 Hung-Yu Chien1†
Hung-Yu Chien1† Wan-Chun Li
Wan-Chun Li- 1Division of Endocrinology and Metabolism, Department of Internal Medicine, Taipei City Hospital, Taipei, Taiwan
- 2Division of Nuclear Medicine, Department of Internal Medicine, Taipei City Hospital, Taipei, Taiwan
- 3Institute of Oral Biology, School of Dentistry, National Yang Ming Chiao Tung University, Taipei, Taiwan
Robust experiment evidence suggests that prolactin can enhance beta-cell proliferation and increase insulin secretion and sensitivity. Apart from acting as an endocrine hormone, it also function as an adipokine and act on adipocytes to modulate adipogenesis, lipid metabolism and inflammation. Several cross-sectional epidemiologic studies consistently showed that circulating prolactin levels positive correlated with increased insulin sensitivity, lower glucose and lipid levels, and lower prevalence of T2D and metabolic syndrome. Bromocriptine, a dopamine receptor agonist used to treat prolactinoma, is approved by Food and Drug Administration for treatment in type 2 diabetes mellitus since 2009. Prolactin lowering suppress insulin secretion and decrease insulin sensitivity, therefore dopamine receptor agonists which act at the pituitary to lower serum prolactin levels are expected to impair glucose tolerance. Making it more complicating, studies exploring the glucose-lowering mechanism of bromocriptine and cabergoline have resulted in contradictory results; while some demonstrated actions independently on prolactin status, others showed glucose lowering partly explained by prolactin level. Previous studies showed that a moderate increase in central intraventricular prolactin levels stimulates hypothalamic dopamine with a decreased serum prolactin level and improved glucose metabolism. Additionally, sharp wave-ripples from the hippocampus modulates peripheral glucose level within 10 minutes, providing evidence for a mechanistic link between hypothalamus and blood glucose control. Central insulin in the mesolimbic system have been shown to suppress dopamine levels thus comprising a feedback control loop. Central dopamine and prolactin levels plays a key role in the glucose homeostasis control, and their dysregulation could lead to the pathognomonic central insulin resistance depicted in the “ominous octet”. This review aims to provide an in-depth discussion on the glucose-lowering mechanism of dopamine receptor agonists and on the diverse prolactin and dopamine actions on metabolism targets.
1 Introduction
Type 2 diabetes mellitus(T2D) is both an endocrine and metabolic disease and the prevalence is increasing exponentially worldwide (1). The pathogenesis of T2D have expanded from dysfunctions of “ominous triumvirate” (beta-cell dysfunction and insulin resistance in muscle, liver) proposed since 1987 by Defronzo to “ominous octet”. The octet includes the fat cell (accelerated lipolysis), gastrointestinal tract (incretin deficiency/resistance), α-cell (hyperglucagonemia), kidney (increased glucose reabsorption), and brain (insulin resistance) (2). T2D develops when the dynamic balance between beta cell (β-cell) proliferation cannot keep up with the rate of β-cell apoptosis. At a point when overt impaired glucose sensing and insufficient β-cell mass ensues, insulin secretion fails to compensate for increased insulin resistance then impaired glucose tolerance and T2D develops (3, 4). In humans β-cell proliferation is exceedingly low, as contrasted to rodents, but is able to expand rapidly under specific situations, such as pregnancy, when prolactin(PRL) and placental lactogen play a pivotal role in stimulating β-cells expansion (5).
Prolactin (PRL) is a polypeptide hormone that is mainly synthesized and secreted by lactotroph cells of the anterior pituitary gland. In humans, PRL is synthesized in the anterior pituitary but also in the hypothalamus, decidua, endometrium, breast, prostate, lymphocytes, leukocytes and adipocytes (6, 7). The well-known role attributed to PRL is to activate the differentiation and proliferation of the mammary cells necessary for lactation. However, as the prolactin receptor (PRLR) is expressed in various tissues and cells (including pancreatic islets, adipocytes, endometrium and the prostate), PRL also modulates and regulates many other physiological processes including metabolism, growth, reproduction, immune regulation, angiogenesis and osmoregulation (8–10). Compelling experimental evidence shows that PRL acts on β-cells to enhance β-cell proliferation and increase insulin secretion and sensitivity (11, 12). It is also an adipokine and act on adipocytes to regulate adipogenesis, lipid metabolism and inflammation (13). Several cross-sectional epidemiologic studies consistently showed that circulating PRL levels positive correlated with increased insulin sensitivity, lower glucose and lipid levels, and lower prevalence of T2D and metabolic syndrome (14, 15). On the other hand, PRL excess in supraphysiologic levels result in increase of HOMA-IR(surrogate index of insulin resistance) (16, 17), in both obese and lean patients. PRL excess induces leptin resistance and inhibition of dopaminergic tone which stimulates food intake and in turn may lead to increased body weight and adiposity in patients with hyperprolactinemia (18–20). Macotela et al. recently defined “homeostatic functionally increased transient prolactinemia” as their study showed the beneficial outcome of PRL on metabolism depends on circulating PRL levels kept within 7 and 100 ng/mL (21). Dopamine agonists bromocriptine and cabergoline have long been the treatment of choice for patients with prolactinomas owing to t+heir good therapeutic response (22–25). In patients with prolactinomas treated with bromocriptine and cabergoline, glucose metabolism and insulin sensitivity significantly improved in most patients but only some patients showed body weight reduction (26, 27). Moreover, in both type 2 diabetic and non-diabetic subjects, bromocriptine treatment lowered glycated hemoglobin (HbA1c), fasting plasma glucose and reduced body weight (25, 28) and had been approved by the Food and Drug Administration for treatment in type 2 diabetes mellitus since 2009. Since PRL lowering mechanistically leads to suppressed insulin secretion and decreased insulin sensitivity, dopamine agonist should be expected to worsen glucose tolerance. Making it even more complicating, studies exploring the glucose-lowering mechanism of bromocriptine and cabergoline have resulted in contradictory results, while some demonstrated significant improvement in glucose metabolism independently on PRL status (25, 29, 30), others showed glucose-lowering in T2DM patients partly explained by PRL level (31). Studies by Park et al. showed that a moderate increase in central intraventricular prolactin (1ug/h chronic infusion into lateral ventricle) levels elevated hypothalamic dopamine levels with a decreased serum PRL level and improved glucose metabolism in diabetic rats (32). It has been shown that animal and human pancreatic β-cells (33) and human adipocytes (34) all express dopamine receptors type 2 (D2R), so both dopamine and PRL acts at peripheral level to modulate β-cells, adipocytes, skeletal muscle and hepatocytes functions. Additionally, recently shown clusters of sharp wave-ripples from the hippocampus lowered peripheral glucose concentrations within 10 minutes by suppressing activity of the lateral septum in rats, which is the major conduit between the hippocampus and the hypothalamus (35). Hippocampal outputs affect endocrine and hormonal systems through hypothalamic hormone-releasing neurons or autonomic control of peripheral organs. This provides a mechanism unrevealing direct central neurohormonal control on β-cell insulin secretion. Central insulin in the mesolimbic system have been shown to suppress dopamine levels thus comprising a feedback control loop (36). It appears that dopamine agonists affect glucose homeostasis by 1) stimulating central PRL that increase ventral medial and arcuate hypothalamic dopamine which act through 2) increased dopaminergic/decreased sympathetic activity signaling. These signal 3) directly act on islet and adipocytes, hepatocytes, skeletal muscle D2R to modulate their metabolic effects and reciprocally insulin feedback inhibits the mesolimbic dopamine system (Figure 1). This review aims to provide an in-depth discussion on the glucose-lowering mechanism of dopamine receptor agonists and on the diverse prolactin and dopamine actions on metabolism targets including β-cells, adipocytes, hepatocytes, skeletal muscle and central nervous system (CNS).
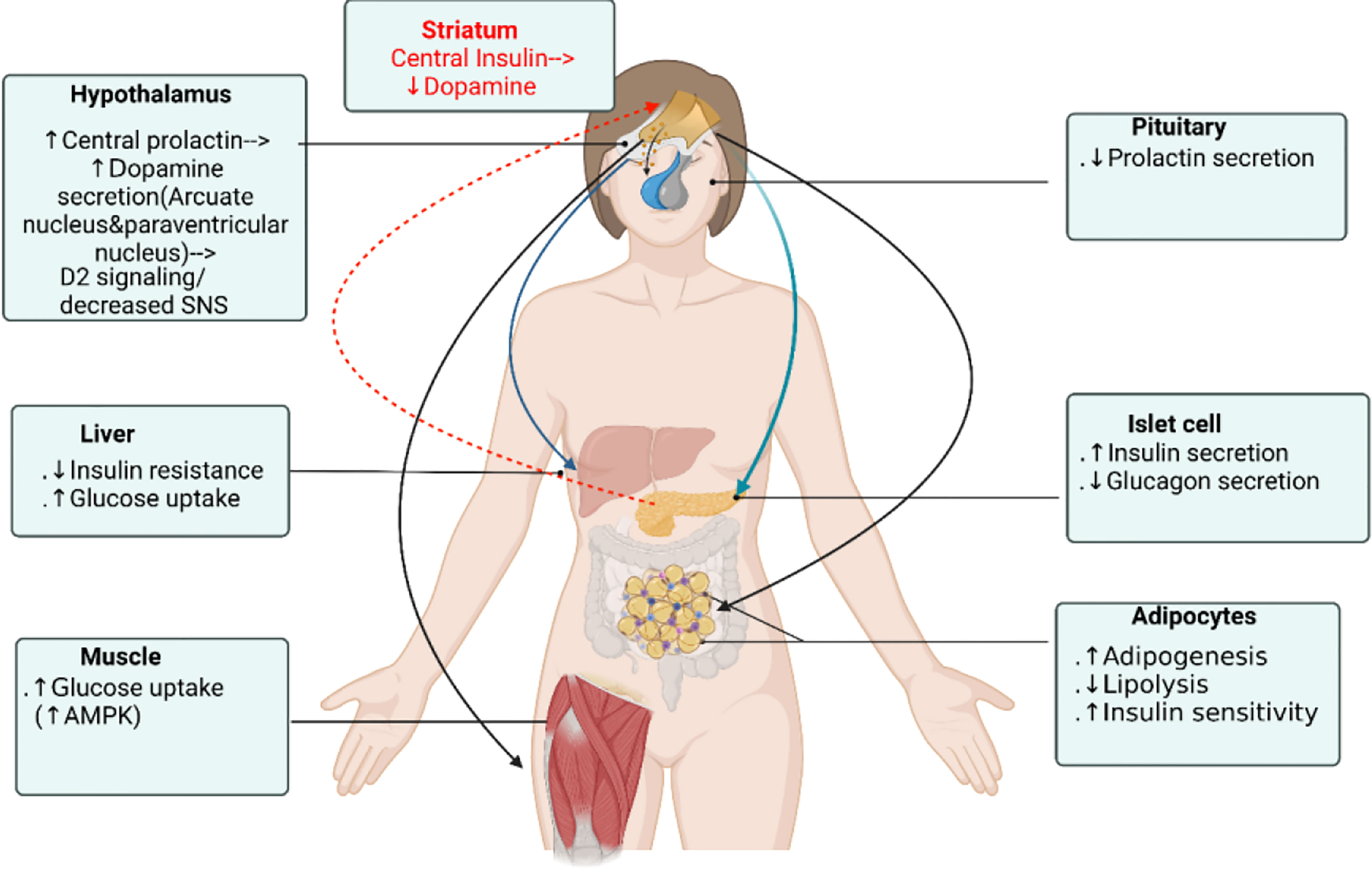
Figure 1 Dopamine receptor agonist glucose-lowering actions on different organs. Dopamine agonists affect glucose homeostasis by 1) stimulating central prolactin that increase ventral medial and arcuate hypothalamic dopamine that act through 2) dopaminergic/decreased sympathetic activity signaling which 3) directly act on islet to stimulate insulin secretion and on adipocyte, hepatocyte, skeletal muscle D2R to modulate their metabolic effects, not by the serum prolactin level. Reciprocally, central insulin suppresses dopamine level in the ventral striatum and mesolimbic dopamine system. Figure created using BioRender (https://biorender.com/).
1.1 Peripheral effects of PRL within the physiologic range
1.1.1 Peripheral PRL effect on β (beta)-cell
Both human islet cell lines and in vitro rodent studies have demonstrated that adding PRL to isolated pancreatic islet enhanced insulin secretion and β-cell proliferation (11), and overexpression of PRL in β-cells resulted in inappropriately increased insulin synthesis and secretion, also enhancing β-cell proliferation. Similarly, overexpression of PL in β-cells resulted in fasting and postprandial hypoglycemia, marked elevated serum insulin levels, increased islet insulin granules and volume and increased β-cell numbers and proliferation (37). In contrast, studies in PRLR knockout (Prlr-) mice demonstrates the fundamental role of PRL and PL during pancreatic islet development. PRLR knockout (Prlr-) mice developed impaired glucose tolerance, decreased insulin secretion, a 20-35% reduction in islet insulin granules, reduced islet density and reduced β-cell mass. Prlr- mice also showed impaired glucose tolerance and blunted insulin secretory response comparing to their wild-type littermates (38).
PRLR is highly expressed on pancreatic β-cell. Both PRL and placental lactogen (PL) that rise during mid-pregnancy lactogenic surge enhance β-cell proliferation and glucose-stimulated insulin hypersecretion, act through PRLR (39, 40). It is well established that the β-cell trophic and metabolic modulation effect of prolactin is not limited to the period of pregnancy (41, 42). These studies provide strong evidence that PRL and PL can enhance β-cell insulin secretion and proliferation. Although numerous evidence showed that lactogenic hormones do regulate β-cell mass during pregnancy, their actions cannot fully explain all of the changes that appears in β-cell mass during gestation. Despite persistent elevated PRL and PL concentrations till the end of pregnancy and even towards the end of lactation, the β-cell proliferation peaks around mid-gestation and drops to a normal rate seen in non-pregnant state and an even lower rate by parturition, leading to rapid normalization of β-cell mass postpartum. Although β-cell numbers and mass increase about 1.4–2.4 times relative to normal (43) during mid-pregnancy lactogenic surge, in patients with gestational diabetes mellitus (GDM), insulin requirements often increase during the third trimester that coincides with the increase in insulin resistance to maintain good glycemic control. Also, insulin requirements double during gestational week 16 to week 37 in type1 DM, which is similar in magnitude to an increase in insulin resistance. Nieuwenhuizen et al. showed that rather than PRL, decreased insulin sensitivity per se during gestation plays a more important role in the regulation of β-cell proliferation (44).
1.1.2 Peripheral PRL effect on adipocyte and insulin resistance
Adipose tissue is the largest organ in the human body and adipokines plays a pivotal role in metabolic and endocrine homeostasis. In obesity, accumulation of excessive fat is distributed either into existing adipocytes (hypertrophy) or into newly recruited adipocytes (hyperplasia) generated by adipogenesis from differentiation of pre-adipocytes. Adipose tissue expansion in the form of hyperplasia is generally regarded as a more metabolic healthy process (45, 46), whereas expansion in the form of excessive adipocyte hypertrophy leads to metabolic disruption and associated with insulin resistance and metabolic syndrome (47). PRL have been shown to promote adipose differentiation and enhance adipocyte hyperplasia and may be a potential therapeutic target against insulin resistance and metabolic syndrome. Adipocytes have been shown to express PRLR (48) and D2R (34), and both PRL and dopamine are produced by human adipose tissue (49). A large body of literature indicates that PRL participates in many aspects of adipose tissue actions, including adipogenesis (50, 51), metabolic enzyme activity, lipolysis, and release of adipokines such as leptin, adiponectin, and inflammatory cytokines such as Il-1, IL-6 and monocyte chemoattractant protein 1 (52, 53). Analogous to the pancreatic islet, adipocytes adapt during pregnancy and lactation, when PRL values may exceed 200 μg/L, including neural (dopaminergic) changes that increase appetite, food intake, and redistribute nutrients, lipid storage from abdominal tissues to mammary glands (54). Physiological levels of PRL is about 10-25 mg/dL in women and 10-20 mg/dL in men and may reach up to 90 mg/dl after exercise, eating, sexual intercourse, general anesthesia, surgical operations, and stress. Recent large cohort studies established that, within normal range, low serum PRL is associated with metabolic syndrome and obesity in children (37), and higher prevalence of impaired glucose intolerance, increased insulin resistance, polycystic ovary syndrome (PCOS), nonalcoholic fatty liver disease (NAFLD), and increased risks of new onset T2D in both men and women (11–18).
In concordance with epidemiologic studies, circulating levels of PRL are reduced in the ob/ob and db/db obese and diabetes mouse models, and in streptozocin-induced (STZ) T2DM rats. The 24 hour secretory pattern of plasma PRL levels is disrupted and reduced in high fat diet(HFD) rats (55), and Ruiz-Herrera et al. showed that raising plasma prolactin levels to 60-80 mg/dL equivalent to those reported in male rats in response to stress (56) counteracts the insulin resistance and adipose tissue dysfunction (57).
On the other hand, PRL is essential for adipose tissue differentiation and Prlr- mice with HFD demonstrated reduced adipocyte hyperplasia, increased adipocyte hypertrophy and increased insulin resistance (Table 1). Prlr- mice showed reduced fat and body weight after 16 weeks which is more prominent in females than males composed with a 29% decrease in fat mass, reduced leptin level and an even more pronounced 49% decrease in VAT (58). The decrease in fat depot is due to a reduced number of adipocytes (59). Aside from the effect on white adipose tissue, PRL have also been demonstrated to play a fundamental role in brown adipose tissue (BAT) differentiation. PRL promotes the generation and expansion of BAT in newborn rodents. BAT mass is significantly reduced in neonate Prlr- mice in comparison to their wild-type littermates with reduced brown adipocyte triglyceride content (60).
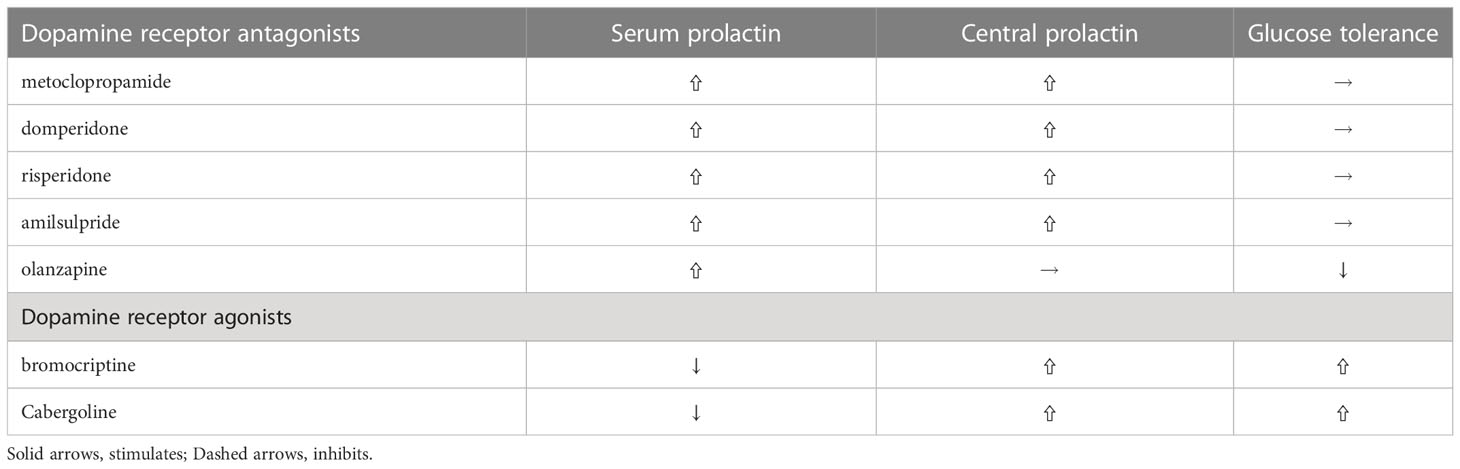
Table 1 Differential effects of various dopamine agonists and antagonists on PRL levels and glucose tolerance.
1.2 Central effects of PRL within the physiologic range
1.2.1 Central PRL effect on β (beta)-cell
Central actions of PRL are very diverse from its peripheral actions. Centrally administered low-dose PRL (0.1ug/h infusion) increased hypothalamus dopamine levels, decreased serum prolactin and improved insulin secretory capacity and sensitivity. In contrast, high-dose central PRL (1ug/h infusion) failed to increase hypothalamic dopamine, increased serum prolactin and decreased insulin secretion and sensitivity. It is suggested that the short-loop regulation of prolactin secretion by dopamine is desensitized and disrupted with continuous high-dose PRL stimulation. PRL acts on specific dopamine neurons in the hypothalamic arcuate nucleus and periventricular nucleus that increase dopamine secretion which tuberoinfundibular pathway inhibit prolactin release from the pituitary gland (61) (Figure 2). These dopaminergic neurons not only modulate PRL secretion, but also regulate the orexigenic and anorexigenic neurons within the arcuate nucleus (62). Central insulin in the dorsal striatum has been shown to reciprocally suppress dopamine levels (36) comprising a feedback control loop. In addition, low-PRL decreased body weight and epididymal fat pads and improved insulin sensitivity in T2DM rats which is consistent with dopamine receptor agonist actions described in paragraph 2.3. Only low-dose PRL stimulated first-phase insulin secretion and improved insulin sensitivity. Despite no increase in serum PRL level, both low and high central PRL dosages enhanced β-cell expansion suggesting that central prolactin could directly regulate β-cell function and proliferation regardless of serum prolactin levels (32). In summary, central prolactin regulates insulin secretion and glucose metabolism through central dopamine release and D2 receptor/sympathetic activity signaling (63). And β-cell stimulatory effects depend more on direct neural dopaminergic signal rather than on serum prolactin level.
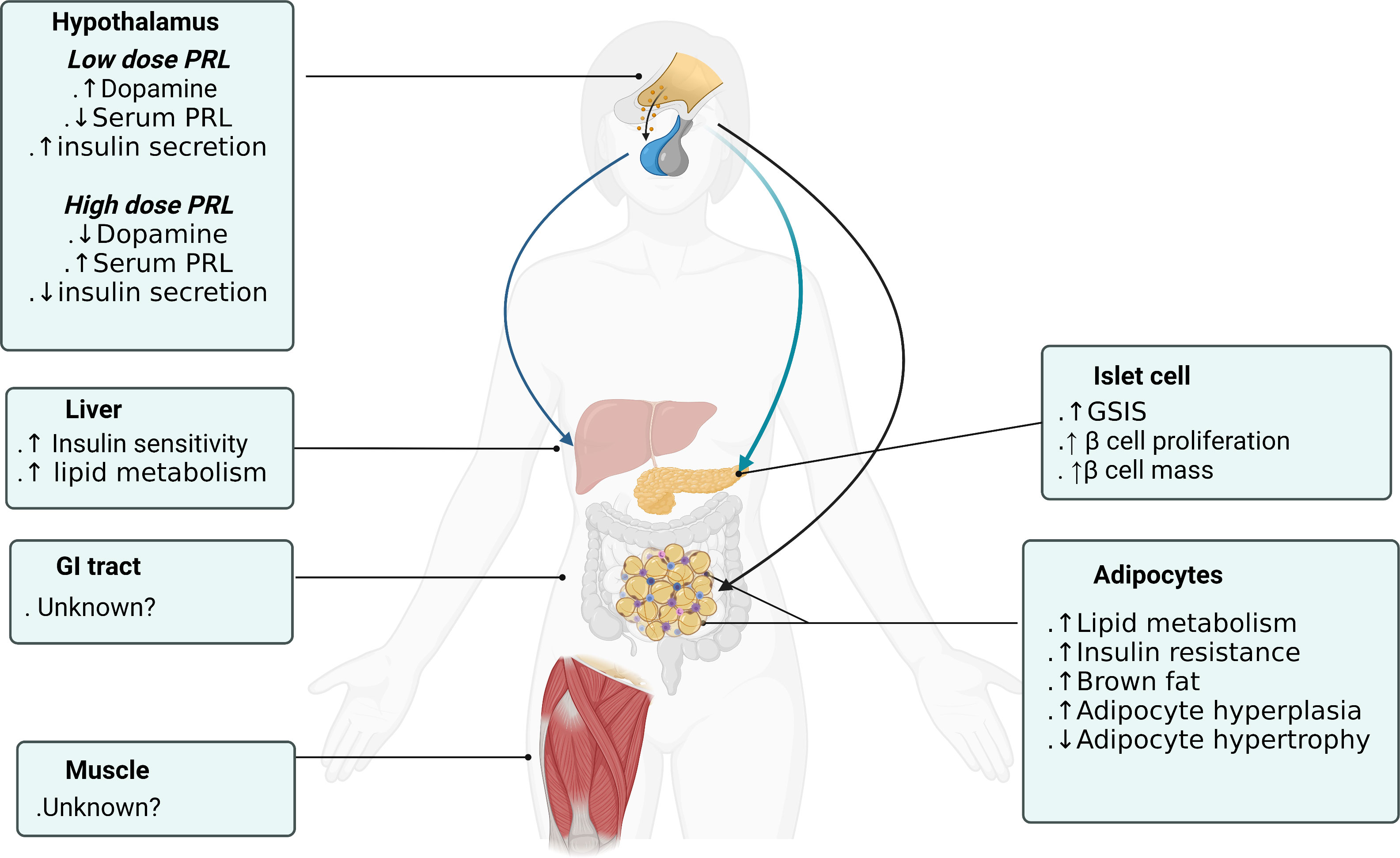
Figure 2 Prolactin actions on different organs to promote metabolic homeostasis. Figure created using BioRender (https://biorender.com/).
1.2.2 Central PRL effect on adipocyte and insulin resistance
No direct effect of central PRL on adipocytes have been discovered. However, dopamine D2/3 receptor inhibition at the ventral striatum results in diminished insulin sensitivity (64) and decreased mesolimbic dopamine activity leads to insulin resistance (65).
1.2.3 Central PRL effect on appetite regulation and hypothalamus
Within normal concentrations, PRL is not considered as a major factor in the occurrence of obesity. As discussed in paragraph 2.1.2, locally produced PRL is involved in the regulation of human adiposity and dysregulation leads to perpetuation of obesity. PRLR shares the same signaling pathways with leptin which is well known involved in regulation of appetite, metabolism and energy balance. In rodent models, increased serum PRL levels provoke leptin resistance that stimulates food intake and results in increased body weight and adiposity (66–68). During pregnancy and lactation, plasma leptin levels reduce by 40% for metabolic adaptions when high PRL levels, leptin resistance, hyperphagia (69), increased body weight coincide to facilitate nutrient shift to offspring (70). Some studies suggested that PRLR activation enhanced expression of suppressor-of-cytokine signaling (SOCS) proteins at hypothalamus including arcuate nucleus, medial preoptic area and the nucleus of the solitary tract which inhibited shared downstream signaling pathways of leptin receptor and leads to increased appetite and body weight (71). In contrast, Prlr- mice treated with leptin antagonist showed Prlr- mice responded to leptin antagonist similarly to the control mice, indicating no interaction between the actions of the two hormones (54). Such discrepancies may occur since insulin co-expression at PRLR is necessary for prolactin mediated effects (72) and central insulin suppresses dopamine levels in the ventral striatum (36). Body weight and adiposity are regulated and coordinated by a variety of factors including brain appetite signals, neuroendocrine signals, nutritional input and energy expenditure. In summary, whether elevated PRL levels in prolactinoma patients results in hyperphagia and overweight is controversial, since lowering PRL levels with D2R agonists do not always associate with reduced body weight and can even result in body weight gain (43, 73), implicating other factors such as dopaminergic signals may play a more dominant role.
1.3 Central effects of PRL excess and its correction with dopamine agonists
Prolactinomas or other causes of pathological hyperprolactinemia are associated with an increased risk of T2D. Mechanistically, pathological hyperprolactinemia has a negative effect on post-prandial insulin sensitivity (69). Despite the afore mentioned action of PRL to stimulate insulin secretion and increase insulin sensitivity, pathologic elevated levels of PRL (>70ng/ml) leads to increased insulin resistance (17, 74). Hyperinsulinemic-euglycemic clamp studies demonstrated post-prandial hyperinsulinemia and a significant decrease in peripheral post-prandial insulin sensitivity in both hyperprolactinemia obese and non-obese subjects (27, 69). In addition, hyperprolactinemia attenuate adiponectin expression in adipose tissue (75), which also decrease insulin sensitivity (76, 77).
Some frequently used dopamine receptor antagonists, such as metoclopramide, domperidone and risperidone, elevates both serum and central (cerebrospinal fluid) PRL levels and by doing so do not disturb energy and glucose metabolism, while other dopamine antagonists, such as olanzapine, induce impaired glucose tolerance and obesity since they do not increase central prolactin levels (78). And another dopamine antagonist, amisulpiride, a preferential antagonist at presynaptic D2/D3 receptors that increase dopaminergic transmission reduces glucose levels in diet-induced obese mice (79) and human (80). Studies with chronic hyperprolactinemia evoked by selective D2R knockout in lactotrophs leads to early glucose intolerance, hyperinsulinemia and subsequent liver steatosis and marked adiposity (81). Whereas studies with global D2R knockout mice develops impaired glucose tolerance, indicating pancreatic D2R participate in the effect of dopamine agonists (82). Recently, Aslanoglou showed that pancreatic hormone (insulin, and glucagon) secretion modulated by dopamine receptor agonists is dependent on D2R expression levels and D2 antagonist significantly increase glucagon as well as insulin secretion in human islets and treatment with D2 antagonists, such as olanzapine, frequently leads to impaired glucose homeostasis (83). Of note, hyperprolactinemia does not disturb glucose homeostasis in global D2R knockout mice (84), indicating that central dopaminergic system is crucial for the metabolic effects of PRL.
On the other hand, dopamine receptor agonists suppressed glucose-stimulated insulin secretion both in rats (85) and in patients with Parkinson’s disease (PD) (86). In prolactinoma patients treated with bromocriptine and cabergoline, glucose metabolism improved with increased insulin sensitivity. Increased insulin sensitivity correlated with bromocriptine dose measured by euglycemic hyperinsulinemic clamp. In studies treated with cabergoline, reduction in fasting insulin and HOMA-IR also correlated with cabergoline dose but not to body weight and BMI, suggesting that directly cabergoline could modulate insulin secretion and sensitivity. These actions are not limited to hyperprolactinemia state and have also been demonstrated in both diabetic and non-diabetic normoprolactinemia subjects (24, 25). Bromocriptine therapy in T2D subjects poorly controlled on high-dose insulin therapy resulted in improved glycemic control and meal tolerance while reducing insulin requirement (87).
Since PRL lowering action is the major mechanism of bromocriptine in prolactinoma treatment, it is also reasonable it contributed to a glucose-lowering effect. However, Framnes-DeBoer et al. demonstrated that bromocriptine improves glucose metabolism in prolactin-deficient, MC4R-deficient, and circadian disrupted mice (88) indicating that the glucose-lowering effect is independent of prolactin, MC4R, or circadian rhythms. In contrast, Furigo et al. using PRL antagonist(G129R-hPrl) treatment showed that bromocriptine effects on glucose metabolism is partly associated with blocked PRL actions, but it appeared only in female mice (31). While the exact glucose-lowering mechanism of bromocriptine remains undefined, it is postulated that bromocriptine may act by decreasing sympathetic activity (89). Decreased dopaminergic signals to the suprachiasmatic nucleus results in impaired glucose intolerance and increased insulin resistance in animal models (90). Studies have shown bromocriptine directly increases brain dopamine levels, most likely at the ventromedial nucleus of hypothalamus that lowers sympathetic activity, which leads to improved glucose homeostasis (91). Low concentrations of bromocriptine, dopamine have been shown to stimulate prolactin release from cultured pituitary tumor cell line GH4ZR7 cells while high concentrations inhibited the release (92). Haloperidol, a D2 receptor antagonist, blocked the inhibitory action, but was unable to block the dopamine-induced stimulatory action (93). Central PRL acts on specific dopaminergic neurons in the arcuate nucleus and paraventricular nucleus of the hypothalamus to increase dopamine secretion (61). Islet insulin secretion is mainly regulated by glucose uptake by glucose transporter-2 (GLUT-2), but it is also controlled by the central nervous system through parasympathetic and sympathetic nerves (94). Recently shown clusters of sharp wave-ripples from the hippocampus lowered peripheral glucose concentrations within 10 minutes by suppressing activity of the lateral septum, which is the major conduit between the hippocampus and the hypothalamus (35). It is suggested that hippocampal outputs affect neuroendocrine systems through hypothalamic hormone-releasing neurons or autonomic control of peripheral organs. Studies in humans showed that central insulin directly suppresses dopamine levels in the ventral striatum (36). And dopamine D2/3 receptor inhibition at the ventral striatum results in diminished insulin sensitivity (64) and in concert decreased mesolimbic dopamine activity leads to insulin resistance (65). In summary, central prolactin and dopamine levels work in coordination to control peripheral insulin secretion, and central insulin levels in turn feedbacks on mesolimbic dopamine system comprising a closed control system.
1.4 Peripheral effects of PRL excess and its correction with dopamine agonists (adipose tissue, skeletal muscle, hepatocyte, insulin resistance)
Ruiz-Herrera et al. showed that raising plasma prolactin levels to 60-80 mg/dL equivalent to those reported in response to stress (56) counteracts the insulin resistance and adipose tissue dysfunction in high fat diet(HFD) rats (57). PRL by increasing circulating adiponectin shows beneficiary effect on metabolism function, and these actions depends on circulating PRL levels kept at a relatively high concentration, but within a physiological range (within 7 and 100 ng/mL), defined as “homeostatic functionally increased transient prolactinemia” by Macotela (21). Mechanistically, PRL treatment in obese, insulin-resistant rats promotes adipocyte hyperplasia in both visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT), as well as reducing adipocyte hypertrophy in VAT. PRL increases adiponectin levels by Xbp1s which induces adipogenesis via the transcriptional activation of CEBP alpha and PPAR gamma (95). In both lean and obese mice, PRL increases adiponectin levels that improved glucose tolerance and insulin sensitivity (96).
In humans, consistent with animal models, Ruiz et al. showed that PRL values are higher in insulin sensitive males than insulin resistant males, regardless of their body mass index(BMI), and that PRL levels are positively associated with adiponectin levels and with the adipocyte fitness makers expressions, including Adiponectin, C1Q And Collagen Domain Containing (ADIPOQ), peroxisome proliferator-activated receptors gamma(PPAR gamma) and glucose transporter 4(GLUT4). In addition, ADIPOQ, PPAR gamma and GLUT4 expression in SAT positively correlated with adipocytes PRL synthesis (57). Recently, Ponce et al. extended these findings showing that circulating PRL levels are associated with VAT adipocyte size in humans (97). Patients with low circulating PRL levels have hypertrophic visceral adipocytes and are associated with increased insulin resistance. Consistent with this, visceral adipocytes from low PRL levels show higher PRLR expression than their subcutaneous counterparts (48). Patients with low circulating PRL have four-fold higher proportion of large adipocytes in only VAT but not SAT comparing to patients with high circulating PRL levels. In contrast, when insulin sensitive and insulin resistant subjects are compared, insulin resistant subjects have an increased proportion of large adipocytes in both VAT and SAT. Consistent with this, visceral adipocytes from insulin resistant subjects show lower PRLR expression and lower expression of adipocyte fitness makers ADIPOQ, PPAR gamma and GLUT4. Insulin resistance together with lower circulating PRL levels may act synergistically to impair beneficial metabolic action of PRL in adipocytes, and thereby exaggerate adipocyte hypertrophy and dysfunction (57). Verboven et al. also showed that adipocyte hypertrophy in VAT but not SAT is associated with increased inflammation and increased accumulation of pro-inflammatory immune cells (98). PRL treatment demonstrate anti-inflammatory effect in adipocytes and increases adiponectin serum levels and decreases IL-1β and monocyte chemoattractant protein 1(MCP 1) (57). However, it appears that exceeding a level of PRL (250 ng/mL) lowers systemic adiponectin values and negatively affect insulin sensitivity in adipocytes cell culture (76) and transgenic female (but not male) mice overexpressing PRL (74), which is discussed in paragraph 2.3. PRL by increasing circulating adiponectin shows beneficiary effect on metabolism function when kept in a range within 7 and 100 ng/mL, defined as “homeostatic functionally increased transient prolactinemia” by Macotela (21). Pathologic elevated levels of PRL (>70ng/ml) leads to increased insulin resistance (17, 74). Hyperinsulinemic-euglycemic clamp studies demonstrated post-prandial hyperinsulinemia and a significant decrease in peripheral post-prandial insulin sensitivity in both hyperprolactinemia obese and non-obese subjects (27, 69). In addition, hyperprolactinemia attenuate adiponectin expression in adipose tissue (75), which also decrease insulin sensitivity (76, 77). PRL and PRLR are related to fat metabolism and promote insulin resistance by activating the Janus kinase 2/signal transducer and activator of transcription 5 (JAK2/STAT5) signaling pathway and increasing triglyceride deposition (99).
Tavares et al. showed peripheral action of bromocriptine directly acts on dopamine receptors at the hepatocytes, skeletal muscle and adipocytes which activates adenosine monophosphate kinase (AMPK) phosphorylation to enhance glucose uptake and modulates lipid metabolism–related proteins in the adipose tissue (100). Bromocriptine treatment in diabetic rats upregulates dopamine receptors at adipose tissue and the liver resulting in higher insulin sensitivity and catabolic function (101).
Taken together, either locally produced PRL (autocrine, paracrine) or circulating PRL (endocrine) plays a principal role to modulate healthy accumulation of excessive fat in adipocytes. PRL promotes PPAR gamma, CEBP alpha and Xbp1s expression to favor adipogenesis, indicating PRL could directly alter adipocyte differentiation and preferentially distribute excessive fat into new adipocytes (increasing adipocyte hyperplasia) and also attenuate adipocyte hypertrophy and adipocyte inflammation (57). PRL are produced and secreted from human adipose tissue and PRL levels in obese adipose tissue are much reduced over lean subjects, indicating diminished autocrine and paracrine actions of PRL in obesity. Exactly how adipose autocrine PRL actions is disrupted under very low and high circulating prolactin level and how they mutually affect another remains for further exploration. In fact, evidence also demonstrate that dopaminergic action is tightly involved in the regulation of peripheral glucose homeostasis and insulin sensitivity discussed in the previous paragraphs. In summary, PRL enhances insulin sensitivity and the facilitate the healthy expansion of adipose tissue. These metabolic favorable mechanisms become disrupted under insulin resistant and obesity conditions, which is associated with low circulating PRL levels and could be counteracted by increasing PRL levels.
1.5 Central and peripheral effects of hypoprolactinemia
Studies in PRLR knockout (Prlr-) mice demonstrates the fundamental role of PRL and PL during pancreatic islet development. PRLR knockout (Prlr-) mice developed impaired glucose tolerance, decreased insulin secretion, a 20-35% reduction in islet insulin granules, reduced islet density and reduced β-cell mass. Prlr- mice also showed impaired glucose tolerance and blunted insulin secretory response comparing to their wild-type littermates (38).
PRL is also essential for adipose tissue differentiation and Prlr- mice with HFD demonstrated reduced adipocyte hyperplasia, increased adipocyte hypertrophy and increased insulin resistance. Prlr- mice showed reduced fat and body weight after 16 weeks which is more prominent in females than males composed with a 29% decrease in fat mass, reduced leptin level and an even more pronounced 49% decrease in VAT (58). The decrease in fat depot is due to a reduced number of adipocytes (59). Aside from the effect on white adipose tissue, PRL have also been demonstrated to play a fundamental role in brown adipose tissue (BAT) differentiation. PRL promotes the generation and expansion of BAT in newborn rodents. BAT mass is significantly reduced in neonate Prlr- mice in comparison to their wild-type littermates with reduced brown adipocyte triglyceride content (60).
However, hypoprolactinemia or a defect in the PRLR is an extraordinary rare clinical condition, with approximately a few dozen cases reported in the literature since 1975. The main clinical findings were excessive amniotic fluid (polyhydramnios) and lactation failure (102). Mutations in the genes encoding PRL or PRLR has not been shown to develop any disorder in non-pregnant condition (103).
2 Conclusion
In the past decade, major progress has emerged in understanding the multifaceted role of prolactin in metabolic homeostasis but is still far from being well understood. With adipocytes, prolactin exerts endocrine and autocrine actions which modulates insulin sensitivity and facilitate the healthy expansion of adipose tissue. However, their regulatory actions become disrupted during obesity and insulin resistance. As on islet cells, prolactin enhance β-cell proliferation and insulin secretion, and these actions depends more on direct neural dopamine signal affecting local prolactin level rather than on serum prolactin level per se. Dopamine receptor agonists which lowers serum prolactin affect glucose homeostasis by 1) affecting central prolactin that increase hypothalamic ventral medial and arcuate nucleus dopamine then through 2) dopaminergic/decreased sympathetic activity signaling, which 3) directly act on islet to stimulate insulin secretion and on adipocyte, hepatocyte, skeletal muscle D2R to modulate their metabolic effects (Figure 1). And reciprocally insulin suppresses mesolimbic dopamine system that comprise a feedback loop. The central dopamine and prolactin levels plays a key role in the glucose homeostasis control, and their dysregulation could lead to the pathognomonic central insulin resistance depicted in the “ominous octet”.
Patients with T2D have a higher risk of developing PD and the presence of T2D is associated with greater PD severity and faster progression. Central prolactin and dopamine actions play crucial roles in both diseases. Robust evidence shows the effects of bromocriptine treatment in type 2 diabetes. This review puts forward the plausible glucose-lowering mechanism of dopamine receptor agonists, however more studies are needed to clarify the specific actions of central dopamine and prolactin and dopaminergic signaling in insulin-sensitive tissues which would identify potential therapeutic strategies for the treatment of metabolic disorders including type 2 diabetes and metabolic syndrome and even Parkinson’s disease.
3 Review criteria
References for this review were identified through searches of PubMed for articles published from January 1970, to October 2022, by use of the terms “prolactin”, “placental lactogens”, “prolactin receptors”, “dopamine”, “dopamine receptor agonist, antagonist”, “insulin”, “insulin resistance”, “hyperprolactinemia”, and “hypoprolactinemia” in combination with the term “diabetes”. Articles resulting from these searches and relevant references cited in those articles were reviewed.
Author contributions
Conceptualization, H-YC. Investigation, H-YC and S-MC. Writing—original draft preparation, H-YC. Writing—review and editing, H-YC and S-MC. Supervision, H-YC and W-CL. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cho NH, Shaw JE, Karuranga S, Huang Y, Fernandes Rocha JD, Ohlrogge AW, et al. IDF diabetes atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract (2018) 138:271–81. doi: 10.1016/j.diabres.2017.03.024
2. Defronzo RA. Banting lecture. from the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes (2009) 58(4):773–95. doi: 10.2337/db09-9028
3. Bonner-Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab (2000) 11(9):375–8. doi: 10.1016/S1043-2760(00)00305-2
4. Sachdeva MM, Stoffers DA. Minireview: Meeting the demand for insulin: Molecular mechanisms of adaptive postnatal ß-cell mass expansion. Mol Endocrinol (2009) 23(6):747–58. doi: 10.1210/me.2008-0400
5. Huang C, Snider F, Cross JC. Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy. Endocrinology (2009) 150(4):1618–26. doi: 10.1210/en.2008-1003
6. Gerlo S, Davis JRE, Mager DL, Kooijman R. Prolactin in man: A tale of two promoters. Bioessays (2006) 28(10):1051–5. doi: 10.1002/bies.20468
7. Ben-Jonathan N, Mershon JL, Allen DL, Steinmetz RW. Extrapituitary prolactin: Distribution, regulation, functions, and clinical aspects*. Endoc Rev (1996) 17(6):639–69. doi: 10.1210/edrv-17-6-639
8. Ben-Jonathan N, LaPensee CR, LaPensee EW. What can we learn from rodents about prolactin in humans? Endoc Rev (2008) 29(1):1–41. doi: 10.1210/er.2007-0017
9. Clapp C, Thebault S, Jeziorski MC, Escalera La De Martínez G. Peptide hormone regulation of angiogenesis. Physiol Rev (2009) 89(4):1177–215. doi: 10.1152/physrev.00024.2009
10. Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: Actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endoc Rev (1998) 19(3):225–68. doi: 10.1210/edrv.19.3.0334
11. Terra LF, Garay-Malpartida MH, Wailemann RAM, Sogayar MC, Labriola L. Recombinant human prolactin promotes human beta cell survival via inhibition of extrinsic and intrinsic apoptosis pathways. Diabetologia (2011) 54(6):1388–97. doi: 10.1007/s00125-011-2102-z
12. Weinhaus AJ, Stout LE, Bhagroo NV, Brelje TC, Sorenson RL. Regulation of glucokinase in pancreatic islets by prolactin: A mechanism for increasing glucose-stimulated insulin secretion during pregnancy. J Endocrinol (2007) 193(3):367–81. doi: 10.1677/JOE-07-0043
13. Costanza M, Binart N, Steinman L, Pedotti R. Prolactin: a versatile regulator of inflammation and autoimmune pathology. Autoimmun Rev (2015) 14(3):223–30. doi: 10.1016/j.autrev.2014.11.005
14. Wagner R, Heni M, Linder K, Ketterer C, Peter A, Böhm A, et al. Age-dependent association of serum prolactin with glycaemia and insulin sensitivity in humans. Acta Diabetol (2014) 51(1):71–8. doi: 10.1007/s00592-013-0493-7
15. Wang T, Lu J, Xu Y, Li M, Sun J, Zhang J, et al. Circulating prolactin associates with diabetes and impaired glucose regulation: A population-based study. Diabetes Care (2013) 36(7):1974–80. doi: 10.2337/dc12-1893
16. Tuzcu A, Bahceci M, Dursun M, Turgut C, Bahceci S. Insulin sensitivity and hyperprolactinemia. J Endocrinol Invest (2003) 26(4):341–6. doi: 10.1007/BF03345182
17. Serri O, Li L, Mamputu J-C, Beauchamp M-C, Maingrette F, Renier G. The influences of hyperprolactinemia and obesity on cardiovascular risk markers: effects of cabergoline therapy. Clin Endocrinol (Oxf) (2006) 64(4):366–70. doi: 10.1111/j.1365-2265.2006.02469.x
18. Baptista T, Lacruz A, Mendoza S, Guillén Mendoza JM, Silvera R, Angeles F, et al. Body weight gain after administration of antipsychotic drugs: correlation with leptin, insulin and reproductive hormones. Pharmacopsychiatry (2000) 33(3):81–8. doi: 10.1055/s-2000-8451
19. Bina KG, Cincotta AH. Dopaminergic agonists normalize elevated hypothalamic neuropeptide y and corticotropin-releasing hormone, body weight gain, and hyperglycemia in ob/ob mice. Neuroendocrinology (2000) 71(1):68–78. doi: 10.1159/000054522
20. Strader AD, Buntin JD. Changes in agouti-related peptide during the ring dove breeding cycle in relation to prolactin and parental hyperphagia. J Neuroendocrinol (2003) 15(11):1046–53. doi: 10.1046/j.1365-2826.2003.01092.x
21. Macotela Y, Triebel J, Clapp C. Time for a new perspective on prolactin in metabolism. Trends Endocrinol Metab (2020) 31(4):276–86. doi: 10.1016/j.tem.2020.01.004
22. Biller BM, Colao A, Petersenn S, Bonert VS, Boscaro M. Prolactinomas, cushing's disease and acromegaly: debating the role of medical therapy for secretory pituitary adenomas. BMC Endocr Disord (2010) 10:10. doi: 10.1186/1472-6823-10-10
23. Holt RI, Barnett AH, Bailey CJ. Bromocriptine: old drug, new formulation and new indication. Diabetes Obes Metab (2010) 12(12):1048–57. doi: 10.1111/j.1463-1326.2010.01304.x
24. Bahar A, Kashi Z, Daneshpour E, Akha O, Ala S. Effects of cabergoline on blood glucose levels in type 2 diabetic patients: A double-blind controlled clinical trial. Med (Baltimore) (2016) 95(40):e4818. doi: 10.1097/MD.0000000000004818
25. Pijl H, Ohashi S, Matsuda M, Miyazaki Y, Mahankali A, Kumar V, et al. Bromocriptine: A novel approach to the treatment of type 2 diabetes. Diabetes Care (2000) 23(8):1154–61. doi: 10.2337/diacare.23.8.1154
26. Greenman Y, Tordjman K, Stern N. Increased body weight associated with prolactin secreting pituitary adenomas: Weight loss with normalization of prolactin levels. Clin Endocrinol (Oxf) (1998) 48(5):547–53. doi: 10.1046/j.1365-2265.1998.00403.x
27. Berinder K, Nyström T, Höybye C, Hall K, Hulting A-L. Insulin sensitivity and lipid profile in prolactinoma patients before and after normalization of prolactin by dopamine agonist therapy. Pituitary (2011) 14(3):199–207. doi: 10.1007/s11102-010-0277-9
28. Kamath V, Jones CN, Yip JC, Varasteh BB, Cincotta AH, Reaven GM, et al. Effects of a quick-release form of bromocriptine (Ergoset) on fasting and postprandial plasma glucose, insulin, lipid, and lipoprotein concentrations in obese nondiabetic hyperinsulinemic women. Diabetes Care (1997) 20(11):1697–701. doi: 10.2337/diacare.20.11.1697
29. Holt RIG, Barnett AH, Bailey CJ. Bromocriptine: old drug, new formulation and new indication. Diabetes Obes Metab (2010) 12:1048–57.
30. Mikhail N. Quick-release bromocriptine for treatment of type 2 diabetes. Curr Drug Deliv (2011) 8(5):511–6. doi: 10.2174/156720111796642255
31. Furigo IC, Suzuki MF, Oliveira JE, Ramos-Lobo AM, Teixeira PDS, Pedroso JA, et al. Suppression of prolactin secretion partially explains the antidiabetic effect of bromocriptine in ob/ob mice. Endocrinology (2019) 160(1):193–204. doi: 10.1210/en.2018-00629
32. Park S, Kang S, Lee H-W, Ko Seob B. Central prolactin modulates insulin sensitivity and insulin secretion in diabetic rats. Neuroendocrinology (2012) 95(4):332–43. doi: 10.1159/000336501
33. Rubí B, Ljubicic S, Pournourmohammadi S, Carobbio S, Armanet M, Bartley C, et al. Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J Biol Chem (2005) 280(44):36824–32. doi: 10.1074/jbc.M505560200
34. Borcherding DC, Hugo ER, Idelman G, Silva De A, Richtand NW, Loftus J, et al. Dopamine receptors in human adipocytes: expression and functions. PloS One (2011) 6(9):e25537. doi: 10.1371/journal.pone.0025537
35. Tingley D, McClain K, Kaya E, Carpenter J, Buzsáki G. A metabolic function of the hippocampal sharp wave-ripple. Nature (2021) 597(7874):82–6. doi: 10.1038/s41586-021-03811-w
36. Kullmann S, Blum D, Jaghutriz BA, Gassenmaier C, Bende B, Häring H-U, et al. Central insulin modulates dopamine signaling in the human striatum. J Clin Endocrinol Metab (2021) 106(10):2949–61. doi: 10.1210/clinem/dgab410
37. Chirico V, Cannavò S, Lacquaniti A, Salpietro V, Mandolfino M, Romeo PD, et al. Prolactin in obese children: A bridge between inflammation and metabolic-endocrine dysfunction. Clin Endocrinol (Oxf) (2013) 79(4):537–44. doi: 10.1111/cen.12183
38. Freemark M, Avril I, Fleenor D, Driscoll P, Petro A, Opara E, et al. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology (2002) 143(4):1378–85. doi: 10.1210/endo.143.4.8722
39. Sorenson RL, Brelje TC. Prolactin receptors are critical to the adaptation of islets to pregnancy. Endocrinology (2009) 150(4):1566–9. doi: 10.1210/en.2008-1710
40. Arumugam R, Fleenor D, Freemark M. Knockdown of prolactin receptors in a pancreatic beta cell line: Effects on DNA synthesis, apoptosis, and gene expression. Endocrine (2014) 46(3):568–76. doi: 10.1007/s12020-013-0073-1
41. Ben-Jonathan N, Hugo ER, Brandebourg TD, LaPensee CR. Focus on prolactin as a metabolic hormone. Trends Endocrinol Metab (2006) 17(3):110–6. doi: 10.1016/j.tem.2006.02.005
42. Labriola L, Montor WR, Krogh K, Lojudice FH, Genzini T, Goldberg AC, et al. Beneficial effects of prolactin and laminin on human pancreatic islet-cell cultures. Mol Cell Endocrinol (2007) 263(1-2):120–33. doi: 10.1016/j.mce.2006.09.011
43. Steele CA, MacFarlane IA, Blair J, Cuthbertson DJ, Didi M, Mallucc C, et al. Pituitary adenomas in childhood, adolescence and young adulthood: presentation, management, endocrine and metabolic outcomes. Eur J Endocrinol (2010) 163(4):515–22. doi: 10.1530/EJE-10-0519
44. Nieuwenhuizen AG, Schuiling GA, Moes H, Koiter TR. Role of increased insulin demand in the adaptation of the endocrine pancreas to pregnancy. J Cell Biol (1997) 159:303–12.
45. Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J Cell Biol (2015) 208(5):501–12. doi: 10.1083/jcb.201409063
46. Rosen ED, Bruce MS. What we talk about when we talk about fat. Cell (2014) 156(1):20–44. doi: 10.1016/j.cell.2013.12.012
47. Gustafson B, Hedjazifar S, Gogg S, Hammarstedt, Smith U. Insulin resistance and impaired adipogenesis. Trends Endocrinol Metab (2015) 26(4):193–200. doi: 10.1016/j.tem.2015.01.006
48. Macotela Y, Emanuelli B, Mori MA, Gesta S, Schulz TJ, Tseng Y-H, et al. Intrinsic differences in adipocyte precursor cells from different white fat depots. Diabetes (2012) 61(7):1691–9. doi: 10.2337/db11-1753
49. Hugo ER, Borcherding DC, Gersin KS, Loftus J, Ben-Jonathan N. Prolactin release by adipose explants, primary adipocytes, and LS14 adipocytes. J Clin Endocrinol Metab (2008) 93(10):4006–12. doi: 10.1210/jc.2008-1172
50. Stewart WC, Baugh JE Jr, Floyd ZE, Stephens JM. STAT 5 activators can replace the requirement of FBS in the adipogenesis of 3T3-L1 cells. Biochem Biophys Res Commun (2004) 324(1):355–9. doi: 10.1016/j.bbrc.2004.09.053
51. Nanbu-Wakao R, Fujitani Y, Masuho Y, Muramatu M, Wakao H. Prolactin enhances CCAAT enhancer-binding protein-beta (C/EBP beta) and peroxisome proliferator-activated receptor gamma (PPAR gamma) messenger RNA expression and stimulates adipogenic conversion of NIH-3T3 cells. Mol Endocrinol (2000) 14(2):307–16. doi: 10.1016/j.bbrc.2007.03.168
52. Brandebourg TD, Bown JL, Ben-Jonathan N. Prolactin upregulates its receptors and inhibits lipolysis and leptin release in male rat adipose tissue. Biochem. Biophys Res Commun (2007) 357(2):408–13. doi: 10.1016/j.bbrc.2007.03.168
53. Combs TP, Berg AH, Rajala MW, Klebanov S, Iyengar P, Jimenez-Chillaron JC. Sexual differentiation, pregnancy, calorie restriction, and aging affect the adipocyte-specific secretory protein adiponectin. Diabetes (2003) 52(2):268–76. doi: 10.2337/diabetes.52.2.268
54. Carré N, Solomon G, Gertler A, Binart N. Effects of high affinity leptin antagonist on prolactin receptor deficient male mouse. PloS One (2014) 9(3):e91422. doi: 10.1371/journal.pone.0091422
55. Lemini M, Ruiz-Herrera X, Ledesma-Colunga MG, Díaz-Lezama N, Ríos Los De EA, López-Barrera F, et al. Prolactin anterior pituitary expression and circulating levels are reduced in obese and diabetic rats: role of TGF-β and TNF-α. Am J Physiol Regul Integr Comp Physiol (2015) 308(9):R792–9. doi: 10.1152/ajpregu.00327.2014
56. Esquifino AI, Chacón F, Jimenez V, Toso Reyes CF, Cardinali DP. d24-hour changes in circulating prolactin, follicle-stimulating hormone, luteinizing hormone and testosterone in male rats subjected to social isolation. J Circadian Rhythms (2004) 2(1):1–1. doi: 10.1186/1740-3391-2-1
57. Ruiz-Herrera X, Ríos Los EA, Díaz JM, Lerma-Alvarado RM, Escalera Martínez L, López-Barrera F, et al. Prolactin promotes adipose tissue fitness and insulin sensitivity in obese males. Endocrinology (2017) 158(1):56–68. doi: 10.1210/en.2016-1444
58. Le JA, Wilson HM, Shehu A, Devi YS, Aguilar T, Gibori G, et al. Prolactin activation of the long form of its cognate receptor causes increased visceral fat and obesity in males as shown in transgenic mice expressing only this receptor subtype. Horm Metab Res (2011) 43(13):931–7. doi: 10.1055/s-0031-1291182
59. Flint DJ, Binart N, Boumard S, Kopchick JJ, Kelly P. Developmental aspects of adipose tissue in GH receptor and prolactin receptor gene disrupted mice: site-specific effects upon proliferation, differentiation and hormone sensitivity. J Endocrinol (2006) 191(1):101–11. doi: 10.1677/joe.1.06939
60. Viengchareun S, Servel N, Fève B, Freemark M, Lombès M, Binart N, et al. Prolactin receptor signaling is essential for perinatal brown adipocyte function: A role for insulin-like growth factor-2. PloS One (2008) 3(2):e1535. doi: 10.1371/journal.pone.0001535
61. Lyons DJ, Broberger C. TIDAL WAVES: Network mechanisms in the neuroendocrine control of prolactin release. Front. Neuroendocrinol (2014) 35(4):420–38. doi: 10.1016/j.yfrne.2014.02.001
62. Zhang X, van den Pol AN. Hypothalamic arcuate nucleus tyrosine hydroxylase neurons play orexigenic role in energy homeostasis. Nat Neurosci (2016) 19(10):1341–7. doi: 10.1038/nn.4372
63. Reis FM, Reis AM, Coimbra CC. Effects of hyperprolactinaemia on glucose tolerance and insulin release in male and female rats. J Endocrinol (1997) 153(3):423–8. doi: 10.1677/joe.0.1530423
64. Caravaggio F, Borlido C, Hahn M, Feng Z, Fervaha G, Gerretsen P, et al. Reduced insulin sensitivity is related to less endogenous dopamine at D2/3 receptors in the ventral striatum of healthy nonobese humans. Int J Neuropsychopharmacol (2015) 18(7):pyv014–4. doi: 10.1093/ijnp/pyv014
65. Volkow ND, Wang GJ, Telang F, Fowler JS, Thanos PK, Logan J, et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: possible contributing factors. NeuroImage (2008) 42(4):1537–43. doi: 10.1016/j.neuroimage.2008.06.002
66. Gerardo-Gettens T, Moore BJ, Stern JS, Horwitz BA. Prolactin stimulates food intake in a dose-dependent manner. Am J Physiol (1989) 256(1 Pt 2):R276–80. doi: 10.1152/ajpregu.1989.256.1.R276
67. Sauvé D, Woodside B. Neuroanatomical specificity of prolactin-induced hyperphagia in virgin female rats. Brain Res (2000) 868(2):306–14. doi: 10.1016/S0006-8993(00)02344-1
68. Naef L, Woodside B. Prolactin/Leptin interactions in the control of food intake in rats. Endocrinology (2007) 148(12):5977–83. doi: 10.1210/en.2007-0442
69. Tuzcu A, Yalaki S, Arikan S, Gokalp D, Bahcec M, Tuzcu S, et al. Evaluation of insulin sensitivity in hyperprolactinemic subjects by euglycemic hyperinsulinemic clamp technique. Pituitary (2009) 12(4):330–4. doi: 10.1007/s11102-009-0183-1
70. Freemark M, Fleenor D, Driscoll P, Binart N, Kelly P. Body weight and fat deposition in prolactin receptor-deficient mice. Endocrinology (2001) 142(2):532–7. doi: 10.1210/endo.142.2.7979
71. Nagaishi VS, Cardinali LI, Zampieri TT, Furigo IC, Metzger M, Donato J Jr, et al. Possible crosstalk between leptin and prolactin during pregnancy. Neuroscience (2014) 259:71–83. doi: 10.1016/j.neuroscience.2013.11.050
72. Bachelot A, Beaufaron J, Servel N, Kedzia C, Monget P, Kellyet PA, et al. Prolactin independent rescue of mouse corpus luteum life span: identification of prolactin and luteinizing hormone target genes. Am J Physiol Endocrinol Metab (2009) 297(3):E676–84. doi: 10.1152/ajpendo.91020.2008
73. Auriemma RS, Granieri L, Galdiero M, Simeoli C, Perone Y, Vitale P, et al. Effect of cabergoline on metabolism in prolactinomas. Neuroendocrinology (2013) 98(4):299–310. doi: 10.1159/000357810
74. dos Santos Silva CM, Barbosa FRP, Lima GAB, Warszawski L, Fontes R, Domingues RC, et al. BMI and metabolic profile in patients with prolactinoma before and after treatment with dopamine agonists. Obes (Silver Spring) (2011) 19(4):800–5. doi: 10.1038/oby.2010.150
75. Zhang XZ, Imachi H, Lyu JY, Fukunaga K, Sato S, Ibata T, et al. Prolactin regulatory element-binding protein is involved in suppression of the adiponectin gene in vivo. J Endocrinol Invest (2017) 40(4):437–45. doi: 10.1007/s40618-016-0589-3
76. Nilsson L, Binart NM, Bohlooly -Y, Bramnert M, Egecioglu E, Kindblom J, et al. Prolactin and growth hormone regulate adiponectin secretion and receptor expression in adipose tissue. Biochem Biophys Res Commun (2005) 331(4):1120–6. doi: 10.1016/j.bbrc.2005.04.026
77. Pala NA, Laway BA, Misgar RA, Shah Amin Z, Gojwari TA, Dar TA, et al. Profile of leptin, adiponectin, and body fat in patients with hyperprolactinemia: Response to treatment with cabergoline. Indian J Endocrinol Metab (2016) 20(2):177–81. doi: 10.4103/2230-8210.176346
78. Park S, Hong SM, Ahn Sung II, Kim Hoon S. Olanzapine, not resperidone, exacerbates β-cell function and mass in ovariectomized diabetic rats and estrogen replacement reverses them. J Psychopharmacol (2010) 24(7):1105–14. doi: 10.1177/0269881109348167
79. Roix JJ, DeCrescenzo GA, Cheung PH, Ciallella JR, Sulpice T, Saha S, et al. Effect of the antipsychotic agent amisulpride on glucose lowering and insulin secretion. Diabetes Obes Metab (2012) 14(4):329–34. doi: 10.1111/j.1463-1326.2011.01529.x
80. Kopf D, Gilles M, Paslakis G, Medlin F, Lederbogen F, Lehnert H, et al. Insulin secretion and sensitivity after single-dose amisulpride, olanzapine or placebo in young male subjects: double blind, cross-over glucose clamp study. Pharmacopsychiatry (2012) 45(6):223–8. doi: 10.1055/s-0031-1301365
81. Luque GM, Lopez-Vicchi F, Ornstein María A, Brie B, Winne De C, Fiore E, et al. Chronic hyperprolactinemia evoked by disruption of lactotrope dopamine D2 receptors impacts on liver and adipocyte genes related to glucose and insulin balance. Am J Physiol Endocrinol Metab (2016) 311(6):E974–88. doi: 10.1152/ajpendo.00200.2016
82. García-Tornadú I, Ornstein AM, Chamson-Reig A, Wheeler MB, Hill DJ, Arany E, et al. Disruption of the dopamine d2 receptor impairs insulin secretion and causes glucose intolerance. Endocrinology (2010) 151(4):1441–50. doi: 10.1210/en.2009-0996
83. Aslanoglou D, Bertera S, Sánchez-Soto M, Free Benjamin R, Lee J, Zong W, et al. Dopamine regulates pancreatic glucagon and insulin secretion via adrenergic and dopaminergic receptors. Transl Psychiatry (2021) 11(1):59. doi: 10.1038/s41398-020-01171-z
84. Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, et al. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron (1997) 19(1):103–13. doi: 10.1016/S0896-6273(00)80351-7
85. Zern RT, Bird JL, Feldman JM. Effect of increased pancreatic islet norepinephrine, dopamine and serotonin concentration on insulin secretion in the golden hamster. Diabetologia (1980) 18(4):341–6. doi: 10.1007/BF00251017
86. Rosati G, Maioli M, Aiello I, Farris A, Agnetti V. Effects of long-term l-dopa therapy on carbohydrate metabolism in patients with parkinson's disease. Eur Neurol (1976) 14(3):229–39. doi: 10.1159/000114744
87. Roe ED, Chamarthi B, Raskin P. Impact of bromocriptine-QR therapy on glycemic control and daily insulin requirement in type 2 diabetes mellitus subjects whose dysglycemia is poorly controlled on high-dose insulin: A pilot study. J Diabetes Res 2015 (2015), 834903. doi: 10.1155/2015/834903
88. Framnes-DeBoer SN, Bakke E, Yalamanchili S, Peterson H, Sandoval DA, Seeley RJ, et al. Bromocriptine improves glucose tolerance independent of circadian timing, prolactin, or the melanocortin-4 receptor. Am J Physiol Endocrinol Metab (2020) 318(1):E62–71. doi: 10.1152/ajpendo.00325.2019
89. Cincotta AH, Meier AH, Cincotta M Jr. Bromocriptine improves glycaemic control and serum lipid profile in obese type 2 diabetic subjects: A new approach in the treatment of diabetes. Expert Opin Investig Drugs (1999) 8(10):1683–707. doi: 10.1517/13543784.8.10.1683
90. Luo S, Luo J, Meier AH, Cincotta AH. Dopaminergic neurotoxin administration to the area of the suprachiasmatic nuclei induces insulin resistance. Neuroreport (1997) 8(16):3495–9. doi: 10.1097/00001756-199711100-00016
91. Garber AJ, Blonde L, Bloomgarden ZT, Handelsman Y, Dagogo-Jack S. The role of bromocriptine-QR in the management of type 2 diabetes expert panel recommendations. Endocr Pract (2013) 19(1):100–6. doi: 10.4158/EP12325.OR
92. Tabak J, Toporikova N, Freeman ME, Bertram R. Low dose of dopamine may stimulate prolactin secretion by increasing fast potassium currents. J Comput Neurosci (2007) 22(2):211–22. doi: 10.1007/s10827-006-0008-4
93. Chang A, Shin SH. Dopamine agonists both stimulate and inhibit prolactin release in GH4ZR7 cells. Eur J Endocrinol (1999) 141(4):387–95. doi: 10.1530/eje.0.1410387
94. Ahrén B. Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia (2000) 43(4):393–410. doi: 10.1007/s001250051322
95. Cho YM, Kwak S-N, Joo N-S, Kim Hun D, Lee A-H, Kim K-S, et al. X-Box binding protein 1 is a novel key regulator of peroxisome proliferator-activated receptor γ2. FEBS J (2014) 281(22):5132–46. doi: 10.1111/febs.13052
96. Ye R, Scherer PE. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab (2013) 2(3):133–41. doi: 10.1016/j.molmet.2013.04.001
97. Ponce AJ, Galván-Salas T, Lerma-Alvarado RM, Ruiz-Herrera X, Hernández-Cortés T, Valencia-Jiménez R, et al. Low prolactin levels are associated with visceral adipocyte hypertrophy and insulin resistance in humans. Endocrine (2020) 67(2):331–43. doi: 10.1007/s12020-019-02170-x
98. Verboven K, Wouters K, Gaens K, Hansen D, Bijnen M, Wetzels S, et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci. Rep (2018) 8(1):4677. doi: 10.1038/s41598-018-22962-x
99. Wang PY, Jin C-C, Liu C, Zhao Z-J, Yang H-Y. PRL/PRLR can promote insulin resistance by activating the JAK2/STAT5 signaling pathway. Comput. Math Methods Med (2022) 2022:1456187. doi: 10.1155/2022/1456187
100. Tavares G, Martins FO, Melo BF, Matafome P, Conde SV. Peripheral dopamine directly acts on insulin-sensitive tissues to regulate insulin signaling and metabolic function. Front Pharmacol (2021) 12:101241. doi: 10.3389/fphar.2021.713418
101. Tavares G, Marques D, Barra C, Rosendo-Silva D, Costa A, Rodrigues T, et al. Dopamine D2 receptor agonist, bromocriptine, remodels adipose tissue dopaminergic signalling and upregulates catabolic pathways, improving metabolic profile in type 2 diabetes. Mol Metab (2021) 51:101241. doi: 10.1016/j.molmet.2021.101241
102. Forsyth IA, Wallis M. Growth hormone and prolactin–molecular and functional evolution. J. Mammary Gland Biol Neoplasia (2002) 7(3):291–312. doi: 10.1023/A:1022804817104
Keywords: central insulin resistance, dopamine receptor agonists, prolactin, insulin resistance, insulin secretion
Citation: Chien H-Y, Chen S-M and Li W-C (2023) Dopamine receptor agonists mechanism of actions on glucose lowering and their connections with prolactin actions. Front. Clin. Diabetes Healthc. 4:935872. doi: 10.3389/fcdhc.2023.935872
Received: 04 May 2022; Accepted: 20 January 2023;
Published: 09 March 2023.
Edited by:
Antonella Amato, University of Palermo, ItalyReviewed by:
Seley Gharanei, University of Warwick, United KingdomRosario Pivonello, University of Naples Federico II, Italy
Copyright © 2023 Chien, Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wan-Chun Li, d2NsaUB5bS5lZHUudHc=
†These authors have contributed equally to this work