr.csail.mit.edu/papers/volume20/18-760/18-760.pdf.
Gentile, F., Agrawal, V., Hsing, M., Ton, A. T., Ban, F., Norinder, U., et al. (2020). Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 6 (6), 939–949. doi:10.1021/acscentsci.0c00229
Heil, B. J., Hoffman, M. M., Markowetz, F., Lee, S. I., Greene, C. S., and Hicks, S. C. (2021). Reproducibility Standards for Machine Learning in the Life Sciences. Nat. Meth. 18 (10), 1132–1135. doi:10.1038/s41592-021-01256-7
Humphreys, I. R., Pei, J., Baek, M., Krishnakumar, A., Anishchenko, I., Ovchinnikov, S., et al. (2021). Computed Structures of Core Eukaryotic Protein Complexes. Science 374, eabm4805. doi:10.1126/science.abm4805
Jorgensen, W. L. (2004). The Many Roles of Computation in Drug Discovery. Science 303 (5665), 1813–1818. doi:10.1126/science.1096361
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly Accurate Protein Structure Prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Kotsias, P. C., Arús-Pous, J., Chen, H., Engkvist, O., Tyrchan, C., and Bjerrum, E. J. (2020). Direct Steering of De Novo Molecular Generation with Descriptor Conditional Recurrent Neural Networks. Nat. Mach. Intell. 2 (1), 254–265. doi:10.1038/s42256-020-0174-5
LeCun, Y., Bengio, Y., and Hinton, G. (2015). Deep Learning. Nature 521 (7553), 436–444. doi:10.1038/nature14539
Leman, J. K., Weitzner, B. D., Lewis, S. M., Adolf-Bryfogle, J., Alam, N., Alford, R. F., et al. (2020). Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nat. Meth. 17 (7), 665–680. doi:10.1038/s41592-020-0848-2
Lundberg, S. M., Erion, G., Chen, H., DeGrave, A., Prutkin, J. M., Nair, B., et al. (2020). From Local Explanations to Global Understanding with Explainable AI for Trees. Nat. Mach. Intell. 2 (1), 56–67. doi:10.1038/s42256-019-0138-9
Maggiora, G., Vogt, M., Stumpfe, D., and Bajorath, J. (2014). Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 57 (8), 3186–3204. doi:10.1021/jm401411z
Marks, D. S., Colwell, L. J., Sheridan, R., Hopf, T. A., Pagnani, A., Zecchina, R., et al. (2011). Protein 3D Structure Computed from Evolutionary Sequence Variation. PLoS One 6 (12), e28766. doi:10.1371/journal.pone.0028766
Matveieva, M., and Polishchuk, P. (2021). Benchmarks for Interpretation of QSAR Models. J. Cheminf. 13 (1), 41. doi:10.1186/s13321-021-00519-x
Murdoch, W. J., Singh, C., Kumbier, K., Abbasi-Asl, R., and Yu, B. (2019). Definitions, Methods, and Applications in Interpretable Machine Learning. Proc. Nat. Acad. Sci. USA 116 (44), 22071–22080. doi:10.1073/pnas.1900654116
Rodríguez-Pérez, R., and Bajorath, J. (2021). Chemistry-Centric Explanation of Machine Learning Models. Artif. Intell. Life Sci. 1 (1), 100009. doi:10.1016/j.ailsci.2021.100009
Segler, M. H. S., Preuss, M., and Waller, M. P. (2018). Planning Chemical Syntheses with Deep Neural Networks and Symbolic AI. Nature 555 (7698), 604–610. doi:10.1038/nature25978
Skinnider, M. A., Stacey, R. G., Wishart, D. S., and Foster, L. J. (2021). Chemical Language Models Enable Navigation in Sparsely Populated Chemical Space. Nat. Mach. Intell. 3 (9), 759–770. doi:10.1038/s42256-021-00368-1
Stanley, M., Bronskill, J. F., Maziarz, K., Misztela, H., Lanini, J., Segler, M., et al. (2021). “FS-mol: A Few-Shot Learning Dataset of Molecules,” in 35th Conference on Neural Information Processing Systems (NeurIPS 2021) (Track on Datasets and Benchmarks). (Round 2)https://openreview.net/pdf?id=701FtuyLlAd.
Struble, T. J., Alvarez, J. C., Brown, S. P., Chytil, M., Cisar, J., DesJarlais, R. L., et al. (2020). Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis. J. Med. Chem. 63 (16), 8667–8682. doi:10.1021/acs.jmedchem.9b02120
Tong, X., Liu, X., Tan, X., Li, X., Jiang, J., Xiong, Z., et al. (2021). Generative Models for De Novo Drug Design. J. Med. Chem. 64 (19), 14011–14027. doi:10.1021/acs.jmedchem.1c00927
Tunyasuvunakool, K., Adler, J., Wu, Z., Green, T., Zielinski, M., Žídek, A., et al. (2021). Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 596 (7873), 590–596. doi:10.1038/s41586-021-03828-1
Walsh, I., Fishman, D., Garcia-Gasulla, D., Titma, T., Pollastri, G., Harrow, J., et al. (2021). DOME: Recommendations for Supervised Machine Learning Validation in Biology. Nat. Meth. 18 (10), 1122–1127. doi:10.1038/s41592-021-01205-4
Walters, W. P., and Murcko, M. (2020). Assessing the Impact of Generative AI on Medicinal Chemistry. Nat. Biotechnol. 38 (2), 143–145. doi:10.1038/s41587-020-0418-2
Wang, L., Wu, Y., Deng, Y., Kim, B., Pierce, L., Krilov, G., et al. (2015). Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 137 (7), 2695–2703. doi:10.1021/ja512751q
Willett, P. (2009). Similarity Methods in Chemoinformatics. Ann. Rev. Inform. Sci. Technol. 43 (1), 3–71. doi:10.1002/aris.2009.1440430108
Keywords: computer-aided drug design, drug discovery, artificial intelligence, ligand-based design, structure-based design, data heterogeneity
Citation: Bajorath J (2022) Deep Machine Learning for Computer-Aided Drug Design. Front. Drug. Discov. 2:829043. doi: 10.3389/fddsv.2022.829043
Received: 04 December 2021; Accepted: 12 January 2022;
Published: 07 February 2022.
Edited by:
Daniel Reker, Duke University, United StatesReviewed by:
David Ryan Koes, University of Pittsburgh, United StatesJosé Jiménez-Luna, ETH Zürich, Switzerland
Copyright © 2022 Bajorath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jürgen Bajorath, YmFqb3JhdGhAYml0LnVuaS1ib25uLmRl