
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Conserv. Sci., 20 February 2024
Sec. Conservation Genetics and Genomics
Volume 5 - 2024 | https://doi.org/10.3389/fcosc.2024.1294358
 Tracy A. Diver1*
Tracy A. Diver1* Steven M. Mussmann1†
Steven M. Mussmann1† Melody J. Saltzgiver1Alexis S. Harrison1†Wade D. Wilson1
Melody J. Saltzgiver1Alexis S. Harrison1†Wade D. Wilson1 Jason A. Coombs2Aaron P. Maloy2
Jason A. Coombs2Aaron P. Maloy2Landscape-scale modifications caused by anthropogenic effects have reshaped global biodiversity and species distributions (Svenning et al., 2015; Boivin et al., 2016; Pecl et al., 2017). For ecologists and environmental managers working to conserve ecosystems, a greater understanding of species’ distributions and community structure is needed to establish conservation priorities that mitigate biodiversity loss (Fordham et al., 2016; Pollock et al., 2020; Harper et al., 2021). Traditional monitoring methods for quantifying biodiversity have typically relied on field observations through visual, physical, or acoustic surveys. Yet, these techniques can be invasive, destructive, prone to bias, or ineffective at detecting rare, elusive, or cryptic species (Thomsen and Willerslev, 2015; Ruppert et al., 2019; Gilbey et al., 2021). While there are limitations, environmental DNA (eDNA) sampling is a powerful tool for monitoring biodiversity and evaluating the impacts of human actions on species and ecosystems that can circumvent many of the aforementioned limitations (Carroll et al., 2018; Beng and Corlett, 2020).
Organisms shed cells containing genetic material as they move through their environments. This extra-organismal DNA can persist in water, soil, or sediment for extended periods of time, and can be collected, extracted, and then amplified to infer detection or non-detection of species without needing to handle any individuals (Freeland, 2017; Garlapati et al., 2019; Beng and Corlett, 2020). The relative ease of collecting environmental samples coupled with the sensitivity of polymerase chain reaction (PCR) has made eDNA instrumental for early detection of biological invasions (Jerde et al., 2013; Nathan et al., 2014; Jo et al., 2021a), non-invasive sampling of imperiled species (Olson et al., 2012; Maruyama et al., 2018; Strickland and Roberts, 2019), and for monitoring community changes and/or ecosystem health (Lee et al., 2020; Wang et al., 2021; Banerjee et al., 2022). However, like any technique, critical considerations for study design are essential, with much attention given to field methods (e.g., contamination prevention, sample collection), benchtop practices (e.g., extraction, PCR inhibition), and assay validation (e.g., sensitivity, specificity; Goldberg et al., 2016).
The efficacy of eDNA applications ultimately hinges on assay design (Freeland, 2017; Schroeter et al., 2020). For targeted detection via end-point PCR, droplet digital PCR (ddPCR), or quantitative PCR (qPCR), species-specific primers and probes are typically designed (Goldberg et al., 2016; Ardura, 2019), whereas for metabarcoding, universal primers are designed to target specific taxonomic groups (Deagle et al., 2014; Miya et al., 2015; Kumar et al., 2022). Regardless of the method used, eDNA assay design begins with sequence alignments of target and sympatric species. This in silico approach allows researchers to identify sequence similarities among taxa for universal primer design or base pair differences across species so that species-specific primers and probes can be developed (Freeland, 2017; Schroeter et al., 2020; Kumar et al., 2022). Whereas additional benchtop (i.e., in vitro) and field (i.e., in situ) testing is required, in silico design remains dependent on in-house sequencing or reference sequences obtained through genomic databases such as National Center for Biotechnology Information (NCBI) or Barcode of Life Data System (BOLD; Ratnasingham and Hebert, 2007). Mitochondrial genes are most frequently targeted for eDNA detections of animals because of their increased coverage in genomic databases relative to nuclear DNA, relatively high copy number per cell, and lower degradation rates that increase detection probability (Taberlet et al., 2012; Bylemans et al., 2018; Jo et al., 2021b). Cytochrome c oxidase subunit I (COI) generally exhibits greater species-level phylogenetic resolution relative to other mitochondrial genes (Hebert et al., 2003; Rach et al., 2017). Consequently, it has become the gene of choice for DNA barcoding, with millions of taxonomically verified sequences available for eDNA assay design (Hebert et al., 2003; Deagle et al., 2014). However, primer design is not always possible within this region, and the COI gene is often too variable to develop universal metabarcoding primers that reliably amplify a broad taxonomic range (Deagle et al., 2014; Kumar et al., 2022). Instead, ribosomal subunits such as 12S and 16S ribosomal RNA (rRNA) are preferred for metabarcoding because they contain both highly conserved regions suitable for universal primer design and variable regions that are taxonomically discriminative (Yang et al., 2014).
While assay design for eDNA monitoring will ultimately be study-specific, access to sequence data that covers multiple gene regions within the mitochondrial genome (herein, mitogenome) is universally needed. Sequence databases that contain mitogenomes are far from complete, with many species lacking publicly available sequence data. For example, of the 34,200 described species of fish (FishBase; Froese and Pauly, 2017), BOLD contains COI sequences for 24,654 species whereas MitoFish, a database of complete and partial fish mitogenomes (Iwasaki et al., 2013), contains only 3,563 complete mitogenomes (accessed March 2023). For common species, access to tissue for sequencing may simply require additional field sampling; however, obtaining tissues from threatened and endangered animals can be difficult due to their rarity and, within the United Sates, the need to obtain handling permits (United States, 1983). Thus, the purpose of this work was to 1) provide complete mitogenomes for many western North American endangered fishes and 2) improve community coverage for the native fishes of New Mexico, USA.
All specimens used for mitogenome sequencing were collected from wild or hatchery populations (Table 1). Caudal fin tissue was collected from 4-10 individuals per species, preserved in 95% ethanol and frozen at -80°C post-sampling. Wild collections were identified by regional experts and confirmed post-sequencing by comparing protein-coding fragments (i.e., COI, ND4) to accessioned sequence data within NCBI or BOLD (Ratnasingham and Hebert, 2007).
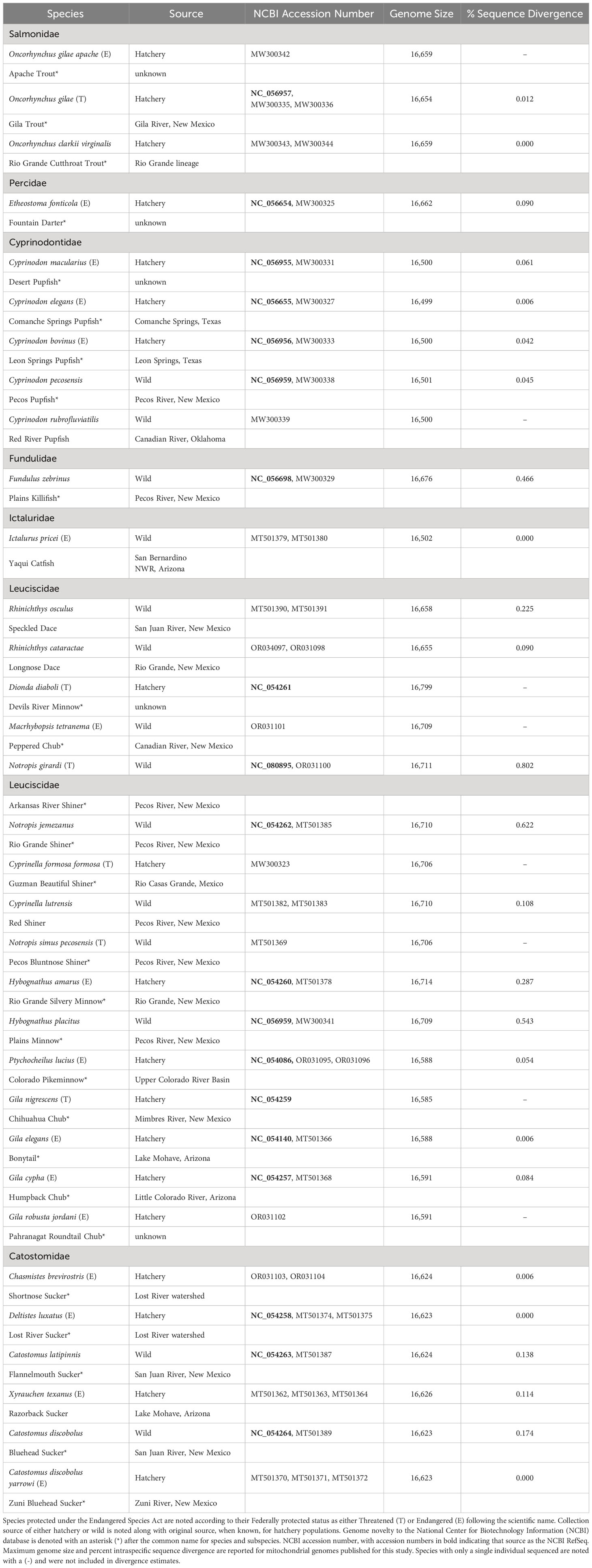
Table 1 Species, with both scientific and common name, with mitochondrial genomes sequenced for this study categorized by Family.
Genomic DNA was extracted using a DNeasy® Blood and Tissue Kit (Qiagen, Inc., Germantown, MD, USA). Long-range PCR was performed using family-specific primers designed by Schroeter et al. (2020). Reactions of 25 µl were conducted using Q5 Hot Start High-Fidelity Master Mix (New England Biolabs, Ipswich, MA, USA) under the following conditions: enzyme activation for 2 min at 98°C, followed by 35 cycles of 20 s denaturing at 98°C, 20 s at primer annealing temperature (Supplementary Table 1; Schroeter et al., 2020) and 3 min at 72°C, followed by a final 7 min elongation at 72°C. Amplification success was confirmed through visualization on a 1.5% agarose gel.
Priming regions that failed to amplify in certain families during the initial PCR screening were redesigned as necessary (Supplementary Table 1). Novel primers were also designed and optimized when primer sequences were unavailable for examined families. Primer design for all families and failed regions followed the methods of Schroeter et al. (2020). Briefly, this included identifying highly conserved regions among NCBI RefSeq (O'Leary et al., 2016) within family groups and designing primers in these regions using the Primer3 tool (Koressaar and Remm, 2007; Untergasser et al., 2012) in Geneious R11.1 software (Biomatters Ltd, Newark, NJ, USA). Three primer pairs in each region were designed, and optimization for each priming region was conducted by running temperature gradients for each primer pair, which were evaluated through visualization on a 1.5% agarose gels. One primer set per region was retained for sequencing.
Amplicons were quantified using a Qubit™ 3.0 fluorometer (Life Technologies, Carlsbad, CA, USA). Only individuals with successful amplification across all four regions were selected for sequencing. PCR products were pooled in equimolar ratios to provide even sequencing coverage. Pooled PCR amplicons were bead purified and diluted to a standard concentration of 0.2 ng/µl. DNA libraries were prepared using the Nextera XT Library Prep Kit following the manufacturer’s instructions (Illumina, Inc., San Diego, CA, USA). Resulting dual-indexed mitogenome libraries were bead purified, sized using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA), and pooled in equimolar ratios for sequencing using the MiSeq Reagent Nano V2 Kit (2×250 paired-end reads). Sequences were demultiplexed into FASTQ files and trimmed using the Illumina FASTQ workflow to remove adaptor and index sequences. Subsequent mitogenome assembly followed Schroeter et al. (2020). This included trimming low quality bases (Q < 20), discarding short reads (< 25 base pairs), and merging overlapping paired reads (merge rate = normal) prior to error correction and normalization (BBNorm v37.25; error correction, default settings; normalization, target coverage 60 and minimum depth = 6). De novo assembly was then conducted allowing a maximum mismatch of 5% (Schroeter et al., 2020). Annotated consensus sequences for complete mitogenomes were submitted to GenBank under BioProject PRJNA633136 (Table 1; Supplementary Table 2).
Thirty-eight complete mitogenomes representing all available native fishes of New Mexico (Supplementary Figure 1; Sublette et al., 1990; Propst, 1999) sequenced outside this study were obtained from the NCBI RefSeq database (O'Leary et al., 2016). These sequences were included with one representative from each species sequenced in this study regardless of geographical origin. Duplicate individuals of a species were only included when RefSeq samples were available (e.g., Razorback Sucker Xyrauchen texanus, Yaqui Catfish Ictalurus pricei); this duplication included the original RefSeq and one individual sequenced here. Multiple sequence alignment of all mitogenomes was conducted in MAFFT v.7.487 using default settings (Katoh and Standley, 2013). A phylogenetic tree was constructed using the maximum likelihood method implemented in IQ-Tree (Nguyen et al., 2015). IQ-Tree used ModelFinder (Kalyaanamoorthy et al., 2017) to find the best-fit model assessed using Bayesian Information Criterion (BIC). Additional run parameters included 100,000 ultrafast bootstraps (Hoang et al., 2017), 1,000,000 maximum iterations, and 100,000 Shimodaira-Hasegawa-like approximate likelihood ratio test (SH-aLRT) replicates. The ape (Paradis and Schliep, 2019), tidytree (Yu, 2022), and ggtree (Yu et al., 2017) packages were used in the R statistical language (R 4.1.2; R Core Team, 2022) to visualize the phylogeny.
The combination of novel primers presented here (Supplementary Table 1) along with those previously published by Schroeter et al. (2020) allowed for successful amplification and sequencing of 63 complete mitogenomes representing 7 families and 33 species/subspecies (Table 1). Primer development for Cyprinodontidae and Fundulidae were unique to this study and built upon those previously developed (Schroeter et al., 2020). Additional primer design was required for fishes we sequenced within the families Leuciscidae, Salmonidae, and Percidae, which were used in combination with published primers to obtain complete mitogenome coverage (Schroeter et al., 2020).
Twenty-seven of the species we sequenced were novel to NCBI, with 19 of those established as the RefSeq (O'Leary et al., 2016). The remaining eight taxa included the following species/subspecies: Apache Trout (Oncorhynchus apache), Red River Pupfish (Cyprinodon rubrofluviatilis), Peppered Chub (Macrohybopsis tetranema), Pahranagat Roundtail Chub (Gila robusta jordani), Zuni Bluehead Sucker (Catostomus discobolus yarrowi), Rio Grande Cutthroat Trout (Oncorhynchus clarkii virginalis), Pecos Bluntnose Shiner (Notropis simus pecosensis), and Guzmán Beautiful Shiner (Cyprinella formosa formosa). Percent pairwise identity among all mitogenomes was high (range = 99.8 – 100%), with only 11 having less than 100%.
Mitogenome length varied among species and ranged between 16,499 to 16,799 base pairs (Table 1). All mitogenomes contained 13 protein coding genes, 22 tRNAs, two rRNAs, and the control region. With the exception of ND6, all protein coding and rRNA genes were coded on the heavy strand. Eight tRNA genes (tRNA-Ala, tRNA-Asn, tRNA-Cys, tRNA-Gln, tRNA-Glu, tRUNA-Pro, tRNA-Ser, tRNA-Tyr) were coded on the light strand; the remaining 14 were coded on the heavy strand. The GC content varied among mitogenomes and ranged from 40.8 – 46.1%, with Peppered Chub and Pecos Bluntnose Shiner having the lowest highest GC content respectively.
We sequenced multiple mitogenomes for most species (N = 25), allowing coarse insight into intraspecific genetic variation. Intraspecific base pair composition varied from 0.000 - 0.802% (Table 1). In general, endangered species had lower sequence variability (mean = 0.104%; range 0.000 - 0.802%) relative to non-listed species (mean = 0.241%; range 0.000 – 0.622%). For Zuni Bluehead Sucker (N = 3), Lost River Sucker (Deltistes luxatus; N = 3), Rio Grande Cutthroat Trout (N = 2), and Yaqui Catfish (N = 2) all individuals sequenced within each species had identical mitogenomes.
A phylogenetic tree was inferred by maximum likelihood using IQ-Tree (Nguyen et al., 2015). ModelFinder (Kalyaanamoorthy et al., 2017) selected GTR+F+R5 as the best-fit model according to BIC. The resultant tree was consistent with previous research (Supplementary Figure 1; Broughton et al., 2013; Schönhuth et al., 2018).
Our work greatly improves mitogenome accessibility for many threatened and endangered North American freshwater fishes and highlights an information gap for species whose management could benefit from non-invasive eDNA monitoring. Of the 33 species/subspecies we sequenced, 22 are currently protected under the Endangered Species Act (ESA) of 1973, as amended (United States, 1983). This represents over 30% of ESA-listed freshwater fishes within the American West (i.e., drainages west of the Mississippi River) and nearly 18% of those listed within the United States (www.ecos.fws.gov, accessed March 2023). Only one-third (N = 41) of all ESA-listed fishes have publicly available mitogenomes (NCBI accessed March 2023), of which our sequencing effort represents nearly 54%.
Community-level mitogenome sampling is an important factor in eDNA monitoring of ESA-listed fishes. Our work greatly improves mitogenome availability for New Mexico, which has five major watersheds (Figure 1) that historically harbored a diverse native fish fauna relative to other arid states in the American West (72 species representing 16 families; Supplementary Table 3). Fish species are frequently listed following declines in population size and range (Mace et al., 2008; Neel et al., 2012), and this is especially true for New Mexico where few fishes occupy all or even large portions of their historic range (Propst, 1999). Additionally, the historical presence of a few species (e.g., Bonytail Gila elegans) remains unclear due to rapid population declines that occurred at the time of their description (Sublette et al., 1990; Propst, 1999). Two additional species, Phantom Shiner (Notropis orca) and Rio Grande Bluntnose Shiner (Notropis simus simus), are now extinct, and seven others are extirpated from the state. An additional fifteen species are currently listed as threatened or endangered (www.ecos.fws.gov), of which seven are limited to New Mexico (Propst, 1999). We sequenced nearly 30% of the extant native community of fishes in New Mexico, of which 16 were novel to NCBI. This effort, in combination with previously published sequences, provides near full coverage of mitogenomes for New Mexico’s native fishes.
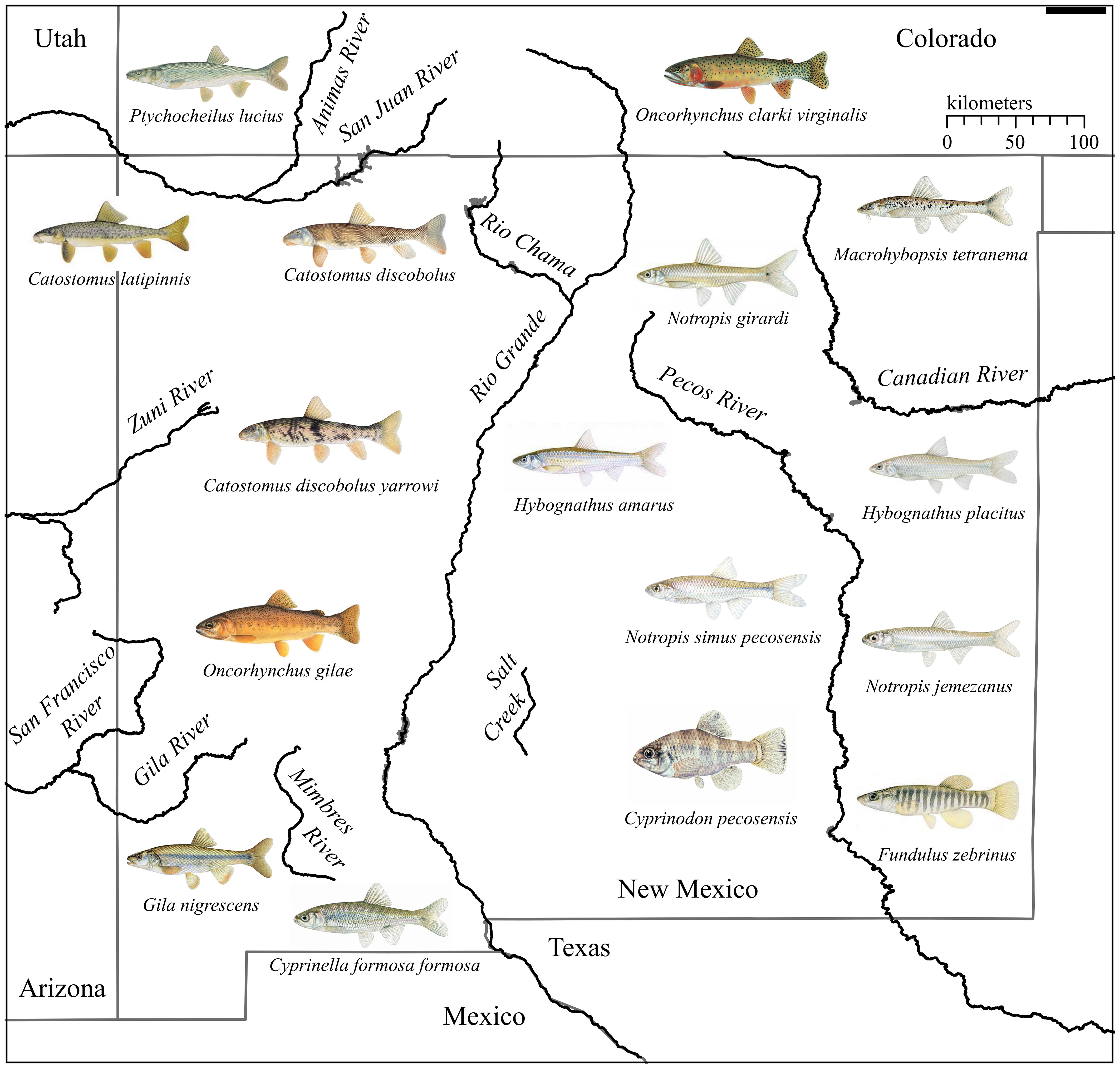
Figure 1 Map of New Mexico, USA, and its major drainages. Illustration of novel species sequenced for this study within the state are shown with copy right permission from © Joseph R. Tomelleri.
Increasing human demand for water in combination with aridification of the American West will likely contribute to further declines of native fishes. The use of eDNA to monitor the effects of these impacts along with management actions may prove useful in maintaining western North American fish diversity. For example, Gila Trout (Oncorhynchus gilae) faces numerous extinction threats including hybridization with nonnative species, limited adaptive potential under current rates of global warming, and population extirpation caused by increases in scale and severity of wildfires (Brown et al., 2001; Kennedy et al., 2009; Propst et al., 2021; U.S. Fish and Wildlife Service, 2021). Management actions that combat these effects include nonnative species removal, fish rescue in response to wildfire threats, hatchery augmentation and reintroduction, and genetic management of distinct lineages (U.S. Fish and Wildlife Service, 2021). Monitoring for environmental impacts and management responses, however, can be difficult due to their remote distributions within the montane region of southwestern New Mexico and southeastern Arizona. Sampling for eDNA offers a comparatively easy alternative to monitoring presence or absence of populations in response to environmental factors and management actions.
While the sequencing efforts presented here will undoubtedly help with future eDNA assay design, phylogeographic considerations are warranted as geographical representation within our sampling is limited. For example, we provide mitogenomes for the native fishes of the San Juan River, with the individuals sequenced here sampled from that community. However, species such as Bluehead Sucker (Catostomus discobolus) and Flannelmouth Sucker (Catostomus latipinnis) are widely distributed throughout the Colorado River Basin, with phylogenetic analyses supporting distinct geographic clades (Unmack et al., 2014; Bangs et al., 2020). Indeed, sequence alignment of our Bluehead Sucker mitogenomes with all available cytb sequences for this species on GenBank revealed up to 6.8% sequence divergence. Thus, while this work can guide locus selection during eDNA assay design, targeted sequencing efforts (i.e., cytb, COI) should be conducted for phylogeographically distinct populations to confirm assay specificity across space. Similarly, absence of sequence variation (e.g., Zuni Bluehead Sucker) identified within this study does not preclude phylogeographic variation, since these samples were obtained from hatchery populations that have likely undergone population bottlenecks (Meffe, 1986; Fordham et al., 2016; Lemopoulos et al., 2019). Checking for primer specificity in situ through genomic resources and/or in silico sequencing of geographically diverse populations should maximize assay utility by identifying and removing potential issues of co-amplification or PCR failure among populations (Thalinger et al., 2021).
Highly biodiverse ecosystems can also challenge eDNA methods. For example, metabarcoding is a preferred approach for biodiversity assessments within speciose congeneric communities since developing numerous species-specific assays can be prohibitively challenging and expensive (e.g., Etheostoma, Notropis; Smart et al., 2016; Kumar et al., 2022). Similarly, there may be greater interest in broader taxonomic coverage with diverse communities to increase understanding of processes that affect community change and species composition. Universal primer design targeting loci such as 12S or 16S rRNA may be possible with a few representatives from each family, but to determine species presence a complete reference database is required for taxonomic assignment of sequences obtained from the environment. Databases such as BOLD, which only contain COI barcodes, are useful for species-specific marker design, but are insufficient for metabarcoding approaches that target highly conserved priming regions, which is often necessary for biomonitoring of highly diverse communities.
Overall, our mitogenome sequencing efforts provide a critical first step towards monitoring species distribution changes while limiting impacts on populations and habitats (Olson et al., 2012; Maruyama et al., 2018; Strickland and Roberts, 2019). We think federal agencies tasked with recovering these species, and whom have greater access to individuals, should lead efforts in providing these resources to the broader scientific community, as access to genomic resources is critical for eDNA assay design. Future development of the mitogenomic resources described in this study through broader geographic and taxonomic sampling will benefit eDNA assessments and conservation actions by providing logistical and financial relief to researchers interested in contributing to conservation actions, minimizing direct handling of ESA-listed populations, and reducing the need for extensive in-house sequencing.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal study was approved by U.S. Fish and Wildlife Service. The study was conducted in accordance with the local legislation and institutional requirements. Endangered fish handling was conducted under U.S. Fish and Wildlife Service permits TE676811-0 and TE676811-9.
TD: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft. SM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. MS: Investigation, Methodology, Resources, Software, Validation, Writing – review & editing. AH: Conceptualization, Funding acquisition, Investigation, Methodology, Resources, Software, Validation, Writing – review & editing. WW: Funding acquisition, Investigation, Project administration, Supervision, Writing – review & editing. JC: Data curation, Formal analysis, Investigation, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. AM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We are sincerely thankful to M. Bartron at the Northeast Fishery Center for assistance with funding acquisition. We are also grateful to the New Mexico Department of Game and Fish (M. Zeigler), the New Mexico Fish and Wildlife Conservation Office (S.R. Davenport, R.E. Grey, and D.W. Furr), and the Southwestern Native Aquatic Resources and Recovery Center (W. Knight and M. Ulibarri) for their assistance with tissue collections. We also thank N.R. Franssen for insightful manuscript review. The views presented here are the authors’ and do not necessarily represent the views of the U.S. Fish and Wildlife Service. Any use of trade, firm, or product names are for descriptive purposes only and do not imply endorsement by the U.S. Government. We thank the U.S. Fish and Wildlife Service for providing the funds needed to conduct this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcosc.2024.1294358/full#supplementary-material
Ardura A. (2019). Species-specific markers for early detection of marine invertebrate invaders through eDNA methods: Gaps and priorities in GenBank as database example. J. Nat. Conserv. 47, 51–57. doi: 10.1016/j.jnc.2018.11.005
Banerjee P., Stewart K. A., Antognazza C. M., Bunholi I. V., Deiner K., Barnes M. A., et al. (2022). Plant–animal interactions in the era of environmental DNA (eDNA)—A review. Environ. DNA 4, 987–999. doi: 10.1002/edn3.308
Bangs M. R., Douglas M. R., Chafin T. K., Douglas M. E. (2020). Gene flow and species delimitation in fishes of Western North America: Flannelmouth (Catostomus latipinnis) and Bluehead sucker (C. Pantosteus discobolus). Ecol. Evol. 10, 6477–6493. doi: 10.1002/ece3.6384
Beng K. C., Corlett R. T. (2020). Applications of environmental DNA (eDNA) in ecology and conservation: opportunities, challenges and prospects. Biodivers. Conserv. 29, 2089–2121. doi: 10.1007/s10531-020-01980-0
Boivin N. L., Zeder M. A., Fuller D. Q., Crowther A., Larson G., Erlandson J. M., et al. (2016). Ecological consequences of human niche construction: Examining long-term anthropogenic shaping of global species distributions. Proc. Natl. Acad. Sci. 113, 6388–6396. doi: 10.1073/pnas.152520011
Broughton R. E., Betancur-R R., Li C., Arratia G., Ortí G. (2013). Multi-locus phylogenetic analysis reveals the pattern and tempo of bony fish evolution. PloS Currents 5. doi: 10.1371/currents.tol.2ca8041495ffafd0c92756e75247483e
Brown D. K., Echelle A. A., Propst D. L., Brooks J. E., Fisher W. L. (2001). Catastrophic wildfire and number of populations as factors influencing risk of extinction for Gila trout (Oncorhynchus gilae). West. North Am. Nat. 61, 139–148.
Bylemans J., Furlan E. M., Gleeson D. M., Hardy C. M., Duncan R. P. (2018). Does size matter? An experimental evaluation of the relative abundance and decay rates of aquatic environmental DNA. Environ. Sci. Technol. 52, 6408–6416. doi: 10.1021/acs.est.8b01071
Carroll E. L., Bruford M. W., DeWoody J. A., Leroy G., Strand A., Waits L., et al. (2018). Genetic and genomic monitoring with minimally invasive sampling methods. Evolution. Appl. 11, 1094–1119. doi: 10.1111/eva.12600
Deagle B. E., Jarman S. N., Coissac E., Pompanon F., Taberlet P. (2014). DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match. Biol. Lett. 10, 20140562. doi: 10.1098/rsbl.2014.0562
Fordham D. A., Akçakaya H. R., Alroy J., Saltré F., Wigley T. M., Brook B. W. (2016). Predicting and mitigating future biodiversity loss using long-term ecological proxies. Nat. Climate Change 6, 909–916. doi: 10.1038/nclimate3086
Freeland J. R. (2017). The importance of molecular markers and primer design when characterizing biodiversity from environmental DNA. Genome 60, 358–374. doi: 10.1139/gen-2016-0100
Froese R., Pauly D. (2017). Fish Base (World Wide Web Electronic Publication). Available at: www.fishbase.org.
Garlapati D., Charankumar B., Ramu K., Madeswaran P., Murthy R. (2019). A review on the applications and recent advances in environmental DNA (eDNA) metagenomics. Rev. Environ. Sci. Bio/Technol. 18, 389–411. doi: 10.1007/s11157-019-09501-4
Gilbey J., Carvalho G., Castilho R., Coscia I., Coulson M. W., Dahle G., et al. (2021). Life in a drop: Sampling environmental DNA for marine fishery management and ecosystem monitoring. Mar. Policy 124, 104331. doi: 10.1016/j.marpol.2020.104331
Goldberg C. S., Turner C. R., Deiner K., Klymus K. E., Thomsen P. F., Murphy M. A., et al. (2016). Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 7, 1299–1307. doi: 10.1111/2041-210X.12595
Harper M., Mejbel H. S., Longert D., Abell R., Beard T. D., Bennett J. R., et al. (2021). Twenty-five essential research questions to inform the protection and restoration of freshwater biodiversity. Aquat. Conserv.: Mar. Freshw. Ecosyst. 31, 2632–2653. doi: 10.1002/aqc.3634
Hebert P. D., Ratnasingham S., de Waard J. R. (2003). Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. London. Ser. B: Biol. Sci. 270, S96–S99. doi: 10.1098/rsbl.2003.0025
Hoang D. T., Chernomor O., von Haeseler A., Minh B. Q., Vinh L. E. (2017). UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Iwasaki W., Fukunaga T., Isagozawa R., Yamada K., Maeda Y., Satoh T. P., et al. (2013). MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 30, 2531–2540. doi: 10.1093/molbev/mst141
Jerde C. L., Chadderton W. L., Mahon A. R., Renshaw M. A., Corush J., Budny M. L., et al. (2013). Detection of Asian carp DNA as part of a Great Lakes basin-wide surveillance program. Can. J. Fish. Aquat. Sci. 70, 522–526. doi: 10.1139/cjfas-2012-0478
Jo T., Ikeda S., Fukuoka A., Inagawa T., Okitsu J., Katano I., et al. (2021a). Utility of environmental DNA analysis for effective monitoring of invasive fish species in reservoirs. Ecosphere 12, e03643. doi: 10.1002/ecs2.3643
Jo T., Takao K., Minamoto T. (2021b). Linking the state of environmental DNA to its application for biomonitoring and stock assessment: Targeting mitochondrial/nuclear genes, and different DNA fragment lengths and particle sizes. Environ. DNA 4, 271–283. doi: 10.1002/edn3.253
Kalyaanamoorthy S., Minh B. Q., Wong T. K. F., von Haeseler A., Jermiin L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kennedy T. L., Gutzler D. S., Leung R. L. (2009). Predicting future threats to the long-term survival of Gila trout using a high-resolution simulation of climate change. Climatic Change 94, 503–515. doi: 10.1007/s10584-008-9503-0
Koressaar T., Remm M. (2007). Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291. doi: 10.1093/bioinformatics/btm091
Kumar G., Reaume A. M., Farrell E., Gaither M. R. (2022). Comparing eDNA metabarcoding primers for assessing fish communities in a biodiverse estuary. PloS One 17, e0266720. doi: 10.1371/journal.pone.0266720
Lee A. H., Lee J., Noh J., Lee C., Hong S., Kwon, et al. (2020). Characteristics of long-term changes in microbial communities from contaminated sediments along the west coast of South Korea: Ecological assessment with eDNA and physicochemical analyses. Mar. pollut. Bull. 160, 111592. doi: 10.1016/j.marpolbul.2020.111592
Lemopoulos A., Prokkola J. M., Uusi-Heikkilä S., Vasemägi A., Huusko A., Hyvärinen P., et al. (2019). Comparing RADseq and microsatellites for estimating genetic diversity and relatedness—Implications for brown trout conservation. Ecol. Evol. 9, 2106–2120. doi: 10.1002/ece3.4905
Mace G. M., Collar N. J., Gaston K. J., Hilton-Taylor C., Akçakaya H. R., Leader-Williams, et al. (2008). Quantification of extinction risk: IUCN's system for classifying threatened species. Conserv. Biol. 22, 1424–1442. doi: 10.1111/j.1523-1739.2008.01044.x
Maruyama A., Sugatani K., Watanabe K., Yamanaka H., Imamura A. (2018). Environmental DNA analysis as a non-invasive quantitative tool for reproductive migration of a threatened endemic fish in rivers. Ecol. Evol. 8, 11964–11974. doi: 10.1002/ece3.4653
Meffe G. K. (1986). Conservation genetics and the management of endangered fishes. Fisheries 11, 14–23. doi: 10.1577/1548-8446(1986)011<0014:CGATMO>2.0.CO;2
Miya M., Sato Y., Fukunaga T., Sado T., Poulsen J. Y., Sato, et al. (2015). MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2, 150088. doi: 10.1098/rsos.150088
Nathan L. M., Simmons M., Wegleitner B. J., Jerde C. L., Mahon A. R. (2014). Quantifying environmental DNA signals for aquatic invasive species across multiple detection platforms. Environ. Sci. Technol. 48, 12800–12806. doi: 10.1021/es5034052
Neel M. C., Leidner A. K., Haines A., Goble D. D., Scott M. J. (2012). By the numbers: how is recovery defined by the US endangered species act? BioScience 62, 646–657. doi: 10.1525/bio.2012.62.7.7
Nguyen L. T., Schmidt H. A., von Haeseler A., Minh B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
O'Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Olson Z. H., Briggler J. T., Williams R. N. (2012). An eDNA approach to detect eastern hellbenders (Cryptobranchus a. alleganiensis) using samples of water. Wildl. Res. 39, 629–636. doi: 10.1071/WR12114
Paradis E., Schliep K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Pecl G. T., Araújo M. B., Bell J. D., Blanchard J., Bonebrake T. C., Chen I. C., et al. (2017). Biodiversity redistribution under climate change: Impacts on ecosystems and human well-being. Science 355, eaai9214. doi: 10.1126/science.aai9214
Pollock L. J., O’connor L. M., Mokany K., Rosauer D. F., Talluto M. V., Thuiller W. (2020). Protecting biodiversity (in all its complexity): new models and methods. Trends Ecol. Evol. 35, 1119–1128. doi: 10.1016/j.tree.2020.08.015
Propst D. L. (1999). Threatened and Endangered Fishes of New Mexico (Santa Fe, NM: Tech. Rpt. No. 1. New Mexico Department of Game and Fish). 84 pp.
Propst D. L., Turner T. F., Monzingo J. A., Brooks J. E., Meyers D. J. (2021). “Ecology, Politics, and Conservation of Gila Trout,” in Standing between Life and Extinction: Ethics and Ecology of Conserving Aquatic Species in North American Deserts. Eds. Propst D. L., Williams J. E., Bestgen K. R., Hoagstrom C. W. (University of Chicago Press, Chicago, IL 60637 USA), 295–315. doi: 10.7208/9780226694504-022
Rach J., Bergmann T., Paknia O., DeSalle R., Schierwater B., Hadrys H. (2017). The marker choice: Unexpected resolving power of an unexplored CO1 region for layered DNA barcoding approaches. PloS One 12, e0174842. doi: 10.1371/journal.pone.0174842
Ratnasingham S., Hebert P. D. (2007). BOLD: The Barcode of Life Data System (http://www. barcodinglife. org). Mol. Ecol. Notes 7, 355–364. doi: 10.1111/j.1471-8286.2007.01678.x
R Core Team (2022). R: A language fand environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available at: https://www.R-project.org/.
Ruppert K. M., Kline R. J., Rahman M. S. (2019). Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA. Global Ecol. Conserv. 17, e00547. doi: 10.1016/j.gecco.2019.e00547
Schönhuth S., Vukić J., Šanda R., Yang L., Mayden R. L. (2018). Phylogenetic relationships and classification of the Holarctic family Leuciscidae (Cypriniformes: Cyprinoidei). Mol. Phylogenet. Evol. 127, 781–799. doi: 10.1016/j.ympev.2018.06.026
Schroeter J. C., Maloy A. P., Rees C. B., Barton M. L. (2020). Fish mitochondrial genome sequencing: expanding genetic resources to support species detection and biodiversity monitoring using environmental DNA. Conserv. Genet. Res. 12, 433–446. doi: 10.1007/s12686-019-01111-0
Smart A. S., Weeks A. R., van Rooyen A. R., Moore A., McCarth M. A., Tingley R. (2016). Assessing the cost-efficiency of environmental DNA sampling. Methods Ecol. Evol. 7, 1291–1298. doi: 10.1111/2041-210X.12598
Strickland G. J., Roberts J. H. (2019). Utility of eDNA and occupancy models for monitoring an endangered fish across diverse riverine habitats. Hydrobiologia 826, 129–144. doi: 10.1007/s10750-018-3723-8
Sublette J. E., Hatch M. D., Sublette M. F. (1990). The fishes of New Mexico. 1st ed (University of New Mexico Press, Albuquerque, NM 87106 USA).
Svenning J. C., Eiserhardt W. L., Normand S., Ordonez A., Sandel B. (2015). The influence of paleoclimate on present-day patterns in biodiversity and ecosystems. Annu. Rev. Ecol. Evol. System. 46, 551–572. doi: 10.1146/annurev-ecolsys-112414-054314
Taberlet P., Coissac E., Hajibabaei M., Rieseberg L. H. (2012). Environmental DNA. Mol. Ecol. 21, 1789–1793. doi: 10.1111/j.1365-294X.2012.05542.x
Thalinger B., Deiner K., Harper L. R., Rees H. C., Blackman R. C., Sint D., et al. (2021). A validation scale to determine the readiness of environmental DNA assays for routine species monitoring. Environ. DNA 3, 823–836. doi: 10.1002/edn3.189
Thomsen P. F., Willerslev E. (2015). Environmental DNA–An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18. doi: 10.1016/j.biocon.2014.11.019
United States (1983). The Endangered Species Act as amended by public law 97–304 (the Endangered Species Act amendments of 1982) (Washington: U.S. G.P.O, U.S. Government Printing Office Washington, D.C. 20401 USA).
Unmack P. J., Dowling T. E., Laitinen N. J., Secor C. L., Mayden R. L., Shiozawa D. K., et al. (2014). Influence of introgression and geological processes on phylogenetic relationships of western North American mountain suckers (Pantosteus, Catostomidae). PloS One 9, e90061. doi: 10.1371/journal.pone.0090061
Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B. C., Remm M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115. doi: 10.1093/nar/gks596
U.S. Fish and Wildlife Service (2021). Draft Revised Recovery Plan for Gila trout (Oncorhynchus gilae). (Albuquerque, New Mexico: U.S. Fish and Wildlife Service, Southwest Region), 172.
Wang S., Yan Z., Hänfling B., Zheng X., Wang P., Fan J., et al. (2021). Methodology of fish eDNA and its applications in ecology and environment. Sci. Total Environ. 755, 142622. doi: 10.1016/j.scitotenv.2020.142622
Yang L., Tan Z., Wang D., Xue L., Guan M. X., Huang T., et al. (2014). Species identification through mitochondrial rRNA genetic analysis. Sci. Rep. 13, 4089. doi: 10.1038/srep04089
Keywords: endangered fishes, mitochondrial genomes, environmental DNA, assay design, genomic resources
Citation: Diver TA, Mussmann SM, Saltzgiver MJ, Harrison AS, Wilson WD, Coombs JA and Maloy AP (2024) Increasing availability of reference mitochondrial genomes for imperiled fishes in western North America for environmental DNA assay design and species monitoring. Front. Conserv. Sci. 5:1294358. doi: 10.3389/fcosc.2024.1294358
Received: 14 September 2023; Accepted: 01 February 2024;
Published: 20 February 2024.
Edited by:
Alison K. S. Wee, University of Nottingham Malaysia Campus, MalaysiaReviewed by:
Derek Houston, Western State Colorado University, United StatesCopyright © 2024 Diver, Mussmann, Saltzgiver, Harrison, Wilson, Coombs and Maloy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracy A. Diver, dHJhY3kuYS5kaXZlckBnbWFpbC5jb20=
†Present addresses: Steven M. Mussmann, Abernathy Fish Technology Center, United States Fish and Wildlife Service, Longview, WA, United States
Alexis S. Harrison, Corvallis Research Lab, Oregon Department of Fish and Wildlife, Corvallis, OR, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.