
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Comput. Neurosci. , 21 December 2012
Volume 6 - 2012 | https://doi.org/10.3389/fncom.2012.00098
This article is part of the Research Topic Biophysically based Computational Models of Astrocyte ~ Neuron Coupling and their Functional Significance View all 11 articles
 Maurizio De Pittà1*
Maurizio De Pittà1* Vladislav Volman2,3,4
Vladislav Volman2,3,4 Hugues Berry5
Hugues Berry5 Vladimir Parpura6,7
Vladimir Parpura6,7 Andrea Volterra8
Andrea Volterra8 Eshel Ben-Jacob1,9,10*
Eshel Ben-Jacob1,9,10*The complexity of the signaling network that underlies astrocyte-synapse interactions may seem discouraging when tackled from a theoretical perspective. Computational modeling is challenged by the fact that many details remain hitherto unknown and conventional approaches to describe synaptic function are unsuitable to explain experimental observations when astrocytic signaling is taken into account. Supported by experimental evidence is the possibility that astrocytes perform genuine information processing by means of their calcium signaling and are players in the physiological setting of the basal tone of synaptic transmission. Here we consider the plausibility of this scenario from a theoretical perspective, focusing on the modulation of synaptic release probability by the astrocyte and its implications on synaptic plasticity. The analysis of the signaling pathways underlying such modulation refines our notion of tripartite synapse and has profound implications on our understanding of brain function.
The simultaneous recognition that astrocytes sense neighboring neuronal activity and release neuroactive agents (or “gliotransmitters”) has been instrumental in the uncovering of the many roles played by these cells in the control of genesis, function and plasticity of synapses (Haydon, 2001; Ullian et al., 2004; Volterra and Meldolesi, 2005; Bains and Oliet, 2007; Santello and Volterra, 2009; Zorec et al., 2012). These findings initiated a conceptual revolution that leads to rethinking how brain communication works since they imply that information travels and is processed not just in the neuronal circuitry but in an expanded neuron-glial network (Haydon, 2001; Volterra and Meldolesi, 2005; Giaume et al., 2010). On the other hand the physiological need for astrocyte signaling in brain information processing and the modes of action of these cells in computational tasks remain largely undefined. This is due, to a large extent, both to the lack of conclusive experimental evidence, and to a substantial lack of a theoretical framework to address modeling and characterization of the many possible astrocyte functions. This review aims at introducing such a perspective providing a framework for future modeling efforts in the field based on preliminary theoretical studies on both astrocytic calcium signaling and gliotransmitter-mediated modulations of synaptic release probability.
Control of synaptic transmission and plasticity by astrocytes subtends a complex signaling network, which involves different biochemical pathways (Volterra and Meldolesi, 2005; Zorec et al., 2012). In general, synaptically-released neurotransmitter can spill out of the synaptic cleft and bind to metabotropic receptors found on the neighboring astrocytic processes triggering there inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ signaling. This was observed at both glutamatergic, cholinergic, noradrenergic, and GABAergic synapses in the hippocampus, in the thalamus and in the cortex (Volterra and Meldolesi, 2005; Haydon and Carmignoto, 2006; Santello and Volterra, 2009; Halassa and Haydon, 2010; Navarrete et al., 2012a,b). Figure 1A summarizes a number of observations made at the level of hippocampal glutamatergic synapses (Bains and Oliet, 2007; Santello and Volterra, 2009). There, synaptic glutamate can trigger Ca2+ signaling in the surrounding astrocytic processes via metabotropic glutamate receptors (mGluRs) (Pasti et al., 1997; Fiacco and McCarthy, 2004; Panatier et al., 2011). There is also evidence in the dentate gyrus that ATP, possibly synaptically-released, triggers astrocytic Ca2+ signaling through the activation of metabotropic purinergic P2Y1 receptors (Jourdain et al., 2007; Di Castro et al., 2011; Larsson et al., 2011; Santello et al., 2011). Following elevation of intracellular Ca2+, astrocytes can release glutamate as well as other chemical transmitters such as D-serine (D-ser) and ATP which can be converted into adenosine (Adn) in the extracellular milieu (Bezzi et al., 2004; Pascual et al., 2005; Montana et al., 2006; Henneberger et al., 2010; Parpura and Zorec, 2010). Astrocyte-released glutamate diffuses in the extrasynaptic space and may bind to glutamate receptors (GluRs), including mGluRs and NMDARs on neighboring presynaptic terminals, modulating the release of neurotransmitter (Fiacco and McCarthy, 2004; Jourdain et al., 2007; Perea and Araque, 2007; Bonansco et al., 2011; Di Castro et al., 2011). An analogous action on synaptic release could also be due to astrocyte-derived ATP and its derivative adenosine through presynaptic purinergic receptors (PRs), including both A1 and A2 receptors (Pascual et al., 2005; Halassa and Haydon, 2010; Panatier et al., 2011). On the postsynaptic side, astrocytic glutamate and D-serine may bind to extrasynaptic NR2B-containing and postsynaptic NMDARs respectively, modulating neuronal firing and participating in the induction of long-term potentiation (Fellin et al., 2004; Bains and Oliet, 2007; Henneberger et al., 2010). Astrocyte could also release tumor necrosis factor-α (TNFα) by Ca2+-dependent activation of TNFα-converting enzyme (TACE) (Bezzi et al., 2001; Santello and Volterra, 2012), which could strengthen excitatory synaptic transmission by promoting surface insertion of AMPA receptors (AMPARs) (Beattie et al., 2002; Stellwagen and Malenka, 2006; Bains and Oliet, 2007). This signaling route could also play a role in pathological states such as post-traumatic epilepsy (Balosso et al., 2009; Volman et al., 2011) or spinal cord injury (Stellwagen et al., 2005; Ferguson et al., 2008). On the other hand, extracellular levels of TNFα control glutamate release from astrocytes, ultimately modulating the astrocytic action on presynaptic function (Domercq et al., 2006; Santello et al., 2011). This intricate signaling network is further complicated by the possibility that astrocyte Ca2+ events are triggered by additional mechanisms, including the action of ATP released extracellularly by astrocytes themselves or of IP3 that diffuses intracellularly, from one astrocyte to another, through gap junction (GJCs) (Kang et al., 2005; Scemes and Giaume, 2006). At Schaffer collateral synapses, astrocytic Ca2+ increases could also be promoted by retrograde endocannabinoid signaling from postsynaptic terminals via activation of endocannabinoid CB1 receptors (Navarrete and Araque, 2008, 2010) (omitted from Figure 1A for clarity). Moreover, the relation between astrocytic Ca2+ and gliotransmitter release is not simple: some of the Ca2+ signals that can be generated in astrocytes are apparently not able to induce gliotransmitter release or its synaptic consequences (Fiacco et al., 2007; Agulhon et al., 2008, 2010; Petravicz et al., 2008; Lovatt et al., 2012), while gliotransmitters can also be released by mechanisms that are independent of Ca2+ signaling (Parpura and Zorec, 2010) (not included in Figure 1A for simplicity).
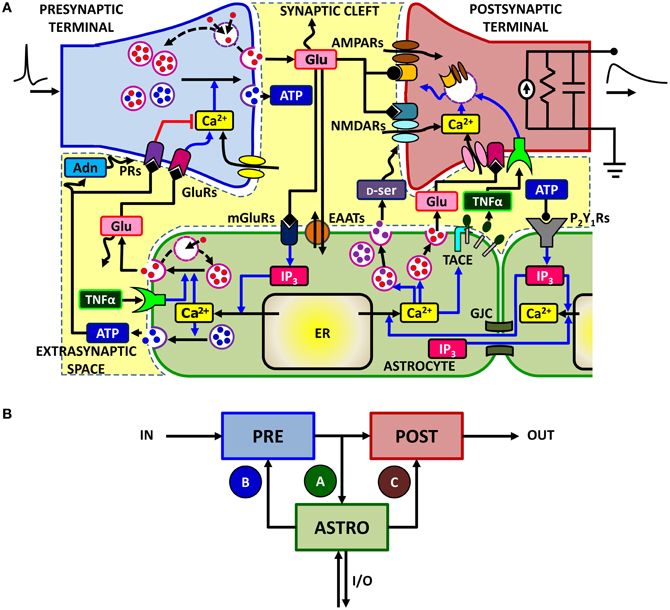
Figure 1. The signaling network of astrocyte-synapse interactions. (A) A simplified scheme of the different signaling pathways between synaptic terminals and astrocytes for the case of excitatory synapses in the hippocampus (see text for a detailed description). Action potentials arriving at the presynaptic terminal trigger release of glutamate, which can spill over from the synaptic cleft. Perisynaptic astrocytes take up glutamate using their plasma membrane transporters (EAATs) while glutamate, by acting on astrocytic metabotropic receptors (mGluRs), triggers Ca2+ signaling in the astrocyte. This signaling pathway includes production of IP3 and causes an increase of cytosolic Ca2+ due to efflux of this ion from the endoplasmic reticulum (ER). At some synapses, such as in the dentate gyrus, the same Ca2+ signaling pathway could also be mediated by astrocytic purinergic P2Y1 receptors, likely activated by synaptically-released ATP (see text for details). Astrocytic Ca2+ excitability can in turn lead to exocytotic release of several neuroactive substances (or “gliotransmitters”) such as glutamate (Glu), D-serine (D-ser) or ATP which can target specific receptors on pre- and post-synaptic terminals and differentially modulate synaptic transmission. Glutamate acting on presynaptic GluRs could enhance synaptic release, whereas ATP and its derivate adenosine (Adn) could depress it (red path) through presynaptic purinergic receptors (PRs). On the postsynaptic spines [depicted here by a standard RC circuit (Ermentrout and Terman, 2010)], the ensuing effect of gliotransmitters could substantially modify postsynaptic currents by enhancing activation of NMDA receptors (D-serine) or by altering expressions of AMPA receptors therein. Astrocytes could also release TNFα by Ca2+-dependent activation of the matrix metalloprotease TNFα-converting enzyme (TACE), while extracellular TNFα could in turn regulate glutamate release from the astrocyte as well as postsynaptic AMPAR expression. Moreover astrocytic Ca2+ could also propagate across different regions of the same cell or to other neighboring astrocytes by intracellular IP3 diffusion through gap junction channels (GJCs) or via extracellular ATP-dependent pathways, extending gliotransmission to some distal sites away from the considered synapse. For clarity both endocannabinoid-mediated Ca2+ signaling (Navarrete and Araque, 2008), retrograde activation of presynaptic glutamate receptors (Navarrete and Araque, 2010), regulation of postsynaptic NMDARs by presynaptic adenosine receptors (Deng et al., 2011), and the possibility for astrocyte-derived adenosine to enhance synaptic release (Panatier et al., 2011) are not included in this scheme. (B) Equivalent modeling scheme for astrocyte-synapse interactions. The astrocyte (ASTRO) constitutes a third active element of the tripartite synapse in addition to the presynaptic (PRE) and postsynaptic (POST) terminals. In its presence, the flow of input (IN) signals to the output (OUT) is no more unidirectional but presynaptically released neurotransmitter can affect astrocyte function through the interaction pathway A. In turn, the astrocyte can regulate both synaptic terminals via pathways B and C. In addition, the astrocyte could receive additional inputs from or send output to remote synapses in a heterosynaptic fashion (I/O).
Despite its apparent complexity, the ensemble of astrocyte-synapse signaling interactions discussed above can be well captured by the modeling scheme of Figure 1B. This scheme shows the three essential components of astrocyte-regulated synapses, also referred to as “tripartite synapses” (Araque et al., 1999; Haydon, 2001): these are the pre- (PRE) and postsynaptic (POST) terminals, and the astrocyte, i.e., an astrocytic process surrounding the synaptic elements (ASTRO) (Araque et al., 1999; Haydon, 2001). Moreover, in addition to the classical neuronal path that leads from input presynaptic action potentials, commonly referred to as input spikes (IN), to the output postsynaptic current (OUT), further input and/or output pathways (I/O) could coexist due to the above-mentioned routes based on astrocytic Ca2+ signaling (Giaume et al., 2010).
Focusing on synapse-astrocyte coupling, three fundamental pathways are identified: one (A) from the synapse to the astrocyte whereby synaptically-released glutamate (or other synaptic agents) promotes Ca2+ signaling in the astrocyte and the other two (B and C) from the astrocyte to synaptic terminals, whereby glutamate or ATP released from the astrocyte affects synaptic function (Volterra and Meldolesi, 2005; Santello and Volterra, 2009). Additional pathways supported by other neuroactive agents such as D-serine or TNFα can also be evoked in parallel to those shown in Figure 1B but they would not alter the essence of the scheme. Moreover, although based on experimental results at excitatory synapses in the hippocampus, (Araque et al., 1998a,b; Fiacco and McCarthy, 2004; Jourdain et al., 2007; Perea and Araque, 2007; Andersson and Hanse, 2010; Santello et al., 2011), the modeling scheme in Figure 1B could also hold for other reported pathways such as GABA-evoked gliotransmission at interneuron-to-pyramidal cell synapses in the hippocampus (Kang et al., 1998; Serrano et al., 2006), or glia-mediated ATP release at hippocampal synapses (Pascual et al., 2005), in the hypothalamus (Gordon et al., 2009), and in the retina (Newman, 2003, 2005), or glial modulation of neuromuscular transmission (Robitaille, 1998; Rousse et al., 2010; Todd et al., 2010) (see Table 1 for a summary of the possible signaling pathways).
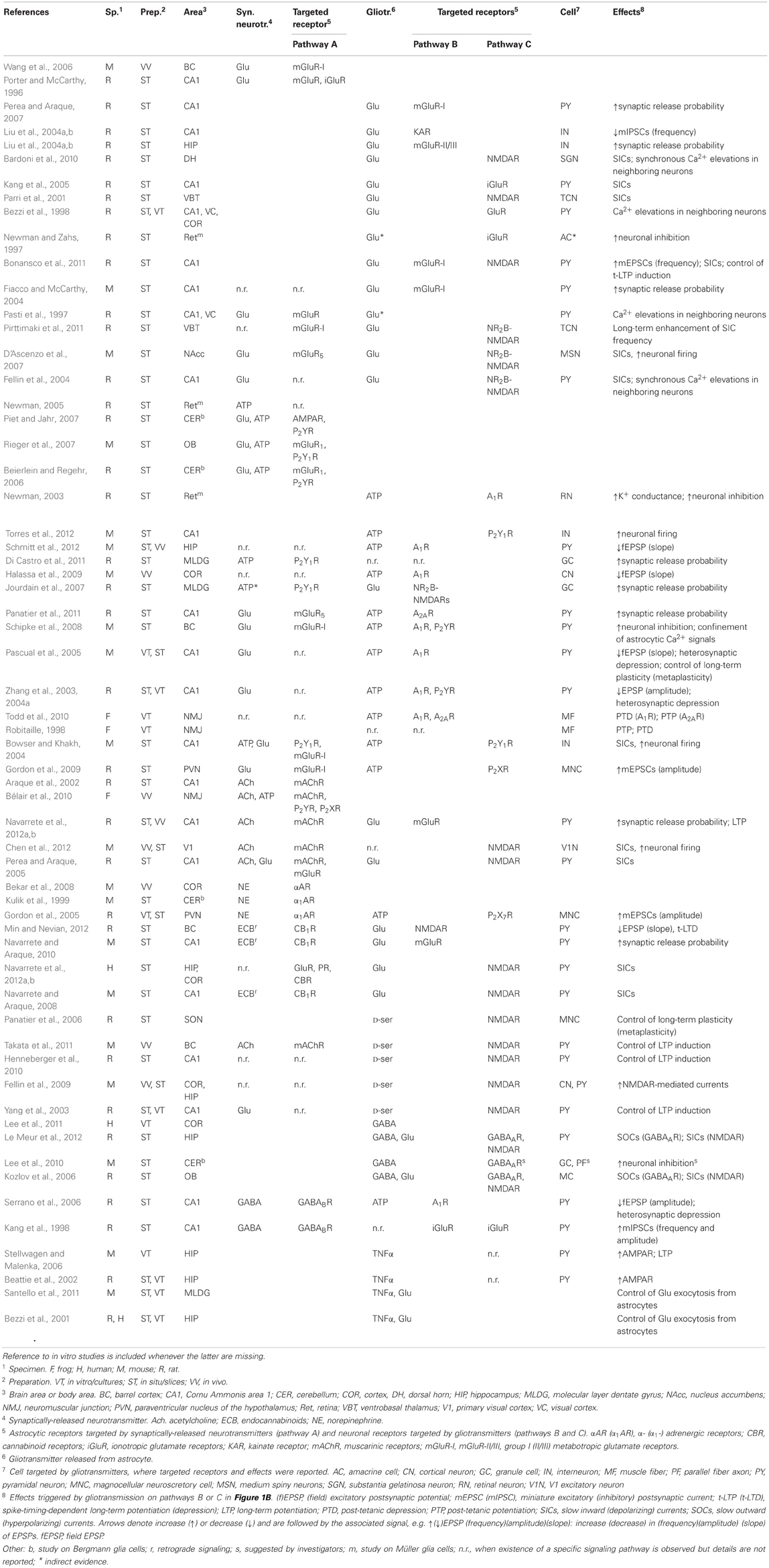
Table 1. Transmitters, targeted receptors, and major effects on synaptic transmission by the signaling pathways A, B, C in Figure 1B (in situ and in vivo studies only).
Analysis of the scheme in Figure 1B reveals that astrocytes mediate two loops in the signal flow from presynaptic to postsynaptic terminal: a feedforward and a feedback one. The feedforward loop ends on the postsynaptic terminal and is activated when synaptic glutamate and/or ATP induces glutamate and/or D-serine release from the astrocyte to the postsynaptic element, i.e. the A–C path in Figure 1B (Bains and Oliet, 2007; Barres, 2008; Santello and Volterra, 2009). The feedback loop ends on the presynaptic terminal (the A–B path in Figure 1B) and is activated when synaptic glutamate or ATP trigger Ca2+-dependent release of glutamate and/or ATP from the astrocyte to the presynaptic terminal, leading to modulation of synaptic release through specific presynaptic receptors (Santello and Volterra, 2009; Halassa and Haydon, 2010).
In principle the two pathways could coexist at the same synapse where they are expected to display different dynamics and respond to different preferred input stimuli. Therefore their coexistence at the same synapse could give rise to complex effects that are hard to quantify when considered altogether. Accordingly, a common approach in experiments is to characterize their effects on synaptic function by separate manipulations of pathways A–C (Figure 1B) using different techniques (Montana et al., 2004; Jourdain et al., 2007; Marchaland et al., 2008; Di Castro et al., 2011; Panatier et al., 2011; Santello et al., 2011). For example, characterization of the feedback pathway on the presynaptic terminal (A–B) can be carried out by opening the feedback loop (for example by inhibiting elements of A or B) and analyzing the signaling components separately. From a theoretical point of view, this approach is put forth by at least three steps of analysis that are: (1) characterization of Ca2+ dynamics in the astrocyte as a function of different synaptic inputs (that is, pathway A); (2) characterization of how gliotransmitter release from the astrocyte depends on different astrocytic intracellular Ca2+ dynamics; and (3) characterization of the effect on synaptic release of Ca2+-dependent gliotransmitter release from the astrocyte (i.e. pathway B). These three aspects are discussed below from a modeling perspective, focusing on their possible roles in synaptic information processing.
Intracellular Ca2+ elevations in the astrocyte are not simple on–off signals (Carmignoto, 2000; Zonta and Carmignoto, 2002; Di Castro et al., 2011). There are multiple and varied spatiotemporal patterns of Ca2+ elevation, which probably underlie different types of function, including generation of diverse output signals (Carmignoto, 2000; Volterra and Meldolesi, 2005; Zorec et al., 2012). Two main types of neuronal activity-dependent Ca2+ responses are observed in astrocytes (Grosche et al., 1999; Codazzi et al., 2001; Matyash et al., 2001; Zonta and Carmignoto, 2002; Scemes and Giaume, 2006): (1) transient Ca2+ increases that are confined to their distal processes (Pasti et al., 1997; Nett et al., 2002; Di Castro et al., 2011) and (2) Ca2+ elevations propagating along these processes as regenerative Ca2+ waves, often eventually reaching the cell soma (Pasti et al., 1997; Sul et al., 2004). This latter kind of response can even propagate to neighboring astrocytes, giving rise to intercellular Ca2+ waves (Tian et al., 2005; Kuga et al., 2011). On the other hand, intercellular Ca2+ propagation does not necessarily need propagation through the cell soma and has been observed across astrocytic processes or from an end foot to an end foot (Mulligan and MacVicar, 2004; Giaume et al., 2010).
The precise signaling cascades underlying the various forms of Ca2+ elevation are not completely understood. In general, Ca2+ signals in astrocytes are determined by an intricate interplay of amplification, buffering, and extrusion pathways linked to cytosolic Ca2+ elevations mediated by influx from the extracellular space (Malarkey et al., 2008; Shigetomi et al., 2011) and/or release from intracellular endoplasmic reticulum (ER) stores (Verkhratsky et al., 2012). IP3-triggered Ca2+-induced Ca2+ release (CICR) from the ER is considered the primary mechanism responsible for intracellular Ca2+ dynamics in astrocytes (Volterra and Meldolesi, 2005; Nimmerjahn, 2009). This mechanism, schematized in Figure 2A, is essentially controlled by the interplay of three fluxes: (1) a Ca2+ transfer from the cytosol to the ER (JP) mediated by endoplasmic-reticulum Ca2+-ATPase (SERCA) pumps which contributes to the maintenance of higher Ca2+ concentrations in the ER stores than in the cytosol; (2) a passive Ca2+ leak (JL) from the ER to the cytosol that is driven by the Ca2+ gradient between the ER and the cytosol; and (3) an efflux (JNL) from the ER to the cytosol through IP3 receptor (IP3R) channels, which depends both on IP3 and Ca2+ concentrations in the cytosol in a nonlinear fashion (Bezprozvanny et al., 1991; Ramos-Franco et al., 2000; Shinohara et al., 2011).
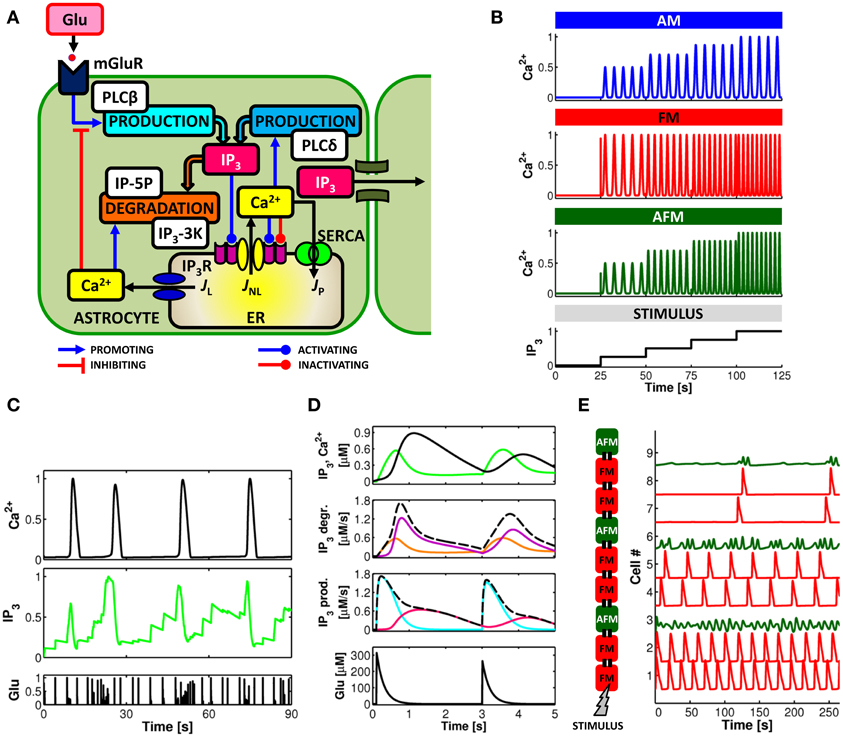
Figure 2. Computational aspects of astrocytic Ca2+ signaling. (A) Scheme of IP3-mediated Ca2+-induced Ca2+ release in the astrocyte. Calcium and IP3 signals are controlled by synaptic glutamate through metabotropic glutamate receptor- (mGluR-) PLCβ-mediated IP3 production (see text for details). (B) IP3 and Ca2+ signals can be envisioned to encode incoming synaptic activity through frequency and amplitude of their oscillations. Astrocytes could thus encode synaptic information either by modulations of the amplitude (AM), the frequency (FM), or both (AFM) of their Ca2+ oscillations. (C) Simulated Ca2+ and IP3 patterns in response to sample synaptic glutamate release (Glu) in a model astrocyte (De Pittà et al., 2009a,b) reveals that IP3 signals could be locked in the AFM-encoding independently of the encoding mode of the associated Ca2+ signals. This feature could allow the astrocyte to optimally integrate synaptic stimuli. (D, top) Simulated Ca2+ (black trace) and IP3 (green trace) signals in the same astrocyte model as in (C), and associated rates of IP3 production (prod.) and degradation (degr.) (middle panels, dashed black lines) in response to two consecutive synaptic glutamate release events (bottom panel, Glu). The analysis of the contributions of different enzymes to IP3 signaling (solid colored traces; cyan: PLCβ ; pink: PLCδ ; orange: IP-5P; and purple: IP3-3K) reveals dynamical regulation by Ca2+ of different mechanisms of IP3 production/degradation which could ultimately underlie dynamical regulation of astrocyte processing of synaptic stimuli. (E) Simulated propagation of Ca2+ waves in a heterogeneous linear chain composed of both FM (red traces) and AFM (green traces) astrocytes reveals that encoding of synaptic activity (STIMULUS) could change according to cell location along the chain. Adapted from Goldberg et al. (2010).
Cytosolic Ca2+ regulates IP3Rs in a biphasic manner: Ca2+ release from the ER is potentiated at low cytosolic Ca2+ concentrations but is inhibited at higher Ca2+ concentrations (Iino, 1990; Bezprozvanny et al., 1991). On the other hand, IP3 monotonically activates IP3R channels at constant Ca2+ concentrations (Watras et al., 1991), but dynamically changes the Ca2+ sensitivity of the channel (Kaftan et al., 1997; Ramos-Franco et al., 2000; Mak et al., 2003). At low, subsaturating IP3 concentrations, the optimal Ca2+ concentration for IP3R modulation becomes lower, whereas at very high IP3 concentrations, channel activity persists at supramicromolar Ca2+ concentrations (Kaftan et al., 1997; Mak et al., 2003). Thus, the level of IP3 determines the dynamics of intracellular Ca2+.
Both production and degradation of IP3 depend on enzymes that are regulated by cytosolic Ca2+ (Berridge et al., 2003; De Pittà et al., 2009a,b). These include Ca2+-dependent PLCδ-mediated IP3 synthesis and Ca2+-dependent IP3 degradation by IP3 3-kinase (IP3-3K) and by inositol polyphosphate 5-phosphatase (IP-5P) (Figure 2A) (Zhang et al., 1993; Sims and Allbritton, 1998; Rebecchi and Pentyala, 2000). However, while the activity of IP3-3K is stimulated by cytosolic Ca2+ (Communi et al., 1997), IP-5P is inhibited instead (Communi et al., 2001). This results in different mechanisms of IP3 degradation depending on the Ca2+ concentration in the cytoplasm (Sims and Allbritton, 1998; Irvine et al., 2006). Thus, for example, for equally-expressed enzymes, at low Ca2+ concentrations, namely lower than 500 nM (Sims and Allbritton, 1998; De Pittà et al., 2009a,b), IP3 degradation is promoted by both IP-5P and IP3-3K, whereas for intermediate-to-high cytosolic Ca2+ concentrations, degradation by IP3-3K becomes predominant (Sims and Allbritton, 1998). Theoretical investigation showed that the interplay of these two regimes is both necessary and sufficient to reproduce Ca2+ oscillations and pulsations observed experimentally (De Pittà et al., 2009a,b).
Intracellular levels of IP3 can also be controlled by gap junction mediated diffusion of IP3 from other regions of the same astrocyte or from neighboring cells (Giaume et al., 2010) (i.e., the I/O pathway in Figure 1B). Moreover, synaptic glutamate (or other synaptic agents) can bind to astrocytic G protein-coupled receptors (GPCRs) like mGluRs that are directly linked to intracellular IP3 production by PLCβ (Zur Nieden and Deitmer, 2006) (pathway A in Figure 1B). In this fashion, astrocytic Ca2+ dynamics triggered by synaptically-controlled IP3 production can be regarded as a form of encoding information about activity in neighboring synapses.
Encoding of synaptic activity by astrocytic Ca2+ is likely multimodal, depending on many possible intracellular properties (De Pittà et al., 2008, 2009a,b; Dupont et al., 2011). A widely adopted classification considers the amplitude and the frequency of Ca2+ increases from resting levels (Berridge, 1997; Falcke, 2004; De Pittà et al., 2008, 2009a,b). In this view, as summarized in Figure 2B, synaptic activity reflected by different intracellular IP3 concentrations (STIMULUS), is encoded by the modulation of Ca2+ oscillations and pulsations either in their amplitude (AM), their frequency (FM), or both (AFM). While available experimental data suggest a preferential FM mode of encoding (Pasti et al., 1997), AM and AFM encoding of synaptic activity are also plausible mechanisms given that the amplitude of Ca2+ response can strongly depend on the stimulation intensity (Wang et al., 2006; Di Castro et al., 2011; Panatier et al., 2011; Torres et al., 2012). This is the case for example, of synaptic inputs that occur rapidly one after the other whereby the ensuing intracellular Ca2+ concentration builds up as a cumulation of such inputs (Perea and Araque, 2005; Torres et al., 2012). In further support of the AM/AFM encoding is the experimental observation that glutamate exocytosis from the astrocyte occurs only when Ca2+ increases beyond a threshold concentration (Newman and Zahs, 1997; Parpura and Haydon, 2000; Pasti et al., 2001; Auld and Robitaille, 2003; Montana et al., 2006). Hence, astrocytic Ca2+ increases in response to synaptic activity would not systematically trigger the release of glutamate or other gliotransmitters from the astrocyte. Acting on the amplitude of astrocytic Ca2+ signals, AM/AFM encodings could constitute a way to regulate astrocytic gliotransmitter release by synaptic activity. Further experiments are needed to elucidate the nature of the Ca2+ threshold for astrocytic exocytosis since this latter might be gliotransmitter-specific (Montana et al., 2006). Accordingly, AM/AFM encoding of Ca2+ dynamics could vary from one gliotransmitter to another.
Experimental evidence suggests that Ca2+ dynamics does not simply mirror synaptic activity but is more complex, to a point that astrocytes have been proposed to perform genuine processing of synaptic information (Perea and Araque, 2005; Perea et al., 2009). This possibility follows from the complex network of IP3 and Ca2+ signaling and subtends a scenario where Ca2+ could be only one of the players in the encoding and processing of synaptic activity by astrocytes (Mishra and Bhalla, 2002). Yet many, if not all, of the other signals underlying the complex cascade of biochemical reactions that link synaptically-released glutamate to CICR, could also carry out encoding and processing (Barlow, 1996; Berridge et al., 2003). Theoretical investigations suggested that IP3 could also encode for the glutamate stimulation levels via a systematic AFM encoding (De Pittà et al., 2009a,b) as shown in Figure 2C. When cytosolic Ca2+ levels are low, close to resting values, IP3 generally increases with ongoing synaptic activity (Glu). With low Ca2+, the activity of IP3-3K is reduced and the resulting IP3 degradation slows down. The contribution to IP3 production by Ca2+-dependent PLCδ is reduced as well, so that intracellular IP3 mostly depends on the frequency of synaptic release. Rapid successions of synaptic release events produce crisp increases of IP3 (essentially proportional to the number of successive synaptic release events) while, between two remote release events, IP3 tends to relax to resting levels. As a result, IP3 dynamics overall evolves as the integral of synaptic activity. If IP3 reaches the CICR-triggering threshold, intracellular Ca2+ increases fast and so does IP3-3K activity. Then, IP3 is rapidly degraded and resting IP3 levels are restored, thus resetting the integral of synaptic activity to initial values.
It is precisely the alternation between these two different phases of IP3 degradation (a high Ca2+—high IP3-3K-activity phase and a low Ca2+—low IP3-3K-activity phase), that endows IP3 signal with high amplitude variability. On the other hand, such AM features still allow fast variations, thus rich spectral content (i.e., FM features), in response to changes in frequency of synaptic release. This enticing possibility could endow the IP3 signal with the necessary properties to function as optimal interface between synaptic stimuli and intracellular Ca2+ signals. Since neural information is carried by the timing of spikes rather than by their amplitude (Sejnowski and Paulsen, 2006), the capability of fast highly-variable amplitude changes corresponding to rich spectral content of IP3 signals, would fulfill this requirement, embedding the essential spectral features of the synaptic signal into the spectrum of the IP3 transduction. On the other hand, because Ca2+ signals are triggered primarily by suprathreshold IP3 elevations (Li et al., 1994; Keizer et al., 1995), the coexistence of AM features within the AFM IP3 signal seems to be a necessary prerequisite in order to trigger CICR.
This could also help elucidate the origin of the integrative properties of Ca2+ signaling in astrocytes (Perea et al., 2009). These properties could result from at least two steps of integration: one step is the transduction of the agonist signal into the IP3 signal; the other step is the cross-talk between IP3 and Ca2+ signals. Hence, AFM-encoding IP3 dynamics could operate a first preliminary integration by smoothing the highly indented synaptic stimulus. The inherent features of CICR would then bring forth a further integration step, yielding Ca2+ patterns that are even smoother than IP3 signals (De Pittà et al., 2009a,b).
The tight dynamical coupling between IP3 and Ca2+ signals also suggests that the way astrocytes process synaptic signals, i.e. their frequency response to synaptic stimuli, is not fixed but rather dynamical and dependent on the history of activation of the astrocyte. This is because different IP3 signaling mechanisms that are dynamically regulated by Ca2+ likely correspond to different frequency responses of the astrocyte with respect to synaptic signals. Figure 2D shows the time course of IP3 production (IP3 prod.) and degradation (IP3 degr.) (dashed black lines) underlying simulated IP3 and Ca2+ signals (top panel, green and black traces, respectively) in response to two events of synaptic glutamate release (bottom panel). While Ca2+-dependent IP3 production by PLCβ (cyan trace) and PLCδ (pink trace) could modulate the threshold frequency of synaptic stimuli that triggers Ca2+ signaling in the astrocyte, existence of different regimes of IP3 degradation within a single Ca2+ oscillation cycle could be responsible for different cutoff frequencies of synaptic release beyond which Ca2+-mediated astrocyte processing of synaptic stimuli ceases. In particular, the cutoff frequency during low IP3-3K activity could be mainly set by the rate of IP3 degradation by IP-5P (orange trace). When Ca2+ is high instead, IP3 degradation by IP3-3K (purple trace) could also become very strong, thus sensibly reducing the cutoff frequency. That is, the cutoff frequency is dependent on the context of underlying Ca2+ signaling which, in turn, depends on the history of activation of the astrocyte by synaptic stimuli.
The subcellular arrangement of the enzymes underlying IP3 signaling could also be responsible for spatial heterogeneity of the frequency response of the astrocyte. Although the subcellular localization of IP3 production and degradation enzymes in astrocytes remains to be elucidated, studies in brain tissue suggest that PLCβ and IP-5P could localize mainly in proximity to the plasma membrane, whereas PLCδ and IP3-3K are preferentially in the cytoplasm (Rebecchi and Pentyala, 2000; Irvine et al., 2006). Given that the ER distribution changes from astrocytic processes to soma (Pivneva et al., 2008), different subcellular regions of the astrocyte could correspond to different cytoplasmic volumes and thus to distinctly different expressions of enzymes mediating IP3 signaling. The ensuing different subcellular arrangement of these enzymes could ultimately provide anatomical specificity to the astrocytic phosphoinositide signaling which underlies CICR-based astrocyte processing of synaptic activity (Fukaya et al., 2008). In this fashion, processing of synaptic stimuli by Ca2+ signaling at astrocytic processes could differ from that carried out in the soma by means of differently expressed IP3 signaling-related enzymes.
Intracellular and intercellular propagation of Ca2+ could contribute new encoding and processing modes, in addition to those depicted in Figure 2B. However, despite the numerous modeling studies developed to account for the rich dynamics of astrocyte Ca2+ signaling (Bennett et al., 2008; Goldberg et al., 2010; Dupont et al., 2011); [for a recent review on calcium modeling see Falcke (2004)], we still lack a comprehensive theoretical framework to link the local Ca2+ signals that are restricted to small regions of the astrocytic processes, to their spatial dynamics and their possible propagation at larger spatial scales: intracellular propagation, global whole-cell signals or cell-to-cell Ca2+ waves.
Propagations at these various scales probably differ by their underlying mechanisms (Falcke, 2004; Scemes and Giaume, 2006). Fast-rising and short-lived local Ca2+ events, observed in response to even a single quantal release from synaptic terminals (Di Castro et al., 2011; Panatier et al., 2011) closely resemble spatially confined Ca2+ puffs or blips in other cell types (Thomas et al., 2000; Bootman et al., 2001) and could depend on spatial clustering of IP3Rs along the ER structures or of mGluRs along the plasma membrane, or on both (Marchaland et al., 2008; Panatier et al., 2011; Arizono et al., 2012). The cumulative recruitment of these Ca2+ puffs could lead to spatially more extended Ca2+ events which could either be still confined within astrocytic processes (Di Castro et al., 2011) or propagate to other cellular regions or to other cells as regenerative Ca2+ waves (Pasti et al., 1997; Kuga et al., 2011).
Calcium could propagate by at least two routes [for a recent review see Scemes and Giaume (2006)]. One is intracellular, through GJCs, involving diffusion of IP3 directly from cytoplasm to cytoplasm. The other route is extracellular, involving release of ATP from the astrocyte which binds to GPCRs of the same cell or neighboring astrocytes, increasing their IP3 levels (Guthrie et al., 1999). The relative contribution of each of these pathways likely depends on developmental, regional and physiological states and could subtend different ranges of propagations as well as different temporal features (Haas et al., 2006; Scemes and Giaume, 2006; Giaume et al., 2010).
The restriction and clustering of mGluRs expression along astrocytic processes to subregions that colocalize with synaptic terminals (van den Pol et al., 1995; Arizono et al., 2012) hints the possibility of a subcellular compartmentalization of Ca2+ signals (Marchaland et al., 2008; Di Castro et al., 2011; Panatier et al., 2011). Local Ca2+ events would be spatially restricted to narrow regions around each mGluRs cluster thus defining independent signals within the same process. In this fashion, astrocytes could carry out parallel integration and processing of synaptic information on different temporal and spatial scales at different processes (Goldberg et al., 2010; Bernardinelli et al., 2011) or even in different subregions of the same process which could constitute separate functional microdomains (Panatier et al., 2011). Furthermore, the resulting neuromodulatory action exerted by astrocytic gliotransmitters on synaptic terminals and their impact on neuronal network activity could vary from one process or microdomain to the other (Navarrete and Araque, 2011).
The spatial scale and the time window of Ca2+ events likely discriminate between different mechanisms of IP3 and Ca2+ signaling. At the level of astrocytic processes and subcellular compartments, Ca2+ propagation could be mediated by fast intracellular IP3 linear (i.e. Fickian) diffusion (Sneyd et al., 1994; Falcke, 2004). On the other hand, when considering intercellular propagation mediated by gap junctions, IP3 transport from one cell to the other could be essentially nonlinear. This scenario was shown to be consistent with the observed variability of Ca2+ wave propagation distance (Goldberg et al., 2010) and could explain the long-range regenerative propagation of Ca2+ waves observed in cortical astrocytes (Scemes and Giaume, 2006; Tian et al., 2006).
Cell heterogeneity likely constitutes an additional critical aspect that substantially affects propagation patterns and extent of propagation of Ca2+ signals (Iacobas et al., 2006; De Pittà et al., 2008; Goldberg et al., 2010). This possibility is illustrated in Figure 2E by a toy example consisting of a heterogeneous linear chain of FM-encoding (red traces) and AFM-encoding (green traces) astrocytes. The synaptic stimulus is restricted to the first cell of the chain (i.e. cell number 1). Only FM-encoding cells guarantee regenerative propagation of Ca2+ signals, whereas AFM cells do not, acting like propagation barriers along the chain. Unlike in AFM cells, Ca2+-dependent IP3 production in FM cells guarantees IP3 diffusion to the next cell in the chain to levels that are beyond the threshold of CICR thus promoting regenerative propagation (Goldberg et al., 2010). Moreover, the shape of the local Ca2+ events in each cell changes along the propagation path: after each AFM cell, the frequency of FM-encoding Ca2+ pulses is reduced, suggesting that different propagation patterns could carry out different processing of synaptic information (Goldberg et al., 2010).
The above scenario hints that the spatial distribution of astrocytes in different brain areas could be made to fulfill specific processing tasks. Indeed neighboring astrocytes in the brain are believed to be distributed in space in a non-random orderly fashion called “contact spacing” (Chan-Ling and Stone, 1991; Volterra and Meldolesi, 2005) or “tiling,” where each astrocyte creates its micro-anatomical domain with its processes overlapping with adjacent astrocytes only at their periphery (Bushong et al., 2002). Such spatial arrangement, combined with the heterogeneity of astrocytic responses, could be important in intercellular Ca2+ wave propagations and the related computational tasks carried out by astrocyte networks. The latter could be relevant in particular for the emergence of astrocytic functional maps observed in several brain areas (Giaume et al., 2010).
Increasing evidence argues in fact for a functional organization of astrocytes, reminiscent of that of cortical neurons (Bernardinelli et al., 2011). In the ferret visual cortex astrocytes, like neurons, respond to visual stimuli, with distinct spatial receptive fields and sharp tuning to visual stimulus features, including orientation and spatial frequency (Schummers et al., 2008). The stimulus-feature preferences of astrocytes there can be mapped across the cortical surface, in close register with neuronal maps (Schummers et al., 2008). Similar observations were also reported for astrocytes in the motor cortex (Haas et al., 2006) and in the somatosensory cortex (Schipke et al., 2008) as well as in the olfactory bulb (De Saint Jan and Westbrook, 2005).
Anatomical compartmentalization of astrocyte networks could underlie such functional organization. Both in the somatosensory barrel cortex and in the olfactory bulb, astrocytes are preferentially connected by gap junctions within the same barrel or olfactory glomerulus rather than between adjacent barrels or glomeruli (Houades et al., 2008; Roux et al., 2011). However, spatial confinement of gap junctions within single barrels/glomeruli might not fully account for the selective activation of astrocyte by electrical stimulation. Indeed, similar Ca2+ signals in response to the same stimulus could be observed in the same barrel yet with pharmacological block of astrocyte gap junctions (Schipke et al., 2008). Thus, additional factors must contribute to the astrocyte tuned response, which likely emerges as a result of the dynamical interactions with surrounding neurons (Rouach et al., 2004) and could ultimately depend on location and nature of activated cells among the other astrocytes in the network (Matyash and Kettenmann, 2010; García-Marqués and Lópes-Mascaraque, 2012). In the case of Figure 2E, for example, stimulated FM-encoding cells could trigger Ca2+ signaling in neighboring cells in a regenerative fashion thus extending their tuned response in space. On the other hand, AFM cells, acting as propagation barriers, could shape the borders of this tuned response, eventually drawing the topographical features of the ensuing functional map (Lallouette and Berry, 2012).
There is a number of possible routes by which astrocytes could release gliotransmitters (Ni et al., 2007; Parpura and Zorec, 2010; Zorec et al., 2012), but Ca2+-dependent exocytosis is likely the major one on a physiological basis (Barres, 2008; Parpura et al., 2011). However, the identity of incoming inputs, the underlying molecular mechanism and the physiological conditions that govern gliotransmitter exocytosis largely remain to be elucidated (Montana et al., 2006; Ni et al., 2007; Santello and Volterra, 2009; Parpura and Zorec, 2010). Calcium-dependent exocytosis of glutamate or ATP from astrocytes, for example, may strictly depend on the nature of the upstream Ca2+ signal (Perea and Araque, 2005; Li et al., 2008; Marchaland et al., 2008; Pryazhnikov and Khiroug, 2008; Malarkey and Parpura, 2011); including the type of neurotransmitter involved and the type of receptor engaged (Enkvist and McCarthy, 1992; Muyderman et al., 2001; Coco et al., 2003; Bezzi et al., 2004; Blomstrand and Giaume, 2006). The influence of astrocytic glutamate or ATP on synaptic activity also likely depends both on the type of incoming stimulus and on the specific localization of the engaged receptor in the astrocyte (Perea and Araque, 2005; Santello and Volterra, 2009).
A large amount of evidence suggests that gliotransmitter exocytosis from astrocytes bears several similarities with its synaptic homologous (Bergersen and Gundersen, 2009; Santello and Volterra, 2009; Bergersen et al., 2012) (Figure 3A). Astrocytes possess vesicular compartments that are competent for the regulated exocytosis of glutamate (Bezzi et al., 2004; Bergersen and Gundersen, 2009) and ATP (Coco et al., 2003; Jaiswal et al., 2007; Zhang et al., 2007). Similarly to synapses, astrocytes express soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) necessary for exocytosis (Parpura et al., 1995; Schubert et al., 2011) as well as proteins responsible for concentrating glutamate or ATP into vesicles (Bezzi et al., 2004; Montana et al., 2004; Zhang et al., 2004b; Sawada et al., 2008). Fusion with the plasma membrane, trafficking and recycling of astrocytic glutamate and ATP secretory vesicles have been observed (Bezzi et al., 2004; Chen et al., 2005; Crippa et al., 2006; Jaiswal et al., 2007; Pangršic et al., 2007; Stenovec et al., 2007), which are indicative of quantal glutamate and ATP release (Del Castillo and Katz, 1954; Pasti et al., 2001; Domercq et al., 2006; Jaiswal et al., 2007; Pangršic et al., 2007; Marchaland et al., 2008; Santello et al., 2011).
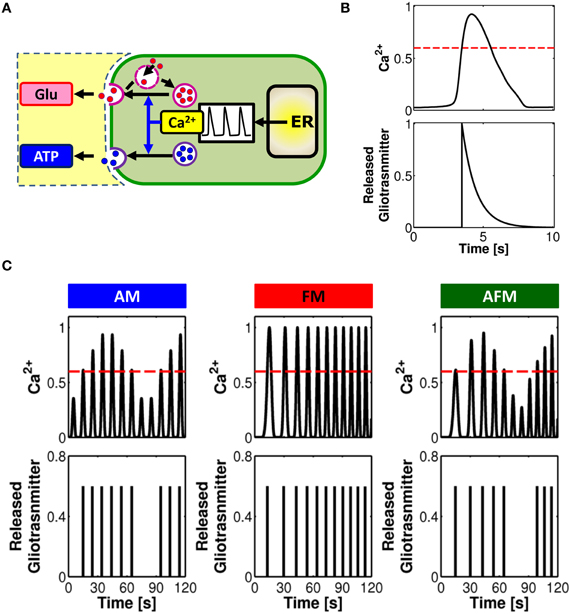
Figure 3. Linking gliotransmitter exocytosis to various Ca2+ encoding modes. (A) Calcium-dependent glutamate and ATP exocytosis from astrocytes are both brought forth by a vesicular compartment in the astrocyte competent for regulated exocytosis. The frequency of exocytotic events is directly controlled by the shape and frequency of Ca2+ oscillations. (B) Modeling concept for an “exocytosis event” from the astrocyte. Calcium (top trace) triggers exocytosis of glutamate or ATP every time it increases beyond a certain threshold concentration value (red dashed line). The overall release can then be approximated, under proper assumptions, by an exponentially-decaying pulse of extracellular concentration of glutamate or ATP (bottom trace). (C) Distinct Ca2+ encoding patterns could translate into distinct rates of gliotransmitter exocytosis events. In this way, synaptic activity encoded by astrocytic Ca2+ signals is linked to the frequency of glutamate/ATP release from the astrocyte in a unique fashion. Adapted from De Pittà et al. (2011).
Glutamate exocytosis from cultured astrocytes evoked by GPCRs is observed with short delay, i.e. 50–100 ms, after the rise in submembrane Ca2+, and is mediated by a rapid succession of fusion events which peaks within ~500 ms from the onset and decays to baseline much more slowly (>1 s), though generally before the recovery of basal Ca2+ levels (Domercq et al., 2006; Marchaland et al., 2008; Santello et al., 2011) (refer also to Table 2). In addition to this “exocytotic burst,” larger delays with slower rate of sustained vesicular fusion events have also been reported (Kreft et al., 2004; Malarkey and Parpura, 2011). The delayed onset with respect to the Ca2+ rise is consistent with a threshold Ca2+ concentration for release (Pasti et al., 1997; Parpura and Haydon, 2000). On the other hand the overall release of glutamate is characterized by a rising phase much faster than its decay and can be approximated by an exponential function like synaptic exocytosis yet with a decay time much slower than the latter (Marchaland et al., 2008; De Pittà et al., 2011; Santello et al., 2011) (Figure 3B). Although much less characterized than its glutamate counterpart, astrocytic ATP exocytosis could occur in a qualitatively similar fashion in spite of different underlying kinetics (Pangršic et al., 2007; Pryazhnikov and Khiroug, 2008; Li et al., 2008). Based on these arguments, it was proposed that astrocytic gliotransmitter release could be modeled using the same mathematical description of synaptic release, although the two mechanisms are likely different in their molecular machinery, with the kinetics of astrocyte release much slower than synaptic release (De Pittà et al., 2011; Schubert et al., 2011).

Table 2. Comparison of time scales of rise, decay and full-width half-maximum (FWHM) duration of changes of intracellular Ca2+, extracellular glutamate (Glu) and ATP in astrocytes and neurons.
How are different modes of Ca2+ encoding translated into glutamate or ATP release? Assuming proper conditions about the rate of clearance of these gliotransmitters with respect to the underlying intracellular Ca2+ dynamics that mediate their exocytosis (Abbracchio et al., 2009), an intriguing theoretical prediction is that various patterns of Ca2+ oscillations could mostly correspond to different rates of gliotransmitter release (De Pittà et al., 2011). This is presented in Figure 3C where, for three stereotypical patterns of Ca2+ oscillations, that is AM, FM, and AFM, the corresponding timing of gliotransmitter release from the astrocyte is shown. In this example, FM-encoding Ca2+ oscillations always cross the threshold for exocytosis (dashed red line), triggering gliotransmitter release every time. Conversely, AM or AFM oscillations may not be large enough to reach such threshold, resulting in some oscillations that fail to trigger gliotransmitter release. In this fashion, while FM Ca2+ oscillations trigger gliotransmitter exocytosis at their own frequency, the amplitude of AM and AFM oscillations could selectively discriminate which Ca2+ pulse triggers exocytosis, eventually dictating the frequency of “measured” glutamate or ATP release events (De Pittà et al., 2011). Further experimental investigations are required to elucidate whether such prediction could effectively mimic reality.
While astrocytic Ca2+ signals could be triggered both by spontaneous and evoked synaptic release, gliotransmitter release might be not (Di Castro et al., 2011; Panatier et al., 2011). Blockade of Ca2+-dependent glutamate release in astrocytes in the dentate gyrus was indeed observed to be effective in reducing the frequency of synaptic release events only when these were evoked by action potentials, but not when they happened spontaneously. This suggests that release of glutamate from astrocytes in this region could effectively occur only in presence of evoked synaptic activity (Di Castro et al., 2011). Interestingly, the Ca2+ elevations in astrocyte processes triggered by action potentials were reported to be more complex, larger in amplitude and more extended in space, than those generated by spontaneous synaptic release events. Since glutamate could be released from several sites along the same astrocyte process and this latter could contact several synapses (Panatier et al., 2011), one of such Ca2+ elevations could then generate multiple spatially-distinct glutamate release events modulating synaptic release at several other synapses. The same Ca2+ elevation however, could bring forth at subcellular regions of the process, different local Ca2+ dynamics, in close analogy to the behavior of the heterogeneous AM/AFM oscillations in the astrocyte chain in Figure 2E. Such different local Ca2+ dynamics could in turn result in different rates of glutamate release and thus in different modulations of synaptic release in a non-random fashion. In this way, the same astrocytic process or a segment of it, could carry out multiple regional modulations of synaptic release, depending both on the temporal and spatial dynamics of synaptic release. It is however possible that the mechanisms underlying Ca2+-dependent gliotransmission differ in different brain areas (Matyash and Kettenmann, 2010; Zhang and Barres, 2010). Indeed, in contrast with the above observations in the dentate gyrus, astrocytes in the stratum radiatum of the hippocampus were reported to release glutamate even in the absence of synaptic activity evoked by action potentials (Bonansco et al., 2011).
Another issue is what makes a single astrocyte release one gliotransmitter rather than another. A possibility is that different gliotransmitters are released in response to different stimuli. In situ studies indeed suggest that purinergic GPCR-mediated astrocytic Ca2+ signals could preferentially trigger glutamate release (Jourdain et al., 2007; Perea and Araque, 2007; Di Castro et al., 2011) (see also Table 1). In contrast, Ca2+ elevations triggered by glutamate could bring forth ATP release (Pascual et al., 2005; Gordon et al., 2009; Panatier et al., 2011). Overall these observations hint that a tight association likely exists between the type of targeted astrocytic receptor and the secretory machinery of gliotransmitters (Zorec et al., 2012). Moreover, in the same astrocyte, different gliotransmitters could be contained in different organelles with different secretory properties in response to Ca2+ signals (Coco et al., 2003). Indeed, while glutamate seems to be preferentially released by synaptic-like microvesicles (Bezzi et al., 2004; Jourdain et al., 2007; Bergersen et al., 2012), ATP is likely released by dense-core granules (Coco et al., 2003), and/or lysosomes (Jaiswal et al., 2007; Zhang et al., 2007; Li et al., 2008). Although the underlying molecular machinery of exocytosis remains to be elucidated, each organelle population is likely secreted in a different fashion (Pryazhnikov and Khiroug, 2008). In vitro evidence showed in fact that Ca2+ signals that triggered release of glutamate-containing vesicles (Marchaland et al., 2008) did not release ATP-filled organelles and vice versa (Coco et al., 2003; Li et al., 2008), ultimately suggesting that glutamate and ATP could be released in response to different Ca2+ signals (Parpura and Zorec, 2010).
Intriguingly, stimulation of astrocyte GPCRs can evoke fast gliotransmitter exocytosis within few hundreds of milliseconds (Bezzi et al., 2004; Domercq et al., 2006; Marchaland et al., 2008; Santello et al., 2011), indicating that the Ca2+-dependent process that couples stimulus with secretion must be fast. Indeed, the peak of GPCR-mediated Ca2+ release from the ER can be as fast as 50–250 ms (Marchaland et al., 2008; Di Castro et al., 2011; Panatier et al., 2011) and Ca2+-dependent exocytosis of single glutamate vesicles can occur within less than 5–20 ms from Ca2+ elevation (Chen et al., 2005; Bowser and Khakh, 2007; Marchaland et al., 2008; Santello et al., 2011). Therefore, to assure fast stimulus-secretion coupling, IP3 diffusion from the site of production by GPCRs at the plasma membrane to IP3 receptors on the ER membrane must also be of the order of tens of milliseconds and so must Ca2+ diffusion from the mouth of IP3Rs to the Ca2+ sensor of exocytosis (Zhang et al., 2003, 2004a; Ni et al., 2007). Given that the diffusion times of IP3 or Ca2+ can be estimated as the half of the square of the diffusion distance from the site of their production in the cytoplasm divided by the respective diffusion constants (Syková and Nicholson, 2008), which can be as high as ~200–300 μm2/s for IP3 (Allbritton et al., 1992; Sneyd et al., 1994) and 30 μm2/s for Ca2+ (Kang and Othmer, 2009), then diffusion times of the order of milliseconds could be obtained only for diffusion lengths at most in the micron range. This prediction is fully supported by the functional evidence of local GPCR-evoked, ER-dependent Ca2+ microdomains in astrocytic processes (Di Castro et al., 2011; Panatier et al., 2011) and by the morphological evidence that at astrocytic processes the ER stores are found at ~200–600 nm distance both from the plasma membrane and gliotransmitter-containing vesicles (Bezzi et al., 2004; Marchaland et al., 2008; Bergersen et al., 2012). Moreover, GPCR agonist-mediated Ca2+ transients were observed restricted areas beneath the plasma membrane, in close proximity to the sites of exocytosis (Marchaland et al., 2008), suggesting that both GPCRs and secretory vesicles must be spatially close to the IP3Rs responsible for Ca2+ release from the ER. This indicates that the location of IP3Rs along the ER membrane in astrocyte processes might be non-random (Blaustein and Golovina, 2001), in tight spatial association with astrocytic GPCRs on the plasma membrane and releasable gliotransmitter organelles (Marchaland et al., 2008; Panatier et al., 2011). Such spatial coupling could ultimately underlie the preferential occurrence of functional gliotransmission from astrocytic processes rather than from the soma (Gordon et al., 2009), entailing strict spatiotemporal requirements for Ca2+ signals to trigger release of gliotransmitters from the astrocyte and modulate synaptic transmission.
Astrocyte-derived glutamate and ATP or adenosine can modulate synaptic transmission, either increasing neurotransmitter release (Araque et al., 1998a,b; Fiacco and McCarthy, 2004; Jourdain et al., 2007; Perea and Araque, 2007; Bonansco et al., 2011; Di Castro et al., 2011; Panatier et al., 2011) or decreasing it (Zhang et al., 2003, 2004a; Pascual et al., 2005; Andersson and Hanse, 2010, 2011) depending on the type of presynaptic receptor involved and the brain area (pathway B in Figure 1B; see also Table 1). At excitatory synapses in the hippocampal dentate gyrus, glutamate is released from surrounding astrocytic processes in close proximity to presynaptic NR2B-containing NMDA receptors (Figure 4A). Activation of these receptors results in increased synaptic release and strengthening of synaptic transmission (Jourdain et al., 2007). At Schaffer collateral synapses in the CA1 hippocampal area a similar effect is mediated by presynaptic mGluRs (Fiacco and McCarthy, 2004; Perea and Araque, 2007). Besides directly targeting presynaptic receptors, astrocyte-released glutamate could also bind ionotropic receptors found along the axons of hippocampal CA3 pyramidal cells, broadening the profile of propagating action potentials (APs) (Sasaki et al., 2011). The broadened APs in turn, trigger larger Ca2+ elevations in presynaptic boutons, resulting in larger synaptic release probability.
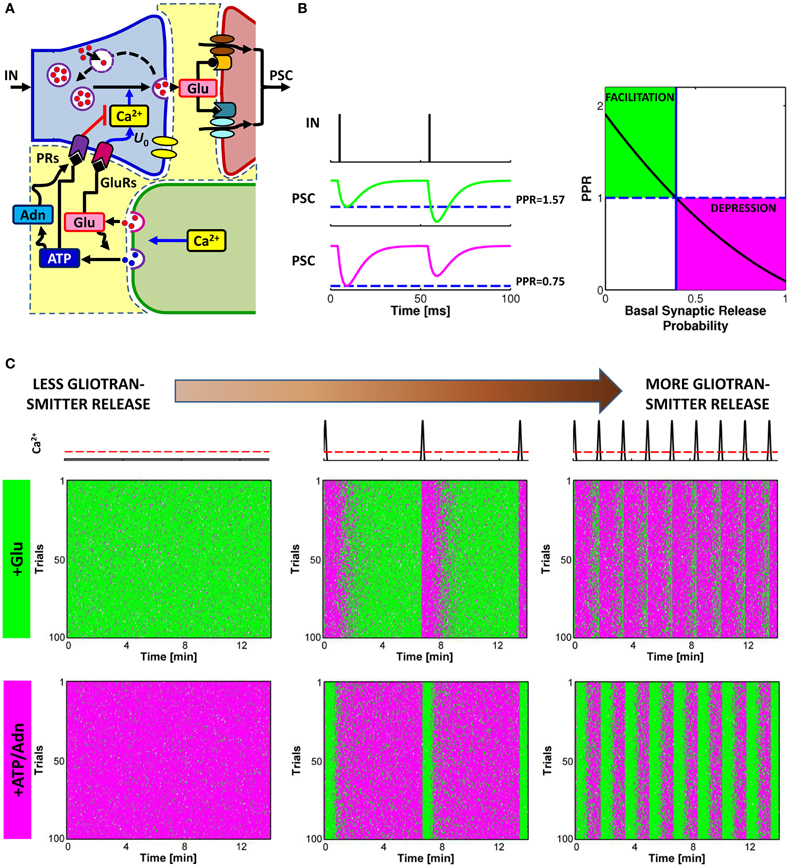
Figure 4. Glutamate or ATP released from astrocytes regulates transitions between facilitation and depression of synaptic transmission. (A) Conceptual framework for the regulation of synaptic release probability at basal conditions by astrocytes. Astrocyte-released glutamate increases basal synaptic release probability (U0), whereas astrocyte-released ATP/Adn generally decreases it. (B) Changes in synaptic release probability due to astrocytic gliotransmitters can be detected by variations of paired-pulse plasticity quantified by paired-pulse ratio (PPR). Paired-pulse facilitation (left, green traces) of postsynaptic currents (PSCs) corresponds to PPR values above 1 (right, green-shaded area) whereas paired-pulse depression (left, magenta traces) are associated with PPR values below 1 (right, magenta-shaded area). (C) Raster plots of simulated PSC pairs for 100 different input spike trains with same statistics colored according to the paired-pulse ratio: green for facilitation, PPR > 1; magenta for depression, PPR < 1. For increasing rates of exocytosis of gliotransmitter from the astrocyte, mimicked by increasing rates of Ca2+ crossing beyond the threshold for exocytosis (top row, red dashed line), synaptic plasticity could be progressively changed to its opposite depending on the type of gliotransmitter. Astrocytic glutamate could thus turn facilitating synapses into depressing (middle row) whereas astrocyte-derived ATP or adenosine could turn depressing synapses into facilitating (bottom row). Simulations are based on a model of astrocyte-regulation of synaptic release introduced in De Pittà et al. (2011). “Basal Synaptic Release Probability” in (B) refers to the probability of synaptic release at rest, that is when synaptic activity is assumed to be very low and the amount of neurotransmitter released upon arrival of an action potential to the presynaptic terminal is essentially independent of previous release events (Zucker and Regehr, 2002; De Pittà et al., 2011).
Conversely, astrocyte-released ATP and its derivative adenosine, bind to presynaptic PRs, i.e. P2Y1Rs or A1Rs, respectively, decreasing synaptic release (Zhang et al., 2003, 2004a; Pascual et al., 2005). However, astrocyte-derived adenosine, could also target A2A receptors which can increase synaptic release (Panatier et al., 2011) (omitted from Figures 1A and 4A for simplicity). Both effects of adenosine—inhibitory via A1Rs and stimulatory via A2ARs—have been described at hippocampal CA3-CA1 synapses (Zhang et al., 2003, 2004a; Pascual et al., 2005; Serrano et al., 2006; Panatier et al., 2011), and the prevalence of one on the other likely depends on the level of synaptic activity (Panatier et al., 2011). This would be in line with observations at the frog's neuromuscular junction, where different stimulations of the tibial nerve differentially activated A1Rs or A2ARs in association with different Ca2+ dynamics in the peri-junctional glial cell (Todd et al., 2010). Intriguingly, at hippocampal synapses different synaptic stimuli could differentially change the morphology of astrocyte perisynaptic processes (Haber et al., 2006; Lavialle et al., 2011) thus reshaping, in an activity-dependent fashion, the extracellular space of interaction between astrocyte-derived adenosine and presynaptic receptors. This could dynamically regulate access of adenosine to one receptor with respect to the other (Haber et al., 2006; Syková and Nicholson, 2008) ultimately modulating synaptic release in a complex fashion. The functional consequences of astrocytic remodeling were indeed demonstrated in the supraoptic nucleus, where dynamic changes in the astrocytic wrapping of synapses during lactation could regulate the extent of synaptic glutamate spillover and thereby control heterosynaptic depression of GABAergic transmission by presynaptic mGluRs (Oliet et al., 2001; Piet et al., 2004).
The opposite effects due to astrocytic glutamate or ATP/adenosine could endow astrocytes with the capacity to exert non-stereotyped bimodal control of synaptic transmission (Volterra and Meldolesi, 2005). On the other hand, the temporal concurrence of both these effects due to co-expression of inhibitory and stimulatory receptors at the same synaptic terminals (Shigemoto et al., 1997; Rebola et al., 2005), could result in occlusion, i.e. no net effect on synaptic release by the astrocyte (De Pittà et al., 2011). Alternatively, balanced activation (and possibly occlusion) of A1 and A2A receptors by astrocyte-derived adenosine could set synaptic release in basal conditions (Panatier et al., 2011). Hence, the ensuing regulation of synaptic transmission triggered by gliotransmitters in response to stimuli, could result instead from an unbalance of activation of these receptors rather than by the distinct activation of one receptor type with respect to the other, ultimately providing a high degree of complexity in the control of synaptic transmission by astrocytes. For the sake of clarity, in the following we will consider only release-decreasing effects of astrocyte ATP or its derivative, adenosine.
The details of the biochemical mechanism underlying modulation of synaptic release by astrocytic glutamate or ATP (or adenosine) likely depend on the type of targeted presynaptic receptors and are not fully understood (Pinheiro and Mulle, 2008). The simplest explanation would be that astrocytic glutamate and ATP lead to a modulation of presynaptic intracellular Ca2+ levels which eventually results in a modulation of synaptic release probability (Zucker and Regehr, 2002; Pinheiro and Mulle, 2008) with significant repercussions on synaptic plasticity, including short-term depression and facilitation.
Short-term facilitation and depression can be assessed by measuring the paired-pulse ratio (PPR), i.e. the ratio between the amplitudes of successive postsynaptic currents (PSCs) recorded in response to a pair of action potentials in rapid succession as illustrated in Figure 4B (Zucker and Regehr, 2002). When the value of the peak postsynaptic current associated to the second incoming spike is larger than the peak current recorded in coincidence with the first spike (green traces), then synaptic release is increasing for incoming spikes, i.e. facilitation occurs, and the corresponding PPR is larger than unity. Conversely, when the second peak is less than the first peak (magenta traces), this marks a decrease of neurotransmitter release from the presynaptic terminal which reflects depression, and the corresponding value of PPR is less than one. It should be noted that, by varying the interval between two pulses, the same synapse can be either depressed or facilitated. For simplicity we omit the interpulse interval as a variable in this description.
In general, in basal conditions, i.e. in response to an individual action potential, the value of synaptic release probability of an individual synapse or of an ensemble of synapses defines the nature of synaptic transmission at that synapse/ensemble, namely whether it is facilitating or depressing, with low values of probability favoring facilitation and high values favoring depression (Abbott and Regehr, 2004) (Figure 4B, right). Thus, any modulation of synaptic release probability by gliotransmitters that changes the PPR from below unity to values above it or vice versa, could switch the mode of synaptic transmission from depressing to facilitating or vice versa. This scenario was theoretically addressed in De Pittà et al. (2011) and it was shown to substantially agree with experiments. Indeed, at hippocampal synapses, the increase of synaptic release probability due to astrocytic glutamate correlates with a decrease of the PPR (Jourdain et al., 2007; Perea and Araque, 2007; Bonansco et al., 2011). Conversely, a decrease of synaptic release due to the action of astrocyte-derived ATP (or adenosine) is accompanied by an increase of the PPR (Zhang et al., 2003, 2004a). The frequency of astrocytic glutamate (Glu) and/or ATP exocytosis are likely crucial in the regulation of the extent of the modulation of synaptic release by astrocytes (De Pittà et al., 2011), a principle illustrated in Figure 4C. This figure shows the simulated peak postsynaptic currents for the same synapse in response to 100 trials of presynaptic spike trains with identical statistics (raster plots). Each column in Figure 4C corresponds to a different frequency of astrocytic Ca2+ pulses (top row), yielding to different frequencies of gliotransmitter release when Ca2+ crosses the threshold for exocytosis (dashed red line) (see also Figure 3B). Colors in the raster plots refer to paired-pulse plasticity quantified by PPR. For two consecutive presynaptic spikes, if the second spike releases synaptic neurotransmitter more than the first spike, then it is PPR > 1, paired-pulse facilitation is observed, and the peak postsynaptic current associated to the second spike is colored in green. Vice versa, if the amount of neurotransmitter released by the second spike is less than that released by the first one, then PPR < 1, that is paired-pulse depression occurs, and the second peak postsynaptic current is colored in magenta.
In absence of astrocytic gliotransmitter (left column), the raster plot of a facilitating synapse (middle row) shows predominant paired-pulse facilitation, i.e. mostly green dots. However, in presence of release of glutamate from the astrocyte (+Glu), as explained above, the stimulatory effect of this gliotransmitter on synaptic release changes paired-pulse plasticity which is marked by the appearance of magenta bands in the raster plots (middle and right columns). These bands locate time intervals where paired-pulse depression becomes prominent (i.e. predominance of magenta dots) on the overall paired-pulse facilitation background (green dots). Notably these bands are almost in coincidence with glutamate release from the astrocyte (top row) and their number increases with the glutamate release frequency. Therefore, for the same time window, the same originally-facilitating synapse gets increasingly depressing as the rate of glutamate release from the astrocyte increases. The opposite is observed for an otherwise depressing synapse (i.e. predominance of paired-pulse depression, thus mostly magenta dots in the bottom left raster plot). In this case, in presence of release of ATP and its derivative adenosine (+ATP/Adn) from the astrocyte, due to the inhibitory effect of astrocyte-derived purines on synaptic release considered in this example, green bands appear in the raster plots which mark the onset of periods of predominant paired-pulse facilitation. The number of these bands grows for higher rates of ATP release from the astrocyte so that the same originally-depressing synapse behaves more akin of a facilitating one as the rate of ATP release from the astrocyte increases.
An intriguing prediction that follows from the above arguments is that the frequency of gliotransmitter release, by modulating synaptic release probability at basal conditions, could dynamically control the nature of synaptic transmission as elucidated in Figure 5A. In particular, under certain conditions, a threshold frequency for gliotransmitter release (blue line) could exist above which the astrocyte can switch the nature of synaptic transmission, turning depressing synapses into facilitating or vice versa, facilitating synapses into depressing (De Pittà et al., 2011). Hence, the plasticity mode at such synapses is not fixed but rather is set by the release rate of gliotransmitters from neighboring astrocytic processes.
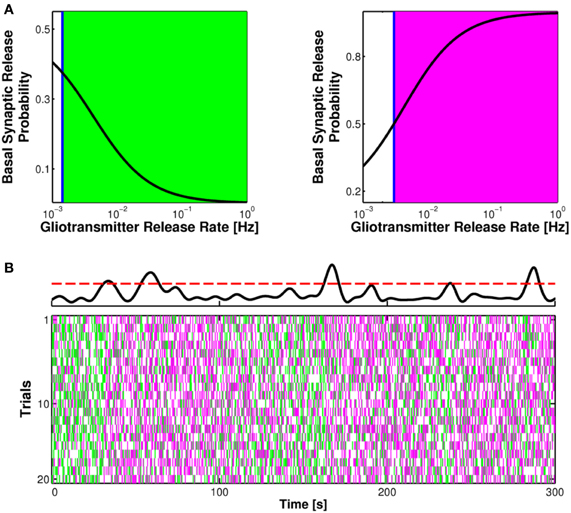
Figure 5. Short-term synaptic plasticity is physiologically set by the rate of gliotransmitter release from the astrocyte. (A) The rate of glutamate or ATP release from the astrocyte could differently affect basal synaptic release probability. In particular, a threshold frequency for the release of these gliotransmitters could exist (blue vertical line) beyond which a depressing synapse could turn into facilitating (right, green area) or vice versa, a facilitating synapse could become depressing (left, magenta area). Adapted from De Pittà et al. (2011). (B) In basal conditions, synaptic release is due to sporadic neuronal network firing and the possible frequency of Ca2+ fluctuations beyond the threshold for exocytosis (dashed red line) in the astrocyte (top trace). In this fashion plastic changes in paired-pulse ratio could be inherently regulated by astrocytic gliotransmitters, as shown here for the case of an originally facilitating synapse under the effect of astrocytic glutamate exocytosis (20 trials with identical input statistics). Adapted from Berry et al. (2011). The color code for the raster plot is the same as in Figure 4C.
Remarkably, the threshold frequency of gliotransmitter release that discriminates between facilitation and depression at one synapse can be as low as 0.05 Hz or less (as in the case of Figure 5A) thus falling within the range of Ca2+ oscillations observed in astrocytes in basal conditions (Parri et al., 2001; Aguado et al., 2002; Bonansco et al., 2011; Di Castro et al., 2011). In such conditions as shown in Figure 5B (top), intracellular Ca2+ levels in astrocytes spontaneously fluctuate in a highly stochastic fashion (Hirase et al., 2004; Di Castro et al., 2011) and can similarly cross the threshold for gliotransmitter release. The ensuing release of gliotransmitter, however, could be sufficient to tonically set the basal tone of synaptic transmission (Figure 5B). Inasmuch as the rate of gliotransmitter release could also correlate with the incoming synaptic stimulus through Ca2+ dynamics in the astrocytes (Aguado et al., 2002; Di Castro et al., 2011), this scenario discloses the possibility that astrocytes act as endogenous regulators of the efficacy of synaptic transmission (Haydon, 2001; Giaume et al., 2010; Halassa and Haydon, 2010; Di Castro et al., 2011; Panatier et al., 2011). That is, through integration of synaptic activity by means of their Ca2+ signals, astrocytes could adapt synaptic strength according to the history of the synapse.
The effect of modulation of synaptic release probability by astrocytic gliotransmitters may decay more slowly than the Ca2+ elevation triggering astrocyte exocytosis (Fiacco and McCarthy, 2004; Serrano et al., 2006), and still be present upon gliotransmitter release by following Ca2+ increases (Volman et al., 2007). Therefore astrocytic Ca2+ activity resulting in high rates of gliotransmitter release, could bring forth long-lasting modulations of synaptic release. Insofar as synaptically-released neurotransmitter could shape postsynaptic signaling at the basis of long-term synaptic plasticity (Bliss and Collingridge, 1993), persistent modulations of synaptic release probability by astrocyte could ultimately contribute to long-term changes of synaptic strength underlying neural processing, memory formation and storage of information. Support to this scenario comes from studies on hippocampal synapses for which the temporal coincidence of postsynaptic depolarization with the increase of Ca2+ elevations in neighboring astrocytes was shown to induce long-term potentiation (LTP) of synaptic transmission (Perea and Araque, 2007; Navarrete and Araque, 2010; Navarrete et al., 2012a,b). Notably, this form of LTP is independent of the activation of postsynaptic NMDARs but rather, it depends on glutamate released from astrocytes, which persistently potentiates synaptic transmitter release through activation of presynaptic mGluRs (Perea and Araque, 2007). On the other hand, at synapses between excitatory neurons in layers 4 and 2/3 in the rat barrel cortex, activity-dependent induction of long-term depression (LTD) also requires astrocyte Ca2+ signaling (Min and Nevian, 2012). At these synapses, postsynaptically-released endocannabinoids mediate Ca2+-dependent release of glutamate from astrocytes which targets presynaptic NMDARs bringing forth LTD (Sjöström et al., 2003; Rodríguez-Moreno and Paulsen, 2008).
The differential induction of LTP and LTD in neighboring synapses has been suggested to determine the size and shape of cortical functional topographic units such as ocular dominance columns in the primary visual cortex and whisker barrels in the primary somatosensory cortex (Feldman and Brecht, 2005; Hensch, 2005; Li et al., 2009). Thus, the possible involvement of astrocytes in LTP and LTD suggests that these cells could contribute to the plasticity of cortical maps and the development of corresponding sensory representations (Rossi, 2012).
Modulation of synaptic release probability by astrocytic gliotransmitters could also alter the temporal order of correlated pre- and postsynaptic spiking that critically dictates spike-timing-dependent plasticity (STDP) (Pascual et al., 2005). Inhibition of spontaneous glutamate release from astrocytes in hippocampal CA1 synapses, consistent with a decrease of synaptic release probability, was indeed reported to modify the threshold for induction of spike-timing-dependent LTP (Bonansco et al., 2011). In this fashion, astrocytes by gliotransmission-mediated regulations of synaptic release probability could control not only different mechanisms of synaptic plasticity but also the threshold of synaptic activity required for their onset, thus playing a role in metaplasticity too, that is the plasticity of synaptic plasticity (Abraham, 2008).
Gliotransmitters do not control synaptic plasticity only via presynaptic actions, but also by actions on postsynaptic receptors. Indeed, the induction of LTP itself appears to be uniquely controlled by astrocytes through the release of D-serine (Santello and Volterra, 2010) (pathway C in Figure 1B). Both at hippocampal and cortical synapses astrocytic D-serine rather than glycine, is the endogenous co-agonist of synaptic NMDA receptors (Henneberger et al., 2010; Takata et al., 2011; Papouin et al., 2012). By controlling the level of co-agonist site occupancy of postsynaptic NMDARs, astrocytic D-serine affects the level of activation of these receptors and thus activity-dependent long-term synaptic changes (Bains and Oliet, 2007). In particular, the additional burst of activation of postsynaptic NMDARs (about 25% more) induced by astrocyte D-serine release seems necessary for LTP induction (Henneberger et al., 2010). Moreover, in conditions of reduced synaptic coverage by astrocytes, such as during lactation in the hypothalamus, higher presynaptic activity is required to obtain LTP while the same level of activity that normally induces LTP results instead in LTD (Panatier et al., 2006). In such conditions, astrocyte-released D-serine is diluted in the larger extracellular space resulting in a reduced number of postsynaptic NMDARs recruited by synaptic activity, which ultimately translates into smaller postsynaptic Ca2+ increases. Therefore, experimental protocols that would be expected to cause LTP, elicit LTD instead. This is in agreement with the Bienenstock-Cooper-Munro (BCM) model for variation of the threshold for LTP, which predicts that the relationship between synaptic activity and persistent changes in synaptic strength can vary according to the number of NMDARs available during synaptic activation (Bienenstock et al., 1982; Abraham and Bear, 1996). Effectively, by adjusting the D-serine occupancy of the NMDAR co-agonist-binding site, astrocytes can shift the relationship between activity and synaptic strength (Panatier et al., 2006).
Besides D-serine, experimental evidence hints that glutamate and ATP released from astrocytes could also directly bind postsynaptically-located receptors and accordingly, play a role in regulation of long-term synaptic plasticity. In the paraventricular nucleus of the hypothalamus for example, ATP released from astrocytes could directly target postsynaptic P2X7 receptors, promoting insertion of postsynaptic AMPARs which results in LTP of synaptic transmission (Gordon et al., 2005). Interestingly, ATP release is mediated by Ca2+ dynamics triggered in astrocytes by noradrenergic afferents which, in the hypothalamus, generally lack direct postsynaptic contacts (Sawyer and Clifton, 1980), thus hinting that signaling in this vital homeostatic circuit may require dynamic neuron-glia interactions.
Glutamate released from astrocyte could also target extrasynaptically-located NR2B-containing NMDA receptors at postsynaptic terminals, triggering slow inward currents (SICs) (Fellin et al., 2004; D'Ascenzo et al., 2007; Navarrete and Araque, 2008; Bardoni et al., 2010; Pirttimaki et al., 2011; Navarrete et al., 2012a,b) mainly mediated by Ca2+ ions (Cull-Candy et al., 2001), whose depolarizing action could affect postsynaptic neuronal firing (D'Ascenzo et al., 2007; Pirttimaki et al., 2011). In the primary visual cortex, nucleus basalis-mediated cholinergic activation of astrocytes mediates an increase of SICs frequency which correlates with a long-lasting increase of firing activity in visual responses of V1 excitatory neurons (Chen et al., 2012). The ensuing modulations of firing activity of these neurons by astrocyte-mediated SICs might ultimately affect STDP at individual synapses controlling orientation-specific responses of V1 neurons to visual stimuli (Jia et al., 2010).
Modulation of synaptic transmission by Ca2+-dependent gliotransmission may not be limited to the very synapses that trigger Ca2+ activity in the astrocyte but it could also affect farther synaptic domains in a multimodal fashion (Kozlov et al., 2006), depending both on the morphology of the sites of astrocyte-synapse reciprocal communication (Ventura and Harris, 1999; Haber et al., 2006) and the functional connectivity of the astrocytic network (Pannasch et al., 2011). High-frequency activity of a Schaffer collateral fiber can trigger the potentiation of synaptic transmission at the same fiber but heterosynaptic suppression of another, adjacent fiber, by inducing ATP release from an astrocyte interposed between the two fibers (Zhang et al., 2003, 2004a; Pascual et al., 2005). In the somatosensory cortex in particular, astrocyte-mediated heterosynaptic suppression could modulate GABAergic inhibitory transmission (Benedetti et al., 2011) which plays a dominant role in the control of cortical neuronal excitability (Swadlow, 2002). Given that both experimental observations and theoretical arguments suggest that excitation and inhibition are globally balanced in cortical circuits (Shadlen and Newsome, 1994; Troyer and Miller, 1997; Shu et al., 2003; Haider et al., 2006), one may speculate that this mechanism could be involved in gating of signal transmission (Buzsáki, 2010). That is, by modulating inhibitory synaptic transmission, astrocytes could favor network excitation resulting in neuronal firing consistent with the transmission, i.e. gating “on,” of specific stimuli rather than others (Vogels and Abbott, 2009).
The latter idea could also bring to a possible role of astrocyte signaling in coherent function of neural networks underlying potential behavioral states (Engel et al., 2001). In cortical slices, for example, stimulation of a single astrocyte was reported to activate large portions of the astrocytic network and to result in an increase of synchronized neuronal depolarizations (Poskanzer and Yuste, 2011). This phenomenon was suggested to modulate the induction of cortical UP and DOWN states, possibly involved in determining the oscillatory activity observed in slow-wave sleep (Fellin et al., 2004; Halassa and Haydon, 2010; Poskanzer and Yuste, 2011), and is consistent with reports of sleep perturbations in mice lacking astrocytic gliotransmitter exocytosis (Fellin et al., 2009; Halassa et al., 2009; Fellin et al., 2012).
A large body of evidence has accumulated over the last years, suggesting an active role of astrocytes in many brain functions. Collectively these data have fuelled the concept that synapses could be tripartite rather than bipartite, since in addition to the pre- and post-synaptic terminals, the astrocyte could be an active element in synaptic transmission (Araque et al., 1999; Haydon, 2001). While the tripartite synapse concept captures well the essence of astrocyte-regulated synapses, the inclusion of astrocytic signaling within our current knowledge of synaptic transmission could add more than just one level of complexity. Existing evidence suggests that astrocytes could produce not just tonic and diffuse modulatory influences on synapses but also engage in more focused, spatially precise and constrained communications with synaptic terminals (Anderson and Swanson, 2000; Jourdain et al., 2007; Santello and Volterra, 2009; Bergersen et al., 2012). This calls to rethink the definition of a functional synapse, to include the contribution from surrounding astrocytes. To conclude, the growing appreciation that astrocytes can regulate synaptic information at many levels, from activity of single synapses to network levels and behavioral states (Fellin et al., 2009; Halassa et al., 2009; Zorec et al., 2012) changes our understanding of brain communication and the role of glial cells in synaptic transmission. This resulting novel scenario offers an enticing platform for future theoretical investigations that we are just beginning to appreciate in its potential far-reaching implications.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Richard Robitaille, Mirko Santello, and Ari Barzilai for insightful conversations and Nicolas Liaudet for critical reading of the manuscript. The authors' research was supported by the Tauber Family Foundation (Maurizio De Pittà and Eshel Ben-Jacob); the Italy-Israel Joint Neuroscience Lab (Maurizio De Pittà); the Maguy-Glass Chair in Physics of Complex Systems at Tel Aviv University (Eshel Ben-Jacob); the Center for Theoretical Biological Physics at Rice University sponsored by the National Science Fundation (NSF) (grant PHY-0822283) (Eshel Ben-Jacob); the NSF program in Physics of Living Systems (PoLS) at the University of Michigan (grant PHY-1058034) (Eshel Ben-Jacob); the National Institute of Health (grants R01 NS059740 and R01 NS060870 to T. Sejnowski and M. Bazhenov) (Vladislav Volman); the High Council for Scientific and Technological Cooperation between France-Israel (Hugues Berry); the National Science Foundation (grant CBET 0943343) (Vladimir Parpura); and the Swiss National Science Foundation (grant 31003A-140999 and NCCRs “Synapsy” and “Transcure”) (Andrea Volterra).
Adn, adenosine; AFM, amplitude and frequency modulation; AM, amplitude modulation; AMPA, 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl) propanoic acid; AMPAR, AMPA glutamate receptor; AP, action potential; ATP, adenosine triphosphate; Ca2+, calcium; CICR, Ca2+-induced Ca2+ release; D-ser, D-serine; EAAT, excitatory aminoacid transporter; ER, endoplasmic reticulum; FM, frequency modulation; GABA, γ-aminobutyric acid; GJC, gap-junction channel; Glu, glutamate; GluR, glutamate receptor; mGluRs, metabotropic glutamate receptor; GPCR, Gq protein-coupled receptor; IP-5P, inositol polyphosphate 5-phosphatase; IP3, inositol 1,4,5-trisphosphate; IP3-3K, IP3 3-kinase; IP3R, IP3 receptor; LTD (LTP), long-term depression (potentiation); NMDA, N-methyl-d-aspartic acid; NMDAR, NMDA glutamate receptor; PLCδ (PLCβ), phospholipase Cδ (Cβ); PPR, paired-pulse ratio; PR, purinergic receptor; PSC, postsynaptic current; SIC, slow inward current; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; STDP, spike-timing-dependent plasticity; TACE, TNFα-converting enzyme; TNFα, tumor necrosis factor-α.
Abbracchio, M. P., Burnstock, G., Verkhratsky, A., and Zimmermann, H. (2009). Purinergic signalling in the nervous system: an overview. Trends Neurosci. 32, 19–29.
Abraham, W. (2008). Metaplasticity: tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 9, 387–387.
Abraham, W., and Bear, M. (1996). Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 19, 126–130.
Aguado, F., Espinosa-Parrilla, J. F., Carmona, M. A., and Soriano, E. (2002). Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J. Neurosci. 22, 9430–9444.
Agulhon, C., Fiacco, T. A., and McCarthy, K. D. (2010). Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signalling. Science 327, 1250–1254.
Agulhon, C., Petravicz, J., McMullen, A. B., Sweger, E. J., Minton, S. K., Taves, S. R., et al. (2008). What is the role of astrocyte calcium in neurophysiology? Neuron 59, 932–946.
Allbritton, N. L., Meyer, T., and Stryer, L. (1992). Range of messenger action of calcium ion and inositol 1 4, 5-trisphosphate. Science 260, 107–260. Abstract.
Andersson, M., and Hanse, E. (2010). Astrocytes impose postburst depression of release probability at hippocampal glutamate synapses. J. Neurosci. 30, 5776–5780.
Anderson, M. C., and Swanson, R. A. (2000). Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32, 1–14.
Andersson, M. S., and Hanse, E. (2011). Astrocyte-mediated short-term synaptic depression in the rat hippocampal CA1 area: two modes of decreasing release probability. BMC Neurosci. 12:87. doi: 10.1186/1471-2202-12-87
Araque, A., Martìn, E., Perea, G., Arellano, J., and Buño, W. (2002). Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J. Neurosci. 22, 2443–2450.
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1998a). Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur. J. Neurosci. 10, 2129–2142.
Araque, A., Sanzgiri, R. P., Parpura, V., and Haydon, P. G. (1998b). Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J. Neurosci. 18, 6822–6829.
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 23, 208–215.
Arizono, M., Bannai, H., Nakamura, K., Niwa, F., Enomoto, M., Matsu-Ura, T., et al. (2012). Receptor-selective diffusion barrier enhances sensitivity of astrocytic processes to metabotropic glutamate receptor stimulation. Sci. Signal. 5, ra27.
Auld, D., and Robitaille, R. (2003). Glial cells and neurotransmission: an inclusive view of synaptic function. Neuron 40, 389–400.
Bains, J. S., and Oliet, S. H. R. (2007). Glia: they make your memories stick! Trends Neurosci. 30, 417–424.
Balosso, S., Ravizza, T., Pierucci, M., Calcagno, E., Invernizzi, R., Di Giovanni, G., et al. (2009). Molecular and functional interactions between tumor necrosis factor-alpha receptors and the glutamatergic system in the mouse hippocampus: implications for seizure susceptibility. Neuroscience 161, 293–300.
Bardoni, R., Ghirri, A., Zonta, M., Betelli, C., Vitale, G., Ruggieri, V., et al. (2010). Glutamate-mediated astrocyte-to-neuron signalling in the rat dorsal horn. J. Physiol. 588, 831–846.
Barlow, H. (1996). Intraneuronal information processing, directional selectivity and memory for spatio-temporal sequences. Network 7, 251–259.
Barres, B. (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440.
Beattie, E., Stellwagen, D., Morishita, W., Bresnahan, J., Ha, B., von Zastrow, M., et al. (2002). Control of synaptic strength by glial TNFα. Science 295, 2282–2285.
Beierlein, M., and Regehr, W. (2006). Brief bursts of parallel fiber activity trigger calcium signals in Bergmann glia. J. Neurosci. 26, 6958–6967.
Bekar, L., He, W., and Nedergaard, M. (2008). Locus coeruleus α-adrenergic–mediated activation of cortical astrocytes in vivo. Cereb. Cortex 18, 2789–2795.
Bélair, E., Vallée, J., and Robitaille, R. (2010). In vivo long-term synaptic plasticity of glial cells. J. Physiol. 588, 1039–1056.
Benedetti, B., Matyash, V., and Kettenmann, H. (2011). Astrocytes control GABAergic inhibition of neurons in the mouse barrel cortex. J. Physiol. 589, 1159–1172.
Bennett, M., Farnell, L., and Gibson, W. (2008). A quantitative model of cortical spreading depression due to purinergic and gap-junction transmission in astrocyte networks. Biophys. J. 95, 5648–5660.
Bergersen, L. H., and Gundersen, V. (2009). Morphological evidence for vesicular glutamate release from astrocytes. Neuroscience 158, 260–265.
Bergersen, L. H., Morland, C., Ormel, L., Rinholm, J. E., Larsson, M., Wold, J. F. H., et al. (2012). Immunogold detection of L-glutamate and D-serine in small synaptic-like microvesicles in adult hippocampal astrocytes. Cereb. Cortex 22, 1690–1697.
Bernardinelli, Y., Salmon, C., Jones, E., Farmer, W., Stellwagen, D., and Murai, K. (2011). Astrocytes display complex and localized calcium responses to single-neuron stimulation in the hippocampus. J. Neurosci. 31, 8905–8919.
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. 4, 517–529.
Berry, H., De Pittà, M., Volman, V., and Ben-Jacob, E. (2011). “Astrocyte regulation of presynaptic plasticity,” in 2011 Neuroscience Meeting Planner (Washington, DC: Society for Neuroscience) [Online].
Bezprozvanny, I., Watras, J., and Ehrlich, B. E. (1991). Bell-shaped calcium-response curves of Ins(1 4, 5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754.
Bezzi, P., Carmignoto, G., Pasti, L., Vesce, S., Rossi, D., Lodi Rizzini, B., et al. (1998). Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391, 281–285.
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., De Clercq, E., et al. (2001). CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710.
Bezzi, P., Gundersen, V., Galbete, J. L., Seifert, G., Steinhäuser, C., Pilati, E., et al. (2004). Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat. Neurosci. 7, 613–620.
Bienenstock, E., Cooper, L., and Munro, P. (1982). Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48.
Blaustein, M., and Golovina, V. (2001). Structural complexity and functional diversity of endoplasmic reticulum Ca2+ stores. Trends Neurosci. 24, 602–608.
Bliss, T., and Collingridge, G. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39.
Blomstrand, F., and Giaume, C. (2006). Kinetics of endothelin-induced inhibition and glucose permeability of astrocyte gap junctions. J. Neurosci. Res. 83, 996–1003.
Bonansco, C., Couve, A., Perea, G., Ferradas, C. A., Roncagliolo, M., and Fuenzalida, M. (2011). Glutamate released spontaneously from astrocytes sets the threshold for synaptic plasticity. Eur. J. Neurosci. 33, 1483–1492.
Bootman, M., Lipp, P., and Berridge, M. (2001). The organisation and functions of local Ca2+ signals. J. Cell Sci. 114, 2213–2222.
Bowser, D., and Khakh, B. (2004). ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J. Neurosci. 24, 8606–8620.
Bowser, D. N., and Khakh, B. S. (2007). Two forms of single-vesicle astrocyte exocytosis imaged with total internal reflection fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 104, 4212–4217.
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocyte in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192.
Carmignoto, G. (2000). Reciprocal communication systems between astrocytes and neurones. Prog. Neurobiol. 62, 561–581.
Chan-Ling, T., and Stone, J. (1991). Factors determining the morphology and distribution of astrocytes in the cat retina: a ‘contact-spacing’ model of astrocyte interaction. J. Comp. Neurol. 303, 387–399.
Chen, N., Sugihara, H., Sharma, J., Perea, G., Petravicz, J., Le, C., et al. (2012). Nucleus basalis enabled stimulus specific plasticity in the visual cortex is mediated by astrocytes. Proc. Natl. Acad. Sci. U.S.A. 109, E2832–E2841.
Chen, X., Wang, L., Zhou, Y., Zheng, L.-H., and Zhou, Z. (2005). “Kiss-and-run” glutamate secretion in cultured and freshly isolated rat hippocampal astrocytes. J. Neurosci. 25, 9236–9243.
Coco, S., Calegari, F., Pravettoni, E., Pozzi, D., Taverna, E., Rosa, P., et al. (2003). Storage and release of ATP from astrocytes in culture. J. Biol. Chem. 278, 1354–1362.
Codazzi, F., Teruel, M. N., and Meyer, T. (2001). Control of astrocyte Ca2+ oscillations and waves by oscillating translocation and activation of protein kinase C. Curr. Biol. 11, 1089–1097.
Communi, D., Gevaert, K., Demol, H., Vandekerckhove, J., and Erneux, C. (2001). A novel receptor-mediated regulation mechanism of type I inositol polyphosphate 5-phosphatase by calcium/calmodulin-dependent protein kinase II phosphorylation. J. Biol. Chem. 276, 38738–38747.
Communi, D., Vanweyenberg, V., and Erneux, C. (1997). d-myo-inositol 1 4, 5-trisphosphate 3-kinase A is activated by receptor activation through a calcium: calmodulin-dependent protein kinase II phosphorylation mechanism. EMBO J. 16, 1943–1952.
Crippa, D., Schenk, U., Francolini, M., Rosa, P., Verderio, C., Zonta, M., et al. (2006). Synaptobrevin2-expressing vesicles in rat astrocytes: insights into molecular characterization, dynamics and exocytosis. J. Physiol. 570, 567–582.
Cull-Candy, S., Brickley, S., and Farrant, M. (2001). NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 11, 327–335.
D'Ascenzo, M., Fellin, T., Terunuma, M., Revilla-Sanchez, R., Meaney, D., Auberson, Y., et al. (2007). mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 104, 1995.
Del Castillo, J., and Katz, B. (1954). Quantal components of the end-plate potential. J. Physiol. 124, 560–573.
Deng, Q., Terunuma, M., Fellin, T., Moss, S. J., and Haydon, P. (2011). Astrocytic activation of A1 receptors regulates the surface expression of NMDA receptors through a src kinase dependent pathway. Glia 59, 1084–1093.
De Pittà, M., Volman, V., Levine, H., and Ben-Jacob, E. (2009a). Multimodal encoding in a simplified model of intracellular calcium signaling. Cogn. Processing 10(Suppl. 1), 55–70.
De Pittà, M., Goldberg, M., Volman, V., Berry, H., and Ben-Jacob, E. (2009b). Glutamate-dependent intracellular calcium and IP3 oscillating and pulsating dynamics in astrocytes. J. Biol. Phys. 35, 383–411.
De Pittà, M., Volman, V., Berry, H., and Ben-Jacob, E. (2011). A tale of two stories: astrocyte regulation of synaptic depression and facilitation. PLoS Comput. Biol. 7:e1002293. doi: 10.1371/journal.pcbi.1002293
De Pittà, M., Volman, V., Levine, H., Pioggia, G., De Rossi, D., and Ben-Jacob, E. (2008). Coexistence of amplitude and frequency modulations in intracellular calcium dynamics. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 77, 030903(R).
De Saint Jan, D., and Westbrook, G. (2005). Detecting activity in olfactory bulb glomeruli with astrocyte recording. J. Neurosci. 25, 2917–2924.
Di Castro, M., Chuquet, J., Liaudet, N., Bhaukaurally, K., Santello, M., Bouvier, D., et al. (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 14, 1276–1284.
Domercq, M., Brambilla, L., Pilati, E., Marchaland, J., Volterra, A., and Bezzi, P. (2006). P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-α and prostaglandins. J. Biol. Chem. 281, 30684–30696.
Dundwiddie, T. V., and Masino, S. A. (2001). The role and regulation of adenosine in the central nervous system. Ann. Rev. Neurosci. 24, 31–55.
Dupont, G., Lokenye, E. F., and Challiss, R. A. (2011). A model for Ca2+ oscillations stimulated by the type 5 metabotropic glutamate receptor: an unusual mechanism based on repetitive, reversible phosphorylation of the receptor. Biochimie 93, 2132–2138.
Emptage, N. J., Reid, C. A., and Fine, A. (2001). Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29, 197–208.
Engel, A., Fries, P., and Singer, W. (2001). Dynamic predictions: oscillations and synchrony in top-down processing. Nat. Rev. Neurosci. 2, 704–716.
Enkvist, M. O., and McCarthy, K. D. (1992). Activation of protein kinase C blocks astroglial gap junction communication and inhibits the spread of calcium waves. J. Neurochem. 59, 519–526.
Ermentrout, G., and Terman, D. (2010). Mathematical Foundations of Neuroscience. New York, NY: Springer Verlag.
Falcke, M. (2004). Reading the patterns in living cells – the physics of Ca2+ signaling. Adv. Phys. 53, 255–440.
Fellin, T., Ellenbogen, J. M., De Pittà, M., Ben-Jacob, E., and Halassa, M. (2012). Astrocyte regulation of sleep circuits: experimental and modeling perspectives. Front. Comput. Neurosci. 6:65. doi: 10.3389/fncom.2012.00065
Fellin, T., Halassa, M., Terunuma, M., Succol, F., Takano, H., Frank, M., et al. (2009). Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 15037–15042.
Fellin, T., Pascual, O., Gobbo, S., Pozzan, T., Haydon, P. G., and Carmignoto, G. (2004). Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43, 729–743.
Ferguson, A., Christensen, R., Gensel, J., Miller, B., Sun, F., Beattie, E., et al. (2008). Cell death after spinal cord injury is exacerbated by rapid TNFα-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J. Neurosci. 28, 11391–11400.
Fiacco, T. A., Agulhon, C., Taves, S. R., Petravicz, J., Casper, K. B., Dong, X., et al. (2007). Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626.
Fiacco, T. A., and McCarthy, K. D. (2004). Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci. 24, 722–732.
Fukaya, M., Uchigashima, M., Nomura, S., Hasegawa, Y., Kikuchi, H., and Watanabe, M. (2008). Predominant expression of phospholipase Cβ 1 in telencephalic principal neurons and cerebellar interneurons, and its close association with related signaling molecules in somatodendritic neuronal elements. Eur. J. Neurosci. 28, 1744–1759.
García-Marqués, J., and Lópes-Mascaraque, L. (2012). Clonal identity determines astrocyte cortical heterogeneity. Cereb. Cortex. doi: 10.1093/cercor/bhs134. [Epub ahead of print].
Giaume, C., Koulakoff, A., Roux, L., Holcman, D., and Rouach, N. (2010). Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 11, 87–99.
Goldberg, M., De Pittà, M., Volman, V., Berry, H., and Ben-Jacob, E. (2010). Nonlinear gap junctions enable long-distance propagation of pulsating calcium waves in astrocyte networks. PLoS Comput. Biol. 6:e1000909. doi: 10.1371/journal.pcbi.1000909
Gordon, G., Baimoukhametova, D., Hewitt, S., Rajapaksha, W., Fisher, T., and Bains, J. (2005). Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat. Neurosci. 8, 1078–1086.
Gordon, G. R. J., Iremonger, K. J., Kantevari, S., Ellis-Davies, G. C. R., MacVicar, B. A., and Bains, J. S. (2009). Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron 64, 391–403.
Grosche, J., Matyash, V., Möller, T., Verkhratsky, A., Reichenbach, A., and Kettenmann, H. (1999). Microdomains for neuron-glia interaction: parallel fiber signaling to bergmann glial cells. Nature 2, 139–143.
Guthrie, P. B., Knappenberger, J., Segal, M., Bennett, M. V., Charles, A. C., and Kater, S. B. (1999). ATP released from astrocytes mediates glial calcium waves. J. Neurosci. 19, 520–528.
Haas, B., Schipke, C. G., Peters, O., Sohl, G., Willecke, K., and Kettenmann, H. (2006). Activity-dependent ATP-waves in the mouse neocortex are independent from astrocytic calcium waves. Cereb. Cortex 16, 237–246.
Haber, M., Zhou, L., and Murai, K. K. (2006). Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J. Neurosci. 26, 8881–8891.
Haider, B., Duque, A., Hasenstaub, A., and McCormick, D. (2006). Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J. Neurosci. 26, 4535–4545.
Halassa, M., Florian, C., Fellin, T., Munoz, J., Lee, S., Abel, T., et al. (2009). Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61, 213–219.
Halassa, M., and Haydon, P. (2010). Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 72, 335–355.
Haydon, P. G., and Carmignoto, G. (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 86, 1009–1031.
Henneberger, C., Papouin, T., Oliet, S. H. R., and Rusakov, D. A. (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature 463, 232–237.
Hensch, T. (2005). Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 6, 877–888.
Herman, M. A., and Jahr, C. E. (2007). Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 27, 9736–9741.
Hirase, H., Qian, L., Barthó, P., and Buzsáki, G. (2004). Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2:E96. doi: 10.1371/journal.pbio.0020096
Houades, V., Koulakoff, A., Ezan, P., Seif, I., and Giaume, C. (2008). Gap junction-mediated astrocytic networks in the mouse barrel cortex. J. Neurosci. 28, 5207–5217.
Iacobas, D. A., Suadicani, S. O., Spray, D. C., and Scemes, E. (2006). A stochastic two-dimensional model of intercellular Ca2+ wave spread in glia. Biophys. J. 90, 24–41.
Iino, M. (1990). Biphasic Ca2+-dependence of inositol 1 4, 5-trisphosphate-induced Ca2+ release in smooth muscle cells of the guinea pig Taenia caeci. J. Gen. Physiol. 95, 1103–1112.
Irvine, R. F., Lloyd-Burton, S. M., Yu, J. C. H., Letcher, A. J., and Schell, M. J. (2006). The regulation and function of inositol 1 4, 5-trisphopshate 3-kinase. Adv. Enzyme Regul. 46, 314–323.
Jaiswal, J., Fix, M., Takano, T., Nedergaard, M., and Simon, S. (2007). Resolving vesicle fusion from lysis to monitor calcium-triggered lysosomal exocytosis in astrocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 14151–14156.
Jia, H., Rochefort, N., Chen, X., and Konnerth, A. (2010). Dendritic organization of sensory input to cortical neurons in vivo. Nature 464, 1307–1312.
Jourdain, P., Bergersen, L. H., Bhaukaurally, K., Bezzi, P., Santello, M., Domercq, M., et al. (2007). Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 10, 331–339.
Kaftan, E. J., Erlich, B. E., and Watras, J. (1997). Inositol 1 4, 5-trisphosphate (InsP3) and calcium interact to increase the dynamic range of InsP3 receptor-dependent calcium signaling. J. Gen. Physiol. 110, 529–538.
Kang, J., Jiang, L., Goldman, S. A., and Nedergaard, M. (1998). Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692.
Kang, M., and Othmer, H. (2009). Spatiotemporal characteristics of calcium dynamics in astrocytes. Chaos 19, 037116.
Kang, N., Xu, J., Xu, Q., Nedergaard, M., and Kang, J. (2005). Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 94, 4121–4130.
Keizer, J., Li, Y., Stojilkovic, S., and Rinzel, J. (1995). InsP3-induced Ca2+ excitability of the endoplasmic reticulum. Mol. Biol. Cell 6, 945–951.
Kozlov, A. S., Angulo, M. C., Audinat, E., and Charpak, S. (2006). Target cell-specific modulation of neuronal activity by astrocytes. Proc. Natl. Acad. Sci. U.S.A. 103, 10058–10063.
Kreft, M., Stenovec, M., Rupnik, M., Grilc, S., KrŽan, M., Potokar, M., et al. (2004). Properties of Ca2+-dependent exocytosis in cultured astrocytes. Glia 46, 437–445.
Kuga, N., Sasaki, T., Takahara, Y., Matsuki, N., and Ikegaya, Y. (2011). Large-scale calcium waves traveling through astrocytic networks in vivo. J. Neurosci. 31, 2607–2614.
Kulik, A., Haentzsch, A., Lüchermann, M., Reichelt, W., and Ballanyi, K. (1999). Neuron-glia singaling via α1 adrenoreceptor-mediated Ca2+ release in Bergmann glial cells in situ. J. Neurosci. 19, 8401–8408.
Lallouette, J., and Berry, H. (2012). “Topology drives calcium wave propagation in 3D astrocyte networks,” in European Conference on Complex Systems 2012, Bruxelles.
Larsson, M., Sawada, K., Morland, C., Hiasa, M., Ormel, L., Moriyama, Y., et al. (2011). Functional and anatomical identification of a vesicular transporter mediating neuronal ATP release. Cereb. Cortex 22, 1203–1214.
Lavialle, M., Aumann, G., Anlauf, E., Pröls, F., Arpin, M., and Derouiche, A. (2011). Structural plasticity of perisynaptic astrocyte processes involves ezrin and metabotropic glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 108, 12915–12919.
Lee, M., McGeer, E., and McGeer, P. (2011). Mechanisms of GABA release from human astrocytes. Glia 59, 1600–1611.
Lee, S., Yoon, B., Berglund, K., Oh, S., Park, H., Shin, H., et al. (2010). Channel-mediated tonic GABA release from glia. Science 330, 790–796.
Le Meur, K., Mendizabal-Zubiaga, J., Grandes, P., and Audinat, E. (2012). GABA release by hippocampal astrocytes. Front. Comput. Neurosci. 6:59. doi: 10.3389/fncom.2012.00059
Li, D., Ropert, N., Koulakoff, A., Giaume, C., and Oheim, M. (2008). Lysosomes are the major vesicular compartment undergoing Ca2+-regulated exocytosis from cortical astrocytes. J. Neurosci. 28, 7648–7658.
Li, L., Bender, K., Drew, P., Jadhav, S., Sylwestrak, E., and Feldman, D. (2009). Endocannabinoid signaling is required for development and critical period plasticity of the whisker map in somatosensory cortex. Neuron 64, 537–549.
Li, Y.-X., Rinzel, J., Keizer, J., and Stojilkovic, S. S. (1994). Calcium oscillations in pituitary gonadotrophs: comparison of experiment and theory. Proc. Natl. Acad. Sci. U.S.A. 91, 58–62.
Liu, Q., Xu, Q., Arcuino, G., Kang, J., and Nedergaard, M. (2004a). Astrocyte-mediated activation of neuronal kainate receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 3172–3177.
Liu, Q.-S., Xu, Q., Kang, J., and Nedergaard, M. (2004b). Astrocyte activation of presynaptic metabotropic glutamate receptors modulates hippocampal inhibitory synaptic transmission. Neuron Glia Biol. 1, 307–316.
Lovatt, D., Xu, Q., Liu, W., Takano, T., Smith, N. A., Schnermann, J., et al. (2012). Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc. Natl. Acad. Sci. U.S.A. 109, 6265–6270.
Mak, D., McBride, S., and Foskett, J. (2003). Spontaneous channel activity of the inositol 1 4, 5-trisphosphate (InsP3) receptor (InsP3R). Application of allosteric modeling to calcium and InsP3 regulation of InsP3R single-channel gating. J. Gen. Physiol. 122, 583–603.
Malarkey, E., Ni, Y., and Parpura, V. (2008). Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia 56, 821–835.
Malarkey, E., and Parpura, V. (2011). Temporal characteristics of vesicular fusion in astrocytes: examination of synaptobrevin 2-laden vesicles at single vesicle resolution. J. Physiol. 589, 4271–4300.
Marchaland, J., Calì, C., Voglmaier, S. M., Li, H., Regazzi, R., Edwards, R. H., et al. (2008). Fast subplasma membrane Ca2+ transients control exo-endocytosis of synaptic-like microvesicles in astrocytes. J. Neurosci. 28, 9122–9132.
Matyash, V., Filippov, V., Mohrhagen, K., and Kettenmann, H. (2001). Nitric oxide signals parallel fiber activity to Bergmann glial cells in the mouse cerebellar slice. Mol. Cell. Neurosci. 18, 664–670. Abstract.
Matyash, V., and Kettenmann, H. (2010). Heterogeneity in astrocyte morphology and physiology. Brain Res. Rev. 63, 2–10.
Min, R., and Nevian, T. (2012). Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat. Neurosci. 15, 746–753.
Mishra, J., and Bhalla, U. S. (2002). Simulations of inositol phosphate metabolism and its interaction with InsP3-mediated calcium release. Biophys. J. 83, 1298–1316.
Montana, V., Malarkey, E. B., Verderio, C., Matteoli, M., and Parpura, V. (2006). Vesicular transmitter release from astrocytes. Glia 54, 700–715.
Montana, V., Ni, Y., Sunjara, V., Hua, X., and Parpura, V. (2004). Vesicular glutamate transporter-dependent glutamate release from astrocytes. J. Neurosci. 24, 2633–2642.
Mulligan, S., and MacVicar, B. (2004). Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431, 195–199.
Muyderman, H., Ängehagen, M., Sandberg, M., Björklund, U., Olsson, T., Hansson, E., et al. (2001). α1-Adrenergic modulation of metabotropic glutamate receptor-induced calcium oscillations and glutamate release in astrocytes. J. Biol. Chem. 276, 46504–46514.
Navarrete, M., and Araque, A. (2008). Endocannabinoids mediate neuron-astrocyte communication. Neuron 57, 883–893.
Navarrete, M., and Araque, A. (2010). Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68, 113–126.
Navarrete, M., and Araque, A. (2011). Basal synaptic transmission: astrocytes rule! Cell 146, 675–677.
Navarrete, M., Perea, G., de Sevilla, D., Gómez-Gonzalo, M., Núñez, A., Martn, E., et al. (2012a). Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 10:e1001259. doi: 10.1371/journal.pbio.1001259
Navarrete, M., Perea, G., Maglio, L., Pastor, J., García de Sola, R., and Araque, A. (2012b). Astrocyte calcium signal and gliotransmission in human brain tissue. Cereb. Cortex. doi: 10.1093/cercor/bhs122. [Epub ahed of print].
Nett, W. J., Oloff, S. H., and McCarthy, K. D. (2002). Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J. Neurophysiol. 87, 528–537.
Newman, E. (2005). Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J. Neurosci. 25, 5502–5510.
Newman, E. A. (2003). Glial cell inhibition of neurons by release of ATP. J. Neurosci. 23, 1659–1666.
Ni, Y., Malarkey, E. B., and Parpura, V. (2007). Vesicular release of glutamate mediates bidirectional signaling between astrocytes and neurons. J. Neurochem. 103, 1273–1284.
Nimmerjahn, A., Kirchhoff, F., Kerr, J. N. D., and Helmchen, F. (2004). Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat. Methods 1, 31–37.
Okubo, Y., Sekiya, H., Namiki, S., Sakamoto, H., Iinuma, S., Yamasaki, M., et al. (2010). Imaging extrasynaptic glutamate dynamics in the brain. Proc. Natl. Acad. Sci. U.S.A. 107, 6526–6531.
Oliet, S. H. R., Piet, R., and Poulain, D. A. (2001). Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292, 923–926.
Panatier, A., Theodosis, D. T., Mothet, J. P., Touquet, B., Pollegioni, L., Poulain, D. A., et al. (2006). Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784.
Panatier, A., Vallée, J., Haber, M., Murai, K., Lacaille, J., and Robitaille, R. (2011). Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146, 785–798.
Pangršic, T., Potokar, M., Stenovec, M., Kreft, M., Fabbretti, E., Nistri, A., et al. (2007). Exocytotic release of ATP from cultured astrocytes. J. Biol. Chem. 282, 28749–28758.
Pankratov, Y., Lalo, U., Verkhratsky, A., and North, R. (2007). Quantal release of ATP in mouse cortex. J. Gen. Physiol. 129, 257–265.
Pannasch, U., Vargová, L., Reingruber, J., Ezan, P., Holcman, D., Giaume, C., et al. (2011). Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 8467–8472.
Papouin, T., Ladépêche, L., Ruel, J., Sacchi, S., Labasque, M., Hanini, M., et al. (2012). Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150, 633–646.
Parpura, V., Fang, Y., Basarsky, T., Jahn, R., and Haydon, P. G. (1995). Expression of synaptobrevin II, cellubrevin and syntaxin but not SNAP-25 in cultured astrocytes. FEBS Lett. 377, 489–492.
Parpura, V., Grubišiæ, V., and Verkhratsky, A. (2011). Ca2+ sources for the exocytotic release of glutamate from astrocytes. Biochim. Biophys. Acta 1813, 984–991.
Parpura, V., and Haydon, P. G. (2000). Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc. Natl. Acad. Sci. U.S.A. 97, 8629–8634.
Parpura, V., and Zorec, R. (2010). Gliotransmission: exocytotic release from astrocytes. Brain Res. Rev. 63, 83–92.
Parri, H. R., Gould, T. M., and Crunelli, V. (2001). Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat. Neurosci. 4, 803–812.
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J. Y., et al. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116.
Pasti, L., Volterra, A., Pozzan, T., and Carmignoto, G. (1997). Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci. 17, 7817–7830.
Pasti, L., Zonta, M., Pozzan, T., Vicini, S., and Carmignoto, G. (2001). Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J. Neurosci. 21, 477–484.
Perea, G., and Araque, A. (2005). Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocyte. J. Neurosci. 25, 2192–2203.
Perea, G., and Araque, A. (2007). Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317, 1083–1086.
Perea, G., Navarrete, M., and Araque, A. (2009). Tripartite synapse: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431.
Petravicz, J., Fiacco, T., and McCarthy, K. (2008). Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J. Neurosci. 28, 4967–4973.
Piet, R., and Jahr, C. (2007). Glutamatergic and purinergic receptor-mediated calcium transients in Bergmann glial cells. J. Neurosci. 27, 4027–4035.
Piet, R., Vargová, L., Syková, E., Poulain, D., and Oliet, S. (2004). Physiological contribution of the astrocytic environment of neurons to intersynaptic crosstalk. Proc. Natl. Acad. Sci. U.S.A. 101, 2151–2155.
Pinheiro, P. S., and Mulle, C. (2008). Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat. Rev. 9, 423–436.
Pirttimaki, T., Hall, S., and Parri, H. (2011). Sustained neuronal activity generated by glial plasticity. J. Neurosci. 31, 7637–7647.
Pivneva, T., Haas, B., Reyes-Haro, D., Laube, G., Veh, R., Nolte, C., et al. (2008). Store-operated Ca2+ entry in astrocytes: different spatial arrangement of endoplasmic reticulum explains functional diversity in vitro and in situ. Cell Calcium 43, 591–601.
Porter, J. T., and McCarthy, K. D. (1996). Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J. Neurosci. 16, 5073–5081.
Poskanzer, K. E., and Yuste, R. (2011). Astrocytic regulation of cortical UP states. Proc. Natl. Acad. Sci. U.S.A. 108, 18453–18458.
Pryazhnikov, E., and Khiroug, L. (2008). Sub-micromolar increase in [Ca2+]i triggers delayed exocytosis of ATP in cultured astrocytes. Glia 56, 38–49.
Raghavachari, S., and Lisman, J. E. (2004). Properties of quantal transmission at CA1 synapses. J. Neurophysiol. 92, 2456–2467.
Ramos-Franco, J., Bare, D., Caenepeel, S., Nani, A., Fill, M., and Mignery, G. (2000). Single-channel function of recombinant type 2 inositol 1,4,5-trisphosphate receptor. Biophys. J. 79, 1388–1399.
Rebecchi, M. J., and Pentyala, S. N. (2000). Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 80, 1291–1335.
Rebola, N., Rodrigues, R., Lopes, L., Richardson, P., Oliveira, C., and Cunha, R. (2005). Adenosine A1 and A2A receptors are co-expressed in pyramidal neurons and co-localized in glutamatergic nerve terminals of the rat hippocampus. Neuroscience 133, 79–83.
Regehr, W. F., Delaney, K. R., and Tank, D. W. (1994). The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J. Neurosci. 14, 523–537.
Rieger, A., Deitmer, J., and Lohr, C. (2007). Axon-glia communication evokes calcium signaling in olfactory ensheathing cells of the developing olfactory bulb. Glia 55, 352–359.
Robitaille, R. (1998). Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron 21, 847–855.
Rodríguez-Moreno, A., and Paulsen, O. (2008). Spike timing–dependent long-term depression requires presynaptic nmda receptors. Nat. Neurosci. 11, 744–745.
Rouach, N., Koulakoff, A., and Giaume, C. (2004). Neurons set the tone of gap junctional communication in astrocytic networks. Neurochem. Int. 45, 265–272.
Rousse, I., St.-Amour, A., Darabid, H., and Robitaille, R. (2010). Synapse-glia interactions are governed by synaptic and intrinsic glial properties. Neuroscience 167, 621–632.
Roux, L., Benchenane, K., Rothstein, J., Bonvento, G., and Giaume, C. (2011). Plasticity of astroglial networks in olfactory glomeruli. Proc. Natl. Acad. Sci. U.S.A. 108, 18442–18446.
Santello, M., Bezzi, P., and Volterra, A. (2011). TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69, 988–1001.
Santello, M., and Volterra, A. (2009). Synaptic modulation by astrocytes via Ca2+-dependent glutamate release. Neuroscience 158, 253–259.
Santello, M., and Volterra, A. (2012). TNFα in synaptic function: switching gears. Trends Neurosci. 35, 638–647.
Sasaki, T., Matsuki, N., and Ikegaya, Y. (2011). Action-potential modulation during axonal conduction. Science 331, 599–601.
Sawada, K., Echigo, N., Juge, N., Miyaji, T., Otsuka, M., Omote, H., et al. (2008). Identification of a vesicular nucleotide transporter. Proc. Natl. Acad. Sci. U.S.A. 105, 5683–5686.
Sawyer, C., and Clifton, D. (1980). Aminergic innervation of the hypothalamus. Fed. Proc. 39, 2889–2895.
Scemes, E., and Giaume, C. (2006). Astrocyte calcium waves: what they are and what they do. Glia 54, 716–725.
Schipke, C., Haas, B., and Kettenmann, H. (2008). Astrocytes discriminate and selectively respond to the activity of a subpopulation of neurons within the barrel cortex. Cereb. Cortex 18, 2450–2459.
Schmitt, L., Sims, R., Dale, N., and Haydon, P. (2012). Wakefulness affects synaptic and network activity by increasing extracellular astrocyte-derived adenosine. J. Neurosci. 32, 4417–4425.
Schubert, V., Bouvier, D., and Volterra, A. (2011). SNARE protein expression in synaptic terminals and astrocytes in the adult hippocampus: a comparative analysis. Glia 59, 1472–1488.
Schummers, J., Yu, H., and Sur, M. (2008). Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science 320, 1638.
Sejnowski, T. J., and Paulsen, O. (2006). Network oscillations: emerging computational principles. J. Neurosci. 26, 1673–1676.
Serrano, A., Haddjeri, N., Lacaille, J., and Robitaille, R. (2006). GABAergic network activation of glial cells underlies heterosynaptic depression. J. Neurosci. 26, 5370–5382.
Shadlen, M., and Newsome, W. (1994). Noise, neural codes and cortical organization. Curr. Opin. Neurobiol. 4, 569–579.
Shigemoto, R., Kinoshita, A., Wada, E., Nomura, S., Ohishi, H., Takada, M., et al. (1997). Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J. Neurosci. 17, 7503–7522.
Shigetomi, E., Tong, X., Kwan, K., Corey, D., and Khakh, B. (2011). TRPA1 channels regulate astrocyte resting calcium levels and inhibitory synapse efficacy via GAT-3. Nat. Neurosci. 15, 70–80.
Shinohara, T., Michikawa, T., Enomoto, M., Goto, J., Iwai, M., Matsu-Ura, T., et al. (2011). Mechanistic basis of bell-shaped dependence of inositol 1 4, 5-trisphosphate receptor gating on cytosolic calcium. Proc. Natl. Acad. Sci. U.S.A. 108, 15486–15491.
Shu, Y., Hasenstaub, A., and McCormick, D. (2003). Turning on and off recurrent balanced cortical activity. Nature 423, 288–293.
Sims, C. E., and Allbritton, N. L. (1998). Metabolism of inositol 1 4, 5-trisphosphate and inositol 1 3 4, 5-tetrakisphosphate by the oocytes of Xenopus laevis. J. Biol. Chem. 273, 4052–4058.
Sjöström, P., Turrigiano, G., and Nelson, S. (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654.
Sneyd, J., Charles, A. C., and Sanderson, M. J. (1994). A model for the propagation of intracellular calcium waves. Am. J. Physiol. 266, C293–C302.
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228.
Stellwagen, D., and Malenka, R. (2006). Synaptic scaling mediated by glial TNF-α. Nature 440, 1054–1059.
Stenovec, M., Kreft, S., Grilc, M., Pokotar, M., Kreft, M. E., Pangršic, T., et al. (2007). Ca2+-dependent mobility of vesicles capturing anti-VGLUT1 antibodies. Exp. Cell Res. 313, 3809–3818.
Sul, J.-Y., Orosz, G., Givens, R. S., and Haydon, P. G. (2004). Astrocytic connectivity in the hippocampus. Neuron Glia Biol. 1, 3–11.
Svoboda, K., Denk, W., Kleinfeld, D., and Tank, D. (1997). In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature 385, 161–165.
Swadlow, H. (2002). Thalamocortical control of feed–forward inhibition in awake somatosensory ‘barrel’ cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 357, 1717–1727.
Syková, E., and Nicholson, C. (2008). Diffusion in brain extracellular space. Physiol. Rev. 88, 1277–1340.
Takata, N., Mishima, T., Hisatsune, C., Nagai, T., Ebisui, E., Mikoshiba, K., et al. (2011). Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 31, 18155–18165.
Thomas, D., Lipp, P., Tovey, S., Berridge, M., Li, W., Tsien, R., et al. (2000). Microscopic properties of elementary Ca2+ release sites in non-excitable cells. Curr. Biol. 10, 8–15.
Tian, G.-F., Azmi, H., Takahiro, T., Xu, Q., Peng, W., Lin, J., et al. (2005). An astrocytic basis of epilepsy. Nat. Med. 11, 973–981.
Tian, G. F., Takano, T., Lin, J. H.-C., Wang, X., Bekar, L., and Nedergaard, M. (2006). Imaging of cortical astrocytes using 2-photon laser scanning microscopy in the intact mouse brain. Adv. Drug Deliv. Rev. 58, 773–787.
Todd, K. J., Darabid, H., and Robitaille, R. (2010). Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J. Neurosci. 30, 11870–11882.
Torres, A., Wang, F., Xu, Q., Fujita, T., Dobrowolski, R., Willecke, K., et al. (2012). Extracellular Ca2+ acts as a mediator of communication from neurons to glia. Sci. Signal. 5, ra8.
Troyer, T., and Miller, K. (1997). Physiological gain leads to high ISI variability in a simple model of a cortical regular spiking cell. Neural Computat. 9, 971–983.
Ullian, E., Christopherson, K., and Barres, B. (2004). Role for glia in synaptogenesis. Glia 47, 209–216.
van den Pol, A. N., Romano, C., and Ghosh, P. (1995). Metabotropic glutamate receptor mGluR5 subcellular distribution and developmental expression in hypothalamus. J. Comp. Neurol. 362, 134–150.
Ventura, R., and Harris, K. M. (1999). Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 19, 6897–6906.
Verkhratsky, A., Rodriguez, R. R., and Parpura, V. (2012). Calcium signalling in astroglia. Mol. Cell. Endocrinol. 353, 45–56.
Vogels, T., and Abbott, L. (2009). Gating multiple signals through detailed balance of excitation and inhibition in spiking networks. Nat. Neurosci. 12, 483–491.
Volman, V., Bazhenov, M., and Sejnowski, T. J. (2011). “A computational model of neuronal and glial homeostatic synaptic plasticity in posttraumatic epileptogenesis,” in Neuroscience Meeting Planner, Number 250.11/V5. Society for Neuroscience. [Online].
Volman, V., Ben-Jacob, E., and Levine, H. (2007). The astrocyte as a gatekeeper of synaptic information transfer. Neural Comput. 19, 303–326.
Volterra, A., and Meldolesi, J. (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640.
Wang, X., Lou, N., Xu, Q., Tian, G.-F., Peng, W. G., Han, X., et al. (2006). Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 9, 816–823.
Waters, J., Larkum, M., Sakmann, B., and Helmchen, F. (2003). Supralinear Ca2+ influx into dendritic tufts of layer 2/3 neocortical pyramidal neurons in vitro and in vivo. J. Neurosci. 23, 8558–8567.
Watras, J., Bezprozvanny, I., and Ehrlich, B. E. (1991). Inositol 1 4, 5-trisphosphate-gated channels in cerebellum: presence of multiple conductance states. J. Neurosci. 11, 3239–3245.
Winship, I., Plaa, N., and Murphy, T. (2007). Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J. Neurosci. 27, 6268–6272.
Yang, Y., Ge, W., Chen, Y., Zhang, Z., Shen, W., Wu, C., et al. (2003). Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc. Natl. Acad. Sci. U.S.A. 100, 15194–15199.
Zhang, B.-X., Zhao, H., and Muallem, S. (1993). Calcium dependent kinase and phosphatase control inositol-1 4, 5-trisphopshate-mediated calcium release: modification by agonist stimulation. J. Biol. Chem. 268, 10997–11001.
Zhang, J.-M., Wang, H.-K., Ye, C.-Q., Ge, W., Chen, Y., Jiang, Z.-L., et al. (2003). ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40, 971–982.
Zhang, Q., Fukuda, M., Van Bockstaele, E., Pascual, O., and Haydon, P. G. (2004a). Synaptotagmin IV regulates glial glutamate release. Proc. Natl. Acad. Sci. U.S.A. 101, 9441–9446.
Zhang, Q., Pangršic, T., Kreft, M., Kržan, M., Li, N., Sul, J.-Y., et al. (2004b). Fusion-related release of glutamate from astrocytes. J. Biol. Chem. 279, 12724–12733.
Zhang, Y., and Barres, B. (2010). Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 20, 588–594.
Zhang, Z., Chen, G., Zhou, W., Song, A., Xu, T., Luo, Q., et al. (2007). Regulated ATP release from astrocytes through lysosome exocytosis. Nat. Cell Biol. 9, 945–953.
Zonta, M., and Carmignoto, G. (2002). Calcium oscillations encoding neuron-to-astrocyte communication. J. Physiol. Paris 96, 193–198.
Zorec, R., Araque, A., Carmignoto, G., Haydon, P., Verkhratsky, A., and Parpura, V. (2012). Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signaling route. ASN Neuro 4, e00080.
Zucker, R. S., and Regehr, W. G. (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405.
Keywords: astrocyte-synapse interactions, astrocyte modeling, calcium signaling, calcium encoding, gliotransmission, synaptic plasticity, metaplasticity, cortical maps
Citation: De Pittà M, Volman V, Berry H, Parpura V, Volterra A, and Ben-Jacob E (2012) Computational quest for understanding the role of astrocyte signaling in synaptic transmission and plasticity. Front. Comput. Neurosci. 6:98. doi: 10.3389/fncom.2012.00098
Received: 23 April 2012; Paper pending published: 28 May 2012;
Accepted: 06 December 2012; Published online: 21 December 2012.
Edited by:
Liam J. McDaid, University of Ulster, UKReviewed by:
Florentin Wörgötter, University Goettingen, GermanyCopyright © 2012 De Pittà, Volman, Berry, Parpura, Volterra, and Ben-Jacob. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Maurizio De Pittà and Eshel Ben-Jacob, School of Physics and Astronomy, Tel Aviv University, 69978 Ramat Aviv, Israel. e-mail:bWF1cml6aW9kZXBpdHRhQGdtYWlsLmNvbQ==;ZXNoZWxiakBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.