 Honghao Zhang
Honghao Zhang Afriti Chinoy
Afriti Chinoy Paymon Mousavi
Paymon Mousavi Ke’ale Louie
Ke’ale Louie Yuji Mishina
Yuji Mishina- Department of Biologic and Materials Sciences & Prosthodontics, School of Dentistry, University of Michigan, Ann Arbor, MI, United States
Ellis-van Creveld (EVC) syndrome is an autosomal recessive chondrodysplasia. The affected individuals bear a series of skeleton defects, congenital heart septum anomalies, midfacial defects, and dental defects. Previous studies using Evc or Evc2 mutant mice have characterized the pathological mechanism leading to various types of congenital defects. Some patients with EVC have supernumerary teeth; however, it is not known yet if supernumerary teeth have formed in Evc or Evc2 mutant mice, and if yes, what is the pathological mechanism associated. In the present study, we used Evc2 mutant mice and analyzed the pattern of molars in Evc2 mutant mice at various stages. Our studies demonstrate that Evc2 loss of function within the dental mesenchymal cells leads to abnormal molar patterning, and that the most anterior molar in the Evc2 mutant mandible represents a supernumerary tooth. Finally, we provide evidence supporting the idea that both compromised Hedgehog signaling and elevated WNT signaling due to Evc2 loss of function contribute to the supernumerary tooth formation.
Introduction
First reported in 1940, Ellis-van Creveld (EVC) syndrome is a rare congenital disease with an estimated occurrence of 1 in 20,000–60,000 births (1, 2). Genetic studies have linked two thirds of EVC syndrome occurrences to genetic mutations in either EVC or EVC2 (aka LIMBIN) (2–5). Typical clinical signs of EVC include dwarfism, postaxial polydactyly, congenital heart defects, and dysplasia of ectoderm-derived tissues such as hair and nails (2, 6). Additionally, genetic mutations or single nucleotide polymorphisms of EVC2 have been identified in Japanese brown cattle, Tyrolean Grey cattle, and penguins, thereby suggesting an evolutionarily conserved function of EVC2/LIMBIN (7–9).
In addition to the aforementioned clinical signs, EVC syndrome also presents with various dental abnormalities including natal teeth, delayed eruption, hypodontia, supernumerary teeth, conical-shaped teeth, taurodontism, and hypoplastic enamel (10, 11). These alterations in tooth eruption, number, and morphology in patients with EVC syndrome suggest that EVC2 plays an important role across multiple stages of tooth development.
Tooth development is a complicated process involving coordinated interactions between epithelium and mesenchymal-derived tissues (12). Organogenesis initiates with thickening of the dental epithelium and progresses through the bud stage, cap stage, and bell stage (12). Next, a single layer of epithelial and mesenchymal cells proliferate and differentiate into ameloblasts and odontoblasts, respectively, to generate dental mineralized tissues in the crown (13). Following the crown formation, the apical epithelial tissues elongate and give to a bilayer structure, Hertwig's epithelial root sheath (HERS). Then continuously growing of HERS guides the growth of dental mesenchymal tissue, which leads to formation of the root and support for further growth of the tooth root (14). Across these stages, multiple signaling pathways including Hedgehog, BMP, WNT, and FGF coordinate to ensure that each tooth achieves the correct morphology (15). In mammals, molars align in a single row. Mouse mandibular molar row development initiates from transiently existing premolars, MS and R2 (16). At E14.5, along with regress of MS and R2, the first molar (M1) formed (16). In vivo and ex vivo reveal a model that anterior molar influences the initiation and development of the next one both in size and shape (16–18). However, full characterization of a detailed molecular signaling network is still needed to provide knowledge fundamental to understand the hypodontia and supernumerary tooth formation.
EVC syndrome is categorized as a ciliopathy due to the ciliary localization of proteins encoded by the causative genes of EVC syndrome, EVC and EVC2/LIMBIN (3). Molecular studies have demonstrated that EVC and EVC2 form a protein complex, which locates to the bottom of primary cilium and is required for full transduction of Hedgehog signaling (3, 19–21). We recently generated Evc2 mutant mouse lines (21) and delineated the pathological mechanisms leading to both dwarfism and midfacial hypoplasia (21–27). We also demonstrated that compromised Hedgehog signaling due to Evc2 loss of function within dental mesenchymal tissue secondarily delays ameloblast differentiation and is responsible for hypoplastic enamel formation in Evc2 mutant mice (26).
Similar to human patients with EVC syndrome (28), Evc mutant mice present with abnormal tooth morphology (29). However, detailed descriptions of molar dysmorphology and supernumerary molar identity in Evc mutant mice have not been determined (29). Here we used Evc2 loss-of-function mutant mice to characterize molar pattern and shape. Our results indicated that Evc2 loss of function leads to abnormal patterning of molars. Several lines of evidence suggest that the most anterior molars are supernumerary teeth. The abnormal molar patterning is due to Evc2 loss of function within dental mesenchyme. In addition to decreased Hedgehog signaling, we found concurrently elevated WNT signaling in molars from Evc2 mutant mice. Our data supports the hypothesis that decreased Hedgehog signaling and elevated WNT signaling contribute to the abnormal molar patterning observed in Evc2 mutant mice.
Materials and methods
Animal models
Mice in the present studies were maintained and used in compliance with the Institutional Animal Care and Use Committee (IACUC) of the University of Michigan in accordance with the National Institutes of Health Guidelines for Care and Use of Animals in research, and all experimental procedures were approved by the IACUC of the University of Michigan (PRO00009613). Generation of Evc2 mutant mice (Evc2 ex12/+) and Evc2 floxed mice (Evc2 fl/+) were as reported previously (21). The Evc2 floxed mice were bred with Sox2-Cre mice (30) to generate mice carrying a Cre-recombined allele (dE). Deletion of Evc2 in a neural crest-specific manner was achieved through crossing Evc2 fl/fl mice with either P0-Cre mice (31) or Wnt1-Cre mice (32). WNT signaling reporter mice were from Jaxson 004623 (33). All mice were maintained in a mixed background of C57BL6/J and 129S6 and were crossed and maintained in our semi-closed mouse colony for at least 7 years. There are no sample exclusions in our studies.
Micro-CT (µCT)
For micro-CT analysis, mouse heads at P28 were fixed in 4% paraformaldehyde in phosphate buffered saline followed by scanning at the University of Michigan using a micro-CT system (µCT100 Scanco Medical, Bassersdorf, Switzerland). Scan settings were as follows: voxel size 12 µm, 55 kVp, 109 µA, 0.5 mm AL filter, and integration time 500 ms.
Image acquisition, segmentation and surface models
To visualize the molars at postnatal day 8 (P8), the surface model for molars was generated based on the micro-CT data, using ITK-SNAP (open-source software developed by grants and contracts from the U.S. National Institutes of Health, www.itksnap.org), according to standard of segmentation using ITK-SNAP and previously described protocols (23, 34).
Tooth germ dissection, mouse embryonic fibroblast (MEF) isolation and in vitro induction of WNT signaling
Mouse tooth germs were dissected from E18.5 mandibles. Under a dissection microscope, tooth germs were separated from surrounding tissues with forceps. For analysis of Hedgehog signaling and WNT signaling, total RNA was isolated from tooth germs homogenized in Trizol (Life Technology) according to standard protocols. Quantitative real-time PCR (Q-RT-PCR) was performed using Applied Biosystems ViiA7, with the following taqman probes: Axin2: Mm00443610_m1, Lef1: Mm00550265_m1, Gli1: Mm00494654_m1, Ptch1: Mm00436026_m1, and Gapdh: Mm99999915_g1. MEFs were prepared from E12.5 embryos and cultured in (DMEM with 10% Fetal bovine serum (FBS) according to previously described method (35). For arresting cell cycles and allowing growth of primary cilia, MEFs were cultured in MEF culture media (DMEM with 0.5% FBS) for at least 18 h. WNT signaling was induced by treating cells with WNT3A in concentration of 100 µg/ml for 18 h. Then total RNA was isolated from non-treated or treated cells. Then, 1 µg of total RNA was reverse transcribed using SuperScript Reverse Transcriptase (Life Technologies, Grand Island, NY, USA). Q-RT-PCR for examining induction of WNT signaling was performed in using Applied Biosystems ViiA7, with the following taqman probes: Axin2: Mm00443610_m1, Lef1: Mm00550265_m1, and Gapdh: Mm99999915_g1.
Histology and immunohistochemistry
Mouse mandibles were dissected out at P8, E18.5, E16.5, E15.5 or E14.5 and fixed in 4% paraformaldehyde (PFA). Subsequently, they were embedded in paraffin, sectioned parasagittally, and stained with hematoxylin and eosin for histologic observations according to standard histology procedure. For immunohistochemistry, dissected mandibles were fixed with 4% PFA and cryo-protected by 30% sucrose in PBS before being embedded parasagittally for cryosection. Sections were incubated overnight at 4 °C with antibody against SHH (Hybridoma Bank, University of Iowa), LEF1 (Cell signaling 2230). Sections were then incubated with secondary antibody (IgG-Alexa fluor 488 (A-32731), IgG-Alexa fluor 594 (A-21203)) at room temperature for 1 h. And subsequently mounted by Prolong Gold antifade mount with DAPI (P36935, Life Technology) and imaged under a confocal microscope (Nikon C1). The intensity of signaling was quantified using Image J. For intensity quantification, at least 40 cells were quantified and the average of all quantified cells were used to represent the biological sample. There were 4 pairs of controls and mutants were used for comparisons.
Statistical analysis
A paired t test was conducted in all studies, performed in SPSS 27.0 (IBM Corp., Armonk, NY, USA). Error bars in the graph indicate standard deviation.
Results
Evc2 mutant mice have abnormal molar patterning
In contrast to alterations in tooth number reported in patients with EvC syndrome, we did not observe different numbers of molars in Evc2 mutant mice (Figures 1A–D) (26). This occurred despite the presence of previously reported dental phenotypes including hypoplastic enamel and incisor shortening (26). However, molar patterning differed between Evc2 mutants and their control counterparts at postnatal day 28 (P28). In control mice at P28, mandibular molars had a gradated pattern of decreasing crown size from anterior-to-posterior with a patterning of M1 as the biggest, then M2, and M3 as the smallest (Figures 1A,B). This is in contrast with the mandibular molars of Evc2 mutant mice at P28 which showed no regular size gradations from anterior-to-posterior, with T2 as the biggest, then T3, and T1 as the smallest (Figures 1C,D). These observations were consistent with previous reports on both Evc and Evc2 mutant mice (26, 29).
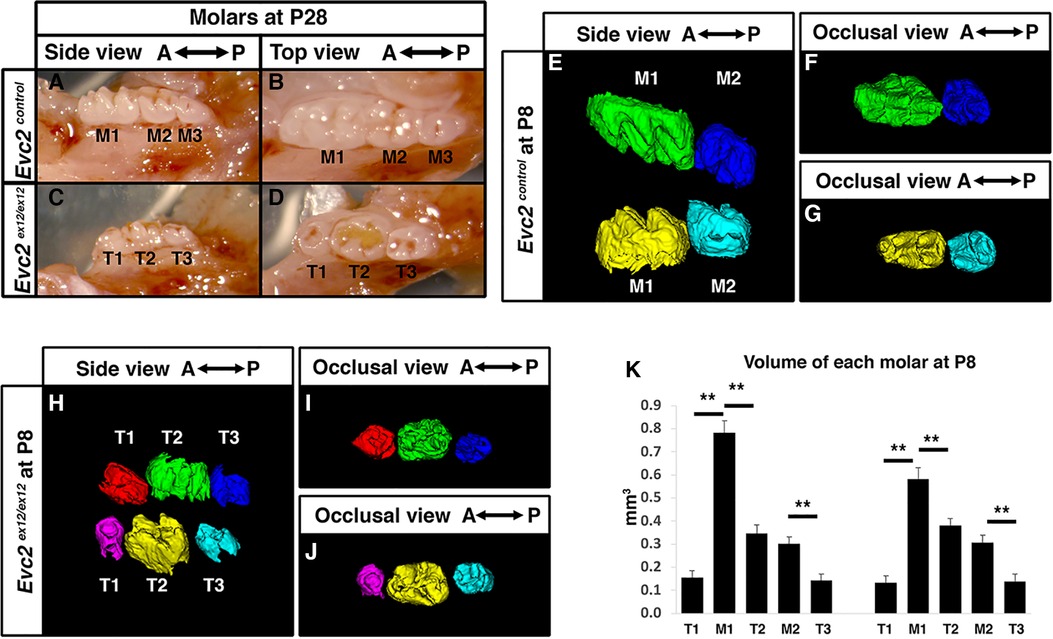
Figure 1 Evc2 mutant mice have abnormal molar morphology. Lateral and occlusal images showing the gross morphology of postnatal day 28 (P28) mandibular molars from control (A,B) and Evc2 mutant (C,D) mice. Molars from controls are marked as M1, M2, and M3 from anterior to posterior. Molars from Evc2 mutants are marked as T1, T2, and T3 from anterior to posterior. CT-reconstructed molars at postnatal day 8 (P8) from control (E–G) and Evc2 mutant (H–J) mice. Comparison of control vs. Evc2 mutant molar volumes at P8 (K). n = 4, for maxilla, M1 vs T1, p < 0.01; M1 vs T2, p < 0.01; M2 vs T3, p < 0.01; for mandible, M1 vs T1, p < 0.01; M1 vs T2, p < 0.01; M2 vs T3, p < 0.01.
To understand the mechanism leading to abnormal patterning of molars in Evc2 mutant mice, we then evaluated the molars at postnatal day 8 (P8), a stage when M1 and M2 are almost ready to erupt, but M3 is still with minimal levels of mineralization in control mice. As expected, in control mice at postnatal day 8 (P8), the M1 and M2 were mineralized, present, and nearing eruption while M3 had minimal levels of mineralization and were not present in scans (Figures 1E–G). In contrast, we observed three mineralized molar bodies, T1, T2, and T3, in the maxillae and mandibles of Evc2 mutants (Figures 1H–J). Volumetric quantification of P8 molars in each arch corroborated P28 molar pattern abnormalities and indicated that M1 was larger than either T1 or T2 and that M2 was larger than T3 (Figure 1K). The observation of 3 molars in Evc2 mutants at P8, however, led us to question the actual identity of each molar and the role of Evc2 on molar pattern and morphology. We henceforth used neural-crest specific Evc2 mutant mice to investigate these questions as these mice presented with identical molar phenotypes (see next section) to Evc2 global mutants while circumventing problems associated with postnatal lethality and delayed growth (21).
Evc2 function within dental mesenchyme is critical for molar patterning
Although tooth development involves coordinated epithelial-mesenchymal interactions, we previously reported that dental epithelia-specific Evc2 deletion did not lead to overt dental abnormalities (26). We therefore investigated the role of Evc2 in molar patterning using two Cre lines, P0-Cre and Wnt1-Cre, that specifically deleted Evc2 within neural crest cells that give rise to dental mesenchymal tissues. At P28, we observed no apparent body weight differences, suggesting that there is no apparent delay in development due to Evc2 loss of function within neural crest derived tissues. P0-Cre mediated deletion of Evc2 (referred to as Evc2 P0 mutant hereafter) resulted in mice that recapitulated the abnormal molar pattern of Evc2 global mutants only 20% of the time (2 out of 10 mutant mice examined) (Figures 2A–C vs. 2D–F). This is in direct contrast to the 100% penetrance (12 out of 12 mutant mice examined) of abnormal molar patterning observed in both P8 (Figures 3A–C vs. 3D–F) and P28 (Figures 3H–J vs. 3K–M) mice with Wnt1-Cre mediated deletion of Evc2 (referred to as Evc2 Wnt1 mutant hereafter).
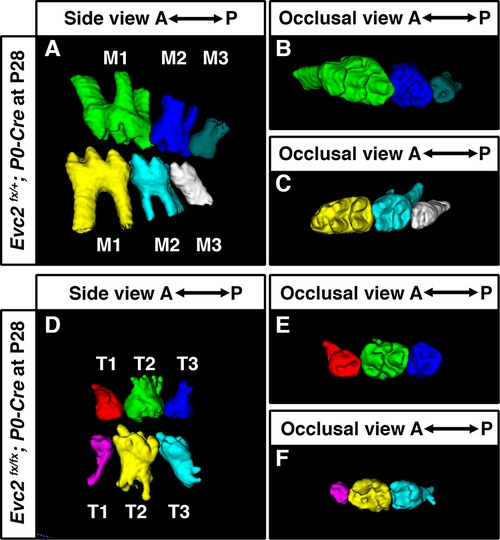
Figure 2 Molars at P28 from controls (A–C) and molars with abnormal pattern in Evc2 P0 mutants (D–F) were reconstructed from micro-CT scans. (Out of 10 Evc2 P0 mutant, 2 with abnormal molar patterning. The mutant with represented abnormal molar patterning is shown.).
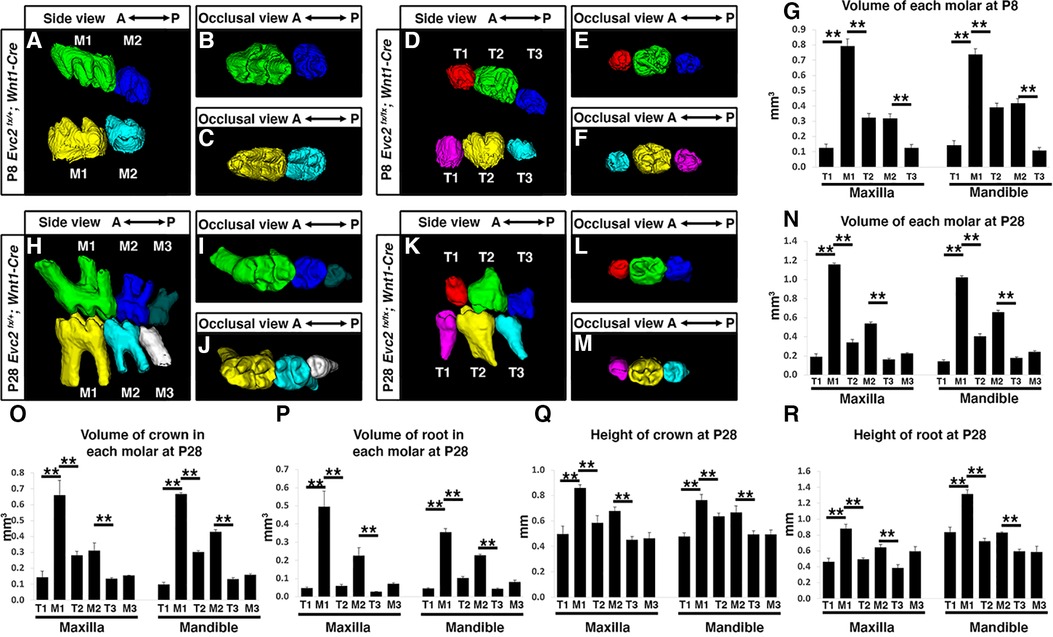
Figure 3 Mandible molars at P8 from controls (A–C) and Evc2 Wnt1- mutants (D–F) were reconstructed from micro-CT scans. The volume of each molar at P8 were quantified and shown in (G). Mandible molars at P28 from controls (H–J) and Evc2 Wnt1 mutants (K–M) were reconstructed from micro-CT scans. The volume of each molar at P28 (N), the volume of crown (O), the volume of root (P), the height of crown (Q), and the height of root (R) were quantified and shown. For molar volumes at P8, n = 4, for maxilla, M1 vs T1, p < 0.01; M1 vs T2, p < 0.01; M2 vs T3, p < 0.01; for mandible, M1 vs T1, p < 0.01; M1 vs T2, p < 0.01; M2 vs T3, p < 0.01. For tooth volume, crown volume, root volume, height of crown, height of root, at P28, for maxilla, M1 vs T1, p< 0.01; M1 vs T2, p < 0.01; M2 vs T3, p < 0.01; for mandible, M1 vs T1, p < 0.01; M1 vs T2, p < 0.01; M2 vs T3, p < 0.01.
Volumetric quantification of maxillary and mandibular molars in Evc2 Wnt1 mutants corroborated molar pattern abnormalities and indicated that M1 was larger than either T1 or T2 and that M2 was larger than T3 at both the P8 and P28 timepoints (Figures 3G,N, respectively). Similar trends were found for crown volume, root volume, crown height, and root height in Evc2 Wnt1 mutants at P28 (Figures 3O–R). Most importantly, phenotypic similarities between mice with mesenchyme-specific loss of Evc2 function and Evc2 global mutants suggest that Evc2 in mesenchymal tissues plays a critical role in determining molar patterning and morphology.
T1 and T2 on Evc2 mutant mandibles are at the same development stage
Crown formation precedes root formation and elongation during tooth development. Because the process of tooth maturation in mice predictably occurs in an anterior to posterior fashion, we therefore investigated the identity of accessory molar buds in Evc2 global mutants via histological means. Roots were fully formed in M1 but rudimentary in M2 of control mice at P8 (Figure 4A brackets and arrows, respectively). By contrast, roots were fully formed in both T1 and T2 but rudimentary in T3 of Evc2 global mutants at P8 (Figure 4B brackets and arrows, respectively). This suggests that molars M1, T1, and T2 were at a similar and more advanced stage of development than molars M2 and T3.
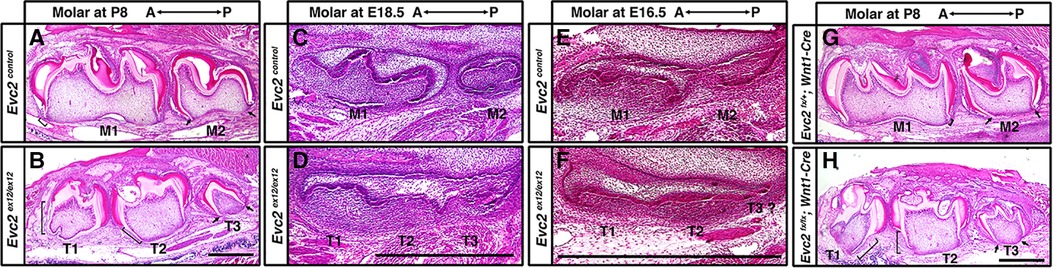
Figure 4 Histologic assessment of molars. Molars from control and Evc2 mutant at P8 (A,B), E18.5 (C,D), E16.5 (E,F), molars from controls and Evc2 Wnt mutant at P8 (H,I) were sectioned, stained for Hemoxylin and Eosin. Bar = 100 µm. (n = 4). Brackets indicate the root and arrows indicate no root formation.
Postnatal observations regarding tooth developmental progression were corroborated via further analyses at embryonic day E18.5. Not only was M1 predictably more developed (bell stage) than M2 (late cup/early bell) in control mice (Figure 4C), but T1 and T2 in Evc2 global mutants were at the same stage, albeit slightly delayed compared to M1 (Figure 4D). The T3 molar in Evc2 global mutants was also at a comparable, though slightly delayed, stage of development to M2 in control mice (Figure 4D). When analyzed at the earlier E16.5 timepoint, M1, which was at late cup stage/early bell stage was still more developed than M2, which was at the cup stage in control mice (Figure 4E). Both T1 and T2 (cup stage) in Evc2 global mutants were a bit delayed than M1, and T3 was a bit delayed than M2 (Figure 4F). Because anterior molars are known inducers of posterior molar formation (36), the presence of similarly staged structures in both Evc2 global and Evc2 Wnt1 mutants (Figures 4G,H) suggests the presence of a supernumerary anterior tooth. We did not see evidence of T4 formation (corresponding to M3 in controls) in Evc2 global mutants or Evc2 Wnt1 mutants. These observations are consistent with micro-CT analysis of Evc2 Wnt1-Cre mutants that Evc2 loss of function within dental mesenchyme phenocopies the molar pattern abnormalities in Evc2 global mutants.
Elevated WNT signaling and compromised Hedgehog signaling were observed in molars in Evc2 mutant mice
Previous studies have indicated that elevated WNT signaling and decreased Hedgehog signaling lead to abnormal molar patterning and supernumerary tooth formation in mice (37, 38). We therefore investigated WNT signaling changes in E16.5 Evc2 global mutant molars using LEF1 as a readout of active WNT signaling. Compared to controls, we observed more cells labeled with LEF1 within the dental mesenchyme of Evc2 global mutant molars (Figures 5A,C,D,F). Additionally, the distribution pattern of SHH corroborates our earlier histologic assessment wherein M1 is in the bell stage, while M2, T1, and T2 are in the cup stage, respectively, at E16.5 (Figures 5B,E). Changes in overall Hedgehog signaling and WNT signaling levels were assessed from dissected M1 and T1 tooth germs at E18.5. qRT-PCR analyses indicated that Hedgehog signaling levels measured by expression of Gli1 and Ptch1 were lower in T1 than M1 (Figure 5G), despite comparable levels of SHH between two genotypes (Figures 5B,E) and that WNT signaling measured by levels of Axin2 and Lef1 was elevated in T1 than M1 (Figure 5H).
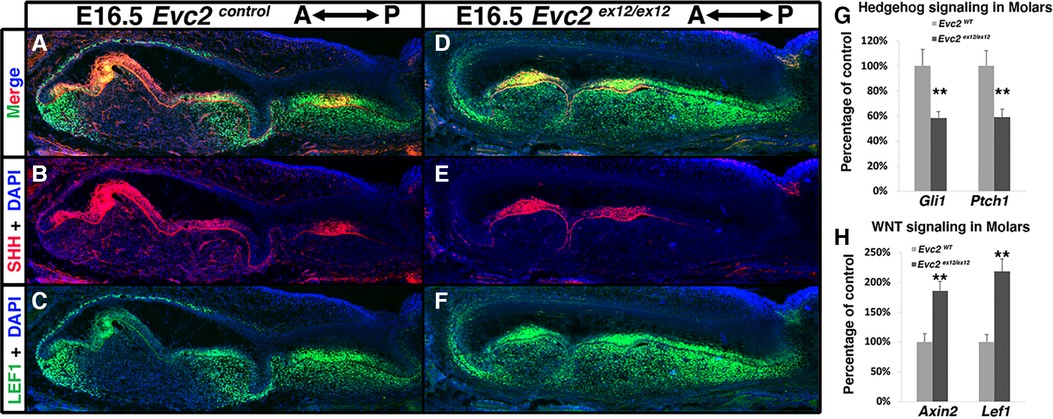
Figure 5 Evc2 mutation leads to decreased Hedgehog signaling and increased WNT signaling at E16.5 and later. Mandible molars of controls (A–C) and Evc2 mutants (D–F) were immunolabeled with LEF (C,F) for reading out WNT signaling and SHH (B,E). Hedgehog signaling (G) and WNT signaling (H) in molars were quantified through assessing the Gli1, Ptch1, Axin2 and Lef1 levels through q-RT-PCR. Percentage of controls were calculated and are shown in G and H. (n = 4; **, p < 0.01).
To understand at which development stage the compromised Hedgehog signaling and elevated WNT signaling leads to supernumerary anterior tooth formation, we then examined the Hedgehog and WNT signaling at E15.5. Comparing to controls, we observed lower intensity of PTCH1 immuno-signals and higher intensity of LEF1 immuno-signals in both dental epithelia and dental mesenchyme (Figures 6A–D,N), suggesting that compromised Hedgehog signaling and elevated WNT signaling are already present at this stage. In contrast to the controls that show only 1 enamel knot at this stage (Figure 6E), we detected two enamel knots in Evc2 mutants (4 out of 4 mutants). In addition to the one found in the middle of tooth germ (Figure 6G), the second one was identified at an anterior location (Figure 6F, F and G are from the same tooth germ but different plain). Associated with the additional enamel knot, we observed abnormal shape of dental epithelia in Evc2 mutant (Figure 6F), suggesting that the supernumerary tooth starts to form anterior to the M1 at E15.5 in Evc2 mutants.
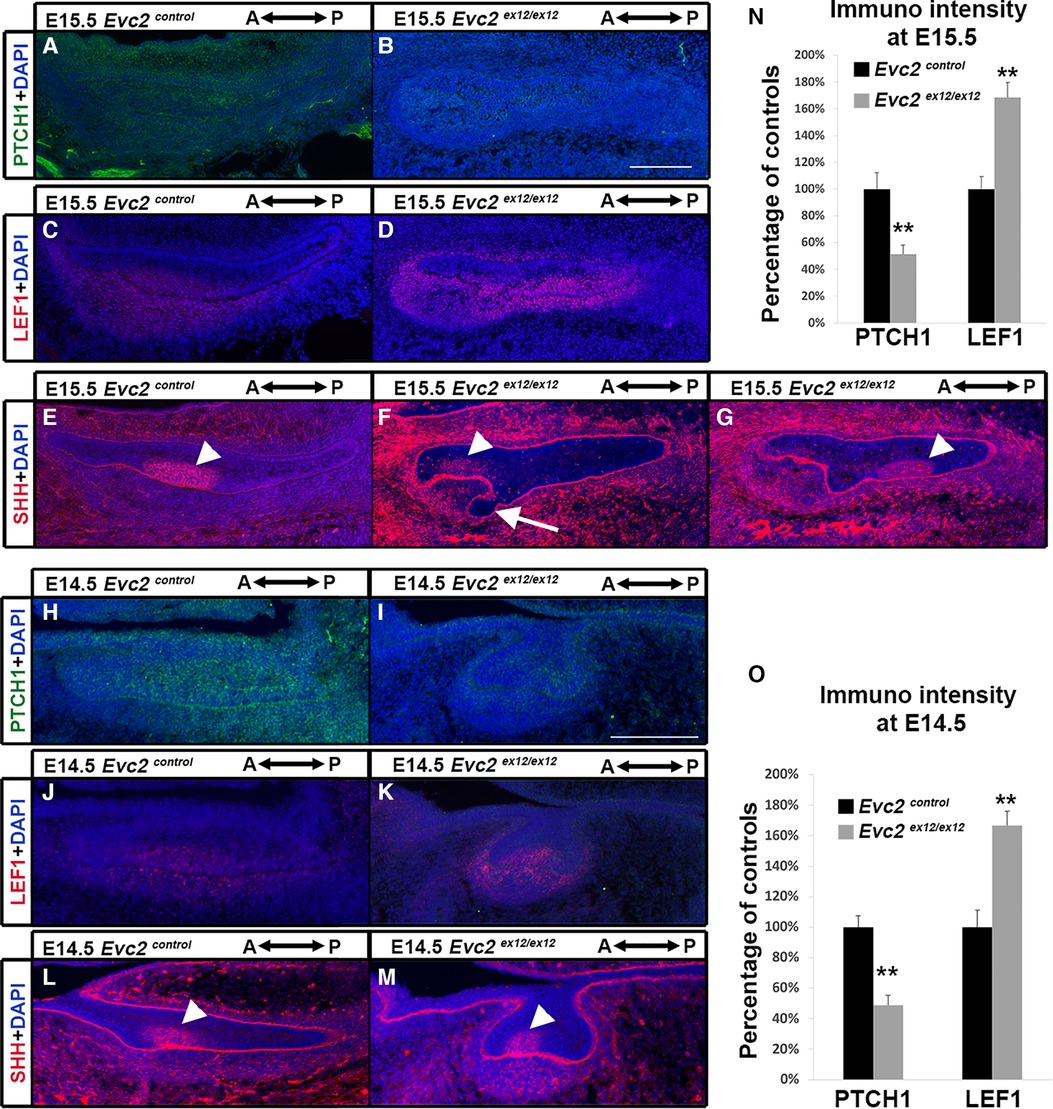
Figure 6 Evc2 mutation leads to decreased Hedgehog signaling and increased WNT signaling at E14.5 and E15.5. Mandible molars from E15.5 (A–G) and E14.5 (H–M) were immunolabeled with PTCH1 (A,B,H,I) for reading out Hedgehog signaling; LEF1 (C,D,J,K) for reading out WNT signaling; and SHH (E,F,G,L,M) for indicating enamel knot. In Evc2 mutants, two enamel knots were not on the same section plain. Thus, two sections with enamel knot were shown in F and G separately. The immunosignal for PTCH1 and LEF1 from E15.5 molars were quantified and shown in N. The immunosignal for PTCH1 and LEF1 from E15.5 molars were quantified and shown in O. (n = 4; **, p < 0.01). White arrow indicates abnormal shaped epithelia. White arrowheads indicate enamel knots.
Similar to E15.5, we detected both compromised Hedgehog signaling and elevated WNT signaling in Evc2 mutants at E14.5 (Figures 6H–K,O). However, we detected only 1 enamel knot in both controls and Evc2 mutants (Figures 6L,M). These observations suggest that compromised Hedgehog signaling and elevated WNT signaling induce abnormal molar patterning between E14.5 and E15.5. The observation of abnormal shaped epithelial tissue at E15.5, not at E14.5 support the idea that initial abnormal patterning occurs between E14.5 and E15.5.
Previous studies have demonstrated that primary cilia play a negative role in regulating WNT signaling (39). Since EVC2 is a ciliary-located protein, we evaluated whether Evc2 loss of function could lead to elevated cellular response to WNT ligands. Using isolated mouse embryonic fibroblasts (MEFs) cultured in a low serum condition to prompt ciliogenesis (ciliated cells), we quantified both Lef1 and Axin2 as readouts of the levels of WNT signaling induced by WNT3A. Under these conditions, we observed elevated WNT signaling in ciliated MEFs with an Evc2 mutation (Figure 7A). In contrast, for cells cultured in under normal serum conditions (non-ciliated cells), we did not observe any changes in response to WNT ligand between Evc2 mutants and controls. These studies indicate that primary cilia function to negatively regulate WNT signaling and that EVC2 protein within primary cilia is responsible, at least in part, for this function.
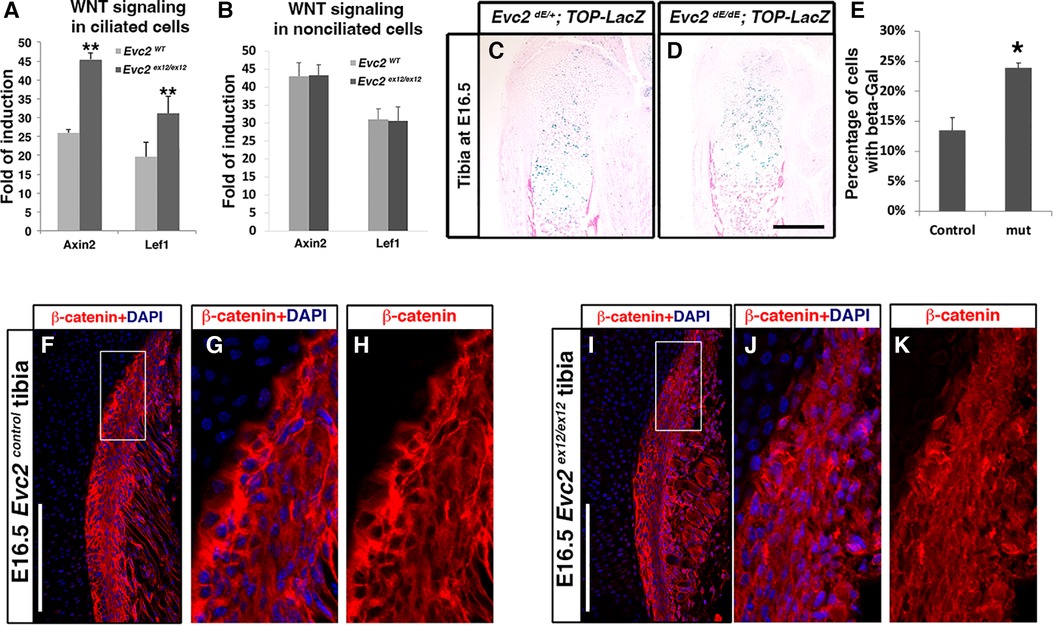
Figure 7 Ciliated (A) and non-ciliated (B) MEFs from controls or Evc2 mutants were treated with WNT3A. The induction of WNT signaling was calculated and shown. WNT signaling within tibia were evaluated with a TOP-gal reporter in controls (C) and Evc2 mutants (D). The number of cells positive for beta-gal were quantified and shown in (E) (n = 3, p < 0.01). E16.5 tibia from controls (F–H) and Evc2 mutants (I–K) were immunolabeled with beta-catenin. White boxes in F and I were shown as enlarged perichondrium for controls (G,H) or Evc2 mutants (J,K). Bar = 100 µm.
Finally, we then examined if there are other tissues in Evc2 mutant mice with elevated WNT signaling. Using TOP-gal reporter mice, we detected an increased number of X-gal-stained cells within Evc2 mutant growth plates compared to controls (Figures 7C–E). Similarly, WNT signaling was elevated within the perichondrium of the growth plates in Evc2 mutants. In E16.5 tibia, we observed an elevated number of cells with nuclear located beta-catenin in the perichondrium of Evc2 mutants (Figures 7F–K). This observation of elevated WNT signaling in multiple tissues of Evc2 mutant mice supports the idea that Evc2 loss of function potentially leads elevated response to WNT ligand in developing tooth, thereby contributing to abnormal molar patterning found in Evc2 mutant mice.
Discussions
Because hypodontia and supernumerary teeth are reported in individuals with EVC syndrome (28), we sought to understand the mechanisms underlying molar pattern alterations using an animal model of disease, especially Evc2 mutant mice we have generated. Our studies indicate that Evc2 within the dental mesenchyme plays a critical role in determining molar patterns. In addition, our in vitro and in vivo studies present evidence of compromised Hedgehog and elevated WNT signaling due to Evc2 loss of function mutation, both of which possibly contribute to the abnormal molar patterns in Evc2 mutant mice.
We utilized two Cre lines to understand the function of Evc2 in leading to the abnormal molar formations. We did so because it was previously reported that Wnt1-Cre transgene influence mid-brain development (40). Though the Wnt1-Cre transgene leads to increased WNT signaling, it is unlikely that ectopically increased WNT signaling by the transgene leads to abnormal tooth formation as we demonstrated in this report, because 1) previous report indicated that the regions affected by Wnt1-Cre transgene is restricted within mid-brain and 2) in our analysis of mice with Wnt1-Cre transgene but no Evc2 mutations do not show abnormal tooth patterning. Our previous studies indicate a largely overlapped but unique regions of neural crest derived tissues targeted by these two lines (41). Relevant to the current studies, Wnt1-Cre leads to almost 100% deletion efficiency in dental mesenchyme; in contrast, in addition to sparsely targeting into the dental epithelia, P0-Cre leads to about 90–95% of deletion efficiency in dental mesenchyme (21). Since Evc2 loss of function only partially compromise Hedgehog signaling, the 90–95% deletion efficiency by P0-Cre may lead to decreased Hedgehog signaling, but just not enough to lead to abnormal tooth patterning.
Although the total number of molars in Evc2 mutants is the same as in controls, we present multiple lines of evidence suggesting that the identity of each molar in Evc2 mutants is different from controls. For example, both histological assessment and micro-CT analysis of Evc2 mutant mandible molars at P8, E18.5, and E16.5 indicate that T1 and T2 are in the same development stage. In contrast, it is known that during molar development, the stage of M2 is around 2 days behind that of M1. The similarity of the development stage of T1 and T2 in Evc2 mutant mandible molars suggests that T1 is a supernumerary tooth. Previous studies suggest that a variety types of abnormal signaling in tooth development leads to abnormal molar patterning. For example, increased FGF signaling due to Spry2 or Spry4 loss of function leads a supernumerary tooth formation anterior of M1 (42). More detailed mechanistic studies suggest that during normal molar development, successful fusion of the enamel knot from the premolar R2 with enamel knot from M1 leads to formation of one tooth germ for presumptive M1. In contrast, a failed fusion of an enamel knot from R2 with an enamel knot from M1 leads to the premolar persistence during development and a supernumerary tooth formation (17, 43–45). In this case, the existence of supernumerary tooth has been reported as early as at E14.5 (42). Additionally, Gas1 loss of function mutant embryos form supernumerary tooth (38). Gene Gas1 encodes a positive regulator for Hedgehog signaling. Gas1 mutant embryos have two molars formed at the same stage during molar development. Based on this, the authors claim that the most anterior tooth is a supernumerary tooth. A similar abnormal molar patterning was reported in studies in Sosdc1 mutant mice (37). Gene Sosdc1 encodes a protein that functions as a negative regulation of both BMP signaling and WNT signaling through competitive bind with both BMP ligands and WNT ligands. However, previous studies indicate that the elevated WNT signaling, but not the elevated BMP signaling, leads to the formation of a supernumerary tooth anterior of M1 (37). In both above cases, the supernumerary tooth is believed as the result of continuous growth of R2 due to abnormal signaling (37, 38).
In Evc2 mutant mandible molars, we detected decreased Hedgehog signaling, elevated WNT signaling. Particularly, the sagittal view of mandible molars is in a distinct shape in comparing to molars in Gas1 mutants or Sosdc1 mutants. The distinct molar shape is likely due to both decreased Hedgehog signaling and elevated WNT signaling, which showcases a unique model to understand the supernumerary tooth formation under pathologic conditions. With the presented data, it is possible that the molar at E14.5 represented a continuously developed R2 and M1 development is delayed till E15.5. Alternatively, it is possible that compromised Hedgehog signaling and increased WNT signaling led to formation of additional enamel knot and abnormal dental epithelia invagination. Characterization of the mechanism leading to supernumerary tooth in Evc2 mutant will rely on future detailed analysis to delineate the identity of molar at E14.5 in Evc2 mutants.
In this study, we provide evidence that elevated WNT signaling potentially contributes to supernumerary tooth formation. Multiple previous studies suggest that elevated WNT signaling within dental epithelia leads to many misshaped ectopic tooth formations (46–48). However, in contrast to previous report, we demonstrated that dental mesenchyme-specific Evc2 disruption results in the abnormal molar patterning and supernumerary tooth formation, because Evc2 loss of function mediated by Wnt1-Cre phenocopies the abnormal tooth patterning in Evc2 global mutants. The impact of moderately elevated WNT signaling within dental mesenchyme on supernumerary tooth formation is an interesting future direction.
The molar patterning in Evc2/Limbin mutant mice phenocopies the pattern of mandible molars in Evc mutant mice (29). This observation is consistent with the findings from molecular studies showing that interaction between EVC and EVC2 are mutually required for localization at the bottom of primary cilia (19, 20). At the bottom of primary cilia, a protein complex formed by EVC, EVC2, and SMO are required for a full activation of Hedgehog signaling. Thus, either EVC loss of function, EVC2 loss of function, or both EVC and EVC2 loss of function lead to absence of EVC-EVC2 complex at the bottom of primary cilia, which leads to compromised induction of Hedgehog signaling. Particularly during molar development in Evc2 mutant embryos, Evc2 loss of function leads to compromised response to Hedgehog ligand and the subsequent decreased Hedgehog signaling, which is a potential reason leading to the abnormally molar patterning in Evc2 mutant embryos. Overall, our findings in the patterning of Evc2 mandible molars are consistent with clinical signs in EVC syndrome and the molecular biological finding of EVC-EVC2 protein complex.
Our in vitro and in vivo studies suggest that loss of Evc2 leads to cell autonomously increased response to WNT ligand. This is primary cilium dependence. In fact, there are multiple studies suggesting that primary cilium negatively regulates the induction of WNT signaling, although the detailed mechanism remains elusive (39, 49). Our studies indicate that EVC-EVC2 protein complex is a part of the protein machinery within primary cilium that fine-tunes the WNT signaling. Future investigation on the function of EVC-EVC2 in inducing WNT signaling will provide an in depth understanding on the primary cilium involved WNT signaling regulation.
Overall, our in vitro and in vivo studies provide evidence of both decreased Hedgehog signaling and WNT signaling in the molars of Evc2 mutant embryos. Both types of abnormal signaling potentially contribute to the abnormal molar patterning in Evc2 mutant embryos. Taken together, our studies implicate the potential pathological mechanisms leading to the supernumerary tooth formation in the rare disease, EVC syndrome.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author/s.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee (IACUC) of the University of Michigan.
Author contributions
Study design: HZ and YM. Study conduct, data collection, and data analysis: HZ, AC, PM, AB, KL, CC and YM. Drafting manuscript: HZ and YM. Approving final version of manuscript: HZ, AC, PM, AB, KL CC, and YM. HZ, AC, PM, AB, KL, CC, and YM took responsibility for the integrity of the data analysis. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Institute of Dental and Craniofacial Research (R01DE020843 to YM, R03DE027456 to HZ and F30DE029667 to KL). The molecular biology core at the School of Dentistry is funded by NIH/P30AR069620. The micro-CT core at the University of Michigan School of Dentistry is funded in part by NIH/NCRR S10RR026475-01.
Acknowledgments
We would like to express our appreciation to Drs. Anshul Kulkarni and Taylor N Snider for their help in generating tooth models from micro-CT scans, and Drs. Yoshiyuki Mochida and Benton Swanson for valuable inputs in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ellis RWB, Van Creveld S. A syndrome characterized by ectodermal dysplasia, polydactyly, chondro-dysplasia and congenital morbus cordis: report of three cases. Arch Dis Child. (1940) 15(82):65–84. doi: 10.1136/adc.15.82.65
2. Baujat G, Merrer ML. Ellis-Van Creveld syndrome. Orphanet J Rare Dis. (2007) 2(27). doi: 10.1186/1750-1172-2-27
3. Louie KW, Mishina Y, Zhang H. Molecular and cellular pathogenesis of Ellis-van Creveld syndrome: lessons from targeted and natural mutations in animal models. J Dev Biol. (2020) 8(4):25. doi: 10.3390/jdb8040025
4. Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. (2000) 24(3):283–6. doi: 10.1038/73508
5. Ruiz-Perez VL, Goodship JA. Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to Hedgehog ligands. Am J Med Genet C Semin Med Genet. (2009) 151(4):341–51. doi: 10.1002/ajmg.c.30226
6. McKusick VA, Egeland JA, Eldridge R, Krusen DE. Dwarfism in the Amish I. The Ellis-Van Creveld syndrome. Bull Johns Hopkins Hosp. (1964) 115:306–36.14217223
7. Takeda H, Takami M, Oguni T, Tsuji T, Yoneda K, Sato H, et al. Positional cloning of the gene LIMBIN responsible for bovine chondrodysplastic dwarfism. Proc Natl Acad Sci U S A. (2002) 99(16):10549–54. doi: 10.1073/pnas.152337899
8. Murgiano L, Jagannathan V, Benazzi C, Bolcato M, Brunetti B, Muscatello LV, et al. Deletion in the EVC2 gene causes chondrodysplastic dwarfism in Tyrolean grey cattle. PLoS ONE. (2014) 9(4). doi: 10.1371/journal.pone.0094861
9. Li C, Zhang Y, Li J, Kong L, Hu H, Pan H, et al. Two Antarctic penguin genomes reveal insights into their evolutionary history and molecular changes related to the Antarctic environment. GigaScience. (2014) 3(1):1–15. doi: 10.1186/2047-217X-3-1
10. Mostafa MI, Temtamy SA, el-Gammal MA, Mazen IM. Unusual pattern of inheritance and orodental changes in the Ellis-van Creveld syndrome. Genet Couns. (2005) 16(1):75–83.15844783
11. Veena KM, Jagadishchandra H, Rao PK, Chatra L. Ellis-van Creveld syndrome in an Indian child: a case report. Imaging Sci Dent. (2011) 41(4):167–70. doi: 10.5624/isd.2011.41.4.167
12. Thesleff I, Hurmerinta K. Tissue interactions in tooth development. Differentiation. (1981) 18(2):75–88. doi: 10.1111/j.1432-0436.1981.tb01107.x
13. Bei M. Molecular genetics of ameloblast cell lineage. J Exp Zool B Mol Dev Evol. (2009) 312B(5):437–44. doi: 10.1002/jez.b.21261
14. Li J, Parada C, Chai Y. Cellular and molecular mechanisms of tooth root development. Development. (2017) 144(3):374–84. doi: 10.1242/dev.137216
15. Jussila M, Thesleff I. Signaling networks regulating tooth organogenesis and regeneration, and the specification of dental mesenchymal and epithelial cell lineages. Cold Spring Harb Perspect Biol. (2012) 4(4):a008425. doi: 10.1101/cshperspect.a008425
16. Sadier A, Jackman WR, Laudet V, Gibert Y. The vertebrate tooth row: is it initiated by a single organizing tooth? Bioessays. (2020) 42(6). doi: 10.1002/bies.201900229
17. Sadier A, Twarogowska M, Steklikova K, Hayden L, Lambert A, Schneider P, et al. Modeling Edar expression reveals the hidden dynamics of tooth signaling center patterning. PLoS Biol. (2019) 17(2). doi: 10.1371/journal.pbio.3000064
18. Kavanagh KD, Evans AR, Jernvall J. Predicting evolutionary patterns of mammalian teeth from development. Nature. (2007) 449(7161):427–32. doi: 10.1038/nature06153
19. Caparros-Martin JA, Valencia M, Reytor E, Pacheco M, Fernandez M, Perez-Aytes A, et al. The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum Mol Genet. (2013) 22(1):124–39. doi: 10.1093/hmg/dds409
20. Dorn KV, Hughes CE, Rohatgi R. A smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev Cell. (2012) 23(4):823–35. doi: 10.1016/j.devcel.2012.07.004
21. Zhang H, Takeda H, Tsuji T, Kamiya N, Rajderkar S, Louie KA, et al. Generation of Evc2/Limbin global and conditional KO mice and its roles during mineralized tissue formation. Genesis. (2015) 53(9):612–26. doi: 10.1002/dvg.22879
22. Zhang H, Kamiya N, Tsuji T, Takeda H, Scott G, Rajderkar S, et al. Elevated fibroblast growth factor signaling is critical for the pathogenesis of the dwarfism in Evc2/limbin mutant mice. PLoS Genet. (2016) 12(12):e1006510. doi: 10.1371/journal.pgen.1006510
23. Kulkarni AK, Louie KaW, Yatabe M, De Oliveira Ruellas AC, Mochida Y, Cevidanes LHS, et al. A ciliary protein EVC2 / LIMBIN plays a critical role in the skull base for mid-facial development. Front Physiol. (2018) 91–13. doi: 10.3389/fphys.2018.01484
24. Badri MK, Zhang H, Ohyama Y, Venkitapathi S, Alamoudi A, Kamiya N, et al. Expression of Evc2 in craniofacial tissues and craniofacial bone defects in Evc2 knockout mouse. Arch Oral Biol. (2016) 68:142–52. doi: 10.1016/j.archoralbio.2016.05.002
25. Badri MK, Zhang H, Ohyama Y, Venkitapathi S, Kamiya N, Takeda H, et al. Ellis Van Creveld2 is required for postnatal craniofacial bone development. Anat Rec. (2016) 299(8):1110–20. doi: 10.1002/ar.23353
26. Zhang H, Takeda H, Tsuji T, Kamiya N, Kunieda T, Mochida Y, et al. Loss of function of Evc2 in dental mesenchyme leads to hypomorphic enamel. J Dent Res. (2017) 96(4):421–9. doi: 10.1177/0022034516683674
27. Zhang HH, Louie KW, Kulkarni AK, Zapien-Guerra K, Yang JW, Mishina Y. The posterior part influences the anterior part of the mouse cranial base development. Jbmr Plus. (2021) 6(2):e10589. doi: 10.1002/jbm4.10589
28. Hattab FN, Yassin OM, Sasa IS. Oral manifestations of ellis-van creveld syndrome: report of two siblings with unusual dental anomalies. J Clin Pediatr Dent. (1998) 22(2):159–65.9643193
29. Nakatomi M, Hovorakova M, Gritli-Linde A, Blair HJ, MacArthur K, Peterka M, et al. Evc regulates a symmetrical response to Shh signaling in molar development. J Dent Res. (2013) 92(3):222–8. doi: 10.1177/0022034512471826
30. Hayashi S, Lewis P, Pevny L, McMahon AP. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Gene Expr Patterns. (2002) 2(1-2):93–7. doi: 10.1016/S0925-4773(02)00292-7
31. Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, et al. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol. (1999) 212(1):191–203. doi: 10.1006/dbio.1999.9323
32. Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. (1998) 8(24):1323–6. doi: 10.1016/S0960-9822(07)00562-3
33. DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. (1999) 126(20):4557–68. doi: 10.1242/dev.126.20.4557
34. Snider TN, Louie KW, Zuzo G, Ruellas ACO, Solem RC, Cevidanes LHS, et al. Quantification of three-dimensional morphology of craniofacial mineralized tissue defects in Tgfbr2/Osx-Cre mice. Oral Sci Int. (2021) 18(3):193–202. doi: 10.1002/osi2.1099
35. Komatsu Y, Kaartinen V, Mishina Y. Cell cycle arrest in node cells governs ciliogenesis at the node to break left-right symmetry. Development. (2011) 138(18):3915–20. doi: 10.1242/dev.068833
36. Cobourne MT, Sharpe PT. Making up the numbers: the molecular control of mammalian dental formula. Semin Cell Dev Biol. (2010) 21(3):314–24. doi: 10.1016/j.semcdb.2010.01.007
37. Ahn Y, Sanderson BW, Klein OD, Krumlauf R. Inhibition of Wnt signaling by Wise (Sostdc1) and negative feedback from Shh controls tooth number and patterning. Development. (2010) 137(19):3221–31. doi: 10.1242/dev.054668
38. Seppala M, Thivichon-Prince B, Xavier GM, Shaffie N, Sangani I, Birjandi AA, et al. Gas1 regulates patterning of the murine and human dentitions through sonic hedgehog. J Dent Res. (2022) 101(4):473–82. doi: 10.1177/00220345211049403
39. Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Reiter JF. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol. (2008) 10(1):70–6. doi: 10.1038/ncb1670
40. Lewis AE, Vasudevan HN, O’Neill AK, Soriano P, Bush JO. The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev Biol. (2013) 379(2):229–34. doi: 10.1016/j.ydbio.2013.04.026
41. Chen GQ, Ishan M, Yang JW, Kishigami S, Fukuda T, Scott G, et al. Specific and spatial labeling of P0-Cre versus Wnt1-Cre in cranial neural crest in early mouse embryos. Genesis. (2017) 55(6). doi: 10.1002/dvg.23034
42. Klein OD, Minowada G, Peterkova R, Kangas A, Yu BD, Lesot H, et al. Sprouty genes control diastema tooth development via bidirectional antagonism of epithelial-mesenchymal FGF signaling. Dev Cell. (2006) 11(2):181–90. doi: 10.1016/j.devcel.2006.05.014
43. Peterkova R, Lesot H, Peterka M. Phylogenetic memory of developing mammalian dentition. J Exp Zool B Mol Dev Evol. (2006) 306(3):234–50. doi: 10.1002/jez.b.21093
44. Prochazka J, Pantalacci S, Churava S, Rothova M, Lambert A, Lesot H, et al. Patterning by heritage in mouse molar row development. Proc Natl Acad Sci USA. (2010) 107(35):15497–502. doi: 10.1073/pnas.1002784107
45. Lochovska K, Peterkova R, Pavlikova Z, Hovorakova M. Sprouty gene dosage influences temporal-spatial dynamics of primary enamel knot formation. BMC Dev Biol. (2015) 15:21. doi: 10.1186/s12861-015-0070-0
46. Jarvinen E, Salazar-Ciudad I, Birchmeier W, Taketo MM, Jernvall J, Thesleff I. Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta-catenin signaling. Proc Natl Acad Sci USA. (2006) 103(49):18627–32. doi: 10.1073/pnas.0607289103
47. Liu F, Chu EY, Watt B, Zhang Y, Gallant NM, Andl T, et al. Wnt/beta-catenin signaling directs multiple stages of tooth morphogenesis. Dev Biol. (2008) 313(1):210–24. doi: 10.1016/j.ydbio.2007.10.016
48. Kim R, Yu T, Li J, Prochazka J, Sharir A, Green JBA, et al. Early perturbation of Wnt signaling reveals patterning and invagination-evagination control points in molar tooth development. Development. (2021) 148(14):dev199685. doi: 10.1242/dev.199685
Keywords: EVC2, limbin, molar, WNT, cilia, supernumerary tooth
Citation: Zhang H, Chinoy A, Mousavi P, Beeler A, Louie K, Collier C and Mishina Y (2022) Elevated WNT signaling and compromised Hedgehog signaling due to Evc2 loss of function contribute to the abnormal molar patterning. Front. Dent. Med 3: 876015. doi: 10.3389/fdmed.2022.876015
Received: 14 February 2022; Accepted: 18 July 2022;
Published: 1 August 2022.
Edited by:
Erin Ealba Bumann, University of Missouri–Kansas City, United StatesReviewed by:
Daniel Graf, University of Alberta, CanadaMaria Hovorakova, Charles University, Czechia
© 2022 Zhang, Chinoy, Mousavi, Beeler, Louie, Collier and Mishina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honghao Zhang, emhhbmdob0B1bWljaC5lZHU=; Yuji Mishina, bWlzaGluYUB1bWljaC5lZHU=
Specialty Section: This article was submitted to Systems Integration, a section of the journal Frontiers in Dental Medicine