 Achamaporn Punnanitinont
Achamaporn Punnanitinont Jill M. Kramer
Jill M. Kramer- Department of Oral Biology, School of Dental Medicine, The University at Buffalo, State University of New York, Buffalo, NY, United States
Many autoimmune diseases show a striking female sex predilection, including primary Sjögren's disease (pSD). Patients with pSD display exocrine gland pathology, such as salivary hypofunction and salivary and lacrimal gland inflammation. Moreover, many serious systemic disease manifestations are well-documented, including interstitial nephritis, hypergammaglobulinemia and neuropathies. Of note, women and men with pSD display distinct clinical phenotypes. While the underlying reasons for these clinical observations were poorly understood for many years, recent studies provide mechanistic insights into the specific regulatory landscapes that mediate female susceptibility to autoimmunity. We will review factors that contribute to the female sex bias, with an emphasis on those that are most relevant to pSD pathogenesis. Specifically, we will focus on sex hormones in disease, genetic alterations that likely contribute to the significant disease prevalence in females, and studies that provide evidence for the role of the gut microbiota in disease. Lastly, we will discuss therapeutics that are in clinical trials for pSD that may be particularly efficacious in targeting signaling networks that mediate inflammation in a sex-specific manner.
1. Introduction
Autoimmune diseases consist of more than 70 chronic disorders, which affect approximately 5% of the US population (1). Many autoimmune diseases preferentially affect females, and the most striking sex differences are observed in Sjögren's disease (pSD), also referred to as Sjögren's syndrome), systemic lupus erythematosus (SLE), autoimmune thyroid disease and scleroderma, which represent a spectrum of diseases in which the patient population is at least 80% women (1). Our understanding of the molecular basis underlying this sexual dimorphism is becoming increasingly sophisticated, and this review will discuss the current knowledge with a focus on sex-specific molecular mechanisms that are particularly relevant to pSD pathogenesis.
SD occurs in 2 forms: primary (pSD) and secondary (sSD). In pSD, SD is the only autoimmune disease present. In contrast, sSD is observed in conjunction with another autoimmune connective tissue disease (2). This review will focus on the primary form of the disease, as pSD shares overlapping features with other autoimmune diseases, and it is often difficult to determine which disease manifestations are caused solely by SD in sSD studies (3). The estimated prevalence of pSD is 0.3–1.0 per 1,000 persons (4). Worldwide, the estimated prevalence of pSD ranges between 0.01 and 0.72% (5). Primary SD exhibits a striking female predilection. Indeed, a recent clinical study estimated that the age- and sex-adjusted prevalence of pSD in the US was 16.3 per 10,000 in women and 3.1 per 10,000 in men (6).
The current classification criteria for pSD were validated jointly by the American College of Rheumatology and the European League against Rheumatism (7). These criteria include the assessment of anti-Ro/SSA autoantibodies, focal lymphocytic sialadenitis (FLS), and evaluation of salivary and ocular function (7). Each of these manifestations is scored and the presence of anti-Ro/SSA antibodies and FLS are weighted most heavily (7). In addition to these disease manifestations, patients may also experience interstitial lung disease and nephritis (8). Moreover, pSD patients display systemic hematopoietic abnormalities including hypergammaglobulinemia and hypocomplementemia, and patients are at increased risk of developing B cell lymphoma (2, 9, 10). Dryness of the mouth and eyes, fatigue and joint pain are common symptoms found in greater than 80% of patients with pSD, and these disease manifestations have a debilitating effect on the patient's quality of life (8). In addition to the significant morbidity associated with the disease, pSD patients also showed increased risk of mortality. A recent meta-analysis that included 14 studies with 14,584 pSD patients reported a total of 902 deaths (11). Compared to the general population, pSD patients had a 1.46-fold increased risk of death (11). Several factors contributed to the increased risk of mortality, including male sex, older age, hypocomplementemia, and interstitial lung disease (11).
Despite the significant morbidity and mortality associated with pSD, the underlying disease mechanisms that govern pSD remain poorly understood and the signaling networks that control sex-specific disease manifestations are not fully elucidated. This review will provide an overview of etiologic factors that likely contribute to the female sex bias in pSD. Herein, we will briefly summarize clinical data from pSD patients that highlight differences in disease presentation between males and females. Next, we will investigate evidence showing the role of sex hormones in pSD and examine findings detailing sex-specific genetic alterations that may contribute to pSD. We will then discuss how microbiota may mediate pSD. Lastly, we will discuss emerging therapeutics that may be tailored to pSD patients in a sex-dependent manner. This topic carries important clinical relevance, as it is crucial to decipher the networks that mediate pSD to identify treatment strategies that are relevant to patient sex.
2. Male and female pSD patients display distinct disease manifestations
For reasons that remain incompletely understood, males and females with pSD tend to display distinct disease manifestations (12). In a recent clinical study that identified comorbidities of pSD patients according to age and sex, female pSD patients had an increased risk of developing numerous conditions, such as fibromyalgia, migraines, hypermobile syndromes, Ehlers-Danlos syndrome and CREST syndrome, while men with pSD were at an increased risk of developing cerebrovascular accidents and cardiac pathologies such as myocardial infarction, atherosclerosis, and congestive heart failure (12). It is important to note that this study did not compare pSD patient data to that from healthy control subjects, and thus it is unclear whether these comorbidities are associated solely with patient sex, or whether they are related specifically to pSD. Further studies to determine whether these comorbidities are increased in pSD patients in a sex-specific manner will be insightful.
Although men tend to develop autoimmune diseases less commonly than women, men often exhibit more severe disease outcomes. In a separate study that included 967 patients with pSD (899 females and 68 males), men with pSD were more frequently diagnosed with interstitial lung disease, lymphadenopathy and lymphoma, while hypothyroidism was more common among females with pSD (13). Similarly, a corroborative study in which medical records of 353 female and 33 male pSD patients were reviewed retrospectively found that pulmonary complications and lymphoma were seen more frequently in males, while females had a higher prevalence of autoimmune thyroid diseases (14).
While sex-specific clinical manifestations tend to be consistent between studies, disparate serological findings are reported. Indeed, while one study reported higher anti-Ro/SSA antibodies in female pSD patients (13), another revealed that males displayed positivity for anti-Ro/SSA, anti-La/SSB and anti-nuclear autoantibodies (ANAs) more frequently than women (14). Such disparities may be explained by the low male prevalence in patient cohorts or by the different methods used to detect serum autoantibodies, as one group used double immunodiffusion while the other employed ELISAs (15). A comprehensive review of the clinical differences between males and females with pSD was recently published and provides an excellent overview of the topic (12).
3. Estrogen mediates a protective effect in exocrine tissue in pSD
Since women are primarily affected by pSD, there has been considerable interest in understanding the role of estrogen and estrogen precursors in disease and several studies have been performed in mouse models to establish a role for sex hormones in pSD. Indeed, healthy ovariectomized C57BL/6 mice exhibited an exocrinopathy with autoimmune characteristics resembling pSD, including lymphocytic infiltration of salivary and lacrimal glands, and anti-Ro/SSA, anti-La/SSB, and anti-α-fodrin autoantibodies (16). Additional elegant work revealed that estrogen deficiency in mice induced the expression of the retinoblastoma-associated protein 48 (RbAp48) in exocrine glands (17). Transgenic (Tg) expression of RbAp48 in the exocrine glands caused estrogen-dependent apoptosis (17). Specifically, overexpression of RbAp48 induced p53-mediated apoptosis in both salivary and lacrimal tissue of mice lacking estrogen, while inhibition of RbAp48 expression inhibited this apoptosis (17).
In a corroborative study, Tg mice overexpressing RbAp48 under the control of exocrine-specific parotid secretory protein promoter exhibited SD-like exocrinopathy characterized by an increase in apoptosis. In addition, lacrimal and salivary gland epithelial cells produced heightened IFNγ and IL-18 in this model (18, 19). Furthermore, salivary gland epithelial cells (SGECs) derived from RbAp48-Tg mice expressed elevated levels of MHC class II and the costimulatory molecules CD86, CD80, and ICAM-1 (18). Importantly, studies in human salivary cells found that both estrogen receptor α (ERα) and ERβ were expressed and functional (20). Accordingly, treatment of SGECs with estrogen resulted in decreased IFNγ-inducible expression of ICAM-1 (20). These data demonstrate that estrogen protects against exocrine gland apoptosis, and diminished estrogen promotes a pro-inflammatory phenotype in salivary tissue.
Corroborative work in the aromatase knockout (ArKO) mouse model also supports a protective role for estrogen in exocrine tissue. Aromatase is an enzyme that converts androgens to estrogens, and thus ArKO mice are deficient in estrogen (21). ArKO mice developed a lymphoproliferative autoimmune disease resembling SD (21, 22), as histological analyses showed inflammatory infiltrates in the lacrimal and salivary glands of ArKO mice that increased with age (21). Administration of an aromatase inhibitor resulted in enhanced lymphocytic infiltration in both salivary and lacrimal tissue derived from the NFS/sld mouse model of SD (21). These data provide further evidence for the protective role of estrogen in the attenuation of immune activation in exocrine tissue.
The role of estrogen in pSD has also been assessed in female patients. A recent case-control study of 1,320 pSD patients and 1,360 sicca controls examined estrogen exposure and disease risk (23). In contrast to murine findings, this study found that greater lifetime estrogen exposure is not necessarily protective against disease. Indeed, composite estrogen scores of pSD patients, which was a score assigned based on estimated lifetime estrogen exposure, did not correlate with unstimulated whole saliva production, Schirmer's test scores, or salivary focus scores (23). The authors also analyzed cumulative menstrual cycling (CMC) as a separate surrogate marker of estrogen exposure, which was calculated as years menstruating minus time pregnant. A higher CMC score was associated with greater unstimulated whole salivary production, although ocular staining score, hypergammaglobulinemia, and salivary focus score were inversely associated with CMC score in pSD patients (23). Thus, the data remain somewhat inconclusive as to whether estrogen exposure is protective or pathogenic in pSD patients, and additional longitudinal studies are needed to demonstrate this conclusively.
4. Altered expression of X-chromosome genes likely contributes to pSD
Although the underlying mechanisms that mediate the female sex predilection in autoimmunity are not completely understood, it is clear that altered expression of X chromosome alleles has a significant impact on disease susceptibility (24). Indeed, males with increased X chromosome copy number are more susceptible to autoimmune disease, as male patients with Klinefelter's syndrome (47, XXY) have a 38-fold increase in the incidence of pSD as compared to healthy men (25). Further evidence of the role of the X chromosome in autoimmunity is provided by one study reporting a 16-year-old Japanese patient with trisomy X (47, XXX) who developed mixed connective tissue disease and SD (26). In addition, a separate study found trisomy X women have a 2.9-fold higher risk of developing pSD as compared to females lacking the trisomy phenotype (46, XX) (27). These studies demonstrate the pathogenic potential of overexpression of X chromosome-related genes in pSD.
Of direct relevance to these observations, X chromosome inactivation (XCI) is the primary regulatory mechanism of X-linked gene dosage compensation between males and females. In XCI, one of the X chromosomes in female cells is transcriptionally silenced by epigenetic mechanisms in a stochastic manner, leading to allele-specific enrichment of epigenetic modifications on the inactive X (Xi). XCI ensures that X-linked genes are expressed at similar levels between females and males (28). In theory, this dosage compensation is stably maintained with each cell division in all female somatic cells (29). It is important to note that many genes associated with immunity are expressed on the X chromosome, including TLR7, TLR8, BTK, and CD40 ligand (CD40LG).
While this mechanism is generally effective at maintaining equivalent expression of X chromosome genes between the sexes, it is estimated that approximately 20% of X-linked human genes escape XCI (30). This inefficiency results in leaky expression of some X-linked genes from the Xi known as XCI escape, and this has important implications for autoimmune diseases, particularly those that display a female predilection. XCI is mediated by the long non-coding RNA termed XIST. It was initially thought that XIST established XCI early in development and then was no longer required to regulate gene expression in the female cells. Recent elegant work, however, established that XIST is crucial in maintaining XCI continually for specific X-linked genes in human B cells, including TLR7 (31).
This has important implications for autoimmunity, as XCI maintenance is altered in both B and T cells of SLE patients and mice, and X-linked genes are upregulated in adaptive immune cells from SLE patients as compared to healthy controls (28, 32). While XCI escape has not been identified in pSD patients thus far to our knowledge, these studies suggest that altered expression of particular X-linked genes could contribute to increased disease prevalence observed in females with pSD. Indeed, numerous genes that reside on the X chromosome are implicated in pSD, and differential expression and methylation of genes associated with immune activation are reported both in salivary tissue and peripheral blood cells in pSD patients (33). An overview of studies related to the expression and function of these genes is provided below:
TLR7: TLR7 is an endosomal TLR that is encoded on the X chromosome. An extensive body of literature demonstrates that TLR7 activation is pathogenic in the context of lupus (34–36), and emerging studies indicate that TLR7 may also mediate pathology in pSD. TLR7 is increased and functional in salivary tissue and in peripheral blood cells of pSD patients (37–41) and data suggest B cells and monocytes are hyper-responsive to TLR7 agonism in pSD (42–45). Indeed, TLR7 stimulation of B cells from pSD patients resulted in enhanced plasma cell differentiation and elevated IFNα secretion (42, 44).
Data from mouse models also provide corroborating evidence that TLR7 agonism mediates organ-specific disease (46–48). Studies in TLR8−/− animals revealed that these mice develop lupus and SD concomitantly, as sialadenitis, autoantibody production, glomerulonephritis, and lung inflammation were observed (46, 49). These disease manifestations were dependent on TLR7, as disease was abrogated in mice lacking both TLR7 and TLR8 (46, 49). Further work in the NOD/ShiLtJ model revealed that TLR7 is required for lacrimal gland inflammation that is characteristic of SD (47). Finally, corroborative work by our group revealed that TLR7 agonism accelerates both local and systemic disease in a mouse model of pSD and drives the expansion of a subset of B cells that are pathogenic in the context of lupus, termed age-associated B cells or ABCs (50).
Alterations in TLR7 expression and function are observed even in healthy 46, XX women (51). Indeed, TLR7 escapes XCI and is overexpressed in plasmacytoid DCs (pDCs), monocytes and B cells from women and biallelic expression of TLR7 was observed in the majority of donors tested (51). Moreover, B cells that showed biallelic expression of TLR7 showed a 2-fold greater likelihood of undergoing class switching as compared to monoallelic B cells (51). Thus, expression of TLR7 from both alleles could result in enhanced plasma cell differentiation. These findings are relevant to pSD, as B cells derived from pSD patients are hyper-responsive to TLR7 ligation and pSD patients often display hypergammaglobulinemia and high autoantibody titers (9, 10, 42, 44). Additionally, TLR7 expression was increased in CD19+ peripheral B cells derived from pSD female patients as compared to sex-matched healthy controls (52). Altogether, these studies suggest TLR7 likely contributes to the female disease predilection observed, although further studies are needed to establish whether high expression of TLR7 in immune cells mediates disease in the context of pSD.
In a corroborative study, XIST was found to be dysregulated in female patients with SLE, and this led to the XCI escape of TLR7 (31). Additionally, TLR7 agonism resulted in the inactivation of XIST and promoted the expansion of ABCs (31). Of note, B cells with features of ABCs are documented in pSD patients as well, although XIST inactivation of the TLR7 locus has not been reported in pSD patients or models to date (53, 54). Altogether, these data suggest that TLR7 dysregulation mediates pSD pathogenesis, and this may account, at least in part, for the striking female disease predilection observed. Table 1 provides a summary of studies that indicate altered expression and/or function of TLR7 in pSD mouse models and patients (37, 39–42, 44–48, 50, 52, 55).
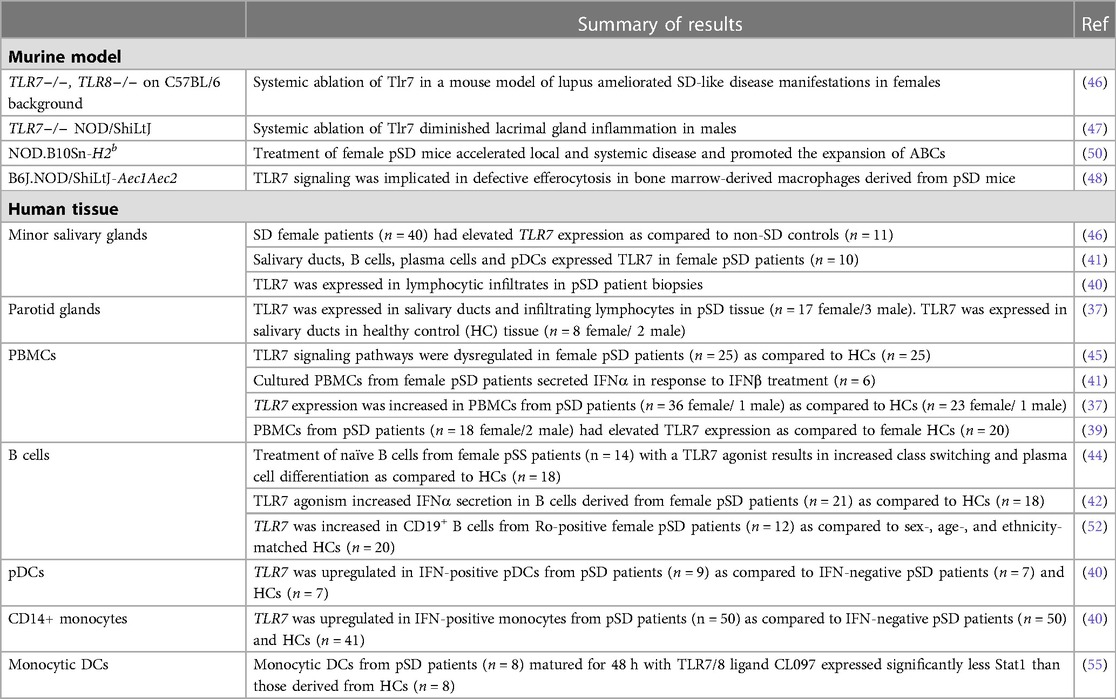
Table 1. Evidence for TLR7 dysregulation in SD mouse models and patients.
CXorf21: Chromosome X open reading frame 21 (CXorf21) (also called TASL) is a risk allele for both SLE and pSD (56). CXorf21 is expressed by pDCs, B cells, and CD14+ and CD16+ monocytes (57, 58). CXorf21 serves as a cytosolic adapter molecule for TLR7, and its activation culminates in the production of IFNα (56). To test the function of CXorf21 in female and male immune cells, knockdown of CXorf21 was performed in primary human heathy monocytes. TLR7 stimulation of female monocytes induced CXorf21 expression, and knockdown of CXorf21 abrogated this increase (58). In contrast, expression of CXorf21 in male monocytes was negligible, and there was no change in relative expression of CXorf21 following knockdown and subsequent TLR7 stimulation (58). Moreover, in healthy female monocytes in which knockdown of CXorf21 was carried out, TLR7 stimulation resulted in a 7-fold decrease in IFNA1 compared with sham-treated control cells (58). Finally, studies in lymphoblastoid cell lines (LCLs) derived from SLE patients revealed expression of CXorf21 was increased in 47,XXX/46,XX women and 47,XXY/46,XY men compared to LCLs derived from healthy males (58).
Additional studies by the same group revealed that CXorf21 escapes XCI and is therefore expressed at higher levels in healthy human female cells as compared to those derived from males, and female monocytes had a more acidic lysosomal pH as compared to males (56). This is significant because TLR7 is cleaved to its active form by a mechanism that is initiated by a low endosomal pH (59). This decreased pH may be of clinical significance because this alteration could contribute to the hyper-responsive endolysosomal-dependent immune response that is seen in females as compared to males (56), and this could contribute to the heightened TLR7 and IFN responses that are observed in women, although further studies are needed to establish whether this mechanism contributes to enhanced immune activation in female pSD patients.
BTK: BTK is expressed on the X chromosome and is also implicated in pSD, although no studies, to our knowledge, have demonstrated that BTK escapes XCI. BTK is activated upon B cell receptor stimulation and thus plays a crucial role in B cell activation and development (60). BTK expression was increased in circulating B cells from pSD patients with high serum autoantibodies and this correlated with T cell infiltration in parotid tissue (61). Overexpression of BTK in B cells specifically drives disease that is reminiscent of both SLE and pSD in mice (62, 63). Finally, BTK was identified as a key driver gene in salivary tissue from pSD patients (64).
CXCR3: CXCR3 is a chemokine receptor that binds CXCL9 and CXCL10. CXCR3 is expressed by activated CD4+ and CD8+ T cells, and functions to promote recruitment of Th1 cells in an immune response (65). CXCR3 exhibits XCI, as T cells derived from female SLE patients show elevated expression of CXCR3 as compared to those from males (66). In pSD patients, elevated levels of CXCL9 and CXCL10 are observed in salivary tissue, and infiltrating CXCR3+ T cells correlate with levels of these chemokines (67). Moreover, CXCR3 is expressed constitutively by salivary epithelial cells derived from both pSD patients and non-SD controls, although it is functionally impaired in tissue derived from pSD patients (68). CXCR3 expression was also increased in the conjunctiva of patients with pSD as compared to controls (69).
CD40LG: CD40LG encodes CD40L, an activation marker that is expressed by T cells. CD40L engages CD40 on B cells and other antigen-presenting cells, and this interaction is required for germinal center formation (70). There is considerable evidence that CD40-CD40L interactions are enhanced in pSD. Recent transcriptome analyses of salivary gland tissue derived from patients with pSD revealed a CD40 signaling gene signature that was enriched in B cells (71). Moreover, CD40L expression was increased on activated CD4+ T cells in patients with pSD and sera derived from pSD patients exhibited elevated levels of soluble CD40L as compared to that from healthy controls (72, 73). Of relevance to pSD, CD40LG was demethylated in T cells derived from female SLE patients, and this correlated with enhanced CD40LG expression in T cells derived from females with lupus as compared to males with the disease (74). While this heightened expression of CD40LG likely contributes to immune activation in SLE and pSD, the functional consequences of this have yet to be established.
In addition to genes encoded by the X chromosome, several genes that show sex-biased expression are implicated in pSD pathogenesis. One such gene is Vestigial-like family member 3 (VGLL3), a cofactor of the TEA-domain containing transcription factor (TEAD) that contributes to diverse inflammatory processes such as autoimmunity (75). While VGLL3 is not located on the X chromosome, this gene is expressed at higher levels in women in both the skin and parotid gland (76). Complement genes are integral to immune function and C4A and C4B display sexual dimorphism (77). Alleles that increase gene dosage of C4A strongly protect against SLE and pSD. Effects of C4 alleles are stronger in men with SLE as compared to female SLE patients, although similar analyses performed in patients with pSD had limited power due to the small numbers of male pSD patient samples available (77). Additional genes display sexual dimorphism in murine salivary tissue in pSD that could contribute to disease, such as those that encode IL-17 and Kallekrein protease family members (78, 79).
A separate study that examined primary B cells from males and females reported that CXCR5 is a pSD-associated SNP that displayed differential eQTL effects in women as compared to men (80). Dysregulation of CXCR5 is well-established in pSD, as CXCR5 expression was increased in lymphocytes within salivary tissue but decreased on peripheral B and T cells (81). Additionally, IRF5 is expressed in a sex-biased manner, as IRF5 gene expression was upregulated in splenic tissue derived from both healthy and lupus-prone females as compared to strain-matched males (82). Of relevance to pSD, polymorphisms in IRF5 are associated with pSD (83, 84) and TLR7 activation of IRF5 mediates the production of type I IFNs (85). Finally, recent data demonstrate that G protein receptor 78 (GPR78) is elevated in minor salivary gland tissue from male pSD patients as compared to females with pSD and male healthy controls (86). Interestingly, a mouse model that overexpressed GPR78 displayed salivary hypofunction and enhanced epithelial apoptosis, and these findings were observed in males but not females (87). Figure 1 contains a summary of genes dysregulated in pSD that are related to the female sex bias observed and a summary of genes implicated in human SLE and pSD is provided in Table 2.
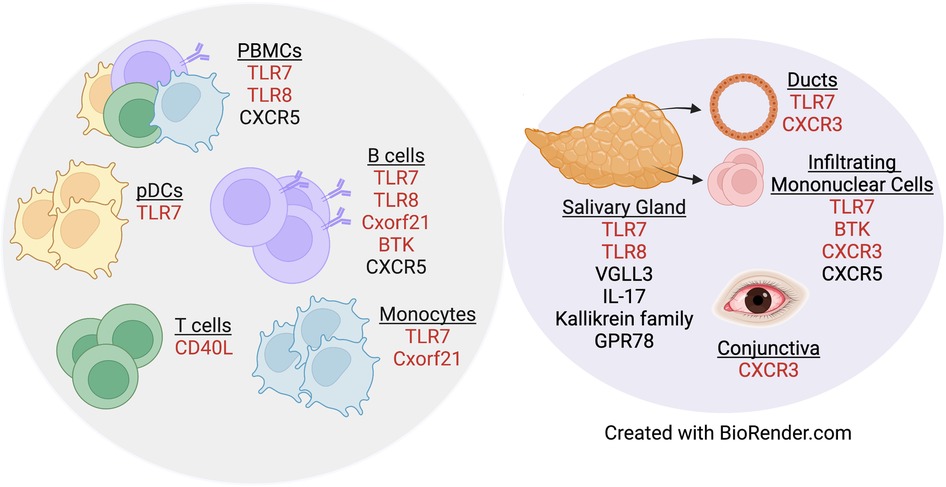
Figure 1. Genes dysregulated in pSD pathogenesis are differentially expressed in a sex-specific manner. Both peripheral (gray circle) and exocrine tissue (purple circle) show alterations in sexually dimorphic genes in pSD. Red text indicates genes that are expressed on the X chromosome.
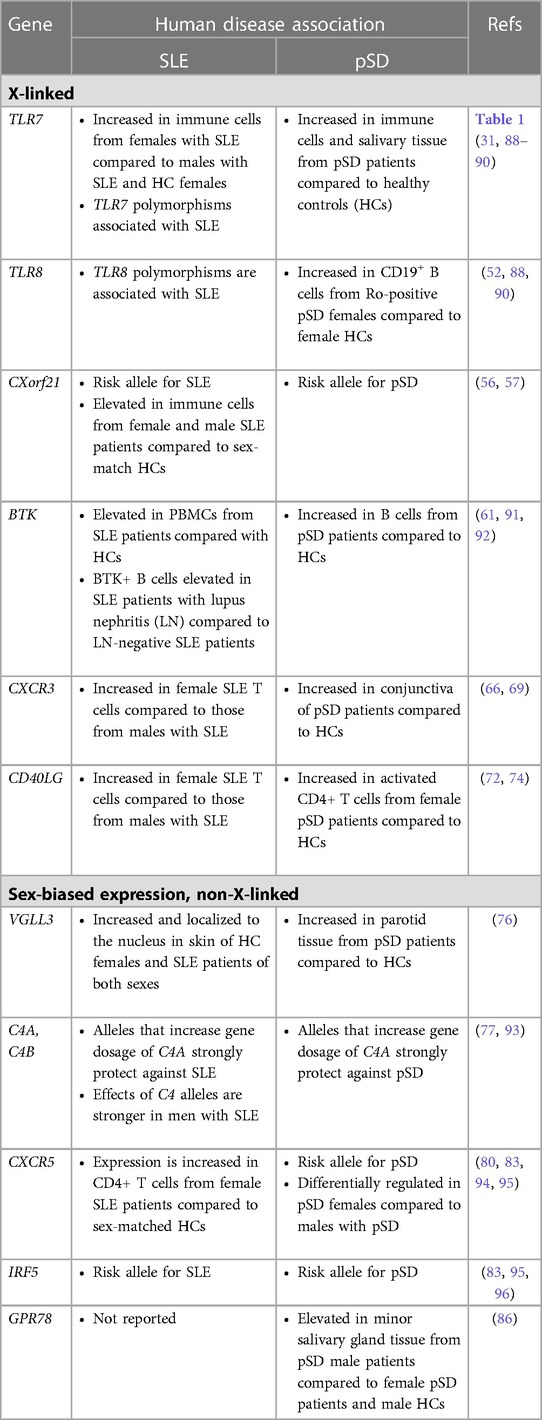
Table 2. A comparison of genes with sex-biased expression in SLE and pSD.
5. Estrogen governs expression of genes implicated in autoimmunity
Recent work highlights the complex regulatory network that exists between estrogen and immune-related genes, including those expressed on the X chromosome. Indeed, elegant studies in a humanized mouse model revealed that TLR7 responses are enhanced in female pDCs as compared to those derived from males (87). Moreover, treatment of post-menopausal women with 17β-estradiol, the main estrogenic hormone, resulted in elevated IFNα secretion by pDCs in a TLR7-dependent manner (87). When ERα was ablated in the DC lineage, this population was rendered unresponsive to TLR7-dependent Type I IFN production (87). Additionally, estrogen stimulation of CD11b+ monocytes derived from C57BL/6 female mice culminated in the enhanced expression of Unc93b1, a molecular chaperone required for TLR7 activity (97) and splenocytes from lupus-prone female mice expressed higher levels of Unc93b1 as compared to C57BL/6 controls (98).
Moreover, splenocytes derived from ERα-deficient mice expressed decreased Irf5 when compared to those from ERα-sufficient controls, and treatment of splenocytes with 17β-estradiol resulted in increased Irf5 levels (82). In addition, mice lacking expression of ERα in either the DC or hematopoietic compartment showed reduced frequency of Type I IFN–producing pDCs following TLR7 stimulation (99). Finally, studies in humans demonstrate levels of IRF5 and ERα correlate in pDCs derived from females but this association is not observed in male pDCs (99). It is important to point out that studies in the peripheral blood suggest that estrogen is pro-inflammatory, while studies in salivary tissue indicate a protective role for this hormone in the context of pSD, and epidemiologic data from pSD patients is somewhat inclusive at present (vide supra). This dichotomous role of estrogen highlights the complex influence of sex hormones in autoimmunity and suggests both pro-inflammatory and protective effects of sex hormones, depending on the cell type and specific microenvironment. Collectively, these data establish mechanisms whereby estrogen exerts potent effects on the expression of genes that are dysregulated in autoimmunity in a sex-dependent manner.
6. Sex-specific differences in microbiota may contribute to pSD
While still an emerging area of study, there are several lines of evidence that suggest sex steroids drive distinct differences in the composition of the intestinal microbiome and this has significant implications for health and disease. In both men and women, levels of sex hormones correlate with microbial diversity, and those with the highest levels of hormones show the greatest diversity (100). Significant differences are reported in the composition of the gut microbiome between pre- and post-menopausal women, as pre-menopausal women had increased levels of Bacteroidetes and Firmicutes species, which was reminiscent of the male microbiota (101). It is important to point out that while the human microbiome is shaped by biological sex and sex steroids, there is considerable variability among studies and the underlying mechanisms governing intestinal dysbiosis in this context remain incompletely understood (101).
While factors that shape the human microbiota are complex, seminal studies established an important sex-specific role for the microbiome in the autoimmune pathogenesis in mice (102, 103). Indeed, these studies revealed that microbial exposure early in life governs sex hormone levels and modifies disease progression in a mouse model of type I diabetes (T1D). Strikingly, when male mice of this strain were colonized with commensal microbiota, serum testosterone was elevated and the animals were protected from disease. When the microbiota from male mice was transferred to females of the same strain, the female recipients showed high testosterone levels and were protected from T1D (102). Transfer of specific species that comprise the microbiome can modulate disease in mice and humans. Indeed, corroborative work found that transfer of the intestinal bacteria Blautia (Ruminococcus) gnavus from SLE patients induced intestinal permeability that was enhanced in female recipient mice as compared to males. These recipient animals developed some features of SLE, such as anti-DNA autoantibodies (104). Of note, this effect was specifically related to patients with SLE, as transfer of the same bacteria derived from healthy donors did not induce this pathology (104). Additional work found that Lactobacillus strains protected against renal damage in the lupus-prone mouse model, MRL/lpr. Treatment of MRL/lpr mice with 5 strains of Lactobacillus resulted in reduced intestinal permeability and diminished IgG2a production, the antibody class that comprises most of the immune deposits observed in glomerulonephritis (105). Moreover, levels of IL-10 were increased, indicating a protective effect of these bacteria (105). Finally, these effects were sex-specific, as they were only observed in females and castrated males (105).
Additional mechanistic studies have uncovered a role for TLR7 activation in gut microbiota interactions in the context of autoimmunity. Indeed, TLR7 activation by intestinal bacteria resulted in lupus-like disease, as Lactobacillus reuteri mediated TLR7-dependent lupus in both conventional and germ-free mice (106). Complimentary studies in different lupus mouse models as well as in other female-biased autoimmune diseases, such as multiple sclerosis and rheumatoid arthritis (RA), provide additional evidence to support a role for sex-driven alterations in intestinal microbiota (107, 108).
While many studies demonstrate alterations in the gut, oral, and vaginal microbiome in patients with pSD (109), there is a paucity of data related to pathogenicity of the microbiota. There is one study, to our knowledge, that provides direct evidence for intestinal dysbiosis in a mouse model of pSD (110). Indeed, in studies in which gut microbiota from pSD patients was transferred to germ-free mice, the recipient animals developed heightened corneal barrier disruption (110). This study, however, only enrolled female pSD patients and microbiota was transferred to female mice, so additional work is needed to determine whether differences exist in male microbiota derived from pSD patients as compared to females, and whether alterations in the clinical phenotype occur between male and female recipient mice.
In addition, a separate study revealed that the bacteria identified in pSD patients were related to clinical disease features (109). Interestingly, treatment of pSD patients with hydroxychloroquine (HCQ) altered the composition of the microbiome in the oral cavity, vagina, and intestine, and this was distinct from that observed in healthy control subjects. Following 6–12 months of treatment, pSD patients who responded to HCQ treatment showed different oral microbiomes as compared to those derived from pSD patients who did not respond to therapy, and the microbiota of the responders was more similar to that of the healthy control subjects as compared to pSD patients who were refractory to treatment (109). Of note, HCQ suppresses inflammation through several mechanisms, including the prevention of endosomal acidification (111). Thus, one of the consequences of HCQ therapy could be the reduction of endosomal TLR activation, including TLR7 (112). However, HCQ has broad effects on many signaling networks (111), and additional studies are needed to determine if TLR7 governs or influences the microbiome in pSD in a sex-biased manner and whether this is of clinical consequence.
7. Implications for therapeutics
Altogether, these studies suggest that therapies targeting proteins derived from genes that are expressed in the X chromosome, particularly those that escape XCI, may be particularly efficacious in 46, XX females, 47, XXX females, and 47, XXY Klinefelter males. Drugs that target pathways that are overexpressed in a sex-biased manner may also have utility in these patient groups. There are several therapeutics in clinical trials that target products of genes that are expressed on the X chromosome.
7.1. Blockade of Cd40/CD40L interactions
Inhibition of CD40/CD40L interactions ameliorates pSD in mouse models, even when delivered at a pre-disease time point (113–115). Two drugs are currently in clinical trials for pSD that block this pathway: Dazodalibep and Iscalimab. Dazodalibep is an Fc-deficient CD40L antagonist fusion human monoclonal antibody. It binds to CD40L on activated T cells and blocks the interaction with CD40. Iscalimab is a humanized IgG1 monoclonal antibody directed against CD40. Both drugs are currently in phase 2 clinical trials for pSD (116–120).
7.2. BTK inhibition
Previous studies in both mice and humans provide a strong rationale for the use of therapies that target BTK in pSD. Indeed, deletion of BTK in a mouse model that has features of both pSD and lupus results in attenuated disease (121). Moreover, genetic analysis of salivary tissue from pSD patients revealed that BTK inhibition was likely to be an effective therapy for pSD patients with certain molecular signatures (64). A selective, covalent BTK inhibitor, Remibrutinib, is currently in phase 2 clinical trial in patients with moderate to severe pSD (122, 123).
7.3. TLR7 and TLR8 inhibition
Phase 2 clinical trials with Enpatoran, a highly selective small molecule inhibitor of TLR7 and TLR8, are ongoing for patients with SLE (124–126). There is considerable evidence in both mice and humans that TLR7 activation mediates immune dysfunction in pSD (Table 1), and thus there is a strong rationale to carry out clinical trials to assess the efficacy of this drug in pSD patients.
7.4. Drug efficacy differs between male and female patients with autoimmune disease
It is important to note that there are several studies with thousands of patients in aggregate that document differences in drug efficacy between males and females with autoimmune disease. This is observed most commonly in studies of male and female patients who receive biologic therapies. In patients with axial spondyloarthritis (315 females, 654 males), females showed a diminished response to first-line TNF inhibitors (TNFi) by the second year of treatment as compared to males (127) and females also exhibited a lower rate of disease remission (104 females, 236 males) (128). Moreover, a lower percentage of female patients with inflammatory arthritis (RA, ankylosing spondylitis, and psoriatic arthritis) achieved disease remission after TNFi therapy as compared to men (138 females, 167 males) (129). Similar findings are reported in patients with moderate to severe psoriasis, as female patients were less likely than males to achieve a therapeutic response after at least one year of TNFi therapy (83 females, 97 males) (130). Finally, a 48-week phase 3 randomized control trial of psoriatic arthritis patients found that women who received methotrexate and the TNFi etanercept had worse treatment outcomes as compared to men (139 females, 144 males) (131).
In addition, a study of RA patients (73 females, 28 males) revealed that in patients receiving the TNFi infliximab as a second-line drug, women were more likely to develop anti-drug antibodies and have lower serum infliximab levels as compared to males (132). Corroborative data were reported in inflammatory bowel disease (IBD) patients who received TNFi therapy for a minimum of 1 year, as females were more likely to discontinue the therapy than males, due to a higher incidence of side effects (265 females, 264 males) (133). Similar results were reported in patients with psoriasis, as females reported experiencing more side effects than males following biologic therapy, and these included fungal and herpes virus infections (127 females, 188 males) (134). Of note, contrasting findings were reported in patients with IBD, as males that received infliximab therapy showed greater anti-drug antibodies and lower concentrations of the drug in sera as compared to females (228 females, 233 males) (135). Interestingly, these observations were specific for infliximab, as there were no differences observed between male and female IBD patients who received the TNFi adalimumab (298 females, 333 males) (135). Altogether, these findings provide evidence that biologic therapies have different efficacies in men and women with autoimmunity, and biological sex is a relevant consideration in the therapeutic management of these patients.
These studies highlight the need for and importance of well-designed clinical trials that have sufficient power to detect differences in therapeutic efficacy that may be present between males and females, as distinct hormonal, genetic and immunological differences are documented between the sexes. It is critical that clinical trials are designed with careful consideration of these underlying sex-specific biological traits, as these likely have important relevance for personalized treatments.
8. Conclusion
In summary, a myriad of inter-related factors drive the female sex-bias that is evident in many autoimmune diseases, including pSD. Studies directed at understanding the underlying pathways that contribute to the profound female sex-bias observed in pSD will allow for the design of targeted therapies that may be tailored in accordance with the patient's biological sex for maximal efficacy.
Author contributions
AP and JK conceived of the project and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Funding for this work was provided by R01DE29472 to JK and AP received support from NIDCR award T32023526. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Xing E, Billi AC, Gudjonsson JE. Sex bias and autoimmune diseases. J Invest Dermatol. (2022) 142(3 Pt B):857–66. doi: 10.1016/j.jid.2021.06.008
2. Mavragani CP, Moutsopoulos HM. Sjögren's syndrome. Annu Rev Pathol. (2014) 9:273–85. doi: 10.1146/annurev-pathol-012513-104728
3. Pasoto SG, Adriano de Oliveira Martins V, Bonfa E. Sjögren's syndrome and systemic lupus erythematosus: links and risks. Open Access Rheumatol. (2019) 11:33–45. doi: 10.2147/OARRR.S167783
4. Qin B, Wang J, Yang Z, Yang M, Ma N, Huang F, et al. Epidemiology of primary Sjögren's syndrome: a systematic review and meta-analysis. Ann Rheum Dis. (2015) 74(11):1983–9. doi: 10.1136/annrheumdis-2014-205375
5. Brito-Zeron P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, et al. Sjögren syndrome. Nat Rev Dis Primers. (2016) 2:16047. doi: 10.1038/nrdp.2016.47
6. Maciel G, Crowson CS, Matteson EL, Cornec D. Prevalence of primary Sjögren's syndrome in a US population-based cohort. Arthritis Care Res (Hoboken). (2017) 69(10):1612–6. doi: 10.1002/acr.23173
7. Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, et al. 2016 American college of rheumatology/European league against rheumatism classification criteria for primary Sjögren's syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis. (2017) 76(1):9–16. doi: 10.1136/annrheumdis-2016-210571
8. Mariette X, Criswell LA. Primary Sjögren's syndrome. N Engl J Med. (2018) 379(1):97. doi: 10.1056/NEJMc1804598
9. Shiboski CH, Baer AN, Shiboski SC, Lam M, Challacombe S, Lanfranchi HE, et al. Natural history and predictors of progression to sjogren's syndrome among participants of the Sjögren's international collaborative clinical alliance registry. Arthritis Care Res (Hoboken). (2018) 70(2):284–94. doi: 10.1002/acr.23264
10. Malladi AS, Sack KE, Shiboski SC, Shiboski CH, Baer AN, Banushree R, et al. Primary sjogren's syndrome as a systemic disease: a study of participants enrolled in an international Sjögren's syndrome registry. Arthritis Care Res (Hoboken). (2012) 64(6):911–8. doi: 10.1002/acr.21610
11. Huang H, Xie W, Geng Y, Fan Y, Zhang Z. Mortality in patients with primary Sjögren's syndrome: a systematic review and meta-analysis. Rheumatology (Oxford). (2021) 60(9):4029–38. doi: 10.1093/rheumatology/keab364
12. Bruno KA, Morales-Lara AC, Bittencourt EB, Siddiqui H, Bommarito G, Patel J, et al. Sex differences in comorbidities associated with Sjögren's disease. Front Med (Lausanne). (2022) 9:958670. doi: 10.3389/fmed.2022.958670
13. Park Y, Lee J, Park SH, Kwok SK. Male patients with primary Sjögren's syndrome: a distinct clinical subgroup? Int J Rheum Dis. (2020) 23(10):1388–95. doi: 10.1111/1756-185X.13940
14. Ramirez Sepulveda JI, Kvarnstrom M, Eriksson P, Mandl T, Norheim KB, Johnsen SJ, et al. Long-term follow-up in primary Sjögren's syndrome reveals differences in clinical presentation between female and male patients. Biol Sex Differ. (2017) 8(1):25. doi: 10.1186/s13293-017-0146-6
15. Chretien P, Soulie E, Johanet C, Abuaf N. Comparisons of double immunodiffusion, ELISA, western blot and CAPE blot for the detection of anti-SSA antibody: study of anti-SSA prevalence in systemic lupus erythematosus. J Autoimmun. (1994) 7(3):379–88. doi: 10.1006/jaut.1994.1027
16. Ishimaru N, Arakaki R, Watanabe M, Kobayashi M, Miyazaki K, Hayashi Y. Development of autoimmune exocrinopathy resembling Sjögren's syndrome in estrogen-deficient mice of healthy background. Am J Pathol. (2003) 163(4):1481–90. doi: 10.1016/S0002-9440(10)63505-5
17. Ishimaru N, Arakaki R, Omotehara F, Yamada K, Mishima K, Saito I, et al. Novel role for RbAp48 in tissue-specific, estrogen deficiency-dependent apoptosis in the exocrine glands. Mol Cell Biol. (2006) 26(8):2924–35. doi: 10.1128/MCB.26.8.2924-2935.2006
18. Ishimaru N, Arakaki R, Yoshida S, Yamada A, Noji S, Hayashi Y. Expression of the retinoblastoma protein RbAp48 in exocrine glands leads to Sjögren's syndrome-like autoimmune exocrinopathy. J Exp Med. (2008) 205(12):2915–27. doi: 10.1084/jem.20080174
19. Mikkelsen TR, Brandt J, Larsen HJ, Larsen BB, Poulsen K, Ingerslev J, et al. Tissue-specific expression in the salivary glands of transgenic mice. Nucleic Acids Res. (1992) 20(9):2249–55. doi: 10.1093/nar/20.9.2249
20. Tsinti M, Kassi E, Korkolopoulou P, Kapsogeorgou E, Moutsatsou P, Patsouris E, et al. Functional estrogen receptors alpha and beta are expressed in normal human salivary gland epithelium and apparently mediate immunomodulatory effects. Eur J Oral Sci. (2009) 117(5):498–505. doi: 10.1111/j.1600-0722.2009.00659.x
21. Iwasa A, Arakaki R, Honma N, Ushio A, Yamada A, Kondo T, et al. Aromatase controls Sjögren syndrome-like lesions through monocyte chemotactic protein-1 in target organ and adipose tissue-associated macrophages. Am J Pathol. (2015) 185(1):151–61. doi: 10.1016/j.ajpath.2014.09.006
22. Shim GJ, Warner M, Kim HJ, Andersson S, Liu L, Ekman J, et al. Aromatase-deficient mice spontaneously develop a lymphoproliferative autoimmune disease resembling Sjögren's syndrome. Proc Natl Acad Sci U S A. (2004) 101(34):12628–33. doi: 10.1073/pnas.0405099101
23. McCoy SS, Sampene E, Baer AN. Sjögren's syndrome is associated with reduced lifetime sex hormone exposure: a case-control study. Arthritis Care Res (Hoboken). (2020) 72(9):1315–22. doi: 10.1002/acr.24014
24. Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. (2010) 10(8):594–604. doi: 10.1038/nri2815
25. Harris VM, Sharma R, Cavett J, Kurien BT, Liu K, Koelsch KA, et al. Klinefelter's syndrome (47,XXY) is in excess among men with Sjögren's syndrome. Clin Immunol. (2016) 168:25–9. doi: 10.1016/j.clim.2016.04.002
26. Fujimoto M, Ikeda K, Nakamura T, Iwamoto T, Furuta S, Nakajima H. Development of mixed connective tissue disease and Sjögren's syndrome in a patient with trisomy X. Lupus. (2015) 24(11):1217–20. doi: 10.1177/0961203315580873
27. Liu K, Kurien BT, Zimmerman SL, Kaufman KM, Taft DH, Kottyan LC, et al. X chromosome dose and sex bias in autoimmune diseases: increased prevalence of 47,XXX in systemic lupus erythematosus and Sjögren's syndrome. Arthritis Rheumatol. (2016) 68(5):1290–300. doi: 10.1002/art.39560
28. Pyfrom S, Paneru B, Knox JJ, Cancro MP, Posso S, Buckner JH, et al. The dynamic epigenetic regulation of the inactive X chromosome in healthy human B cells is dysregulated in lupus patients. Proc Natl Acad Sci U S A. (2021) 118(24). doi: 10.1073/pnas.2024624118
29. Csankovszki G, Nagy A, Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol. (2001) 153(4):773–84. doi: 10.1083/jcb.153.4.773
30. Souyris M, Mejia JE, Chaumeil J, Guery JC. Female predisposition to TLR7-driven autoimmunity: gene dosage and the escape from X chromosome inactivation. Semin Immunopathol. (2019) 41(2):153–64. doi: 10.1007/s00281-018-0712-y
31. Yu B, Qi Y, Li R, Shi Q, Satpathy AT, Chang HY. B cell-specific XIST complex enforces X-inactivation and restrains atypical B cells. Cell. (2021) 184(7):1790–803.e17. doi: 10.1016/j.cell.2021.02.015
32. Syrett CM, Paneru B, Sandoval-Heglund D, Wang J, Banerjee S, Sindhava V, et al. Altered X-chromosome inactivation in T cells may promote sex-biased autoimmune diseases. JCI Insight. (2019) 4(7). doi: 10.1172/jci.insight.126751
33. Mougeot JL, Noll BD, Bahrani Mougeot FK. Sjögren's syndrome X-chromosome dose effect: an epigenetic perspective. Oral Dis. (2019) 25(2):372–84. doi: 10.1111/odi.12825
34. Satterthwaite AB. TLR7 Signaling in lupus B cells: new insights into synergizing factors and downstream signals. Curr Rheumatol Rep. (2021) 23(11):80. doi: 10.1007/s11926-021-01047-1
35. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. (2006) 312(5780):1669–72. doi: 10.1126/science.1124978
36. Deane JA, Bolland S. Nucleic acid-sensing TLRs as modifiers of autoimmunity. J Immunol. (2006) 177(10):6573–8. doi: 10.4049/jimmunol.177.10.6573
37. Zheng L, Zhang Z, Yu C, Yang C. Expression of toll-like receptors 7, 8, and 9 in primary Sjögren's syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (2010) 109(6):844–50. doi: 10.1016/j.tripleo.2010.01.006
38. Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren's syndrome. Proc Natl Acad Sci U S A. (2006) 103(8):2770–5. doi: 10.1073/pnas.0510837103
39. Karlsen M, Jakobsen K, Jonsson R, Hammenfors D, Hansen T, Appel S. Expression of toll-like receptors in peripheral blood mononuclear cells of patients with primary Sjögren's syndrome. Scand J Immunol. (2017) 85(3):220–26. doi: 10.1111/sji.12520
40. Maria NI, Steenwijk EC, Ijpma AS, van Helden-Meeuwsen CG, Vogelsang P, Beumer W, et al. Contrasting expression pattern of RNA-sensing receptors TLR7, RIG-I and MDA5 in interferon-positive and interferon-negative patients with primary Sjögren's syndrome. Ann Rheum Dis. (2017) 76(4):721–30. doi: 10.1136/annrheumdis-2016-209589
41. Shimizu T, Nakamura H, Takatani A, Umeda M, Horai Y, Kurushima S, et al. Activation of toll-like receptor 7 signaling in labial salivary glands of primary Sjögren's syndrome patients. Clin Exp Immunol. (2019) 196(1):39–51. doi: 10.1111/cei.13242
42. Karlsen M, Jonsson R, Brun JG, Appel S, Hansen T. TLR-7 and -9 stimulation of peripheral blood B cells indicate altered TLR signalling in primary Sjögren's syndrome patients by increased secretion of cytokines. Scand J Immunol. (2015) 82(6):523–31. doi: 10.1111/sji.12368
43. Vogelsang P, Jonsson MV, Dalvin ST, Appel S. Role of dendritic cells in Sjögren's syndrome. Scand J Immunol. (2006) 64(3):219–26. doi: 10.1111/j.1365-3083.2006.01811.x
44. Brauner S, Folkersen L, Kvarnstrom M, Meisgen S, Petersen S, Franzen-Malmros M, et al. H1n1 vaccination in Sjögren's syndrome triggers polyclonal B cell activation and promotes autoantibody production. Ann Rheum Dis. (2017) 76(10):1755–63. doi: 10.1136/annrheumdis-2016-210509
45. Davies R, Sarkar I, Hammenfors D, Bergum B, Vogelsang P, Solberg SM, et al. Single cell based phosphorylation profiling identifies alterations in toll-like receptor 7 and 9 signaling in patients with primary Sjögren's syndrome. Front Immunol. (2019) 10:281. doi: 10.3389/fimmu.2019.00281
46. Wang Y, Roussel-Queval A, Chasson L, Hanna Kazazian N, Marcadet L, Nezos A, et al. TLR7 Signaling drives the development of Sjögren's syndrome. Front Immunol. (2021) 12:676010. doi: 10.3389/fimmu.2021.676010
47. Debreceni IL, Chimenti MS, Serreze DV, Geurts AM, Chen YG, Lieberman SM. Toll-Like receptor 7 is required for lacrimal gland autoimmunity and type 1 diabetes development in male nonobese diabetic mice. Int J Mol Sci. (2020) 21(24):9478. doi: 10.3390/ijms21249478
48. Witas R, Shen Y, Nguyen CQ. Bone marrow-derived macrophages from a murine model of Sjögren's syndrome demonstrate an aberrant, inflammatory response to apoptotic cells. Sci Rep. (2022) 12(1):8593. doi: 10.1038/s41598-022-12608-4
49. Demaria O, Pagni PP, Traub S, de Gassart A, Branzk N, Murphy AJ, et al. TLR8 Deficiency leads to autoimmunity in mice. J Clin Invest. (2010) 120(10):3651–62. doi: 10.1172/JCI42081
50. Punnanitinont A, Kasperek EM, Kiripolsky J, Zhu C, Miecznikowski JC, Kramer JM. TLR7 Agonism accelerates disease in a mouse model of primary Sjögren's syndrome and drives expansion of T-bet(+) B cells. Front Immunol. (2022) 13:1034336. doi: 10.3389/fimmu.2022.1034336
51. Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, et al. TLR7 Escapes X chromosome inactivation in immune cells. Sci Immunol. (2018) 3(19). doi: 10.1126/sciimmunol.aap8855
52. Imgenberg-Kreuz J, Sandling JK, Bjork A, Nordlund J, Kvarnstrom M, Eloranta ML, et al. Transcription profiling of peripheral B cells in antibody-positive primary Sjögren's syndrome reveals upregulated expression of CX3CR1 and a type I and type II interferon signature. Scand J Immunol. (2018) 87(5):e12662. doi: 10.1111/sji.12662
53. Saadoun D, Terrier B, Bannock J, Vazquez T, Massad C, Kang I, et al. Expansion of autoreactive unresponsive CD21-/low B cells in Sjögren's syndrome-associated lymphoproliferation. Arthritis Rheum. (2013) 65(4):1085–96. doi: 10.1002/art.37828
54. Glauzy S, Boccitto M, Bannock JM, Delmotte FR, Saadoun D, Cacoub P, et al. Accumulation of antigen-driven lymphoproliferations in complement receptor 2/CD21(-/low) B cells from patients with Sjögren's syndrome. Arthritis Rheumatol. (2018) 70(2):298–307. doi: 10.1002/art.40352
55. Vogelsang P, Karlsen M, Brun JG, Jonsson R, Appel S. Altered phenotype and Stat1 expression in toll-like receptor 7/8 stimulated monocyte-derived dendritic cells from patients with primary Sjögren's syndrome. Arthritis Res Ther. (2014) 16(4):R166. doi: 10.1186/ar4682
56. Harris VM, Harley ITW, Kurien BT, Koelsch KA, Scofield RH. Lysosomal pH is regulated in a sex dependent manner in immune cells expressing CXorf21. Front Immunol. (2019) 10:578. doi: 10.3389/fimmu.2019.00578
57. Heinz LX, Lee J, Kapoor U, Kartnig F, Sedlyarov V, Papakostas K, et al. TASL Is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature. (2020) 581(7808):316–22. doi: 10.1038/s41586-020-2282-0
58. Harris VM, Koelsch KA, Kurien BT, Harley ITW, Wren JD, Harley JB, et al. Characterization of cxorf21 provides molecular insight into female-bias immune response in SLE pathogenesis. Front Immunol. (2019) 10:2160. doi: 10.3389/fimmu.2019.02160
59. Petes C, Odoardi N, Gee K. The toll for trafficking: toll-like receptor 7 delivery to the endosome. Front Immunol. (2017) 8:1075. doi: 10.3389/fimmu.2017.01075
60. Kurosaki T. Molecular mechanisms in B cell antigen receptor signaling. Curr Opin Immunol. (1997) 9(3):309–18. doi: 10.1016/s0952-7915(97)80075-1
61. Corneth OBJ, Verstappen GMP, Paulissen SMJ, de Bruijn MJW, Rip J, Lukkes M, et al. Enhanced bruton's tyrosine kinase activity in peripheral blood B lymphocytes from patients with autoimmune disease. Arthritis Rheumatol. (2017) 69(6):1313–24. doi: 10.1002/art.40059
62. Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood. (2012) 119(16):3744–56. doi: 10.1182/blood-2011-12-397919
63. Corneth OB, de Bruijn MJ, Rip J, Asmawidjaja PS, Kil LP, Hendriks RW. Enhanced expression of bruton's tyrosine kinase in B cells drives systemic autoimmunity by disrupting T cell homeostasis. J Immunol. (2016) 197(1):58–67. doi: 10.4049/jimmunol.1600208
64. Jung SM, Baek IW, Park KS, Kim KJ. De novo molecular subtyping of salivary gland tissue in the context of Sjögren's syndrome heterogeneity. Clin Immunol. (2022) 245:109171. doi: 10.1016/j.clim.2022.109171
65. Groom JR, Luster AD. CXCR3 Ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. (2011) 89(2):207–15. doi: 10.1038/icb.2010.158
66. Hewagama A, Gorelik G, Patel D, Liyanarachchi P, McCune WJ, Somers E, et al. Overexpression of X-linked genes in T cells from women with lupus. J Autoimmun. (2013) 41:60–71. doi: 10.1016/j.jaut.2012.12.006
67. Ogawa N, Ping L, Zhenjun L, Takada Y, Sugai S. Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible 10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with Sjögren's syndrome. Arthritis Rheum. (2002) 46(10):2730–41. doi: 10.1002/art.10577
68. Sfriso P, Oliviero F, Calabrese F, Miorin M, Facco M, Contri A, et al. Epithelial CXCR3-B regulates chemokines bioavailability in normal, but not in Sjögren's syndrome, salivary glands. J Immunol. (2006) 176(4):2581–9. doi: 10.4049/jimmunol.176.4.2581
69. Yoon KC, Park CS, You IC, Choi HJ, Lee KH, Im SK, et al. Expression of CXCL9, -10, -11, and CXCR3 in the tear film and ocular surface of patients with dry eye syndrome. Invest Ophthalmol Vis Sci. (2010) 51(2):643–50. doi: 10.1167/iovs.09-3425
70. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. (2015) 15(3):137–48. doi: 10.1038/nri3804
71. Verstappen GM, Gao L, Pringle S, Haacke EA, van der Vegt B, Liefers SC, et al. The transcriptome of paired Major and Minor salivary gland tissue in patients with primary Sjögren's syndrome. Front Immunol. (2021) 12:681941. doi: 10.3389/fimmu.2021.681941
72. Belkhir R, Gestermann N, Koutero M, Seror R, Tost J, Mariette X, et al. Upregulation of membrane-bound CD40L on CD4+ T cells in women with primary Sjögren's syndrome. Scand J Immunol. (2014) 79(1):37–42. doi: 10.1111/sji.12121
73. Goules A, Tzioufas AG, Manousakis MN, Kirou KA, Crow MK, Routsias JG. Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J Autoimmun. (2006) 26(3):165–71. doi: 10.1016/j.jaut.2006.02.002
74. Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol. (2007) 179(9):6352–8. doi: 10.4049/jimmunol.179.9.6352
75. Takakura Y, Hori N, Terada N, Machida M, Yamaguchi N, Takano H, et al. VGLL3 Activates inflammatory responses by inducing interleukin-1alpha secretion. FASEB J. (2021) 35(11):e21996. doi: 10.1096/fj.202100679RR
76. Liang Y, Tsoi LC, Xing X, Beamer MA, Swindell WR, Sarkar MK, et al. A gene network regulated by the transcription factor VGLL3 as a promoter of sex-biased autoimmune diseases. Nat Immunol. (2017) 18(2):152–60. doi: 10.1038/ni.3643
77. Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, et al. Complement genes contribute sex-biased vulnerability in diverse disorders. Nature. (2020) 582(7813):577–81. doi: 10.1038/s41586-020-2277-x
78. Voigt A, Esfandiary L, Wanchoo A, Glenton P, Donate A, Craft WF, et al. Sexual dimorphic function of IL-17 in salivary gland dysfunction of the C57BL/6.NOD-Aec1Aec2 model of Sjögren's syndrome. Sci Rep. (2016) 6:38717. doi: 10.1038/srep38717
79. Moustardas P, Yamada-Fowler N, Apostolou E, Tzioufas AG, Turkina MV, Spyrou G. Deregulation of the kallikrein protease family in the salivary glands of the Sjögren's syndrome ERdj5 knockout mouse model. Front Immunol. (2021) 12:693911. doi: 10.3389/fimmu.2021.693911
80. Linden M, Ramirez Sepulveda JI, James T, Thorlacius GE, Brauner S, Gomez-Cabrero D, et al. Sex influences eQTL effects of SLE and Sjögren's syndrome-associated genetic polymorphisms. Biol Sex Differ. (2017) 8(1):34. doi: 10.1186/s13293-017-0153-7
81. Aqrawi LA, Ivanchenko M, Bjork A, Ramirez Sepulveda JI, Imgenberg-Kreuz J, Kvarnstrom M, et al. Diminished CXCR5 expression in peripheral blood of patients with Sjögren's syndrome may relate to both genotype and salivary gland homing. Clin Exp Immunol. (2018) 192(3):259–70. doi: 10.1111/cei.13118
82. Shen H, Panchanathan R, Rajavelu P, Duan X, Gould KA, Choubey D. Gender-dependent expression of murine Irf5 gene: implications for sex bias in autoimmunity. J Mol Cell Biol. (2010) 2(5):284–90. doi: 10.1093/jmcb/mjq023
83. Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren's syndrome. Nat Genet. (2013) 45(11):1284–92. doi: 10.1038/ng.2792
84. Taylor KE, Wong Q, Levine DM, McHugh C, Laurie C, Doheny K, et al. Genome-Wide association analysis reveals genetic heterogeneity of Sjögren's syndrome according to ancestry. Arthritis Rheumatol. (2017) 69(6):1294–305. doi: 10.1002/art.40040
85. Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, et al. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem. (2005) 280(17):17005–12. doi: 10.1074/jbc.M412584200
86. Tanaka T, Guimaro MC, Nakamura H, Perez P, Ji Y, Michael DG, et al. Association of G protein-coupled receptor 78 with salivary dysfunction in male Sjögren's patients. Oral Dis. (2023). doi: 10.1111/odi.14506
87. Seillet C, Laffont S, Tremollieres F, Rouquie N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood. (2012) 119(2):454–64. doi: 10.1182/blood-2011-08-371831
88. dos Santos BP, Valverde JV, Rohr P, Monticielo OA, Brenol JC, Xavier RM, et al. TLR7/8/9 Polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus. (2012) 21(3):302–9. doi: 10.1177/0961203311425522
89. Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, et al. Sex-specific association of X-linked toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A. (2010) 107(36):15838–43. doi: 10.1073/pnas.1001337107
90. Wang CM, Chang SW, Wu YJ, Lin JC, Ho HH, Chou TC, et al. Genetic variations in toll-like receptors (TLRs 3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population. Sci Rep. (2014) 4:3792. doi: 10.1038/srep03792
91. Liao HT, Tung HY, Tsai CY, Chiang BL, Yu CL. Bruton's tyrosine kinase in systemic lupus erythematosus. Joint Bone Spine. (2020) 87(6):670–2. doi: 10.1016/j.jbspin.2020.05.004
92. Kong W, Deng W, Sun Y, Huang S, Zhang Z, Shi B, et al. Increased expression of bruton's tyrosine kinase in peripheral blood is associated with lupus nephritis. Clin Rheumatol. (2018) 37(1):43–9. doi: 10.1007/s10067-017-3717-3
93. Lundtoft C, Sjowall C, Rantapaa-Dahlqvist S, Bengtsson AA, Jonsen A, Pucholt P, et al. Strong association of combined genetic deficiencies in the classical complement pathway with risk of systemic lupus erythematosus and primary Sjögren's syndrome. Arthritis Rheumatol. (2022) 74(11):1842–50. doi: 10.1002/art.42270
94. Liao J, Luo S, Yang M, Lu Q. Overexpression of CXCR5 in CD4(+) T cells of SLE patients caused by excessive SETD3. Clin Immunol. (2020) 214:108406. doi: 10.1016/j.clim.2020.108406
95. Khatri B, Tessneer KL, Rasmussen A, Aghakhanian F, Reksten TR, Adler A, et al. Genome-wide association study identifies Sjögren's risk loci with functional implications in immune and glandular cells. Nat Commun. (2022) 13(1):4287. doi: 10.1038/s41467-022-30773-y
96. Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. (2007) 104(16):6758–63. doi: 10.1073/pnas.0701266104
97. Fukui R, Saitoh S, Kanno A, Onji M, Shibata T, Ito A, et al. Unc93B1 restricts systemic lethal inflammation by orchestrating toll-like receptor 7 and 9 trafficking. Immunity. (2011) 35(1):69–81. doi: 10.1016/j.immuni.2011.05.010
98. Panchanathan R, Liu H, Choubey D. Expression of murine Unc93b1 is up-regulated by interferon and estrogen signaling: implications for sex bias in the development of autoimmunity. Int Immunol. (2013) 25(9):521–9. doi: 10.1093/intimm/dxt015
99. Griesbeck M, Ziegler S, Laffont S, Smith N, Chauveau L, Tomezsko P, et al. Sex differences in plasmacytoid dendritic cell levels of IRF5 drive higher IFN-alpha production in women. J Immunol. (2015) 195(11):5327–36. doi: 10.4049/jimmunol.1501684
100. Shin JH, Park YH, Sim M, Kim SA, Joung H, Shin DM. Serum level of sex steroid hormone is associated with diversity and profiles of human gut microbiome. Res Microbiol. (2019) 170(4-5):192–201. doi: 10.1016/j.resmic.2019.03.003
101. Pace F, Watnick PI. The interplay of sex steroids, the immune response, and the intestinal Microbiota. Trends Microbiol. (2021) 29(9):849–59. doi: 10.1016/j.tim.2020.11.001
102. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. (2013) 339(6123):1084–8. doi: 10.1126/science.1233521
103. Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity. (2013) 39(2):400–12. doi: 10.1016/j.immuni.2013.08.013
104. Silverman GJ, Deng J, Azzouz DF. Sex-dependent lupus Blautia (Ruminococcus) gnavus strain induction of zonulin-mediated intestinal permeability and autoimmunity. Front Immunol. (2022) 13:897971. doi: 10.3389/fimmu.2022.897971
105. Mu Q, Zhang H, Liao X, Lin K, Liu H, Edwards MR, et al. Control of lupus nephritis by changes of gut microbiota. Microbiome. (2017) 5(1):73. doi: 10.1186/s40168-017-0300-8
106. Zegarra-Ruiz DF, El Beidaq A, Iniguez AJ, Lubrano Di Ricco M, Manfredo Vieira S, Ruff WE, et al. A diet-sensitive commensal Lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe. (2019) 25(1):113–27.e6. doi: 10.1016/j.chom.2018.11.009
107. Rosser EC, de Gruijter NM, Matei DE. Mini-Review: gut-Microbiota and the sex-bias in autoimmunity—lessons learnt from animal models. Front Med (Lausanne). (2022) 9:910561. doi: 10.3389/fmed.2022.910561
108. Bae HR, Leung PSC, Hodge DL, Fenimore JM, Jeon SM, Thovarai V, et al. Multi-omics: differential expression of IFN-gamma results in distinctive mechanistic features linking chronic inflammation, gut dysbiosis, and autoimmune diseases. J Autoimmun. (2020) 111:102436. doi: 10.1016/j.jaut.2020.102436
109. Wang X, Pang K, Wang J, Zhang B, Liu Z, Lu S, et al. Microbiota dysbiosis in primary Sjögren's syndrome and the ameliorative effect of hydroxychloroquine. Cell Rep. (2022) 40(11):111352. doi: 10.1016/j.celrep.2022.111352
110. Schaefer L, Trujillo-Vargas CM, Midani FS, Pflugfelder SC, Britton RA, de Paiva CS. Gut Microbiota from Sjögren syndrome patients causes decreased T regulatory cells in the lymphoid organs and desiccation-induced corneal barrier disruption in mice. Front Med (Lausanne). (2022) 9:852918. doi: 10.3389/fmed.2022.852918
111. Niemann B, Puleo A, Stout C, Markel J, Boone BA. Biologic functions of hydroxychloroquine in disease: from COVID-19 to cancer. Pharmaceutics. (2022) 14(12):2551. doi: 10.3390/pharmaceutics14122551
112. Costedoat-Chalumeau N, Dunogue B, Morel N, Le Guern V, Guettrot-Imbert G. Hydroxychloroquine: a multifaceted treatment in lupus. Presse Med. (2014) 43(6 Pt 2):e167–80. doi: 10.1016/j.lpm.2014.03.007
113. Mahmoud TI, Wang J, Karnell JL, Wang Q, Wang S, Naiman B, et al. Autoimmune manifestations in aged mice arise from early-life immune dysregulation. Sci Transl Med. (2016) 8(361):361ra137. doi: 10.1126/scitranslmed.aag0367
114. Wieczorek G, Bigaud M, Pfister S, Ceci M, McMichael K, Afatsawo C, et al. Blockade of CD40-CD154 pathway interactions suppresses ectopic lymphoid structures and inhibits pathology in the NOD/ShiLtJ mouse model of sjogren's syndrome. Ann Rheum Dis. (2019) 78(7):974–8. doi: 10.1136/annrheumdis-2018-213929
115. Voynova E, Mahmoud T, Woods LT, Weisman GA, Ettinger R, Braley-Mullen H. Requirement for CD40/CD40L interactions for development of autoimmunity differs depending on specific checkpoint and costimulatory pathways. Immunohorizons. (2018) 2(1):54–66. doi: 10.4049/immunohorizons.1700069
116. A Study to Evaluate the Efficacy and Safety of VIB4920 in Participants With Sjögren's Syndrome (SS) (2023). Available at: https://www.clinicaltrials.gov/ct2/show/NCT04129164?term=NCT04129164&draw=2&rank=1 (Accessed February 17, 2023).
117. Singh AK, Goerlich CE, Zhang T, Lewis BGT, Hershfeld A, Mohiuddin MM. CD40-CD40L Blockade: update on novel investigational therapeutics for transplantation. Transplantation. (2022). doi: 10.1097/TP.0000000000004469
118. Viklicky O, Slatinska J, Novotny M, Hruba P. Developments in immunosuppression. Curr Opin Organ Transplant. (2021) 26(1):91–6. doi: 10.1097/MOT.0000000000000844. .33332922
119. Wang B, Chen S, Li Y, Xuan J, Liu Y, Shi G. Targeted therapy for primary Sjögren's syndrome: where are we now? BioDrugs. (2021) 35(6):593–610. doi: 10.1007/s40259-021-00505-7
120. Study of Safety and Tolerability of CFZ533 in Patients With Sjögren's Syndrome (TWINSS Extn) (2023). Available at: https://www.clinicaltrials.gov/ct2/show/NCT04541589?term=CD40&cond=Sj%C3%B6gren&draw=2&rank=3 (Accessed February 17, 2023).
121. Wright JA, Bazile C, Clark ES, Carlesso G, Boucher J, Kleiman E, et al. Impaired B cell apoptosis results in autoimmunity that is alleviated by ablation of btk. Front Immunol. (2021) 12:705307. doi: 10.3389/fimmu.2021.705307
122. A Phase 2 Study to Evaluate the Safety and Efficacy of LOU064 in Patients With Moderate to Severe Sjögren's Syndrome (LOUiSSe) (2023). Available at: https://www.clinicaltrials.gov/ct2/show/NCT04035668?term=Remibrutinib&cond=Sjogren%27s&draw=2&rank=1 (Accessed February 17, 2023).
123. Robak E, Robak T. Bruton's kinase inhibitors for the treatment of immunological diseases: current Status and perspectives. J Clin Med. (2022) 11(10):2807. doi: 10.3390/jcm11102807
124. The WILLOW Study With M5049 in SLE and CLE (SCLE and/or DLE) (WILLOW) (2023). Available at: https://clinicaltrials.gov/ct2/show/NCT05162586?term=M5049&draw=2&rank=7 (Accessed February 17, 2023).
125. Klopp-Schulze L, Shaw JV, Dong JQ, Khandelwal A, Vazquez-Mateo C, Goteti K. Applying modeling and simulations for rational dose selection of novel toll-like receptor 7/8 inhibitor enpatoran for indications of high medical need. Clin Pharmacol Ther. (2022) 112(2):297–306. doi: 10.1002/cpt.2606
126. Port A, Shaw JV, Klopp-Schulze L, Bytyqi A, Vetter C, Hussey E, et al. Phase 1 study in healthy participants of the safety, pharmacokinetics, and pharmacodynamics of enpatoran (M5049), a dual antagonist of toll-like receptors 7 and 8. Pharmacol Res Perspect. (2021) 9(5):e00842. doi: 10.1002/prp2.842
127. Fernandez-Carballido C, Sanchez-Piedra C, Valls R, Garg K, Sanchez-Alonso F, Artigas L, et al. Female sex, age, and unfavorable response to tumor necrosis factor inhibitors in patients with axial spondyloarthritis: results of statistical and artificial intelligence-based data analyses of a national multicenter prospective registry. Arthritis Care Res (Hoboken). (2023) 75(1):115–24. doi: 10.1002/acr.25048
128. Lubrano E, Perrotta FM, Manara M, D'Angelo S, Addimanda O, Ramonda R, et al. The sex influence on response to tumor necrosis factor-alpha inhibitors and remission in axial spondyloarthritis. J Rheumatol. (2018) 45(2):195–201. doi: 10.3899/jrheum.170666
129. Michelsen B, Berget KT, Loge JH, Kavanaugh A, Haugeberg G. Sex difference in disease burden of inflammatory arthritis patients treated with tumor necrosis factor inhibitors as part of standard care. PLoS One. (2022) 17(5):e0266816. doi: 10.1371/journal.pone.0266816
130. Van Voorhees AS, Mason MA, Harrold LR, Guo N, Guana A, Tian H, et al. Characterization of insufficient responders to anti-tumor necrosis factor therapies in patients with moderate to severe psoriasis: real-world data from the US corrona psoriasis registry. J Dermatolog Treat. (2021) 32(3):302–9. doi: 10.1080/09546634.2019.1656797
131. Mease PJ, Gladman DD, Merola JF, Deodhar A, Ogdie A, Collier DH, et al. Potential impact of sex and BMI on response to therapy in psoriatic arthritis: post hoc analysis of results from the SEAM-PsA trial. J Rheumatol. (2022) 49(8):885–93. doi: 10.3899/jrheum.211037
132. Hambardzumyan K, Hermanrud C, Marits P, Vivar N, Ernestam S, Wallman JK, et al. Association of female sex and positive rheumatoid factor with low serum infliximab and anti-drug antibodies, related to treatment failure in early rheumatoid arthritis: results from the SWEFOT trial population. Scand J Rheumatol. (2019) 48(5):362–6. doi: 10.1080/03009742.2019.1602670
133. Schultheiss JPD, Brand EC, Lamers E, van den Berg WCM, van Schaik FDM, Oldenburg B, et al. Earlier discontinuation of TNF-alpha inhibitor therapy in female patients with inflammatory bowel disease is related to a greater risk of side effects. Aliment Pharmacol Ther. (2019) 50(4):386–96. doi: 10.1111/apt.15380
134. van der Schoot LS, van den Reek J, Groenewoud JMM, Otero ME, Njoo MD, Ossenkoppele PM, et al. Female patients are less satisfied with biological treatment for psoriasis and experience more side-effects than male patients: results from the prospective BioCAPTURE registry. J Eur Acad Dermatol Venereol. (2019) 33(10):1913–20. doi: 10.1111/jdv.15733
135. Shehab M, Alasfour H, Abdullah I, Alhendi G, Alhadab A, Alfadhli A, et al. Relationship between patient sex and serum tumor necrosis factor antagonist drug and anti-drug antibody concentrations in inflammatory bowel disease; a nationwide cohort study. Front Med (Lausanne). (2021) 8:801532. doi: 10.3389/fmed.2021.801532
Keywords: X chromosome, estrogen, X chromosome inactivation (XCI), microbiome, salivary glands
Citation: Punnanitinont A and Kramer JM (2023) Sex-specific differences in primary Sjögren's disease. Front. Dent. Med 4:1168645. doi: 10.3389/fdmed.2023.1168645
Received: 17 February 2023; Accepted: 28 April 2023;
Published: 16 May 2023.
Edited by:
Ariadne Letra, University of Pittsburgh, United StatesReviewed by:
Fabian Flores-Borja, Queen Mary University of London, United KingdomLinda Sangalli, University of Kentucky, United States
© 2023 Punnanitinont and Kramer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jill M. Kramer, amtyYW1lckBidWZmYWxvLmVkdQ==