 Henrique Ballassini Abdalla
Henrique Ballassini Abdalla Thomas E. Van Dyke
Thomas E. Van Dyke
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Dent. Med. , 01 February 2023
Sec. Systems Integration
Volume 4 - 2023 | https://doi.org/10.3389/fdmed.2023.1129371
This article is part of the Research Topic Bone and Metabolic Activities View all 10 articles
Periodontitis is a chronic inflammatory disease with complex pathogenesis. Uncontrolled inflammation is driven by the immune system in response to accumulation of oral biofilm that leads to alveolar bone loss, bleeding, increased periodontal probing depth with loss of attachment of the connective tissues to the tooth, and ultimately, tooth loss. Soluble epoxide hydrolase (sEH) is an enzyme that converts epoxy fatty acids (EpFAs) produced by cytochrome P450 (CYP450) to an inactive diol. It has been shown that EpFAs display important features to counteract an exaggerated inflammatory process. Based upon this observation, inhibitors of sEH have been developed and are being proposed as a strategy to regulate proinflammatory lipid mediator production and the chronicity of inflammation. This mini review focuses on the impact of sEH inhibition on periodontal tissues focusing on the mechanisms involved. The interaction between Specialized Pro-Resolving Mediators and sEH inhibition emerges as a significant mechanism of action of sEH inhibitors that was not formerly appreciated and provides new insights into the role SPMs may play in prevention and treatment of periodontitis.
Periodontitis is a chronic inflammatory disease with a complex pathogenesis that encompasses the host immune system and oral microbiome dysbiosis (1–3). The uncontrolled inflammation in the periodontium leads to the destruction of hard and soft tissues and, eventually, tooth loss (4). The unwanted excessive inflammatory reaction in periodontitis is due to the failure of endogenous inflammation resolution pathway activation (5). The cessation of the inflammatory process occurs when a balance between pro-inflammatory and pro-resolution mediators is achieved that determines health or disease (6, 7).
Inflammation is a natural and physiological reaction to injury or infection in all biological systems. This biochemical response is finely orchestrated and well-organized to fight pathogens and to restore homeostasis. It is generally accepted as a vital process for our existence. In an ideal scenario, an inflammatory reaction is self-limiting, characterized by a local increase of protein mediators (cytokines, chemokines) and lipid mediators (LMs) (e.g., prostaglandins and leukotrienes), vascular dilation and enhanced capillary permeability, and leukocyte trafficking and activation (8). The initiation or resolution of inflammation is dictated in large part by the metabolism of polyunsaturated fatty acids (PUFA) by cyclooxygenases (COX), lipoxygenases (LOX), or cytochrome P450 (CYP450) (9, 10).
Eicosanoids, a group of LMs, are oxidized derivates from the metabolism of arachidonic acid (ARA) by oxidative pathways, the COXs, LOXs, or CYP450 enzymes (8, 11). The resulting bioactive molecules, prostanoids, leukotrienes, hydroxyeicosatetraenoic acids (HETEs), epoxyeicosatrienoic acids (EETs), and hydroperoxyeicosatetraenoic acids (HPETEs) are largely generated in inflammation, with distinct biological functions (12). Although much is known about the metabolism of polyunsaturated fatty acids by the cyclooxygenases and lipoxygenases enzymatic pathways and the activities of their downstream metabolites (13), the cytochrome P450 pathways are less understood, and are the center of this mini review. Notably, the EETs, as well as epoxides of other long-chain polyunsaturated fatty acids (EpFA) generated by the cytochrome P450 pathway, are important bioactive lipids with immunomodulatory actions in inflammation (14, 15). Most of these LMs are short-lived due to their rapid metabolization into inactive diols in the presence of soluble epoxide hydrolase (sEH), losing their ability to resolve inflammation (16). Worst, some of their diols contribute to inflammatory cytokine storm and block the initiation of the resolution phase (17). The sEH enzymes are largely found in the liver, brain, spleen, kidney, intestine, and joints (18–20), and high sEH expression was detected in chronic osteolytic inflammatory disorders, such as periodontitis and arthritis (19, 21–23).
Here, this mini review dissects the mechanisms uncovered to date explaining how sEH inhibition impacts the inflammatory process in periodontal tissues, protects against alveolar bone resorption, and speculates possible interactions/synergism between metabolites derived from sEH inhibition and the resolvent lipid mediators (lipoxins, resolvins) in periodontal tissues.
Periodontitis is a chronic inflammatory and infectious disease culminating in a dysbiotic dental biofilm that disrupts the homeostasis of the subgingival environment (24). It is the sixth most prevalent disease among inflammatory osteolytic disorders worldwide, representing a significant public health problem (1, 2, 25). Clinically, periodontitis manifests as loss of clinical attachment, alveolar bone resorption, bleeding on probing, and periodontal pockets, and unlike gingivitis, these clinical symptoms are usually permanent. Individual periodontal susceptibility encompasses genetic, behavioral, and environmental factors that regulate the host immune response and generate ideal conditions for pathogenic biofilm microbial colonization (24, 26). Although microbial pathogens are associated with disease progression and severity, the molecular and biological basis of periodontitis is now realized to be the result of an excessive and uncontrolled inflammatory response rather than a classic infection with an exogenous organism(s) (7, 27). This shift in the periodontal disease paradigm began when increased levels of prostaglandin E2 (PGE2) were found in crevicular fluid of children and adults, and the levels of PGE2 were correlated with disease severity. What caught the researcher's attention was that children had higher levels of PGE2 than adults, and the capacity of PGE2 to provoke periodontal tissue destruction (28–30). In this sense, the inflammation is an essential component of periodontal disease genesis; the tissues are destroyed by the host, not the bacteria.
As a chronic inflammatory disease, periodontitis stimulates a wide range of immune cells, from residents to infiltrating and patrolling cells, that disrupt tissue homeostasis and is characterized by a change in the immune cell composition (31). Additionally, the communication between the osseous and immune systems are intimately interconnected and responsible for bone destruction or remodeling (32–34). Alvarez and colleagues elegantly describe the spatiotemporal profile of the main gingival immune cell composition in ligature induced experimental periodontitis (35). Initially, neutrophils (CD45+LY6GhighLY6CmidCD11b+/−) are the most abundant leukocyte cells in the gingiva, reaching their peak 24 h after ligature placement, indicating the activation of the innate immune response. This intense infiltration is accompanied by an over-expression of inflammatory cytokines (IL-1β, IL-6, IL-8, IL-12, and TNF-α), giving birth to a hyper-inflammatory phenomenon (36, 37). The transition from innate immunity to adaptive immune response begins on day 3 when tissue-resident macrophages are expanded, and circulating monocytes are recruited to be differentiated into M1-like macrophages (CD45+CD64+CD11b+MHCII+) (35). Macrophages are highly plastic cells that can exhibit dual roles in tissue repair or destruction, depending on their microenvironment (36, 38). Particularly, macrophage phenotypes, M1-like (pro-inflammatory subtype) or M2-like subsets (pro-resolving), are temporally associated with the different stages of experimental periodontitis progression (39). Although M1 macrophages are usually associated with an exacerbated inflammatory response, their presence and activation are needed to fight against pathogen invasion during the acute phase. They are implicated in producing several protein and lipid mediators (cytokines, chemokines, lipids mediators), which are fundamental to orchestrating the inflammatory response and guiding the return to tissue homeostasis, in a normal, self-limiting acute inflammatory response (40). On the other hand, resolving macrophages (M2-like) coordinate the resolution process of inflammation by removing dead cells through efferocytosis, producing anti-inflammatory cytokines (e.g., IL-10, IL-4, and TGF-β), counteracting osteoclast activity and boosting osteoblastic functions with augmented cystatin C (39, 40). Moreover, resolving macrophages are well-known synthesizers of Specialized Pro-resolving Mediators (SPMs), a fundamental lipid mediator class switching that defines inflammation termination and resolution stimulation (9, 41).
Failure of the acute response to resolve normally leads to chronicity and chronic inflammatory diseases which include periodontal disease. In experimental periodontitis, T cells (CD45+CD3+) represent roughly 70% of all cell populations in the gingiva, reaching the peak at day 10 post-ligature (35). Specifically, alveolar bone resorption relies on the imbalance between T-helper type 17 and regulatory T cells (Treg) (40, 42). Although Th17 cells have a physiological immune-protective role in the oral mucosa, their exaggerated activation establishes an interaction with the osteoclast by directly inducing RANKL expression by osteoblasts and periodontal ligament fibroblasts through IL-17A and IL-17F synthesis, ultimately leading to bone loss (34). The CD4+ Th17 cells were first described in the early 2000s (43–45). This abnormal reaction is associated with augmented IL-23 levels, from the IL-12 cytokine family (46). Further, transforming growth factor-beta (TGF-beta) primes IL-23R, enhancing the Th17 responsiveness to IL-23 (45), culminating in intense neutrophil transmigration to inflamed sites and RANK/RANKL axis incitement (47). To the contrary, another subset of T cells, Tregs, are regulators of exaggerated inflammatory reactions, maintaining humoral tolerance and reestablishing homeostasis (48). The mainly immunosuppressive Treg features are linked with the release of inhibitory cytokines, such as IL-10, TGF-beta (48), and IL-35 (49), and by dampening dendritic cells via the interaction between cytotoxic T-lymphocyte antigen 4 (CTLA4) and cluster of differentiation (CD) 80/86 (48). Curiously, in experimental periodontitis, Tregs from cervical lymph nodes lose their capacity to counteract osteoclastogenic activity, presenting lower expression of Foxp3, and show a Th17-type response (increased IL-17 gene expression) without fully transdifferentiating into Th17-like cells (50).
Endorsing the immunological aspects of periodontal disease progression, inflammatory lipid mediators are dramatically elevated in periodontal tissues and crevicular fluid, such as leukotriene B4 (LTB4) and prostaglandin E2 (PGE2) (28–30). Apart from inflammatory lipid mediators, differences in the Specialized Pro-Resolving Mediators (SPMs) and other lipid mediator profiles are associated with the stages of periodontal inflammation (51, 52). Gingival samples from healthy and periodontitis subjects showed distinct lipid profiles in PCA (Principal Component Analysis) of metabolipidomics (51). Notably, none of the SPMs were found to be higher in periodontitis than in healthy subjects, although several pathway markers for omega-6 driven SPMs (e.g., 5-HETE and 15-HETE), D-series resolvins (e.g., 4-HDHA and 7-HDHA), and E-series resolvins [15(S)-HEPE] were higher in periodontitis. Moreover, the resolvin E1 receptor (BLT1) was lower in periodontitis than in healthy subjects’ samples (51). These findings suggest that in periodontitis, there is an effort by the body to re-establish homeostasis and initiate the resolution process through SPM synthesis; however, even though essential pathways seem to be activated, none of the final SPM metabolites were found at physiological levels, enough to exert cell function, and SPM receptor expression was decreased.
John Casida's group led the discovery of soluble epoxide hydrolase in the 1970s, when they described an unknown epoxide hydrolase activity in the soluble fraction of liver homogenates (53–55). Interestingly, the fundamental biological role of sEH is proved by its conservation among species, from chordates to mammalians (56), and it is mostly expressed in the liver, kidney, intestine, brain, and endothelial cells (57).
Soluble epoxide hydrolase was found to be essential for the hydrolysis of the epoxy fatty acids. The epoxy fatty acids are generated by polyunsaturated fatty acid metabolism [including ARA, linoleic acid (LA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), docosapentaenoic acid (DPA)] through the enzymatic activity of cytochrome P450, resulting in lipid mediators with a broad spectrum of biological functions at the systemic and cellular levels (58). The epoxidized metabolites are primarily anti-inflammatory and resolution lipid mediators, such as epoxyeicosatrienoic acids (EETs) from omega-6 ARA, epoxyeicosatrienoic acids (EEQs) from omega-3 EPA, and epoxydocosapentaenoic acids (EDPs) from omega-3 DHA (Wagner et al., 2017). However, in the presence of sEH (their principal regulatory enzyme), these epoxy metabolites are rapidly transformed into inactive diols, which could also possess pro-inflammatory functions (59).
In this regard, targeted inhibition of sEH during the inflammatory process, and consequently, enhancement of epoxy fatty acid bioavailability, offers an attractive strategy for inflammation control. The first inhibitors designed were too unstable for in vivo experiments (60). With the advent of crystallographic studies and the discovery of dicyclohexyl urea as a reversible inhibitor of soluble epoxide hydrolase (61), the next generation of inhibitors was produced with higher efficacy, stability, pharmacokinetics, and minor off-target activity (62). Since then, many studies have been carried out in several inflammatory models with promising results. Below, we summarize the findings on soluble epoxide hydrolase inhibition in periodontal tissues.
The pharmacological inhibition of soluble epoxide hydrolase and its impact on inflammatory, autoimmune, and pain disorders has been widely explored (63–66). Nevertheless, its application in periodontitis or other orofacial conditions is new (18, 21–23, 67). There are only a few studies involving the inhibition of the soluble epoxide hydrolase enzyme in periodontal disease (21–23); therefore, we will address them in detail. Still, in our bibliographic search, we found only one article that shows the impact of EETs on osteoclasts (68) and another on fibroblasts (69), although both are not focused on oral tissues.
Trindade-da-Silva and colleagues initially demonstrated the protective effect of soluble epoxide hydrolase inhibitors (TPPU) on alveolar bone resorption in experimental periodontitis induced by Aggregatibacter actinomycetemcomitans (Aa), as exemplified in Figure 1 in a ligature-induced periodontitis model (21). The potential bacteriostatic effect of the sEH inhibitor was discarded when no changes in Aa’ growth were found in the presence of TPPU. Subsequently, by measuring the distance between the cemento–enamel junction and the alveolar bone crest, the researchers showed that by inhibiting soluble epoxide hydrolase, lower bone loss in infected animals was detected, altering the phenotype of experimental periodontitis. Interestingly, treatment with EETs, one of the CYP450 metabolite branches that is inactivated by sEH, did not prevent bone loss. Additionally, the treatment with sEH and EETs concomitantly, did not result in a greater prevention of bone loss. Corroborating evidence was provided by genetic inhibition of soluble epoxide hydrolase by gene KO, which showed reduced bone loss, recapitulating the previous observations from the pharmacological inhibition by TPPU (21). Mechanistically, pharmacological inhibition and genetic ablation decreased activation of the RANK/RANKL/OPG axis in gingival tissue. In agreement, the reduced protein expression of MCP-1 (monocyte chemotactic protein 1), a vital monocyte recruiter associated with lower levels of F4/80 (EGF-like module-containing mucin-like hormone receptor-like 1) in the gingiva, endorses that the protective effect of sEH inhibition is related to the regulation of the exaggerated inflammation and the immune system response (21). The decreased inflammatory process was tracked by two essential downstream stress kinases, mitogen-activated protein kinase phosphorylation (p38 and JNK 1/2), which ultimately led to nuclear factor kappa B (NFκB) activation (70). Animals treated with TPPU, TPPU and EETs, or in sEH KOs, showed greatly reduced phosphorylation of p38 and JNK 1/2. Finally, pharmacological sEH inhibition and knockout animals (sEH−/−) showed inhibition of phosphorylation of the ER stress sensor (PERK, protein kinase RNA-like ER kinase); eIF2α, eukaryotic initiation factor 2α; IRE1, inositol-requiring enzyme 1; sXBP1, spliced X-box binding protein 1 and associated apoptosis (c-Caspase-3 and immunoglobulin binding protein) (21).
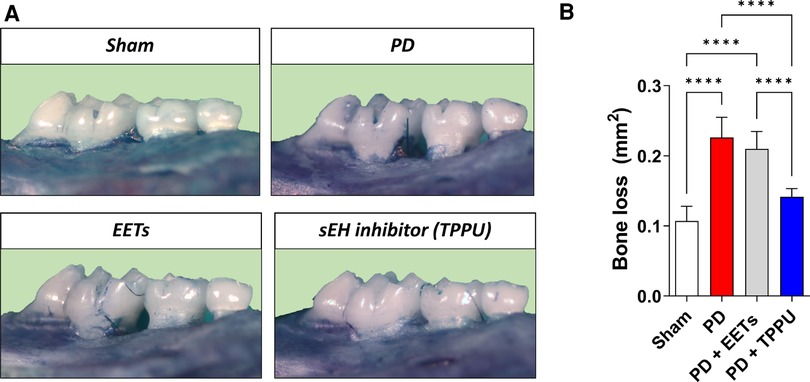
Figure 1 Inhibition of soluble epoxide hydrolase prevents alveolar bone loss in experimental periodontitis in mice. (A) Representative images from a palatal view of maxillary molars. TPPU was used as the soluble epoxide hydrolase inhibitor. (B) Bone loss was quantified as the area between the cementum-enamel junction and the alveolar bone. PD, periodontal disease; EETs, epoxyeicosatrienoic acids. ****P < 0.0001. The data are expressed as mean ± S.D; n = 5 animals per group.
Napimoga and collaborators, in a succeeding study by the same research group, showed that inflamed gingival tissue induced by experimental periodontitis expressed higher levels of sEH than control animals. Pharmacological inhibition of sEH dampened this expression, and correlated with lessening disease severity (22). Using an RNA array to explore the innate and adaptive immune systems, sEH inhibition diminished the expression of toll-like receptors 1 and 9 (Trl1 and Trl9), which play a crucial role in inflammatory cytokine release upon triacylated lipopeptide recognition (71) and activation of osteoclastic functions (72). T cells were also affected. The expression of Cd8 and Cd4 was diminished, as well as Cd40l, interferon-alpha2 (Ifnα2), and interferon-beta (Ifnβ) (21). Downregulation of Cd40l impairs B-cell activation and, therefore, the production of IL-2, IL-6, and TNF-alpha (73). The signal transducer and activator of transcription 4 (Stat4) is a factor that contributes to IL-12, IL-23, and IFN-1 production, in addition to differentiating Th1 and Th17 cells (74), which was also reduced by sEH inhibition. These findings reinforce the concept that by inhibiting sEH, the unwanted lymphocyte response is managed, as also demonstrated in a collagen-induced model of arthritis (19), preventing osteoclastogenic activity in the periodontium (21) and knee joint (19).
Recently, Abdalla and coworkers thoroughly characterized the impact of the sEH/EET axis on gingival macrophage plasticity in experimental periodontitis in mice. The work revealed for the first time that pharmacological inhibition of sEH fosters communication between epoxy fatty acid metabolites, increasing the levels of Specialized Pro-Resolving Mediators [e.g., resolvin (Rv) E-series and lipoxins] in saliva, as well as their respective receptors in the gingival tissues (23).
Mechanistically, pharmacological sEH inhibition suppressed alveolar bone loss via actions on inflammatory osteolytic factors, such as Il17a and RANKL. In metabolipidomic analyses, soluble epoxide hydrolase inhibitor treated animals showed lipid profiles that were distinct from experimental periodontitis and control animals in two-dimensional and three-dimensional Principal Component Analyses. The foremost lipid mediators enhanced by sEH inhibition were RvE1, RvE2, and LXA4, well-known SPMs with robust immunoresolvent features that guide healing. Moreover, 20-hydroxy LTB4 was enhanced, inferring an inactivation of LTB4, a critical inflammatory lipid mediator. Further, the Specialized Pro-Resolving Mediator receptors (LTB4R1, CMKLR1/ChemR23, and ALX/FPR2) were also found to increase in gingival tissue, suggesting greater effectiveness of SPM activity at the site of inflammation. In macrophages, the pharmacological inhibition of soluble epoxide hydrolase stimulated a dynamic transcriptional reprogramming of inflammatory macrophages toward resolving macrophages (characterized by CD11c+/CD206+ double-positive cells in the CD45+/CD11b+/CD64+ macrophage population), associated with reduced expression of Il1β, TNFα, Il12, and Nos2. Finally, in vitro assays revealed that sEH inhibition and EET treatment triggered SPM release in bone marrow derived macrophages (BMDMs) in both inflammatory and resolvent macrophages (23). These findings are summarized in Figure 2.
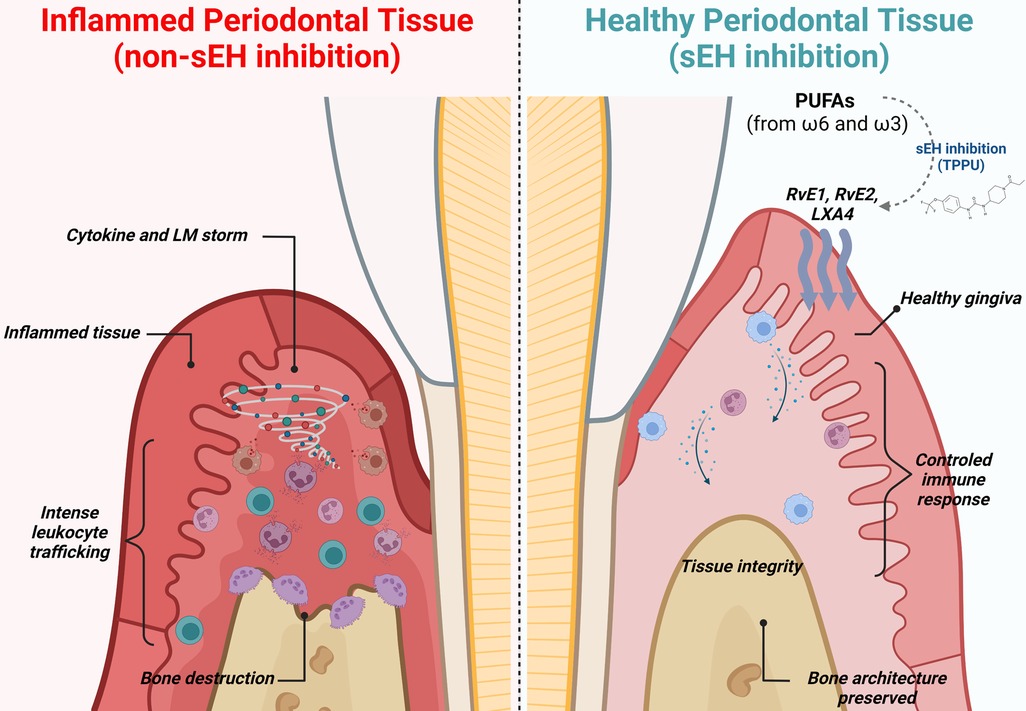
Figure 2 Immune modulation and lipid mediator synthesis induced by sEH inhibition in experimental periodontitis. During periodontitis, the immune system drives an unwanted and uncontrolled inflammatory reaction, leading to an intense release of inflammatory cytokines, chemokines, and lipid mediators, ultimately leading to alveolar bone loss, gingival tissue damage, and increased probing depth (left panel). Pharmacological inhibition of sEH improves the bioavailability of epoxy fatty acids (EpFAs), shifting polyunsaturated fatty acid (PUFA) metabolism and favoring production of Specialized Pro-Resolving Mediators. Further, macrophages undergo phenotypic reprogramming towards resolving and repairing features associated with releasing anti-inflammatory cytokines. Innate immunity is controlled and well-orchestrated. Finally, inhibition of sEH prevents osteoclastic activation, preventing alveolar bone loss.
The direct influence of the EETs/sEH/DHET axis on osteoclast differentiation and activity was explored in vitro using BMMCs (Bone marrow mononuclear cells) and RAW264.7 murine cells (68). Authors showed that DHETs, the inactive diol form of EETs, could not reduce TRAP-positive cells, but increased their number. Differently, treatment with EETs or sEH inhibitors (TPPU) significantly diminished the number of multinucleated TRAP-positive cells. Likewise, bone resorption pits were hardly impaired by EETs and sEH inhibition, as well as expression of RANK, TRAP, cathepsin K (CK), and matrix metalloproteinase (MMP)-9. Further, in an osteoblast precursor cell line (MC3T3), EETs reduced the ratio between RANKL:OPG (68). In TGF-β1-induced activation of murine fibroblasts (NIH3T3), EETs attenuate cell activation by impairing the expression of collagen, smooth muscle alpha-actin (α-SMA), and proliferating cell nuclear antigen (PCNA) in a peroxisome proliferation activated receptor γ (PPARγ) dependent-manner (69).
The pharmacological inhibition of sEH has shown impressive results in inflammatory diseases and has been the subject of extensive research. Concerning the dental medicine area, including painful orofacial conditions and periodontal disease, a few studies have been conducted, revealing promising findings. As a note, sEH inhibitors are in the clinical development phase, making them a promising forthcoming therapeutic strategy. Nevertheless, a profound molecular mechanistic analysis of how sEH inhibition acts through the immune system must be carried out. Future research should deeply analyze the impact of sEH inhibition on immune system cells and how they respond in its absence. Nevertheless, recent findings demonstrate that the inhibition of sEH influences the production of SPMs (omega-3 and -6 fatty acids metabolites from CYP450), which paves the way for a new perspective on its mechanism of action, as well as pharmacological implications, as they boost resolution pathways of inflammation rather than silencing them.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported in part by USPHS grant DE025020.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. (2021) 21(7):426–40. doi: 10.1038/s41577-020-00488-6
2. Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. (2015) 21(3):172–83. doi: 10.1016/j.molmed.2014.11.004
3. Alvarez C, Monasterio G, Cavalla F, Córdova LA, Hernández M, Heymann D, et al. Osteoimmunology of oral and maxillofacial diseases: translational applications based on biological mechanisms. Front Immunol. (2019) 10:1664. doi: 10.3389/fimmu.2019.01664
4. Van Dyke TE, Sima C. Understanding resolution of inflammation in periodontal diseases: is chronic inflammatory periodontitis a failure to resolve? Periodontol 2000. (2020) 82(1):205–13. doi: 10.1111/prd.12317
5. Van Dyke TE. Proresolving lipid mediators: potential for prevention and treatment of periodontitis. J Clin Periodontol. (2011) 38(Suppl 11):119–25. doi: 10.1111/j.1600-051X.2010.01662.x
6. Balta MG, Papathanasiou E, Blix IJ, Van Dyke TE. Host modulation and treatment of periodontal disease. J Dent Res. (2021) 100(8):798–809. doi: 10.1177/0022034521995157
7. Van Dyke TE. Shifting the paradigm from inhibitors of inflammation to resolvers of inflammation in periodontitis. J Periodontol. (2020) 91(Suppl 1):S19–25. doi: 10.1002/JPER.20-0088
8. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510(7503):92–101. doi: 10.1038/nature13479
9. Panigrahy D, Gilligan MM, Serhan CN, Kashfi K. Resolution of inflammation: an organizing principle in biology and medicine. Pharmacol Ther. (2021) 227:107879. doi: 10.1016/j.pharmthera.2021.107879
10. Maddipati KR. Non-inflammatory physiology of “inflammatory” mediators - unalamation, a new paradigm. Front Immunol. (2020) 11:580117. doi: 10.3389/fimmu.2020.580117
11. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. (2010) 10(3):181–93. doi: 10.1038/nrc2809
12. Haeggström JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. (2011) 111(10):5866–98. doi: 10.1021/cr200246d
13. Hoxha M, Zappacosta B. CYP-derived eicosanoids: implications for rheumatoid arthritis. Prostaglandins Other Lipid Mediat. (2020) 146:106405. doi: 10.1016/j.prostaglandins.2019.106405
14. Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. (2001) 276(39):36059–62. doi: 10.1074/jbc.R100030200
15. Hammock BD, Wang W, Gilligan MM, Panigrahy D. Eicosanoids: the overlooked storm in coronavirus disease 2019 (COVID-19)? Am J Pathol. (2020) 190(9):1782–8. doi: 10.1016/j.ajpath.2020.06.010
16. Wang B, Wu L, Chen J, Dong L, Chen C, Wen Z, et al. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Signal Transduct Target Ther. (2021) 6(1):94. doi: 10.1038/s41392-020-00443-w
17. Kodani SD, Hammock BD. The 2014 bernard B. Brodie award lecture-epoxide hydrolases: drug metabolism to therapeutics for chronic pain. Drug Metab Dispos. (2015) 43(5):788–802. doi: 10.1124/dmd.115.063339
18. Abdalla HB, Napimoga MH, Teixeira JM, Trindade-da-Silva CA, Pieroni VL, Dos Santos Araújo FSM, et al. Soluble epoxide hydrolase inhibition avoid formalin-induced inflammatory hyperalgesia in the temporomandibular joint. Inflammopharmacology. (2022) 30(3):981–90. doi: 10.1007/s10787-022-00965-5
19. Trindade-da-Silva CA, Clemente-Napimoga JT, Abdalla HB, Rosa SM, Ueira-Vieira C, Morisseau C, et al. Soluble epoxide hydrolase inhibitor, TPPU, increases regulatory T cells pathway in an arthritis model. FASEB J. (2020) 34(7):9074–86. doi: 10.1096/fj.202000415R
20. Norwood S, Liao J, Hammock BD, Yang GY. Epoxyeicosatrienoic acids and soluble epoxide hydrolase: potential therapeutic targets for inflammation and its induced carcinogenesis. Am J Transl Res. (2010) 2(4):447–57. PMID: 20733953; PMCID: PMC2923867
21. Trindade-da-Silva CA, Bettaieb A, Napimoga MH, Lee KSS, Inceoglu B, Ueira-Vieira C, et al. Soluble epoxide hydrolase pharmacological inhibition decreases alveolar bone loss by modulating host inflammatory response, RANK-related signaling, endoplasmic Reticulum stress, and apoptosis. J Pharmacol Exp Ther. (2017) 361(3):408–16. doi: 10.1124/jpet.116.238113
22. Napimoga MH, Rocha EP, Trindade-da-Silva CA, Demasi APD, Martinez EF, Macedo CG, et al. Soluble epoxide hydrolase inhibitor promotes immunomodulation to inhibit bone resorption. J Periodontal Res. (2018) 53(5):743–9. doi: 10.1111/jre.12559
23. Abdalla HB, Alvarez C, Wu YC, Rojas P, Hammock BD, Maddipati KR, et al. Soluble epoxide hydrolase inhibition enhances specialized pro-resolving lipid mediator production and promotes macrophage plasticity. Br J Pharmacol. (2022). doi: 10.1111/bph.16009. [Epub ahead of print]
24. Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, et al. Periodontitis: consensus report of workgroup 2 of the 2017 world workshop on the classification of periodontal and peri-implant diseases and conditions. J Periodontol. (2018) 89(Suppl 1):S173–82. doi: 10.1002/JPER.17-0721
25. Kassebaum NJ, Smith AGC, Bernabé E, Fleming TD, Reynolds AE, Vos T, et al. Global, regional, and national prevalence, incidence, and disability-adjusted life years for oral conditions for 195 countries, 1990-2015: a systematic analysis for the global burden of diseases, injuries, and risk factors. J Dent Res. (2017) 96(4):380–7. doi: 10.1177/0022034517693566
26. Kurgan S, Kantarci A. Molecular basis for immunohistochemical and inflammatory changes during progression of gingivitis to periodontitis. Periodontol 2000. (2018) 76(1):51–67. doi: 10.1111/prd.12146
27. Van Dyke TE, Bartold PM, Reynolds EC. The nexus between periodontal inflammation and dysbiosis. Front Immunol. (2020) 11:511. doi: 10.3389/fimmu.2020.00511
28. Offenbacher S, Odle BM, Gray RC, Van Dyke TE. Crevicular fluid prostaglandin E levels as a measure of the periodontal disease status of adult and juvenile periodontitis patients. J Periodontal Res. (1984) 19(1):1–13. doi: 10.1111/j.1600-0765.1984.tb01190.x
29. Offenbacher S, Odle BM, Van Dyke TE. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J Periodontal Res. (1986) 21(2):101–12. doi: 10.1111/j.1600-0765.1986.tb01443.x
30. Van Dyke TE, Lester MA, Shapira L. The role of the host response in periodontal disease progression: implications for future treatment strategies. J Periodontol. (1993) 64(Suppl 8S):792–806. doi: 10.1902/jop.1993.64.8s.792
31. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. (2015) 15(1):30–44. doi: 10.1038/nri3785
32. Arron JR, Choi Y. Bone versus immune system. Nature. (2000) 408(6812):535–6. doi: 10.1038/35046196
33. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. (2000) 408(6812):600–5. doi: 10.1038/35046102
34. Ikeuchi T, Moutsopoulos NM. Osteoimmunology in periodontitis; a paradigm for Th17/IL-17 inflammatory bone loss. Bone. (2022) 163:116500. doi: 10.1016/j.bone.2022.116500
35. Alvarez C, Abdalla H, Sulliman S, Rojas P, Wu YC, Almarhoumi R, et al. Rve1 impacts the gingival inflammatory infiltrate by inhibiting the T cell response in experimental periodontitis. Front Immunol. (2021) 12:664756. doi: 10.3389/fimmu.2021.664756
36. Ling MR, Chapple IL, Matthews JB. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun. (2015) 21(7):714–25. doi: 10.1177/1753425915589387
37. Dutzan N, Konkel JE, Greenwell-Wild T, Moutsopoulos NM. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. (2016) 9(5):1163–72. doi: 10.1038/mi.2015.136
38. Chazaud B. Macrophages: supportive cells for tissue repair and regeneration. Immunobiology. (2014) 219(3):172–8. doi: 10.1016/j.imbio.2013.09.001
39. Viniegra A, Goldberg H, Çil Ç, Fine N, Sheikh Z, Galli M, et al. Resolving macrophages counter osteolysis by anabolic actions on bone cells. J Dent Res. (2018) 97(10):1160–9. doi: 10.1177/0022034518777973
40. Garlet GP, Giannobile WV. Macrophages: the bridge between inflammation resolution and tissue repair? J Dent Res. (2018) 97(10):1079–81. doi: 10.1177/0022034518785857
41. Dalli J, Serhan C. Macrophage proresolving mediators-the when and where. Microbiol Spectr. (2016) 4(3):10.1128/microbiolspec.MCHD-0001-2014. doi: 10.1128/microbiolspec.MCHD-0001-2014
42. Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, et al. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. (2018) 10(463):eaat0797. doi: 10.1126/scitranslmed.aat0797
43. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
44. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
45. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. (2006) 441(7090):231–4. doi: 10.1038/nature04754
46. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. (2000) 13(5):715–25. doi: 10.1016/S1074-7613(00)00070-4
47. Pan W, Wang Q, Chen Q. The cytokine network involved in the host immune response to periodontitis. Int J Oral Sci. (2019) 11(3):30. doi: 10.1038/s41368-019-0064-z
48. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8(7):523–32. doi: 10.1038/nri2343
49. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450(7169):566–9. doi: 10.1038/nature06306
50. Alvarez C, Suliman S, Almarhoumi R, Vega ME, Rojas C, Monasterio G, et al. Regulatory T cell phenotype and anti-osteoclastogenic function in experimental periodontitis. Sci Rep. (2020) 10(1):19018. doi: 10.1038/s41598-020-76038-w
51. Ferguson B, Bokka NR, Maddipati KR, Ayilavarapu S, Weltman R, Zhu L, et al. Distinct profiles of specialized pro-resolving lipid mediators and corresponding receptor gene expression in periodontal inflammation. Front Immunol. (2020) 11:1307. doi: 10.3389/fimmu.2020.01307
52. Elabdeen HR, Mustafa M, Szklenar M, Rühl R, Ali R, Bolstad AI. Ratio of pro-resolving and pro-inflammatory lipid mediator precursors as potential markers for aggressive periodontitis. PLoS One. (2013) 8(8):e70838. doi: 10.1371/journal.pone.0070838
53. Gill SS, Hammock BD, Yamamoto I, Casida JE. Preliminary chromatographic studies on the metabolites and photodecomposition products of the juvenoid 1-(4'-ethylphenoxy)-6,7-epoxy-3,7-dimethyl-2-octene. In: Menn JJ, Beroza M, editors. Insect juvenile hormones: Chemistry and action. New York: Academic Press (1972). p. 177–89.
54. Gill SS, Hammock BD, Casida JE. Mammalian metabolism and environmental degradation of the juvenoid 1-(4'-ethylphenoxy)-3,7-dimethyl-6,7-epoxy-trans-2-octene and related compounds. J Agric Food Chem. (1974) 22(3):386–95. doi: 10.1021/jf60193a058
55. Hammock BD, Gill SS, Stamoudis V, Gilbert LI. Soluble mammalian epoxide hydratase: action on juvenile hormone and other terpenoid epoxides. Comp Biochem Physiol B. (1976) 53(2):263–5. doi: 10.1016/0305-0491(76)90045-6
56. Harris TR, Hammock BD. Soluble epoxide hydrolase: gene structure, expression and deletion. Gene. (2013) 526(2):61–74. doi: 10.1016/j.gene.2013.05.008
57. Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem. (2004) 52(4):447–54. doi: 10.1177/002215540405200403
58. Dyall SC, Balas L, Bazan NG, Brenna JT, Chiang N, da Costa Souza F, et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: recent advances in the understanding of their biosynthesis, structures, and functions. Prog Lipid Res. (2022) 86:101165. doi: 10.1016/j.plipres.2022.101165
59. Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. (2009) 50:S52–6. doi: 10.1194/jlr.R800038-JLR200
60. Mullin CA, Hammock BD. Chalcone oxides–potent selective inhibitors of cytosolic epoxide hydrolase. Arch Biochem Biophys. (1982) 216(2):423–39. doi: 10.1016/0003-9861(82)90231-4
61. Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, et al. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci U S A. (1999) 96(16):8849–54. doi: 10.1073/pnas.96.16.8849
62. Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. (2013) 53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244
63. Wagner KM, McReynolds CB, Schmidt WK, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther. (2017) 180:62–76. doi: 10.1016/j.pharmthera.2017.06.006
64. Fishbein A, Hammock BD, Serhan CN, Panigrahy D. Carcinogenesis: failure of resolution of inflammation? Pharmacol Ther. (2021) 218:107670. doi: 10.1016/j.pharmthera.2020.107670
65. Wang Y, Wagner KM, Morisseau C, Hammock BD. Inhibition of the soluble epoxide hydrolase as an analgesic strategy: a review of preclinical evidence. J Pain Res. (2021) 14:61–72. doi: 10.2147/JPR.S241893
66. Shan J, Hashimoto K. Soluble epoxide hydrolase as a therapeutic target for neuropsychiatric disorders. Int J Mol Sci. (2022) 23(9):4951. doi: 10.3390/ijms23094951
67. Teixeira JM, Abdalla HB, Basting RT, Hammock BD, Napimoga MH, Clemente-Napimoga JT. Peripheral soluble epoxide hydrolase inhibition reduces hypernociception and inflammation in albumin-induced arthritis in temporomandibular joint of rats. Int Immunopharmacol. (2020) 87:106841. doi: 10.1016/j.intimp.2020.106841
68. Guan H, Zhao L, Cao H, Chen A, Xiao J. Epoxyeicosanoids suppress osteoclastogenesis and prevent ovariectomy-induced bone loss. FASEB J. (2015) 29(3):1092–101. doi: 10.1096/fj.14-262055
69. Tao JH, Liu T, Zhang CY, Zu C, Yang HH, Liu YB, et al. Epoxyeicosatrienoic acids inhibit the activation of murine fibroblasts by blocking the TGF-β1-Smad2/3 signaling in a PPARγ-dependent manner. Oxid Med Cell Longev. (2022) 2022:7265486. doi: 10.1155/2022/7265486
70. Nandipati KC, Subramanian S, Agrawal DK. Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol Cell Biochem. (2017) 426(1-2):27–45. doi: 10.1007/s11010-016-2878-8
71. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. (2007) 130(6):1071–82. doi: 10.1016/j.cell.2007.09.008
72. Matsumoto C, Oda T, Yokoyama S, Tominari T, Hirata M, Miyaura C, et al. Toll-like receptor 2 heterodimers, TLR2/6 and TLR2/1 induce prostaglandin E production by osteoblasts, osteoclast formation and inflammatory periodontitis. Biochem Biophys Res Commun. (2012) 428(1):110–5. doi: 10.1016/j.bbrc.2012.10.016
73. Figueredo CM, Lira-Junior R, Love RM. T and B cells in periodontal disease: new functions in A complex scenario. Int J Mol Sci. (2019) 20(16):3949. doi: 10.3390/ijms20163949
Keywords: periodontitis, inflammation, lipid mediator, soluble epoxide hydrolase (sEH), soluble epoxide hydrolase (sEH) inhibitors
Citation: Abdalla HB and Van Dyke TE (2023) The impact of the soluble epoxide hydrolase cascade on periodontal tissues. Front. Dent. Med 4:1129371. doi: 10.3389/fdmed.2023.1129371
Received: 21 December 2022; Accepted: 9 January 2023;
Published: 1 February 2023.
Edited by:
Paulo Sergio Cerri, Universidade Estadual Paulista, BrazilReviewed by:
Vincent Everts, VU Amsterdam, Netherlands© 2023 Abdalla and Van Dyke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas E. Van Dyke, dHZhbmR5a2VAZm9yc3l0aC5vcmc=
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Systems Integration, a section of the journal Frontiers in Dental Medicine
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.