 Nuno Jorge Lamas1,2,3*
Nuno Jorge Lamas1,2,3* Laurent Roybon4,5*
Laurent Roybon4,5*- 1Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Braga, Portugal
- 2ICVS/3B’s, PT Government Associate Laboratory, Braga, Portugal
- 3Anatomic Pathology Service, Pathology Department, Centro Hospitalar Universitário do Porto, Porto, Portugal
- 4Department of Experimental Medical Science, BMC D10, Faculty of Medicine, Lund University, Lund, Sweden
- 5MultiPark and Lund Stem Cell Center, Faculty of Medicine, Lund University, Lund, Sweden
Amyotrophic Lateral Sclerosis (ALS) is a motor neurodegenerative disorder whose cellular hallmarks are the progressive death of motor neurons (MNs) located in the anterior horn of the spinal cord, brainstem and motor cortex, and the formation of intracellular protein aggregates. Over the course of the disease, progressive paralysis takes place, leading to patient death within 3–5 years after the diagnosis. Despite decades of intensive research, only a few therapeutic options exist, with a limited benefit on the disease progression. Preclinical animal models have been very useful to decipher some aspects of the mechanisms underlying ALS. However, discoveries made using transgenic animal models have failed to translate into clinically meaningful therapeutic strategies. Thus, there is an urgent need to find solutions to discover drugs that could impact on the course of the disease, with the ultimate goal to extend the life of patients and improve their quality of life. Induced pluripotent stem cells (iPSCs), similarly to embryonic stem cells (ESCs), have the capacity to differentiate into all three embryonic germ layers, which offers the unprecedented opportunity to access patient-specific central nervous system cells in an inexhaustible manner. Human MNs generated from ALS patient iPSCs are an exciting tool for disease modelling and drug discovery projects, since they display ALS-specific phenotypes. Here, we attempted to review almost 2 decades of research in the field, first highlighting the steps required to efficiently generate MNs from human ESCs and iPSCs. Then, we address relevant ALS studies which employed human ESCs and iPSC-derived MNs that led to the identification of compounds currently being tested in clinical trials for ALS. Finally, we discuss the potential and caveats of using patient iPSC-derived MNs as a platform for drug screening, and anticipate ongoing and future challenges in ALS drug discovery.
Introduction
Neurodegenerative disorders (NDs) are a tremendous public health challenge worldwide. Besides having a devastating impact on the quality of life of patients, NDs are a heavy load for caregivers and they pose a tremendous financial burden for health-care systems (Collaborators, 2019; Deuschl et al., 2020). Across the globe, with special emphasis on Western countries, as the population is increasingly ageing, problems are expected to loom bigger in the coming decades, with dramatic increases in the number of diagnosis of NDs (Collaborators, 2019). In Europe, the annual costs for brain disorders including neurodegenerative diseases and brain injury related with stroke are close to a trillion Euros, representing an average yearly cost per inhabitant of € 5,555, which equals 3.7 months of average yearly salary (Olesen et al., 2012). A delay of 5 years in the occurrence of NDs coupled to a better recovery, would effectively reduce the costs associated with a potential saving of one third (Olesen et al., 2012).
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease that is characterized by the progressive loss of motor neurons (MNs) that locate in the motor cortex, brainstem and anterior horn of the spinal cord (Figure 1) (Rowland and Shneider, 2001; Brown and Al-Chalabi, 2017). Moreover, in addition to being the most common motor neuron degenerative disorder, ALS is the most common neurodegenerative disorder in mid-life and amongst the most rapidly fatal, with death occurring nearly 2–3 years after symptom onset (Kiernan et al., 2011; Brown and Al-Chalabi, 2017). Motor neurons are a type of specialized neurons of the central nervous system (CNS), which have their cell bodies located in the motor cortex, in the mid- and hind-brain nuclei and in columns throughout the ventral horns of the spinal cord (Figure 1) (Kanning et al., 2010). Motor neurons transport information from the brain to the periphery and, thus, permit the magnificently and delicately orchestrated contraction of skeletal muscles (Figure 1) (Grillner and Jessell, 2009; Kanning et al., 2010). The nearly 300 bilateral pairs of muscles present in the body are innervated by nearly 120,000 MNs in the spinal cord (Kanning et al., 2010). However, even though MNs have well-known common functions, they constitute in fact a complex and heterogeneous population of CNS cells (Kanning et al., 2010; Nijssen et al., 2017). Several actions on which life depends are critically controlled by MNs, including swallowing and breathing (Grillner and Jessell, 2009; Jessell et al., 2011). The death of MNs leads to progressive motor impairment and ultimately death (Talbot and Marsden, 2008; Talbot and Oxford University Press., 2010; Brown and Al-Chalabi, 2017). The group of diseases known as MN disorders can be fundamentally divided into three categories: those with exclusive upper MN degeneration [e.g., Primary Lateral Sclerosis (PLS)], those with specific lower MN involvement [e.g., Spinal Muscular Atrophy (SMA)] and MN diseases with combined upper and lower MN involvement [e.g., ALS] (McDermott and Shaw, 2008; Talbot and Marsden, 2008; Goutman, 2017; Takeda et al., 2020). Regarding ALS, despite decades of research, the available therapeutic options have a limited impact on patients’ prognosis and the great majority of ALS patients still die due to respiratory failure, which show us that our knowledge on the disease remains in its initial stages (Rowland and Shneider, 2001; Brown and Al-Chalabi, 2017).
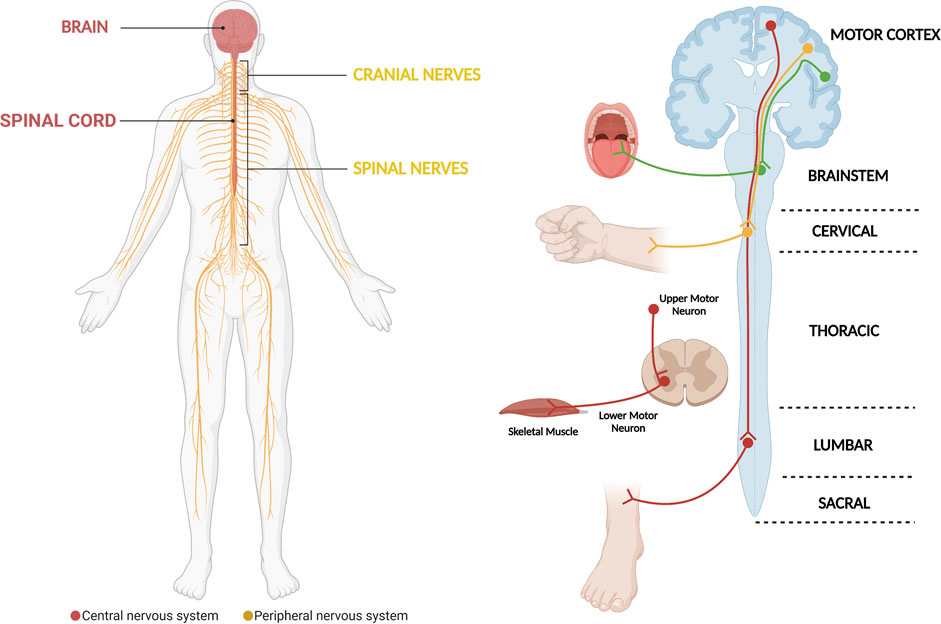
FIGURE 1. The motor system, which is critically affected in ALS. The motor system comprises motor neurons whose cell bodies are located in the central nervous system (brain and spinal cord). They exit the central nervous system assembled in cranial and peripheral nerves, which will be in contact with and carry the electric signals that elicit skeletal muscle contraction. In general, the different motor circuits comprehend upper cortical motor neurons and lower bulbar or spinal cord motor neurons. The motor neurons influence the actions of skeletal muscle through a special type of synapse, the neuromuscular junction. The demise of motor neurons in ALS leads to impairment of different motor circuits, with patients ultimately developing muscle weakness and paralysis (Diagram created with BioRender.com).
Human induced pluripotent stem cells (hiPSCs) are a type of human pluripotent stem cells (hPSCs) that can be generated by the reprogramming of somatic cells of an individual (Yamanaka, 2020). These cells, like human embryonic stem cells (hESCs), are able to differentiate into all three germ layers and they can give rise to virtually all cell types of the body. The use of hiPSCs has opened up new fields of studies and exceptional possibilities to generate in vitro, nearly inexhaustible sources of cells which are normally inaccessible to study in the human body (Nizzardo et al., 2010; Han et al., 2011; Karagiannis et al., 2019; Yamanaka, 2020). Indeed, numerous ethical and technical constraints prevent the use of MNs obtained from patients in laboratory studies and, thus, hPSCs have become an extraordinary resource to study NDs like ALS (Silani et al., 1998; Palmer et al., 2001; Nizzardo et al., 2010; Han et al., 2011). In the past 2 decades, hiPSCs and hESCs have represented a powerful tool for studying human development, modelling diseases, performing drug screening campaigns, and evaluating the future of cell replacement therapy (Avior et al., 2016; Karagiannis et al., 2019; Rowe and Daley, 2019; Chang et al., 2020; Yamanaka, 2020). Importantly, it is also anticipated that these cells could be used to meaningfully and robustly stratify patients based on prediction of drug therapy response (Haston and Finkbeiner, 2016; Holmqvist et al., 2016; Stern et al., 2018a; Pasteuning-Vuhman et al., 2020).
In the present review, we first appraise the main clinical and neuropathological aspects of ALS. Next, we review the steps undertaken to generate MNs from hESCs and hiPSCs, and summarize how these cells were employed for ALS-directed drug discovery efforts, which have led to the identification of novel drug candidates and translation into a few recent clinical trials for ALS. Finally, we discuss the current challenges in the field, and novel avenues for drug discovery and drug repurposing using these models.
Clinical and Epidemiological Features of Amyotrophic Lateral Sclerosis
Also known as Lou Gehrig’s disease, ALS was initially described in the scientific literature in 1869 by the French neurologist and anatomical pathologist Jean-Martin Charcot (Rowland, 2001; Kiernan et al., 2011; Katz et al., 2015). Clinically, ALS presents more commonly as a progressive muscular weakness leading to paralysis and death (Wijesekera and Leigh, 2009; Kiernan et al., 2011) due to the progressive degeneration of MNs in the motor cortex, brainstem and anterior horn of the spinal cord (Figure 1) (Rowland and Shneider, 2001; Kiernan et al., 2011; Leblond et al., 2014; Brown and Al-Chalabi, 2017). The ALS diagnosis is fundamentally clinical due to the lack of disease biomarkers, being common a 1–2 years delay between the initial symptoms and the final diagnosis (Rowland and Shneider, 2001; Ludolph, 2011; Brown and Al-Chalabi, 2017; Masrori and Van Damme, 2020). ALS seems to affect women and men equally and the risk of the disease increases with age, with an average age of onset around 62 years (Orrell, 2007; Chio et al., 2013). The incidence is roughly 2 per 100,000 persons per year, and the prevalence is around 6 per 100,000 persons (Worms, 2001; Dunckley et al., 2007; Orrell, 2007; Chio et al., 2013). The average cumulative risk of developing ALS during lifetime is nearly 1 to 350 in men and 1 to 400 in women (Hardiman et al., 2011; Ryan et al., 2019; Masrori and Van Damme, 2020). The time course of ALS can be markedly heterogeneous and complex, but the prognosis is poor for all patients, with death occurring within 3–5 years after the initial diagnosis (Rowland and Shneider, 2001; Wijesekera and Leigh, 2009; Brown and Al-Chalabi, 2017). ALS is an invariably fatal disease, with the median survival after symptom onset of 27.5 months and the 4-years survival rate is close to 40% (Hardiman et al., 2011; Su et al., 2014). Unfortunately, only nearly 20% of patients survive longer than 5 years (Cooper-Knock et al., 2014) and no more than 10% of patients are able to survive for more than 8 years (Hardiman et al., 2011; Kiernan et al., 2011).
For most of the ALS patients, the disease process starts in one area and spreads in an anatomically contiguous manner throughout the motor system (Ravits and La Spada, 2009; Kanouchi et al., 2012; Ravits, 2014; Takeda et al., 2020). This involves insidious progression to paralysis that initially starts in one limb (arm or leg) as progressive weakness (limb-onset) or the bulbar muscles, with speech and swallowing problems (bulbar-onset) (Ravits et al., 2007; Ravits and La Spada, 2009; Kanouchi et al., 2012; Brown and Al-Chalabi, 2017; Takeda et al., 2020). In extremely rare cases, the disease starts simultaneously in multiple areas or in the respiratory muscles (Ravits and La Spada, 2009; Kanouchi et al., 2012; Ravits, 2014; Takeda et al., 2020). Other unusual initial disease manifestations include weight loss, cramps, fasciculations without muscle weakness, emotional fluctuation and cognitive abnormalities, as well as, isolated respiratory failure (Brown and Al-Chalabi, 2017; Masrori and Van Damme, 2020; Norris et al., 2020). Upper MNs injury causes spasticity and brisk deep reflexes, whereas dysfunction of lower MNs leads initially to fasciculations (spontaneous muscle twitching); and, with ensuing degeneration, loss of synaptic connectivity with target muscles, which causes muscle wasting, weakness and atrophy (Brown and Al-Chalabi, 2017; Masrori and Van Damme, 2020; Norris et al., 2020). Numerous studies have demonstrated that different groups of MNs show differential vulnerability to neurodegeneration in ALS (Kanning et al., 2010; Saxena and Caroni, 2011; Ravits et al., 2013; Kaplan et al., 2014; Nijssen et al., 2017). The large alpha-MNs are the first to degenerate, and fast-twitch motor units are preferentially affected (Pun et al., 2006; Hegedus et al., 2007; Gordon et al., 2010; Kanning et al., 2010; Nijssen et al., 2017). Apparently, there is a gradient of vulnerability in the spinal cord motor neurons, since slower motor units become affected later in the disease comparatively to fast motor units (Nijssen et al., 2017). Consequently, the fast glycolytic muscles become paralyzed first, followed by the slow oxidative muscles (Nijssen et al., 2017). The presence of enhanced oxidative stress or higher energetic demands that are not fulfilled could help explaining the selective vulnerability of large MNs to neurodegeneration (Schmitt et al., 2014; Ioannides et al., 2016; Vandoorne et al., 2018). Interestingly, the MNs in the oculomotor (the third cranial nerve), trochlear (the fourth cranial nerve) and abducens (the sixth cranial nerve) nerves, which regulate eye movements; and those of the Onuf’s nucleus are resistant to degeneration in ALS (Mannen et al., 1977, 1982; Schroder and Reske-Nielsen, 1984; Gizzi et al., 1992; Kaplan et al., 2014; Nijssen et al., 2017). Studies employing omics analyses of MNs with distinct susceptibility to degeneration in ALS allowed to identify candidate genes/proteins that protect susceptible MNs. Thus, a protein signature for resistant oculomotor MNs has been established; it includes insulin-like growth factor 2 (IGF-2), GABAA receptor α1 (Gabra1), guanylate cyclase soluble subunit alpha-3 (Gucy1a3) and parvalbumin, whose selective expression in these unique neurons was consistently demonstrated through immunohistochemistry in both human and rodent samples (Hedlund et al., 2010; Comley et al., 2015). Candidate molecules selectively expressed by vulnerable MNs are numerous and include neuronal matrix metalloproteinase-9 (MMP-9) (Kaplan et al., 2014). Interestingly, the reduction of neuronal MMP-9 levels delayed muscle denervation and extended the survival of ALS mice, while the introduction of MMP9 was demonstrated to be sufficient to induce degeneration of fast MNs (Kaplan et al., 2014). In contrast, MMP-9 overexpression in resistant MNs did not enhance neuronal death, even in the presence of SOD1 (Kaplan et al., 2014). Together, these results highlight the role of MMP-9 in selective MN degeneration, through a mechanism initially involving activation of ER stress; and set forth MMP-9 inhibition as a promising therapeutic target in ALS (Kaplan et al., 2014). A recent study also suggests that the mechanism underlying MNs subtype vulnerability may be linked to proteostatic stress and ability to efficiently degrade SOD1 protein aggregates (An et al., 2019). The ongoing research efforts aiming at understanding more in-depth the selective vulnerability of MNs in ALS will generate invaluable knowledge on the requirements of MNs to keep themselves alive, and have the prospect to open novel avenues in therapeutic discovery for ALS.
The current lack of a definitive diagnostic test for ALS is one of the major hurdles in the field. Therefore, the ALS diagnosis is mainly based on the presence of indicative clinical signs coupled with negative laboratory tests and imaging studies to rule out other differential diagnosis (Baumer et al., 2014; Vucic et al., 2014; Brown and Al-Chalabi, 2017; Norris et al., 2020). Electromyography studies help to confirm the extent of denervation and constitute a relevant and helpful diagnostic tool (Ludolph et al., 2015; Brown and Al-Chalabi, 2017; Masrori and Van Damme, 2020). In order to make the diagnostic process less problematic and also to more accurately stratify patients for clinical research studies and clinical drug trials, the scientific community has put enormous efforts in the past decades to unify patient symptoms and signs, into a well-defined clinically entity (Wijesekera and Leigh, 2009). To this end, ALS researchers developed the El Escorial diagnostic criteria and the Arlie House criteria, which have been updated over time (Ludolph et al., 2015; Hardiman et al., 2017; van den Berg et al., 2019). In addition, based on comprehensive clinical information numerous ALS staging systems have been proposed. The Milano-Torino (MiToS) functional staging and King’s clinical staging systems are the most widely studied and previous studies demonstrated they could be complementary (Roche et al., 2012; Chio et al., 2015; Hardiman et al., 2017; Kiernan et al., 2020). The King’s clinical staging system is capable to differentiate early to mid-disease well, whereas the MiToS staging allows detailed differentiation in late ALS stages (Fang et al., 2017; Hardiman et al., 2017; Kiernan et al., 2020). Thus, researchers have proposed to use both in the evaluation of the ALS disease stage (Fang et al., 2017; Kiernan et al., 2020). Importantly, the ALS community has also attempted to develop robust models to better estimate the individual prognosis for ALS patients. In one recent successful attempt, a group of European researchers developed a solid prognostic model which was validated across 14 European ALS centers, and which was made freely available online for the usage by medical doctors (Mitsumoto, 2018; Westeneng et al., 2018). Eight powerful prognostic factors were identified and a comprehensive model was developed leading to five main different prognostic categories (very long, long, intermediate, short, and very short times) to the composite outcome (survival without tracheostomy or non-invasive ventilation for more than 23 h per day) were proposed (Westeneng et al., 2018). Robust and accurate models for disease prognosis prediction are of fundamental application in personalized ALS patient management and in the optimized design of clinical studies (Mitsumoto, 2018; Kiernan et al., 2020).
Less than 10% of ALS cases are considered ‘‘familial’’ (fALS) or inherited, with a familial genetic cause underlying (Brown and Al-Chalabi, 2017; Chia et al., 2018; Kim G. et al., 2020). These result from mutations inherited in a dominant monogenetic manner, but they can also be dominant with incomplete penetrance, recessive, and X-linked (Chia et al., 2018; Kim G. et al., 2020). The remaining majority of ALS cases have an idiopathic origin, hence they are considered ‘‘sporadic’’ (sALS), or presenting without a clear familial history (Chia et al., 2018; Kim G. et al., 2020; Shatunov and Al-Chalabi, 2021). Remarkably, the courses of fALS and sALS cannot be distinguished (Rowland and Shneider, 2001; Brown and Al-Chalabi, 2017). The extraordinary advances in genetics research over the past decades allowed the identification of mutations in nearly 30 genes which are linked with familial ALS, sporadic ALS, or both (Chia et al., 2018; Kim G. et al., 2020; Shatunov and Al-Chalabi, 2021). Generically, they can be grouped into three main categories: genes coding for proteins implicated in protein homeostasis [for example, superoxide dismutase 1 (SOD1) (Rosen et al., 1993), vesicle-associated membrane protein B (VAPB) (Nishimura et al., 2004), optineurin (OPTN) (Maruyama et al., 2010), valosin-containing protein (VCP) (Johnson et al., 2010), ubiquilin 2 (UBQLN2) (Deng et al., 2011) and sequestosome 1 (SQSTM1) (Rubino et al., 2012; Teyssou et al., 2013)], those involved in altered RNA homeostasis and trafficking [fused in sarcoma (FUS) (Kwiatkowski et al., 2009; Vance et al., 2009), TAR DNA binding protein (TARDBP/TDP-43) (Sreedharan et al., 2008) and Chromosome 9 open reading frame 72 (C9ORF72) (DeJesus-Hernandez et al., 2011; Renton et al., 2011)] and genes coding for cytoskeletal-related proteins [dynactin (DCTN1) (Puls et al., 2003), profilin (PFN1) (Wu et al., 2012) tubulin alpha 4a (TUBA4A) (Smith et al., 2014), and kinesin family member 5A (KIF5A) (Nicolas et al., 2018)], which further highlights the disease complexity and heterogeneity of potential pathophysiological mechanisms at play (Figure 2) (Grad et al., 2017; Chia et al., 2018; Kim G. et al., 2020).
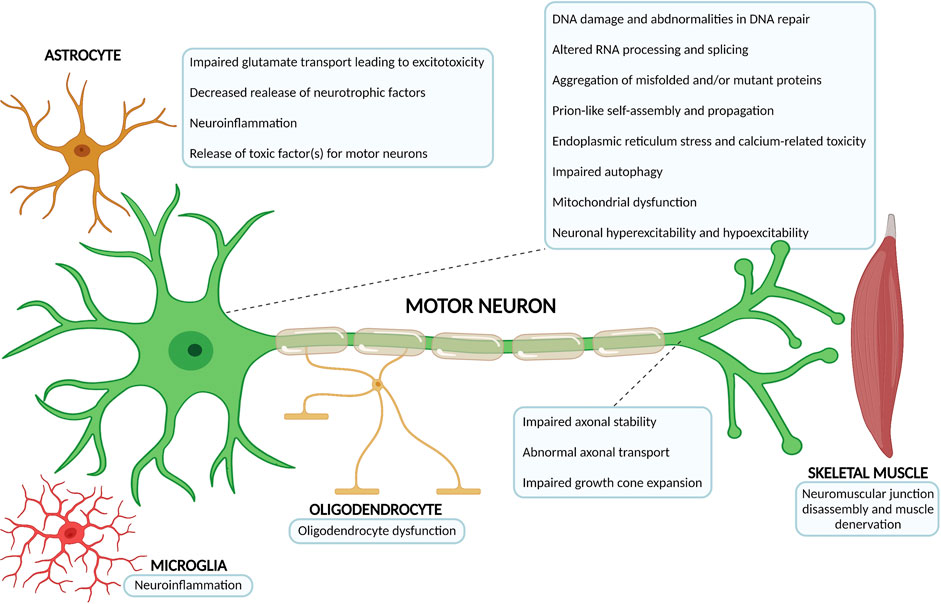
FIGURE 2. Overview of the most relevant ALS pathophysiological mechanisms. The complex and multifactorial neurodegenerative process that is at the core of ALS not only involves mechanisms of disease occuring within the motor neuron, but also pathophysiological processes that arise from the interaction with neighbouring partners, namely astrocytes, microglia, oligodendrocytes and skeletal muscle. The number of possible mechanisms and proteins associated with ALS onset has been growing considerably over the past decades (Diagram created with BioRender.com).
The SOD1 gene and the GGGGCC (G4C2) hexanucleotide repeat expansion in the C9ORF72 gene are the two most common genetic alterations linked with ALS (Chia et al., 2018; Mejzini et al., 2019). The SOD1 gene on chromosome 21 was the first gene whose mutations were linked with ALS, in 1993 (Rosen et al., 1993). Mutations in SOD1 are present in nearly 12% of the fALS cases and 2% of sALS cases (Chia et al., 2018; Kim G. et al., 2020). SOD1, which is an enzyme ubiquitously expressed, localizes to different cellular compartments, protecting cells from toxic reactive oxygen species (Kim G. et al., 2020). Over the past 30 years more than 180 mutations in the SOD1 gene have been identified (Mejzini et al., 2019). These are associated with diverse ALS clinical phenotypes and disease courses (Su et al., 2014; Mejzini et al., 2019). For example, the D90A (aspartic acid to alanine substitution in codon 90) SOD1 mutation is recessive and patients homozygous for this SOD1 variant are only mildly affected by the disease, with patient survival usually greater than 10 years (Andersen et al., 1996). Contrarily, the A4V (alanine to valine substitution in codon 4) SOD1 dominant mutation leads to a rapidly progressive form of ALS, with an average survival of only 1.4 years after the initial symptoms (Juneja et al., 1997; Su et al., 2014). The majority of studies in SOD1-ALS have demonstrated the presence of gain-of-function mutations (Chia et al., 2018; Kim G. et al., 2020). The discovery of SOD1 mutations in ALS originated the generation of the first transgenic animal models and in vitro cellular models of ALS, which have been instrumental to gain a more in-depth knowledge of the disease (Turner and Talbot, 2008; McGoldrick et al., 2013).
In the last decade, a novel major genetic ALS breakthrough was reported: the identification of a GGGGCC (G4C2) hexanucleotide repeat expansion in the C9ORF72 gene (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Chia et al., 2018; Kim G. et al., 2020). This unique mutation is present in nearly 40% of fALS cases and approximately 8–10% of sALS cases (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Chia et al., 2018; Kim G. et al., 2020). In addition, C9ORF72 expansions are also directly related to frontotemporal dementia (FTD) (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Kim G. et al., 2020; Yang et al., 2020). This helped to explain the recently acknowledged clinical overlap between ALS and FTD, which is present in 25% of familial FTD patients and up to 88% in familial ALS/FTD patients (Ji et al., 2017; Strong et al., 2017; Chia et al., 2018). In fact, up to 50% of ALS patients develop progressive cognitive abnormalities and behavioral changes, which ultimately lead to FTD, with autopsy evidence of neuronal loss in the frontal and temporal lobes (Saberi et al., 2015; Ji et al., 2017; Strong et al., 2017). Consequently, the understanding of ALS pathogenesis has increased tremendously with the discovery of C9ORF72 hexanucleotide expansions, since they are linked not only to ALS, but also to FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). ALS is a very complex neurodegenerative disorder, which possibly affects multiple organic systems, besides the neuromuscular axis (Cooper-Knock et al., 2014; Strong et al., 2017; Kim G. et al., 2020). However, the exact function of the C9ORF72 protein remains to be firmly established (Kim G. et al., 2020). Furthermore, the detailed mechanisms by which the C9ORF72 gene expansions lead to neurodegeneration are also not entirely understood, with three main prospective disease mechanisms currently being considered: gain-of-function mechanisms linked with repeat-RNA-mediated toxicity in the form of either RNA foci or other aberrant RNAs; production of toxic homo-polymeric dipeptide repeat proteins (DPRs) through RNA translation and loss of function of the C9ORF72 protein (Donnelly et al., 2013; Balendra and Isaacs, 2018; Kim G. et al., 2020). Further extensive studies are needed to better elucidate the disease mechanisms of C9ORF72 gene expansions in ALS and ALS/FTD (Balendra and Isaacs, 2018; Kim G. et al., 2020).
The ALS phenotype is highly heterogeneous, and to add further to the complexity of disease mechanisms (Figure 2), the interplay between genetic risks and the exposure to different environmental risk factors (for example, pesticides, lead or smoking, among others) cannot be ignored (Malek et al., 2014; Oskarsson et al., 2015; Koeman et al., 2017). For example, a recent study showed that there is also a complex interaction between exercise and ALS pathology, which could involve the Fibroblast Growth Factor (FGF) pathway, as a result of oxidative stress and hypoxia (Julian et al., 2021). Interestingly, modulation of the FGF pathway is important to lower glial reactivity (Savchenko et al., 2019) observed in ALS, and increase levels and activity of glutamate transporter GLT1/EAAT2, important to buffer glutamate-induced excitotoxicity (Figure 2) (Rothstein et al., 2005; Roybon et al., 2013). FGFs secretion by injured MNs could be a natural process to counteract glial reactivity and modulate glutamate transporter activity. Interestingly, FGF2 and FGF receptor 1 expression is preserved in different parts of the motor system in post-mortem tissue of ALS patients (Petri et al., 2009).
The complexity of ALS is also highlighted by the fact that different gene mutations can result in the same clinical phenotype, and different clinical phenotypes can be evident in the presence of the same gene mutation (Al-Chalabi et al., 2012; Grad et al., 2017; Takeda et al., 2020). The survival of ALS patients is also influenced by a myriad of factors, including the clinical phenotype; rate of disease progression; appropriate interventions to manage symptoms, including use of nasogastric feeding; nutritional status; prevention of aspiration (control of salivary secretions and application of cough-assist devices), and the specialized management of respiratory impairment (Jenkins et al., 2014; Vucic et al., 2014; Dorst et al., 2018; Chio et al., 2020; Norris et al., 2020). As far as pharmacological therapy is concerned, only two drugs are currently used for the treatment of ALS. The first one, Riluzole, started being used in ALS patients in 1995 (Bensimon et al., 1994; Norris et al., 2020). The drug prolongs the life of the patients by about 2–3 months when used in the standard dose of 2 × 50 mg per day (Miller et al., 2007; Miller et al., 2012; Dorst et al., 2018). Riluzole is approved by both the Food and Drug Administration (FDA) and European Medicines Agency (EMA). Even though Riluzole has been used for more than 2 decades, its precise mechanism of action remains elusive, with current evidence suggesting that the drug elicits the reduction of presynaptic glutamate release (anti-glutamatergic effect), which may be due to blockage of sodium or calcium channels, as well as, a decrease of persistent sodium currents (Bellingham, 2011; Dorst et al., 2018). The second drug recently approved is Edaravone. This drug was initially used in Japan to treat acute ischemic stroke (Edaravone Acute Infarction Study, 2003). Edaravone is an anti-oxidative stress agent currently in use in ALS patients in United States, Canada and Japan, but which is only available as a compassionate medication in the European Union (Dorst et al., 2018; Chio et al., 2020). In a selected group of ALS patients in early disease stage, the drug demonstrated a significantly smaller decline of the scores of the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) after 6 months of treatment (Writing and Edaravone, 2017). The long-term effects of Edaravone on ALS patients have not yet been fully evaluated; however, a Japanese study showed that the survival rate until the first endpoint (tracheostomy-free survival or death) was significantly improved in the Edaravone group (Okada et al., 2018). Therefore, despite decades of knowledge on the disease and extensive pre-clinical and clinical research, ALS is still incurable and the development of new disease-modifying therapeutic strategies is vital.
Neuropathological Features of Amyotrophic Lateral Sclerosis
The loss of anterior horn cells and sclerosis in the lateral columns of the spinal cord, which are the key macroscopic ALS neuropathological features, were initially described by Charcot in 1860s (Rowland, 2001; Katz et al., 2015). Indeed, there is characteristically atrophy of the anterior nerve roots and a reduction of the white matter especially in the corticospinal tract (Saberi et al., 2015). Furthermore, the degeneration of the corticospinal axons leads to thinning and scarring (sclerosis) of the lateral elements of the spinal cord (Rowland, 2001; Katz et al., 2015; Grad et al., 2017; Takeda et al., 2020). The progressive death of brain stem and spinal motor neurons will eventually give rise to the denervation-induced atrophy of the muscles (amyotrophy) (Figure 2) (Rowland, 2001; Katz et al., 2015; Takeda et al., 2020). Macroscopically, the brain of the majority of ALS patients is unremarkable, except for some cases which have atrophy of the precentral gyrus (Saberi et al., 2015). However, if the ALS patient also had dementia, it will be possible to observe atrophy of the frontal or temporal cortex, with these atrophic features being more pronounced in ALS/FTD cases (Boxer et al., 2011; Saberi et al., 2015).
Microscopically, the most distinctive features are a marked neuronal and axonal loss (Saberi et al., 2015; Takeda et al., 2020). Upon observation of routine hematoxylin-eosin (H&E) sections, depletion of large MNs in the anterior horn of the spinal cord, lower cranial motor brainstem nuclei, and Betz cells in the motor cortex (aspect originally described by Broadman, in 1909) can easily be identified (Saberi et al., 2015; Hardiman et al., 2017; Takeda et al., 2020). Moreover, with stains highlighting myelin (for example, luxol fast blue), reduction of myelinated axons in the lateral and anterior columns of the spinal cord, as well as, decreases in the dimension of the anterior horn of the spinal cord can be observed (Saberi et al., 2015; Spencer et al., 2020; Takeda et al., 2020). Remarkably, the neuropathological changes observed in the central nervous system of ALS patients go beyond these generic features. In fact, it also possible to observe in representative sections a sponge-like appearance given by neuronal vacuolization, empty spaces near neurons, microscopic holes and spongiosis (Saberi et al., 2015; Takeda et al., 2020). Another frequent and distinctive feature is the presence of 3–6 mm round to oval eosinophilic intracellular inclusions in the cytoplasm of MNs in the spinal cord and brain stem, which were initially described in the 1960s (Bunina, 1962; Saberi et al., 2015). These are Bunina bodies, which are best seen on H&E-stained tissue samples, and are present in both fALS and sALS patients (Saberi et al., 2015). Their presence in Betz cells, oculomotor nuclei neurons and Onuf nuclei has scarcely been reported (Saberi et al., 2015). The demise of MNs is paralleled by a neuroinflammatory process, with significant proliferation and activation of astroglia, microglia and oligodendroglia, along with the increasingly recognized altered function of immune cells in the CNS and peripherally (Figure 2) (Philips and Robberecht, 2011; Thonhoff et al., 2018; Chiot et al., 2019; Spencer et al., 2020).
A distinct neuropathological feature of ALS which has only more recently been unraveled, thanks to the advent of immunohistochemistry and other techniques, is the presence of ubiquitin-positive cytoplasmic inclusions (Saberi et al., 2015; Takeda et al., 2020). These were initially described in the 1980s, and they were later shown to be composed primarily by TDP-43 protein (Saberi et al., 2015; Takeda et al., 2020). The misfolding of TDP-43 protein is a significant ALS neuropathological feature, and aggregates are present in nearly 97% of the cases (Saberi et al., 2015; Hardiman et al., 2017; Takeda et al., 2020). For both ALS and ALS/FTD, TDP-43 aggregates are commonly found in neurons of the frontal cortex, temporal cortex, hippocampus and striatum, but they can occasionally been seen in glial cells (Arai et al., 2003). There are different kinds of TDP-43 inclusions, which include fine skeins, coarse skeins, dot-like and dense round inclusions (Saberi et al., 2015; Hardiman et al., 2017). Nevertheless, TDP-43 inclusions are far from being pathognomonic for ALS, since they can also be seen in other neurodegenerative diseases including Alzheimer’s disease (McAleese et al., 2017; Montalbano et al., 2020; Tome et al., 2020).
For each genetic form of ALS there are also additional and distinctive molecular features, which have begun to emerge and will be briefly mentioned here. For example, in SOD1-ALS, the anterior horn MNs also show inclusions composed by a hyalinized, poorly stainable substance, known as Lewy body–like inclusions (LBLIs) (Saberi et al., 2015). Using immunohistochemistry LBLIs were shown to stain for SOD1, ubiquitin, phosphorylated neurofilaments and different chaperone proteins, but which are negative for TDP-43 or phosphorylated TDP-43 (Okamoto et al., 2011; Saberi et al., 2015). In the C9ORF72 cases, most of the observed ubiquitinated inclusions are p62 positive, but negative for TDP-43 (Al-Sarraj et al., 2011; Saberi et al., 2015). Interestingly, most of these proteins (for example, p62, ubiquitin and HSP70) were found co-localized in LBLIs in a human cortical neuron-based model of alpha-synucleinopathy (Gribaudo et al., 2019). Furthermore, another signature of C9ORF72-ALS is the presence of foci of RNA of the expanded repeats, which is a feature of several of the repeat expansion diseases and can be detected by fluorescent in situ hybridization (FISH) (Zu et al., 2013; Cooper-Knock et al., 2015). These repeat expansions can hopefully be mitigated experimentally by antisense intervention (Donnelly et al., 2013; Riboldi et al., 2014). The ongoing and future studies in the field will help to elucidate the pathophysiological mechanisms leading to the neuropathological changes observed in ALS patients.
Drug Discovery Efforts in Amyotrophic Lateral Sclerosis Have Failed to Translate Into Clinically Applicable Therapeutic Strategies for Patients
The discovery of ALS pathophysiological mechanisms has been accelerated by the generation of several animal and in vitro cellular models ALS-linked, a great majority based on the ALS mutations identified over the past decades. These models have also allowed the testing of promising novel drugs that might change the course of the disease (Lanka and Cudkowicz, 2008; Turner and Talbot, 2008; Su et al., 2014; Philips and Rothstein, 2015; Lutz, 2018). Despite extensive research in the field over the years, an unifying model of the molecular mechanisms accounting for MN degeneration is still lacking, which explains in part why few therapeutic advances have been achieved so far, rendering ALS still incurable.
The most widely studied animal models of ALS are transgenic mice overexpressing mutant forms of the human SOD1 gene (Jackson et al., 2002; Pasinelli and Brown, 2006; Turner and Talbot, 2008; Van Den Bosch, 2011; Philips and Rothstein, 2015; Browne and Abbott, 2016; Lutz, 2018). In the past decade, several other animal models were developed, with mutations in C9ORF72, TARDBP, FUS, among other genes (Philips and Rothstein, 2015; Sharma et al., 2016; Batra and Lee, 2017; Lutz, 2018; Ebstein et al., 2019). Much knowledge on ALS pathology has been gained from studying transgenic mouse models. The mouse models have also served as the best accessible benchmark preclinical platforms to test numerous promising drug candidates. Over the past decades anti-epileptic compounds, antibiotics, anti-oxidants, anti-inflammatory drugs, anti-apoptotic small molecules and neurotrophic factors, among others were pre-clinically tested as promising ALS drugs (Aggarwal and Cudkowicz, 2008; Lanka and Cudkowicz, 2008; Turner and Talbot, 2008; Vincent et al., 2008; Mitsumoto et al., 2014; Philips and Rothstein, 2015). Unvaryingly, over the years, a myriad of promising drug candidates discovered using those animal models have failed to translate into relevant ALS therapies in human clinical trials (Ludolph and Sperfeld, 2005; Aggarwal and Cudkowicz, 2008; Turner and Talbot, 2008; Berry and Cudkowicz, 2011; Mitsumoto et al., 2014; Su et al., 2014; Petrov et al., 2017). This demonstrates a discrepancy between promising animal-based studies and a lack of therapies that are effectively translated into ALS patients (Benatar, 2007; Lanka and Cudkowicz, 2008; Turner and Talbot, 2008; Vincent et al., 2008; Mitsumoto et al., 2014; Petrov et al., 2017). Numerous possible explanations can elucidate this scenario. For example, it remains to be determined whether those animal models that pathocopy and phenocopy ALS can truthfully recapitulate both fALS and sALS pathogenesis, or whether they can only model certain features of the disease. It is also important to note that the great majority of ALS mouse models are created through expression of high copy numbers of a mutated gene (Benatar, 2007; Turner and Talbot, 2008; van der Worp et al., 2010; Mitsumoto et al., 2014; Moujalled and White, 2016). Indeed, robust animal models of sALS are very much needed. Furthermore, in most of the animal studies, the drugs are given before disease onset, a strategy which is not feasible in human clinical trials since relevant biological markers to identify patients with a high risk of developing ALS have not been found (Benatar, 2007; Aggarwal and Cudkowicz, 2008; Berry and Cudkowicz, 2011; Otto et al., 2012; Mitsumoto et al., 2014; Imamura et al., 2017; Petrov et al., 2017; Kiernan et al., 2020). In addition, there are important pharmacokinetic differences between rodents and humans, which makes it challenging to directly extrapolate the mouse dosages and pharmacokinetics to the ALS patients (Benatar, 2007; Lanka and Cudkowicz, 2008; Kiernan et al., 2020). Interestingly, the great majority of animal studies published until today have important methodological weaknesses and did not involve randomization and blindness to treatment (Benatar, 2007; Vincent et al., 2008; Kiernan et al., 2020). These are standard obligatory conditions for a well-conducted human clinical trial (Benatar, 2007; Vincent et al., 2008; Kiernan et al., 2020). Furthermore, there are important differences in the outcome measures in animal versus human studies and species-specific responses to cellular damage (Benatar, 2007; Kiernan et al., 2020). To curb the problem and improve the conduction and interpretation of animal model-based pre-clinical research in ALS, important guidelines were established in the last decade (Ludolph et al., 2010).
Lastly, the lack of success in the translation of novel ALS therapies into the clinic has also been linked with errors in clinical trial design, patient recruitment due to the lack of meaningful stratification, optimal drug dosage, control of the confounding effects of prescription and non-prescription drugs taken by ALS patients when undergoing a clinical trial, among others (Benatar, 2007; Berry and Cudkowicz, 2011; Mitsumoto et al., 2014; Petrov et al., 2017; Kiernan et al., 2020). In line with this, there are fundamental concerns regarding the potential interaction between a candidate drug undergoing a clinical trial and the concomitant usage of the standard Riluzole therapy (Mitsumoto et al., 2014). In addition, the clinical heterogeneity of ALS has also been neglected in clinical trial designs, leading to underpowered studies (Beghi et al., 2011; Goyal et al., 2020; Kiernan et al., 2020). Undeniably, studies involving a heterogeneous patient population in a context of a heterogeneous disease may mask the efficacy of certain drugs on a specific subset of patients, such as genetic forms of the disease or restricted phenotypes (Petrov et al., 2017; Goyal et al., 2020; Kiernan et al., 2020). For example, the positive effects of Edaravone were only initially demonstrated in a restricted group of patients (Writing and Edaravone, 2017). Interestingly, a post-hoc meta-analysis on the data gathered from ALS clinical trials involving lithium carbonate demonstrated that this drug was able to enhance the survival of ALS patients carrying UNC13A mutations, while it was not efficacious in the global ALS population (van Eijk et al., 2017). Furthermore, only SOD1-ALS patients seem to benefit from SOD1 antisense oligonucleotide therapy, arimoclomol and pyrimethamine (Lange et al., 2017; Benatar et al., 2018; Miller et al., 2020). Thus, better stratification of patients will help to efficiently direct therapeutics to the adequate ALS patient groups and disease subtypes, since certain therapeutics may only work on a given genetic or pathophysiologic ALS form rather than others (Haston and Finkbeiner, 2016; Goyal et al., 2020; Kiernan et al., 2020). In this respect, the development of accurate models for ALS diagnosis, progression prediction, patient stratification and treatment are much needed (Vihinen, 2017; Vihinen, 2020). To treat the constellation of different ALS patients, multiple different precision medicine approaches might also be required (Morgan et al., 2018; Goyal et al., 2020; Morello et al., 2020). Indeed, similarly to other diseases (for example, hypertension, cardiac insufficiency, chronic obstructive pulmonary disease, cancer, among others), it might be necessary to combine diverse medications to address different pathological mechanisms, in order to obtain meaningful ALS-modifying strategies. Importantly, those efficacious therapeutics will have to reach the CNS by crossing the blood-CNS barrier, which appears to be impaired in ALS patients, posing challenges to efficiently deliver drugs into the CNS (Garbuzova-Davis et al., 2016; Saul et al., 2020).
Altogether, these aspects have incited the search for optimized strategies to improve clinical trial outcomes and biomarkers in ALS, which shall lead to more personalized medicine approaches (Mitsumoto et al., 2014; Goyal et al., 2020; Kiernan et al., 2020). Accordingly, the EMA (EMA/531686/2015) and the FDA (FDA-2013-N-0035) proposed in the last years new guidelines for the conduction of clinical trials in ALS. Hopefully, we will witness in the coming years improved therapeutic effects and improved success rates of treatments in better stratified ALS clinical trials.
Generation of Human Motor Neurons From Pluripotent Stem Cells
The study of human MNs is a valid and invaluable alternative to the classical animal-based ALS studies (Han et al., 2011; Haston and Finkbeiner, 2016; Karagiannis et al., 2019). Though, for ethical and technical reasons studies involving human MNs retrieved from patients are not feasible (Palmer et al., 2001; Han et al., 2011). Besides, until recently, human MNs from ALS patients were only accessible post-mortem when pathological and adaptive cellular and molecular mechanisms are advanced (Han et al., 2011; Sances et al., 2016). Thus, the outstanding capacity to generate relevant human neural cell types from pluripotent stem cells – both ESCs and iPSCs, has opened unprecedented paths towards the understanding of ALS and other MN disorders (Han et al., 2011; Bellin et al., 2012; Haston and Finkbeiner, 2016). Regarding therapeutic development, it is also anticipated that by studying human cells and performing drug tests directly on them, the time of translation of interesting pre-clinical research findings towards clinical applicable strategies could be considerably shortened (Figure 3) (Ebert and Svendsen, 2010; Engle and Puppala, 2013; Engle and Vincent, 2014; Haston and Finkbeiner, 2016; Lee et al., 2018).
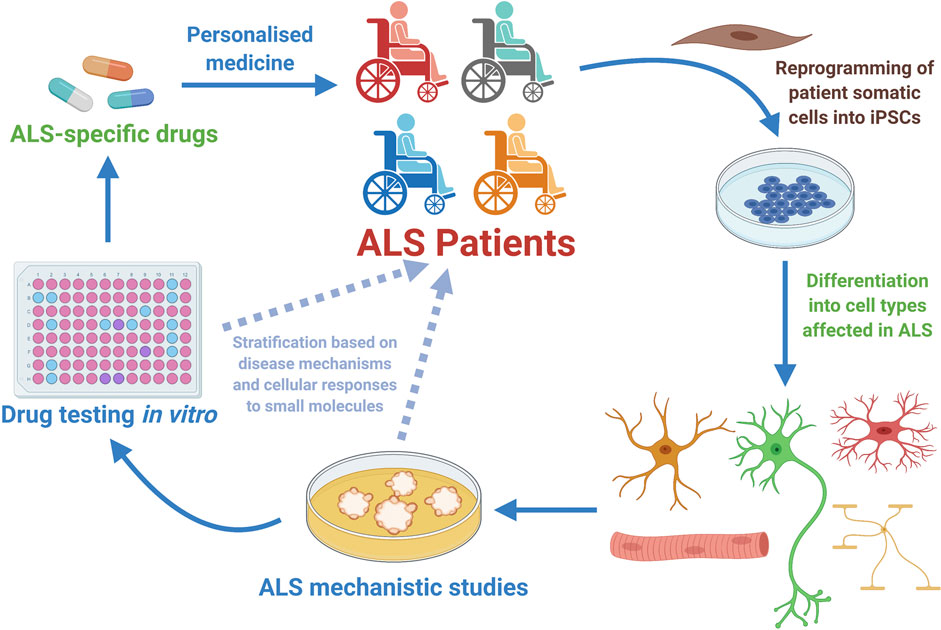
FIGURE 3. Human motor neurons generated from Amyotrophic Lateral Sclerosis (ALS) patients have allowed disease mechanistic studies and in vitro drug screening campaigns, with already some candidate compounds being identified and tested in the clinical setting (Table 1). ALS patient-specific iPSCs can be generated through reprogramming of somatic cells harvested from patients. Once generated, the ALS hiPSCs can be efficiently differentiated into the specific cell types involved in the disease (MNs, glial cells or sketelal muscle). Those cells can be utilized in vitro for disease mechanistic studies and massive efforts of drug testing. In the particular case of hiPSC-derived MNs, it is anticipated that these cells might as well be employed in patient stratification strategies, and also in better selecting at the laboratory stage the list of most promising small molecules that will be further tested in clinical studies involving ALS patients. Therefore, hiPSC-derived MNs constitute a powerful tool for personalized medicine approaches that start at the clinic, being then further developed in the laboratory and hopefully translated back to the ALS patient as a personalized effective therapeutic strategy (Diagram created with BioRender.com).
The development of studies involving human MNs generated from pluripotent stem cells is directly linked with the advances in the successful utilization of ESCs for medical research. The ESCs are a unique group of cells isolated from the inner cell mass (ICM) of developing blastocysts which exhibit unique properties of self-renewal (capacity to generate identical stem cells by cell division) and pluripotency [the ability to differentiate into all three embryonic germ layers (endoderm, ectoderm and mesoderm)] (Evans, 2005; Zwaka and Thomson, 2005; Yu and Thomson, 2008; Evans, 2011). Since their initial use in research, because they can potentially originate any mature cell type in the body, they have been viewed as an exciting tool to unravel the mechanisms of developmental biology, for prospective cell replacement therapies and for drug discovery studies (Evans and Kaufman, 1981; Martin, 1981; Lerou and Daley, 2005; Gokhale and Andrews, 2009; Evans, 2011). The first ESCs were derived from mouse embryos in the early 1980s (Evans and Kaufman, 1981; Martin, 1981). The first meaningful attempts to isolate and culture in vitro hESCs was reported more than 1 decade later, with human fallopian tube cells being used as system to grow them undifferentiated (Bongso et al., 1994). However, the cells could maintain a pluripotent state for only two passages (Bongso et al., 1994). In 1998, Thomson and collaborators made a long-desired breakthrough in the field of stem cell biology, by reporting a novel robust strategy to isolate and culture hESCs that involved the usage of a supporting monolayer of mitotically inactive mouse embryonic fibroblast feeders, to guarantee proliferation in undifferentiated state for up to 4 months in vitro (Thomson et al., 1998). The hESC lines were generated after isolation of the inner cell mass of blastocysts, a procedure which almost inevitably causes the destruction of the fertilized human embryo, leading to unending ethical discussions (Thomson et al., 1998; Engels, 2002; de Wert and Mummery, 2003; Landry and Zucker, 2004; Walters, 2004; Gavrilov et al., 2009; Taylor, 2011). Although mouse ESCs and hESCs are considered fundamentally comparable in their unlimited capacity to originate any cell type of the three embryonic germ layers, they have relevant differences in colony morphology, expression of surface markers, growth factor requirements for self-renewal and pluripotency maintenance, epigenetic profile and resistance to apoptosis upon single cell dissociation (Sato et al., 2003; Ginis et al., 2004; Gokhale and Andrews, 2009; Schnerch et al., 2010; Ohgushi and Sasai, 2011).
For the first time, in early 2000’s, Wichterle and collaborators demonstrated that mouse ESCs could be robustly differentiated towards a specific spinal cord MN fate using the well-known spinal cord developmental cues retinoic acid (RA) and sonic hedgehog (SHH) (Wichterle et al., 2002). After an initial period of expansion, the mESCs were allowed to differentiate by growing as free-floating aggregates designated as embryoid bodies (EBs) (Wichterle et al., 2002). To mimic the established in vivo mouse motor neurogenic period, both RA and SHH were applied in vitro in a logical and defined temporal window (Wichterle et al., 2002). The RA was initially used to drive neuroectodermal cells towards a spinal cord identity (neuralization followed by caudalization) (Wichterle et al., 2002). Then, SHH was used to commit the previously caudalized prospective progenitor cells towards a MN lineage (ventralization) (Wichterle et al., 2002). A culture period of 7 days was sufficient to obtain an enriched population of neuronal cells positive for MN and pancreas homeobox 1 (MNX1), also known as Homeobox HB9 (HLXB9); the insulin gene enhancer protein ISL-1, also known as the ISL LIM Homeobox 1 (ISL1); and choline acetyltransferase (ChAT) (Wichterle et al., 2002). These mESCs-derived MNs (mESC-MNs) shared many of the well-known molecular characteristics of spinal MNs (Wichterle et al., 2002). Interestingly, once they were successfully transplanted into the developing chick embryonic neural tube, they were capable to both integrate in the ventral horn of the spinal cord and also to project axons to muscle targets (Wichterle et al., 2002). Additional studies also allowed to demonstrate that in vitro-generated mESC-MNs could recapitulate functional properties displayed by embryonic MNs in vivo (Miles et al., 2004). In addition, the mESC-MNs were shown to contain properly functioning receptors for excitatory and inhibitory neurotransmitters and, thus, develop adequate electrophysiological properties by producing the typical firing patterns (Miles et al., 2004). Finally, when cultured in vitro with C2C12 myotubes, they were able to establish functional cholinergic synapses (Miles et al., 2004).
This work inspired the development of protocols to efficiently generate MNs from hESCs with the first successful attempts being reported in 2005 (Li et al., 2005; Singh Roy et al., 2005). However, the time required to generate hESC-MNs expressing HB9, ISL1 and ChAT induced by RA and SHH was five times longer when compared to mESC-MNs (Restagno et al., 2005; Singh Roy et al., 2005). Similarly to mESC-MNs, the hESC-MNs were shown to harbour normal electrophysiological activity and, once co-cultured with muscle cells in vitro, develop functional synapses (Restagno et al., 2005; Singh Roy et al., 2005). Since these early days, numerous protocols to improve the efficiency of MN generation from pluripotent stem cells were developed and published (Faravelli et al., 2014; Sances et al., 2016). In the majority of the proposed protocols, three fundamental steps in MN differentiation comply with the initial methodology established by Wictherle and colleagues: neuralization, followed by caudalization and then ventralization (Wichterle et al., 2002). Neuralization is currently most commonly performed by dual-SMAD signalling pathway inhibition, using the chemical compound SB431542, a potent inhibitor of transforming growth factor (TGF)-beta type I receptor/ALK5, ALK4 and ALK7; and either the noggin protein [inhibitor of several bone morphogenic proteins (BMPs)] or the small molecule LDN193189, a derivative of dorsomorphin which is a highly selective antagonist of BMP receptor ALK2 and ALK3 (Chambers et al., 2009; Roybon et al., 2013; Sances et al., 2016). The caudalization stage is directed by RA and together with Wnt activation via inhibition of glycogen synthase kinase 3 (GSK-3) using the compound CHIR-99021 (Maury et al., 2015; Sances et al., 2016). The ventralization process relies on the adequate hedgehog signalling, using the recombinant SHH protein or agonists of the hedgehog receptor smoothened (SMO), purmorphamine and/or SAG (Faravelli et al., 2014; Sances et al., 2016). Nevertheless, the exact mechanisms that lead to the generation of different subtypes of MNs have only recently started to be understood (Peljto et al., 2010; Amoroso et al., 2013; Maury et al., 2015; Allodi et al., 2019; An et al., 2019). A more in-depth knowledge of these mechanisms will allow us to robustly generate in vitro all the different subtypes of MNs present in the human spinal cord, especially the entire set of thoracic and lumbar MNs (Sances et al., 2016). In line with this, another fundamental challenge is that we are still unable to consistently generate pure populations of MNs from pluripotent stem cells (Sances et al., 2016). Indeed, these MN cultures also contain a myriad of other neural related cells including glia (Lamas et al., 2014; Sances et al., 2016). This has led to enormous challenges using these cultures, especially in MN survival studies, since ongoing neurogenesis occurs, leading to important confounding effects (Lamas et al., 2014). Using laborious and expensive methodologies this problem can, however, be efficiently circumvented using cell sorting strategies [for example, employing fluorescence activated cell sorting (FACS)] and adequate culturing conditions (Lamas et al., 2014; Garcia-Diaz et al., 2020).
The stem cell scientific community always aimed to robustly generate patient-specific pluripotent stem cells, due to the far-reaching therapeutic and regenerative possibilities offered by cells with such unique characteristics (Kondo et al., 2017; Readhead et al., 2018; Laperle et al., 2020; Schweitzer et al., 2020; Yamanaka, 2020). On one hand, the chances of rejection would be theoretically decreased if regenerative strategies were attempted using cells and tissues that immunologically match the donor by being generated from personalized stem cells (Hallett et al., 2015; Karagiannis et al., 2019; Schweitzer et al., 2020; Yamanaka, 2020). On the other hand, personalized stem cells could permit the study of any cell type with the genetic background of the donor, opening novel avenues towards in vitro patient-specific studies of disease mechanisms and also patient-directed drug therapies, making the vision of personalized medicine a closer reality (Figure 3) (Bellin et al., 2012; Haston and Finkbeiner, 2016; Stern et al., 2018b; Karagiannis et al., 2019; Yamanaka, 2020). In 2006, this long-aimed scenario became a real possibility with the breakthrough successful generation of iPSCs by the team of Shinya Yamanaka (Takahashi and Yamanaka, 2006). In a set of well-thought and planned experiments, they initially screened 24 genes that were linked to pluripotency, to demonstrate later that the combined overexpression of only four factors [octamer 3/4 (OCT3/4), sex determining region Y-box 2 (SOX2), kruppel-like factor 4 (KLF4) and cellular myelocytomatosis oncogene (c-MYC)] was capable to transform a fibroblast into a pluripotent stem-cell like cell that could be efficiently and endlessly grown in vitro as small colonies (Takahashi and Yamanaka, 2006). The newly generated iPSCs not only annihilated one of the core dogmas of cell biology, which was that a cell could not go back to a pluripotent stem cell stage once differentiated; but also paved the way for the development of easy-to-use and robust methods to generate the long-aimed-for individual-specific pluripotent stem cells (Takahashi and Yamanaka, 2006; Yamanaka, 2020). One year later, this methodological approach was promptly applied to human fibroblasts leading to the historic generation of hiPSCs by two independent research teams (Takahashi et al., 2007; Yu et al., 2007). While the Yamanaka team followed an analogous methodology and reprogrammed human adult dermal fibroblasts by applying the same four factors (Takahashi et al., 2007); that of Thomson reported the generation of hiPSCs from embryonic fibroblasts and postnatal foreskin fibroblasts by using a lentiviral system to overexpress the transcription factors OCT4, SOX2, NANOG and LIN28 (Yu et al., 2007). A new era in stem cell biology was initiated with these two milestone studies, which further triggered a myriad of follow-up studies. First, researchers aimed to demonstrate that other human cell types could as well be reprogrammed into iPSCs (Stadtfeld and Hochedlinger, 2010; Robinton and Daley, 2012). Soon, numerous studies were reporting the generation of hiPSCs from peripheral blood monocytes, keratinocytes, adipose-derived stem cells, hepatocytes, urothelial cells, among others (Stadtfeld and Hochedlinger, 2010; Pan et al., 2012; Robinton and Daley, 2012; Liu et al., 2020). Second, numerous studies have also aimed to investigate novel methods to generate hiPSC lines in a more efficient and safer manner because the initially proposed reprogramming methods had low efficiency and involved the usage of two oncogenic transcription factors (c-MYC and KLF4), and transduction using viruses integrating the genome to overexpress the four transcription factors (Gonzalez et al., 2011; Robinton and Daley, 2012; Liu et al., 2020). In the meantime, others researchers have compared hESCs and hiPSCs properties to confirm or refute their biological equivalence, whereas other groups have studied the mechanisms underlying reprogramming (Amabile and Meissner, 2009; Narsinh et al., 2011; Bilic and Izpisua Belmonte, 2012; Christodoulou and Kotton, 2012; Puri and Nagy, 2012; Robinton and Daley, 2012; Cahan and Daley, 2013). Unsurprisingly, the advent of hiPSCs was also followed by a scientific boom in research applied to specific diseases, aiming to develop in vitro models of those diseases (Grskovic et al., 2011; Han et al., 2011; Tiscornia et al., 2011; Robinton and Daley, 2012; Karagiannis et al., 2019; Chang et al., 2020). Regarding NDs, hiPSCs have made possible the generation of the diverse neuronal cell types, which were previously not easily accessible, carrying the different genetic traits linked with the deterioration of the CNS environment of a given individual (Han et al., 2011; Faravelli et al., 2014; Sances et al., 2016; Chang et al., 2020).
The first hiPSCs derived from an ALS patient were originally reported in 2008 by Dimos and collaborators (Dimos et al., 2008). In their milestone study, not only the authors demonstrated for the first time the differentiation of MNs from hiPSCs, but also they successfully reprogrammed fibroblasts from aged ALS patients into iPSC with the capacity to differentiate into spinal cord MNs using a protocol that was very similar to the one previously described to generate hESC-MNs (Dimos et al., 2008). Accordingly, the generation of patient-specific ALS-hiPSCs and the ability to differentiate in vitro human MNs harbouring the genetic background of the original patient, immediately opened the prospect to generate models of human MN diseases in vitro, despite their embryonic features (Dimos et al., 2008; Ho et al., 2016; Sances et al., 2016). Since the pioneer study of Dimos and colleagues, numerous research teams have generated different ALS patient-derived hiPSC lines, taking advantages of the more efficient protocols to generate human MNs (Boulting et al., 2011; Faravelli et al., 2014; Li et al., 2015; Sances et al., 2016). This allowed to conduct relevant in vitro disease modelling studies to reveal early and late phenotypic alterations in the MNs, many of them mimicking those identified in ALS patient post-mortem tissue (Vasques et al., 2020). The most significant cellular changes reported in ALS patient iPSCs-derived MNs (from SOD1, C9ORF72, TARDBP, FUS, VAPB and sporadic cases) comprise DNA damage and abnormalities in DNA repair, reduced cell viability, increased susceptibility to different stressors, abnormalities in neuronal morphology, presence of protein aggregates, mitochondrial alterations and electrophysiological changes, among others (Figure 2) (Mitne-Neto et al., 2011; Burkhardt et al., 2013; Sareen et al., 2013; Chen et al., 2014; Kiskinis et al., 2014; Wainger et al., 2014; Devlin et al., 2015; Lopez-Gonzalez et al., 2016; Naujock et al., 2016; Bhinge et al., 2017; Imamura et al., 2017; Wang et al., 2017; Fujimori et al., 2018; Naumann et al., 2018; Selvaraj et al., 2018; Seminary et al., 2018; Shi et al., 2018; Bursch et al., 2019; Keskin et al., 2019; Kim B. W. et al., 2020; Hawrot et al., 2020; Vasques et al., 2020). For example, the hiPSC-derived MNs have been instrumental to better understand the role of DNA damage in ALS pathogenesis, which is triggered by different routes, including oxidative stress linked with impaired anti-oxidative mechanisms (Kim B. W. et al., 2020). In a recent study, different forms of DNA damage were observed in postmortem CNS tissue from ALS patients, both in upper MNs of the motor cortex and lower spinal cord MNs (Kim B. W. et al., 2020). Through the study of some DNA damage repair (DDR) mechanisms, it was possible to demonstrate an apparently intact DDR response, also evident in human ALS iPSC-derived motor neurons harbouring SOD1 mutations, which displayed a robust DDR response equivalent to wild-type MNs (Kim B. W. et al., 2020). Mechanisms of DNA damage have also been studied in FUS ALS-hiPSC derived MNs, which were shown to have mislocalization of the cytoplasmic RNA/DNA-binding protein FUS and to develop FUS-positive inclusions, which correlated well with ALS severity (Higelin et al., 2016). FUS ALS-hiPSC derived MNs were shown to accumulate foci of DNA damage (Higelin et al., 2016) and have abnormalities in DNA nick ligation and oxidative damage repair (Wang et al., 2018). New experiments are needed to further assess the mechanisms of DNA damage and altered DNA repair related with MN degeneration and also evaluate the quality and effectiveness of repair mechanisms evidenced by ALS hiPSC-derived MNs.
Hence, hiPSCs allow the generation in vitro of infinite quantities of different neuronal cell types that are patient-specific, enabling the study of early cellular dysfunction and other neurodegenerative processes (Figure 2), otherwise difficult to observe in post-mortem tissues. Finally, these patient cell-based innovative in vitro ALS models have the prospect to help unravel novel pathogenic mechanisms, and to evaluate the utility of new therapies, opening promising avenues towards the discovery of efficacious pharmacological agents to halt the progression or even cure ALS (Figure 3) (Sances et al., 2016; Lee et al., 2018; Hawrot et al., 2020; Okano et al., 2020; Vasques et al., 2020).
Using Human Motor Neurons Generated From Pluripotent Stem Cells to Perform in Vitro Drug Testing
The usage of patient-specific ALS hiPSC-derived MNs has led to innovative in vitro disease models and also to drug screening campaigns, some of which involve drug repurposing (Haston and Finkbeiner, 2016; Lee et al., 2018; Hawrot et al., 2020). In the last decade, hiPSC-derived MNs have allowed the identification of several promising compounds to tackle ALS (Wainger et al., 2014; McNeish et al., 2015; Imamura et al., 2017; Fujimori et al., 2018; Lee et al., 2018; Okano et al., 2020). Among the candidate compounds identified, a few have already been translated into drug testing involving ALS patients, in well-designed clinical trials (Table 1). The first of these therapeutic clinical candidates, identified in 2015, was the Kv7 channel activator Retigabine/Ezogabine, a known anti-epileptic drug (Wainger et al., 2014; Wainger et al., 2020). Neuronal hyperexcitability is a significant pathophysiological mechanism in ALS (Vucic et al., 2008; Fogarty, 2018; Huang et al., 2021). Through electrophysiological analysis using multielectrode arrays, the Eggan team demonstrated that Retigabine/Ezogabine was capable of suppressing the hyperexcitability of ALS iPSC-derived MNs (Wainger et al., 2014). In brief, spontaneous neuronal excitability of MNs derived from fALS-SOD1A4V hiPSCs was initially demonstrated to be increased (Wainger et al., 2014). This was linked with reduced delayed-rectifier K+ current amplitudes in patient-derived MNs comparatively to control MNs, leading possibly to neuronal hyperexcitability (Wainger et al., 2014). The application of Retigabine/Ezogabine, which is a potent K+ channel activator that causes membrane hyperpolarization, blocked hyperexcitability and enhanced the in vitro survival of hiPSC-derived MNs from fALS patients with mutations in SOD1 or FUS, and C9ORF72 repeat expansions (Wainger et al., 2014). On this basis, a phase 2 randomized clinical trial involving 65 patients was conducted to assess the efficacy of Retigabine/Ezogabine on central and peripheral nerve excitability in ALS (ClinicalTrials.gov: NCT02450552) (Wainger et al., 2020). Participants were treated with 600 mg/day or 900 mg/day of Retigabine/Ezogabine or a matched placebo for 10 weeks (Wainger et al., 2020). The drug was well-tolerated, similarly to studies involving epilepsy patients; and decreased cortical and spinal MN excitability in the involved ALS patients was demonstrated (Wainger et al., 2020). However, it remains to be determined whether a similar treatment for a longer period can endure the effects on excitability and halt disease progression (Wainger et al., 2020). Interestingly, the data obtained in this clinical trial are in line with the findings gathered during pre-clinical studies involving ALS hiPSC-derived MNs, reinforcing the idea that patient hiPSC-based in vitro models are suitable to identify novel disease relevant targets and to quickly help translating basic research findings into clinically testable strategies (McNeish et al., 2015; Hawrot et al., 2020; Okano et al., 2020).
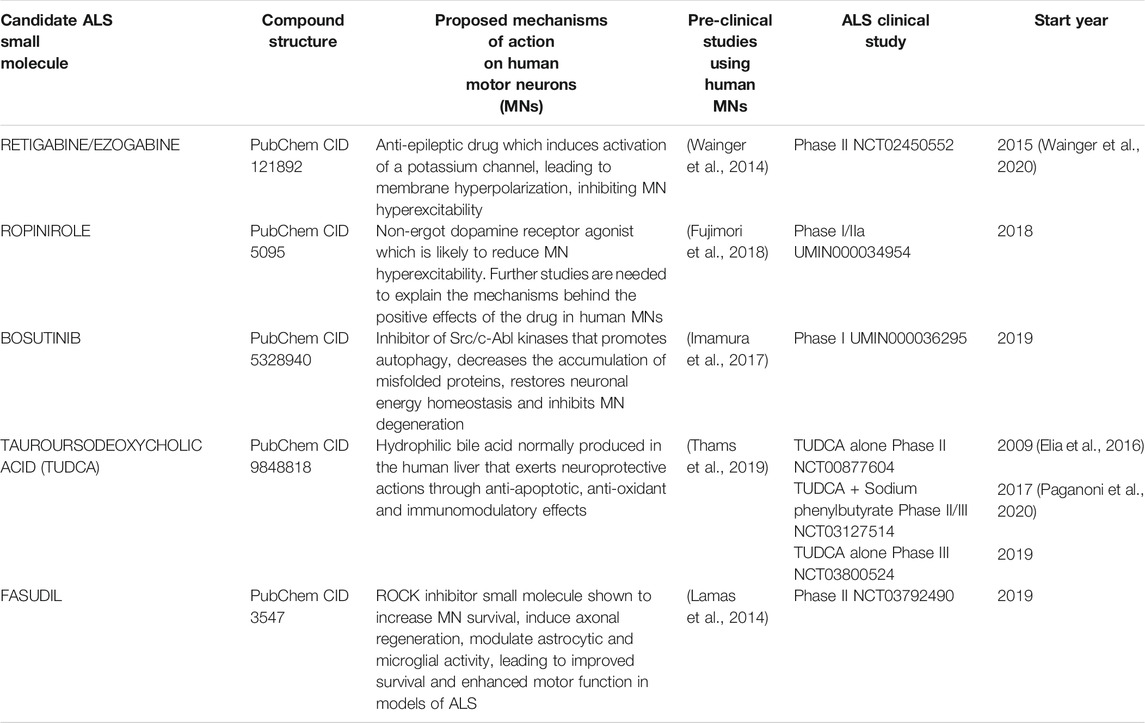
TABLE 1. Summary table of clinical studies involving ALS patients and employing small molecules with pre-clinical supportive data comprising drug testing in human pluripotent stem cell-derived motor neurons (hPSC-MNs).
A second drug entering ALS clinical testing following its discovery after drug screening involving ALS hiPSC models is Ropinirole, which was identified by the Okano group (Fujimori et al., 2018). A panel of 1232 FDA-approved drugs was tested in vitro in FUS and TDP-43 (TARDBP) fALS iPSC-derived MNs, for their capacity to revert established ALS-related phenotypes, namely MN death/damage, neurite retraction, mislocalization of FUS/TDP-43 and stress granule formation (Fujimori et al., 2018). All the compounds were tested on selected fALS models and nine drugs were identified as top candidates (Fujimori et al., 2018). Following enriched gene ontology terms and transcripts pathways analysis, detailed drug information regarding permeation through the blood-brain barrier (BBB), presence of serious side effects, and dose-response relationships, the authors selected Ropinirole as the most promising small molecule candidate (Fujimori et al., 2018). Ropinirole is a non-ergot dopamine receptor agonist employed in the treatment of Parkinson’s disease (Okano et al., 2020). The positive action of Ropinirole in ALS MNs is not fully understood yet, but it was proposed to be linked with the reduction of toxic neuronal hyperexcitability via Dopamine D2R activation (Okano et al., 2020). Other studies have also recently identified D2 dopamine receptors as significant modulators of ALS MN excitability (Huang et al., 2021). Interestingly, the beneficial effects of Ropinirole were also identified in non-SOD1 sALS MNs, but not in SOD1-mutant ALS models (Fujimori et al., 2018). Following these promising results, the ROPALS phase I/IIa clinical trial [UMIN Clinical Trials Registry (UMIN-CTR): UMIN000034954] started in Japan in 2018, involving 15 ALS patients treated with oral Ropinirole up to 16 mg/day and five ALS patients submitted to placebo (Okano et al., 2020). This first clinical trial aims to evaluate the safety, tolerability and efficacy of Ropinirole, as measured by delay in the progression of ALS, for an initial 24 weeks (double-blind phase) and ensuing 24 weeks (open-label continuation phase) (Okano et al., 2020). The results of this trial have not been published yet.
The third drug identified in a high-throughput screening campaign involving ALS hiPSC-derived MNs is Bosutinib, a BCR-ABL and src tyrosine kinase inhibitor employed to treat chronic myelogenous leukemia patients (Imamura et al., 2017). The study was conducted by the Inoue research team and involved the testing of 1,416 compounds (including several FDA and EMA approved drugs), at 10 µM concentration, in fALS-SOD1 patient iPSC-derived MNs to assess their ability to enhance neuronal survival beyond 7 days in culture (Imamura et al., 2017). The screening led to the initial identification of 27 hit compounds, with 14 of them targeting the Src/c-Abl signalling pathway (Imamura et al., 2017). Further experiments showed Bosutinib as the most promising compound (Imamura et al., 2017). The inhibition of Src/c-Abl kinases was demonstrated to promote autophagy, reduce the amount of misfolded SOD1 protein, restore energy homeostasis and rescue ALS MN degeneration (Imamura et al., 2017). Furthermore, Bosutinib also had a positive effect on TDP-43 ALS and C9ORF72 ALS hiPSC-derived MNs (Imamura et al., 2017). Interestingly, Bosutinib was also tested in an ALS animal model, prolonging their survival by 7–8 days (Imamura et al., 2017). After these encouraging results a phase I clinical trial of the drug Bosutinib for ALS started in Japan, in 2019 [UMIN Clinical Trials Registry (UMIN-CTR): UMIN000036295]. The main objective of the study is to evaluate the safety and tolerability of Bosutinib (100 mg/day, 200 mg/day, 300 mg/day, or 400 mg/day) to define the maximum tolerated dose (MTD) and a recommended phase 2 dose to treat ALS patients (Okano et al., 2020). Three to six ALS patients will be enrolled in each of the four planned Bosutinib dose levels (Okano et al., 2020). The study involves a 12-weeks observation period, a 1-week (5–9 days) transitional period, a 12-weeks study treatment period, and a 4-weeks follow-up period (Okano et al., 2020). The results of this trial are not yet publicly available.
Other studies of the past decade captured the attention of the scientific community. Among them are three studies, independently led by the research teams of Rubin (Yang et al., 2013) and those of Wichterle and Henderson (Lamas et al., 2014; Thams et al., 2019). In the first study, researchers employed wild-type mESCs and mESC harbouring the SOD1G93A transgene, also carrying the expression of the green fluorescent protein (GFP) under the control of the MN-specific promoter HB9. The produced HB9::GFP-positive MNs were used to develop a MN survival assay based on neurotrophic factor deprivation in line with previous studies (Bordet et al., 2007). The survival assay was employed in a drug screening campaign involving nearly 5,000 compounds (Yang et al., 2013). Out of the several hits identified, the small molecule Kenpaullone had the most neuroprotective effect (Yang et al., 2013). Kenpaullone, which is an ATP-competitive inhibitor of glycogen synthase kinase 3β, besides inhibiting other kinases, was further explored in that landmark study (Kim and Lee, 2013; Yang et al., 2013). The drug demonstrated to enhance the survival of wild-type and ALS hiPSC-derived MNs, leading the authors to propose the HGK-Tak1-MKK4-JNK-c-Jun cell death signalling cascade as a potential ALS therapeutic target (Yang et al., 2013). This pioneer study represented one of the first successful attempts to use patient iPSC-derived MNs to validate candidate drugs to treat MN disorders (Yang et al., 2013). Despite not showing ALS disease specific phenotypes in human MNs, their approach was also a remarkable example of preclinical testing using human MNs “in the dish” (Figure 3), since they tested other candidate drugs like Dexpramipexole, which had promising results in ALS mouse models, but that later failed in ALS clinical trials (Cudkowicz et al., 2011; Yang et al., 2013). In line with the clinical trial results, Dexpramipexole was unable to increase the survival of human MNs carrying SOD1 mutations, further suggesting that in vitro studies involving hiPSC-derived MNs could be used to conduct patient stratification and also refine the list of candidate drugs that will be tested in ALS clinical trials (Figure 3) (Yang et al., 2013; McNeish et al., 2015; Haston and Finkbeiner, 2016; Fujimori et al., 2018). The study by Yang and colleagues has stimulated follow up studies which aimed to identify blockers of the HGK-Tak1-MKK4-JNK-c-Jun pathway (Bos et al., 2019).
The second study employed an innovative dual-color mESC-derived MN co-culture assay to evaluate 1,300 compounds (Thams et al., 2019). The co-culture assay [with equal numbers of HB9::red fluorescent protein (RFP)-hSOD1 WT MNs displaying red colour under fluorescent light and HB9::green fluorescent protein (GFP)-hSOD1 G93A MNs displaying green colour under fluorescent light] allowed the researchers to identify Cyclopiazonic acid (CPA), an inducer of endoplasmic reticulum stress, as a molecule prompting preferential accelerated degeneration of hSOD1 G93A mutant MNs, comparatively to hSOD1 WT MNs (Thams et al., 2019). Later, in a secondary screening effort to find compounds that protected MNs against CPA-induced degeneration, the authors identified numerous candidate neuroprotective compounds, including Kenpaullone and Tauroursodeoxycholic acid (TUDCA) (Thams et al., 2019). TUDCA is a drug in clinical use that has also captured the attention of the ALS community in the past decade. TUDCA is a hydrophilic bile acid derivate normally produced in the human liver by conjugation of Taurine to Ursodeoxycholic acid (UDCA) (Kusaczuk, 2019). It is widely used clinically for the treatment of chronic cholestatic liver diseases and gallstones and, thus, it is well tolerated and safe (Vang et al., 2014; Kusaczuk, 2019). TUDCA has revealed anti-apoptotic, anti-inflammatory and anti-oxidant effects in various models of NDs, including ALS (Vang et al., 2014; Cortez et al., 2015; Elia et al., 2016; Daruich et al., 2019). Subsequent studies employing hiPSCSOD1A4V MNs validated the neuroprotective properties of TUDCA on MNs (Thams et al., 2019). Besides, TUDCA supplementation led to reduced muscle denervation in a transgenic SOD1G93A mouse model of ALS (Thams et al., 2019). This study helped to gather further information on the mechanisms leading to the selective vulnerability of MNs in ALS, and also highlighted the relevance of using pluripotent stem cell-derived MNs in the discovery of meaningful neurotoxic and neuroprotective small molecules (Thams et al., 2019). In a phase IIb clinical trial involving a small series of ALS patients (ClinicalTrials.gov: NCT00877604), TUDCA was well tolerated and the data showed a slower ALS progression in the TUDCA-treated group when compared to placebo-treated control group (Elia et al., 2016). A larger phase 3 randomized clinical trial to establish the efficacy of the TUDCA in ALS patients is currently underway in multiple ALS centers across Europe, with the results expected to be available late in the year 2021 (ClinicalTrials.gov: NCT03800524). In another recently finished ALS clinical trial TUDCA was combined with Sodium Phenylbutyrate (ClinicalTrials.gov: NCT03127514) (Paganoni et al., 2020). Both drugs were shown to significantly decrease neuronal death in previous preclinical studies (Ryu et al., 2005; Castro-Caldas et al., 2012; Roy et al., 2012; Gaspar et al., 2013; Cortez et al., 2015; Gomez-Vicente et al., 2015). In the multicenter, randomized, double-blinded clinical trial that comprised 177 definite ALS patients, it was demonstrated that the drug combination resulted in a gentler decline of the ALSFRS-R score comparatively to the placebo group over a period of 24 weeks (Paganoni et al., 2020). Even though the results are promising, it remains to be determined if this drug combination is able to induce benefits in larger populations of ALS patients and for extended periods.
Finally, the Rho kinase (ROCK) inhibitor Fasudil, which has been in clinical use in Japan since 1995 with encouraging results in the prevention of the vasospasm associated with subarachnoid haemorrhage (Zhao et al., 2006; Zhao et al., 2011), recently emerged as another promising neuroprotective compound for MNs in ALS and other motor neuron disorders (Takata et al., 2013; Coque et al., 2014; Tonges et al., 2014). In a SOD1G93A mouse model of ALS, treatment with Fasudil was shown to reduce MNs loss, slow disease progression, improve motor function and increase lifespan (Takata et al., 2013; Tonges et al., 2014). Fasudil was also shown to influence the astroglial activity and to modulate the phenotype of microglia from “M1” (pro-inflammatory, with release of pro-inflammatory cytokines and chemokines) to “M2” (anti-inflammatory, with release of anti-inflammatory cytokines and growth factors) (Tonges et al., 2014). Additionally, ROCK inhibition up-regulates astrocytic glutamate transport, which could also help to explain the beneficial effects of the drug in ALS models since glutamate transporters are of fundamental value in the maintenance of the CNS homeostasis by countering cell death due to excitotoxicity (Lau et al., 2011; Lau et al., 2012). Moreover, Fasudil supplementation helps astrocytes to adopt a pro-survival phenotype (Lau et al., 2012). Furthermore, in a previous work using a MN survival assay based on FACS-purified hESC-MNs submitted to neurotrophic deprivation for 7 days, we showed that Y-27632, another small molecule in the ROCK inhibitor family, significantly increased the survival of FACS-purified human MNs in vitro (Lamas et al., 2014). Besides, Y-27632 also stimulated neuronal outgrowth, similarly to what was initially described in mouse studies (Lamas et al., unpublished data) (Dergham et al., 2002; Fournier et al., 2003). When tested along with Y-27632 and other ROCK inhibitor compounds, Fasudil was able to induce axonal growth, but could not significantly increase the survival of hESC-MNs deprived of neurotrophic factors for 7 days (Lamas et al., unpublished data). Fasudil has already been used in a compassionate regimen in 3 ALS patients, and it was well tolerated (Koch et al., 2020). A randomized phase IIa clinical trial of Fasudil in ALS patients (EudraCT: 2017–003,676–31; ClinicalTrials.gov: NCT03792490) started in early 2019 (Lingor et al., 2019). The primary endpoints are safety and tolerability, whereas efficacy is a secondary endpoint [assessed by the change in ALSFRS-R, slow vital capacity (SVC) and survival, among other parameters] (Lingor et al., 2019). A total of 120 patients will be recruited and randomized to receive a daily dose of either 30 mg or 60 mg Fasudil, or placebo in two intravenous applications for a total of 20 days (Lingor et al., 2019). A follow-up period of 180 days will ensue (Lingor et al., 2019). It remains to be established whether Y-27632 has a similar safety profile as Fasudil, but our observation that Y-27632 is also capable of promoting the survival of hESC-derived MNs deprived of neurotrophic factors for nearly 1 week (Lamas et al., 2014) along with previous studies in animal models, raise the possibility that Y-27632, and perhaps novel ROCK inhibitors in the market, similarly to Fasudil, merit a more in-depth pre-clinical and clinical assessment as promising disease-modifying drugs for ALS patients.
Challenges and Future Perspectives
The comprehensive work carried out in the field of pluripotent stem cells over the past decade has led to the development of numerous protocols for generating hiPSCs, producing neurons, glia and many other cell types from them, as well as enhanced conditions for in vitro culture (Figure 4) (Roybon et al., 2013; Sances et al., 2016; Tao and Zhang, 2016; Gonzalez et al., 2017; Costamagna et al., 2019; Logan et al., 2019; Savchenko et al., 2019; Suga et al., 2019; Karagiannis and Inoue, 2020; Li and Shi, 2020). Even though we still lack a comprehensive knowledge on the survival requirements of human MNs in vitro, patient-specific MNs can still be used to gain insights into the underlying mechanisms of ALS and to perform screenings to identify drug candidates (Figure 3). However, hurdles still remain. Different methodological approaches lead to numerous sources of variation, which help to explain the differences in viability and in ALS hiPSC-derived MN-based phenotypes observed across numerous studies. The choice of the human iPSC lines employed to produce MNs is also key, as clonal variability exists and it may affect the yield of MNs produced from them and their response to compounds. This is even more important in the case iPSC lines carry different ALS-associated gene variants. If diseased human iPSC lines are employed for screening, one will need to ascertain that the lines exhibit accurate phenotypes, as these may slightly vary between gene variants due to different altered cellular pathways and networks present in the individuals they are derived from. Resistance to drug compounds can also be identified using iPSCs (Vadodaria et al., 2019). Ultimately, small compound screens should be designed to employ several patient iPSC lines. Thus, it will be important to keep on developing more efficient protocols and lines displaying accurate and robust phenotypes, as well as implementing minimal standards across laboratories worldwide, so that results can be more easily compared and, ultimately, more easily translated into clinical applicable solutions for ALS patients (Sances et al., 2016).
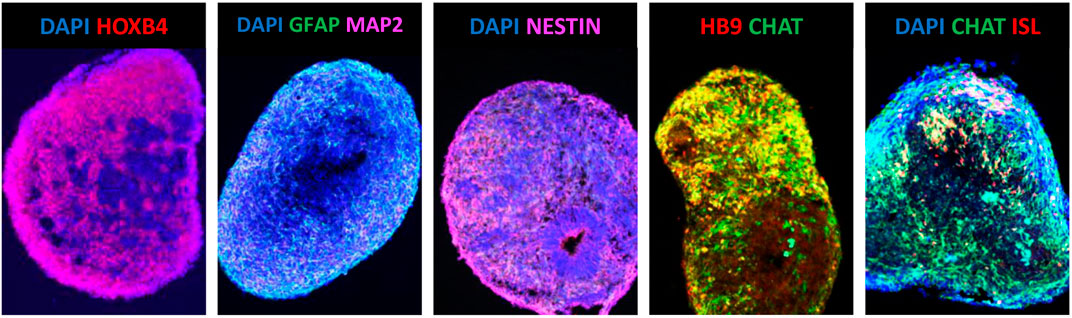
FIGURE 4. Human iPSC-derived spinal cord spheroids aged 30 days in vitro. The central nervous system-like structures were generated from hiPSCs by the sequential addition of LDN + SB431542 and RA + SAG + BDNF + GDNF (Roybon et al., unplublished data). The panels are representative fluorescence images of sectioned immunostained cervical HOXB4+ human spinal cord spheroids composed of NESTIN + neural progenitors, GFAP + astroglia and MAP2+, HB9+, ISL+, CHAT + MNs.
In a translational perspective, MNs generated from different forms of ALS patient-specific iPSCs could be useful to help stratify patients for clinical studies according to their genetic background, drug response, etc (Figure 3) (Bellin et al., 2012; Haston and Finkbeiner, 2016; Shinde et al., 2016; Fermini et al., 2018a; Silva and Haggarty, 2020). In line with this, for example, the study conducted by Fujimori and collaborators, besides demonstrating that Ropinirole is a promising drug against ALS, also showed that ALS iPSC-based disease modelling might help to subdivide genetically and clinically heterogeneous sALS cases based on the observed in vitro MN features (Fujimori et al., 2018). Those sub-classifications were correlated with both clinical features and disease progression, enabling the generation of complex disease models that might accurately reflect the evolution of the disease in ALS patients (Fujimori et al., 2018). This type of approach, if validated by other research groups, has the prospect to change dramatically the pre-clinical studies and clinical trials based on putative drug responders vs non-responders, with far reaching positive implications in ALS drug development (Fujimori et al., 2018). The fact that Ropinirole did not impact positively in SOD1 mutant ALS models illustrates the heterogeneity and complexity of the disease (Fujimori et al., 2018) and highlights the need for a more targeted and sound approach for studying pathophysiological mechanisms and in efficient drug testing.
In the present review, we have mainly focused on MNs, the cell type that is ultimately affected in ALS patients. However, over the past decade, a growing number of studies have implicated other players of the CNS environment in the development of ALS (Figure 2) (Lee et al., 2016; Serio and Patani, 2018; Valori et al., 2019; Crabe et al., 2020). Indeed, astrocytes, microglia and oligodendrocytes have all been implicated in MN death either through the release of yet to be identified toxic factors or through the lack of neuronal support (Figure 2) (Di Giorgio et al., 2007; Di Giorgio et al., 2008; Ilieva et al., 2009; Phatnani and Maniatis, 2015; Lee et al., 2016; Serio and Patani, 2018; Valori et al., 2019; Crabe et al., 2020; Izrael et al., 2020). Therefore, multi-cellular culture systems, involving neuronal, glial cells and muscle cells, grown as 2D monolayers or in 3D structures, may more closely mimic the brain and spinal cord environment (Figure 4), hence allowing the access to pathogenic processes that are at play in the spinal cord, the brainstem or the motor cortex, which are critically important to help understand the pathophysiological mechanisms underlying ALS and also in drug testing (Roybon et al., 2013; de Boer and Eggan, 2015; Richard and Maragakis, 2015; Costamagna et al., 2019; Halpern et al., 2019; Logan et al., 2019; Rowe and Daley, 2019; de Jongh et al., 2020).
The recently 3D cortico-motor assembloids model developed by the Paşca group bring hiPSCs into another dimension as this innovative approach allows the efficient combination of 3D structures analogous to the cerebral cortex and/or the hindbrain/spinal cord with human skeletal muscle spheroids, creating a functional nervous circuit in vitro (Andersen et al., 2020). This offers unprecedented opportunities in terms of ALS disease modelling and for drug discovery (Andersen et al., 2020; Panoutsopoulos, 2020).
Most of the drug testing and drug repurposing strategies based on hiPSCs described so far involved limited numbers of compounds. Through collaboration with pharma companies and academic-based drug discovery centers, it will be of priceless value and solid potential to test larger arrays of compounds. For example, we previously screened 50,400 small molecules for their ability to promote MN axonal growth on inhibitory substrata (MAG-expressing CHO cells) and found different hit compounds, among which the most relevant hits were the clinically used cholesterol lowering drugs statins (Li et al., 2016). Statins strongly stimulated the neurite outgrowth of MNs, both in vitro and in vivo, even under marked growth inhibitory conditions (Li et al., 2016). Accordingly, in vitro drug discovery studies based on hiPSCs should aim to test larger collections of small molecules, so that more disease-relevant hits can be identified.
Regarding the clinical setting, given the underlying molecular complexity of ALS and in light of the progress in precision medicine, a push for genetically targeted or patient-tailored therapies is emerging. In addition, it will be of fundamental value to identify biomarkers oriented towards specific ALS-patient clusters. Besides, the translation of novel therapies into clinical applicable strategies will also benefit from better methods for patient stratification and more efficient clinical trial designs (Haston and Finkbeiner, 2016; Fermini et al., 2018b; Kiernan et al., 2020). One recent appealing proposal is the multi-arm multi-stage (MAMS) strategy, in which the sample size is not fixed in advance (Ghosh et al., 2020; Kiernan et al., 2020). It is a strategy already used in cancer research which is a cost-effective approach for testing several drugs in parallel with a single placebo arm (Kiernan et al., 2020; Millen and Yap, 2020). In fact, eligible patients are initially randomly assigned to one of several sub-studies (Ghosh et al., 2020; Kiernan et al., 2020; Millen and Yap, 2020). The data are then sequentially analyzed, with pre-determined futility or superiority analyses built in, which enables treatment arms to be discontinued owing to a lack of efficacy (Kiernan et al., 2020; Millen and Yap, 2020). Subsequently, in the next phase of the study, patients are randomly assigned to receive active treatment or placebo (Kiernan et al., 2020; Millen and Yap, 2020). New arms can be added over time (Kiernan et al., 2020). The results of future ALS clinical trials might also greatly benefit from incorporating genotypic information into patient selection. This fact is specially highlighted in the post-hoc meta-analysis of 3 clinical trials that demonstrated that lithium carbonate, although overall ineffective, could have a positive effect in the subgroup of ALS patients carrying a UNC13A polymorphism (van Eijk et al., 2017; Kiernan et al., 2020).
Finally, the ALS community should stimulate the widespread sharing of pre-clinical and clinical data and the assembly of relevant databases like the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) database, which is the largest worldwide publicly available database of ALS patients, comprising data from 10,723 ALS patients involved in ALS clinical trials since 1990 (Atassi et al., 2014; Zach et al., 2015). This database was kindly donated by the members of the Pooled Resource Open-Access ALS Clinical Trials Consortium, an organization created in 2011 by Prize4Life, in collaboration with the Northeast ALS Consortium, and with funding from the ALS Therapy Alliance (Atassi et al., 2014; Zach et al., 2015). Taking advantage of databases like the PRO-ACT database, together with genetic information and pre-clinical data, coupled with artificial intelligence (AI)-based strategies, it will be possible to develop novel artificial neural network-based systems to confidently predict patient survival and stratify ALS patients for enrolment in clinical trials, perhaps, improving the efficiency of the drug discovery process (Atassi et al., 2014; Ko et al., 2014; Zach et al., 2015; Zhou and Manser, 2020). For example, similar systems have already been employed clinically in the estimation of survival prognosis in patients diagnosed with eye melanoma (Damato et al., 2008), thyroid cancer (Mourad et al., 2020), glioblastoma multiforme (Hao et al., 2018), among other diseases (Kourou et al., 2015; Zhu et al., 2020). Indeed, AI-based technologies can be wisely applied to compile, digest and interpret “hidden knowledge” in large datasets and make the data usable to researchers in the ALS field and beyond (Ko et al., 2014; Kusumoto and Yuasa, 2019; Cota-Coronado et al., 2020; Zhou and Manser, 2020).
Together, all these aspects have the prospect to impact positively on the natural course of ALS, which is a deadly heterogeneous ND most likely triggered by the complex interaction of different genetic traits with a myriad of environmental factors.
Conclusion
Neurodegenerative diseases have a devastating impact on the quality of life of patients. In the case of ALS, MNs gradually perish, resulting in an early and rapid death due to respiratory failure. ALS is a complex ND, with diverse pathological mechanisms connected with disease onset and progression, which appears to be mediated by, yet to be explained, composite interactions between different genes and multiple environmental factors. Despite extensive research over the past 4 decades and the hundreds of promising compounds to treat ALS that emerged from preclinical animal-based studies, only Riluzole and Edaravone seem to have changed the course of the disease, albeit with a very modest increase in lifespan and quality of life. The continuous efforts to develop novel therapeutic drug candidates for ALS are of vital importance. Human pluripotent stem cells have opened unparalleled opportunities to study previously inaccessible neuronal and glial cell populations, as well as muscular cells from ALS patients (and healthy individuals). In fact, hESC- and hiPSC-derived MNs have been a robust platform that allows a more in-depth understanding of the mechanisms involved in the death and survival of human MNs. In addition, the possibility of studying human MNs and glial cells that capture the genetic background of patients opens novel avenues towards a thorough understanding of MN degeneration and ultimately might lead to the development of more effective ALS therapeutic strategies. In this regard, there have been several encouraging success stories in drug discovery in ALS, through drug screening or drug repurposing, using easily accessible human MNs specified from hESCs and hiPSCs, which were highlighted in the present review. Yet, similarly to other neurodegenerative diseases (e.g., Parkinson’s disease, Alzheimer’s disease, etc) and in light of a personalized medicine approach, it is becoming clear that a single therapeutic agent will not be a panacea for all ALS patients. In fact, given the heterogeneity of the ALS cases and the complexity of the disease, together with advances in genetics and biomarker development, we should envision a personalized ALS-patient approach, based on the specific clinical and biological characteristics of homogeneous subgroups of patients. Despite its predicted high cost, this personalized strategy is expected to be paramount for the discovery of more effective ALS treatments. The advent of novel technologies such as 3D organoids (Lancaster et al., 2013; Kawada et al., 2017), spheroids (Chumarina et al., 2019; Pomeshchik et al., 2020) and assembloids (Sloan et al., 2018; Andersen et al., 2020; Miura et al., 2020) has the prospect to give further insights into the pathogenesis of neurodegenerative diseases including ALS, and also lead to successful drug discovery campaigns. These developments, coupled with genetic studies, big data analysis and AI platforms will help to better stratify ALS patients and build more robust clinical trials. Altogether, in the near future, we may be able to significantly accelerate the process of drug development, and considerably shorten the time and reduce the costs needed to translate preclinical research into clinically applicable drug therapies for ALS patients, hopefully, halting disease progression or even cure ALS.
Author Contributions
NJL and LR designed and wrote the manuscript. Both authors approved the final version of the manuscript.
Funding
The authors would very much like to thank the Olle Engkvist Byggmästare Foundation in Sweden and the AFM-Téléthon in France for supporting their projects on ALS research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aggarwal, S., and Cudkowicz, M. (2008). ALS Drug Development: Reflections from the Past and a Way Forward. Neurotherapeutics 5 (4), 516–527. doi:10.1016/j.nurt.2008.08.002
Al-Chalabi, A., Jones, A., Troakes, C., King, A., Al-Sarraj, S., and van den Berg, L. H. (2012). The Genetics and Neuropathology of Amyotrophic Lateral Sclerosis. Acta Neuropathol. 124 (3), 339–352. doi:10.1007/s00401-012-1022-4
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 Positive, TDP-43 Negative, Neuronal Cytoplasmic and Intranuclear Inclusions in the Cerebellum and hippocampus Define the Pathology of C9orf72-Linked FTLD and MND/ALS. Acta Neuropathol. 122 (6), 691–702. doi:10.1007/s00401-011-0911-2
Allodi, I., Nijssen, J., Benitez, J. A., Schweingruber, C., Fuchs, A., Bonvicini, G., et al. (2019). Modeling Motor Neuron Resilience in ALS Using Stem Cells. Stem Cel Rep. 12 (6), 1329–1341. doi:10.1016/j.stemcr.2019.04.009
Amabile, G., and Meissner, A. (2009). Induced Pluripotent Stem Cells: Current Progress and Potential for Regenerative Medicine. Trends Mol. Med. 15 (2), 59–68. doi:10.1016/j.molmed.2008.12.003
Amoroso, M. W., Croft, G. F., Williams, D. J., O'Keeffe, S., Carrasco, M. A., Davis, A. R., et al. (2013). Accelerated High-Yield Generation of Limb-Innervating Motor Neurons from Human Stem Cells. J. Neurosci. 33 (2), 574–586. doi:10.1523/JNEUROSCI.0906-12.2013
An, D., Fujiki, R., Iannitelli, D. E., Smerdon, J. W., Maity, S., Rose, M. F., et al. (2019). Stem Cell-Derived Cranial and Spinal Motor Neurons Reveal Proteostatic Differences between ALS Resistant and Sensitive Motor Neurons. Elife 8. doi:10.7554/eLife.44423
Andersen, J., Revah, O., Miura, Y., Thom, N., Amin, N. D., Kelley, K. W., et al. (2020). Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 183 (7), 1913–1929. doi:10.1016/j.cell.2020.11.017
Andersen, P. M., Forsgren, L., Binzer, M., Nilsson, P., Ala-Hurula, V., Keränen, M. L., et al. (1996). Autosomal Recessive Adult-Onset Amyotrophic Lateral Sclerosis Associated with Homozygosity for Asp90Ala CuZn-Superoxide Dismutase Mutation. A Clinical and Genealogical Study of 36 Patients. Brain 119 ( Pt 4) (Pt 4), 1153–1172. doi:10.1093/brain/119.4.1153
Arai, T., Nonaka, T., Hasegawa, M., Akiyama, H., Yoshida, M., Hashizume, Y., et al. (2003). Neuronal and Glial Inclusions in Frontotemporal Dementia with or without Motor Neuron Disease Are Immunopositive for P62. Neurosci. Lett. 342 (1-2), 41–44. doi:10.1016/s0304-3940(03)00216-7
Atassi, N., Berry, J., Shui, A., Zach, N., Sherman, A., Sinani, E., et al. (2014). The PRO-ACT Database: Design, Initial Analyses, and Predictive Features. Neurology 83 (19), 1719–1725. doi:10.1212/WNL.0000000000000951
Avior, Y., Sagi, I., and Benvenisty, N. (2016). Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Mol. Cel Biol 17 (3), 170–182. doi:10.1038/nrm.2015.27
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 14 (9), 544–558. doi:10.1038/s41582-018-0047-2
Batra, R., and Lee, C. W. (2017). Mouse Models of C9orf72 Hexanucleotide Repeat Expansion in Amyotrophic Lateral Sclerosis/Frontotemporal Dementia. Front. Cel. Neurosci. 11, 196. doi:10.3389/fncel.2017.00196
Bäumer, D., Talbot, K., and Turner, M. R. (2014). Advances in Motor Neurone Disease. J. R. Soc. Med. 107 (1), 14–21. doi:10.1177/0141076813511451
Beghi, E., Chiò, A., Couratier, P., Esteban, J., Hardiman, O., Logroscino, G., et al. (2011). The Epidemiology and Treatment of ALS: Focus on the Heterogeneity of the Disease and Critical Appraisal of Therapeutic Trials. Amyotroph. Lateral Scler. 12 (1), 1–10. doi:10.3109/17482968.2010.502940
Bellin, M., Marchetto, M. C., Gage, F. H., and Mummery, C. L. (2012). Induced Pluripotent Stem Cells: the New Patient? Nat. Rev. Mol. Cel Biol 13 (11), 713–726. doi:10.1038/nrm3448
Bellingham, M. C. (2011). A Review of the Neural Mechanisms of Action and Clinical Efficiency of Riluzole in Treating Amyotrophic Lateral Sclerosis: what Have We Learned in the Last Decade? CNS Neurosci. Ther. 17 (1), 4–31. doi:10.1111/j.1755-5949.2009.00116.x
Benatar, M. (2007). Lost in Translation: Treatment Trials in the SOD1 Mouse and in Human ALS. Neurobiol. Dis. 26 (1), 1–13. doi:10.1016/j.nbd.2006.12.015
Benatar, M., Wuu, J., Andersen, P. M., Atassi, N., David, W., Cudkowicz, M., et al. (2018). Randomized, Double-Blind, Placebo-Controlled Trial of Arimoclomol in Rapidly Progressive SOD1 ALS. Neurology 90 (7), e565–e574. doi:10.1212/WNL.0000000000004960
Bensimon, G., Lacomblez, L., and Meininger, V. (1994). A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 330 (9), 585–591. doi:10.1056/NEJM199403033300901
Berry, J. D., and Cudkowicz, M. E. (2011). New Considerations in the Design of Clinical Trials for Amyotrophic Lateral Sclerosis. Clin. Invest. 1 (10), 1375–1389. doi:10.4155/cli.11.127
Bhinge, A., Namboori, S. C., Zhang, X., VanDongen, A. M. J., and Stanton, L. W. (2017). Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cel Rep. 8 (4), 856–869. doi:10.1016/j.stemcr.2017.02.019
Bilic, J., and Belmonte, J. C. I. (2012). Concise Review: Induced Pluripotent Stem Cells versus Embryonic Stem Cells: Close Enough or yet Too Far Apart? Stem Cells 30 (1), 33–41. doi:10.1002/stem.700
Bongso, A., Fong, C.-Y., Ng, S.-C., and Ratnam, S. (1994). Fertilization and Early Embryology: Isolation and Culture of Inner Cell Mass Cells from Human Blastocysts. Hum. Reprod. 9 (11), 2110–2117. doi:10.1093/oxfordjournals.humrep.a138401
Bordet, T., Buisson, B., Michaud, M., Drouot, C., Galéa, P., Delaage, P., et al. (2007). Identification and Characterization of Cholest-4-En-3-One, Oxime (TRO19622), a Novel Drug Candidate for Amyotrophic Lateral Sclerosis. J. Pharmacol. Exp. Ther. 322 (2), 709–720. doi:10.1124/jpet.107.123000
Bos, P. H., Lowry, E. R., Costa, J., Thams, S., Garcia-Diaz, A., Zask, A., et al. (2019). Development of MAP4 Kinase Inhibitors as Motor Neuron-Protecting Agents. Cel Chem. Biol. 26 (12), 1703–1715. doi:10.1016/j.chembiol.2019.10.005
Boulting, G. L., Kiskinis, E., Croft, G. F., Amoroso, M. W., Oakley, D. H., Wainger, B. J., et al. (2011). A Functionally Characterized Test Set of Human Induced Pluripotent Stem Cells. Nat. Biotechnol. 29 (3), 279–286. doi:10.1038/nbt.1783
Boxer, A. L., Mackenzie, I. R., Boeve, B. F., Baker, M., Seeley, W. W., Crook, R., et al. (2011). Clinical, Neuroimaging and Neuropathological Features of a New Chromosome 9p-Linked FTD-ALS Family. J. Neurol. Neurosurg. Psychiatry 82 (2), 196–203. doi:10.1136/jnnp.2009.204081
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 377 (2), 162–172. doi:10.1056/NEJMra1603471
Browne, E. C., and Abbott, B. M. (2016). Recent Progress towards an Effective Treatment of Amyotrophic Lateral Sclerosis Using the SOD1 Mouse Model in a Preclinical Setting. Eur. J. Med. Chem. 121, 918–925. doi:10.1016/j.ejmech.2016.02.048
Bunina, T. L. (1962). On Intracellular Inclusions in Familial Amyotrophic Lateral Sclerosis. Zh Nevropatol Psikhiatr Im S S Korsakova 62, 1293–1299.
Burkhardt, M. F., Martinez, F. J., Wright, S., Ramos, C., Volfson, D., Mason, M., et al. (2013). A Cellular Model for Sporadic ALS Using Patient-Derived Induced Pluripotent Stem Cells. Mol. Cell Neurosci. 56, 355–364. doi:10.1016/j.mcn.2013.07.007
Bursch, F., Kalmbach, N., Naujock, M., Staege, S., Eggenschwiler, R., Abo-Rady, M., et al. (2019). Altered Calcium Dynamics and Glutamate Receptor Properties in iPSC-Derived Motor Neurons from ALS Patients with C9orf72, FUS, SOD1 or TDP43 Mutations. Hum. Mol. Genet. 28 (17), 2835–2850. doi:10.1093/hmg/ddz107
Cahan, P., and Daley, G. Q. (2013). Origins and Implications of Pluripotent Stem Cell Variability and Heterogeneity. Nat. Rev. Mol. Cel Biol 14 (6), 357–368. doi:10.1038/nrm3584
Castro-Caldas, M., Carvalho, A. N., Rodrigues, E., Henderson, C. J., Wolf, C. R., Rodrigues, C. M. P., et al. (2012). Tauroursodeoxycholic Acid Prevents MPTP-Induced Dopaminergic Cell Death in a Mouse Model of Parkinson's Disease. Mol. Neurobiol. 46 (2), 475–486. doi:10.1007/s12035-012-8295-4
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly Efficient Neural Conversion of Human ES and iPS Cells by Dual Inhibition of SMAD Signaling. Nat. Biotechnol. 27 (3), 275–280. doi:10.1038/nbt.1529
Chang, C.-Y., Ting, H.-C., Liu, C.-A., Su, H.-L., Chiou, T.-W., Lin, S.-Z., et al. (2020). Induced Pluripotent Stem Cell (iPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 25 (8), 2000. doi:10.3390/molecules25082000
Chen, H., Qian, K., Du, Z., Cao, J., Petersen, A., Liu, H., et al. (2014). Modeling ALS with iPSCs Reveals that Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell 14 (6), 796–809. doi:10.1016/j.stem.2014.02.004
Chia, R., Chiò, A., and Traynor, B. J. (2018). Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet Neurol. 17 (1), 94–102. doi:10.1016/S1474-4422(17)30401-5
Chiò, A., Hammond, E. R., Mora, G., Bonito, V., and Filippini, G. (2015). Development and Evaluation of a Clinical Staging System for Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 86 (1), 38–44. doi:10.1136/jnnp-2013-306589
Chiò, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., et al. (2013). Global Epidemiology of Amyotrophic Lateral Sclerosis: a Systematic Review of the Published Literature. Neuroepidemiology 41 (2), 118–130. doi:10.1159/000351153
Chiò, A., Mazzini, L., and Mora, G. (2020). Disease-modifying Therapies in Amyotrophic Lateral Sclerosis. Neuropharmacology 167, 107986. doi:10.1016/j.neuropharm.2020.107986
Chiot, A., Lobsiger, C. S., and Boillée, S. (2019). New Insights on the Disease Contribution of Neuroinflammation in Amyotrophic Lateral Sclerosis. Curr. Opin. Neurol. 32 (5), 764–770. doi:10.1097/WCO.0000000000000729
Christodoulou, C., and Kotton, D. N. (2012). Are Embryonic Stem and Induced Pluripotent Stem Cells the Same or Different? Implications for Their Potential Therapeutic Use. Cell Cycle 11 (1), 5–6. doi:10.4161/cc.11.1.18655
Chumarina, M., Russ, K., Azevedo, C., Heuer, A., Pihl, M., Collin, A., et al. (2019). Cellular Alterations Identified in Pluripotent Stem Cell-Derived Midbrain Spheroids Generated from a Female Patient with Progressive External Ophthalmoplegia and Parkinsonism Who Carries a Novel Variation (p.Q811R) in the POLG1 Gene. Acta Neuropathol. Commun. 7 (1), 208. doi:10.1186/s40478-019-0863-7
Collaborators, G. B. D. N. (2019). Global, Regional, and National burden of Neurological Disorders, 1990-2016: a Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18 (5), 459–480. doi:10.1016/S1474-4422(18)30499-X
Comley, L., Allodi, I., Nichterwitz, S., Nizzardo, M., Simone, C., Corti, S., et al. (2015). Motor Neurons with Differential Vulnerability to Degeneration Show Distinct Protein Signatures in Health and ALS. Neuroscience 291, 216–229. doi:10.1016/j.neuroscience.2015.02.013
Cooper-Knock, J., Higginbottom, A., Stopford, M. J., Highley, J. R., Ince, P. G., Wharton, S. B., et al. (2015). Antisense RNA Foci in the Motor Neurons of C9ORF72-ALS Patients Are Associated with TDP-43 Proteinopathy. Acta Neuropathol. 130 (1), 63–75. doi:10.1007/s00401-015-1429-9
Cooper-Knock, J., Shaw, P. J., and Kirby, J. (2014). The Widening Spectrum of C9ORF72-Related Disease; Genotype/phenotype Correlations and Potential Modifiers of Clinical Phenotype. Acta Neuropathol. 127 (3), 333–345. doi:10.1007/s00401-014-1251-9
Coque, E., Raoul, C. d., and Bowerman, M. l. (2014). ROCK Inhibition as a Therapy for Spinal Muscular Atrophy: Understanding the Repercussions on Multiple Cellular Targets. Front. Neurosci. 8, 271. doi:10.3389/fnins.2014.00271
Cortez, L. M., Campeau, J., Norman, G., Kalayil, M., Van der Merwe, J., McKenzie, D., et al. (2015). Bile Acids Reduce Prion Conversion, Reduce Neuronal Loss, and Prolong Male Survival in Models of Prion Disease. J. Virol. 89 (15), 7660–7672. doi:10.1128/JVI.01165-15
Costamagna, G., Andreoli, L., Corti, S., and Faravelli, I. (2019). iPSCs-Based Neural 3D Systems: A Multidimensional Approach for Disease Modeling and Drug Discovery. Cells 8 (11), 1438. doi:10.3390/cells8111438
Cota-Coronado, A., Durnall, J. C., Díaz, N. F., Thompson, L. H., and Díaz-Martínez, N. E. (2020). Unprecedented Potential for Neural Drug Discovery Based on Self-Organizing hiPSC Platforms. Molecules 25 (5), 1150. doi:10.3390/molecules25051150
Crabé, R., Aimond, F., Gosset, P., Scamps, F., and Raoul, C. (2020). How Degeneration of Cells Surrounding Motoneurons Contributes to Amyotrophic Lateral Sclerosis. Cells 9 (12), 2550. doi:10.3390/cells9122550
Cudkowicz, M., Bozik, M. E., Ingersoll, E. W., Miller, R., Mitsumoto, H., Shefner, J., et al. (2011). The Effects of Dexpramipexole (KNS-760704) in Individuals with Amyotrophic Lateral Sclerosis. Nat. Med. 17 (12), 1652–1656. doi:10.1038/nm.2579
Damato, B., Eleuteri, A., Fisher, A. C., Coupland, S. E., and Taktak, A. F. G. (2008). Artificial Neural Networks Estimating Survival Probability after Treatment of Choroidal Melanoma. Ophthalmology 115 (9), 1598–1607. doi:10.1016/j.ophtha.2008.01.032
Daruich, A., Picard, E., Boatright, J. H., and Behar-Cohen, F. (2019). Review: The Bile Acids Urso- and Tauroursodeoxycholic Acid as Neuroprotective Therapies in Retinal Disease. Mol. Vis. 25, 610–624.
de Boer, A. S., and Eggan, K. (2015). A Perspective on Stem Cell Modeling of Amyotrophic Lateral Sclerosis. Cell Cycle 14 (23), 3679–3688. doi:10.1080/15384101.2015.1093712
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72 (2), 245–256. doi:10.1016/j.neuron.2011.09.011
Deng, H.-X., Chen, W., Hong, S.-T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/dementia. Nature 477 (7363), 211–215. doi:10.1038/nature10353
Dergham, P., Ellezam, B., Essagian, C., Avedissian, H., Lubell, W. D., and McKerracher, L. (2002). Rho Signaling Pathway Targeted to Promote Spinal Cord Repair. J. Neurosci. 22 (15), 6570–6577. doi:10.1523/jneurosci.22-15-06570.2002
Deuschl, G., Beghi, E., Fazekas, F., Varga, T., Christoforidi, K. A., Sipido, E., et al. (2020). The burden of Neurological Diseases in Europe: an Analysis for the Global Burden of Disease Study 2017. The Lancet Public Health 5 (10), e551–e567. doi:10.1016/S2468-2667(20)30190-0
Devlin, A.-C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-Derived Motoneurons Harbouring TARDBP or C9ORF72 ALS Mutations Are Dysfunctional Despite Maintaining Viability. Nat. Commun. 6, 5999. doi:10.1038/ncomms6999
Di Giorgio, F. P., Boulting, G. L., Bobrowicz, S., and Eggan, K. C. (2008). Human Embryonic Stem Cell-Derived Motor Neurons Are Sensitive to the Toxic Effect of Glial Cells Carrying an ALS-Causing Mutation. Cell Stem Cell 3 (6), 637–648. doi:10.1016/j.stem.2008.09.017
Di Giorgio, F. P., Carrasco, M. A., Siao, M. C., Maniatis, T., and Eggan, K. (2007). Non-cell Autonomous Effect of Glia on Motor Neurons in an Embryonic Stem Cell-Based ALS Model. Nat. Neurosci. 10 (5), 608–614. doi:10.1038/nn1885
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced Pluripotent Stem Cells Generated from Patients with ALS Can Be Differentiated into Motor Neurons. Science 321 (5893), 1218–1221. doi:10.1126/science.1158799
Donnelly, C. J., Zhang, P.-W., Pham, J. T., Haeusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron 80 (2), 415–428. doi:10.1016/j.neuron.2013.10.015
Dorst, J., Ludolph, A. C., and Huebers, A. (2018). Disease-modifying and Symptomatic Treatment of Amyotrophic Lateral Sclerosis. Ther. Adv. Neurol. Disord. 11, 175628561773473. doi:10.1177/1756285617734734
Dunckley, T., Huentelman, M. J., Craig, D. W., Pearson, J. V., Szelinger, S., Joshipura, K., et al. (2007). Whole-genome Analysis of Sporadic Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 357 (8), 775–788. doi:10.1056/NEJMoa070174
Ebert, A. D., and Svendsen, C. N. (2010). Human Stem Cells and Drug Screening: Opportunities and Challenges. Nat. Rev. Drug Discov. 9 (5), 367–372. doi:10.1038/nrd3000
Ebstein, S. Y., Yagudayeva, I., and Shneider, N. A. (2019). Mutant TDP-43 Causes Early-Stage Dose-dependent Motor Neuron Degeneration in a TARDBP Knockin Mouse Model of ALS. Cel Rep. 26 (2), 364–373. doi:10.1016/j.celrep.2018.12.045
Edaravone Acute Infarction Study, G. (2003). Effect of a Novel Free Radical Scavenger, Edaravone (MCI-186), on Acute Brain Infarction. Cerebrovasc. Dis. 15 (3), 222–229. doi:10.1159/000069318
Elia, A. E., Lalli, S., Monsurrò, M. R., Sagnelli, A., Taiello, A. C., Reggiori, B., et al. (2016). Tauroursodeoxycholic Acid in the Treatment of Patients with Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 23 (1), 45–52. doi:10.1111/ene.12664
Engels, E.-M. (2002). Human Embryonic Stem Cells - the German Debate. Nat. Rev. Genet. 3 (8), 636–641. doi:10.1038/nrg871
Engle, S. J., and Puppala, D. (2013). Integrating Human Pluripotent Stem Cells into Drug Development. Cell Stem Cell 12 (6), 669–677. doi:10.1016/j.stem.2013.05.011
Engle, S. J., and Vincent, F. (2014). Small Molecule Screening in Human Induced Pluripotent Stem Cell-Derived Terminal Cell Types. J. Biol. Chem. 289 (8), 4562–4570. doi:10.1074/jbc.R113.529156
Evans, M. (2005). Embryonic Stem Cells: a Perspective. Novartis Found. Symp. 265, 98–8. doi:10.2174/1566523211313020003
Evans, M. (2011). Discovering Pluripotency: 30 Years of Mouse Embryonic Stem Cells. Nat. Rev. Mol. Cel Biol 12 (10), 680–686. doi:10.1038/nrm3190
Evans, M. J., and Kaufman, M. H. (1981). Establishment in Culture of Pluripotential Cells from Mouse Embryos. Nature 292 (5819), 154–156. doi:10.1038/292154a0
Fang, T., Al Khleifat, A., Stahl, D. R., Lazo La Torre, C., Murphy, C., Young, C., et al. (2017). Comparison of the King's and MiToS Staging Systems for ALS. Amyotroph. Lateral Scler. Frontotemporal Degeneration 18 (3-4), 227–232. doi:10.1080/21678421.2016.1265565
Faravelli, I., Bucchia, M., Rinchetti, P., Nizzardo, M., Simone, C., Frattini, E., et al. (2014). Motor Neuron Derivation from Human Embryonic and Induced Pluripotent Stem Cells: Experimental Approaches and Clinical Perspectives. Stem Cel Res. Ther. 5 (4), 87. doi:10.1186/scrt476
Fermini, B., Coyne, K. P., and Coyne, S. T. (2018a). Challenges in Designing and Executing Clinical Trials in a Dish Studies. J. Pharmacol. Toxicol. Methods 94 (Pt 2), 73–82. doi:10.1016/j.vascn.2018.09.002
Fermini, B., Coyne, S. T., and Coyne, K. P. (2018b). Clinical Trials in a Dish: A Perspective on the Coming Revolution in Drug Development. SLAS DISCOVERY: Advancing Sci. Drug Discov. 23 (8), 765–776. doi:10.1177/2472555218775028
Fogarty, M. J. (2018). Driven to Decay: Excitability and Synaptic Abnormalities in Amyotrophic Lateral Sclerosis. Brain Res. Bull. 140, 318–333. doi:10.1016/j.brainresbull.2018.05.023
Fournier, A. E., Takizawa, B. T., and Strittmatter, S. M. (2003). Rho Kinase Inhibition Enhances Axonal Regeneration in the Injured CNS. J. Neurosci. 23 (4), 1416–1423. doi:10.1523/jneurosci.23-04-01416.2003
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling Sporadic ALS in iPSC-Derived Motor Neurons Identifies a Potential Therapeutic Agent. Nat. Med. 24 (10), 1579–1589. doi:10.1038/s41591-018-0140-5
Garbuzova-Davis, S., Thomson, A., Kurien, C., Shytle, R. D., and Sanberg, P. R. (2016). Potential New Complication in Drug Therapy Development for Amyotrophic Lateral Sclerosis. Expert Rev. Neurotherapeutics 16 (12), 1397–1405. doi:10.1080/14737175.2016.1207530
Garcia-Diaz, A., Efe, G., Kabra, K., Patel, A., Lowry, E. R., Shneider, N. A., et al. (2020). Standardized Reporter Systems for Purification and Imaging of Human Pluripotent Stem Cell-Derived Motor Neurons and Other Cholinergic Cells. Neuroscience 450, 48–56. doi:10.1016/j.neuroscience.2020.06.028
Gaspar, J. M., Martins, A., Cruz, R., Rodrigues, C. M. P., Ambrósio, A. F., and Santiago, A. R. (2013). Tauroursodeoxycholic Acid Protects Retinal Neural Cells from Cell Death Induced by Prolonged Exposure to Elevated Glucose. Neuroscience 253, 380–388. doi:10.1016/j.neuroscience.2013.08.053
Gavrilov, S., Papaioannou, V., and Landry, D. (2009). Alternative Strategies for the Derivation of Human Embryonic Stem Cell Lines and the Role of Dead Embryos. Cscr 4 (1), 81–86. doi:10.2174/157488809787169057
Ghosh, P., Liu, L., and Mehta, C. (2020). Adaptive Multiarm Multistage Clinical Trials. Stat. Med. 39 (8), 1084–1102. doi:10.1002/sim.8464
Ginis, I., Luo, Y., Miura, T., Thies, S., Brandenberger, R., Gerecht-Nir, S., et al. (2004). Differences between Human and Mouse Embryonic Stem Cells. Developmental Biol. 269 (2), 360–380. doi:10.1016/j.ydbio.2003.12.034
Gizzi, M., DiRocco, A., Sivak, M., and Cohen, B. (1992). Ocular Motor Function in Motor Neuron Disease. Neurology 42 (5), 1037. doi:10.1212/wnl.42.5.1037
Gokhale, P. J., and Andrews, P. W. (2009). Human Embryonic Stem Cells: 10 Years on. Lab. Invest. 89 (3), 259–262. doi:10.1038/labinvest.2008.162
Gómez-Vicente, V., Lax, P., Fernández-Sánchez, L., Rondón, N., Esquiva, G., Germain, F., et al. (2015). Neuroprotective Effect of Tauroursodeoxycholic Acid on N-Methyl-D-Aspartate-Induced Retinal Ganglion Cell Degeneration. PLoS One 10 (9), e0137826. doi:10.1371/journal.pone.0137826
Gonzalez, D. M., Gregory, J., and Brennand, K. J. (2017). The Importance of Non-neuronal Cell Types in hiPSC-Based Disease Modeling and Drug Screening. Front. Cel Dev. Biol. 5, 117. doi:10.3389/fcell.2017.00117
González, F., Boué, S., and Belmonte, J. C. I. (2011). Methods for making induced pluripotent stem cells: reprogramming à la carte. Nat. Rev. Genet. 12 (4), 231–242. doi:10.1038/nrg2937
Gordon, T., Tyreman, N., Li, S., Putman, C. T., and Hegedus, J. (2010). Functional Over-load Saves Motor Units in the SOD1-G93a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 37 (2), 412–422. doi:10.1016/j.nbd.2009.10.021
Goutman, S. A. (2017). Diagnosis and Clinical Management of Amyotrophic Lateral Sclerosis and Other Motor Neuron DisordersPeripheral Nerve and Motor Neuron Disorders. CONTINUUM: Lifelong Learn. Neurol. 23 (5), 1332–1359. doi:10.1212/CON.0000000000000535
Goyal, N. A., Berry, J. D., Windebank, A., Staff, N. P., Maragakis, N. J., Berg, L. H., et al. (2020). Addressing Heterogeneity in Amyotrophic Lateral Sclerosis CLINICAL TRIALS. Muscle Nerve 62 (2), 156–166. doi:10.1002/mus.26801
Grad, L. I., Rouleau, G. A., Ravits, J., and Cashman, N. R. (2017). Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb Perspect. Med. 7 (8), a024117. doi:10.1101/cshperspect.a024117
Gribaudo, S., Tixador, P., Bousset, L., Fenyi, A., Lino, P., Melki, R., et al. (2019). Propagation of α-Synuclein Strains within Human Reconstructed Neuronal Network. Stem Cel Rep. 12 (2), 230–244. doi:10.1016/j.stemcr.2018.12.007
Grillner, S., and Jessell, T. M. (2009). Measured Motion: Searching for Simplicity in Spinal Locomotor Networks. Curr. Opin. Neurobiol. 19 (6), 572–586. doi:10.1016/j.conb.2009.10.011
Grskovic, M., Javaherian, A., Strulovici, B., and Daley, G. Q. (2011). Induced Pluripotent Stem Cells - Opportunities for Disease Modelling and Drug Discovery. Nat. Rev. Drug Discov. 10 (12), 915–929. doi:10.1038/nrd3577
Hallett, P. J., Deleidi, M., Astradsson, A., Smith, G. A., Cooper, O., Osborn, T. M., et al. (2015). Successful Function of Autologous iPSC-Derived Dopamine Neurons Following Transplantation in a Non-human Primate Model of Parkinson's Disease. Cell Stem Cell 16 (3), 269–274. doi:10.1016/j.stem.2015.01.018
Halpern, M., Brennand, K. J., and Gregory, J. (2019). Examining the Relationship between Astrocyte Dysfunction and Neurodegeneration in ALS Using hiPSCs. Neurobiol. Dis. 132, 104562. doi:10.1016/j.nbd.2019.104562
Han, S. S. W., Williams, L. A., and Eggan, K. C. (2011). Constructing and Deconstructing Stem Cell Models of Neurological Disease. Neuron 70 (4), 626–644. doi:10.1016/j.neuron.2011.05.003
Hao, J., Kim, Y., Kim, T.-K., and Kang, M. (2018). PASNet: Pathway-Associated Sparse Deep Neural Network for Prognosis Prediction from High-Throughput Data. BMC Bioinformatics 19 (1), 510. doi:10.1186/s12859-018-2500-z
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 3, 17071. doi:10.1038/nrdp.2017.71
Hardiman, O., van den Berg, L. H., and Kiernan, M. C. (2011). Clinical Diagnosis and Management of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 7 (11), 639–649. doi:10.1038/nrneurol.2011.153
Haston, K. M., and Finkbeiner, S. (2016). Clinical Trials in a Dish: The Potential of Pluripotent Stem Cells to Develop Therapies for Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 56, 489–510. doi:10.1146/annurev-pharmtox-010715-103548
Hawrot, J., Imhof, S., and Wainger, B. J. (2020). Modeling Cell-Autonomous Motor Neuron Phenotypes in ALS Using iPSCs. Neurobiol. Dis. 134, 104680. doi:10.1016/j.nbd.2019.104680
Hedlund, E., Karlsson, M., Osborn, T., Ludwig, W., and Isacson, O. (2010). Global Gene Expression Profiling of Somatic Motor Neuron Populations with Different Vulnerability Identify Molecules and Pathways of Degeneration and protection. Brain 133 (Pt 8), 2313–2330. doi:10.1093/brain/awq167
Hegedus, J., Putman, C. T., and Gordon, T. (2007). Time Course of Preferential Motor Unit Loss in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 28 (2), 154–164. doi:10.1016/j.nbd.2007.07.003
Higelin, J., Demestre, M., Putz, S., Delling, J. P., Jacob, C., Lutz, A.-K., et al. (2016). FUS Mislocalization and Vulnerability to DNA Damage in ALS Patients Derived hiPSCs and Aging Motoneurons. Front. Cel. Neurosci. 10, 290. doi:10.3389/fncel.2016.00290
Ho, R., Sances, S., Gowing, G., Amoroso, M. W., O'Rourke, J. G., Sahabian, A., et al. (2016). ALS Disrupts Spinal Motor Neuron Maturation and Aging Pathways within Gene Co-expression Networks. Nat. Neurosci. 19 (9), 1256–1267. doi:10.1038/nn.4345
Holmqvist, S., Lehtonen, Š., Chumarina, M., Puttonen, K. A., Azevedo, C., Lebedeva, O., et al. (2016). Creation of a Library of Induced Pluripotent Stem Cells from Parkinsonian Patients. npj Parkinson's Dis. 2, 16009. doi:10.1038/npjparkd.2016.9
Huang, X., Roet, K. C. D., Zhang, L., Brault, A., Berg, A. P., Jefferson, A. B., et al. (2021). Human Amyotrophic Lateral Sclerosis Excitability Phenotype Screen: Target Discovery and Validation. Cel Rep. 35 (10), 109224. doi:10.1016/j.celrep.2021.109224
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell Autonomous Toxicity in Neurodegenerative Disorders: ALS and beyond. J. Cel Biol 187 (6), 761–772. doi:10.1083/jcb.200908164
Imamura, K., Izumi, Y., Watanabe, A., Tsukita, K., Woltjen, K., Yamamoto, T., et al. (2017). The Src/c-Abl Pathway Is a Potential Therapeutic Target in Amyotrophic Lateral Sclerosis. Sci. Transl. Med. 9 (391). doi:10.1126/scitranslmed.aaf3962
Ioannides, Z. A., Ngo, S. T., Henderson, R. D., McCombe, P. A., and Steyn, F. J. (2016). Altered Metabolic Homeostasis in Amyotrophic Lateral Sclerosis: Mechanisms of Energy Imbalance and Contribution to Disease Progression. Neurodegener Dis. 16 (5-6), 382–397. doi:10.1159/000446502
Izrael, M., Slutsky, S. G., and Revel, M. (2020). Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease. Front. Neurosci. 14, 824. doi:10.3389/fnins.2020.00824
Jackson, M., Ganel, R., and Rothstein, J. D. (2002). Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Neurosci. 20, Unit 9 13. doi:10.1002/0471142301.ns0913s20
Jenkins, T. M., Hollinger, H., and McDermott, C. J. (2014). The Evidence for Symptomatic Treatments in Amyotrophic Lateral Sclerosis. Curr. Opin. Neurol. 27 (5), 524–531. doi:10.1097/WCO.0000000000000135
Jessell, T. M., Sürmeli, G., and Kelly, J. S. (2011). Motor Neurons and the Sense of Place. Neuron 72 (3), 419–424. doi:10.1016/j.neuron.2011.10.021
Ji, A.-L., Zhang, X., Chen, W.-W., and Huang, W.-J. (2017). Genetics Insight into the Amyotrophic Lateral Sclerosis/frontotemporal Dementia Spectrum. J. Med. Genet. 54 (3), 145–154. doi:10.1136/jmedgenet-2016-104271
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 68 (5), 857–864. doi:10.1016/j.neuron.2010.11.036
Jongh, R., Spijkers, X. M., Pasteuning‐Vuhman, S., Vulto, P., and Pasterkamp, R. J. (2021). Neuromuscular junction‐on‐a‐chip: ALS Disease Modeling and Read‐out Development in Microfluidic Devices. J. Neurochem. 157, 393–412. doi:10.1111/jnc.15289
Julian, T. H., Glascow, N., Barry, A. D. F., Moll, T., Harvey, C., Klimentidis, Y. C., et al. (2021). Physical Exercise Is a Risk Factor for Amyotrophic Lateral Sclerosis: Convergent Evidence from Mendelian Randomisation, Transcriptomics and Risk Genotypes. EBioMedicine 68, 103397. doi:10.1016/j.ebiom.2021.103397
Juneja, T., Pericak-Vance, M. A., Laing, N. G., Dave, S., and Siddique, T. (1997). Prognosis in Familial Amyotrophic Lateral Sclerosis: Progression and Survival in Patients with Glu100gly and Ala4val Mutations in Cu,Zn Superoxide Dismutase. Neurology 48 (1), 55–57. doi:10.1212/wnl.48.1.55
Kanning, K. C., Kaplan, A., and Henderson, C. E. (2010). Motor Neuron Diversity in Development and Disease. Annu. Rev. Neurosci. 33, 409–440. doi:10.1146/annurev.neuro.051508.135722
Kanouchi, T., Ohkubo, T., and Yokota, T. (2012). Can Regional Spreading of Amyotrophic Lateral Sclerosis Motor Symptoms Be Explained by Prion-like Propagation? J. Neurol. Neurosurg. Psychiatry 83 (7), 739–745. doi:10.1136/jnnp-2011-301826
Kaplan, A., Spiller, K. J., Towne, C., Kanning, K. C., Choe, G. T., Geber, A., et al. (2014). Neuronal Matrix Metalloproteinase-9 Is a Determinant of Selective Neurodegeneration. Neuron 81 (2), 333–348. doi:10.1016/j.neuron.2013.12.009
Karagiannis, P., and Inoue, H. (2020). ALS, a Cellular Whodunit on Motor Neuron Degeneration. Mol. Cell Neurosci. 107, 103524. doi:10.1016/j.mcn.2020.103524
Karagiannis, P., Takahashi, K., Saito, M., Yoshida, Y., Okita, K., Watanabe, A., et al. (2019). Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 99 (1), 79–114. doi:10.1152/physrev.00039.2017
Katz, J. S., Dimachkie, M. M., and Barohn, R. J. (2015). Amyotrophic Lateral Sclerosis. Neurol. Clin. 33 (4), 727–734. doi:10.1016/j.ncl.2015.07.013
Kawada, J., Kaneda, S., Kirihara, T., Maroof, A., Levi, T., Eggan, K., et al. (2017). Generation of a Motor Nerve Organoid with Human Stem Cell-Derived Neurons. Stem Cel Rep. 9 (5), 1441–1449. doi:10.1016/j.stemcr.2017.09.021
Keskin, I., Forsgren, E., Lehmann, M., Andersen, P. M., Brännström, T., Lange, D. J., et al. (2019). The Molecular Pathogenesis of Superoxide Dismutase 1-linked ALS Is Promoted by Low Oxygen Tension. Acta Neuropathol. 138 (1), 85–101. doi:10.1007/s00401-019-01986-1
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic Lateral Sclerosis. The Lancet 377 (9769), 942–955. doi:10.1016/S0140-6736(10)61156-7
Kiernan, M. C., Vucic, S., Talbot, K., McDermott, C. J., Hardiman, O., Shefner, J. M., et al. (2020). Improving Clinical Trial Outcomes in Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 17, 104–118. doi:10.1038/s41582-020-00434-z
Kim, B. W., Jeong, Y. E., Wong, M., and Martin, L. J. (2020a). DNA Damage Accumulates and Responses Are Engaged in Human ALS Brain and Spinal Motor Neurons and DNA Repair Is Activatable in iPSC-Derived Motor Neurons with SOD1 Mutations. Acta Neuropathol. Commun. 8 (1), 7. doi:10.1186/s40478-019-0874-4
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R., and Gitler, A. D. (2020b). ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 108 (5), 822–842. doi:10.1016/j.neuron.2020.08.022
Kim, Y. J., and Lee, G. (2013). Candidate ALS Therapeutics Motor toward "In Vitro Clinical Trials". Cell Stem Cell 12 (6), 633–634. doi:10.1016/j.stem.2013.05.009
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways Disrupted in Human ALS Motor Neurons Identified through Genetic Correction of Mutant SOD1. Cell Stem Cell 14 (6), 781–795. doi:10.1016/j.stem.2014.03.004
Ko, K. D., El-Ghazawi, T., Kim, D., Morizono, H., and Pooled Resource Open-Access, A. L. S. C. T. C. (2014). Predicting the Severity of Motor Neuron Disease Progression Using Electronic Health Record Data with a Cloud Computing Big Data Approach. IEEE Symp. Comput. Intell. Bioinforma Comput. Biol. Proc. 2014. doi:10.1109/CIBCB.2014.6845506
Koch, J. C., Kuttler, J., Maass, F., Lengenfeld, T., Zielke, E., Bähr, M., et al. (2020). Compassionate Use of the ROCK Inhibitor Fasudil in Three Patients with Amyotrophic Lateral Sclerosis. Front. Neurol. 11, 173. doi:10.3389/fneur.2020.00173
Koeman, T., Slottje, P., Schouten, L. J., Peters, S., Huss, A., Veldink, J. H., et al. (2017). Occupational Exposure and Amyotrophic Lateral Sclerosis in a Prospective Cohort. Occup. Environ. Med. 74 (8), 578–585. doi:10.1136/oemed-2016-103780
Kondo, T., Imamura, K., Funayama, M., Tsukita, K., Miyake, M., Ohta, A., et al. (2017). iPSC-Based Compound Screening and In Vitro Trials Identify a Synergistic Anti-amyloid β Combination for Alzheimer's Disease. Cel Rep. 21 (8), 2304–2312. doi:10.1016/j.celrep.2017.10.109
Kourou, K., Exarchos, T. P., Exarchos, K. P., Karamouzis, M. V., and Fotiadis, D. I. (2015). Machine Learning Applications in Cancer Prognosis and Prediction. Comput. Struct. Biotechnol. J. 13, 8–17. doi:10.1016/j.csbj.2014.11.005
Kusaczuk, M. (2019). Tauroursodeoxycholate-Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 8 (12), 1471. doi:10.3390/cells8121471
Kusumoto, D., and Yuasa, S. (2019). The Application of Convolutional Neural Network to Stem Cell Biology. Inflamm. Regener 39, 14. doi:10.1186/s41232-019-0103-3
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 323 (5918), 1205–1208. doi:10.1126/science.1166066
Lamas, N. J., Johnson-Kerner, B., Roybon, L., Kim, Y. A., Garcia-Diaz, A., Wichterle, H., et al. (2014). Neurotrophic Requirements of Human Motor Neurons Defined Using Amplified and Purified Stem Cell-Derived Cultures. PLoS One 9 (10), e110324. doi:10.1371/journal.pone.0110324
Lancaster, M. A., Renner, M., Martin, C.-A., Wenzel, D., Bicknell, L. S., Hurles, M. E., et al. (2013). Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 501 (7467), 373–379. doi:10.1038/nature12517
Landry, D. W., and Zucker, H. A. (2004). Embryonic Death and the Creation of Human Embryonic Stem Cells. J. Clin. Invest. 114 (9), 1184–1186. doi:10.1172/JCI23065
Lange, D. J., Shahbazi, M., Silani, V., Ludolph, A. C., Weishaupt, J. H., Ajroud-Driss, S., et al. (2017). Pyrimethamine Significantly Lowers Cerebrospinal Fluid Cu/Zn Superoxide Dismutase in Amyotrophic Lateral Sclerosis Patients with SOD1 Mutations. Ann. Neurol. 81 (6), 837–848. doi:10.1002/ana.24950
Lanka, V., and Cudkowicz, M. (2008). Therapy Development for ALS: Lessons Learned and Path Forward. Amyotroph. Lateral Scler. 9 (3), 131–140. doi:10.1080/17482960802112819
Laperle, A. H., Sances, S., Yucer, N., Dardov, V. J., Garcia, V. J., Ho, R., et al. (2020). iPSC Modeling of Young-Onset Parkinson's Disease Reveals a Molecular Signature of Disease and Novel Therapeutic Candidates. Nat. Med. 26 (2), 289–299. doi:10.1038/s41591-019-0739-1
Lau, C. L., Perreau, V. M., Chen, M. J., Cate, H. S., Merlo, D., Cheung, N. S., et al. (2012). Transcriptomic Profiling of Astrocytes Treated with the Rho Kinase Inhibitor Fasudil Reveals Cytoskeletal and Pro-survival Responses. J. Cel. Physiol. 227 (3), 1199–1211. doi:10.1002/jcp.22838
Lau, C., O'Shea, R., Broberg, B., Bischof, L., and Beart, P. (2011). The Rho Kinase Inhibitor Fasudil Up-Regulates Astrocytic Glutamate Transport Subsequent to Actin Remodelling in Murine Cultured Astrocytes. Br. J. Pharmacol. 163 (3), 533–545. doi:10.1111/j.1476-5381.2011.01259.x
Leblond, C. S., Kaneb, H. M., Dion, P. A., and Rouleau, G. A. (2014). Dissection of Genetic Factors Associated with Amyotrophic Lateral Sclerosis. Exp. Neurol. 262, 91–101. doi:10.1016/j.expneurol.2014.04.013
Lee, J.-H., Liu, J.-W., Lin, S.-Z., Harn, H.-J., and Chiou, T.-W. (2018). Advances in Patient-specific Induced Pluripotent Stem Cells Shed Light on Drug Discovery for Amyotrophic Lateral Sclerosis. Cel Transpl. 27 (9), 1301–1312. doi:10.1177/0963689718785154
Lee, J., Hyeon, S. J., Im, H., Ryu, H., Kim, Y., and Ryu, H. (2016). Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 25 (5), 233–240. doi:10.5607/en.2016.25.5.233
Lerou, P., and Daley, G. (2005). Therapeutic Potential of Embryonic Stem Cells. Blood Rev. 19 (6), 321–331. doi:10.1016/j.blre.2005.01.005
Li, H., Kuwajima, T., Oakley, D., Nikulina, E., Hou, J., Yang, W. S., et al. (2016). Protein Prenylation Constitutes an Endogenous Brake on Axonal Growth. Cel Rep. 16 (2), 545–558. doi:10.1016/j.celrep.2016.06.013
Li, L., and Shi, Y. (2020). When Glia Meet Induced Pluripotent Stem Cells (iPSCs). Mol. Cell Neurosci. 109, 103565. doi:10.1016/j.mcn.2020.103565
Li, X.-J., Du, Z.-W., Zarnowska, E. D., Pankratz, M., Hansen, L. O., Pearce, R. A., et al. (2005). Specification of Motoneurons from Human Embryonic Stem Cells. Nat. Biotechnol. 23 (2), 215–221. doi:10.1038/nbt1063
Li, Y., Balasubramanian, U., Cohen, D., Zhang, P.-W., Mosmiller, E., Sattler, R., et al. (2015). A Comprehensive Library of Familial Human Amyotrophic Lateral Sclerosis Induced Pluripotent Stem Cells. PLoS One 10 (3), e0118266. doi:10.1371/journal.pone.0118266
Lingor, P., Weber, M., Camu, W., Friede, T., Hilgers, R., Leha, A., et al. (2019). ROCK-ALS: Protocol for a Randomized, Placebo-Controlled, Double-Blind Phase IIa Trial of Safety, Tolerability and Efficacy of the Rho Kinase (ROCK) Inhibitor Fasudil in Amyotrophic Lateral Sclerosis. Front. Neurol. 10, 293. doi:10.3389/fneur.2019.00293
Liu, G., David, B. T., Trawczynski, M., and Fessler, R. G. (2020). Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cel Rev Rep 16 (1), 3–32. doi:10.1007/s12015-019-09935-x
Logan, S., Arzua, T., Canfield, S. G., Seminary, E. R., Sison, S. L., Ebert, A. D., et al. (2019). Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Compr. Physiol. 9 (2), 565–611. doi:10.1002/cphy.c180025
Lopez-Gonzalez, R., Lu, Y., Gendron, T. F., Karydas, A., Tran, H., Yang, D., et al. (2016). Poly(GR) in C9ORF72 -Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 92 (2), 383–391. doi:10.1016/j.neuron.2016.09.015
Ludolph, A. C., Bendotti, C., Blaugrund, E., Chio, A., Greensmith, L., Loeffler, J.-P., et al. (2010). Guidelines for Preclinical Animal Research in ALS/MND: A Consensus Meeting. Amyotroph. Lateral Scler. 11 (1-2), 38–45. doi:10.3109/17482960903545334
Ludolph, A. C., and Sperfeld, A.-D. (2005). Preclinical Trials - an Update on Translational Research in ALS. Neurodegener Dis. 2 (3-4), 215–219. doi:10.1159/000089628
Ludolph, A. C. (2011). Urgently Needed-Biomarkers for Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 7 (1), 13–14. doi:10.1038/nrneurol.2010.196
Ludolph, A., Drory, V., Hardiman, O., Nakano, I., Ravits, J., Robberecht, W., et al. (2015). A Revision of the El Escorial Criteria - 2015. Amyotroph. Lateral Scler. Frontotemporal Degeneration 16 (5-6), 291–292. doi:10.3109/21678421.2015.1049183
Lutz, C. (2018). Mouse Models of ALS: Past, Present and Future. Brain Res. 1693 (Pt A), 1–10. doi:10.1016/j.brainres.2018.03.024
Malek, A. M., Barchowsky, A., Bowser, R., Heiman-Patterson, T., Lacomis, D., Rana, S., et al. (2014). Environmental and Occupational Risk Factors for Amyotrophic Lateral Sclerosis: a Case-Control Study. Neurodegener Dis. 14 (1), 31–38. doi:10.1159/000355344
Mannen, T., Iwata, M., Toyokura, Y., and Nagashima, K. (1977). Preservation of a Certain Motoneurone Group of the Sacral Cord in Amyotrophic Lateral Sclerosis: its Clinical Significance. J. Neurol. Neurosurg. Psychiatry 40 (5), 464–469. doi:10.1136/jnnp.40.5.464
Mannen, T., Iwata, M., Toyokura, Y., and Nagashima, K. (1982). The Onuf's Nucleus and the External Anal Sphincter Muscles in Amyotrophic Lateral Sclerosis and Shy-Drager Syndrome. Acta Neuropathol. 58 (4), 255–260. doi:10.1007/bf00688606
Martin, G. R. (1981). Isolation of a Pluripotent Cell Line from Early Mouse Embryos Cultured in Medium Conditioned by Teratocarcinoma Stem Cells. Proc. Natl. Acad. Sci. 78 (12), 7634–7638. doi:10.1073/pnas.78.12.7634
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature 465 (7295), 223–226. doi:10.1038/nature08971
Masrori, P., and Van Damme, P. (2020). Amyotrophic Lateral Sclerosis: a Clinical Review. Eur. J. Neurol. 27 (10), 1918–1929. doi:10.1111/ene.14393
Maury, Y., Côme, J., Piskorowski, R. A., Salah-Mohellibi, N., Chevaleyre, V., Peschanski, M., et al. (2015). Combinatorial Analysis of Developmental Cues Efficiently Converts Human Pluripotent Stem Cells into Multiple Neuronal Subtypes. Nat. Biotechnol. 33 (1), 89–96. doi:10.1038/nbt.3049
McAleese, K. E., Walker, L., Erskine, D., Thomas, A. J., McKeith, I. G., and Attems, J. (2017). TDP-43 Pathology in Alzheimer's Disease, Dementia with Lewy Bodies and Ageing. Brain Pathol. 27 (4), 472–479. doi:10.1111/bpa.12424
McDermott, C. J., and Shaw, P. J. (2008). Diagnosis and Management of Motor Neurone Disease. BMJ 336 (7645), 658–662. doi:10.1136/bmj.39493.511759.BE
McGoldrick, P., Joyce, P. I., Fisher, E. M. C., and Greensmith, L. (2013). Rodent Models of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1832 (9), 1421–1436. doi:10.1016/j.bbadis.2013.03.012
McNeish, J., Gardner, J. P., Wainger, B. J., Woolf, C. J., and Eggan, K. (2015). From Dish to Bedside: Lessons Learned while Translating Findings from a Stem Cell Model of Disease to a Clinical Trial. Cell Stem Cell 17 (1), 8–10. doi:10.1016/j.stem.2015.06.013
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 13, 1310. doi:10.3389/fnins.2019.01310
Miles, G. B., Yohn, D. C., Wichterle, H., Jessell, T. M., Rafuse, V. F., and Brownstone, R. M. (2004). Functional Properties of Motoneurons Derived from Mouse Embryonic Stem Cells. J. Neurosci. 24 (36), 7848–7858. doi:10.1523/JNEUROSCI.1972-04.2004
Millen, G. C., and Yap, C. (2020). Adaptive Trial Designs: what Are Multiarm, Multistage Trials? Arch. Dis. Child. Educ. Pract. Ed. 105 (6), edpract–2019. doi:10.1136/archdischild-2019-317826
Miller, R. G., Mitchell, J. D., Lyon, M., and Moore, D. H. (2007). Riluzole for Amyotrophic Lateral Sclerosis (ALS)/motor Neuron Disease (MND). Cochrane Database Syst. Rev. 1, CD001447. doi:10.1002/14651858.CD001447.pub2
Miller, R. G., Mitchell, J. D., and Moore, D. H. (2012). Riluzole for Amyotrophic Lateral Sclerosis (ALS)/motor Neuron Disease (MND). Cochrane Database Syst. Rev. 3, CD001447. doi:10.1002/14651858.CD001447.pub3
Miller, T., Cudkowicz, M., Shaw, P. J., Andersen, P. M., Atassi, N., Bucelli, R. C., et al. (2020). Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 383 (2), 109–119. doi:10.1056/NEJMoa2003715
Mitne-Neto, M., Machado-Costa, M., Marchetto, M. C. N., Bengtson, M. H., Joazeiro, C. A., Tsuda, H., et al. (2011). Downregulation of VAPB Expression in Motor Neurons Derived from Induced Pluripotent Stem Cells of ALS8 Patients. Hum. Mol. Genet. 20 (18), 3642–3652. doi:10.1093/hmg/ddr284
Mitsumoto, H., Brooks, B. R., and Silani, V. (2014). Clinical Trials in Amyotrophic Lateral Sclerosis: Why So many Negative Trials and How Can Trials Be Improved? Lancet Neurol. 13 (11), 1127–1138. doi:10.1016/S1474-4422(14)70129-2
Mitsumoto, H. (2018). What if You Knew the Prognosis of Your Patients with ALS? Lancet Neurol. 17 (5), 386–388. doi:10.1016/S1474-4422(18)30111-X
Miura, Y., Li, M.-Y., Birey, F., Ikeda, K., Revah, O., Thete, M. V., et al. (2020). Generation of Human Striatal Organoids and Cortico-Striatal Assembloids from Human Pluripotent Stem Cells. Nat. Biotechnol. 38 (12), 1421–1430. doi:10.1038/s41587-020-00763-w
Montalbano, M., McAllen, S., Cascio, F. L., Sengupta, U., Garcia, S., Bhatt, N., et al. (2020). TDP-43 and Tau Oligomers in Alzheimer's Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 146, 105130. doi:10.1016/j.nbd.2020.105130
Morello, G., Salomone, S., D’Agata, V., Conforti, F. L., and Cavallaro, S. (2020). From Multi-Omics Approaches to Precision Medicine in Amyotrophic Lateral Sclerosis. Front. Neurosci. 14, 577755. doi:10.3389/fnins.2020.577755
Morgan, S., Duguez, S., and Duddy, W. (2018). Personalized Medicine and Molecular Interaction Networks in Amyotrophic Lateral Sclerosis (ALS): Current Knowledge. Jpm 8 (4), 44. doi:10.3390/jpm8040044
Moujalled, D., and White, A. R. (2016). Advances in the Development of Disease-Modifying Treatments for Amyotrophic Lateral Sclerosis. CNS Drugs 30 (3), 227–243. doi:10.1007/s40263-016-0317-8
Mourad, M., Moubayed, S., Dezube, A., Mourad, Y., Park, K., Torreblanca-Zanca, A., et al. (2020). Machine Learning and Feature Selection Applied to SEER Data to Reliably Assess Thyroid Cancer Prognosis. Sci. Rep. 10 (1), 5176. doi:10.1038/s41598-020-62023-w
Narsinh, K. H., Plews, J., and Wu, J. C. (2011). Comparison of Human Induced Pluripotent and Embryonic Stem Cells: Fraternal or Identical Twins? Mol. Ther. 19 (4), 635–638. doi:10.1038/mt.2011.41
Naujock, M., Stanslowsky, N., Bufler, S., Naumann, M., Reinhardt, P., Sterneckert, J., et al. (2016). 4-Aminopyridine Induced Activity Rescues Hypoexcitable Motor Neurons from Amyotrophic Lateral Sclerosis Patient-Derived Induced Pluripotent Stem Cells. Stem Cells 34 (6), 1563–1575. doi:10.1002/stem.2354
Naumann, M., Pal, A., Goswami, A., Lojewski, X., Japtok, J., Vehlow, A., et al. (2018). Impaired DNA Damage Response Signaling by FUS-NLS Mutations Leads to Neurodegeneration and FUS Aggregate Formation. Nat. Commun. 9 (1), 335. doi:10.1038/s41467-017-02299-1
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 97 (6), 1268–e1266. doi:10.1016/j.neuron.2018.02.027
Nijssen, J., Comley, L. H., and Hedlund, E. (2017). Motor Neuron Vulnerability and Resistance in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 133 (6), 863–885. doi:10.1007/s00401-017-1708-8
Nishimura, A. L., Mitne-Neto, M., Silva, H. C. A., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A Mutation in the Vesicle-Trafficking Protein VAPB Causes Late-Onset Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis. Am. J. Hum. Genet. 75 (5), 822–831. doi:10.1086/425287
Nizzardo, M., Simone, C., Falcone, M., Locatelli, F., Riboldi, G., Comi, G. P., et al. (2010). Human Motor Neuron Generation from Embryonic Stem Cells and Induced Pluripotent Stem Cells. Cell. Mol. Life Sci. 67 (22), 3837–3847. doi:10.1007/s00018-010-0463-y
Norris, S. P., Likanje, M.-F. N., and Andrews, J. A. (2020). Amyotrophic Lateral Sclerosis: Update on Clinical Management. Curr. Opin. Neurol. 33 (5), 641–648. doi:10.1097/WCO.0000000000000864
Ohgushi, M., and Sasai, Y. (2011). Lonely Death Dance of Human Pluripotent Stem Cells: ROCKing between Metastable Cell States. Trends Cel Biol. 21 (5), 274–282. doi:10.1016/j.tcb.2011.02.004
Okada, M., Yamashita, S., Ueyama, H., Ishizaki, M., Maeda, Y., and Ando, Y. (2018). Long-term Effects of Edaravone on Survival of Patients with Amyotrophic Lateral Sclerosis. eNeurologicalSci 11, 11–14. doi:10.1016/j.ensci.2018.05.001
Okamoto, Y., Shirakashi, Y., Ihara, M., Urushitani, M., Oono, M., Kawamoto, Y., et al. (2011). Colocalization of 14-3-3 Proteins with SOD1 in Lewy Body-like Hyaline Inclusions in Familial Amyotrophic Lateral Sclerosis Cases and the Animal Model. PLoS One 6 (5), e20427. doi:10.1371/journal.pone.0020427
Okano, H., Yasuda, D., Fujimori, K., Morimoto, S., and Takahashi, S. (2020). Ropinirole, a New ALS Drug Candidate Developed Using iPSCs. Trends Pharmacol. Sci. 41 (2), 99–109. doi:10.1016/j.tips.2019.12.002
Olesen, J., Gustavsson, A., Svensson, M., Wittchen, H.-U., Jönsson, B., group, C. s., et al. (2012). The Economic Cost of Brain Disorders in Europe. Eur. J. Neurol. 19 (1), 155–162. doi:10.1111/j.1468-1331.2011.03590.x
Orrell, R. W. (2007). Understanding the Causes of Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 357 (8), 822–823. doi:10.1056/NEJMe078146
Oskarsson, B., Horton, D. K., and Mitsumoto, H. (2015). Potential Environmental Factors in Amyotrophic Lateral Sclerosis. Neurol. Clin. 33 (4), 877–888. doi:10.1016/j.ncl.2015.07.009
Otto, M., Bowser, R., Turner, M., Berry, J., Brettschneider, J., Connor, J., et al. (2012). Roadmap and Standard Operating Procedures for Biobanking and Discovery of Neurochemical Markers in ALS. Amyotroph. Lateral Scler. 13 (1), 1–10. doi:10.3109/17482968.2011.627589
Paganoni, S., Macklin, E. A., Hendrix, S., Berry, J. D., Elliott, M. A., Maiser, S., et al. (2020). Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 383 (10), 919–930. doi:10.1056/NEJMoa1916945
Palmer, T. D., Schwartz, P. H., Taupin, P., Kaspar, B., Stein, S. A., and Gage, F. H. (2001). Progenitor Cells from Human Brain after Death. Nature 411 (6833), 42–43. doi:10.1038/35075141
Pan, G., Wang, T., Yao, H., and Pei, D. (2012). Somatic Cell Reprogramming for Regenerative Medicine: SCNT vs. iPS Cells. Bioessays 34 (6), 472–476. doi:10.1002/bies.201100174
Panoutsopoulos, A. A. (2020). Organoids, Assembloids, and Novel Biotechnology: Steps Forward in Developmental and Disease-Related Neuroscience. Neuroscientist 27, 463–472. doi:10.1177/1073858420960112
Pasinelli, P., and Brown, R. H. (2006). Molecular Biology of Amyotrophic Lateral Sclerosis: Insights from Genetics. Nat. Rev. Neurosci. 7 (9), 710–723. doi:10.1038/nrn1971
Pasteuning-Vuhman, S., de Jongh, R., Timmers, A., and Pasterkamp, R. J. (2021). Towards Advanced iPSC-Based Drug Development for Neurodegenerative Disease. Trends Mol. Med. 27, 263–279. doi:10.1016/j.molmed.2020.09.013
Peljto, M., Dasen, J. S., Mazzoni, E. O., Jessell, T. M., and Wichterle, H. (2010). Functional Diversity of ESC-Derived Motor Neuron Subtypes Revealed through Intraspinal Transplantation. Cell Stem Cell 7 (3), 355–366. doi:10.1016/j.stem.2010.07.013
Petri, S., Krampfl, K., Kuhlemann, K., Dengler, R., and Grothe, C. (2009). Preserved Expression of Fibroblast Growth Factor (FGF)-2 and FGF Receptor 1 in Brain and Spinal Cord of Amyotrophic Lateral Sclerosis Patients. Histochem. Cel Biol 131 (4), 509–519. doi:10.1007/s00418-008-0549-x
Petrov, D., Mansfield, C., Moussy, A., and Hermine, O. (2017). ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 9, 68. doi:10.3389/fnagi.2017.00068
Phatnani, H., and Maniatis, T. (2015). Astrocytes in Neurodegenerative Disease: Table 1. Cold Spring Harb Perspect. Biol. 7 (6), a020628. doi:10.1101/cshperspect.a020628
Philips, T., and Robberecht, W. (2011). Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Glial Activation in Motor Neuron Disease. Lancet Neurol. 10 (3), 253–263. doi:10.1016/S1474-4422(11)70015-1
Philips, T., and Rothstein, J. D. (2015). Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 69, 67 61–65 67 21. doi:10.1002/0471141755.ph0567s69
Pomeshchik, Y., Klementieva, O., Gil, J., Martinsson, I., Hansen, M. G., de Vries, T., et al. (2020). Human iPSC-Derived Hippocampal Spheroids: An Innovative Tool for Stratifying Alzheimer Disease Patient-specific Cellular Phenotypes and Developing Therapies. Stem Cel Rep. 15 (1), 256–273. doi:10.1016/j.stemcr.2020.06.001
Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L. F., Tokito, M., Mann, E., et al. (2003). Mutant Dynactin in Motor Neuron Disease. Nat. Genet. 33 (4), 455–456. doi:10.1038/ng1123
Pun, S., Santos, A. F., Saxena, S., Xu, L., and Caroni, P. (2006). Selective Vulnerability and Pruning of Phasic Motoneuron Axons in Motoneuron Disease Alleviated by CNTF. Nat. Neurosci. 9 (3), 408–419. doi:10.1038/nn1653
Puri, M. C., and Nagy, A. (2012). Concise Review: Embryonic Stem Cells versus Induced Pluripotent Stem Cells: the Game Is on. Stem Cells 30 (1), 10–14. doi:10.1002/stem.788
Ravits, J., Appel, S., Baloh, R. H., Barohn, R., Rix Brooks, B., Elman, L., et al. (2013). Deciphering Amyotrophic Lateral Sclerosis: what Phenotype, Neuropathology and Genetics Are Telling Us about Pathogenesis. Amyotroph. Lateral Scler. Frontotemporal Degeneration 14 (Suppl. 1), 5–18. doi:10.3109/21678421.2013.778548
Ravits, J. (2014). Focality, Stochasticity and Neuroanatomic Propagation in ALS Pathogenesis. Exp. Neurol. 262, 121–126. doi:10.1016/j.expneurol.2014.07.021
Ravits, J. M., and La Spada, A. R. (2009). ALS Motor Phenotype Heterogeneity, Focality, and Spread: Deconstructing Motor Neuron Degeneration. Neurology 73 (10), 805–811. doi:10.1212/WNL.0b013e3181b6bbbd
Ravits, J., Paul, P., and Jorg, C. (2007). Focality of Upper and Lower Motor Neuron Degeneration at the Clinical Onset of ALS. Neurology 68 (19), 1571–1575. doi:10.1212/01.wnl.0000260965.20021.47
Readhead, B., Hartley, B. J., Eastwood, B. J., Collier, D. A., Evans, D., Farias, R., et al. (2018). Expression-based Drug Screening of Neural Progenitor Cells from Individuals with Schizophrenia. Nat. Commun. 9 (1), 4412. doi:10.1038/s41467-018-06515-4
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 72 (2), 257–268. doi:10.1016/j.neuron.2011.09.010
Restagno, G., Gomez, A., Lombardo, F., Cocco, E., Calvo, A., Ghiglione, P., et al. (2005). The IVS1 +319 T>a of SOD1 Gene Is Not an ALS Causing Mutation. Amyotroph. Lateral Scler. 6 (1), 45–49. doi:10.1080/14660820410021276
Riboldi, G., Zanetta, C., Ranieri, M., Nizzardo, M., Simone, C., Magri, F., et al. (2014). Antisense Oligonucleotide Therapy for the Treatment of C9ORF72 ALS/FTD Diseases. Mol. Neurobiol. 50 (3), 721–732. doi:10.1007/s12035-014-8724-7
Richard, J.-P., and Maragakis, N. J. (2015). Induced Pluripotent Stem Cells from ALS Patients for Disease Modeling. Brain Res. 1607, 15–25. doi:10.1016/j.brainres.2014.09.017
Robinton, D. A., and Daley, G. Q. (2012). The Promise of Induced Pluripotent Stem Cells in Research and Therapy. Nature 481 (7381), 295–305. doi:10.1038/nature10761
Roche, J. C., Rojas-Garcia, R., Scott, K. M., Scotton, W., Ellis, C. E., Burman, R., et al. (2012). A Proposed Staging System for Amyotrophic Lateral Sclerosis. Brain 135 (Pt 3), 847–852. doi:10.1093/brain/awr351
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 362 (6415), 59–62. doi:10.1038/362059a0
Rothstein, J. D., Patel, S., Regan, M. R., Haenggeli, C., Huang, Y. H., Bergles, D. E., et al. (2005). β-Lactam Antibiotics Offer Neuroprotection by Increasing Glutamate Transporter Expression. Nature 433 (7021), 73–77. doi:10.1038/nature03180
Rowe, R. G., and Daley, G. Q. (2019). Induced Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Genet. 20 (7), 377–388. doi:10.1038/s41576-019-0100-z
Rowland, L. P. (2001). How Amyotrophic Lateral Sclerosis Got its Name. Arch. Neurol. 58 (3), 512–515. doi:10.1001/archneur.58.3.512
Rowland, L. P., and Shneider, N. A. (2001). Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 344 (22), 1688–1700. doi:10.1056/NEJM200105313442207
Roy, A., Ghosh, A., Jana, A., Liu, X., Brahmachari, S., Gendelman, H. E., et al. (2012). Sodium Phenylbutyrate Controls Neuroinflammatory and Antioxidant Activities and Protects Dopaminergic Neurons in Mouse Models of Parkinson's Disease. PLoS One 7 (6), e38113. doi:10.1371/journal.pone.0038113
Roybon, L., Lamas, N. J., Garcia-Diaz, A., Yang, E. J., Sattler, R., Jackson-Lewis, V., et al. (2013). Human Stem Cell-Derived Spinal Cord Astrocytes with Defined Mature or Reactive Phenotypes. Cel Rep. 4 (5), 1035–1048. doi:10.1016/j.celrep.2013.06.021
Rubino, E., Rainero, I., Chio, A., Rogaeva, E., Galimberti, D., Fenoglio, P., et al. (2012). SQSTM1 Mutations in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Neurology 79 (15), 1556–1562. doi:10.1212/WNL.0b013e31826e25df
Ryan, M., Heverin, M., McLaughlin, R. L., and Hardiman, O. (2019). Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 76, 1367. doi:10.1001/jamaneurol.2019.2044
Ryu, H., Smith, K., Camelo, S. I., Carreras, I., Lee, J., Iglesias, A. H., et al. (2005). Sodium Phenylbutyrate Prolongs Survival and Regulates Expression of Anti-apoptotic Genes in Transgenic Amyotrophic Lateral Sclerosis Mice. J. Neurochem. 93 (5), 1087–1098. doi:10.1111/j.1471-4159.2005.03077.x
Saberi, S., Stauffer, J. E., Schulte, D. J., and Ravits, J. (2015). Neuropathology of Amyotrophic Lateral Sclerosis and its Variants. Neurol. Clin. 33 (4), 855–876. doi:10.1016/j.ncl.2015.07.012
Sances, S., Bruijn, L. I., Chandran, S., Eggan, K., Ho, R., Klim, J. R., et al. (2016). Modeling ALS with Motor Neurons Derived from Human Induced Pluripotent Stem Cells. Nat. Neurosci. 19 (4), 542–553. doi:10.1038/nn.4273
Sareen, D., O’Rourke, J. G., Meera, P., Muhammad, A. K. M. G., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA Foci in iPSC-Derived Motor Neurons from ALS Patients with a C9ORF72 Repeat Expansion. Sci. Transl. Med. 5 (208), 208ra149. doi:10.1126/scitranslmed.3007529
Sato, N., Sanjuan, I. M., Heke, M., Uchida, M., Naef, F., and Brivanlou, A. H. (2003). Molecular Signature of Human Embryonic Stem Cells and its Comparison with the Mouse. Developmental Biol. 260 (2), 404–413. doi:10.1016/s0012-1606(03)00256-2
Saul, J., Hutchins, E., Reiman, R., Saul, M., Ostrow, L. W., Harris, B. T., et al. (2020). Global Alterations to the Choroid Plexus Blood-CSF Barrier in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 8 (1), 92. doi:10.1186/s40478-020-00968-9
Savchenko, E., Teku, G. N., Boza-Serrano, A., Russ, K., Berns, M., Deierborg, T., et al. (2019). FGF Family Members Differentially Regulate Maturation and Proliferation of Stem Cell-Derived Astrocytes. Sci. Rep. 9 (1), 9610. doi:10.1038/s41598-019-46110-1
Saxena, S., and Caroni, P. (2011). Selective Neuronal Vulnerability in Neurodegenerative Diseases: from Stressor Thresholds to Degeneration. Neuron 71 (1), 35–48. doi:10.1016/j.neuron.2011.06.031
Schmitt, F., Hussain, G., Dupuis, L., Loeffler, J.-P., and Henriques, A. (2014). A Plural Role for Lipids in Motor Neuron Diseases: Energy, Signaling and Structure. Front. Cel. Neurosci. 8, 25. doi:10.3389/fncel.2014.00025
Schnerch, A., Cerdan, C., and Bhatia, M. (2010). Distinguishing between Mouse and Human Pluripotent Stem Cell Regulation: the Best Laid Plans of Mice and Men. Stem Cells 28 (3), A–N. doi:10.1002/stem.298
Schrøder, H. D., and Reske-Nielsen, E. (1984). Preservation of the Nucleus X-Pelvic Floor Motosystem in Amyotrophic Lateral Sclerosis. Clin. Neuropathol. 3 (5), 210–216.
Schweitzer, J. S., Song, B., Herrington, T. M., Park, T.-Y., Lee, N., Ko, S., et al. (2020). Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson's Disease. N. Engl. J. Med. 382 (20), 1926–1932. doi:10.1056/NEJMoa1915872
Selvaraj, B. T., Livesey, M. R., Zhao, C., Gregory, J. M., James, O. T., Cleary, E. M., et al. (2018). C9ORF72 Repeat Expansion Causes Vulnerability of Motor Neurons to Ca2+-Permeable AMPA Receptor-Mediated Excitotoxicity. Nat. Commun. 9 (1), 347. doi:10.1038/s41467-017-02729-0
Seminary, E. R., Sison, S. L., and Ebert, A. D. (2018). Modeling Protein Aggregation and the Heat Shock Response in ALS iPSC-Derived Motor Neurons. Front. Neurosci. 12, 86. doi:10.3389/fnins.2018.00086
Serio, A., and Patani, R. (2018). Concise Review: The Cellular Conspiracy of Amyotrophic Lateral Sclerosis. Stem Cells 36 (3), 293–303. doi:10.1002/stem.2758
Sharma, A., Lyashchenko, A. K., Lu, L., Nasrabady, S. E., Elmaleh, M., Mendelsohn, M., et al. (2016). ALS-associated Mutant FUS Induces Selective Motor Neuron Degeneration through Toxic Gain of Function. Nat. Commun. 7, 10465. doi:10.1038/ncomms10465
Shatunov, A., and Al-Chalabi, A. (2021). The Genetic Architecture of ALS. Neurobiol. Dis. 147, 105156. doi:10.1016/j.nbd.2020.105156
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W.-H., Hung, S.-T., et al. (2018). Haploinsufficiency Leads to Neurodegeneration in C9ORF72 ALS/FTD Human Induced Motor Neurons. Nat. Med. 24 (3), 313–325. doi:10.1038/nm.4490
Shinde, V., Sureshkumar, P., Sotiriadou, I., Hescheler, J., and Sachinidis, A. (2016). Human Embryonic and Induced Pluripotent Stem Cell Based Toxicity Testing Models: Future Applications in New Drug Discovery. Cmc 23 (30), 3495–3509. doi:10.2174/0929867323666160627113436
Silani, V., Brioschi, A., Braga, M., Ciammola, A., Zhou, F. C., Bonifati, C., et al. (1998). Immunomagnetic Isolation of Human Developing Motor Neurons. Neuroreport 9 (6), 1143–1147. doi:10.1097/00001756-199804200-00034
Silva, M. C., and Haggarty, S. J. (2020). Human Pluripotent Stem Cell-Derived Models and Drug Screening in CNS Precision Medicine. Ann. N.Y. Acad. Sci. 1471 (1), 18–56. doi:10.1111/nyas.14012
Singh Roy, N., Nakano, T., Xuing, L., Kang, J., Nedergaard, M., and Goldman, S. A. (2005). Enhancer-specified GFP-Based FACS Purification of Human Spinal Motor Neurons from Embryonic Stem Cells. Exp. Neurol. 196 (2), 224–234. doi:10.1016/j.expneurol.2005.06.021
Sloan, S. A., Andersen, J., Pașca, A. M., Birey, F., and Pașca, S. P. (2018). Generation and Assembly of Human Brain Region-specific Three-Dimensional Cultures. Nat. Protoc. 13 (9), 2062–2085. doi:10.1038/s41596-018-0032-7
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide Rare Variant Analysis Identifies TUBA4A Mutations Associated with Familial ALS. Neuron 84 (2), 324–331. doi:10.1016/j.neuron.2014.09.027
Spencer, K. R., Foster, Z. W., Rauf, N. A., Guilderson, L., Collins, D., Averill, J. G., et al. (2020). Neuropathological Profile of Long‐duration Amyotrophic Lateral Sclerosis in Military Veterans. Brain Pathol. 30 (6), 1028–1040. doi:10.1111/bpa.12876
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 319 (5870), 1668–1672. doi:10.1126/science.1154584
Stadtfeld, M., and Hochedlinger, K. (2010). Induced Pluripotency: History, Mechanisms, and Applications. Genes Development 24 (20), 2239–2263. doi:10.1101/gad.1963910
Stern, S., Linker, S., Vadodaria, K. C., Marchetto, M. C., and Gage, F. H. (2018a). Prediction of Response to Drug Therapy in Psychiatric Disorders. Open Biol. 8 (5), 180031. doi:10.1098/rsob.180031
Stern, S., Santos, R., Marchetto, M. C., Mendes, A. P. D., Rouleau, G. A., Biesmans, S., et al. (2018b). Neurons Derived from Patients with Bipolar Disorder divide into Intrinsically Different Sub-populations of Neurons, Predicting the Patients' Responsiveness to Lithium. Mol. Psychiatry 23 (6), 1453–1465. doi:10.1038/mp.2016.260
Strong, M. J., Abrahams, S., Goldstein, L. H., Woolley, S., McLaughlin, P., Snowden, J., et al. (2017). Amyotrophic Lateral Sclerosis - Frontotemporal Spectrum Disorder (ALS-FTSD): Revised Diagnostic Criteria. Amyotroph. Lateral Scler. Frontotemporal Degeneration 18 (3-4), 153–174. doi:10.1080/21678421.2016.1267768
Su, X. W., Broach, J. R., Connor, J. R., Gerhard, G. S., and Simmons, Z. (2014). Genetic Heterogeneity of Amyotrophic Lateral Sclerosis: Implications for Clinical Practice and Research. Muscle Nerve 49 (6), 786–803. doi:10.1002/mus.24198
Suga, M., Kondo, T., and Inoue, H. (2019). Modeling Neurological Disorders with Human Pluripotent Stem Cell-Derived Astrocytes. Ijms 20 (16), 3862. doi:10.3390/ijms20163862
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 131 (5), 861–872. doi:10.1016/j.cell.2007.11.019
Takahashi, K., and Yamanaka, S. (2006). Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126 (4), 663–676. doi:10.1016/j.cell.2006.07.024
Takata, M., Tanaka, H., Kimura, M., Nagahara, Y., Tanaka, K., Kawasaki, K., et al. (2013). Fasudil, a Rho Kinase Inhibitor, Limits Motor Neuron Loss in Experimental Models of Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 170 (2), 341–351. doi:10.1111/bph.12277
Takeda, T., Kitagawa, K., and Arai, K. (2020). Phenotypic Variability and its Pathological Basis in Amyotrophic Lateral Sclerosis. Neuropathology 40 (1), 40–56. doi:10.1111/neup.12606
Talbot, K., and Oxford University Press., (2010). in Motor Neuron Disease a Practical manualOxford Care Manuals (Oxford: Oxford University Press).
Tao, Y., and Zhang, S.-C. (2016). Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell 19 (5), 573–586. doi:10.1016/j.stem.2016.10.015
Taylor, P. L. (2011). Responsibility Rewarded: Ethics, Engagement, and Scientific Autonomy in the Labyrinth of the Minotaur. Neuron 70 (4), 577–581. doi:10.1016/j.neuron.2011.05.009
Teyssou, E., Takeda, T., Lebon, V., Boillée, S., Doukouré, B., Bataillon, G., et al. (2013). Mutations in SQSTM1 Encoding P62 in Amyotrophic Lateral Sclerosis: Genetics and Neuropathology. Acta Neuropathol. 125 (4), 511–522. doi:10.1007/s00401-013-1090-0
Thams, S., Lowry, E. R., Larraufie, M.-H., Spiller, K. J., Li, H., Williams, D. J., et al. (2019). A Stem Cell-Based Screening Platform Identifies Compounds that Desensitize Motor Neurons to Endoplasmic Reticulum Stress. Mol. Ther. 27 (1), 87–101. doi:10.1016/j.ymthe.2018.10.010
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 282 (5391), 1145–1147. doi:10.1126/science.282.5391.1145
Thonhoff, J. R., Simpson, E. P., and Appel, S. H. (2018). Neuroinflammatory Mechanisms in Amyotrophic Lateral Sclerosis Pathogenesis. Curr. Opin. Neurol. 31 (5), 635–639. doi:10.1097/WCO.0000000000000599
Tiscornia, G., Vivas, E. L., and Belmonte, J. C. I. (2011). Diseases in a Dish: Modeling Human Genetic Disorders Using Induced Pluripotent Cells. Nat. Med. 17 (12), 1570–1576. doi:10.1038/nm.2504
Tomé, S. O., Vandenberghe, R., Ospitalieri, S., Van Schoor, E., Tousseyn, T., Otto, M., et al. (2020). Distinct Molecular Patterns of TDP-43 Pathology in Alzheimer's Disease: Relationship with Clinical Phenotypes. Acta Neuropathol. Commun. 8 (1), 61. doi:10.1186/s40478-020-00934-5
Tönges, L., Günther, R., Suhr, M., Jansen, J., Balck, A., Saal, K.-A., et al. (2014). Rho Kinase Inhibition Modulates Microglia Activation and Improves Survival in a Model of Amyotrophic Lateral Sclerosis. Glia 62 (2), 217–232. doi:10.1002/glia.22601
Turner, B., and Talbot, K. (2008). Transgenics, Toxicity and Therapeutics in Rodent Models of Mutant SOD1-Mediated Familial ALS. Prog. Neurobiol. 85 (1), 94–134. doi:10.1016/j.pneurobio.2008.01.001
Vadodaria, K. C., Ji, Y., Skime, M., Paquola, A. C., Nelson, T., Hall-Flavin, D., et al. (2019). Altered Serotonergic Circuitry in SSRI-Resistant Major Depressive Disorder Patient-Derived Neurons. Mol. Psychiatry 24 (6), 808–818. doi:10.1038/s41380-019-0377-5
Valori, C. F., Guidotti, G., Brambilla, L., and Rossi, D. (2019). Astrocytes in Motor Neuron Diseases. Adv. Exp. Med. Biol. 1175, 227–272. doi:10.1007/978-981-13-9913-8_10
van den Berg, L. H., Sorenson, E., Gronseth, G., Macklin, E. A., Andrews, J., Baloh, R. H., et al. (2019). Revised Airlie House Consensus Guidelines for Design and Implementation of ALS Clinical Trials. Neurology 92 (14), e1610–e1623. doi:10.1212/WNL.0000000000007242
Van Den Bosch, L. (2011). Genetic Rodent Models of Amyotrophic Lateral Sclerosis. J. Biomed. Biotechnol. 2011, 1–11. doi:10.1155/2011/348765
van der Worp, H. B., Howells, D. W., Sena, E. S., Porritt, M. J., Rewell, S., O'Collins, V., et al. (2010). Can Animal Models of Disease Reliably Inform Human Studies? Plos Med. 7 (3), e1000245. doi:10.1371/journal.pmed.1000245
van Eijk, R. P. A., Jones, A. R., Sproviero, W., Shatunov, A., Shaw, P. J., Leigh, P. N., et al. (2017). Meta-analysis of Pharmacogenetic Interactions in Amyotrophic Lateral Sclerosis Clinical Trials. Neurology 89 (18), 1915–1922. doi:10.1212/WNL.0000000000004606
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 323 (5918), 1208–1211. doi:10.1126/science.1165942
Vandoorne, T., De Bock, K., and Van Den Bosch, L. (2018). Energy Metabolism in ALS: an Underappreciated Opportunity? Acta Neuropathol. 135 (4), 489–509. doi:10.1007/s00401-018-1835-x
Vang, S., Longley, K., Steer, C. J., and Low, W. C. (2014). The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Health Med. 3 (3), 58–69. doi:10.7453/gahmj.2014.017
Vasques, J. F., Mendez-Otero, R., and Gubert, F. (2020). Modeling ALS Using iPSCs: Is it Possible to Reproduce the Phenotypic Variations Observed in Patients In Vitro? Regenerative Med. 15 (7), 1919–1933. doi:10.2217/rme-2020-0067
Vihinen, M. (2017). How to Define Pathogenicity, Health, and Disease? Hum. Mutat. 38 (2), 129–136. doi:10.1002/humu.23144
Vihinen, M. (2020). Strategy for Disease Diagnosis, Progression Prediction, Risk Group Stratification and Treatment-Case of COVID-19. Front. Med. 7, 294. doi:10.3389/fmed.2020.00294
Vincent, A., Sakowski, S. A., Schuyler, A., and Feldman, E. L. (2008). Strategic Approaches to Developing Drug Treatments for ALS. Drug Discov. Today 13 (1-2), 67–72. doi:10.1016/j.drudis.2007.10.011
Vucic, S., Nicholson, G. A., and Kiernan, M. C. (2008). Cortical Hyperexcitability May Precede the Onset of Familial Amyotrophic Lateral Sclerosis. Brain 131 (Pt 6), 1540–1550. doi:10.1093/brain/awn071
Vucic, S., Rothstein, J. D., and Kiernan, M. C. (2014). Advances in Treating Amyotrophic Lateral Sclerosis: Insights from Pathophysiological Studies. Trends Neurosciences 37 (8), 433–442. doi:10.1016/j.tins.2014.05.006
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S. W., Sandoe, J., et al. (2014). Intrinsic Membrane Hyperexcitability of Amyotrophic Lateral Sclerosis Patient-Derived Motor Neurons. Cel Rep. 7 (1), 1–11. doi:10.1016/j.celrep.2014.03.019
Wainger, B. J., Macklin, E. A., Vucic, S., McIlduff, C. E., Paganoni, S., Maragakis, N. J., et al. (2021). Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis. JAMA Neurol. 78, 186. doi:10.1001/jamaneurol.2020.4300
Walters, L. (2004). Human Embryonic Stem Cell Research: an Intercultural Perspective. Kennedy Inst. Ethics J. 14 (1), 3–38. doi:10.1353/ken.2004.0019
Wang, H., Guo, W., Mitra, J., Hegde, P. M., Vandoorne, T., Eckelmann, B. J., et al. (2018). Mutant FUS Causes DNA Ligation Defects to Inhibit Oxidative Damage Repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 9 (1), 3683. doi:10.1038/s41467-018-06111-6
Wang, L., Yi, F., Fu, L., Yang, J., Wang, S., Wang, Z., et al. (2017). CRISPR/Cas9-mediated Targeted Gene Correction in Amyotrophic Lateral Sclerosis Patient iPSCs. Protein Cell 8 (5), 365–378. doi:10.1007/s13238-017-0397-3
Wert, G. d., and Mummery, C. (2003). Human Embryonic Stem Cells: Research, Ethics and Policy. Hum. Reprod. 18 (4), 672–682. doi:10.1093/humrep/deg143
Westeneng, H.-J., Debray, T. P. A., Visser, A. E., van Eijk, R. P. A., Rooney, J. P. K., Calvo, A., et al. (2018). Prognosis for Patients with Amyotrophic Lateral Sclerosis: Development and Validation of a Personalised Prediction Model. Lancet Neurol. 17 (5), 423–433. doi:10.1016/S1474-4422(18)30089-9
Wichterle, H., Lieberam, I., Porter, J. A., and Jessell, T. M. (2002). Directed Differentiation of Embryonic Stem Cells into Motor Neurons. Cell 110 (3), 385–397. doi:10.1016/s0092-8674(02)00835-8
Wijesekera, L. C., and Nigel Leigh, P. (2009). Amyotrophic Lateral Sclerosis. Orphanet J. Rare Dis. 4, 3. doi:10.1186/1750-1172-4-3
Worms, P. M. (2001). The Epidemiology of Motor Neuron Diseases: a Review of Recent Studies. J. Neurol. Sci. 191 (1-2), 3–9. doi:10.1016/s0022-510x(01)00630-x
Writing, G., and Edaravone, A. L. S. S. G. (2017). Safety and Efficacy of Edaravone in Well Defined Patients with Amyotrophic Lateral Sclerosis: a Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 16 (7), 505–512. doi:10.1016/S1474-4422(17)30115-1
Wu, C.-H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the Profilin 1 Gene Cause Familial Amyotrophic Lateral Sclerosis. Nature 488 (7412), 499–503. doi:10.1038/nature11280
Yamanaka, S. (2020). Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 27 (4), 523–531. doi:10.1016/j.stem.2020.09.014
Yang, Q., Jiao, B., and Shen, L. (2020). The Development of C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Disorders. Front. Genet. 11, 562758. doi:10.3389/fgene.2020.562758
Yang, Y. M., Gupta, S. K., Kim, K. J., Powers, B. E., Cerqueira, A., Wainger, B. J., et al. (2013). A Small Molecule Screen in Stem-Cell-Derived Motor Neurons Identifies a Kinase Inhibitor as a Candidate Therapeutic for ALS. Cell Stem Cell 12 (6), 713–726. doi:10.1016/j.stem.2013.04.003
Yu, J., and Thomson, J. A. (2008). Pluripotent Stem Cell Lines. Genes Development 22 (15), 1987–1997. doi:10.1101/gad.1689808
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., et al. (2007). Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 318 (5858), 1917–1920. doi:10.1126/science.1151526
Zach, N., Ennist, D. L., Taylor, A. A., Alon, H., Sherman, A., Kueffner, R., et al. (2015). Being PRO-ACTive: What Can a Clinical Trial Database Reveal about ALS? Neurotherapeutics 12 (2), 417–423. doi:10.1007/s13311-015-0336-z
Zhao, J., Zhou, D., Guo, J., Ren, Z., Zhou, L., Wang, S., et al. (2006). Effect of Fasudil Hydrochloride, a Protein Kinase Inhibitor, on Cerebral Vasospasm and Delayed Cerebral Ischemic Symptoms after Aneurysmal Subarachnoid Hemorrhage-Results of a Randomized Trial of Fasudil Hydrochloride versus Nimodipine-. Neurol. Med. Chir.(Tokyo) 46 (9), 421–428. doi:10.2176/nmc.46.421
Zhao, J., Zhou, D., Guo, J., Ren, Z., Zhou, L., Wang, S., et al. (2011). Efficacy and Safety of Fasudil in Patients with Subarachnoid Hemorrhage: Final Results of a Randomized Trial of Fasudil versus Nimodipine. Neurol. Med. Chir.(Tokyo) 51 (10), 679–683. doi:10.2176/nmc.51.679
Zhou, N., and Manser, P. (2020). Does Including Machine Learning Predictions in ALS Clinical Trial Analysis Improve Statistical Power? Ann. Clin. Transl Neurol. 7 (10), 1756–1765. doi:10.1002/acn3.51140
Zhu, W., Xie, L., Han, J., and Guo, X. (2020). The Application of Deep Learning in Cancer Prognosis Prediction. Cancers 12 (3), 603. doi:10.3390/cancers12030603
Zu, T., Liu, Y., Banez-Coronel, M., Reid, T., Pletnikova, O., Lewis, J., et al. (2013). RAN Proteins and RNA Foci from Antisense Transcripts in C9ORF72 ALS and Frontotemporal Dementia. Proc. Natl. Acad. Sci. 110 (51), E4968–E4977. doi:10.1073/pnas.1315438110
Keywords: human induced pluripotent cells, drug screening, ALS, clinical trials, motor neuron (MN)
Citation: Lamas NJ and Roybon L (2021) Harnessing the Potential of Human Pluripotent Stem Cell-Derived Motor Neurons for Drug Discovery in Amyotrophic Lateral Sclerosis: From the Clinic to the Laboratory and Back to the Patient. Front. Drug. Discov. 1:773424. doi: 10.3389/fddsv.2021.773424
Received: 09 September 2021; Accepted: 26 October 2021;
Published: 11 November 2021.
Edited by:
Robin Polt, University of Arizona, United StatesReviewed by:
Werner J. Geldenhuys, West Virginia University, United StatesMahmoud Al-Khrasani, Semmelweis University, Hungary
Copyright © 2021 Lamas and Roybon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nuno Jorge Lamas, bnVub2psYW1hc0BtZWQudW1pbmhvLnB0; Laurent Roybon, bGF1cmVudC5yb3lib25AbWVkLmx1LnNl