 Ludwig Triest
Ludwig Triest Tim Sierens
Tim Sierens Tom Van der Stocken
Tom Van der Stocken- Ecology and Biodiversity Research Group, Biology Department, Vrije Universiteit Brussel, Brussels, Belgium
Historical processes of long-distance migration and ocean-wide expansion feature the global biogeographic pattern of Rhizophora species. Throughout the Indian Ocean, Rhizophora stylosa and Rhizophora mucronata seem to be a young phylogenetic group with an expansion of R. mucronata toward the Western Indian Ocean (WIO) driven by the South Equatorial Current (SEC). Nuclear microsatellites revealed genetic patterns and breaks; however, the estimation of propagule dispersal routes requires maternally inherited cytoplasmic markers. Here, we examine the phylogeography of 21 R. mucronata provenances across a >4,200 km coastal stretch in the WIO using R. stylosa as an outgroup. Full-length chloroplast genome (164,474 bp) and nuclear ribosomal RNA cistron (8,033 bp) sequences were assembled. The boundaries, junction point, sequence orientation, and stretch between LSC/IRb/SSC/IRa/LSC showed no differences with R. stylosa chloroplast genome. A total of 58 mutations in R. mucronata encompassing transitions/transversions, insertions-deletions, and mononucleotide repeats revealed three major haplogroups. Haplonetwork, Bayesian maximum likelihood (ML), and approximate Bayesian computation (ABC) analyses supported discrete historical migration events. An ancient haplogroup A in the Seychelles and eastern Madagascar was as different from other haplogroups as from R. stylosa. A star-like haplonetwork referred as the recent range expansion of haplogroup B from northern Madagascar toward the African mainland coastline, including a single variant spanning >1,800 km across the Mozambique Channel area (MCA). Populations in the south of Delagoa Bight contained haplogroup C and was originated from a unique bottleneck dispersal event. Divergence estimates of pre- and post-Last Glacial Maximum (LGM) illustrated the recent emergence of Rhizophora mangroves in the WIO compared to other oceans. Connectivity patterns could be aligned with the directionality of major ocean currents. Madagascar and the Seychelles each harbored haplogroups A and B, albeit among spatially separated populations, explained from a different migration era. Likewise, the Aldabra Atoll harbored spatially distinct haplotypes. Nuclear ribosomal cistron (8,033 bp) variants corresponded to haplogroups and confirmed admixtures in the Seychelles and Aldabra. These findings shed new light on the origins and dispersal routes of R. mucronata lineages that have shaped their contemporary populations in large regions of the WIO, which may be the important information for defining marine conservation units both at ocean scale and at the level of small islands.
Introduction
Mangroves represent characteristic intertidal forests along tropical, subtropical, and some warm-temperate coasts (Tomlinson, 2016). Sea-faring propagules as dispersal units allow mangrove tree species to be globally distributed and for connectivity between populations in different ocean basins (Van der Stocken et al., 2019a). Their origin and history over geological time offer an interesting multidisciplinary field involving continental drift, phylogenetic reconstruction, and molecular evolutionary genetics (Duke et al., 2002; Triest, 2008; Lo et al., 2014; Yan et al., 2016; Tomizawa et al., 2017; Takayama et al., 2021).
Unraveling the formation history of geographic distribution, the divergence time estimates of pantropical Rhizophora species by means of chloroplast DNA (cpDNA) phylogeny gave a robust time scale of mangrove diversification followed by ancestral range estimation (Takayama et al., 2021). Nucleotide sequences of chloroplast and nuclear DNA as well as nuclear microsatellites were informative to estimate the relationships and ocean-wide evolutionary history of all Rhizophora species (Chen et al., 2015; Ng et al., 2015; Wee et al., 2015; Takayama et al., 2021). Global phylogenetic and comparative phylogeographic studies on Rhizophora revealed a clear genetic differentiation of cpDNA haplotypes between Atlantic and Pacific populations of R. mangle L. and R. racemosa G. Mey explained by the Central American Isthmus as a dispersal barrier, whereas R. mangle experienced recent and frequent transatlantic propagule dispersal (Takayama et al., 2013; Cerón-Souza et al., 2015). A multispecies approach of comparative large-scale phylogeography using chloroplast sequences gained importance to reveal the importance and effect of species-specific propagule traits, including those of Rhizophora species that are capable to disperse over long distances. Ocean currents and isolation by distance (IBD) are the significant predictors of the genetic structure in R. mangle from the Caribbean area though the historical factors or changes in ocean current patterns could interfere (Hodel et al., 2018).
Major ocean currents may connect different seas and ocean basins globally, however, not necessarily in a continuous or perpetual way as the success of long-distance connectivity depends on the processes that are inherently stochastic in nature, such as ocean currents and seedling establishment (Siegel et al., 2008). Ocean currents and oceanographic features such as eddies or zones of convergence may limit or modify gene flow, thereby making an IBD model too simplistic to explain the observed genetic differentiation (Thomas et al., 2017; Hodel et al., 2018). Combining the evidence from nuclear microsatellites between R. racemosa populations and a Lagrangian particle-tracking model allowed to explain strong genetic discontinuity from their positioning on both sides of an oceanic convergence zone in the Gulf of Guinea (Ngeve et al., 2016). Likewise, the genetic structure of R. mucronata Lam. populations along the Malay Peninsula could be explained by prevailing ocean currents and dispersal potential of the species, and not necessarily by a geographical distance (Wee et al., 2014, 2020). Overall, ocean currents explain much of the patterns, both in the case of barriers caused by land masses (Triest, 2008) or opposing currents (Wee et al., 2014, 2015, 2020), as in the case of the bifurcation of equatorial currents (Mori et al., 2015; Ngeve et al., 2017; Triest et al., 2021b).
The amount of mangrove genetic and phylogeographic data is rapidly increasing, with many studies focusing on the populations of Avicennia and Rhizophora species in Southeast Asia (Ng et al., 2013; Guo et al., 2016; Yan et al., 2016; Triest et al., 2018; Do et al., 2019; He et al., 2019), Central and South America (Pil et al., 2011; Sandoval-Castro et al., 2014; Cerón-Souza et al., 2015; Kennedy et al., 2017; Francisco et al., 2018; Hodel et al., 2018; Mori et al., 2021), and East Atlantic (Ngeve et al., 2016, 2017) with recent attention for mangrove populations in the Western Indian Ocean (WIO) (De Ryck et al., 2016; Triest et al., 2021b,c). Several large-scale mangrove genetic studies have considered a limited number of samples from the WIO region of Bruguiera (Urashi et al., 2013; Wee et al., 2015), Rhizophora (Lo et al., 2014; Yan et al., 2016; Takayama et al., 2021), Xylocarpus (Tomizawa et al., 2017), and Heritiera (Banerjee et al., 2020) with a majority of locations and focus on relationships and connectivity between the Eastern Indian Ocean (EIO) and Western Pacific region.
In the Indo-West Pacific (IWP) region, the mangrove species R. apiculata Blume, Rhizophora stylosa Griff., and R. mucronata might form their present distribution through different demographic histories in each species despite possessing sympatric distributions nowadays (Ng et al., 2015; Wee et al., 2015). A disjunction between R. stylosa and R. mucronata remains scattered and unclear over different regions (Lo et al., 2014; Chen et al., 2015; Takayama et al., 2021). Rhizophora mucronata has a sympatric distribution with other IWP Rhizophora species (R. apiculata and R. stylosa) in the Western Pacific. However, it is the only Rhizophora species in the WIO region (Duke et al., 2002) and has among the widest ranges of all mangrove species. The distributional range of R. mucronata stretches across the IWP region, from the East African shorelines to the Western Pacific (Duke et al., 2002), with a southern limit in southeast Africa and a northern limit in the Persian Gulf (Spalding et al., 2010). Cycles of isolation and gene flow between the two ocean basins, such as the Pacific Ocean and Indian Ocean through the Strait of Malacca, were responsible for repeated mixing during several hundred thousand of years and shaped the speciation of abovementioned three Rhizophora species with an estimated divergence time of 1.20–6.44 Ma (He et al., 2019). Rhizophora mucronata and R. stylosa were reported as very closely related species based on the cpDNA sequences that lack a clear bootstrap support to be separated, unless as a separate cluster from R. apiculata (Takayama et al., 2013; Ng et al., 2015). A model of speciation with a repeated gene flow involved was supported to explain the divergence of R. mucronata vs. R. stylosa based on whole-genome nuclear markers (He et al., 2019). The identity of both species in the IWP was strongly influenced by vicariance processes since the Oligocene–Miocene boundary, and chloroplast and internal transcribed spacer (ITS) markers separated conspecific lineages and regions such that the potential long-distance dispersal (LDD) routes of R. mucronata could be proposed between, e.g., Northern Australia and Eastern Africa (Lo et al., 2014). Similarly, microsatellites clustered individuals from Australia and Eastern Africa apart from Asian populations (Yan et al., 2016). Rhizophora mucronata showed a low genetic differentiation in cpDNA and nuclear sequences between the East and West Malay Peninsula (Ng et al., 2015), whereas nuclear microsatellites also revealed a higher proportion of recent migration along the Malacca Strait corresponding to contemporary gene flow (Wee et al., 2020).
Rhizophora mucronata exhibits low levels of nuclear microsatellite diversity throughout its range despite the separation of WIO populations from other regions (Wee et al., 2015). The population genetic structure and estimates of gene flow and migration directionality of R. mucronata in the WIO using nuclear microsatellites revealed several genetic breaks and patterns of connectivity aligned with major ocean currents (Triest et al., 2021b). Along the African mainland coast, the bifurcation of the South Equatorial Current (SEC) separated an allelically more diverse region (East Africa) from the Mozambique Channel Area (MCA) with a genetic break located south of Delagoa Bight and characterized by monomorphism and strong inbreeding (Triest et al., 2021b). The Aldabra Atoll populations strongly differs genetically from the population of the African mainland with a less genetic differentiation from North Madagascar populations. The granitic Seychelles of Madagascar seemed to be less differentiated from those of Eastern Africa. Altogether, private alleles of microsatellite loci did not explain much of the obtained genetic structure that was rather shaped by the differences in shared allele frequencies or by elevated inbreeding levels (Triest et al., 2021b).
Comparative studies of mangrove species of different genera indicated an overall lower genetic differentiation of Rhizophora populations than for the mangrove trees of other genera, hence the genetic patterns that resulted from rather recent dispersal events could be detected, to be explained from successful dispersal events of their water-buoyant propagules (Do et al., 2019; Wee et al., 2020). The migration models of Rhizophora and Avicennia species usually indicate a unidirectional stepping-stone model (Wee et al., 2020; Triest et al., 2021a,b,c). An approximate Bayesian computation (ABC) simulation supported that R. mucronata populations from East Africa were more recent than those of the islands (Madagascar and Seychelles) with divergence times of the former being roughly estimated to have occurred within the Holocene (Triest et al., 2021b).
Direct measurements of dispersal pathways and connectivity estimates can be challenging. Connectivity determines the resulting genetic identity and (eventually also the diversity in case of multiple sources and repeated dispersal events) overall genetic structure. The genetic structure and diversity of mangrove populations are determined by the cumulative effect of insect, wind, and bird pollination (Hermansen et al., 2014; Wee et al., 2015) and by the transport, establishment, and survivorship of water-buoyant propagules (Rabinowitz, 1978). The propagules with germinating embryos are the diploid vectors of LDD migration (i.e., combined influence of egg cell and pollen genomes), whereas pollination occurs at local scales and transmit only paternal haploid nuclear genomes. On the other hand, maternally inherited cytoplasmic molecular markers such as cpDNA and mtDNA contain direct phylogenetic and phylogeographic information about the origin of similar or related haplotype variants (Triest, 2008). Ocean circulation regimes are the main abiotic vector of LDD in mangroves (Stieglitz and Ridd, 2001; Van der Stocken et al., 2015, 2019b) and cause these variants to disperse and distribute over time. The accumulation of mutations in the cpDNA genome gives a way to microevolutionary traces at the within-species level. This could not be detected from nuclear microsatellite markers, and therefore additional chloroplast sequence information could provide valuable information for unraveling the patterns of connectivity between the Seychelles, Madagascar, and other African populations (Triest et al., 2021b). For example, nuclear genes and chloroplast sequences of Bruguiera gymnorrhiza (L.) Lamk. indicated a relatedness of Madagascar populations to those of the EIO (Urashi et al., 2013), and cpDNA of the widespread Xylocarpus granatum J. Koenig showed the most common South China Sea haplotype in several WIO populations (Tomizawa et al., 2017).
In this study, our overall aim was to use full-genome cpDNA data to unravel the dispersal history of R. mucronata on WIO islands and along the East African mainland coastline. This oceanic region features the SEC and a complex of eddies in the MCA. Previously, a null-hypothesis stated that “over a broad geographical scale, R. mucronata populations in the WIO would comprise a single evolutionary unit” was already rejected (Triest et al., 2021b) and can be replaced by more precise research questions based on the reported genetic breaks and nuclear-based connectivity patterns. Therefore, we hypothesize that for complete genome cpDNA variants of R. mucronata of the WIO: (1) a phylogeographical analysis reflects the historical processes of different haplotype lineages; (2) LDD routes align with ocean surface circulation patterns; and (3) the divergence time estimation reveals their origin on continental and remote island coasts. We considered 21 provenances of R. mucronata of which 13 were taken from the populations used in our previous study of nuclear microsatellites (Triest et al., 2021b) and 8 sites could be added to enlarge the geographic range and obtain more island representatives within the WIO (namely from Madagascar and Seychelles and previously discarded from the microsatellite analysis due to too small sample sizes of N <5). To ensure the highest possible resolution, we de novo assembled full sequences of the cpDNA genome for the use of all detectable mutations: transitions/transversions, insertions-deletions, and mononucleotide repeats. A fully annotated cpDNA genome of R. stylosa was available in Genbank (Li et al., 2019) and served for comparison as an outgroup to R. mucronata. Overall insights into population identity and connectivity patterns are discussed within a context of essential knowledge for effective conservation management.
Materials and Methods
Study Area
We considered Rhizophora samples from 21 mangrove populations distributed across the WIO (Table 1). About 10 populations were located along a >4,200 km stretch of the eastern African continent and 11 populations in the WIO islands. Samples were from remote islands of the Seychelles (both sides of Mahé and Isle Curieuse) and Aldabra Atoll (3 sites), from Madagascar (2 sites on eastern coastline, 2 at northern tip, and 1 along western coastline), and on the East African mainland coastline from Kenya, Mozambique, and the Republic of South Africa (10 sites). These were collected or provided and morphologically identified as R. mucronata according to Tomlinson (2016). For each sample site, fresh leaves were collected from individual adult trees and stored in individual bags with silica gel for transport. For comparative reasons, an outgroup for maximum likelihood (ML) analysis, and for the construction of a haplonetwork, we included three samples (Table 1) of closely related R. stylosa from Vietnam (two sites taken from Do et al., 2019) and from China (Hainan; Genbank accession number MK070169).
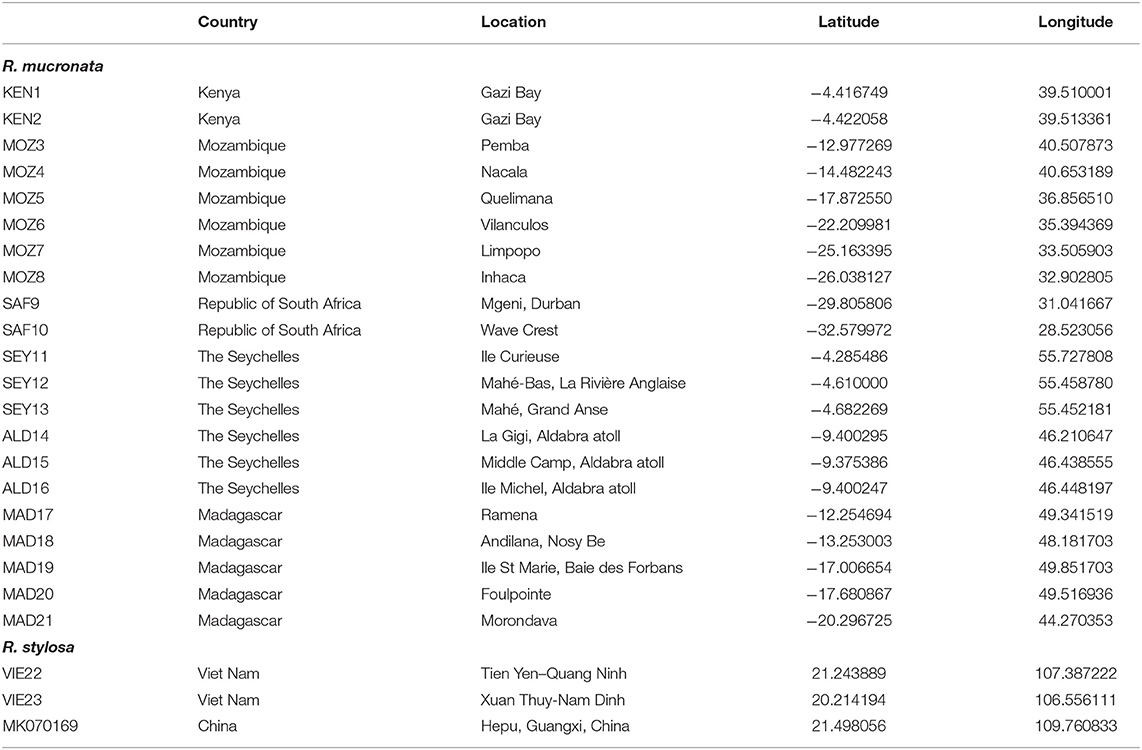
Table 1. Location details of 21 Rhizophora mucronata provenances in the Western Indian Ocean (WIO) region and of 3 Rhizophora stylosa samples from South East Asia that were used as an outgroup.
DNA Extraction and Next Generation Sequencing
Genomic DNA extracts of 23 samples were made at the Plant Biology and Nature Management (APNA) lab of the Vrije Universiteit Brussel (VUB) and processed for next generation sequencing (NGS) analysis using the E.Z.N.A. SP plant DNA Mini Kit (Omega Biotek, Norcross, GA, USA). The quantity and purity (260/280 and 260/230 ratios) of the DNA were determined using a nanodrop one spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Extractions were repeated for samples with a 260/280 ratio of <1.8 and/or a concentration lower than 5 ng/μl. If necessary, multiple DNA extractions of one sample were pooled and concentrated by ethanol precipitation. An Illumina paired-end library was constructed using the TruSeq nano DNA Kit (Illumina, San Diego, CA, USA). After passing quality inspection (DNA concentration between 5 and 15 ng/μl), the constructed library (TruSeq Nano DNA Kit, Illumina, San Diego, CA, USA) was sequenced by 300 × 2 bp paired-end sequencing in an Illumina MiSeq platform (Macrogen, Seoul, South Korea).
Chloroplast Genome Assembly, Alignment, and Comparative Analysis
Raw data were filtered out to remove the joint sequence and low-quality reads to obtain high-quality clean data. The Illumina pair-end NGS product is used as the input file for de novo chloroplast assemblies. The de novo chloroplast assemblies were done at first for the three chloroplast genomes using the NOVOPlasty assembly at Kmer = 33 (Dierckxsens et al., 2017). All assemblies were executed by taking a single read from the data set that originates from the targeted plastid as seed (rbcL) and taking 30% as a subsample from the FASTA file with default parameters. These were compared to the existing annotated R. stylosa chloroplast genome (GenBank accession number MK070169 by Li et al., 2019), and seemed to be similar in the genome structure and perfectly aligned with R. stylosa despite not aligning well with GenBank accession number MN307165 (R. mucronata, Wu, unpublished) or MW387538 (R. apiculata, Jiang, unpublished). Therefore, the remaining samples were assembled using an “assemble to reference” function in the Geneious software. Illumina 2 bp × 300 bp paired-end was processed in Geneious Prime v 2019.2.1 (2005–2019 Biomatters Ltd.) to obtain complete chloroplast genome sequences. All assemblies used MK070169 (R. stylosa) as a reference genome, and the mapping of reads of 23 samples was performed. This approach yielded identical results as obtained with Novoplasty and the assemblages using R. stylosa as a reference to map every sample, and averaged 66,892–454,099 reads with a mean depth of reads ranging from 118 to 829 coverage. This approach facilitated the analysis without the need to describe de novo chloroplast genome features and gene annotations. The 23 consensus sequences and MK070169 were aligned with MAFFT v7.388 (Katoh et al., 2002; Katoh and Standley, 2013).
The %G~C content and length of the inverted repeat (IR) region, large single-copy (LSC) region, and small single-copy (SSC) region of R. stylosa and of three haplogroups of R. mucronata (MAD20, MAD17, and MOZ7) were obtained from statistics generated by Geneious. Because all R. mucronata samples aligned well to the closely related R. stylosa with an exactly similar number and position of genes, CDS, and non-coding transfer RNA (tRNA) and ribosomal RNA (rRNA), we refer to the circular visualization of the annotated cp genome of MK070169 in Genbank and in Li et al. (2019). Given the similarity of R. mucronata with R. stylosa, we decided to use the MK070169 annotated genome as a reference for the comparative cp genome. The alignments of the complete chloroplast genome sequences of three selected R. mucronata haplogroups A, B, and C with reference to R. stylosa were visualized using mVISTA (Mayor et al., 2000; Frazer et al., 2004) to show the patterns of variation between species and major haplogroup lineages. The four boundaries between IR, LSC, and SSC were characterized and measured because in the process of genome evolution, these may expand or contract the IR boundary, pushing some genes into either the IR region or a single-copy region. We calculated the bp distance between boundaries and the nearest gene to unambiguously document any junction sequence divergence among three haplogroups with reference to R. stylosa chloroplasts. A simple sequence repeat (SSR) analysis, including mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide SSRs, was conducted using the MISA v2.1 (MIcroSAtellite identification tool; http://pgrc.ipkgatersleben.de/misa/misa.html) software (Beier et al., 2017). The minimum repetition times of these SSRs were set to 10, 6, 5, 5, 5, and 5, respectively.
Nuclear rRNA Cistron Assembly
The nuclear ribosomal cistron (18S, ITS1, 5.8S, ITS2, and 26S) was assembled from a R. mucronata (Genbank KJ194220.1, Lo et al., 2014) 639-bp sequence containing an ITS 1 (partial sequence), 5.8S ribosomal RNA gene (complete sequence), and ITS 2 (partial sequence), and was subsequently used three times as a seed in Geneious Prime v 2019.2.1 to progressively enlarge the flanking regions. More than 8,000 -bp nuclear ribosomal cistron was obtained for one sample (SAF9) and further used as a reference to map every sample, averaging 8,694–54,438 reads with a mean depth of reads ranging from 292 to 1,827 coverage. The generated consensus sequences of 8,033-bp length for a total of 23 samples (21 WIO R. mucronata and 2 R. stylosa from Vietnam) were aligned for a comparative description of the mutated positions using MAFFT v7.388 (Katoh et al., 2002; Katoh and Standley, 2013).
Phylogeographical Analyses of Full cpDNA Genome
The MAFFT alignment of full-genome cp sequences were used to infer the phylogenetic trees. The plastid phylogeny was produced using a total of 24 cp sequences, including R. stylosa as an outgroup. The phylogenetic reconstruction (ML tree) was performed with MrBayes 3.2.6 (Huelsenbeck and Ronquist, 2001) plugin option in Geneious (general time reversible nucleotide substitution model with a gamma distributed rate variation among sites (GTR+G), the ML estimate of alpha parameter, 1,100,000 MCMC chain length, and 100,000 burn-in length). The tree was visualized using Geneious version 2019.2 created by Biomatters available from https://www.geneious.com. For comparative purposes, additional analysis based on the variable sites of coding sequences only was done after an alignment with ClustalW in MEGA 7.026 (Kumar et al., 2016). The data contained 23 variable sites (18 with parsimony and 5 singletons). For phylogenetic interpretation, a Bayesian ML tree with partial deletion option was performed. The best model (general time reversible) was followed with the Bayesian information criterion (BIC) = 614.627, Akaike information criterion, corrected (AICc) = 375.098, and ML value – lnL = −130.639 using the MEGA software with 1,000 bootstrap replicates (Nei and Kumar, 2000). Rhizophora stylosa (NCBI GenBank MK070169) was taken as an outgroup. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor-join and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach, and then selecting the topology with a superior log-likelihood value.
From the aligned cpDNA sequences of all 24 Rhizophora samples (21 R. mucronata and 3 R. stylosa), we further adjusted manually at sites with mononucleotide repeats and insertions/deletions (indels). All mutational steps were considered for a haplotype definition on the basis of transitions, transversions, indels, and mononucleotide repeats. Indels were considered as a single event regardless of their size and recoded as proposed by Müller (2006). A minimum spanning network using NETWORK 10.1 (Fluxus Engineering) served for a haplotype definition on the basis of 96 characters. In this paper, we use the term “haplogroups” for the three major clusters A, B, and C and designate their minor variants as “haplotypes.”
The demographic history of divergence between R. mucronata populations of the WIO was carried out on all mutations, including indels and mononucleotide repeats considered as 58 binary haploid data, using the ABC approach implemented in DIYABC ver. 2.0 (Cornuet et al., 2014). We narrowed down the ABC scenario testing with subsequent demographic (ABC1), origin (ABC2), and bottleneck (ABC3) models (Supplementary Information ABC and—also see further in section Results—Figure 4). We first tested demographic event models (ABC1 = demographic model with four scenarios): (1) constant population size, (2) population decline, (3) population expansion, and (4) population bottleneck followed by an expansion hereafter simply referred to as “population bottleneck.” For the WIO region, we considered all 21 R. mucronata provenances to conduct these conceptualized scenarios of demographic history. However, this implies that the cpDNA mutations reflect demographic changes, and are not biased by past admixture that might be a problem (in fact, we a posteriori came to know—see section Results—that on the Seychelles and on Madagascar both haplogroups A and B occur, whereas on Aldabra and the East African mainland coast only one haplogroup, either B or C). Because of the poor resolution (see results), an additional demographic model was conducted, however thereby keeping separately the different regions that harbor haplogroup B. This allowed to test whether Northern Madagascar could be a source area for expansion to other regions. For the demographic model at the WIO level (ABC1-WIO), we used broad time priors t1 and tb (10–100,000) and effective size priors on Ne and N1 (10–100,000) despite the restriction for the bottleneck N1b (10–2,000). For the demographic model zooming at the geographical regions with haplogroup B (ABC1-HaploB), we used 14 loci and 4 summary statistics of genic diversity parameters. We considered shorter time priors t1 (10–10,000) and tb (10–2,000) and smaller effective size priors on Ne (2,000–100,000), N1 (2,000–20,000), and on the bottlenecked N1b (10–2,000). These shorter time priors can be argued from the similar or only slightly differing sequences within haplogroup B. Then, the timing of inferred demographic events could be estimated from the computed posterior distribution of time parameters from the 1% best simulated data sets (mode value and 95% CI).
Further analysis of origin models (ABC2 = origin model with seven scenarios; Supplementary Information ABC) with an aim to answer main phylogeographic questions that were raised from the phylogenetic outcome and geographical distribution of the haplotypes, namely, about (1) an ancestral origin of haplogroup A of eastern WIO island populations (granitic Seychelles and East Madagascar); (2) the origin of coastal African populations sharing haplogroup B; and (3) about the origin of haplogroup C (south of Delagoa Bay), either from an ancestral group (haplogroup A) or derived from haplogroup B. For the origin model at the WIO level (ABC2), we used broad time priors t1 and td (10–100,000) and effective size priors on NA and N1-N3 (10–100,000). Then, the timing of inferred demographic events could be estimated from the computed posterior distribution of time parameters from the 1% best simulated data sets (mode value and 95% CI).
On the basis of the most likely origin model (the outcome of ABC2), we tested for different scenarios of the “expansion after bottleneck” model using the three geographical groups: (1) the islands of Seychelles and Madagascar; (2) the Aldabra Atoll and East African mainland coast from Kenya to Mozambique (MOZ6); and (3) the populations along African coast situated south of the Delagoa Bight (MOZ7, MOZ8, SAF9, and SAF10). These groups were analyzed in an ABC3, a bottleneck model with seven scenarios, namely all possible combinations of one, two, or three bottlenecked groups (Supplementary Information ABC). For the bottleneck model (ABC3), we considered broad time priors for t1, td, and db (10–100,000) and on effective size priors NA, N1, N2, N3 (10–100,000) despite the size being smaller for the bottlenecks of N1b, N2b, and N3b (10–2,000). Then, the timing of inferred demographic events could be estimated from the computed posterior distribution of time parameters from the 1% best simulated data sets (mode value and 95% CI).
Thus, the three ABC models were allowed to infer (1) the overall demographic history (ABC1) for the entire WIO (ABC1-WIO) and for haplogroup B alone (ABC1-HaploB); (2) the most likely origin model among the options of geographical grouping of populations (ABC2); and (3) the most likely bottlenecked group (ABC3). In all scenarios, t# represents the time scale measured in number of generations (largest numbers refer to the oldest events) and N# represents the effective size of the corresponding populations during the indicated time period. We used default prior values for all parameters, except for ABC1-HaploB and for ABC3 (as mentioned earlier) based on the outcome of the preliminary test runs of prior distributions. Summary statistics of 4 variables (ABC1) and 36 variables (ABC2 and ABC3) were considered for each population or pairwise comparison of the three samples: one-sample summary statistics were genic diversities and two-sample summary statistics were FST distances and Nei's distances, all considering the proportion of zero values; mean of non-zero values; variance of non-zero values; and mean of complete distribution. About 4 million simulation data sets were run for the prior distribution of ABC1. About 7 million simulation data sets were run for the prior distribution of ABC2 and ABC3. The most likely scenario was obtained from a comparative assessment of their posterior probabilities. About 1 million simulated data sets were used to compute posterior, of which 10,000 were used in the local regression. From the posterior, 1,000 data sets were simulated. The goodness-of-fit was checked through a principal component analysis (PCA) using the “model checking” option. The posterior distribution of parameters (N# and t#) was estimated for the most likely supported scenarios.
Results
Chloroplast Genome Features of R. mucronata in Comparison to R. stylosa
The nucleotide sequences of the three chloroplast genomes of R. mucronata obtained from the NOVOPlasty assembler of MAD20 (East Madagascar, haplotype A), MAD17 (Northern Madagascar, haplotype B), and MOZ7 (Southern Mozambique, haplotype C) were 164,438, 164,474, and 164,464 bp, respectively (Table 2). The overall %G~C content was 34.9 for all Rhizophora chloroplast genomes (Table 2). The chloroplast genome of R. mucronata from WIO contained 131 genes, including 85 protein-coding genes, 38 tRNA genes, and 8 rRNA genes. Among the protein-coding genes, 10 genes contain a single intron while 4 genes possess 2 introns. Most genes occurred in a single copy, whereas eight protein-coding genes, eight tRNA genes, and the four rRNA genes in IR regions were duplicated. A comparison of the gene orientation and stretches of four junction points at LSC, SSC, and IR boundaries showed an exactly similar organization for R. mucronata of WIO and R. stylosa (Figure 1; Table 2). A sequence identity plot comparing the three haplotypes of R. mucronata with R. stylosa as the annotated reference genome using mVISTA revealed that mutational changes occurred mainly in the non-coding sequences of the LSC region (Figure 1). For a fully annotated genome and codon usage, we refer to Li et al. (2019) as R. mucronata of the WIO shared the same gene content and organization with R. stylosa. Rhizophora mucronata haplotypes A, B, and C possess a pair of IRs with a size of IR (all 26,324 bp) each separated by an SSC (19,242; 19,252; and 19,241 bp) and an LSC (92,449; 92,575; and 92,576 bp), respectively (Table 2). Rhizophora stylosa possess a pair of IRs with a size of IR (26,325 bp) each separated by an SSC (19,243 bp) and an LSC (92,580 bp) (Table 2). There were 96 changes detected between R. mucronata and R. stylosa, namely 22 transitions, 26 transversions, 18 insertion-deletions, and 30 mononucleotide repeats (Table 3; Supplementary Table 1). Overall amount of 126 SSRs was present in haplogroup A (116 mono-, 8 di-, and 2 tri-repeats) of which 112 were A- and T-repeats; 125 SSRs in haplogroups B and C (115 mono repeats) of which 111 were A- and T-repeats. A very comparable level of microsatellites was found in R. stylosa and 3 R. mucronata haplogroups with the majority of up to 11 length repeats and only minor fluctuations due to mononucleotide repeat differences (Supplementary Figure 1). In the IRa and IRb, only a single mutational change was observed (mononucleotide T-repeat or the reverse complement as A-repeat, considered just once) and was different between R. mucronata and R. stylosa (Supplementary Table 1).
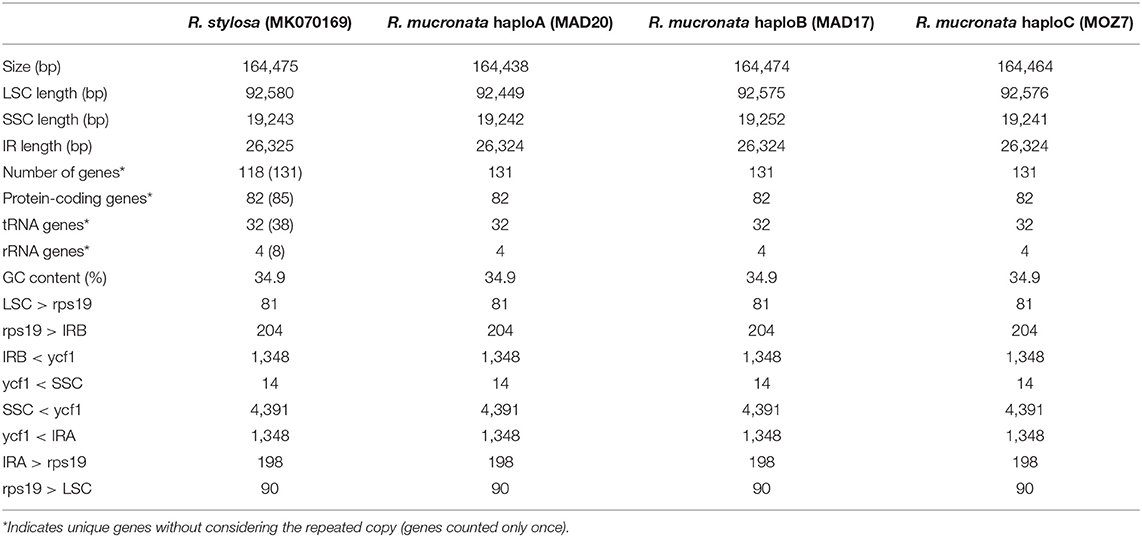
Table 2. Comparison of chloroplast genome characteristics in three haplogroups of R. mucronata with reference to R. stylosa.
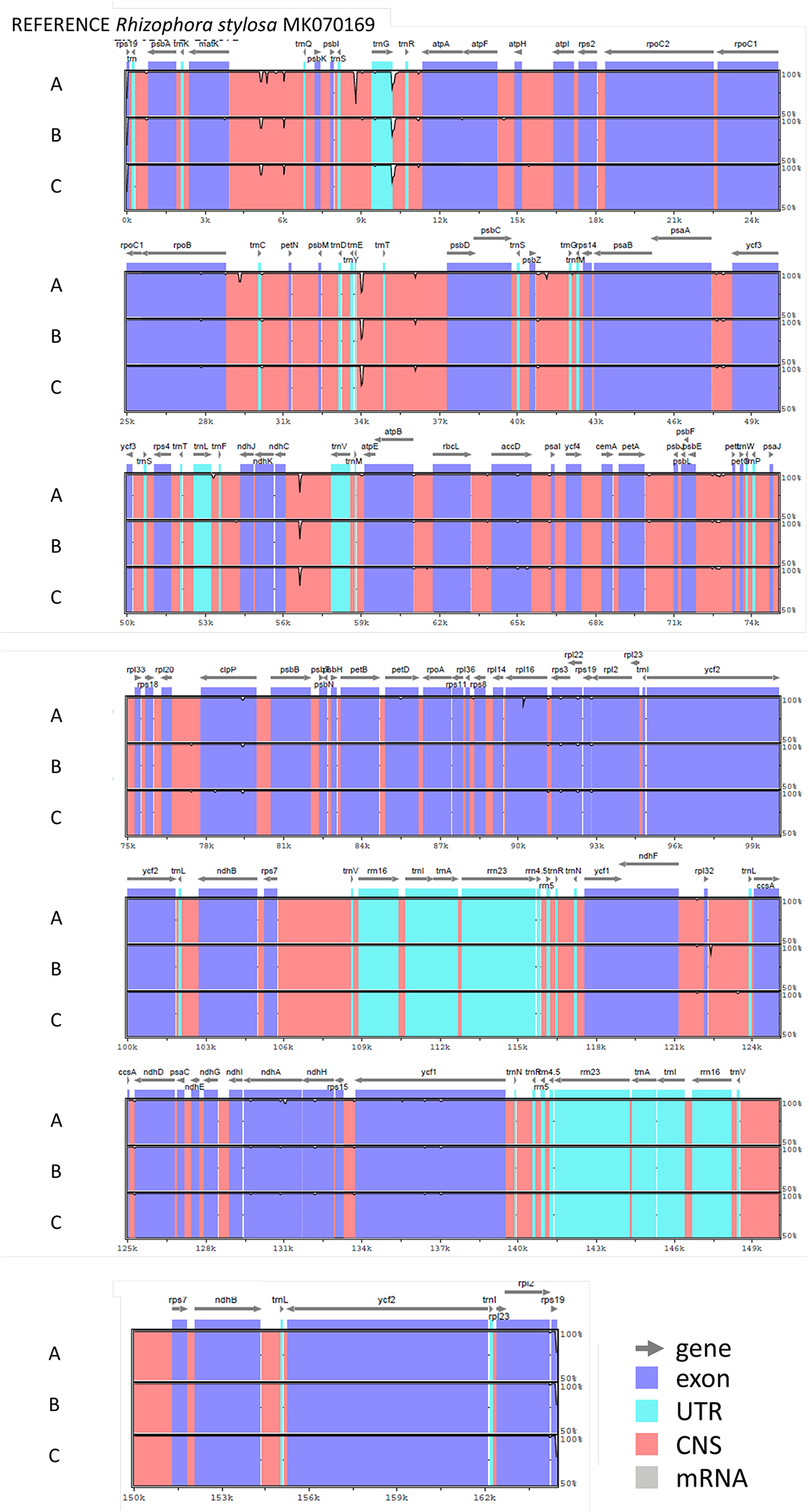
Figure 1. Sequence identity plot comparing three haplogroups (A, B, and C) of Rhizophora mucronata with Rhizophora stylosa as the annotated reference genome using mVISTA. The location and orientation of the genes are indicated on the top. Exons and untranslated regions (UTRs) are in purple and turquoise, respectively. Conserved non-coding regions are in orange. Y-axis ranges from 100% (top) to 50% identity between each sequence and the reference. The order of the haplogroups used and their codes of origin from the top to bottom is: haplogroup A (MAD20, eastern Madagascar), haplogroup B (MAD17, northern Madagascar), and haplogroup C (MOZ7, southern Mozambique).
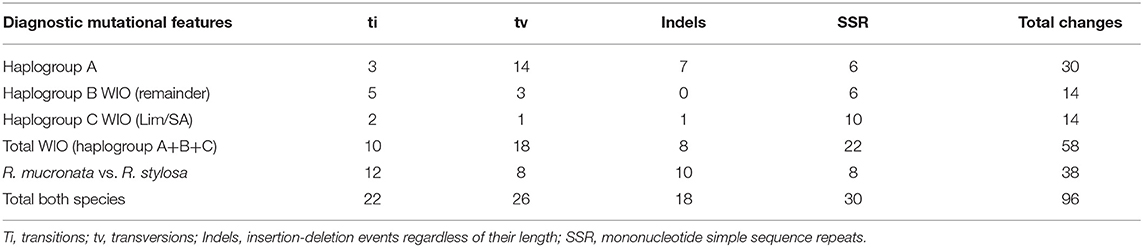
Table 3. Number of diagnostic features of mutational changes to distinguish R. stylosa and three haplogroups of R. mucronata.
Chloroplast Genome Variants of WIO R. mucronata
Within R. mucronata from the WIO, we detected 58 mutational changes, namely 10 transitions, 18 transversions, 8 insertion-deletions, and 22 mononucleotide repeats (Supplementary Table 1). These changes occurred as three distinguishable haplogroups A, B, and C. Haplogroup A with 30 unique changes (3 transitions, 14 transversions, 7 insertions-deletions, and 6 mononucleotide repeats) was mostly different from haplogroup B with 14 unique changes (5 transitions, 3 transversions, 0 insertions-deletions, and 6 mononucleotide repeats) and haplogroup C with 14 unique changes (2 transitions, 1 transversions, 1 insertions-deletions, and 10 mononucleotide repeats).
Nuclear rRNA Cistron Variants
The nuclear ribosomal cistron of 8,033 bp length was composed of an external transcribed spacer upstream (1 > 1,548, 1,548 bp), the 18S small subunit rRNA (1,549 > 3,335, 1,787 bp), the ITS region ITS1 (3,336 > 3,580, 245 bp), the 5.8S rRNA subunit (3,581 > 3,744, 164 bp), the ITS spacer region ITS2 (3,745 > 3,947, 203 bp), the 26S large subunit rRNA (3,948 > 7,321, 3,374 bp), and the external transcribed spacer downstream (7,322 > 8,033, 712 bp). This nuclear ribosomal cistron revealed a substantial number of 37 variable sites between R. mucronata and R. stylosa despite the presence of only 4 mutated positions among the 21 R. mucronata WIO samples. A combination of these four mutations (Table 4) showed one to three bases, which were not identical between populations and revealed four different sequence variants corresponding to a cistron-A (all sites of the Seychelles, East Madagascar, and Aldabra); cistron-B (the North and West Madagascar, East Africa from KEN 1 to MOZ6); cistron-C (MOZ7, SAF9, and SAF10, the South of Delagoa Bight); and a singleton cistron-B′ (MOZ8). Cistron-A featured transversions (T > C at 301 bp and A > G at 970 bp) of the externally transcribed spacer. Cistron-C shared the latter A > G transversion at 970 bp of the externally transcribed spacer and featured a unique T > C transversion at 4,728 bp of the large subunit rRNA. Cistron-B featured the abovementioned T (at 301 bp), A (at 970 bp), and T (at 4,728 bp), whereas the cistron-B′ showed a single transversion (A > G at 3,507 bp) of the ITS1 region.
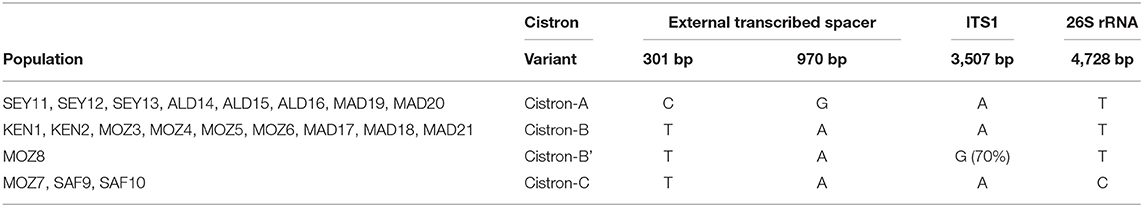
Table 4. Four mutations and their position in assembled sequences of the nuclear ribosomal cistron (8,023 bp length) providing four variants (A, B, B′, and C) of R. mucronata populations of the WIO.
Phylogeographical Analyses of Full cpDNA Genome
The Bayesian ML revealed and supported (with 100% bootstrap values) the R. mucronata from the WIO as three different haplogroups of which haplogroup A was most differentiated, as much as the difference with the outgroup R. stylosa (Figure 2). Haplogroup A contained both samples from the East Madagascar (MAD20 and MAD21) and one site on the Seychelles (SEY13, Mahé-Bas) despite possessing the divergence significantly among each other. Haplogroup B was mostly common throughout the WIO and indicated a star-like radiation of recent variants. Haplogroup C was rare and occurred in the southern part of the African coast though it was well-supported as a separate lineage.
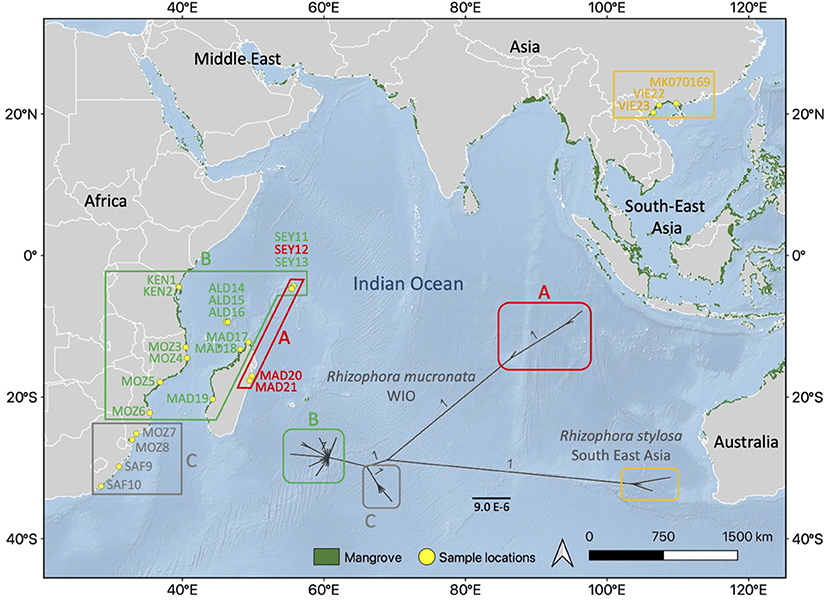
Figure 2. Maximum likelihood (ML) tree showing phylogenetic relationship between the three haplogroups (A, B, and C) of R. mucronata with R. stylosa as an outgroup using chloroplast genome (1 refers to 100% support). Similar colors indicate geographical range of each chloroplast haplogroup within the Western Indian Ocean (WIO) region. The background map was created by the QGIS 3.10.10 software (www.qgis.org), using detailed (1:10 m) ocean bottom relief, land, and island polygons, as well as country administrative boundaries provided by Natural Earth (www.naturalearthdata.com). Mangrove distribution data for 2016 from the Global Mangrove Watch (Bunting et al., 2018).
A haplonetwork was concordant with the ML tree in terms of the same samples clustered as three haplogroups but incorporated all mutational changes and indicated all stepwise modifications within R. mucronata (Figure 3). Haplogroup A seemed to be most divergent from all other samples due to transversions (Table 3), whereas haplogroup C was separated mainly by mononucleotide microsatellite repeats (Table 3).
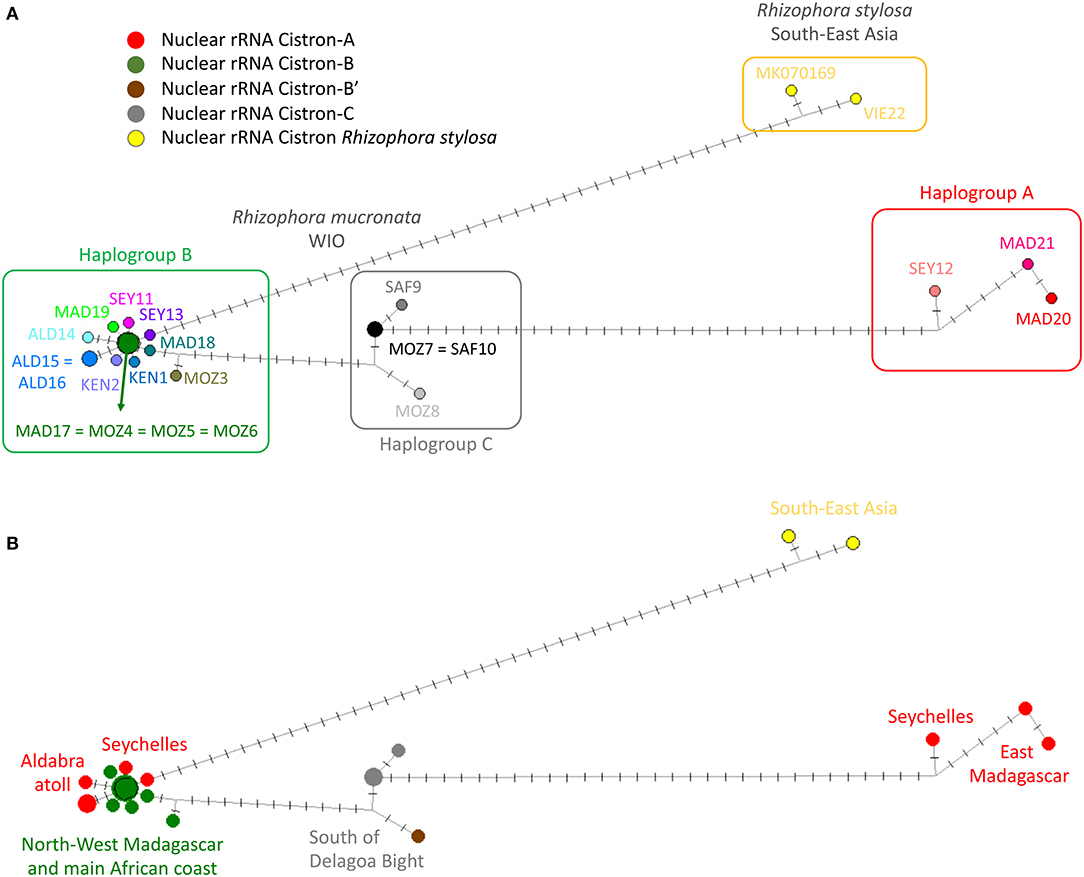
Figure 3. Haplotype network showing mutational steps between the three haplogroups (A, B, and C) of R. mucronata with R. stylosa as an outgroup using chloroplast genome. The length of branches and number of crossed bars are proportional to the number of mutational steps between all samples. (A) Three haplogroups and all haplotypes encountered in populations (colors as in Figure 5A); (B) Nuclear ribosomal RNA (rRNA) cistron variants superposed on the haplotype network showing an admixture on the Seychelles and Aldabra (colors as in Figure 5B).
Within haplogroup B, a common haplotype was found in northern Madagascar (MAD17) and shared with locations along the East African mainland coast (MOZ4, MOZ5, and MOZ6). The star-like network indicates that on several occasions only one or few mutations were derived from this common variant (Figure 3). Haplogroup B was mostly widespread with representatives on the Seychelles, Aldabra, northern and western Madagascar, and along a major part of the African coast (Figures 1, 3). On the Seychelles, the populations on Mahé having haplogroup B (SEY12) and the one with haplogroup A (SEY13) were spatially separated on either sides of the island. SEY12 (Grand Anse) was oriented/exposed to the West and SEY13 (Mahé-Bas) to the East. The isolated SEY14 (Isle Curieuse) differed in a transition and a mononucleotide repeat from SEY12 (Figure 3). Aldabra populations showed two lineages that were spatially segregated in the western part (ALD15) vs. the eastern part (ALD16 and ALD17) of the inner lagoon of the atoll. These haplogroup lineages accumulated six mutational changes. Haplogroup C was restricted to the populations of the southern part of Africa (south of Delagoa Bight) and was similar over the largest distance between MOZ7 and SAF10, indicating recent migration events from source populations in the Delagoa Bight (Figure 3). MOZ8 differed in four mononucleotide repeats and SAF9 in one transition from the putative common haplotype (Figure 3). The ML phylogenetic tree of the same samples but considering only 23 mutated positions of coding sequences gave a similar outcome when compared to the abovementioned analysis using all 58 mutational changes regardless of their position along the chloroplast genome. Three major clades corresponding to the abovementioned haplogroups A, B, and C were obtained. Haplogroup A was well-separated and consisted of East Madagascar (bootstrap value 97) and Seychelles (bootstrap value 95) samples. Haplogroup B (most common throughout the WIO; bootstrap value 69) and haplogroup C (four sites south of Delagoa Bight; bootstrap value 69) appeared as separate lineages (Supplementary Figure 2). The overall mean distance was 0.20 (including R. mucronata and R. stylosa) and 0.11 for WIO R. mucronata samples.
Estimation of Demographic History (ABC Models)
For the whole WIO (21 populations), the demographic analysis (ABC1; Supplementary Information ABC1-WIO) showed a support for scenario 1 (population constancy), however, with all four scenarios having probabilities around 25% (Table 5; Figure 4). This unclear result when pooling all populations at the WIO level, however, is possible due to the cpDNA mutations, which do not reflect demographic changes but are biased by past admixture. This problem when pooling at the WIO level was created by the fact that both divergent haplogroups A and B occur on the Seychelles and Madagascar.
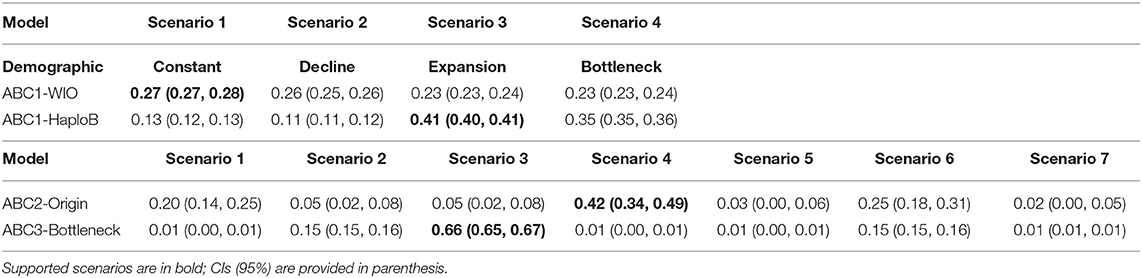
Table 5. Posterior probabilities of the tested scenarios under a demographic model at level of WIO (ABC1-WIO), at level of haplogroup B (ABC1-HaploB); under an origin model (ABC2), and a bottleneck model (ABC3).
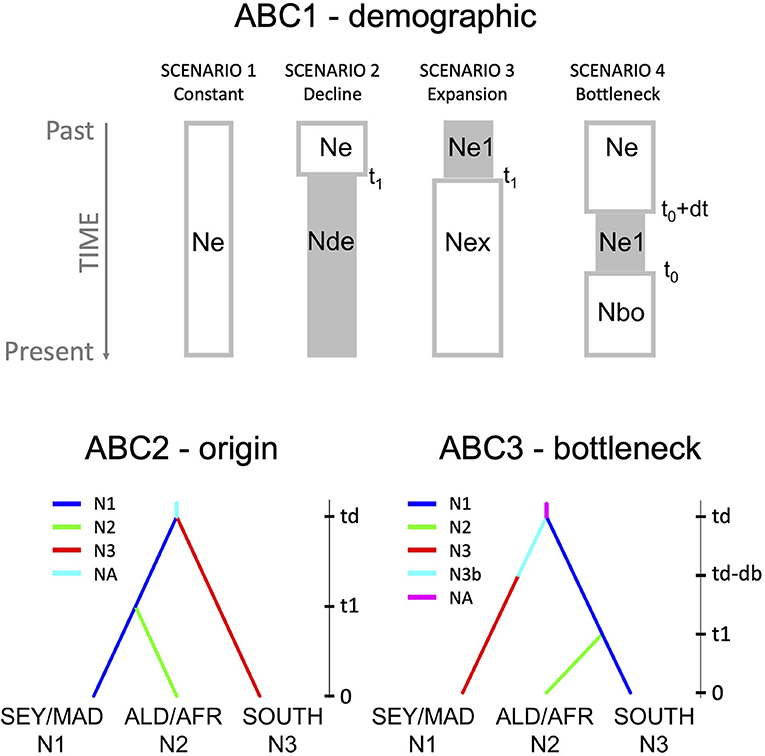
Figure 4. The design of demographic tests (ABC1) applied on the whole WIO region and on the haplogroup B for the four scenarios of population constancy, decline, expansion, and expansion after bottleneck; supported scenario of origin model (ABC2) and bottleneck model (ABC3) for biogeographical groups N1 (the Seychelles and Madagascar), N2 (Aldabra and African mainland coast), and N3 (populations in the south of Delagoa Bight) using DIYABC. Posterior probabilities are given in Table 4. An overview of schemes and a comparison of all scenarios, the PCA of prior and posterior distribution of the selected scenario, the posterior distribution of parameters, and the posterior value of summary statistics of supported scenario are provided as Supplementary Information.
A more appropriate demographic analysis (ABC1; Supplementary Information ABC1-haploB) restricted to 14 populations of haplogroup B, thereby avoiding the abovementioned bias, supported a scenario involving a population expansion (posterior probability of 41%) and rejected scenarios involving population constancy, decline, or bottleneck (Table 5; Figure 4). The expansion of haplogroup B at t1 was estimated at a generation time of 1,970 (mode) to 3,240 (mean) with posterior distribution of effective sizes of ancestral Ne = 10 and current effective size N1 = 20,000 (Supplementary Information ABC1-haploB). The expansion of haplogroup B aligns with the finding of a star-like haplonetwork and few mutational changes involved. The number of samples within haplotype A and haplotype C was too small to verify their demographic model.
Among the seven origin scenarios (ABC2), scenario 4 (Table 5; Figure 4; Supplementary Information ABC2) was best supported (posterior probability of 42%) with populations considered under the following biogeographical groups: (N1) the Seychelles and Madagascar; (N2) Aldabra and African coast; and (N3) south of Delagoa Bight. The outcome suggests an oldest origin of the Seychelles and Madagascar (N1) with the split of populations located south of Delagoa Bight (N3) estimated at td = 52,100 (mode) to 58,300 (mean) generations, whereas a 5-fold younger split of N1 with the populations of Aldabra and East African mainland coast (N2) was estimated at a generation time t1 of 11,100 (mode) to 16,700 (mean) with posterior distribution of effective sizes of ancestral NA = 98,700 and current effective size N1 = 94,300, N2 = 41,700, and N3 = 12,500 (Supplementary Information ABC2). Current effective sizes are different in ABC1 and ABC2, giving higher estimates of current effective sizes and larger generation time in ABC2. The expansion scenario of haplogroup B as supported by ABC1 included the populations from both biogeographical groups N1 and N2 as considered under the origin model ABC2.
The bottleneck model analysis (ABC3; Supplementary Information ABC3) with seven scenarios of either one, two, or three bottlenecks in the biogeographical groups N1, N2, and N3 (cf. ABC2 outcome) indicated that a bottleneck of the haplogroup C (populations in the south of Delagoa Bight) stood out for its best fit to the observed data with a posterior probability of 66% (Table 5; Figure 4). The outcome suggests a bottleneck origin (i.e., then followed by expansion) of the populations in the south of Delagoa Bight (N3) estimated at td-db = 18,700 (mode) to 39,300 (mean) generations, about two times as long as the split of N1 with the populations of Aldabra and East African mainland coast (N2) that was estimated at a generation time t1of 9,480 (mode) to 16,800 (mean) with posterior distribution of effective sizes of ancestral NA = 98,700 of bottleneck N3b = 1,970 and current effective size N1 = 97,900, N2 = 43,600, and N3 = 24,800 (Supplementary Information ABC3).
As a whole, the approach of narrowing down ABC models subsequently has indicated that the demography of haplogroup B lineages, widely distributed on the WIO islands and a large part of the African coast underwent a range expansion, and this represents a most recent lineage. The outcome of origin models clearly suggests an oldest origin of the Seychelles and Madagascar (N1) with the divergence of populations located south of Delagoa Bight (N3) estimated at t = 52,100–52,800 (mode in ABC2 and ABC3) generations, followed by a bottleneck period of the latter haplogroup C, and this prior to the period of divergence and expansion of the haplogroup B toward Aldabra and the East African mainland coast (N2). The split of N2 from N1 occurred in <5-fold at t = 9,480–11,100 (the mode as obtained in ABC2 and ABC3). As mentioned earlier, this split divergence was followed by the recent range expansion of haplogroup B (N2 + part of N1), estimated at t = 1,970 and a current Ne = 20,000 (ABC1) when considering the distribution of haplogroup over the WIO (thus also including the Seychelles and Madagascar populations with their variants of haplogroup B). Current effective population sizes of the Seychelles and Madagascar populations (N1, containing haplogroups A and B variants) are about two times larger than the Aldabra Atoll and African coast populations (N3, only haplogroup B variants) and four times larger than the populations in the south of Delagoa Bight (N2, only haplogroup C variants).
Discussion
In this study, we used the full genome cpDNA sequences as an ultimate source of mutational changes to unravel the maternal dispersal history of R. mucronata on WIO islands and along the East African mainland coastline. Prior to this approach, a previously mentioned null hypothesis of a single evolutionary unit was already rejected based on nuclear microsatellite-based connectivity patterns and genetic breaks (Triest et al., 2021b) such that a search for maternally inherited markers would give complementary information. The genomic cpDNA variants revealed three major haplogroups and many haplotype variants though there were also fully similar >164,000 bp lengthy sequences between distant populations. Such the highest possible resolution could be ensured because we considered all detectable mutations (transitions/transversions, insertions-deletions, and mononucleotide repeats). A phylogeographic analysis elucidated novel aspects on the historical processes of different haplotype lineages. ABC model testing and divergence time estimation revealed discrete origins of biogeographical entities on continental coasts and around remote islands. High-quality full chloroplast sequences delivered ample mutational changes that aid to reconstruct LDD routes, which are basically aligned with contemporary ocean currents. From all these evidence, an overall conceptually integrated connectivity pattern could be proposed (Figure 5), thereby raising new hypotheses. The characteristics of the ocean surface circulation of the WIO region and the detectable genetic patterns of microsatellite diversity have been discussed previously in much detail (Triest et al., 2021b). Hence, for those hydrodynamic aspects, we refer to Triest et al. (2021b) such that we can focus here on the findings that are in agreement between microsatellite and the current cpDNA genome analyses. We will highlight these cpDNA genomic findings that are novel and bring clarity in any previous interpretation on R. mucronata demographic history and LDD routes in the WIO.
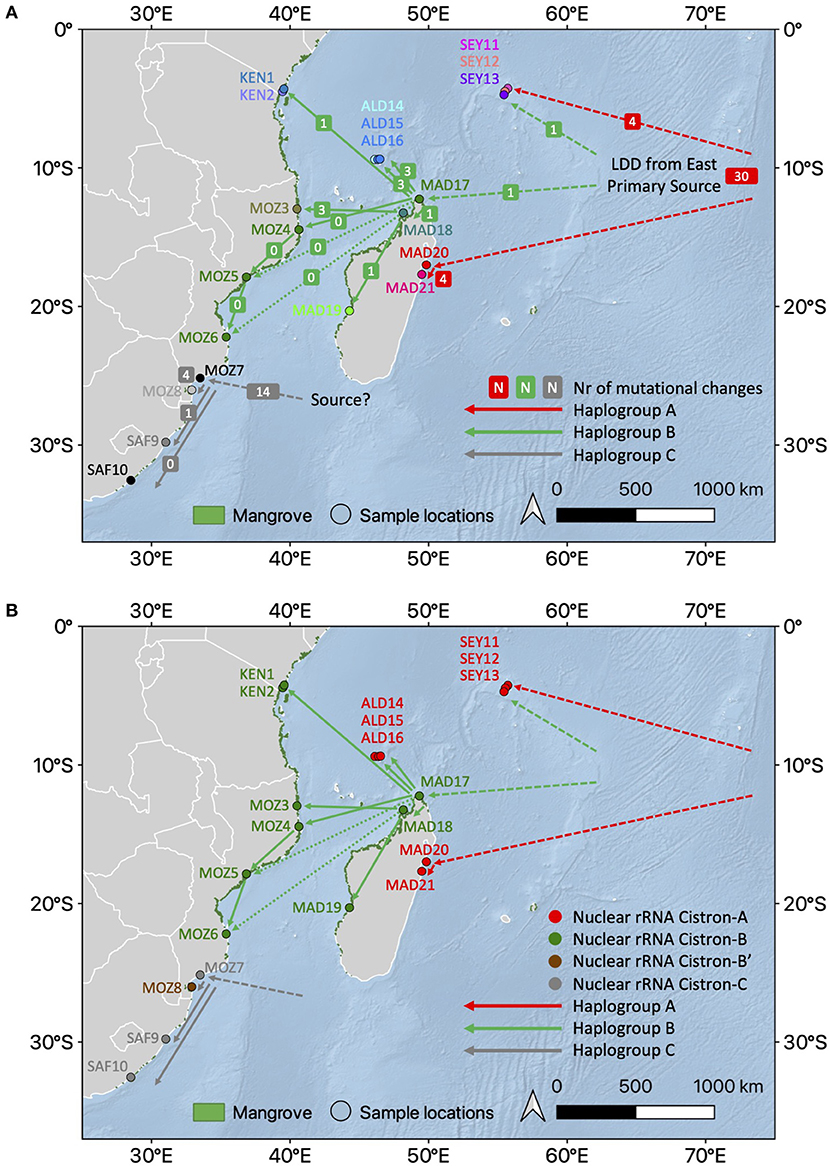
Figure 5. Conceptual and integrative connectivity scenario based on haplotype network and prevailing ocean currents between R. mucronata populations. (A) Haplogroups A, B, and C with an indication of the number of mutational steps involved between haplotypes of the populations. Note the similarity of haplogroup B from northern Madagascar and the African mainland coast populations. Dashed lines refer to alternative or unknown routes (colors as in Figure 3A). (B) Nuclear rRNA cistron variants superposed on the haplotype network showing an admixture on The Seychelles and Aldabra (colors as in Figure 3B). The background map was created using the QGIS 3.10.10 software (www.qgis.org), using detailed (1:10 m) ocean bottom relief, land, and island polygons, as well as country administrative boundaries provided by Natural Earth (www.naturalearthdata.com). Mangrove distribution data for 2016 from the Global Mangrove Watch (Bunting et al., 2018).
Overall, a haplonetwork, a Bayesian ML tree, and the approach of narrowing down ABC models subsequently indicated that the demography of haplogroup B lineages, widely distributed on the WIO islands and a large part of the East African mainland coast, undergoes a recent range expansion estimated at t = 1,970 generations. When considering a generation time of 10–20 years for Rhizophora (though overlapping generations occur), this very likely refers to 1,000 of years ago corresponding to the post-Last Glacial Maximum (LGM) period and Holocene for the settlement of vast areas of Rhizophora mangroves along the western Madagascar, the East African mainland coast, and islands in the MCA region, following a rise in the sea level. Origin models pointed out that some populations of the Seychelles and Madagascar were most ancient, whereas bottleneck models strongly supported a divergence of haplogroup C populations, the south of Delagoa Bight, estimated at t = 52,100–52,800 generations, hence in the pre-LGM period of Pleistocene glacial–interglacial cycles. A further divergence of haplogroup B from haplogroup A occurred within about <5-fold at t = 9,480–11,100 generations, referring to a period during the LGM (when considering the generation times of 10–20 years). From the Pleistocene until the LGM period (100,000–20,000 years ago), the Cargados Carajos Shoals (Hansen et al., 2017) might have served as an important refugium for the settlement of propagules arriving from the east along with the SEC (Australia). We hypothesize that haplogroups B and C might have encountered this elevated archipelago and land mass, whereas haplogroup A is supposed to be even older (cf. complex of R. mucronata with R. stylosa) and experiences the Pleistocene climatic cycles and associated sea level fluctuations.
An ABC analysis previously conducted on microsatellite data (Triest et al., 2021b) was in fact done mainly for populations of haplogroup B (as we can document from the current study) except for the two southern populations of haplogroup C (and a mixed A+B on the Seychelles), and therefore the unidirectional stepping-stone model with northern Madagascar and the Seychelles as ancestral divergence still holds. In addition, even older lineages of haplogroups C and A compared to this common and widespread haplogroup B were revealed in the current study. The ML analysis indicated a nearly large divergence of haplogroup A with haplogroup B than from the nearest species R. stylosa.
Relationship of R. mucronata in WIO With R. stylosa
Rhizophora mucronata has a sympatric distribution with R. apiculata and R. stylosa in the Western Pacific. The phylogenetic relationship between R. stylosa and R. mucronata remains unresolved whereas a R. apiculata clade is well-supported (Lo et al., 2014; Chen et al., 2015; Ng et al., 2015; Takayama et al., 2021). Large-scale Rhizophora genetic studies considered only a limited number of samples from the WIO region (Lo et al., 2014; Yan et al., 2016; Takayama et al., 2021) and indicated a large similarity of WIO and EIO R. mucronata for a fairly limited number of nuclear or chloroplast genes used. Rhizophora stylosa and R. mucronata apparently are very related in their nuclear genome because full sets of microsatellite loci could be cross-amplified and revealed polymorphism (Wee et al., 2015; Do et al., 2019). Complete R. mucronata chloroplast genome sequences (164,474 bp) assembled in our study showed no differences with R. stylosa chloroplast genome (from Genbank) in the boundaries, junction point, sequence orientation, and stretch between LSC/IRb/SSC/IRa/LSC. Even though the total gene content, gene arrangements, and GC content among the studied Rhizophora cpDNA genomes were similar, 38 diagnostic mutational changes between both species, encompassing all transitions/transversions, insertions-deletions, and mononucleotide repeats revealed that R. stylosa is not phylogenetically very distant from the R. mucronata haplogroups. A hypothesis of mixing–isolation–mixing (MIM), rather than allopatric speciation during isolation as raised by He et al. (2019), could as well be interesting to explain this albeit close relationship between R. mucronata and R. stylosa, maintained over their large sympatric distribution area. Speciation of the three Rhizophora species from the West Pacific and EIO occurred within an estimated divergence time of 1.20–6.44 Ma. The divergence time of evolutionary significant units (ESU) of the WIO is thus expected to be younger and indicated by the cpDNA genome divergence of R. stylosa with R. mucronata in the WIO that is only about as large as for the different ESU (haplogroups A, B, and C) within R. mucronata. Rhizophora mucronata exhibits low levels of nuclear microsatellite diversity throughout its range despite the separation of WIO populations from other regions (Wee et al., 2015).
Evidence of an origin of WIO mangrove populations lying in the EIO and brought along with the SEC from Australia or Southeast Asia is increasing. The potential LDD routes of R. mucronata could be proposed between Northern Australia and East Africa (Lo et al., 2014). Comparably, microsatellites grouped individuals from Australia and East Africa apart from Asian populations (Yan et al., 2016). On the other hand, cpDNA haplotypes of X. granatum indicated similar sequences in populations of Southeast Asia and the WIO (Tomizawa et al., 2017). However, one must remain critical because the occurrence of similarity in a few chloroplast genes is not an indication of LDD, thereby omitting all possible mutational changes of the introns. Similarity can only be proven by the comparison of a full chloroplast genome. Combined conservative cpDNA sequences (trnG-trnS and trnH-rpl2 introns of total 1,395 bp length) and nuclear ITS data indicated a deeper Australian origin of R. mucronata (Lo et al., 2014). We verified these cpDNA genes for our samples of the WIO, and these showed three diagnostic indel differences for R. stylosa and two indels for haplogroup A but were invariable for haplogroups B and C.
The question remains open whether three (or more if yet undetected) dispersal and establishment events happened for the haplogroups A, B, and C, and in which time frame. We recommend testing of this hypothesis of an Australian origin of R. mucronata haplogroups, thereby illustrating the young nature of WIO Rhizophora mangroves in comparison to Rhizophora from other ocean basins. Our ABC analysis supported a history of discrete invasions during pre-LGM times for the most ancient haplogroup A. This lineage was in fact rare because thus far only detected in a single site on the Seychelles and two sites within close vicinity on East Madagascar. Based on nuclear microsatellites, we recently hypothesized the existence of at least two separate ancestral gene pools, namely on the granitic Seychelles and on northern Madagascar (Triest et al., 2021b). Unfortunately, we had no sufficient samples from East Madagascar for conducting microsatellite studies. Nonetheless, our current study clarified that these gene pools are associated with the maternal haplogroups A (Seychelles) and B (northern Madagascar). Haplogroup A seems to be most divergent from all other samples due to a large proportion of transversions as substitutions and should be regarded as a separate ESU given that both nuclear and cytoplasmic genetic information are concordant.
One may question whether the use of all mutated positions or the position only restricted to coding genes would be preferred because of the presence of plenty microsatellite regions. The Bayes ML tree obtained for either the full genome or for coding sequences only was similar for R. mucronata of the WIO. Chloroplast microsatellites, or simple sequence repeats (cpSSRs), are typically mononucleotide tandem repeats, and these markers were proven as useful genetic markers in gene flow analysis and population genetics (George et al., 2015). Nevertheless, homoplasy may lead to the overestimation of relatedness when samples share the similar fragment sizes despite experiencing different evolutionary histories. The latter may be detectable in the underlying sequence, a problem of which the severity will be the greatest at higher taxonomic levels and in large-scale phylogeographic studies (Provan et al., 2001). Size homoplasy in cpSSR loci can be troublesome but does not hamper its usefulness when the underlying sequence of a microsatellite locus can be determined, then the deviations from the standard assumptions of mutational patterns can be taken into account (Wheeler et al., 2014). Obviously, criticism about the use of cpSSR is raised when this type of marker solely is used, when only fragment lengths instead of sequences are considered, and when applied for phylogeny at the species level and over large distances. In our R. mucronata chloroplast genome study, the microsatellite variations (22 mononucleotide repeats of 58 variable sites) were plenty and usually found in the non-coding regions despite their presence in a full sequenced and verified form for their true length variance from read coverages in the assemblage of contigs. Based on our analysis, we adhere the opinion that, at the population level within the same species, cpSSR is informative and that minor mutational steps in mononucleotide repeat number should be considered. In complete genomes, these cpSSR do not stand alone as markers but are always to be used in combination with substitutions and indels. One should remain cautious if ancient demographic events or speciation is involved such that mononucleotide repeats might become an issue and show homoplasy; however, detecting recent historical demographic events will clearly benefit from cpSSR phylogeographic information.
Recent LDD Across MCA of haplogroup B
A star-like haplonetwork referred to an expansion of haplogroup B from northern Madagascar toward Aldabra and substantially along the East African mainland coastline. A genetic break between Aldabra and northern Madagascar as revealed from nuclear microsatellites (Triest et al., 2021b) was also apparent from the haplonetwork showing the largest number of mutational steps of cpDNA variants between Aldabra provenances and MAD17 in comparison to the most other haplotype B variants, except for MOZ1. It can be hypothesized that populations with increased mutational changes have migrated earlier, and this would make sense because both regions, namely Aldabra and northern Mozambique (MOZ1), are in close vicinity to northern Madagascar when crossing the MCA. We previously also suggested that the Aldabra Atoll was populated from nearby sources on northern Madagascar (Triest et al., 2021b), which now could be confirmed from their position in the star-like haplonetwork of haplogroup B.
The historical role of northern Madagascar as a secondary source area for dispersal and rapid expansion throughout the main part of the WIO could have been more pronounced in the post-LGM period with a rise in the sea level and the flooding of the extended shelf area along northern but even more western Madagascar. These patterns of inferred connectivity could align with the directionality of major ocean currents. We hypothesize that historical mangroves along this shelf area comprised a much more important source area for propagule dispersal across the MCA compared to the contemporary engulfed bays. Transportation across the MCA was estimated to take only 19–30 days (Hancke et al., 2014), which roughly is within a month and within the reach of reported buoyancy and viability periods of R. mucronata propagules (Drexler, 2001).
The population genetic structure and estimates of gene flow and migration directionality of R. mucronata in the WIO using nuclear microsatellites revealed several genetic breaks and patterns of connectivity aligned with the direction of major ocean currents (Triest et al., 2021b). Along the East African mainland coast, the bifurcation of the SEC separated an allelically more diverse region (East Africa) from the MCA. The most diverse region of nuclear microsatellite allelic richness was observed in East Africa (Kenya to Mozambique) (Triest et al., 2021b). Such a diversity is reflected also in the number of cpDNA variants (a star-like network), which allows to hypothesize that multiple/repeated migrations toward the East African mainland coast experienced a most direct effect of the SEC oriented toward the “core region” of mangrove populations in the WIO. The genetic break found with nuclear microsatellites along the East African mainland coast and caused by a bifurcation of the SEC sorting out different cohorts of migrants, could also be confirmed with cpDNA in this study, namely the single variant toward the south of the SEC bifurcation. This can be explained and interpreted as multiple dispersal events over time that accumulate allelic diversity in nuclear microsatellites but having similar maternal cpDNA when arriving from the same source area (or have arrived in historical times from northern and western Madagascar) sharing a common cpDNA haplotype.
A single cpDNA variant of haplogroup B spanned more than 1,800 km from northern Madagascar further across the MCA and meaningfully reflects most recent migration events through LDD with the range expansion. The presence of exactly or nearly similar full genomic sequences of the haplogroup B between northern Madagascar and a large stretch of the East African mainland coastline evokes that geographically distant populations across the MCA show a pattern deviating from IBD due to recent or repeated dispersal of Rhizophora propagules. As a comparable phenomenon, the population decline of R. mangle around early Holocene followed by the recent range expansion was indicated by low genetic diversity over hundreds of kilometers along each side of the Central American Isthmus (Cerón-Souza et al., 2015). The detection of similar haplogroups over large distances also may explain why no evolutionary signal in RST (i.e., considering differentiation from repeated mutational steps) of nuclear microsatellites was found in the WIO (Triest et al., 2021b). The latter was interpreted as an indication of recent colonization of populations in the WIO. Based on microsatellites, we emphasized that a genetic connectivity from the eastern part of the MCA toward Mozambique could not be detected with certainty but only indirectly inferred (Triest et al., 2021b) though from our current study we can confirm such recent colonization based on fully identical (or only 1–3 mutational changes) haplotypes. As a whole, the MCA features poorly differentiated populations that share a same gene pool for nuclear microsatellites (Triest et al., 2021b) as well as complete chloroplast sequences that can be maintained over long distances by a generally southward flowing current along the Mozambican coast (Hancke et al., 2014). Migration models of Rhizophora and Avicennia species usually indicated a unidirectional stepping-stone model over large distance (Wee et al., 2020; Triest et al., 2021a,b,c).
Based on microsatellite nuclear markers, an ABC simulation indicated that R. mucronata populations from the East African mainland coast were more recent than those of the islands (Madagascar and Seychelles) with divergence times of the former roughly estimated to have occurred at t = 999–1,980 generations, very likely within the Holocene (Triest et al., 2021b). Recent history of R. mucronata along the East African mainland coast certainly holds and was confirmed from cpDNA genome sequences. The demographic ABC simulation supported a recent range expansion of haplogroup B estimated at t = 1,970 generations and a current Ne = 20,000 over the WIO. This concordance between nuclear microsatellites and genomic chloroplast sequences evidences a Holocene or at least post-LGM migration of R. mucronata shaping core mangrove areas along the East African mainland coast.
Genetic Break in the South of Delagoa Bight
Populations located south of Delagoa Bight contained haplogroup C, and the ABC analysis supported a discrete origin and bottleneck migration. The nuclear microsatellite loci previously indicated an obvious genetic break located south of Delagoa Bight and featured monomorphism and strong inbreeding (Triest et al., 2021b). These microsatellites also revealed that kinships dropped to zero between populations on either sides of the Delagoa Bight and in fact did belong to distinct gene pools of which populations located south of Delagoa Bight exhibited high inbreeding levels (Triest et al., 2021b). Previously, we explained this genetic break between populations based on a jet current passing the Delagoa Bight without entering it (Hancke et al., 2014) and based on a nearly permanent lee eddy in the area (Quartly and Srokosz, 2004) that may retain propagules. However, our current findings shed another light on the demographic history of this southernmost gene pool as a discrete haplogroup C with evidence of a different and an older origin. The contemporary hydrography of the region is still considered to keep this gene pool spatially separated. We need to revise our idea that this southern gene pool was assigned to a separate cluster due to the higher inbreeding levels of less variable microsatellite loci. More importantly, the ABC bottleneck model strongly supported a bottleneck origin of these populations, and this is concordant with their large number of fixed loci, high inbreeding levels, and lower allelic richness of nuclear microsatellites. Whereas, haplogroup A appears as most divergent from all other samples in its transversions, the haplogroup C genome differs mainly in mononucleotide microsatellite repeats. It was raised that fast-evolving nuclear SSR markers could be more suitable for detecting the contemporary patterns of genetic structure and not for vicariant groups that diverged (Yan et al., 2016). However, when using full cpDNA genome, these contain slow- as well as fast-evolving regions. Indeed, we found that mononucleotide repeats and indels largely gave information as shaped from recent dispersal events within a particular oceanic region such as near this southern part of Africa.
Migration models of Rhizophora and Avicennia species usually indicate a unidirectional stepping-stone model (Wee et al., 2020; Triest et al., 2021a,b,c) and therefore, we launch a new hypothesis to be tested. Considering the Rhizophora populations of southern Madagascar, Reunion, or Mauritius (located at the southern part of Cargados Carajos Shoals during LGM) as potential sources of origin for this southern lineage, Haplotype C can be regarded as an ESU because of concordance of separate gene pools for both nuclear microsatellites and the complete cpDNA. We can assume that the distribution area of this ESU is limited and thus of conservation interest. Populations of haplogroup C are restricted to this southern part of Africa because of the prevailing southward coastal currents. Interestingly, when considering the Delagoa Bight population as a potential source area, only a few mutations have accumulated in the more southward located populations. Namely, a few mutations occurred in nearby populations (MOZ8 differed in three mononucleotide repeats and SAF9 in one transition from the putative common haplotype), and similar cpDNA genomes were observed over the largest distance between MOZ7 and SAF10. This indicates recent LDD migration patterns from source populations in the Delagoa Bight despite experiencing repeated bottlenecks regardless of their historical origin. However, the coastal landform along this coastal stretch of Africa is not ideal for the massive release and capture of mangrove propagules (Triest and Van der Stocken, 2021; Triest et al., 2021b). Important pressures on mangrove areas are the estuary mouth dynamics causing a change in mangrove population structure (Adams and Rajkaran, 2020). South African estuarine populations maintain high inbreeding levels because of recurrent closing of river mouths, hence lowering the chance of external propagule input (De Ryck et al., 2016) and increases in freshwater inflow and siltation (Adams and Rajkaran, 2020). Temporal interaction of river flow and tides during open-mouth conditions may be sufficient to occasionally allow external propagule establishment.
Small Islands as Unique “Filters” Witnessing Different Eras
It is sensible to assume that, compared to mainland coastal areas, drifting propagules are less likely to reach and establish on islands with relatively short coastline and limited expanses of suitable habitat, resulting in the absence of IBD due to large genetic differences over small distances (Hodel et al., 2018). On Aldabra and the granitic Seychelles, we can explain a different origin from their haplotype variants and put forward that small islands undergo only rare founder events after LDD, but that populations may persist on the island for a long time as was indicated by the most ancient haplogroup A. However, Madagascar and small islands of the Seychelles each harbored multiple haplogroups A and B though in spatially separated populations, which can be explained from a different era of migration.
Based on the multiple alleles for nuclear microsatellites of WIO Rhizophora populations, we previously argued that at least more than a single propagule reached the granitic Seychelles, possibly during different cycles of colonization (Triest et al., 2021b). This could be confirmed from cpDNA sequences. Haplogroups A and B are found on Mahé Island despite their presence in spatially separated mangroves on the different sides of the island. Unfortunately, in our previous study, we analyzed Mahé as a single pooled population, which should have been avoided. Rhizophora mucronata from Bas-Mahé and from Grand Anse represent two totally different maternal lineages (one may argue ESU's). Bas-Mahé is the ancient haplogroup A, whereas Grand Anse and Isle Curieuse (SEY14 differed in a transition and a mononucleotide repeat) are the more recent haplogroup B, which is common throughout the WIO. A potential role of the eastward flowing South Equatorial Counter Current during the winter monsoon (Schott et al., 2009) might enable the transport of propagule transport from the East African mainland mangrove populations toward the Seychelles. The populations of the granitic Seychelles seemed less distinct from the populations along the East African mainland coast than from the populations of Madagascar. Altogether, private alleles of microsatellite loci did not explain much of the obtained genetic structure that was rather shaped by differences in shared allele frequencies or by elevated inbreeding levels (Triest et al., 2021b). From genomic cpDNA sequences, we cannot be conclusive about this possibility of eastward “mainland-to-island” migration because of the star-like network that indicates very recent radiation for all sites except Aldabra. Whether the Seychelles populations on Mahé (Grand Anse) and Isle Curieuse originate from direct and parallel westward SEC input of haplogroup B propagules during a same era as northern Madagascar populations, or whether these arrived via the eastward flowing South Equatorial Counter Current cannot be concluded. Based on the star-like network, we could also postulate Madagascar as a provenance if one assumes that ocean currents could reach the Seychelles at the advent of post-LGM, at that time different from contemporary ocean currents.
Our study revealed a strong genetic barrier between populations of the Aldabra Atoll and the East African mainland, but with a less pronounced barrier between populations of the Aldabra Atoll and northern Madagascar. We previously suggested that the Aldabra Atoll was populated from nearby sources of northern Madagascar (Triest et al., 2021b), which now could be confirmed from their position in the star-like haplonetwork of haplogroup B. However, despite the relatively short distance of 420 km in Madagascar, Aldabra populations showed two well-separated lineages that also were spatially segregated in the inner lagoon of the western part (ALD15) vs. the eastern part (ALD16 and ALD17) of the atoll. The Aldabra Atoll experienced a minimum of two separate invasions unless an alternative explanation would be that a single invasion event happened and locally evolved separately as “unreachable” cohorts within the atoll. Each mangrove area within the atoll appears to be isolated given the larger number of mutational changes. The elevated outer rim of the atoll is contemporary unfavorable for colonization. One may assume that at periods of higher flooding (e.g., During warmer Atlantic period of Holocene?) the lagoon could be entered occasionally, whereas most of the time the outer rim poses a barrier. Biogeographical “barriers,” therefore, can be more permeable than previously thought and act as filters rather than impenetrable barriers (Lessios and Robertson, 2006).
Substantial differences in maternal cpDNA of populations found within close vicinity on the Aldabra atoll indicate the absence or restriction of propagule flow despite their similarity in nuclear microsatellites (Triest et al., 2020). This could be explained from a local wind-mediated pollen flow, thereby mixing the nuclear gene pool. In red mangroves, the pollen-mediated transmission of genetic material was considered as an important factor for connectivity over long distances due to wind pollination (Hodel et al., 2018). Likewise, on the granitic Seychelles, the pollen flow on Mahé Island could be responsible for connecting nuclear genetic diversity despite large differences in their maternal cpDNA referring to either haplogroup A or B. Future research should include more samples of each population to either confirm such a separation or reveal a mixture of cpDNA haplotypes.
Comparison of cpDNA and Nuclear rRNA Evidence
Overall, the nuclear ribosomal cistrons matched well to the chloroplast haplogroups despite revealing the following extra information when combining both markers: (1) The nuclear markers of all Seychelles and Aldabra populations resembled those of East Madagascar, revealing a homogenization of the nuclear ribosomal cistron. An admixture of R. mucronata populations on the Seychelles and on Aldabra thus have occurred, whereas the traces of their maternal chloroplast divergence and origin were kept. One may assume a chloroplast capture of haplogroup B on Aldabra, thereby keeping the nuclear genome cistron-A. The ancient cpDNA haplogroup A in combination with nuclear cistron-A was most restricted and detected in only one population of the Seychelles, whereas the other two Seychelles populations showed an admixture comparable to Aldabra atoll populations, namely chloroplast capture of haplogroup B despite maintaining a nuclear cistron-A. This provides another interesting perspective, namely, that both the Seychelles and Aldabra were colonized more than once at different time periods and that the R. mucronata propagules on Aldabra arrived from the similar sources as (or directly from) East Madagascar and the Seychelles as shown by their shared nuclear ribosomal cistron-A; (2) The nuclear cistron-A was only detected on the WIO islands, not (yet) along the African mainland, indicating a low probability to spread from eastern Madagascar or the Seychelles and Aldabra toward the continent. (3) Both the nuclear cistron-B and chloroplast haplogroup B were encountered on northern and western Madagascar and on a large part of the African coastline from Kenya to central Mozambique, confirming their shared origin and possibility of expansion. (4) South of the Delagoa Bight, the cistron-C was uniquely combined with haplogroup C in three southern African populations, thereby confirming the unicity and separate origin of R. mucronata in that region. However, an admixture of the nuclear cistron-B′ (very similar to the common cistron B along African coast) with a maternal haplogroup C (similarly as found in the other three southernmost populations) was noted for MOZ8. The verification of the reads for the latter MOZ8 revealed a mixture of G (70%) and A (30%) pointing at an incomplete homogenization of that ITS variant across the nuclear rRNA tandem repeats. Connectivity with northward located populations thus has been possible.
We remain however critical that on the basis of our sampling design, where we considered as a first approach a single sample per site and a total of 21 populations of R. mucronata across a part of the WIO, only most obvious patterns became apparent and allowed to put forward additional and more targeted hypotheses for future research. Obtaining a full picture of diverse historical migration routes from the EIO to the WIO, and recent connectivity patterns within the WIO will require NGS-based data of both complete chloroplast genomes and the nuclear ribosomal cistron, preferably in several replicate samples per site and covering a larger geographic zone of the Indian Ocean. It is hypothesized that besides the patterns revealed from our current study that additional variants and more cases of admixture would be present in R. mucronata. Complementarity vs. admixtures of these maternal and nuclear genomic markers will also allow to better understand the allelic and genetic diversity of nuclear microsatellites at the population level. The microsatellite results of southern African populations previously were interpreted as high levels of inbreeding, however, the current uniqueness of both chloroplast and ribosomal cistron point at evidence of a different origin instead of merely a lack of heterozygous conditions (Triest et al., 2021b). This is a first report of the full-length nuclear ribosomal cistron of R. mucronata and will offer opportunities to be widely utilized for identification, taxonomic relationships, admixtures, hybrids, and phylogenies in Rhizophora species of oceans worldwide.
Conservation Relevance of a Common and Widespread Species
Even though R. mucronata has to be regarded as a widespread common species throughout the WIO and a historically recent colonization in comparison to the EIO, this species shows unique and discrete units as evidenced from those three chloroplast genome haplogroups and variants therein, and from nuclear microsatellites such that we may propose to introduce the concept of ESUs especially on the islands and on Madagascar. The evidence of a recent and widespread ESU gene pool with—most likely repeatedly—migration over long distances from northern or western Madagascar across the MCA and along the African coastline itself should not be the only criterion for regarding R. mucronata as a common species without conservation status. On the contrary, in this study we documented on a rare and ancient ESU haplogroup A to consider on the Seychelles and East Madagascar (phylogenetically as different from the common haplogroup B than from R. stylosa) that are hypothesized to be remnants from Pleistocene colonization events that happened at lower sea levels and an exposed Cargados bank ridge and archipelago that expanded from the granitic Seychelles southwards until La Réunion (Hansen et al., 2017). Therefore, it would be worth to investigate Rhizophora from East and South Madagascar, La Réunion and Mauritius, and verify its status as either of the haplogroups A, B, C, or even a different one. Additionally, Rhizophora from the southern part of Africa should also be regarded as a separate ESU and the high level of inbreeding maybe not only associated with past bottleneck events but might also be related to a different flowering or pollination strategy that allows more selfing in this deme. Understanding the physical and biological processes that structure their dispersal and connectivity patterns is relevant not only because such knowledge provides insights into the ecology and biogeographical ranges but also can be used to document on local conservation (Balbar and Metaxas, 2019), as genetic diversity and unicity may offer the potential of local adaptation. Rhizophora mucronata is not threatened nor declining though the existence of different evolutionary units within close vicinity on islands or along different stretches of a coastline warrants attention for mangrove restoration and management, as Rhizophora species due to the great ease of propagule collection and planting are most frequently considered in reforestation initiatives. Our findings shed another light on the origin and dispersal routes of R. mucronata lineages that have shaped their contemporary populations throughout major regions of the WIO, which can be the important information for defining marine conservation units, at large spatial scales as well as at the level of small islands.
Data Availability Statement
The original contributions presented in the study are publicly available in Dryad. This data of Rhizophora complete chloroplast genome sequences (fasta format) can be found at https://doi.org/10.5061/dryad.vmcvdnctd. Further inquiries can be directed to the corresponding author.
Author Contributions
LT: conceptualization (lead), data curation (lead), formal analysis (lead), funding acquisition (lead), investigation (lead), methodology (lead), resources (lead), visualization (lead), writing-original draft (lead), and writing-review and editing (lead). TS: conceptualization (supporting), data curation (lead), formal analysis (supporting), funding acquisition (supporting), investigation (lead), methodology (lead), resources (supporting), visualization (supporting), writing-original draft (supporting), and writing-review and editing (supporting). TV: conceptualization (lead), investigation (lead), methodology (supporting), visualization (supporting), writing-original draft (supporting), and writing-review and editing (supporting). All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the VUB (Grant No. BAS42), the Marie-Curie International Research Staff Exchange Scheme Coastal Research Network on Environmental Changes – CREC (EC Grant Agreement No. 247514) and a mobility scholarship from the Leopold III-fonds voor natuuronderzoek en natuurbehoud vzw. TV was supported by the EU Horizon 2020 Framework Programme for Research and Innovation under the Marie Skłodowska-Curie actions Individual Fellowship (MSCA-IF) with Grant Agreement No. 896888 (GLOMAC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors sincerely thank Rosa van der Ven (VUB), Dennis De Ryck (VUB), Hajaniaina Andrianavalonarivo Ratsimbazafy (VUB), and François Fromard (CNRS) for collecting materials.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcosc.2021.726676/full#supplementary-material
Abbreviations
ABC, Approximate Bayesian computation; cpDNA, chloroplast DNA; EIO, Eastern Indian Ocean; IR, inverted repeat region; IWP, Indo-West Pacific; LDD, long-distance dispersal; LGM, last glacial maximum; LSC, large single-copy region; MCA, Mozambique Channel Area; ML, maximum likelihood; NGS, next generation sequencing; SEC, South Equatorial Current; SSC, small single-copy region; SSR, simple sequence repeats; WIO, Western Indian Ocean.
References
Adams, J. B., and Rajkaran, A. (2020). Changes in mangroves at their southernmost African distribution limit. Estuar. Coast. Shelf Sci. 247:862. doi: 10.1016/j.ecss.2020.106862
Balbar, A. C., and Metaxas, A. (2019). The current application of ecological connectivity in the design of marine protected areas. Glob. Ecol. Conserv. 17:e00569. doi: 10.1016/j.gecco.2019.e00569
Banerjee, A. K., Guo, W., Qiao, S., Li, W., Xing, F., Lin, Y., et al. (2020). Land masses and oceanic currents drive population structure of Heritiera littoralis, a widespread mangrove in the Indo-West Pacific. Ecol. Evol. 10, 7349–7363. doi: 10.1002/ece3.6460
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bunting, P., Rosenqvist, A., Lucas, R., Rebelo, L.-M., Hilarides, L., Thomas, N., et al. (2018). The Global mangrove watch - a new 2010 global baseline of mangrove extent. Remote Sens. 10:1669. doi: 10.3390/rs10101669
Cerón-Souza, I., Gonzalez, E. G., Schwarzbach, A. E., Salas-Leiva, D. E., Rivera-Ocasio, E., Toro-Perea, N., et al. (2015). Contrasting demographic history and gene flow patterns of two mangrove species on either side of the Central American Isthmus. Ecol. Evol. 5, 3486–3499. doi: 10.1002/ece3.1569
Chen, Y., Hou, Y., Guo, Z., Wang, W., Zhong, C., Zhou, R., et al. (2015). Applications of multiple nuclear genes to the molecular phylogeny, population genetics and hybrid identification in the mangrove genus rhizophora. PLoS ONE 10:e0145058. doi: 10.1371/journal.pone.0145058
Cornuet, J. M., Veyssier, J., Pudlo, P., Dehne-Garcia, A., Gautier, M., Leblois, R., et al. (2014). DIYABC v2.0: a software to make Approximate Bayesian Computation inferences about population history using Single nucleotide Polymorphism, DNA sequence and microsatellite data. Bioinformatics 30, 1187–1189. doi: 10.1093/bioinformatics/btt763
De Ryck, D. J. R., Koedam, N., Van der Stocken, T., van der Ven, R. M., Adams, J., and Triest, L. (2016). Dispersal limitation of the mangrove Avicennia marina at its South African range limit in strong contrast to connectivity in its core East African region. Mar. Ecol. Prog. Ser. 545, 123–134. doi: 10.3354/meps11581
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Do, B. T. N., Koedam, N., and Triest, L. (2019). Avicennia marina maintains genetic structure whereas Rhizophora stylosa connects mangroves in a flooded, former inner sea (Vietnam). Estuar. Coast. Shelf Sci. 222, 195–204. doi: 10.1016/j.ecss.2019.04.005
Drexler, J. Z. (2001). Maximum longevities of Rhizophora apiculata and R. mucronata propagules. Pac. Sci. 55, 17–22. doi: 10.1353/psc.2001.0004
Duke, N. C., Lo, E. Y. Y., and Sun, M. (2002). Global distribution and genetic discontinuities of mangroves - emerging patterns in the evolution of Rhizophora. Trees Struct. Funct. 16, 65–79 doi: 10.1007/s00468-001-0141-7
Francisco, P. M., Mori, G. M., Alves, F. A., Tambarussi, E. V., and de Souza, A. P. (2018). Population genetic structure, introgression, and hybridization in the genus Rhizophora along the Brazilian coast. Ecol. Evol. 8, 3491–3504. doi: 10.1002/ece3.3900
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32:W273-9. doi: 10.1093/nar/gkh458
George, B., Bhatt, B. S., Awasthi, M., George, B., and Singh, A. K. (2015). Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 61, 665–677. doi: 10.1007/s00294-015-0495-9
Guo, Z., Huang, Y., Chen, Y., Duke, N. C., Zhong, C., and Shi, S. (2016). Genetic discontinuities in a dominant mangrove Rhizophora apiculata (Rhizophoraceae) in the Indo-Malaysian region. J. Biogeogr. 43, 1856–1868. doi: 10.1111/jbi.12770
Hancke, L., Roberts, M. J., and Ternon, J. F. (2014). Surface drifter trajectories highlight flow pathways in the Mozambique Channel. Deep-Sea Res. II 100, 27–37 doi: 10.1016/j.dsr2.2013.10.014
Hansen, D. M., Austin, J. J., Baxter, R. H., de Boer, E. J., Falcón, W., Norder, S. J., et al. (2017). Origins of endemic island tortoises in the western Indian Ocean: a critique of the human-translocation hypothesis. J. Biogeogr. 44, 1430–1435. doi: 10.1111/jbi.12893
He, Z., Li, X., Yang, M., Wang, X., Zhong, C., Duke, N. C., et al. (2019). Speciation with gene flow via cycles of isolation and migration: insights from multiple mangrove taxa. Natl. Sci. Rev. 6, 275–288. doi: 10.1093/nsr/nwy078
Hermansen, T. D., Britton, D. R., Ayre, D. J., and Minchonton, T. E. (2014). Identifying the real pollinators? Exotic honeybees are the dominant flower visitors and only effective pollinators of Avicennia marina in Australian temperate mangroves. Estuaries Coast. 37, 621–635. doi: 10.1007/s12237-013-9711-3
Hodel, R., Knowles, L., McDaniel, S., Payton, A., Dunaway, J., Soltis, P., et al. (2018). Terrestrial species adapted to sea dispersal: differences in propagule dispersal of two Caribbean mangroves. Mol. Ecol. 27, 4612–4626. doi: 10.1111/mec.14894
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kennedy, J. P., Garavelli, L., Truelove, N. K., Devlin, D. J., Box, S. J., Chérubin, L. M., et al. (2017). Contrasting genetic effects of red mangrove (Rhizophora mangle L.) range expansion along West and East Florida. J. Biogeogr. 44, 335–347. doi: 10.1111/jbi.12813
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lessios, H. A., and Robertson, D. R. (2006). Crossing the impassable: genetic connections in 20 reef fishes across the eastern Pacific barrier. Proc. Royal Soc. B Biol. Sci. 273, 2201–2208. doi: 10.1098/rspb.2006.3543
Li, C.-T., Guo, P., Huang, H.-R., Pei, N.-C., Shi, M.-M., and Yan, H.-F. (2019). The complete chloroplast genome of Rhizophora stylosa and its phylogenetic implications. Mitochondrial DNA B Resour. 4, 374–375. doi: 10.1080/23802359.2018.1547167
Lo, E. Y., Duke, N. C., and Sun, M. (2014). Phylogeographic pattern of Rhizophora (Rhizophoraceae) reveals the importance of both vicariance and long-distance oceanic dispersal to modern mangrove distribution. BMC Evol. Biol. 14, 83. doi: 10.1186/1471-2148-14-83
Mayor, C., Brudno, M., Schwartz, J. R., Poliakov, A., Rubin, E. M., Frazer, K. A., et al. (2000). VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16:1046. doi: 10.1093/bioinformatics/16.11.1046
Mori, G. M., Madeira, A. G., Cruz, M. V., Tsuda, Y., Takayama, K., Matsuki, Y., et al. (2021). Testing species hypotheses in the mangrove genus Rhizophora from the Western hemisphere and South Pacific islands, Estuarine, Coastal and Shelf. Science 248:106948. doi: 10.1016/j.ecss.2020.106948
Mori, G. M., Zucchi, M. I., and Souza, A. P. (2015). Multiple-geographic-scale genetic structure of two mangrove tree species: the roles of mating system, hybridization, limited dispersal and extrinsic factors. PLoS ONE 10:e0118710. doi: 10.1371/journal.pone.0118710
Müller, K. (2006). Incorporating information from length-mutational events into phylogenetic analysis. Mol. Phylogen. Evol. 38, 667–676. doi: 10.1016/j.ympev.2005.07.011
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. New York, NY: Oxford University Press.
Ng, W. L., Chan, H. T., and Szmidt, A. E. (2013). Molecular identification of natural mangrove hybrids of Rhizophora in Peninsular Malaysia. Tree Genet. Genomes 9, 1151–1160. doi: 10.1007/s11295-013-0619-7
Ng, W. L., Onishi, Y., Inomata, N. K. M., Teshima, N., Chan, H., Baba, S., et al. (2015). Closely related and sympatric but not all the same: genetic variation of Indo-West Pacific Rhizophora mangroves across the Malay Peninsula. Conserv. Genet. 16, 137–150. doi: 10.1007/s10592-014-0647-3
Ngeve, M., Van der Stocken, T., Menemenlis, D., Koedam, N., and Triest, L. (2017). Hidden founders? Strong bottlenecks and fine-scale genetic structure in mangrove populations of the Cameroon Estuary complex. Hydrobiologia 803, 189–207. doi: 10.1007/s10750-017-3369-y
Ngeve, M. N., Van der Stocken, T., Menemenlis, D., Koedam, N., and Triest, L. (2016). Contrasting effects of historical sea level rise and contemporary ocean currents on regional gene flow of Rhizophora racemosa in eastern Atlantic mangroves. PLoS ONE 11:e0150950. doi: 10.1371/journal.pone.0150950
Pil, M., Boeger, M., Muschner, V., Pie, M., Ostrensky, A., and Boeger, W. (2011). Postglacial north-south expansion of populations of Rhizophora mangle (Rhizophoraceae) along the Brazilian coast revealed by microsatellite analysis. Am. J. Bot. 98, 1031–1039 doi: 10.3732/ajb.1000392
Provan, J. W., Powell, W., and Hollingsworth, P. M. (2001). Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol. Evol. 16, 142–147. doi: 10.1016/S0169-5347(00)02097-8
Quartly, G. D., and Srokosz, M. A. (2004). Eddies in the southern Mozambique Channel. Deep-Sea Res. II Top. Stud. Oceanogr. 51, 69–83 doi: 10.1016/j.dsr2.2003.03.001
Rabinowitz, D. (1978). Dispersal properties of mangrove propagules. Biotropica 10, 47–57. doi: 10.2307/2388105
Sandoval-Castro, E., Dodd, R. S., Riosmena-Rodriguez, R., Enriquez-Paredes, L. M., Tovilla-Hernandez, C., Lopez-Vivas, J., et al. (2014). Post-glacial expansion and population genetic divergence of mangrove species Avicennia germinans (L.) Stearn and Rhizophora mangle L. along the Mexican coast. PLoS ONE 9:e93358. doi: 10.1371/journal.pone.0093358
Schott, F. A., Shang-Ping, X., and McCreary, J. P. Jr. (2009). Indian Ocean circulation and climate variability. Rev. Geophys. 47:RG1002. doi: 10.1029/2007RG000245
Siegel, D. A., Mitarai, S., Costello, C. J., Gaines, S. D., Kendall, B. E., Warner, R. R., et al. (2008). The stochastic nature of larval connectivity among nearshore marine populations. Proc. Natl. Acad. Sci. USA. 105, 8974–8979. doi: 10.1073/pnas.0802544105
Spalding, M., Kainuma, M., and Collins, L. (2010). World Atlas of Mangroves. London: Earthscan and James and James.
Stieglitz, T., and Ridd, P. V. (2001). Trapping of mangrove propagules due to density-driven secondary circulation in the Normanby River estuary, NE Australia. Mar. Ecol. Prog. Ser. 211, 131–142. doi: 10.3354/meps211131
Takayama, K., Tamura, M., Tateshi, Y., Webb, E. L., and Kajita, T. (2013). Strong genetic structure over the American continents and transoceanic dispersal in red mangroves Rhizophora (Rhizophoraceae), revealed by broad-scale nuclear and chloroplast DNA analysis. Am. J. Bot. 100, 1191–1201. doi: 10.3732/ajb.1200567
Takayama, K., Tateishi, Y., and Kajita, T. (2021). Global phylogeography of a pantropical mangrove genus Rhizophora. Sci. Rep. 11:7228. doi: 10.1038/s41598-021-85844-9
Thomas, N., Lucas, R., Bunting, P., Hardy, A., Rosenqvist, A., and Simard, M. (2017). Distribution and drivers of global mangrove forest change, 1996-2010. PLoS ONE 12:e0179302. doi: 10.1371/journal.pone.0179302
Tomizawa, Y., Tsuda, Y., Saleh, M., Wee, A., Takayama, K., Yamamoto, T., et al. (2017). Genetic structure and population demographic history of a widespread mangrove plant Xylocarpus granatum. J. Koenig across the Indo-West Pacific Region. Forests 8:480. doi: 10.3390/f8120480
Triest, L. (2008). Molecular ecology and biogeography of mangrove trees towards conceptual insights on gene flow and barriers: a review. Aquat. Bot. 89, 138–154. doi: 10.1016/j.aquabot.2007.12.013
Triest, L., Del Socorro, A., Gado, V. J., Mazo, A. M., and Sierens, T. (2021a). Avicennia genetic diversity and fine-scaled structure influenced by coastal proximity of mangrove fragments. Front. Mar. Sci. 8:643982. doi: 10.3389/fmars.2021.643982
Triest, L., Hasan, S., Mitro, P., De Ryck, D., and Van der Stocken, T. (2018). Geographical distance and large rivers shape genetic structure of avicennia officinalis in the highly dynamic sundarbans mangrove forest and ganges delta region. Estuar. Coasts, 41, 908–920. doi: 10.1007/s12237-017-0309-z
Triest, L., and Van der Stocken, T. (2021). Coastal landform constrains dispersal in mangroves. Front. Mar. Sci. 8:617855. doi: 10.3389/fmars.2021.617855
Triest, L., Van der Stocken, T., Akinyi, A. A., Sierens, T., Kairo, J., and Koedam, N. (2020). Channel network structure determines genetic connectivity of landward-seaward Avicennia marina populations in a tropical bay. Ecol Evol. 11:4987. doi: 10.1002/ece3.6829
Triest, L., Van der Stocken, T., De Ryck, D., Kochzius, M., Lorent, S., Ngeve, M., et al. (2021b). Expansion of the mangrove species Rhizophora mucronata in the Western Indian Ocean launched contrasting genetic patterns. Sci. Rep. 11:4987. doi: 10.1038/s41598-021-84304-8
Triest, L., Van der Stocken, T., Sierens, T., Deus, E. K., Mangora, M. M., and Koedam, N. (2021c). Connectivity of Avicennia marina populations within a proposed marine transboundary conservation area between Kenya and Tanzania. Biol. Cons. 256:109040. doi: 10.1016/j.biocon.2021.109040
Urashi, C., Teshima, K. M., Minobe, S., Koizumi, O., and Inomata, N. (2013). Inferences of evolutionary history of a widely distributed mangrove species, Bruguiera gymnorrhiza, in the Indo-West Pacific region. Ecol. Evol. 3, 2251–2261. doi: 10.1002/ece3.624
Van der Stocken, T., Carroll, D., Menemenlis, D., Simard, M., and Koedam, N. (2019a). Global-scale dispersal and connectivity in mangroves. Proc. Natl. Acad. Sci. USA. 116, 915–922. doi: 10.1073/pnas.1812470116
Van der Stocken, T., Vanschoenwinkel, B., De Ryck, D. J. R., Bouma, T. J., Dahdouh-Guebas, F., and Koedam, N. (2015). Interaction between water and wind as a driver of passive dispersal in mangroves. PLoS ONE 10:e0121593. doi: 10.1371/journal.pone.0121593
Van der Stocken, T., Wee, A. K. S., De Ryck, D. J. R., Vanschoenwinkel, B., Friess, D. A., Dahdouh-Guebas, F., et al. (2019b). A general framework for propagule dispersal in mangroves. Biol. Rev. 94, 1547–1575. doi: 10.1111/brv.12514
Wee, A. K. S., Low, S. Y., and Webb, E. L. (2015). Pollen limitation affects reproductive outcome in the bird-pollinated mangrove Bruguiera gymnorrhiza (Lam.) in a highly urbanized environment. Aquat. Bot. 120, 240–243. doi: 10.1016/j.aquabot.2014.09.001
Wee, A. K. S., Noreen, A. M. E., Ono, J., Takayama, K., Kumar, P. P., Tan, H. T. W., et al. (2020). Genetic structure across a biogeographical barrier reflect dispersal potential of four Southeast Asian mangrove plant species. J. Biogeogr. 47, 1258–1271. doi: 10.1111/jbi.13813
Wee, A. K. S., Takayama, K., Asakawa, T., Thompson, B., Onrizal, S. S., Tung, N. X., et al. (2014). Oceanic currents, not land masses, maintain the genetic structure of the mangrove Rhizophora mucronata Lam. (Rhizophoraceae) in Southeast Asia. J. Biogeogr. 41, 954–964. doi: 10.1111/jbi.12263
Wheeler, G. L., Dorman, H. E., Buchanan, A., Challagundla, L., and Wallace, L. E. (2014). A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl. Plant Sci. 2:59. doi: 10.3732/apps.1400059
Keywords: Rhizophora, chloroplast genome, haplotype network, dispersal, Western Indian Ocean
Citation: Triest L, Sierens T and Van der Stocken T (2021) Complete Chloroplast Genome Variants Reveal Discrete Long-Distance Dispersal Routes of Rhizophora in the Western Indian Ocean. Front. Conserv. Sci. 2:726676. doi: 10.3389/fcosc.2021.726676
Received: 17 June 2021; Accepted: 23 August 2021;
Published: 23 September 2021.
Edited by:
Gustavo Maruyama Mori, São Paulo State University, BrazilReviewed by:
Koji Takayama, Museum of Natural and Environmental History, Shizuoka, JapanDamien Daniel Hinsinger, INRA Centre Versailles-Grignon, France
Copyright © 2021 Triest, Sierens and Van der Stocken. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludwig Triest, bHRyaWVzdEB2dWIuYmU=