 Cristóbal Valenzuela-Turner
Cristóbal Valenzuela-Turner José Horacio Grau2
José Horacio Grau2 Jörns Fickel
Jörns Fickel Daniel W. Förster
Daniel W. Förster- 1Dept. of Evolutionary Genetics, Leibniz Institute for Zoo and Wildlife Research, Berlin, Germany
- 2Smithsonian Conservation Biology Institute, Center for Species Survival, Washington, DC, United States
- 3Institute for Biochemistry and Biology, University of Potsdam, Potsdam, Germany
Advances in high-throughput sequencing (HTS) have made it a powerful resource for the conservation of threatened species, providing information at both population and individual levels to inform management decisions. In South America, however, the application of HTS in conservation has been limited, primarily due to challenges in funding and access to advanced genomic equipment and analytical expertise. Darwin’s fox (Lycalopex fulvipes), endemic to Chile’s Valdivian Temperate Rainforest, is the most endangered canid in South America with a small and declining population estimated at less than 1000 mature individuals. Despite its endangered status, significant knowledge gaps remain. Here we highlight the potential of HTS to address these challenges, such as clarifying its taxonomy, demographic history, geographic distribution, population structure, genetic diversity, and pathogen exposure. Integrating molecular data into conservation planning will be pivotal in ensuring the long-term survival of Darwin’s fox by identifying priorities for targeted management interventions, highlighting areas of critical habitat for conservation, and guiding genetic rescue efforts to enhance genetic diversity and resilience.
1 Introduction
The Chilean endemic Darwin’s fox (Lycalopex fulvipes) is the most endangered canid in South America (Silva-Rodríguez et al., 2016). This small, solitary, omnivorous species is obligate to forest habitats and primarily confined to the dense understory of the Valdivian Temperate Rainforest in southern Chile, which is recognized as a biodiversity hotspot threatened by unsustainable commercial logging and large-scale deforestation (Moreira-Arce et al., 2016). Darwin’s fox populations persist in native forest remnants within the Nahuelbuta mountain range, where fewer than 100 mature individuals remain (Silva-Rodríguez et al., 2016), on Chiloé Island, home to fewer than 500 mature individuals (Silva-Rodríguez et al., 2018), as well as in the Valdivian coastal range (Vilà et al., 2004; Farias et al., 2014), and Gorbea (D’elía et al., 2013) dominated by agricultural land and some remaining native forest. These populations exhibit slight ecological, behavioural and phenotypic differences. Mainland foxes primarily inhabit dense forests and are predominantly nocturnal, while Chiloé foxes are more habitat-flexible, exhibiting coastal foraging and more diurnal activity. Additionally, Chiloé individuals are slightly smaller on average (Sillero-Zubiri E by et al., 2004).
Additional unpublished sightings raise the question about the true distribution and the existence of undiscovered populations. Beyond habitat loss and human-induced disturbances, Darwin’s fox faces significant threats from feral and free-ranging domestic dogs which attack them (D’elía et al., 2013), disrupt their behaviour (Jiménez, 2007), and expose them to pathogens, posing the risk of disease spillover (Acosta-Jamett et al., 2015; Hidalgo-Hermoso et al., 2020).
Until the mid 1990’s, Darwin’s fox was considered a subspecies of the South American grey fox (Lycalopex griseus). However, the use of mitochondrial DNA (mtDNA) markers led to its classification as a distinct species (Yahnke et al., 1996), though further studies disagree on its phylogenetic position within the genus Lycalopex (Tchaicka et al., 2016; Chavez et al., 2022; Favarini et al., 2022).
Despite its Endangered status on the IUCN Red List of Threatened Species (Silva-Rodríguez et al., 2016) and in Chilean legislation (DS 151/2007 MINSEGPRES), there has been no comprehensive assessment of population structure or consistent evaluation of intraspecific variation, and genetic monitoring for the species remains absent. However, studies have suggested that genetic diversity among Darwin’s foxes on Chiloé Island is lower compared to their mainland counterparts in Nahuelbuta (Yahnke et al., 1996; Vilà et al., 2004; Cabello and Dávila, 2014; Chavez et al., 2022). Additionally, Darwin’s fox exhibits extremely low genome-wide heterozygosity, with a significant portion of its autosomal genome characterised by extensive runs of homozygosity (ROH) (Chavez et al., 2022).
In September 2023, the “Plan for the Recovery, Conservation and Management of Darwin’s fox” (Chiloé Silvestre, 2023) was submitted to the Chilean Ministry of Environment. This recovery plan underscores the importance of closing key knowledge gaps about Darwin’s fox, which are critical for shaping effective conservation management strategies and actions. Molecular data are expected to play a pivotal role in their success, as genetic markers can provide insights at the population or individual level that are otherwise difficult to obtain. Some major knowledge gaps that still need to be addressed include: (i) What is the phylogenetic position of Darwin’s fox within the genus Lycalopex? (ii) What is the evolutionary and phylogeographic history of the species? (iii) What is the current distribution of Darwin’s foxes? (iv) To what extent are remnant populations connected? (v) How extensive and widespread is inbreeding? (vi) What pathogens are Darwin’s foxes exposed to? While some of these questions can be answered using traditional methods (e.g. camera trap surveillance, parasite egg counts from faecal samples, non-invasive sample screening), many can only be adequately addressed through molecular approaches, particularly through high-throughput sequencing (HTS) techniques.
Here we focus on how HTS approaches can help to address critical, immediate, and conservation-relevant issues. Other topics that can be studied using the same or similar techniques fall outside the scope of this review.
2 Use of high-throughput sequencing in defining strategies for Darwin’s fox conservation management
The main HTS technologies are provided by Illumina, Pacific BioScience (PacBio), and Oxford Nanopore Technologies (ONT). Illumina platforms generate short, high-accuracy sequences ranging from 50 to 300 base pairs (bp) in length, either as single or paired-end reads. These sequences are applicable to a wide range of experimental designs, from whole genome sequencing (WGS) to metagenomics. In contrast, PacBio platforms produce long reads with an average length of 20 kilobases (kb), which are advantageous for resolving complex genomic regions and detecting structural variants. ONT can produce even longer reads, with some kits capable of generating sequences exceeding 50 kb, but has lower base-calling accuracy compared with PacBio. Additionally, the portability of certain ONT devices makes this technology suitable for field-based, on-site sequencing.
Three main HTS approaches can be followed: sequencing of the whole genome (whole-genome sequencing, WGS), sequencing only parts of the genome (reduced representation approach, RRA), or sequencing environmental or invertebrate-derived DNA (eDNA/iDNA).
i. WGS (Table 1) provides complete genetic information of a specimen and thus unravels its complete genetic landscape, including genetic diversity, inbreeding levels, evolutionary and demographic history, and even gene-environment associations via whole genome bisulfite sequencing (WGBS), which is useful to detect epigenetically modified (methylated) sites. To date, only two Darwin’s foxes have been sequenced through WGS (Chavez et al., 2022), and a chromosome level assembly has not yet been generated.
ii. RRAs such as RNA sequencing (RNA-seq), Restriction Site Associated DNA Sequencing (RAD-seq), Targeted Capture and SNP Arrays (Table 1) retrieve sequence information from a subset of the genome, utilising methods designed to obtain/target specific regions of the genome, which enables comparison amongst samples. These methods allow the cost-effective study of genetic diversity within and between populations.
iii. Environmental genomics (eDNA/iDNA) utilizes genetic material shed by organisms into their surroundings, such as water, soil, or air, or from blood consumed by invertebrates (“invertebrate-derived DNA”) (Carvalho et al., 2022). These methods enable non-invasive species detection, biodiversity assessment, and population monitoring.
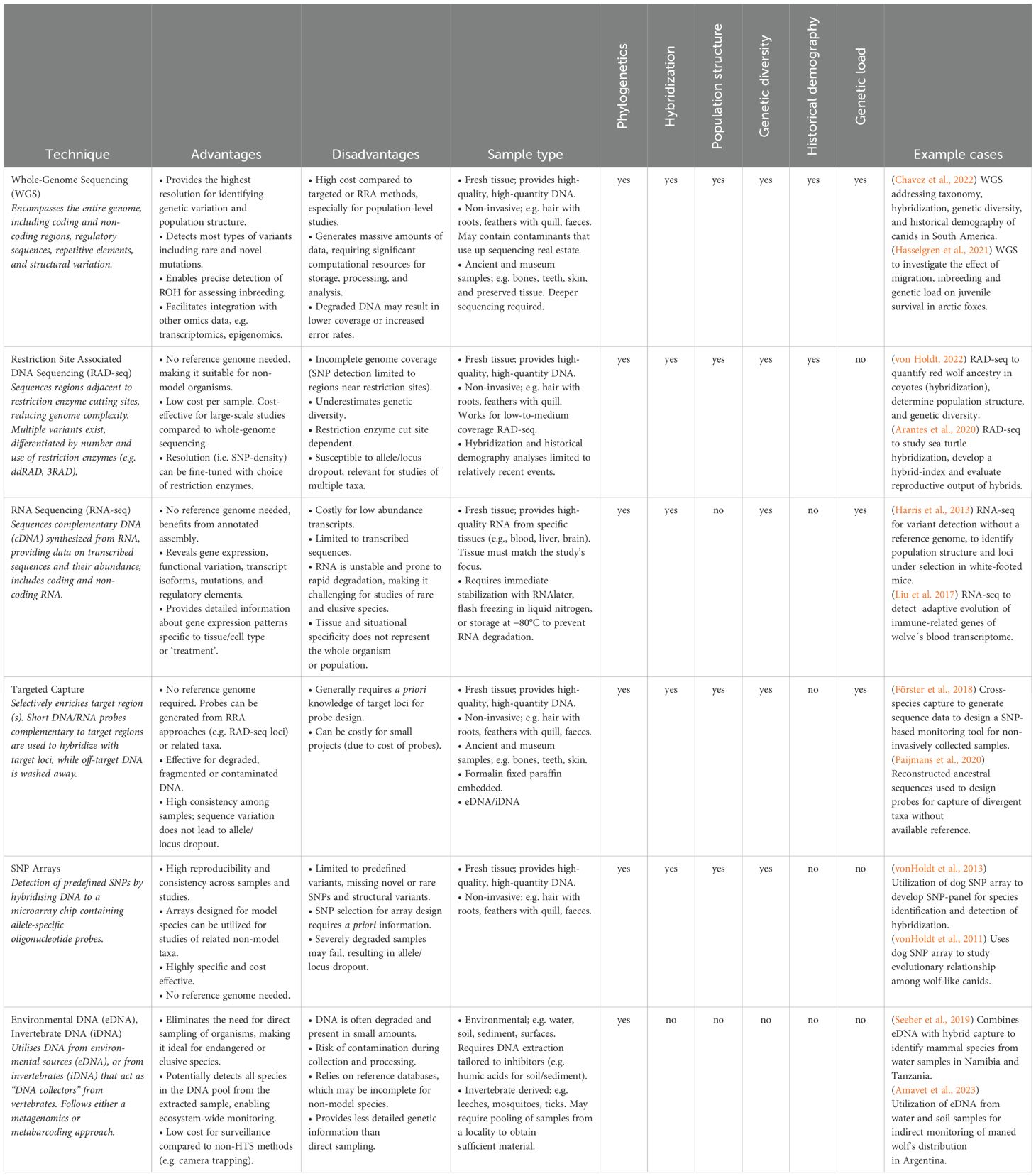
Table 1. Brief summary of high-throughput sequencing techniques and their potential usage for conservation related analyses.
3 Taxonomic uncertainty
Poor taxonomic assessment can lead to species misidentification, misallocation of resources, and ineffective protection measures, ultimately hindering conservation efforts (Morrison WR et al., 2009). While the species status of Darwin’s fox is undisputed, its precise phylogenetic placement within the genus Lycalopex remains unresolved. The rapid divergence of Lycalopex taxa began 1.3 million years ago (Mya), and started between 0.7 and 0.27 Mya for Darwin’s fox (Yahnke et al., 1996; Perini et al., 2010; Tchaicka et al., 2016; Favarini et al., 2022), complicating phylogenetic reconstruction. This is primarily due to the retention of ancestral polymorphisms (i.e. incomplete lineage sorting) and hybridization among various Lycalopex species (Tchaicka et al., 2016; Chavez et al., 2022; Pizarro et al., 2023; Garcez et al., 2024). Recent studies examining taxonomic relationships within the Lycalopex genus using various genetic markers (Yahnke et al., 1996; Vilà et al., 2004; Perini et al., 2010; Tchaicka et al., 2016; Chemisquy et al., 2019; Favarini et al., 2022), and whole genomes (Chavez et al., 2022) disagree on species relationships within Lycalopex (Figure 1).
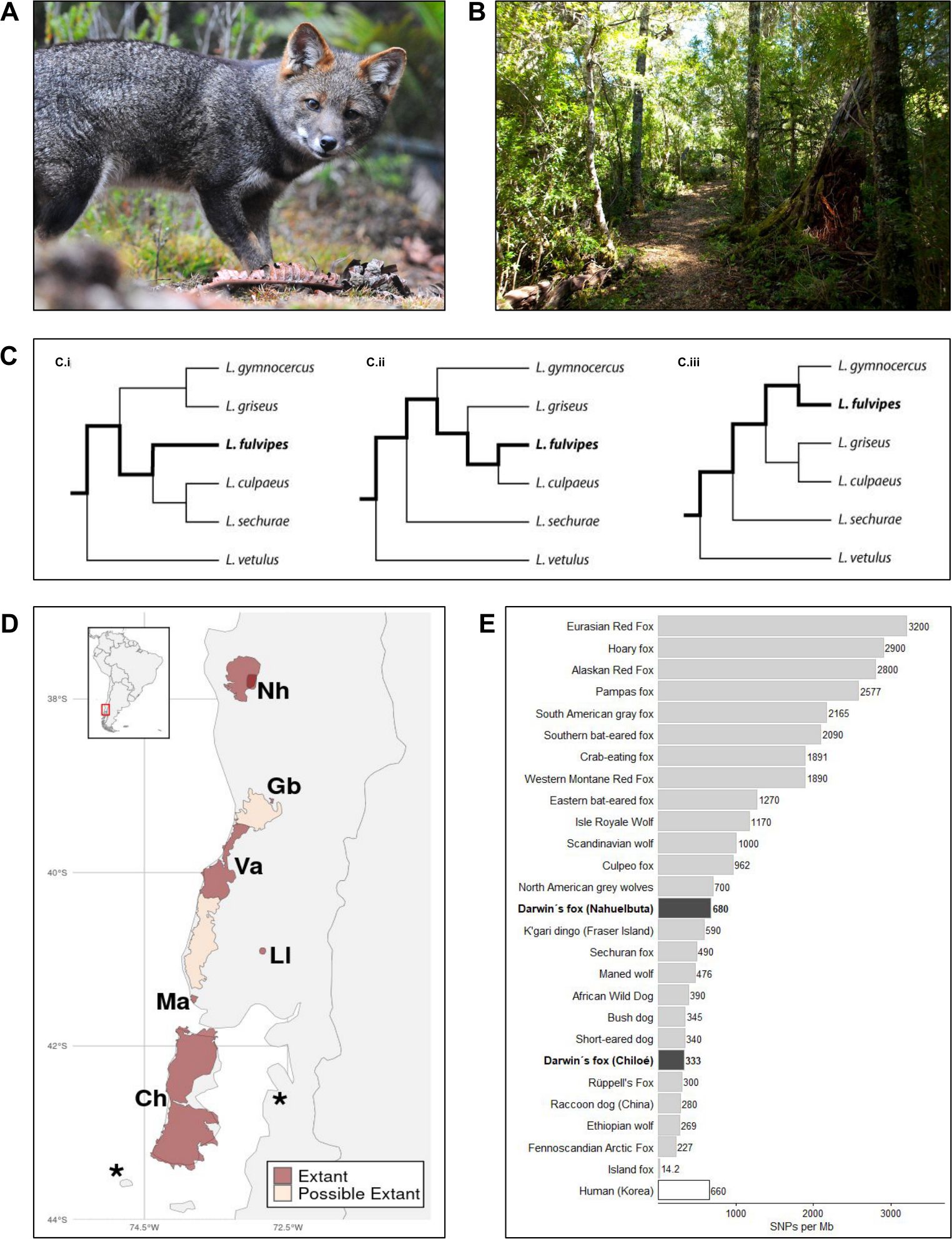
Figure 1. Overview of conservation related topics to be addressed using high-throughput sequencing. (A) Darwin’s fox (B) Habitat in Nahuelbuta. (C) Phylogenetic relationships reconstructed in previous studies (i: Chavez et al., 2022; ii: Favarini et al., 2022; iii: Tchaicka et al., 2016). (D) Distribution according to Silva-Rodríguez et al. (2016); known distribution and possible distribution indicated. Nh, Nahuelbuta; Ch, Chiloé; Gb, Gorbea; Ma, Maullín; Ll, Llanquihue; Va, Valdivia; *, proposed survey areas. (E) Genetic diversity measured as heterozygosity across genomes (SNPs/Mb). (A) Copyright Thomas Kramer Hepp, Fundación Alerce 3000 (thomaskramerhepp@gmail.com), used with permission. (B) Copyright Cristóbal Valenzuela-Turner.
Darwin’s fox is sympatric with two other Lycalopex species (L. culpaeus and L. griseus) in parts of its range, but it remains unknown whether hybridization occurs among these taxa. From a conservation perspective, hybridization can be a double-edged sword: it may threaten endangered species by diluting their gene pool (genetic swamping) or, conversely, increase genetic diversity and adaptability, thereby enhancing resilience (Howard-McCombe et al., 2023). Developing diagnostic markers for species identification is essential for detecting hybridization, distinguishing species, and guiding conservation strategies. Resolving the Lycalopex species tree using genome-wide data is an important step towards this goal.
WGS is likely the most effective method for reconstructing the Lycalopex species tree, as it enables a comprehensive evaluation of phylogenetic incongruences by sampling across both coding and non-coding regions, detecting rare variants, and (potentially) incorporating structural variants in analyses (Rakotoarivelo et al., 2024). RRA approaches, such as RNA-seq (Tomasco I et al., 2022) or RAD-seq (Andrews et al., 2016) are viable alternatives, especially under financial constraints, enabling greater sample sizes at lower cost. However, these methods have lower resolution and may suffer from locus or allele dropout in interspecific studies (Andrews et al., 2016).
Broad sampling across the full geographic distribution of all Lycalopex species is advisable, as introgression may be geographically localized or restricted to specific lineages. Ideally, samples with uncertain provenance or heritage, such as those from zoos, should be avoided to prevent confounding results.
4 Uncertain distribution, abundance and connectivity
The extent of Darwin’s fox geographic distribution is unknown (Figure 1), as are population numbers, their sizes, and potential connectivity among populations, rendering effective conservation measures difficult. Underestimating the species’ range risks neglecting populations that could serve as critical genetic reservoirs, leading to a loss of genetic diversity. Maintaining that diversity, however, is essential for the species’ adaptive potential and long-term survival. The oldest documented and the most thoroughly studied population, first recorded by Darwin in 1840, is located on Chiloé Island, which represents the southernmost edge of the known distribution range of the species. The northernmost population resides in the Nahuelbuta area (Medel et al., 1990). Among the other known or suspected mainland populations, only the one in the Valdivian Coastal Range has been confirmed using camera traps (Farias et al., 2014). The existence of a population living at Punta Chanchán is based on a L. fulvipes-like mtDNA control region, sequenced from a skin stored in a nearby household (Vilà et al., 2004), while a population in Gorbea is suggested by the identification of a L. fulvipes-like mtDNA haplotype from a fox apparently killed during a dog attack (D’elía et al., 2013). Additional populations may exist based on observations near the Maullín River and north of Lake Llanquihue (Silva-Rodríguez et al., 2016). Along the coastal mountain range, suitable forest habitat exists, but camera trapping efforts to verify Darwin’s fox presence have not yet been successful (Silva-Rodríguez et al., 2018).
Detecting and surveying an elusive species in dense rainforests using traditional methods can be costly, logistically challenging and time-intensive. Non-invasive approaches, such as use of eDNA (Beng and Corlett, 2020) and iDNA (Abrams et al., 2019), offer informative, time- and cost-effective alternative or complementary strategies to detect species presence in a given area. In the absence of observational or population genetic data, this can also provide evidence of population connectivity, which is crucial for guiding conservation strategies aimed at preserving or restoring habitat corridors to prevent genetic isolation. Furthermore, advancements in eDNA/iDNA methods are expected to provide insights beyond taxonomic identification, including estimates of species abundance, allele or haplotype frequencies, and eventually individual-level data (Andres et al., 2023). Additionally, these techniques can also help monitor other species, including invasive competitors such as domestic dogs or the American mink.
Promising areas for eDNA/iDNA surveillance include regions predicted as suitable habitat by niche modelling (Escobar et al., 2018; Molina et al., 2018), such as the mainland east of Chiloé Island and the islands of Guafo and Guamblin, alongside areas already under camera trap surveillance. However, successful implementation of eDNA/iDNA will require robust, species-specific diagnostic markers that can distinguish L. fulvipes from sympatric Lycalopex species and potential hybrids. These markers should be validated through genomic and mitochondrial comparisons to ensure accurate identification (Beng and Corlett, 2020).
5 Genetic diversity and population structure
Surveying Darwin’s foxes’ intraspecific genetic variation is essential for evaluating population structure, genetic differentiation, isolation times, and detecting bottlenecks or signs of genomic erosion. It helps determine whether geographical distances or barriers contribute to genetic divergence among populations. Preserving remaining genetic diversity is critical, as signs of inbreeding are already present in Darwin’s foxes (Chavez et al., 2022). The combined effects of inbreeding and genetic drift can drive small populations into an “extinction vortex”, where accelerated genetic diversity loss compromises adaptive potential (Stange et al., 2021).
Early research on Darwin’s fox genetic variability focused on mtDNA control-region sequences, revealing that foxes on Chiloé Island shared the same haplotype, while mainland foxes had distinct haplotypes, suggesting differentiation between these populations (Yahnke et al., 1996; Vilà et al., 2004; D’elía et al., 2013). Microsatellite markers also revealed low variability compared to other canids, with only 2 to 4 alleles per locus and observed heterozygosity ranging from 0.041 to 0.608 in Chiloé Island foxes, underscoring the limited genetic diversity in this population (Cabello and Dávila, 2014). The only WGS study including Darwin’s foxes revealed extensive runs of homozygosity across a high proportion of the genome, and a demographic decline in both regions sampled. Of particular note was that the Nahuelbuta fox had long ROH (>10 Mb) spanning 5% of the genome, and that the Chiloé fox had medium-length ROH (1-10 Mb) spanning 37% of the genome. Heterozygosity levels were also very low, averaging 0.680 SNPs per kb in Nahuelbuta and 0.333 SNPs per kb in Chiloé (Figure 1), which is among the lowest values in South American canids (Chavez et al., 2022).
Although these pioneering studies have provided valuable insights, their limited sample and/or marker numbers limit the generalizability of the findings regarding genetic variability. Future research should aim for broader sampling across and within populations to better capture intraspecific variation. WGS offers the most comprehensive data on genetic diversity (e.g. SNPs, indels, runs of homozygosity, and structural variants), enabling detailed analyses of population structure, demography, connectivity, kinship, divergence times, and more (Cockerill et al., 2022). Reduced representation approaches, like RAD-seq, offer a cost-effective, high-throughput option for genetic diversity and population structure assessment, but may have lower resolution in highly inbred species such as Darwin’s fox (Escoda et al., 2022). SNP arrays are also cost-effective, though they can miss rare SNPs and structural variants unless specifically targeted (Balagué-Dobón et al., 2022). Such arrays are available for canids (Cairns et al., 2018). RNA-seq can provide insight into functional differences between individuals and populations, which is relevant for the allocation of conservation resources. However, the tissue-specific nature of RNA-seq poses a challenge and may not capture the neutral variation necessary for some genetic analyses (Perry et al., 2012).
An important factor in selecting a HTS approach is its ability to assess genetic load, which is vital for understanding inbreeding depression and the population dynamics of deleterious alleles. Genetic load arises from the accumulation of harmful variants that reduce fitness by increasing expression of recessive deleterious alleles and potentially fixing them through genetic drift; expression of these harmful variants can negatively affect health, adaptability, and reproduction (Robinson et al., 2023). Studies on the Iberian lynx (Kleinman-Ruiz et al., 2022), Arctic fox (Cockerill et al., 2022), and Isle Royale wolves (Robinson et al., 2019) underscore the importance of accounting for genetic load in conservation efforts. WGS is likely the most effective approach for identifying potentially deleterious variants across the genome.
6 Pathogen community and adaptive immune system
Diseases are an important, yet often underestimated factor influencing species demography, particularly when new pathogens are introduced into naive populations. Dogs roaming close to and within protected areas of Darwin’s fox distribution range, unvaccinated and untreated for parasites (Silva-Rodríguez et al., 2018), can be a source of disease transmission, potentially having devastating effects (Cleaveland et al., 2007).
Several bacterial pathogens have been detected in Darwin’s foxes, including Toxoplasma gondii, Leptospira sp., Mycoplasma haemocanis (Hidalgo-Hermoso et al., 2022) and Mycoplasma haematoparvum (Di Cataldo et al., 2020). RNA-seq revealed higher genetic diversity of M. haematoparvum in foxes than in dogs, suggesting transmission among foxes. Viral diseases, such as canine distemper virus (CDV) and parvovirus (CPV), present in dogs near Nahuelbuta, also pose significant risks due to the foxes’ lack of immunity (Acosta-Jamett et al., 2015; Hidalgo-Hermoso et al., 2022). On Chiloé Island, gammaherpesvirus is prevalent in foxes but has not shown pathogenic effects to date (Cabello et al., 2013). Gastrointestinal parasites, including trematodes, cestodes, nematodes, and protozoa, are prevalent in foxes from Chiloé and Nahuelbuta and are often shared with domestic species, suggesting possible cross-species transmission (Acosta-Jamett et al., 2018).
HTS is revolutionising pathogen detection by enabling broad-spectrum analysis, integrating data from hosts, vectors, and environmental samples to provide a comprehensive understanding of pathogen transmission (Bass et al., 2023). Non-invasive sampling techniques, such as the use of faecal samples, eDNA, and iDNA are becoming ubiquitous for pathogen detection and surveillance in wildlife (Bass et al., 2023). While unbiased metagenomic deep sequencing provides a comprehensive view of microbial communities, it can be cost-prohibitive. More affordable alternatives, such as metabarcoding (PCR-based) and targeted capture (RNA/DNA-probes), allow for extensive multiplexing but require prior knowledge to design primers or probes, such as 16S rRNA for bacterial studies (Blanchong et al., 2016). Characterising viral communities is more complex due to a lack of conserved markers, though resources for targeting viral sequences exist (e.g. Virochip microarray (Wang et al., 2002), with RNA viruses requiring additional laboratory steps (Bass et al., 2023). Direct sampling from living or deceased animals may be necessary to diagnose tissue-specific bacterial (Kim et al., 2023) and viral (Van Borm et al., 2015) pathogens of conservation concern, such as rabies, CDV, CPV, and intracellular parasites. RNA-seq of tissue samples can also provide insights into the host’s immune response to pathogens (Michel et al., 2021).
Genomic regions like the Major Histocompatibility Complex (MHC), or Dog Leukocyte Antigen (DLA) in canids, are essential for adaptive immunity and pathogen response (Yuhki et al., 2007). Reduced diversity at these loci can increase susceptibility to disease, while introgressive hybridization with other canids may enhance variation and resilience to pathogens. The uncharacterized diversity of DLA genes in Darwin’s foxes raises uncertainty about whether populations exhibit reduced variation at these loci, which is detrimental for developing strategies to maintain functional diversity and enhance the species’ resilience to disease (Sommer, 2005). Long-read sequencing can resolve these complex genomic regions with structural rearrangements and duplications, which short-read methods struggle to resolve (Plasil et al., 2022).
7 Discussion and future perspectives
High-throughput sequencing has emerged as an increasingly valuable tool for conservationists, gaining prominence in wildlife management due to its enhanced accessibility and effectiveness. It generates highly informative data that can support critical decision-making, as evidenced by efforts to conserve species on the brink of extinction, such as the Iberian lynx (Kleinman-Ruiz et al., 2019), Tasmanian devil (Wright et al., 2020), Montane red foxes (Quinn et al., 2024), Black-footed ferret (Wisely et al., 2015), Florida panther (Onorato et al., 2024), and Cuvier’s gazelle (Alvarez-Estape et al., 2022). Despite South America’s rich endemic biodiversity and its urgent needs for conservation measures, the application of genomics in conservation within the region remains limited. This is largely attributable to challenges in securing funding, alongside restricted access to specialised professionals and laboratory infrastructure (Napolitano et al., 2024). In Chile, conservation genomics is still a novelty, thus the allocation of limited resources must be strategically prioritised for key species such as Darwin’s fox. Above we have detailed HTS approaches that could help to address important, immediate, and conservation relevant issues. These efforts would generate essential baseline data on Darwin’s fox distribution, abundance, genetic diversity, number of distinct genetic lineages and population health. Such data would establish a foundation for the development of species-specific genetic markers, long-term genetic monitoring, and the design of targeted management strategies. Molecular data from HTS can guide targeted management interventions to mitigate threats and prioritize conservation actions by providing information about population connectivity (i.e. gene flow), highlighting critical areas for conservation, identifying at-risk populations that are genetically impoverished, designating potential source populations for translocations/reintroductions, and providing a genetic basis to define conservation management units. Implementing genetic rescue strategies, such as breeding programs and translocations (Wright et al., 2020) may be necessary to mitigate inbreeding depression and prevent local extinction. However, without robust genetic data, these efforts may be counterproductive, potentially leading to outbreeding depression, despite the well-intentioned effort (Bell et al., 2019). In addition, biobanking gametes would ensure the preservation of genetic lineages and safeguard germplasm for future breeding and population recovery efforts (Comizzoli, 2017).
The integration of genomic data into conservation decision-making is indispensable in shaping effective conservation policies, allocation of resources and designing management plans, like the one proposed to Chilean authorities by the N.G.O. Chiloé Silvestre (2023). Collaborative initiatives, such as the ongoing 1000 Genomes Project Chile (https://1000genomas.cl/), aim to sequence the genomes of endemic species and create a comprehensive genomic database to support their conservation. This effort leverages community and institutional networks, opening avenues for further research in areas such as assembling reference genomes, hologenomics, epigenetics, transcriptomics, and adaptation. The benefit of sequencing Darwin’s foxes’ genomes extends to the study of the whole genus Lycalopex, by helping to resolve its evolutionary history, the history of their dispersal across South America, identify local adaptive variation and measure current degrees of hybridization.
Finally, as a flagship species for the Valdivian Temperate Rainforest, efforts to protect Darwin’s foxes would also contribute to the conservation of the whole biodiversity of this unique ecosystem, which harbours many threatened and emblematic species, such as the Pudu (Pudu puda), Southern River Otter (Lontra provocax), Darwin’s frog (Rhinoderma darwinii), Monito del monte (Dromiciops gliroides), Alerce tree (Fitzroya cupressoides) and the Long-nosed shrew opossum (Rhyncholestes raphanurus).
Author contributions
CV: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. JG: Writing – original draft, Writing – review & editing. JF: Supervision, Writing – review & editing. DF: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Leibniz Institut für Zoo und Wildtierforschung (IZW), as well as through the scholarship grant provided by the Agencia Nacional de Investigación y Desarrollo (ANID) (grant n° 62210037) and the Deutscher Akademischer Austauschdienst (DAAD) (grant n° 91825331). Open Access funding was provided by the IZW's Institutional partnership with Frontiers within the Open Access Publishing Framework Agreement.
Acknowledgments
We would like to thank Dr. Juliana Vianna, Dr. Francisco Fonturbel, Felipe Osorio, Juan José Saez, and Eduardo Pizarro for their valuable feedback on early drafts of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcosc.2025.1512531/full#supplementary-material
References
Abrams J. F., Hörig L. A., Brozovic R., Axtner J., Crampton-Platt A., Mohamed A., et al. (2019). Shifting up a gear with iDNA: From mammal detection events to standardised surveys. J. Appl. Ecol. 56, 1637–1648. doi: 10.1111/jpe.2019.56.issue-7
Acosta-Jamett G., Contreras S., Muñoz P., Briceño C., Chirgwin C., Hernández F. (2018). Description of gastrointestinal parasitism through coprologic survey in Darwin’s fox, Lycalopex fulvipes (Martin 1837), and kodkod, Leopardus guigna (Molina 1782), in Chiloé island, Chile. Gayana 82, 160–165. doi: 10.4067/S0717-65382018000200160
Acosta-Jamett G., Surot D., Cortés M., Marambio V., Valenzuela C., Vallverdu A., et al. (2015). Epidemiology of canine distemper and canine parvovirus in domestic dogs in urban and rural areas of the Araucanía region in Chile. Vet. Microbiol. 178, 260–264. doi: 10.1016/j.vetmic.2015.05.012
Alvarez-Estape M., Fontsere C., Serres-Armero A., Kuderna L. F. K., Dobrynin P., Guidara H., et al. (2022). Insights from the rescue and breeding management of Cuvier’s gazelle (Gazella cuvieri) through whole-genome sequencing. Evolutionary Appl. 15, 351–364. doi: 10.1111/eva.13336
Amavet P., Poletta G., Odetti L., Marcó M. V. P., Siroski P. (2023). Detection of the maned wolf, a cryptic and vulnerable species, through environmental DNA studies. J. Nat. Conserv. 74, 126439. doi: 10.1016/j.jnc.2023.126439
Andres K. J., Lodge D. M., Sethi S. A., Andrés J. (2023). Detecting and analysing intraspecific genetic variation with eDNA: From population genetics to species abundance. Mol. Ecol. 32, 4118–4132. doi: 10.1111/mec.v32.15
Andrews K. R., Good J. M., Miller M. R., Luikart G., Hohenlohe P. A. (2016). Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 17, 81–92. doi: 10.1038/nrg.2015.28
Arantes L. S., Ferreira L. C. L., Driller M., Filho F. P. M., Mazzoni C., Santos F. R. (2020). Genomic evidence of recent hybridization between sea turtles at Abrolhos Archipelago and its association to low reproductive output. Sci. Rep. 10, 12847. doi: 10.1038/s41598-020-69613-8
Balagué-Dobón L., Cáceres A., González J. R. (2022). Fully exploiting SNP arrays: a systematic review on the tools to extract underlying genomic structure. Brief Bioinform. 23, bbac043. doi: 10.1093/bib/bbac043
Bass D., Christison K. W., Stentiford G. D., Cook L. S. J., Hartikainen H. (2023). Environmental DNA/RNA for pathogen and parasite detection, surveillance, and ecology. Trends Parasitol. 39, 285–304. doi: 10.1016/j.pt.2022.12.010
Bell D. A., Robinson Z. L., Funk W. C., Fitzpatrick S. W., Allendorf F. W., Tallmon D. A., et al. (2019). The exciting potential and remaining uncertainties of genetic rescue. Trends Ecol. Evol. 34, 1070–1079. doi: 10.1016/j.tree.2019.06.006
Beng K. C., Corlett R. T. (2020). Applications of environmental DNA (eDNA) in ecology and conservation: opportunities, challenges and prospects. Biodivers Conserv. 29, 2089–2121. doi: 10.1007/s10531-020-01980-0
Blanchong J. A., Robinson S. J., Samuel M. D., Foster J. T. (2016). Application of genetics and genomics to wildlife epidemiology: Genetics and Wildlife Epidemiology. J. Wildl Manage 80, 593–608. doi: 10.1002/jwmg.v80.4
Cabello J. E., Dávila J. A. (2014). Isolation and characterization of microsatellite loci in Darwin’s fox (Lycalopex fulvipes) and cross-amplification in other canid species. Conserv. Genet. Resour 6, 759–761. doi: 10.1007/s12686-014-0208-6
Cabello J., Esperón F., Napolitano C., Hidalgo E., Dávila J. A., Millán J. (2013). Molecular identification of a novel gammaherpesvirus in the endangered Darwin’s fox (Lycalopex fulvipes). J. Gen. Virol. 94, 2745–2749. doi: 10.1099/vir.0.057851-0
Cairns K. M., Shannon L. M., Koler-Matznick J., Ballard J. W. O., Boyko A. R. (2018). Elucidating biogeographical patterns in Australian native canids using genome wide SNPs. PLoS One 13, e0198754. doi: 10.1371/journal.pone.0198754
Carvalho C. S., de Oliveira M. E., Rodriguez-Castro K. G., Saranholi B. H., Galetti P. M. Jr. (2022). Efficiency of eDNA and iDNA in assessing vertebrate diversity and its abundance. Mol. Ecol. Resour. 22, 1262–1273. doi: 10.1111/1755-0998.13543
Chavez D. E., Gronau I., Hains T., Dikow R. B., Frandsen P. B., Figueiró H. V., et al. (2022). Comparative genomics uncovers the evolutionary history, demography, and molecular adaptations of South American canids. Proc. Natl. Acad. Sci. U.S.A. 119, e2205986119. doi: 10.1073/pnas.2205986119
Chemisquy M. A., Prevosti F. J., Martínez P., Raimondi V., Stom J. E. C., Acosta-Jamett G., et al. (2019). How many species of grey foxes (Canidae, Carnivora) are there in southern South America? Mastozool Neotrop 26, 81–97. doi: 10.31687/saremMN.19.26.1.0.16
Chiloé Silvestre O. N. G. (2023). Plan de Recuperación, Conservación y Gestión del Zorro Chilote O de Darwin (Lycalopex fulvipes). Chile: Ministerio del Medio Ambiente. Available online at: https://consultasciudadanas.mma.gob.cl/storage/consultation/YBOBEjvOGhxEd2VNvPpBgKcV71lrPOHRlwHc5eIa.pdf.
Cleaveland S., Mlengeya T., Kaare M., Haydon D., Lembo T., Laurenson M. K., et al. (2007). The conservation relevance of epidemiological research into carnivore viral diseases in the serengeti. Conserv. Biol. 21, 612–622. doi: 10.1111/j.1523-1739.2007.00701.x
Cockerill C. A., Hasselgren M., Dussex N., Dalén L., von Seth J., Angerbjörn A., et al. (2022). Genomic consequences of fragmentation in the endangered Fennoscandian arctic fox (Vulpes lagopus). Genes 13 (11), 2124. doi: 10.3390/genes13112124
Comizzoli P. (2017). Biobanking and fertility preservation for rare and endangered species. Anim. Reprod. 14, 30–33. doi: 10.21451/1984-3143-AR889
D’elía, Ortloff, Sanchez, Guinez (2013). A new geographic record of the endangered Darwin’s fox Lycalopex fulvipes (Carnivora: Canidae): filling the distributional gap. Rev. Chil Anat. 86 (4), 485–488. doi: 10.4067/S0716-078X2013000400010
Di Cataldo S., Hidalgo-Hermoso E., Sacristán I., Cevidanes A., Napolitano C., Hernández C. V., et al. (2020). Hemoplasmas are endemic and cause asymptomatic infection in the endangered Darwin’s fox (Lycalopex fulvipes). Appl. Environ. Microbiol. 86 (12), e00779-20. doi: 10.1128/AEM.00779-20
Escobar L. E., Qiao H., Cabello J., Peterson A. T. (2018). Ecological niche modeling re-examined: A case study with the Darwin’s fox. Ecol. Evol. 8, 4757–4770. doi: 10.1002/ece3.2018.8.issue-10
Escoda L., Hawlitschek O., González-Esteban J., Castresana J. (2022). Methodological challenges in the genomic analysis of an endangered mammal population with low genetic diversity. Sci. Rep. 12, 21390. doi: 10.1038/s41598-022-25619-y
Farias A. A., Sepúlveda M. A., Silva-Rodríguez E. A., Eguren A., González D., Jordán N. I., et al. (2014). A new population of Darwin’s fox (Lycalopex fulvipes) in the Valdivian Coastal Range. Rev. Chil Hist Nat. 87, 3. doi: 10.1186/0717-6317-87-3
Favarini M. O., Simão T. L. L., Macedo G. S., Garcez F. S., Oliveira L. R., Cárdenas-Alayza S., et al. (2022). Complex evolutionary history of the South American fox genus lycalopex (Mammalia, Carnivora, Canidae) inferred from multiple mitochondrial and nuclear markers. Diversity 14, 642. doi: 10.3390/d14080642
Förster D. W., Bull J. K., Lenz D., Autenrieth M., Paijmans J. L. A., Kraus R. H. S., et al. (2018). Targeted resequencing of coding DNA sequences for SNP discovery in nonmodel species. Mol. Ecol. Resour 18, 1356–1373. doi: 10.1111/1755-0998.12924
Garcez F. S., Tchaicka L., Lemos F. G., Kasper C. B., Dalponte J. C., Eizirik E. (2024). Phylogeographic analyses of an endemic Neotropical fox (Lycalopex vetulus) reveal evidence of hybridization with a different canid species (L. gymnocercus). J. Hered. 115 (4), 399–410. doi: 10.1093/jhered/esae012
Harris S. E., Munshi-South J., Obergfell C., O’Neill R. (2013). Signatures of rapid evolution in urban and rural transcriptomes of white-footed mice (Peromyscus leucopus) in the New York metropolitan area. PLoS One 8, e74938. doi: 10.1371/journal.pone.0074938
Hasselgren M., Dussex N., von Seth J., Angerbjörn A., Olsen R.-A., Dalén L., et al. (2021). Genomic and fitness consequences of inbreeding in an endangered carnivore. Mol. Ecol. 30, 2790–2799. doi: 10.1111/mec.v30.12
Hidalgo-Hermoso E., Cabello J., Vega C., Kroeger-Gómez H., Moreira-Arce D., Napolitano C., et al. (2020). An eight-year survey for canine distemper virus indicates lack of exposure in the endangered Darwin’s fox (Lycalopex fulvipes). J. Wildl Dis. 56, 482–485. doi: 10.7589/2019-08-195
Hidalgo-Hermoso E., Cabello J., Verasay J., Moreira-Arce D., Hidalgo M., Abalos P., et al. (2022). Serosurvey for selected parasitic and bacterial pathogens in Darwin’s fox (Lycalopex fulvipes): Not only dog diseases are a threat. J. Wildl Dis. 58, 76–85. doi: 10.7589/JWD-D-21-00024
Howard-McCombe J., Jamieson A., Carmagnini A., Russo I.-R. M., Ghazali M., Campbell R., et al. (2023). Genetic swamping of the critically endangered Scottish wildcat was recent and accelerated by disease. Curr. Biol. 33, 4761–9.e5. doi: 10.1016/j.cub.2023.10.026
Jiménez J. E. (2007). Ecology of a coastal population of the critically endangered Darwin’s fox (Pseudalopex fulvipes) on Chiloé Island, southern Chile. J. Zool 271, 63–77. doi: 10.1111/j.1469-7998.2006.00218.x
Kim M., Bae J., Oh B., Rhim H., Yang M.-S., Yang S., et al. (2023). Surveillance of wild animals carrying infectious agents based on high-throughput screening platform in the Republic of Korea. BMC Vet. Res. 19, 158. doi: 10.1186/s12917-023-03714-0
Kleinman-Ruiz D., Lucena-Perez M., Villanueva B., Fernández J., Saveljev A. P., Ratkiewicz M., et al. (2022). Purging of deleterious burden in the endangered Iberian lynx. Proc. Natl. Acad. Sci. 119, e2110614119. doi: 10.1073/pnas.2110614119
Kleinman-Ruiz D., Soriano L., Casas-Marce M., Szychta C., Sánchez I., Fernández J., et al. (2019). Genetic evaluation of the Iberian lynx ex situ conservation programme. Heredity 123, 647–661. doi: 10.1038/s41437-019-0217-z
Liu G., Zhang H., Sun G., Zhao C., Shang S., Gao X., et al. (2017). Characterization of the peripheral blood transcriptome and adaptive evolution of the MHC I and TLR gene families in the wolf (Canis lupus). BMC Genomics 18, 584. doi: 10.1186/s12864-017-3983-0
Medel R. G., Jiménez J. E., Jaksić F. M., Yáñez J., Armesto J. J. (1990). Discovery of a continental population of the rare Darwin’s fox, Dusicyon fulvipes (Martin, 1837) in Chile. Biol. Conserv. 51, 71–77. doi: 10.1016/0006-3207(90)90033-L
Michel A. L., Van Heerden H., Crossley B. M., Al Dahouk S., Prasse D., Rutten V. (2021). Pathogen detection and disease diagnosis in wildlife: challenges and opportunities: -EN- -FR- Détection des agents pathogènes et diagnostic des maladies dans la faune sauvage: difficultés spécifiques et perspectives -ES- Detección de patógenos y diagnóstico de enfermedades en la fauna silvestre: dificultades y oportunidades. Rev. Sci. Tech 40, 105–118. doi: 10.20506/rst.issue.40.1.3205
Molina C., Castillo A., Samaniego H. (2018). Evaluación del nicho ambiental de Lycalopex fulvipes (zorro de Darwin) y la incidencia del cambio climático sobre su distribución geográfica. Gayana 82, 65–78. doi: 10.4067/S0717-65382018000100065
Moreira-Arce D., Vergara P. M., Boutin S., Carrasco G., Briones R., Soto G. E., et al. (2016). Mesocarnivores respond to fine-grain habitat structure in a mosaic landscape comprised by commercial forest plantations in southern Chile. For Ecol. Manage 369, 135–143. doi: 10.1016/j.foreco.2016.03.024
Morrison WR I. I. I., Lohr J. L., Duchen P., Wilches R., Trujillo D., Mair M., et al. (2009). The impact of taxonomic change on conservation: Does it kill, can it save, or is it just irrelevant? Biol. Conserv. 142, 3201–3206. doi: 10.1016/j.biocon.2009.07.019
Napolitano C., Clavijo C., Rojas-Bonzi V., Miño C. I., González-Maya J. F., Bou N., et al. (2024). Understanding the conservation-genetics gap in Latin America: challenges and opportunities to integrate genetics into conservation practices. Front. Genet. 15, 1425531. doi: 10.3389/fgene.2024.1425531
Onorato D. P., Cunningham M. W., Lotz M., Criffield M., Shindle D., Johnson A., et al. (2024). Multi-generational benefits of genetic rescue. Sci. Rep. 14, 17519. doi: 10.1038/s41598-024-67033-6
Paijmans J. L. A., Barlow A., Henneberger K., Fickel J., Hofreiter M., Foerster D. W. G. (2020). Ancestral mitogenome capture of the Southeast Asian banded linsang. PloS One 15, e0234385. doi: 10.1371/journal.pone.0234385
Perini F. A., Russo C. A. M., Schrago C. G. (2010). The evolution of South American endemic canids: a history of rapid diversification and morphological parallelism. J. Evol. Biol. 23, 311–322. doi: 10.1111/j.1420-9101.2009.01901.x
Perry G. H., Melsted P., Marioni J. C., Wang Y., Bainer R., Pickrell J. K., et al. (2012). Comparative RNA sequencing reveals substantial genetic variation in endangered primates. Genome Res. 22, 602–610. doi: 10.1101/gr.130468.111
Pizarro E. J., Julio-Kalajžić B., Sallaberry-Pincheira N., Muñoz V., González-Acuña D., Cabello J., et al. (2023). Species delimitation and intraspecific diversification in recently diverged South American foxes. Mamm Res. 69, 71–87. doi: 10.1007/s13364-023-00717-y
Plasil M., Futas J., Jelinek A., Burger P. A., Horin P. (2022). Comparative genomics of the major histocompatibility complex (MHC) of felids. Front. Genet. 13, 829891. doi: 10.3389/fgene.2022.829891
Quinn C. B., Preckler-Quisquater S., Buchalski M. R., Sacks B. N. (2024). Whole genomes inform genetic rescue strategy for Montane red foxes in north America. Mol. Biol. Evol. 41, msae193. doi: 10.1093/molbev/msae193
Rakotoarivelo A. R., Rambuda T., Taron U. H., Stalder G., O’Donoghue P., Robovský J., et al. (2024). Complex patterns of gene flow and convergence in the evolutionary history of the spiral-horned antelopes (Tragelaphini). Mol. Phylogenet Evol. 198, 108131. doi: 10.1016/j.ympev.2024.108131
Robinson J., Kyriazis C. C., Yuan S. C., Lohmueller K. E. (2023). Deleterious variation in natural populations and implications for conservation genetics. Annu. Rev. Anim. Biosci. 11, 93–114. doi: 10.1146/annurev-animal-080522-093311
Robinson J. A., Räikkönen J., Vucetich L. M., Vucetich J. A., Peterson R. O., Lohmueller K. E., et al. (2019). Genomic signatures of extensive inbreeding in Isle Royale wolves, a population on the threshold of extinction. Sci. Adv. 5, eaau0757. doi: 10.1126/sciadv.aau0757
Seeber P. A., McEwen G. K., Löber U., Förster D. W., East M. L., Melzheimer J., et al. (2019). Terrestrial mammal surveillance using hybridization capture of environmental DNA from African waterholes. Mol. Ecol. Resour 19, 1486–1496. doi: 10.1111/1755-0998.13069
Sillero-Zubiri E. by C., Hoffmann M., Macdonald D. W. (2004). Canids: Foxes, Wolves, Jackals and Dogs. Sillero-Zubiri C, Hoffmann M, Macdonald DW. (IUCN, Gland, Switzerland and Cambridge, UK).
Silva-Rodríguez E., Farias A., Moreira-Arce D., Cabello J, Hidalgo-Hermoso E., Lucherini M., et al. (2016). Lycalopex fulvipes (errata version published in 2016). The IUCN Red List of Threatened Species 2016: e.T41586A107263066. doi: 10.2305/IUCN.UK.2016-1.RLTS.T41586A85370871.en
Silva-Rodríguez E. A., Ovando E., González D., Zambrano B., Sepúlveda M. A., Svensson G. L., et al. (2018). Large-scale assessment of the presence of Darwin’s fox across its newly discovered range. Mamm Biol. 92, 45–53. doi: 10.1016/j.mambio.2018.04.003
Sommer S. (2005). The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool 2, 16. doi: 10.1186/1742-9994-2-16
Stange M., Barrett R. D. H., Hendry A. P. (2021). The importance of genomic variation for biodiversity, ecosystems and people. Nat. Rev. Genet. 22, 89–105. doi: 10.1038/s41576-020-00288-7
Tchaicka L., de Freitas T. R. O., Bager A., Vidal S. L., Lucherini M., Iriarte A., et al. (2016). Molecular assessment of the phylogeny and biogeography of a recently diversified endemic group of South American canids (Mammalia: Carnivora: Canidae). Genet. Mol. Biol. 39, 442–451. doi: 10.1590/1678-4685-GMB-2015-0189
Tomasco I H., Giorello F. M., Boullosa N., Feijoo M., Lanzone C., Lessa E. P. (2022). The contribution of incomplete lineage sorting and introgression to the evolutionary history of the fast-evolving genus Ctenomys (Rodentia, Ctenomyidae). Mol. Phylogenet Evol. 176, 107593. doi: 10.1016/j.ympev.2022.107593
Van Borm S., Belák S., Freimanis G., Fusaro A., Granberg F., Höper D., et al. (2015). “Next-generation sequencing in veterinary medicine: how can the massive amount of information arising from high-throughput technologies improve diagnosis, control, and management of infectious diseases?,” in Veterinary Infection Biology: Molecular Diagnostics and High-Throughput Strategies, vol. 1247 . Eds. Cunha M. V., Inácio J. (Springer New York, New York, NY), 415–436.
Vilà C., Leonard J. A., Iriarte A., O’Brien S. J., Johnson W. E., Wayne R. K. (2004). Detecting the vanishing populations of the highly endangered Darwin’s fox, Pseudalopex fulvipes. Anim. Conserv. Forum 7, 147–153. doi: 10.1017/S1367943004001271
von Holdt K. (2022). “Conclusion: towards a southern sociology,” in Critical Engagement with Public Sociology, vol. p . Eds. Bezuidenhout A., Sonwabile M., von Holdt K. (Bristol University Press), 265–279.
vonHoldt B. M., Pollinger J. P., Earl D. A., Knowles J. C., Boyko A. R., Parker H., et al. (2011). A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 21, 1294–1305. doi: 10.1101/gr.116301.110
vonHoldt B. M., Pollinger J. P., Earl D. A., Parker H. G., Ostrander E. A., Wayne R. K. (2013). Identification of recent hybridization between gray wolves and domesticated dogs by SNP genotyping. Mamm Genome 24, 80–88. doi: 10.1007/s00335-012-9432-0
Wang D., Coscoy L., Zylberberg M., Avila P. C., Boushey H. A., Ganem D., et al. (2002). Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. U.S.A. 99, 15687–15692. doi: 10.1073/pnas.242579699
Wisely S. M., Ryder O. A., Santymire R. M., Engelhardt J. F., Novak B. J. (2015). A road map for 21st century genetic restoration: gene pool enrichment of the black-footed ferret. J. Heredity 106, 581–592. doi: 10.1093/jhered/esv041
Wright B. R., Farquharson K. A., McLennan E. A., Belov K., Hogg C. J., Grueber C. E. (2020). A demonstration of conservation genomics for threatened species management. Mol. Ecol. Resour 20, 1526–1541. doi: 10.1111/1755-0998.13211
Yahnke C. J., Johnson W. E., Geffen E., Smith D., Hertel F., Roy M. S., et al. (1996). Darwin’s fox: A distinct endangered species in a vanishing habitat. Conserv. Biol. 10, 366–375. doi: 10.1046/j.1523-1739.1996.10020366.x
Keywords: conservation genomics, high-throughput sequencing, Lycalopex fulvipes, genetic diversity, Chile
Citation: Valenzuela-Turner C, Grau JH, Fickel J and Förster DW (2025) Preserving Darwin’s fox: genomic tools for the conservation of South America’s most endangered canid. Front. Conserv. Sci. 6:1512531. doi: 10.3389/fcosc.2025.1512531
Received: 16 October 2024; Accepted: 06 January 2025;
Published: 23 January 2025.
Edited by:
Juan Pablo Jaramillo-Correa, National Autonomous University of Mexico, MexicoReviewed by:
Jaime Gasca-Pineda, Posdoctoral Fellow, MexicoGustavo P. Lorenzana, Universidad de la Sierra, Mexico
Copyright © 2025 Valenzuela-Turner, Grau, Fickel and Förster. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristóbal Valenzuela-Turner, dmFsZW56dWVsYUBpenctYmVybGluLmRl