
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Drug Discov., 12 January 2023
Sec. Technologies and Strategies to Enable Drug Discovery
Volume 2 - 2022 | https://doi.org/10.3389/fddsv.2022.1093153
 Maria A. Argiriadi1*†
Maria A. Argiriadi1*† Kangwen Deng1†David Egan2Lei Gao1
Kangwen Deng1†David Egan2Lei Gao1 Farid Gizatullin1
Farid Gizatullin1 John Harlan2Denise Karaoglu Hanzatian1Wei Qiu2
John Harlan2Denise Karaoglu Hanzatian1Wei Qiu2 Ruth Villanueva1Andrew Goodearl1
Ruth Villanueva1Andrew Goodearl1LRP8 is a member of the LDLR-like protein family. It is a transport receptor, which can be used in the design of antibodies specific for investigating increasing exposure to therapeutics with respect to the blood brain barrier (BBB). In this study, a LRP8 peptide immunization strategy was implemented to generate antibodies to a specific epitope of the CR1 domain of LRP8 that could enable transport function and cross-react in mice, cynomolgus monkeys and humans. Additionally, a cyclized peptide immunogen was designed to conserve the structural β-hairpin element observed in a previously solved crystal structure of a related CR domain. As a result of this structure-based antigenic design, an LRP8 specific antibody, 11H1, was selected and characterized in ligand binding assays and crystallographic structure determination. The high-resolution structure of the 11H1 Fab complexed to the cyclized CR1 peptide revealed key interactions driving epitope recognition that were confirmed using a site-directed mutagenesis approach. A critical observation was that the identified structural CR1 epitope of 11H1 did not compete with reelin’s recognition of CR1 allowing for simultaneous binding. This was predicted by an in silico ternary model and confirmed by reelin binding data. These simultaneous binding events (11H1/CR1/reelin) could therefore enable the CR1 domain of LRP8, 11H1 and reelin to be used as a “BBB transporter” ternary complex in the design of therapeutic proteins. More importantly, 11H1 showed enhanced brain penetration after systemic intravenous dosing in a mouse study, which confirmed its potential function as BBB transporter for therapeutic proteins.
There are many antibody drugs currently being clinically evaluated for neurological diseases including targeting misfolded protein clearance in Alzheimer’s disease and Parkinson’s disease as well as other neurological indications, including stroke and broader classes of taopathies and dementia (Alpaugh and Cicchetti, 2019). A common challenge to achieve full efficacy amongst these therapies is the presence of the blood brain barrier (BBB) that limits exposure of systemically dosed protein therapeutics in the parenchymal space of the brain where many target cells such as neurons and glia reside and where their pharmacological mechanisms are in play (Terstappen et al., 2021). The BBB is formed primarily by endothelial cells surrounding the lumen of brain forming microvessels that overlap each other and are connected by robust tight junctions, together with pericytes and astrocytic endfeet. This multicellular structure serves to limit the paracellular flow from the blood vessel lumen of water and solutes including metal ions, lipids, hormones, serum proteins and antibodies. Many nutrients vital to cells in the parenchyma are transported via specific cellular receptors on endothelial cells that can bind to their cognate ligands on the luminal (apical) surface and after internalization and intracellular transport, release the ligand on the parenchymal (basolateral) surface through the process of transcytosis. The notion of engineering therapeutic biologic drugs to bind transport receptors in brain endothelial cells to affect elevated parenchymal exposure (receptor-mediated transcytosis, RMT) was first demonstrated in vivo using antibodies specific for Transferrin receptors (Pardridge et al., 1991). Since then, optimized transferrin receptor transport properties have been characterized (Yu et al., 2011; Karaoglu Hanzatian et al., 2018; Chang et al., 2021) and enhanced in vivo brain uptake of targeted bi-specific biologics has been demonstrated for additional transport receptors including insulin receptor, (Pardridge et al., 1995), CD98, (Farrington et al., 2014; Zuchero et al., 2016), IGF1R, (Stanimirovic et al., 2015), FC5, (Abulrob et al., 2005), and the Folate Receptor (Wu and Pardridge 1999; Grapp et al., 2013). Some of these shuttle targets have been used in the clinic with success mostly in the enzyme replacement therapy space (Terstappen et al., 2021). Many of these receptors are broadly expressed in many cell types and tissues, including peripheral endothelial cells, which might limit their use as BBB transport receptors. One exception to this is LRP8, also known as ApoE Receptor 2, which, other than on platelets, is selectively expressed in the periphery only by brain endothelial cells. Based on this unique expression profile, we decided to generate a panel of antibodies to LRP8 to better characterize its potential to act as a BBB transporter for therapeutic proteins.
LRP8 is member of the LDLR-like protein family and is a single pass transmembrane protein whose N-terminal extracellular domain (ECD) is comprised of multiple modules including seven LDLR Class A (LA) repeat domains, three EGF repeats with an embedded β-propeller domain and an O-linked glycosylation domain (Bu, 2009). LRP8 is subject to alternative splicing with a total of nine splice variants identified at the mRNA level. In addition to acting as a receptor for Apolipoprotein E, further LRP8 ECD-binding ligands have been identified including Reelin, (D'Arcangelo et al., 1999), Selenoprotein P, (Kurokawa et al., 2014), Clusterin (ApoJ), Thrombospondin (Blake et al., 2008) and F-spondin (Hoe et al., 2005). Binding epitopes in LRP8 for some of these ligands have been established, including LA1 (Reelin) and the β-propeller domain (Selenoprotein P). Genetic knockout of LRP8 in mice is non-lethal, leading to lissencephaly, a condition with changes in the folding structure of the cortex in brain and cerebellum during development resulting in an impaired movement (Hack et al., 2007). This phenotype is highly similar to the reelin knockout, suggesting a role for LRP8 as the major reelin receptor during development. The adult function of LRP8 is less clear, although it may include a possible role in ApoE metabolism in common with other LRP family members.
Immunogen options for cell surface proteins such as LRP8 include cDNA plasmids, high expressing recombinant cell lines, recombinant extracellular domains and peptides. As part of parallel immunization strategies, we evaluated the potential of peptide immunogens. Peptide immunogens are alternative antigenic forms, which can be advantageous over proteins because they can be reliably prepared in large quantities with high purity and stability. Further, sequences can be selected for a specific epitope, for example by selecting a homologous sequence region to generate antibodies that cross-react with target protein in different species. In order to improve the chances of cross reactivity to native folded protein from which the peptide sequence is derived, three-dimensional protein structure can be used in order to mimic the natural 3-dimensional conformation of the target protein in specific regions. Structural and thermodynamic studies have predicted success of immunizations based on the stability of linear peptides (Camacho et al., 2008). For example, certain secondary structural elements can be helpful in making a peptide more antigenic including β-turns and/or helices (Lee et al., 2016). While longer peptides can adopt such structural elements, cyclic peptides have been used to conserve these structural features in a predictable fashion in shorter peptides during immunizations and antigen-antibody recognition (Misumi et al., 2003; Jakab et al., 2009). In this study we describe the use of a cyclic peptide immunization strategy that resulted in a mouse/cynomolgus monkey/human cross-reactive LRP8 specific antibody, 11H1. We also describe the characterization of the binding epitope using ligand binding assays, crystallographic structure determination and modeling analysis with wild type and mutant peptides.
LRP8 is a large multidomain protein. In order to choose a peptide segment to serve as a robust antigen, we initially referred to the previously solved high-resolution structure (1.8 Å PDB code 1J8E) of CR7 from human Low-density lipoprotein receptor-related protein (LRP) (Simonovic et al., 2001). Retrospectively, we validated this approach by examining the CR1 domain in complex with murine reelin (PDB code: 3A7Q) (Yasui et al., 2010). After closely examining the structure and sequence in LRP8, a peptide segment was chosen to mimic a β-turn motif consisting of 2 beta strands with a loop in between. In order to stabilize this motif, peptide cyclization was considered. An “unpaired” cysteine within the sequence was mutated to Ser to prevent aggregation. This tactic was used to improve protein aggregation as with a recently reported example (Saetang et al., 2022). The resulting peptide was synthesized by New England Peptide (Gardner, MA) was [Cyc (1.130] H2N-CEKDQFQSRNERCIPSVWR (KAoa)-amide. A similar peptide for CR2 was also created based on the structural alignment [Cyc (1.130)] H2N-CADSDFTSDNGHCIHERWK (KAoa)-amide.
KLH-conjugated CR1 and CR2 peptides were synthesized by New England Peptide (Gardner, MA). Equal amounts of CR1 and CR2 (50 µg) were mixed for immunization and were injected in mice subcutaneously every 3 weeks for four times before the mouse spleens were harvested. Lymphocytes were isolated and fused with NS0 cells with a well-established protocol. Hybridoma supernatant (SN) was used for cell-based FACS with hLRP-8-HEK293 stable cells and HEK293 parental cells. Supernatants (SN) that bind to hLRP-8-HEK293 stable cells, but not to HEK293 parental cells were selected.
HEK293H cells are cultured in T25 culture flasks and incubated at 37°C, 5% CO2, and cells were passaged every 4–5 days. On the day before transfection, cells were diluted to 2 × 10*5 cells in a 6 well plate at 99% cell viability. Homo sapiens (human) Low-density lipoprotein receptor-related protein 8 (LRP8) (isoform3) (Accession # NP_059992) sequence was identified from GenBank. Mus musculus (mouse) Low-density lipoprotein receptor-related protein 8 (LRP8) (isoform2) (Accession # NP_001074395) sequence was identified from GenBank. Cynomolgus LRP8 was identified in house by de novo cloning. LRP8 genes were cloned into the pCMV vector. The mixture of 2.5 μg plasmid DNA and 10 μL Lipofectamine 2000 (Invitrogen) in 500 μL Opti-MEM were incubated at room temperature for 20 min, and then added into cells. The cells were incubated at 37°C 5% CO2 for 4 h. After 4 h, the cells were incubated in culture medium at 37°C 5% CO2 overnight. On the day after transfection, 2 mL of selection media with 5 mg/mL G-418 (final concentration) was added. Growth media was changed on the transfected cells every 4–5 days. Clonal stable cell lines were generated by serial dilution of the parental cells and subsequent expansion of isolated single cell colonies. For characterization of cell lines by FACS, HEK293 transfected cells were dissociated cells using growth medium, washed, and re-suspended in cold PBS (pH7.2)/2% FBS (FACS Buffer) to 1 × 10e6 cells/mL, incubated 1 h at 4°C with the primary antibodies and analyzed by an Accuri C6.
Peptide or recombinant protein were coated to MSD 96-well (MSD Cat# L15XB-3/ L11XB-3) plates and incubated at 4°C overnight. Plates were washed and blocked using 15% FBS (Hyclone, Thermo Scientific Cat# SH300700.03) at room temperature for 30 min with mild agitation, plates were washed with DPBS 3 times and antibodies were added. After 1 h of incubation at room temperature, plates were washed with DPBS and goat anti-human (MSD Cat#R32AJ-1) or goat anti-mouse Sulfo TAG (MSD Cat#R32AC-1) was added. Plates were incubated at room temperature for 1 h, washed with DPBS and immersed in MSD read buffer (MSD Cat#R92TC-2) before reading on an MSD SECTOR Imager 6000. EC50 values were obtained using the Xlfit4 software package.
HEK293 cells overexpressing human, monkey, or mouse LRP8 were added onto MSD 96-well plates (MSD Cat# L15XB-3/L11XB-3) and incubated at 37°C for 1 h. Cells were blocked using 15% FBS (Hyclone, Thermo Scientific Cat# SH300700.03) at room temperature for 30 min with mild agitation, plates were washed with DPBS 3 times and antibodies were added. After 1 h incubation at room temperature, plates were washed with DPBS and goat anti-human (MSD Cat#R32AJ-1) or goat anti-mouse Sulfo TAG (MSD Cat#R32AC-1) was added. Plates were incubated at room temperature for 1 h, washed with DPBS and immersed in MSD read buffer (MSD Cat#R92TD-2) before reading on an MSD SECTOR Imager 6000. EC50 values were obtained using the Xlfit4 software package.
LRP8 stable cells and parental HEK293 cells were collected and incubated in FACS buffer (1xPBS +2%FCS), and an aliquot was removed for control wells. Parental cells were labelled with CFSE (5 (6)-Carboxyfluorescein N-hydroxysuccinimidyl ester). 50,000 cells/well of equal numbers of CFSE labeled parental cells and un-labeled LRP8 stable cells were mixed and centrifuged for 10 min at 1200 rpm. After adding FACS buffer, cells at 5 × 104 cells per well were spun 2000 rpm for 3 min and then incubated for 15 min at 4°C with mAb from 30 μg/mL and 3-fold across the plate. After three washes with FACS buffer, cells were incubated 15 min at 4°C with 50 μL of secondary Thermo-scientific APC antibody diluted 1:500. Unbounded secondary antibodies were removed by three washes with FACS buffer, then cells were resuspended in 50 μL of FACS buffer and analyzed with CANTO (BD FACSCanto).
Based on the aligned sequence of CR1 and CR2 peptide, the sequence FxSxN appeared to be common in both binding peptides, and therefore likely important for the binding epitope. Note that the serine in the CR1 and CR2 peptide sequences was already changed from the parent LRP-8 protein sequence, removing an unpaired cysteine. Modified forms of cyclic peptide CR1, containing the changes illustrated in Figure 8A were synthesized, and examined for binding to chimeric anti-LRP-8-11H1.5B2 antibody [hu IgG1/k] LALA by direct ELISA assays (Figure 8B). In both assays, peptides CR1.2 and CR1.3 bound with affinity similar that of the unmodified CR1, whereas peptides CR1.1 & CR1.4, both containing the F- > A substitution, did not bind. This indicated that the phenylalanine in the sequence–FQSRN- was required for antibody binding, and thus a key residue in the epitope.
Fab fragment of LRP-8 11H1.5B2 was prepared by papain cleavage of the parent antibody, anti-LRP-8.11H1.5B2 [mu/hu IgG1/k] LALA chimeric antibody. Papain was activated with 50 mM cysteine in PBS, pH 7.4 buffer. Anti-LRP-8.11H1.5B2 chimeric antibody in PBS, pH 7.4 buffer was mixed with papain at 1:100 weight ratio of papain to the antibody and incubated for 1 h at 37°C. The reaction was quenched with 5 mM iodoacetamide. The mixture was purified on 5 ml Mab SelectSure resin (GE Healthcare) where the Fab fragment was collected as flow through. The flow through was concentrated using an Ultrafree-15 Biomax 10 kDa molecular weight cut-off (MWCO) centrifugal device (Millipore). The concentrated mixture was purified on 2.6 cm × 60 cm Sephacryl 200 HiPrep column (GE Healthcare) pre-equilibrated in 50 mM HEPES, 50 mM NaCl, pH 7.5 buffer.
The cyclic peptide was dissolved with the protein buffer (50 mM NaCl, 50 mM HEPES, pH7.5) to a final concentration of 100 mM. The peptide was added to the protein (29.4 mg/mL) to a final molar ratio (peptide over protein) of 8:1. The stacked thin plate crystals were initially observed after 2–3 days. They grew to their full size within 1 week under the conditions of 25% PEG 4000, 0.2M Ammonium Sulfate, 0.1M Sodium acetate/HCl, pH 4.6. Plate crystals were then separated, and flash frozen into liquid nitrogen using 20% propylene glycol plus the reservoir solution as the cryo-protectant. Diffraction data were collected at a temperature of 100 K using beamline XALOC (BL13) at ALBA synchrotron, Spain.
X-ray diffraction data for the 11H1/CR1 peptide complex crystals were collected at the ALBA synchrotron beamline to 1.72 Å resolution. The crystals were maintained at 100K with an Oxford Cryosystems Cryostream cooler during data collection. The data were processed using the program AUTOPROC from Global Phasing (Vonrhein et al., 2011). The x-ray diffraction data and refinement statistics are summarized in Table 1. The following lists indexing for the crystal form: space group P21, a = 41.3 Å, b = 79.8 Å, c = 67.1 Å β = 95.5.
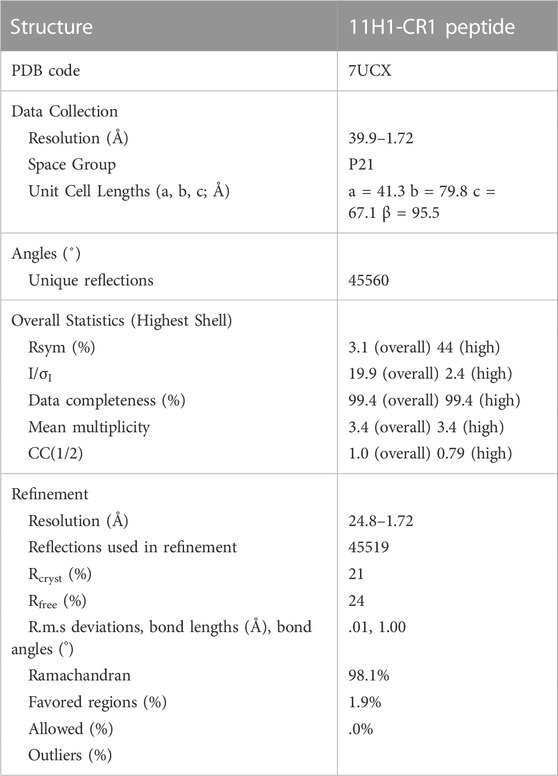
TABLE 1. Crystallographic statistics of 11H1/CR1 peptide complex structure.
A maximum likelihood molecular replacement solution was determined using the program PHASER (McCoy et al., 2007) using a Fab search model reported previously (Protein Data Bank entry 1VPO, (Valjakka et al., 2002). Coordinates were generated based on the molecular replacement solution. Preliminary refinement of the resulting solution was conducted using REFMAC (Murshudov et al., 2011) and the program BUSTER (Blanc et al., 2004). Iterative protein model building employed the use of COOT (Emsley and Cowtan, 2004) by examination of 2Fo-Fc and Fo-Fc electron-density maps. The CR1 peptide was manually built into electron density. The last five residues of the peptide were not seen in electron density likely due to flexibility in this region. Additionally, the interaction between Cys1 and Cys13 was modeled as a disulfide due to close proximity of protein backbone, despite weaker density in this region. Refinement concluded with the addition of water molecules using BUSTER and analysis in the program suite PHENIX (Adams et al., 2010). Final refinement statistics are shown in Table 1.
Molecular dynamics simulations were conducted in the program Desmond (Schrödinger Release 2021–1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2021. Schrodinger/DE Shaw). A 20 ns simulation was conducted at 300K using the OPLS3e forcefield. Trajectories were examined to determine r.m.s.d ranges for side chain and backbone movements.
HEK293 cells overexpressing cynomolgus monkey LRP-8 were added to the MSD 96-well plate (MSD Cat# L15XB-3/L11XB-3) and incubated at 37°C for 1 h. Cells were blocked using 15% FBS (Hyclone, Thermo Scientific Cat# SH300700.03) at room temperature for 30 min with mild agitation, plates were washed with DPBS 3 times. Anti-LRP-8 antibody with a human Fc and reelin-His-FLAG were used in two competition combinations:
Competition 1 (Competitor: Anti-LRP8 Ab): a 1:1 mixture of fixed concentrated reelin-HIS-FLAG and a titer of competitor LRP-8.11H1 or control antibody were added to the plate and incubated at room temperature for 1 h. After washing, reelin binding signal were detected with anti-his Ab (Novex). Competition 2 (Competitor: Reelin): a 1:1 mixture of fixed concentrated anti-LRP8.11H1 Ab and a titer of competitor reelin were added to the plate and incubated at room temperature for 1 h. After washing, an anti-human SULFO-TAG Ab was added to the plate and incubated for 1 h. The plates were washed with DPBS and immersed in MSD read buffer T surfactant free (MSD Cat# R92TD-2) before reading on an MSD SECTOR Imager 6000. Data were obtained and analyzed using a GraphPad Prism 6 software package (GraphPad Software, Inc., La Jolla, CA).
Wild-type female C57Bl/6N mice (6–8 weeks old) were purchased from Taconic Bioscience, Inc. Mice were maintained and handled according to the following Institutional Animal Care and User Committee (IACUC) approved protocol. Four animals per group were injected with 50 mg/kg of antibody via intravenous route. After the indicated time (24 h), animals were euthanized with an overdose of Ketamine-Xylazine (Fort Dodge, Anased) administered via intraperitoneal injection. The right atrium was incised, and animals were transcardially perfused with cold Dulbeccos’s phosphate buffered saline containing Heparin (1000 units/L) at a rate of 2 mL/min for 10 min via programmable peristaltic pump (NE-1000). Serum and Tissue were collected.
Brain was dissected from each perfused mouse, vertically divided into equal halves; one-half was saved for immunohistology, and the other half was homogenized using Bullet Blender Blue (NextAdvance, BBX24B) and zirconium beads (NextAdvance, ZROB05/ZROB10) in 1% NP-40 (Thermo Scientific Cat# 28324) in PBS containing protease inhibitors (Roche Diagnostics Complete Mini, EDTA-Free Ref# 11836170001). Homogenized brain samples were rotated at 4°C for 1 h before spinning at 14,000 rpm for 20 min. Supernatant was isolated and antibody measurement in brain was made using an ECL-MSD assay.
Whole blood was collected from cardiac puncture at the terminal time point. Whole blood from cardiac puncture was collected in serum separator BD microtainer™ tubes (BD Diagnostics, Ref# 365956), allowed to clot for 30 min, and spun down at 13,000 rpm at room temperature for 8 min. Supernatant was isolated and an antibody measurement in serum was made using an ECL-MSD assay.
Antibody concentrations in mouse serum and tissue samples were measured with an ECL-MSD assay. MSD 96-well plates (MSD Cat# L15XB-3/L11XB-3) were coated with an F (ab’)2 fragment of donkey anti-human IgG Fc fragment-specific polyclonal antibody (Jackson ImmunoResearch Code# 709-006-098) at 2 μg/mL overnight at 4°C. Plates were blocked with 3% MSD blocking buffer (MSD Cat# R93BA-04) for 1 h at 25°C. Plates were washed three times with 1X Tween-Tris buffered saline with a microplate washer (ELx45 Bio-Tek Instruments Inc.). Standards were made by serial dilution in 1% MSD assay buffer or 1% MSD assay buffer containing .1% serum. Tissue samples were diluted to 1:2 or 1:4 in 1% MSD assay buffer and serum samples were serially diluted starting at 1:10 in 1% MSD assay buffer and 25 μL (in duplicates) were added per well. Each antibody was used as an internal standard to quantify respective antibody concentrations. Plates were incubated for 2 h at 25°C and bound antibody was detected with goat anti-human Sulfo-TAG (MSD Cat# R32AJ-1). Plates were read on an MSD SECTOR Imager 6000. Concentration was determined from the standard curve with a Four-Parameter Logistic (4PL) non-linear regression program from IDBS XLfit® an add-in of Microsoft® software. The ECL-MSD assay lower limit of quantitation values ranged from .05–.46 ng/mL in serum and tissue samples. Molar concentration was calculated by the MSD quantification value (in ng/ml) that best fit its corresponding standard curve with a coefficient of variance ≤20% and within acceptable recovery of 80%–120% divided by the respective antibody molecular weight. For tissue samples, multiplication of tissue homogenate dilution factor was considered. Data was expressed as means ± standard deviation (SD) and statistics were assessed by unpaired, two-tailed t-tests. Total protein concentrations in brain extracts were measured using the BCA protein assay kit (Thermo Scientific, Cat#23225) and were consistently found to be within 15% of coefficient of variation (CV). Samples that did not meet these criteria were not used for analysis.
Half brains from perfused antibody-treated mice were immersion fixed in 4% paraformaldehyde for 6 h. Following fixation, tissues were processed through a graded series RUSH protocol (Leica TP1050 Tissue Processor) of alcohol to xylene and then embedded in paraffin (Leica EG1150H). Brain sections (5 μm) were cut with a microtome (Microm, HM355S). Sections were de-paraffinized and rehydrated with water and placed into Tris with Tween-20 buffer (Teknova Cat# T5155). Staining was then performed on a Dako autostainer links 48 system. Briefly, the sections were blocked with 3% hydrogen peroxide plus methanol for 30 min, washed with 10x Tris with Tween-20 buffer (Teknova Cat# T5155) then incubated for 8 min with protease I (Ventana Ref# 760–2018). Sections were blocked with a streptavidin and biotin blocking kit (Vector Laboratories Cat# SP-2002) for 8 min each, followed by Dako protein block for 30 min. Next, the sections were incubated for 1 h at room temperature with a biotinylated donkey anti-human IgG (H + L) F (ab’) (Jackson ImmunoResearch Code# 709-066-149) at 15 μg/mL followed by an incubation with peroxidase conjugated avidin for 30 min at room temperature [R.T.U ABC Kit (Vector PK-7100)]. The sections were then reacted with diaminobenzidine (DAB) chromogen (Dako Ref# K3468) for 3 min to form a brown precipitate, washed with water, counterstained with Gill Modified Hematoxylin (EMD Harleco Ref# 65065) for 30 s and bluing reagent by dipping slides 5–6 times in a reservoir (Richard-Allan Scientific Ref#7301), dehydrated and mounted for microscopy observation. Sections from four different brain regions (forebrain, midbrain, hindbrain, cerebellum) from the four animals per group were stained. Representative staining images were captured by Olympus BX43, or slides were scanned with Panoramic 250 Slide Scanner. All settings (filters and light levels) for each image were kept constant throughout the experiment.
LRP8 is a large multidomain protein containing eight ligand binding regions and an EGF-like domain containing cysteine rich repeats. In order to choose an LRP8 peptide segment to serve as a robust antigen, available LRP structural information was examined. A previously solved structure (1.8 Å PDB code 1J8E) of CR7 from human low-density lipoprotein receptor-related protein (LRP) became a starting point for peptide engineering (Simonovic et al., 2001). A sequence alignment of LRP8’s CR1, CR2 and the x-ray structure of 1J8E was performed (Figure 1A) to enable the design of a peptide segment which would meet several criteria: 1) a peptide not exceeding 20 residues 2) a peptide sequence which was preserved in multiple orthologs such as mouse and cyno 3) a peptide with adequate secondary structure for antibody recognition.
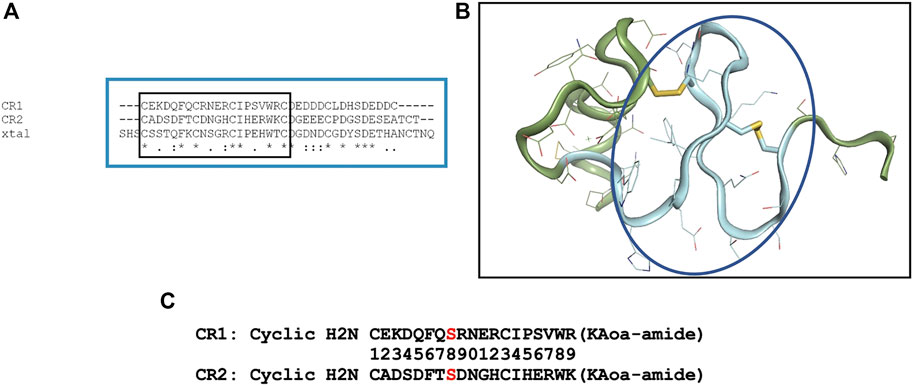
FIGURE 1. Design of cyclic CR1 and cyclic CR2 peptide. (A) Sequence alignment of the complement-like repeat region (CR1/2) domains of LRP8 with the crystal structure of CR7 of LRP (PDB code 1J8E). (B) Crystal structure of CR7 from LRP (PDB code 1J8E). The backbone colored and circled in blue is the sequence chosen for the antigenic peptide. (C) Sequences of the two CR peptides synthesized for immunization with specific C → S mutation shown in red.
After closely examining the 1J8E structure (Figure 1B) and sequence in LRP8, a peptide segment was chosen for CR1 to mimic a β-turn motif where the loop in between beta strands could be available for antibody recognition. In order to stabilize this motif, peptide cyclization was recommended. Additionally, an “unpaired” cysteine within the sequence was changed to Ser to prevent aggregation during production of the peptide. Based on alignment, a CR2 peptide was also designed. The resulting synthesized peptides are shown in Figure 1C.
4 Balb/c and 4 SJL mice were immunized with subcutaneous injection of equal amounts of KLH-conjugated CR1 and CR2 peptides (50 µg) mixture. Sera from mice were tested by FACS using HEK293-LRP8-HA stable cell line and exhibited some specificity (data not shown). 2 SJL mice were fused. Hybridoma supernatants (SNs) were screened for three rounds by CR1, CR2 peptide ELISA (data not shown). Selected SNs, 11H1, 20E8 and 12F6, from peptide ELISA were diluted 3 to 81-fold and tested in a cell-based FACS binding assay using LRP8 expressing HEK293 cells and HEK293 parental cells (Figure 2). SN 11H1 showed good binding to LRP8 expressing HEK293 cells (Figure 2A), but not HEK293 parental cells (Figure 2B). 11H1 was the best binder and was subcloned to obtain clone 11H1.5B2. Anti-LRP8.11H1.5B2 was a mouse IgG1 with a kappa light chain. The yield of 11H1.5B2 by hybridoma reached 198 mg/L. Purity of this molecule was 100% monomer by size exclusion chromatography (SEC).
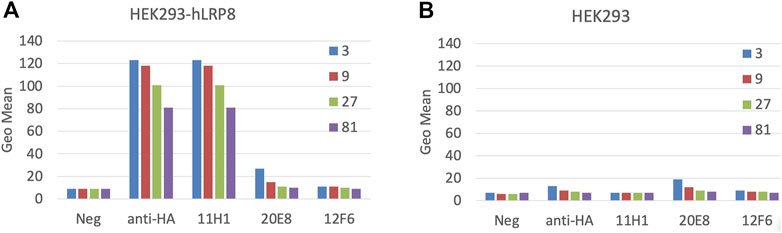
FIGURE 2. Mice immunization using cyclic CR1 and CR2 peptide. Mice were immunized with equal amounts of KLH-conjugated CR1 and CR2 peptides. Cell-based FACS binding of a 1:3 serial dilution of Hybridoma supernatant (SN) to (A) hLRP-8-HEK293 stable cells and (B) HEK293 parental cells was shown. Anti-HA is a positive control antibody. Color represents the serial dilution factor.
Variable domain sequences of 11H1.5B2 were obtained by cDNA cloning. Chimeric anti-LRP8.11H1.5B2 with mouse variable domain and human constant domains were constructed and produced in transiently transfected HEK293 cells. Purified anti-LRP8.11H1.5B2 m/hIgG bound to CR1 and CR2 peptides with 1.5 nM and 2.1 nM affinity, respectively (Figure 3). Sequence homologies between mouse/cyno/human LRP8 are the following: human versus mouse 89%, human versus cyno 86%. Sequence homology between the CR1 and CR2 domains is 50%. In a cell-based MSD binding assay using LRP8-overexpressing HEK293 cells, 11H1 bound to mouse (.27 nM), cyno (.46 nM) and human (.32 nM) LRP8 with high and comparable affinity (Figure 3B). The difference in maximal binding signal is due to the level of LRP8 expression in these cell lines. Control hIgG did not bind. Additionally, 11H1 demonstrated no binding to the C2 and C3 domains of LRP1, which is another member of the LRP receptor family that contains related CR domains (Figure 3C).
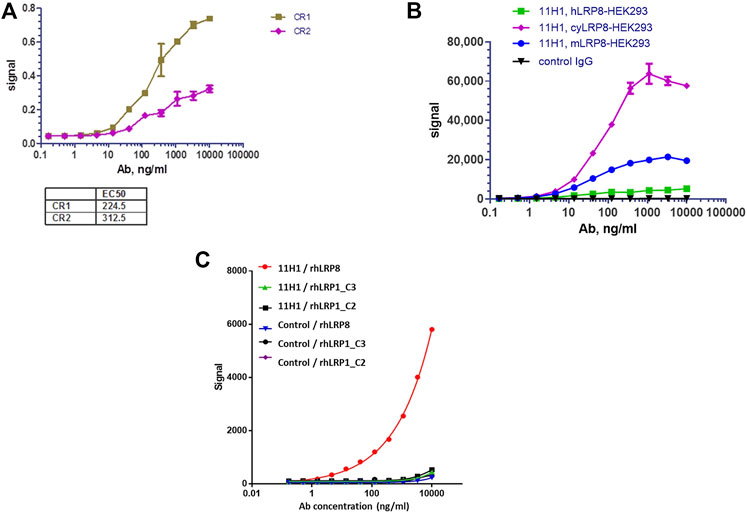
FIGURE 3. Anti-LRP8.11H1 binds both CR1, CR2 peptides and LRP8 expressing cells. (A) Purified anti-LRP8.11H1 antibody binds well with both CR1 and CR2 peptides. (B) Anti-LRP8.11H1 antibody or control IgG binds to HEK293 cells that overexpress human LRP8, cyno LRP8 or mouse LRP8 in cell-based MSD assay. (C) Anti-LRP8.11H1 binds specifically to recombinant human LRP8 (rhLRP8) and does not bind to rhLRP1_C2 and rhLRP1_C3 domains. Isotype control antibody showed minimal binding.
The crystal structure of 11H1 Fab complexed to CR1 peptide was solved to a 1.7 Å resolution (Figure 4). The structure was solved using molecular replacement with a previously reported crystal structure (PDB: 1VPO) (Valjakka et al., 2002). The electron density for the majority of the Fab and the peptide was unambiguous. Only five of the C-terminal peptide residues were not fit into density due to protein flexibility. The peptide was found bound to the 11H1 Fab in between the light and heavy chains with multiple contacts at the Fab interface. Additionally, the CR1 peptide assumed a cyclic conformation, which was stabilized by a modeled disulfide bridge as predicted by close proximity of Cys1 and Cys13 (Figure 5A). This disulfide bridge was also observed in the previously described x-ray structure, 1J8E (Simonovic et al., 2001). When overlaying the peptide as seen in the Fab structure with a recently reported CR1 structure complexed to reelin (PDB code: 3A7Q, Figure 5B) (Yasui et al., 2010), the disulfide linked loop structures were similar in cyclic architecture. However, the engineered peptide assumed two helical turns in contrast to the beta strand observed in the 3A7Q structure. The unexpected helical turns enabled several key residues to project into the Fab light and heavy chain interface. Several critical interactions participated in the 11H1 Fab-LRP8 CR1 epitope. The peptide side chains from Phe6 and Gln5 inserted into the pocket to engage in multiple interactions as illustrated in the Molecular Dynamics 2D output (Figure 6). When analyzing the x-ray structure, CR1 peptide Gln5 interacted with surrounding water molecules which in turn hydrogen bonded to the side chains of (L)Tyr41 and (L)Ser94. This Gln was also proximal to the sidechains of (H)Thr50 and (H)Ser35 but not directly engaged in hydrogen bond interactions. The backbone NH of the peptide Gln5 made a direct interaction with (H)Asp99. CR1 peptide Phe6 made an edge-to-face π−π interaction with (H)Trp47 in addition to aromatic packing from the sidechain of (H)Tyr59 (Figure 7A). It is important to note that CR2 also contained a Phe at this position, which also likely packed with these residues. Additional key hydrogen bond interactions were observed between the CR1 peptide Glu2 and (H)Ser52 and CR1 peptide Asp4 and (H)Tyr59 (Figure 7B).
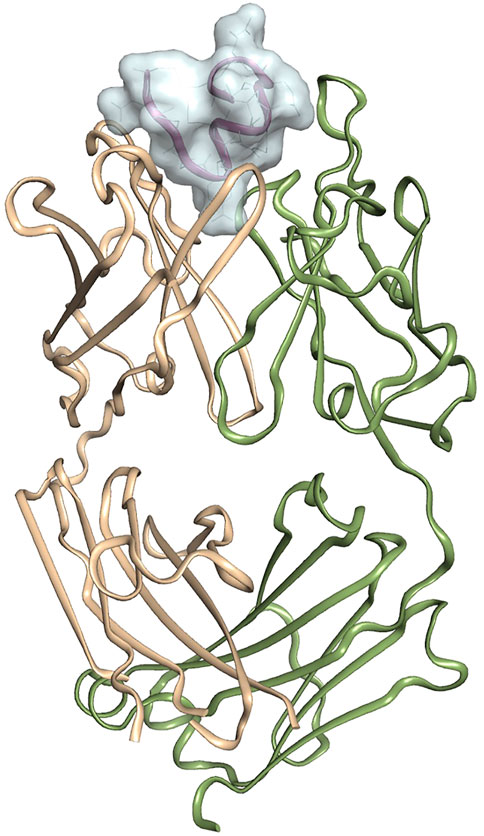
FIGURE 4. Crystal structure of 11H1 Fab complexed to CR1 peptide.The light chain is colored green (left protein chain), and the heavy chain is colored tan (right protein chain). The peptide is located in between light and heavy chain CDR’s shown and represented by surface.
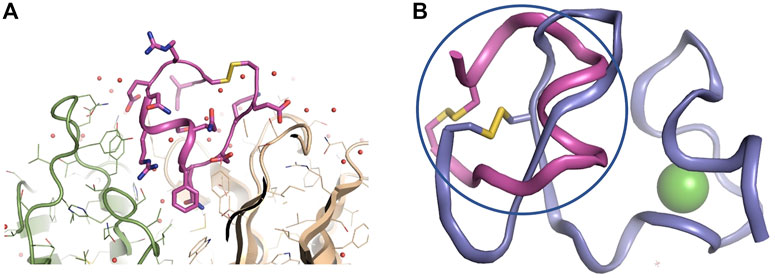
FIGURE 5. Zoomed in view of 11H1 Fab complexed to CR1 peptide. Overlay of x-ray peptide conformation and CR1 of LRP8. (A). The light chain is colored green (left protein chain), and the heavy chain is colored tan (right protein chain). Peptide assumes a cyclic conformation (shown in center) via disulfide bond from terminal ends of the peptide. (B) The cyclic peptide (in magenta and circled) assumes a conformation that resembles the cyclic nature of the LRP8 CR1 scaffold (from PDB code 3A7Q in purple), however the peptide assumes a unique helical conformation in the x-ray structure.
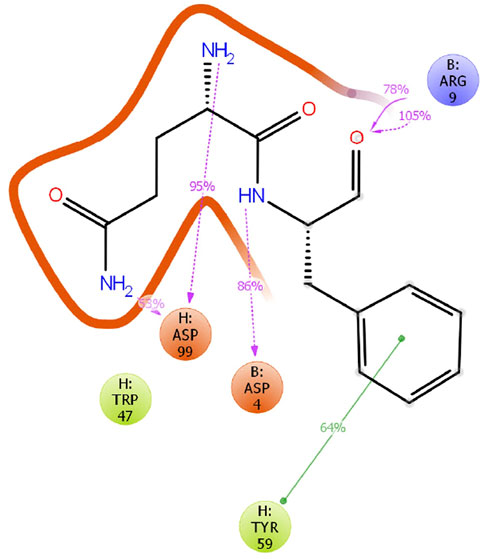
FIGURE 6. MD analysis of CR1 bound to antibody. A 20 ns Molecular Dynamics simulation was conducted for the 11H1/CR1 peptide structure in the program Desmond on this model at 300 K and pH 7.0 using the OPLS3e forcefield. Interactions were observed as consistent in the time course for the critical Phe6 and Gln5 peptide residues in the Fab cleft.
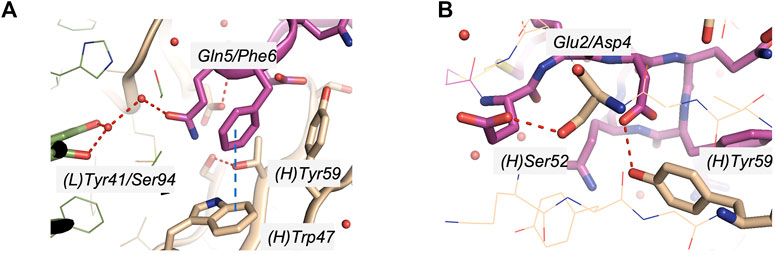
FIGURE 7. Specific interactions of CR1 peptides to the light and heavy chain interface of 11H1. (A) CR1 peptide Gln5 (magenta) interacted with water molecules which in turn hydrogen bonded to the side chains of (L)Tyr41 and (L)Ser94. CR1 peptide residue Phe6 (magenta) demonstrated a π−π aromatic interaction with (H)Trp47 in addition to showing proximity to (H)Tyr59. (B) Additional key interactions were observed between the CR1 peptide Glu2 and (H)Ser52 and CR1 peptide Asp4 and (H)Tyr59.
To confirm the importance of the peptide Phe6 residue, an orthogonal site-directed mutagenesis approach was conducted. Four mutants of the CR1 peptide were synthesized by mutating several potential 11H1 interacting amino acids Phenylalanine (F) Serine (S) and Asparagine (N) to Alanine (A) (Figure 8A). 11H1 binding to peptide was lost in the Phenylalanine (F) mutants CR1.1 and CR1.4 and was unaffected by the S and N mutations CR1.2 and CR1.3 (Figure 8B).
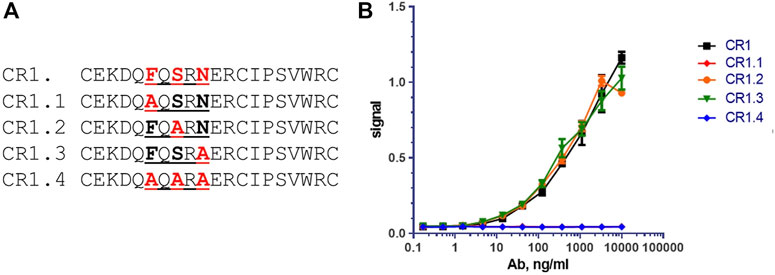
FIGURE 8. Mutational analysis reveals that Phenylalanine (F) is key amino acid for binding. (A) Peptides CR1.1, CR1.2, CR1.3 and CR1.4 are alanine mutants of CR1. (B) CR1.2 and CR1.3 bind with affinity similar that of the unmodified CR1, whereas peptides CR1.1 & CR1.4, both containing the F→A substitution, did not bind to anti-LRP8.11H1. These two curves are directly overlapped.
When comparing CR1 and CR2 in sequence, it was postulated that the key disulfide bridges were preserved as noted by the conserved cysteines in both sequences. Figure 1A shows the sequence alignment of both domains. Proximal to the proposed antigenic Phe, the critical Gln in CR1 was an Asp in CR2, which presented a repelling charge to nearby (H)Asp99 in 11H1. This might account for why 11H1 was not as tight of a binder to CR2 as for CR1. Additionally, in CR1, the Asp which preceded Gln in CR1 made a key interaction to (H)Tyr59. This position was a Ser in CR2, which may have been too short to make this interaction to the (H)CDR2.
In order to confirm the stability of the peptide binding within the antibody binding cleft, a 20 ns molecular dynamics simulation was conducted in the program Desmond (Schrödinger Release 2021–1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2021). The model was solvated with water molecules, and the simulation was conducted at 300K at pH 7.0 using the forcefield OPLS3e. Over the course of the simulation most interactions of the CR1 peptide and 11H1 were maintained as seen through the clustered trajectories. Interestingly, the MD output demonstrated the potential importance of an aromatic side chain interaction between the peptide Phe6 and (H)Tyr59. The disulfide linkage, which formed the cyclic peptide structure, was also maintained in the structure. Additionally, the Cα backbone displayed stability with a RMSF range between .6–2.0 Å.
Reelin 5,6 fragment comprises the reelin LRP binding domain (Yasui et al., 2010). When comparing the interaction surface of LRP8 CR1 with reelin and the interaction surface of CR1 peptide with 11H1.5B2 Fab, the critical 11H1.5B2 peptide recognition residues Asp-Gln-Phe were not used for reelin recognition, which suggested that a reelin-CR1-1H11.5B2 ternary could be achieved. Figure 9A shows the generated model of this ternary complex, which was created in Maestro using the previously determined 3A7Q structure and the new 11H1 complex structure as templates. The ternary complex was further minimized using a protein preparation workflow in Maestro. As observed in the model, the light and heavy chains of the Fab antibody did not sterically clash with the reelin scaffold allowing for simultaneous protein-protein interactions using the CR1 domain as a dual “transporter” binding partner. This model agreed with the reelin binding data in Figures 9B,C. Specifically, this data showed reelin R5,6-His-FLAG bound to LRP8 expressing HEK293 cells. In addition, anti-LRP8.11H1 showed no competition with reelin for binding to LRP8 in a cell-based competition assay with either fixed 365 nM of reelin and titration of 11H1 up to 10 μg/mL, or fixed 2 nM of 11H1 and titration of reelin up to 365 nM.
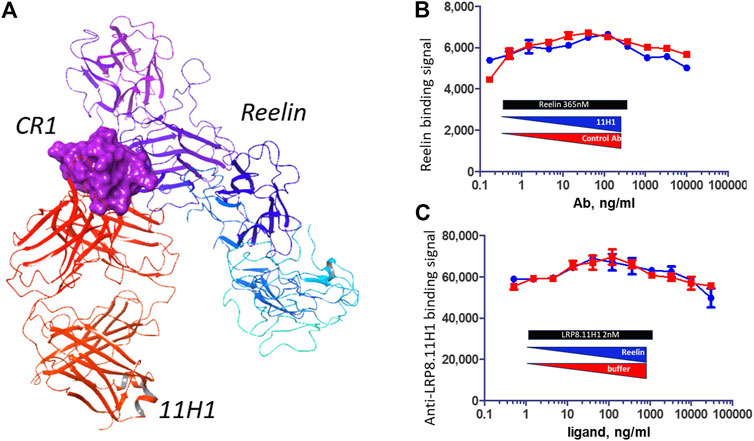
FIGURE 9. 11H1 and Reelin bind to opposite side of CR1. (A) A ternary model of Reelin complexed to CR1 and 11H1. (B,C) Cell-based competition assay with LRP8 expressing HEK293 cells coated to wells and incubated with mixture of different ratio of reelin and 11H1. 11H1 does not interfere with reelin binding, which support the model.
In order to confirm the brain penetration function of anti-LRP8.11H1 antibody, 50 mg/kg of anti-LRP8.11H1 antibody or isotype control antibody was intravenously injected into C57BL/6 mice. 11H1 concentration in serum was 2379 ± 473 nM, which was comparable to the isotype control antibody (1741 ± 924 nM). The level of mice brain uptake of 11H1 was 2-fold higher compared to the isotype control antibody. Immunohistochemistry staining of mouse brain tissue indicated good vasculature staining in all brain regions. Enhanced parenchyma staining in all brain regions compared to control IgG was observed. Neuronal staining in pons, medulla and spinal cord were also observed (Figure 10).
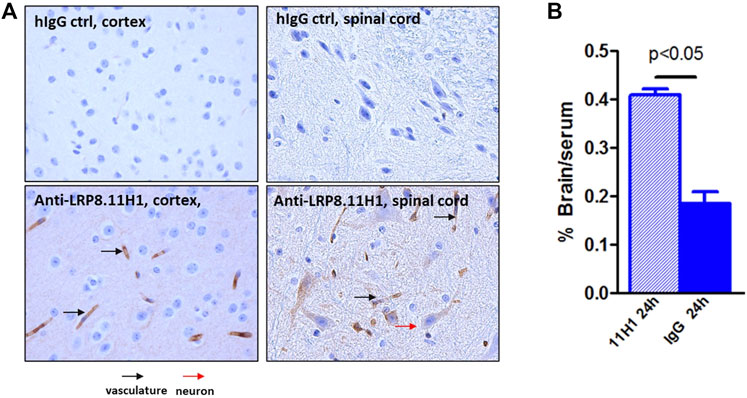
FIGURE 10. Enhanced mouse brain uptake of anti-LRP8.11H1 antibody compared to control IgG. (A) Immunohistology staining of mouse brain sections 24 h after intravenous antibody infusion. Cortex section with vasculature (black arrow) and parenchymal staining and spinal cord section with neuronal staining (red arrow) was shown. (B) Antibody concentration at 24 h post single intravenous injection of 50 mg/kg showed significantly enhanced brain uptake compared to isotype control antibody. Data was expressed as % Brain/serum, n = 4 mice per group.
In this study, an antibody was generated, which recognized both a cyclic CR1 peptide and the full length LRP8 CR1 domain. This suggested that 1) this domain of the protein, which was chosen for design, was accessible for antibody binding and 2) the peptide assumed a cyclic conformation which mimicked an antigenic loop region in the native protein. The antigenic peptide design used a structure-based approach coupled with close inspection of CR domain sequences to meet the necessary criteria to achieve a robust LRP8 epitope. Ortholog cross-reactivity and stable structural motifs were top considerations when examining an early CR structure. Despite the small size of the CR domain, there were several regions that were examined, and it was a disulfide stabilized region of this domain which was prioritized for a cyclic peptide design in support of an LRP8 hybridoma campaign. As a result, a selective LRP8 reagent, 11H1, was identified and confirmed in binding assays across species. Structural studies confirmed the original hypothesis that an isolated structural motif could be used as a means for antigenic recognition. Additionally, site-directed mutagenesis confirmed the importance of hot-spot residues on the antigenic peptide. Molecular modeling and dynamics experiments shed light on, not only the stability of Fab/CR1 recognition, but also solidified the requirement that LRP8 could simultaneously bind reelin and antibody. Therefore, a combination of experimental and in silico peptide design proved to be an extremely successful exercise for a large protein such as LRP8. Through this workflow, we were able to focus on a small yet robust domain of the protein to pursue the ortholog cross-reactivity and dual reelin binding needed for BBB transport. Generally speaking, structural approaches should be implemented routinely when designing stable, unique, secondary structural elements of a target protein and is a powerful way to create antigens for hybridoma campaigns. There are additional techniques that could be considered for this type of design approach. When thinking of our results retrospectively, there are possible considerations for improvement of the strategy.
At the time of peptide design, the reelin complex structure was not available for inspection. One consideration would have been to confirm our initial modeling/design hypothesis and choice of peptide by using site directed mutagenesis of the entire CR1 domain with respect to reelin binding data. Variant positions, which would not affect reelin binding, would be prioritized for peptide incorporation. A second consideration would have been to confirm the secondary structure with orthogonal techniques such as circular dichroism (CD). This technique is known to identify helix-coil transitions of peptides (Bakshi et al., 2014). Our structure reveals that the CR1 peptide assumed two helical turns in the 11H1 cleft, which is different from the conformation of the unbound form of CR1. Monitoring secondary structure through CD with different variations of peptide might have been a means for optimization. An additional consideration would have been to solve the co-crystal structure of the entire CR1 domain with 11H1 Fab, which would 1) confirm the helical antigenic secondary structure in the context of the full domain and 2) provide additional structural information to optimize the design of the peptide.
In addition to disulfide bridging for peptide stabilization, there are additional tactics which could have been used to drive secondary structure: cross-linking using lactam bridging or thiol-reactive alkyne-based cross-linkers (Bracken et al., 1994; Zhang et al., 2007). These approaches have been used for several protein targets. After observing the helical recognition of 11H1, a stapled peptide approach might have been an appropriate follow-on strategy. For example, hydrocarbon stapling was demonstrated with gp41 specific peptides, in an effort to develop HIV-1 immunogens for vaccination. This stapling method aided in preserving the bioactive a-helical conformation of the peptide (Bird et al., 2014). Paired with CD analysis, all of these different linking methods might have resulted in a further optimized bioactive peptide.
A final consideration would have been to compare LRP8 CR1 sequence to other family members to consider cross-reactivity and to alternate desired binding profiles. For example, when examining sequence and structural model comparisons, LRP1 does have an identical phenylalanine, which is critical at the interface of the LRP8 CR1-11H1 structure (Figures 11A,B). However as mentioned earlier, the periphery of the LRP1 peptide contains different charged residues, which alter the electrostatic surface profile of the peptide when assuming a cyclic conformation (Figure 11C). If the desired outcome was to create an antibody to cross-react to LRP1 and other isoforms in the family, electrostatic profiling in addition to sequence comparisons with the designed peptides would be necessary. By preliminary sequence inspection, higher sequence identity between LRP1 CR1 and LRP8 CR1 is observed in the C-terminal part of the sequences. A peptide designed in this part of the sequence might result in higher cross-reactivity.
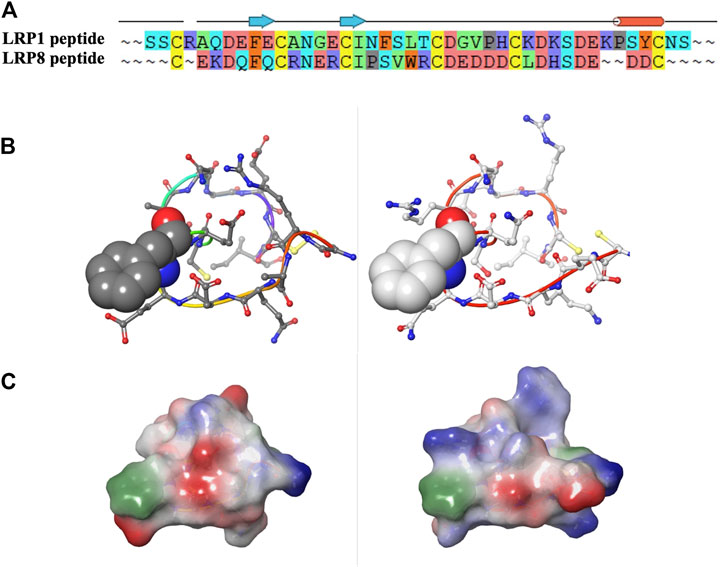
FIGURE 11. (A–C) Sequence, structural and electrostatic comparisons between CR1 peptides of LRP1 (left) and LRP8 (right).
Although it had previously been inferred from sequence homology that LRP8 might have transport properties, in this report we confirmed experimentally that LRP8 can be a BBB target for enhancing brain uptake of bound protein utilizing the RMT pathway. The increased brain uptake of the anti-LRP8.11H1tool antibody is modest (2-3 fold), but it is important to note that the binding affinity might not be optimal, possibly due to its relatively high affinity to LRP8. For other BBB RMT targets, it has been shown that brain uptake of a high affinity antibody could be improved by decreasing its binding affinity to BBB target (Yu et al., 2011; Karaoglu Hanzatian et al., 2018). Demonstration of functional BBB shuttle properties leveraging the brain endothelial cell specific expression of LRP8 (Zhang et al., 2014; Munji et al., 2019; Zhang et al., 2020) awaits further study with affinity optimized LRP8 bi-specific molecules.
To conclude, we have used a structure-based method to design a peptide antigen for the BBB transport target LRP8. The resulting antibody is a selective reagent with respect to the other LRP family members due to key interactions in the epitope recognition. Additionally, the LRP8 CR1 domain can simultaneously bind to this antibody and its binding partner, reelin, which enables a “transport” ternary complex between reelin, LRP8 and 11H1. Overall, this exploration highlights how structure-based design can enhance the creation of antigenic peptides and increase success in identifying robust biologic reagents for target proteins of interest.
The PDB data presented in this study can be found in the online repository https://www.rcsb.org with the following accession code: 7UCX.
The animal study was reviewed and approved by Institutional Animal Care and User Committee (IACUC).
MA, KD, and AG prepared the manuscript. MA, KD, JH, WQ, AG, DE, LG, FG, RV, and DKH all participated in the review of the manuscript. MA, KD, JH, WQ, DE, LG, RV, and FG contributed experimental data supporting this manuscript.
The authors thank Eve Barlow (former AbbVie employee) for valuable scientific discussions regarding the LRP8 11H1 project.
MAA, KD, LG, FG, DKH, RV and AG were employed by AbbVie Bioresearch Center. DE, JH and WQ were employed by AbbVie Inc.
The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abulrob, A., Sprong, H., Van Bergen en Henegouwen, P., and Stanimirovic, D. (2005). The blood-brain barrier transmigrating single domain antibody: Mechanisms of transport and antigenic epitopes in human brain endothelial cells. J. Neurochem. 95 (4), 1201–1214. doi:10.1111/j.1471-4159.2005.03463.x
Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66 (2), 213–221. doi:10.1107/S0907444909052925
Alpaugh, M., and Cicchetti, F. (2019). A brief history of antibody-based therapy. Neurobiol. Dis. 130, 104504. doi:10.1016/j.nbd.2019.104504
Bakshi, K., Liyanage, M. R., Volkin, D. B., and Middaugh, C. R. (2014). Circular dichroism of peptides. Methods Mol. Biol. 1088, 247–253. doi:10.1007/978-1-62703-673-3_17
Bird, G. H., Irimia, A., Ofek, G., Kwong, P. D., Wilson, I. A., and Walensky, L. D. (2014). Stapled HIV-1 peptides recapitulate antigenic structures and engage broadly neutralizing antibodies. Nat. Struct. Mol. Biol. 21 (12), 1058–1067. doi:10.1038/nsmb.2922
Blake, S. M., Strasser, V., Andrade, N., Duit, S., Hofbauer, R., Schneider, W. J., et al. (2008). Thrombospondin-1 binds to ApoER2 and VLDL receptor and functions in postnatal neuronal migration. EMBO J. 27 (22), 3069–3080. doi:10.1038/emboj.2008.223
Blanc, E., Roversi, P., Vonrhein, C., Flensburg, C., Lea, S. M., and Bricogne, G. (2004). Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D. Biol. Crystallogr. 60 (1), 2210–2221. doi:10.1107/S0907444904016427
Bracken, C., Gulyas, J., Taylor, J. W., and Baum, J. (1994). Synthesis and nuclear magnetic resonance structure determination of an .alpha.-Helical, bicyclic, lactam-bridged hexapeptide. J. Am. Chem. Soc. 116 (14), 6431–6432. doi:10.1021/ja00093a052
Bu, G. (2009). Apolipoprotein E and its receptors in alzheimer's disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10 (5), 333–344. doi:10.1038/nrn2620
Camacho, C. J., Katsumata, Y., and Ascherman, D. P. (2008). Structural and thermodynamic approach to peptide immunogenicity. PLoS Comput. Biol. 4 (11), e1000231. doi:10.1371/journal.pcbi.1000231
Chang, H. Y., Wu, S., Li, Y., Zhang, W., Burrell, M., Webster, C. I., et al. (2021). Brain pharmacokinetics of anti-transferrin receptor antibody affinity variants in rats determined using microdialysis. MAbs 13 (1), 1874121. doi:10.1080/19420862.2021.1874121
D'Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D. S., Sheldon, M., and Curran, T. (1999). Reelin is a ligand for lipoprotein receptors. Neuron 24 (2), 471–479. doi:10.1016/s0896-6273(00)80860-0
Emsley, P., and Cowtan, K. (2004). Coot: Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60 (1), 2126–2132. doi:10.1107/S0907444904019158
Farrington, G. K., Caram-Salas, N., Haqqani, A. S., Brunette, E., Eldredge, J., Pepinsky, B., et al. (2014). A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 28 (11), 4764–4778. doi:10.1096/fj.14-253369
Grapp, M., Wrede, A., Schweizer, M., Huwel, S., Galla, H. J., Snaidero, N., et al. (2013). Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 4, 2123. doi:10.1038/ncomms3123
Hack, I., Hellwig, S., Junghans, D., Brunne, B., Bock, H. H., Zhao, S., et al. (2007). Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development 134 (21), 3883–3891. doi:10.1242/dev.005447
Hoe, H. S., Wessner, D., Beffert, U., Becker, A. G., Matsuoka, Y., and Rebeck, G. W. (2005). F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol. Cell Biol. 25 (21), 9259–9268. doi:10.1128/MCB.25.21.9259-9268.2005
Jakab, A., Schlosser, G., Feijlbrief, M., Welling-Wester, S., Manea, M., Vila-Perello, M., et al. (2009). Synthesis and antibody recognition of cyclic epitope peptides, together with their dimer and conjugated derivatives based on residues 9-22 of herpes simplex virus type 1 glycoprotein D. Bioconjug Chem. 20 (4), 683–692. doi:10.1021/bc800324g
Karaoglu Hanzatian, D., Schwartz, A., Gizatullin, F., Erickson, J., Deng, K., Villanueva, R., et al. (2018). Brain uptake of multivalent and multi-specific DVD-Ig proteins after systemic administration. MAbs 10 (5), 765–777. doi:10.1080/19420862.2018.1465159
Kurokawa, S., Bellinger, F. P., Hill, K. E., Burk, R. F., and Berry, M. J. (2014). Isoform-specific binding of selenoprotein P to the beta-propeller domain of apolipoprotein E receptor 2 mediates selenium supply. J. Biol. Chem. 289 (13), 9195–9207. doi:10.1074/jbc.M114.549014
Lee, B. S., Huang, J. S., Jayathilaka, L. P., Lee, J., and Gupta, S. (2016). Antibody production with synthetic peptides. Methods Mol. Biol. 1474, 25–47. doi:10.1007/978-1-4939-6352-2_2
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40 (4), 658–674. doi:10.1107/S0021889807021206
Misumi, S., Endo, M., Mukai, R., Tachibana, K., Umeda, M., Honda, T., et al. (2003). A novel cyclic peptide immunization strategy for preventing HIV-1/AIDS infection and progression. J. Biol. Chem. 278 (34), 32335–32343. doi:10.1074/jbc.M301209200
Munji, R. N., Soung, A. L., Weiner, G. A., Sohet, F., Semple, B. D., Trivedi, A., et al. (2019). Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat. Neurosci. 22 (11), 1892–1902. doi:10.1038/s41593-019-0497-x
Murshudov, G. N., Skubak, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., et al. (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 67 (4), 355–367. doi:10.1107/S0907444911001314
Pardridge, W. M., Buciak, J. L., and Friden, P. M. (1991). Selective transport of an anti-transferrin receptor antibody through the blood-brain barrier in vivo. J. Pharmacol. Exp. Ther. 259 (1), 66–70.
Pardridge, W. M., Kang, Y. S., Buciak, J. L., and Yang, J. (1995). Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood-brain barrier in vivo in the primate. Pharm. Res. 12 (6), 807–816. doi:10.1023/a:1016244500596
Saetang, J., Roongsawang, N., Sangkhathat, S., Voravuthikunchai, S. P., Sangkaew, N., Prompat, N., et al. (2022). Surface cysteine to serine substitutions in IL-18 reduce aggregation and enhance activity. PeerJ 10, e13626. doi:10.7717/peerj.13626
Simonovic, M., Dolmer, K., Huang, W., Strickland, D. K., Volz, K., and Gettins, P. G. (2001). Calcium coordination and pH dependence of the calcium affinity of ligand-binding repeat CR7 from the LRP. Comparison with related domains from the LRP and the LDL receptor. Biochemistry 40 (50), 15127–15134. doi:10.1021/bi015688m
Stanimirovic, D., Kemmerich, K., Haqqani, A. S., Sulea, T., Arbabi-Ghahroudi, M., Massie, B., et al. (2015). Insulin-like growth factor 1 receptor -specific antibodies and uses thereof. Patent WO2015131256A1. N. R. C. O. Canada. Canada.
Terstappen, G. C., Meyer, A. H., Bell, R. D., and Zhang, W. (2021). Strategies for delivering therapeutics across the blood-brain barrier. Nat. Rev. Drug Discov. 20 (5), 362–383. doi:10.1038/s41573-021-00139-y
Valjakka, J., Hemminki, A., Niemi, S., Soderlund, H., Takkinen, K., and Rouvinen, J. (2002). Crystal structure of an in vitro affinity- and specificity-matured anti-testosterone Fab in complex with testosterone. Improved affinity results from small structural changes within the variable domains. J. Biol. Chem. 277 (46), 44021–44027. doi:10.1074/jbc.M208392200
Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., et al. (2011). Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D. Biol. Crystallogr. 67 (4), 293–302. doi:10.1107/S0907444911007773
Wu, D., and Pardridge, W. M. (1999). Blood-brain barrier transport of reduced folic acid. Pharm. Res. 16 (3), 415–419. doi:10.1023/a:1018829920158
Yasui, N., Nogi, T., and Takagi, J. (2010). Structural basis for specific recognition of reelin by its receptors. Structure 18 (3), 320–331. doi:10.1016/j.str.2010.01.010
Yu, Y. J., Zhang, Y., Kenrick, M., Hoyte, K., Luk, W., Lu, Y., et al. (2011). Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 3 (84), 84ra44. doi:10.1126/scitranslmed.3002230
Zhang, F., Sadovski, O., Xin, S. J., and Woolley, G. A. (2007). Stabilization of folded peptide and protein structures via distance matching with a long, rigid cross-linker. J. Am. Chem. Soc. 129 (46), 14154–14155. doi:10.1021/ja075829t
Zhang, W., Liu, Q. Y., Haqqani, A. S., Leclerc, S., Liu, Z., Fauteux, F., et al. (2020). Differential expression of receptors mediating receptor-mediated transcytosis (RMT) in brain microvessels, brain parenchyma and peripheral tissues of the mouse and the human. Fluids Barriers CNS 17 (1), 47. doi:10.1186/s12987-020-00209-0
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O'Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34 (36), 11929–11947. doi:10.1523/JNEUROSCI.1860-14.2014
Keywords: LRP8, crystal structure, cyclic peptide, BBB, structure-based antigen design, ApoER2
Citation: Argiriadi MA, Deng K, Egan D, Gao L, Gizatullin F, Harlan J, Karaoglu Hanzatian D, Qiu W, Villanueva R and Goodearl A (2023) The use of cyclic peptide antigens to generate LRP8 specific antibodies. Front. Drug. Discov. 2:1093153. doi: 10.3389/fddsv.2022.1093153
Received: 08 November 2022; Accepted: 14 December 2022;
Published: 12 January 2023.
Edited by:
Michael Tranter, University of Cincinnati, United StatesReviewed by:
Amber Onorato, Northern Kentucky University, United StatesCopyright © 2023 Argiriadi, Deng, Egan, Gao, Gizatullin, Harlan, Karaoglu Hanzatian, Qiu, Villanueva and Goodearl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria A. Argiriadi, bWFyaWEuYXJnaXJpYWRpQGFiYnZpZS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.