 Sohaib Mohammed
Sohaib Mohammed Ajay Krishna Sunkara
Ajay Krishna Sunkara Casey Elizabeth Walike1
Casey Elizabeth Walike1 Greeshma Gadikota
Greeshma Gadikota
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Clim. , 02 August 2021
Sec. Carbon Dioxide Removal
Volume 3 - 2021 | https://doi.org/10.3389/fclim.2021.713708
This article is part of the Research Topic Reservoir Processes and Global Practices in Geologic Carbon Sequestration View all 5 articles
Advancing a portfolio of technologies that range from the storage of excess renewable natural gas for distributed use to the capture and storage of CO2 in geological formation are essential for meeting our energy needs while responding to challenges associated with climate change. Delineating the surface interactions and the organization of these gases in nanoporous environments is one of the less explored approaches to ground advances in novel materials for gas storage or predict the fate of stored gases in subsurface environments. To this end, the molecular scale interactions underlying the organization and transport behavior of CO2 and CH4 molecules in silica nanopores need to be investigated. To probe the influence of hydrophobic surfaces, a series of classical molecular dynamics (MD) simulations are performed to investigate the structure and dynamics of CO2 and CH4 confined in OH-terminated and CH3-terminated silica pores with diameters of 2, 4, 6, 8, and 10 nm at 298 K and 10 MPa. Higher adsorption extents of CO2 compared to CH4 are noted on OH-terminated and CH3-terminated pores. The adsorbed extents increase with the pore diameter. Further, the interfacial CO2 and CH4 molecules reside closer to the surface of OH-terminated pores compared to CH3-terminated pores. The lower adsorption extents of CH4 on OH-terminated and CH3-terminated pores result in higher diffusion coefficients compared to CO2 molecules. The diffusivities of both gases in OH-terminated and CH3-terminated pores increase systematically with the pore diameter. The higher adsorption extents of CO2 on OH-terminated and CH3-terminated pores are driven by higher van der Waals and electrostatic interactions with the pore surfaces, while CH4 adsorption is mainly due to van der Waals interactions with the pore walls. These findings provide the interfacial chemical basis underlying the organization and transport behavior of pressurized CO2 and CH4 gases in confinement.
Scientific advancements that enable us to harness renewable energy resources and facilitate the removal and storage of greenhouse gas emissions are essential for meeting our energy and resource needs in a sustainable manner while responding to challenges associated with climate change. One approach in the portfolio of strategies needed to address this challenge is the storage of renewable energy carriers such as hydrogen and biomethane. Storage of excess renewable energy carriers in natural and engineered environments is critical to meeting our energy needs on demand. In this context, there is an emerging interest in exploring subsurface environments (Pfeiffer and Bauer, 2015; Berta et al., 2018; Shi et al., 2020) and engineered materials (Yun et al., 2002; Düren et al., 2004; Dündar-Tekkaya and Yürüm, 2016) to store clean energy carriers such as biomethane and hydrogen. Further, safe and permanent storage of CO2 in geologic formations at the gigaton scale is essential if renewable and non-renewable carbon-bearing fuels continue to be used (Bachu et al., 2007; Aydin et al., 2010; Michael et al., 2010; Jiang, 2011; Zhang and Bachu, 2011). Resolving the physical and chemical adsorption of carbon dioxide (CO2) and methane (CH4) on porous silica is an area of increasing interest due to its relevance in various energy and environmental fields, including CO2 capture and CO2 utilization and storage coupled with enhanced oil and gas recovery from subsurface environments (Kuuskraa et al., 2013; Huo et al., 2017; Mohammed and Gadikota, 2019a; Farajzadeh et al., 2020; Klewiah et al., 2020).
While prior studies reported the link between functionalized silica surfaces and the structure of CO2 and CH4 for applications related to gas separations (Wiheeb et al., 2015; Mafra et al., 2018), the coupled effects of confinement and surface hydrophobicity have not been systematically probed. The surfaces of silica nanopores and nanoparticles can be either hydrophilic or hydrophobic depending on the surface polarity and the chemistry of the functional groups on the surface (Jin et al., 2019). The hydrophobicity of silica surfaces has significant impacts on the organization and flow of confined fluids, including water (Jin et al., 2021), hydrocarbons (Ghoufi et al., 2013) and gases. Given the interest in storing gases in subsurface environments, delineating the mechanisms underlying the storage of gases in oil-wet vs. water-wet environments is essential. Further, the influence of nano-scale confinement on the organization of these gases needs to be further delineated given the abundance of nanoporous-rich environments in subsurface environments and the anomalous thermodynamics and transport behavior of nano-confined fluids.
Extensive progress has already been made toward explaining the physical and chemical interactions of CO2 with amine-functionalized porous silica using a wide range of experimental and computational characterization techniques (Chaikittisilp et al., 2011; Liu et al., 2014; Cogswell et al., 2015). Amine-functionalized porous silica provides the advantage of chemically binding CO2 with the impregnated amines in the porous silica that enhance the gas uptake (Serna-Guerrero et al., 2008), as opposed to OH-functionalized porous silica in which CO2 adsorbs physically through van der Waals interactions, electrostatic interactions, and hydrogen bonding. The chemical binding of CO2 with the amine groups results in high CO2 adsorption capacity, good selectivity, fast adsorption and desorption rates, and low energy consumption when compared to the porous materials that capture CO2 through physical adsorption (Li et al., 2014). Despite these advancements in developing functional materials, there have been no efforts to contrast the organization of CO2 and CH4 in pores with hydrophobic and hydrophilic silica surfaces.
The structure, dynamics and flow of CH4 in organic and inorganic confinement have also been studied using a range of experimental and computational tools, including small-angle neutron scattering (Chiang et al., 2016a,b; Mohammed et al., 2020b) and molecular simulations (Siderius et al., 2017; Xiong et al., 2017; Mohammed and Gadikota, 2020; Wang et al., 2020). The core-shell organization of pressurized CH4 molecules in confinement and their associated dimensions can be successfully determined using small-angle neutron scattering (Chiang et al., 2016a,b; Mohammed et al., 2020b), and these measurements can be used to validate molecular simulations. The differences in the spatial organization of confined gases contribute to anisotropic variations in their diffusivities (Wu et al., 2015; Xiong et al., 2016, 2017; Mohammed and Gadikota, 2019a). Confined effects influence the desorption of bound species such as water and CO2 (Baumgartner et al., 2019; Knight et al., 2019).
Prior work by Qin et al. (2008) used classical molecular dynamics (MD) simulations to show that supercritical CO2 is preferentially adsorbed on hydroxylated silica surfaces [-Si(OH)] instead of silylated [-Si(CH3)3] silica pores due to the hydrogen bonding interactions between the confined CO2 and -OH groups on the pore. Although these insights provided by Qin and co-workers provide a crucial link to the fate of confined CO2 and the surface chemistry of silica pores, the effects of pore diameter remain unresolved.
To address this knowledge gap, we performed a series of classical MD simulations to investigate the structure, organization, dynamics, and energetics of CO2 and CH4 confined in OH-functionalized and CH3-functionalized silica pores with diameters of 2, 4, 6, 8, and 10 nm. The hypothesis that the organization of CO2 and CH4 molecules is influenced by the hydrophobicity of the surfaces is investigated. The simulations are performed at 10 MPa and 298 K to mimic the mechanical and thermal conditions of subsurface environments.
CO2, CH4, and β-cristobalite silica unit cells are built using Avogadro software. The structure of isolated cristobalite unit cell is optimized using density functional theory implemented in Quantum Espresso (Giannozzi et al., 2017). A kinetic energy cutoff for wavefunctions of 36 Ry and K-points mesh of (6 × 6 × 6) are used to optimize the β-cristobalite silica unit cell. These values are chosen based on the convergence of the total energy of the silica structure. Ultrasoft pseudopotentials are implemented in which the generalized gradient approximation (GGA) (Perdew et al., 1996) is selected for the exchange correlation functional. The Broyden-Fletcher-Goldfarn-Shanno (BFGS) (Head and Zerner, 1985) algorithm is utilized to perform the structural optimization of the silica unit cell. BFGS has been widely used to optimize the electronic structure of a wide range of materials in DFT calculations (Han et al., 2019; Sharma et al., 2019; Neupane and Adhikari, 2020).
Silica surfaces are constructed by replicating the optimized β-cristobalite unit cell in x, y, and z directions. Slit-shaped pores with heights of 2, 4, 6, 8, and 10 nm are cleaved in the constructed surfaces. The non-bridging oxygens on the interior pore surfaces are functionalized with -OH and -CH3 groups to tune the surface hydrophobicity of the constructed pores. Each pore has a length of 9.95 nm along the X-axis and a depth of 3.98 nm along the Y-axis, while the pore height varies from 2 to 10 nm along the Z-axis.
The pressures of CO2 and CH4 inside the pores is set to 10 MPa. The number of CO2 and CH4 molecules are 193, 385, 578, 771, and 964 in 2, 4, 6, 8, and 10 nm pores, respectively (see Figure 1). Silica pores are modeled using ClayFF forcefield (Cygan et al., 2004). The TraPPE forcefield (Potoff and Siepmann, 2001) is used to model -CH3 functional group. The CO2 molecules are modeled using TraPPE forcefield, while CH4 molecules are modeled using OPLS/AA forcefield (Jorgensen et al., 1996). These forcefields have been widely used to study the properties of CO2 and CH4 on silica surfaces because the governed adsorption extents, organizations and dynamics agree with the experimental data (Phan et al., 2014; Le et al., 2015; Mohammed et al., 2020b, 2021).
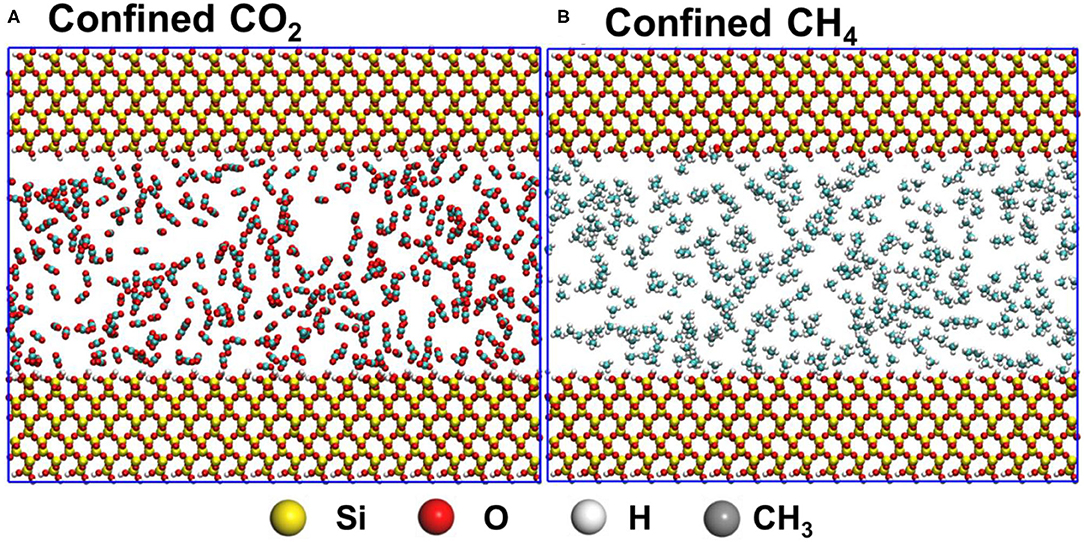
Figure 1. The initial configurations of confined CO2 and CH4 in silica pores with diameters 4 nm are shown in panels (A,B), respectively. Atoms in silica pores and confined gases molecules shown in CPK and VDW drawing methods, respectively, are implemented in VMD software.
Energy minimization is performed on the initial configurations for 50,000 steps using the steepest descent method to remove inappropriate geometries and reduce the high energy of the randomly distributed gas molecules in the pore space. Constant number of molecules, constant volume and constant temperature (NVT) ensemble are performed on the optimized cells for 50 ns under 298 K. The temperature is controlled using Nose-Hoover thermostat with a relaxation time of 1 ps (Evans and Holian, 1985). The short-range interaction is calculated in a cutoff of 1.4 nm, while the long-range electrostatic interactions are treated using Particle Mesh Ewald (PME). The equation of motion is integrated by leapfrog integrator with a time step of 2 fs. Non-bonding interactions are accounted for van der Waals and electrostatic potentials. Bonding interactions are calculated from bond stretching, angle bending, and dihedrals in CO2 and CH4 molecules while only O-H and O-CH3 bond stretching are considered in silica pores. MD simulations are performed and analyzed using GROMACS 2018 code (Abraham et al., 2015) and the visualization of the simulations inputs and outputs are performed using VDM software (Humphrey et al., 1996). Further information on the simulation details can be found in Mohammed and Gadikota (2018, 2019a,b); Mohammed and Gadikota (2020) and Mohammed et al. (2020a,b).
Insights into the organization of CO2 and CH4 molecules in silica nanopores are obtained from the density profiles of these gases. The density profiles of confined CO2 and CH4 along the pore height (z-axis) in OH-terminated and CH3-terminated pores with diameter of 2 nm is shown in Figure 2 as a representative for the distribution of confined gases in slit pores with different pore hydrophobicity. The density profiles show a preferential adsorption of CO2 and CH4 on the pore surfaces in both OH-terminated and CH3-terminated pores. The density profile of the confined gases decreases sharply on approaching the pore center where the minimum density values are observed for both gases. However, the number density of CO2 molecules confined in OH-terminated pores is higher at the pore surface while the number density of CH4 molecules is higher in the center of the pore (see Figures 2A,B). These observations suggest that CO2 adsorbs preferentially on the OH-terminated silica pores compared to CH4 at similar conditions of temperature, pressure and pore size. Prior studies probing the competitive adsorption of CO2 on OH-terminated silica pores showed that CO2 adsorbs preferentially over nonpolar aliphatic hydrocarbons (paraffins) such as ethane (C2H6) (Elola and Rodriguez, 2019), propane (C3H8) (Mohammed and Gadikota, 2019a), butane (C4H10) (Le et al., 2015) and octane (C8H18) (Le et al., 2016).
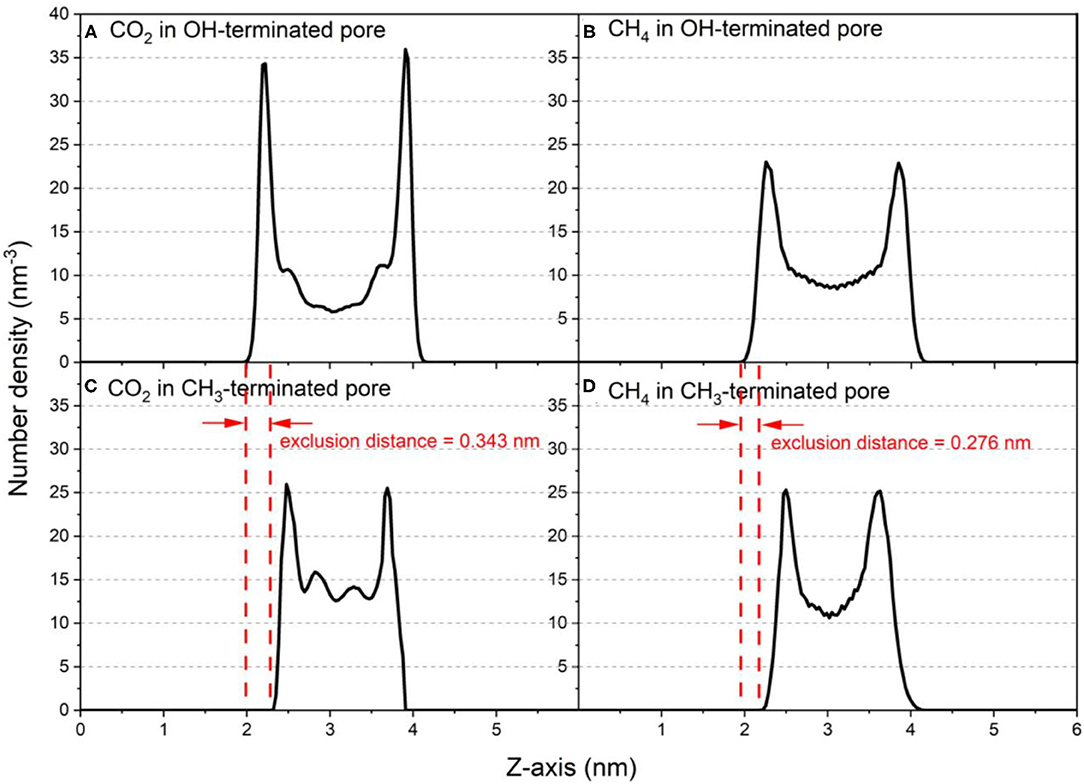
Figure 2. Number density profiles of confined CO2 and CH4 molecules along the vector normal to the pore surface (z-axis) in (A,B) OH-terminated pores and (C,D) CH3-terminated pores in pore diameter of 2 nm. The density profiles are averaged over the last 10 ns of the simulation time.
The preferential adsorption of CO2 and CH4 on the surfaces of the OH-terminated and CH3-terminated pores compared to the pore center is evident from the 2D density maps of the gases' distribution in the pore space (see Figures 3, 4). The density maps are averaged over the last 10 ns of the simulation time to ensure an equilibrium state. CO2 showed higher density on the pore surfaces compared to CH4 in OH-terminated and CH3-terminated surfaces in all pore sizes. Further, the interactions with the functional groups on the pore surface influence the distribution of the interfacial molecules such that higher CO2 densities are observed at the OH groups and higher CH4 densities are noted at CH3 groups.
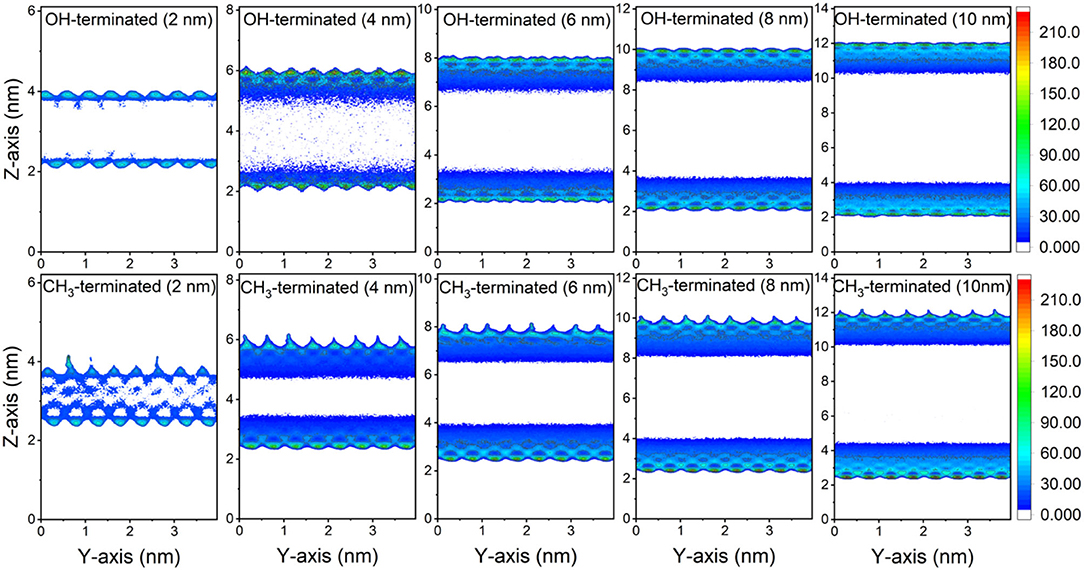
Figure 3. 2D density maps of CO2 confined in OH-terminated and CH3-terminated pores as a function of the pore diameter. The density maps are averaged over the last 10 ns of the simulation times.
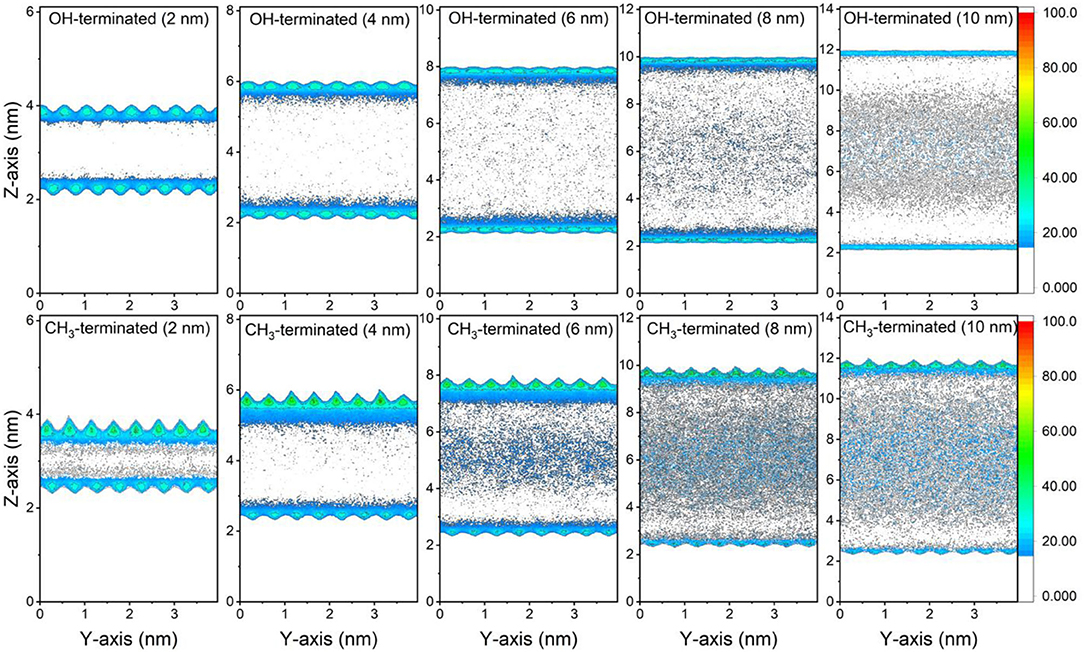
Figure 4. 2D density maps of CH4 confined in OH-terminated and CH3-terminated pores as a function of the pore diameter. The density maps are averaged over the last 10 ns of the simulation times.
Interestingly, the location of the adsorbed CO2 and CH4 peaks on the CH3-terminated pores are shifted toward the pore center by about 0.34 and 0.27 nm, respectively, in 2 nm sized pores (see Table 1). The shift in the adsorbed peaks “exclusion region” in hydrophobic silica surfaces is evident for water (Jin et al., 2019), but studies on gases confined in CH3-teriminated silica are scarce, making it difficult to compare such observations with previous studies. The exclusion of CO2 and CH4 from the CH3-terminated pore surface is also shown by the snapshots taken from the silica-CO2 and silica-CH4 interfaces (see Figure 5). Figure 5 shows that the interfacial CO2 and CH4 molecules can reside between the dangling OH groups on the OH-terminated surfaces, while there is a finite distance between the dangling CH3 groups and the interfacial CO2 and CH4 molecules.
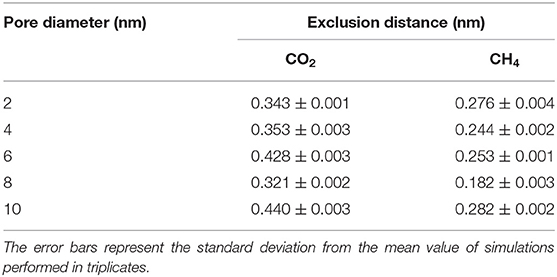
Table 1. The exclusion distance (nm) of CO2 and CH4 in CH3-terminated pores as a function of the pore diameter.
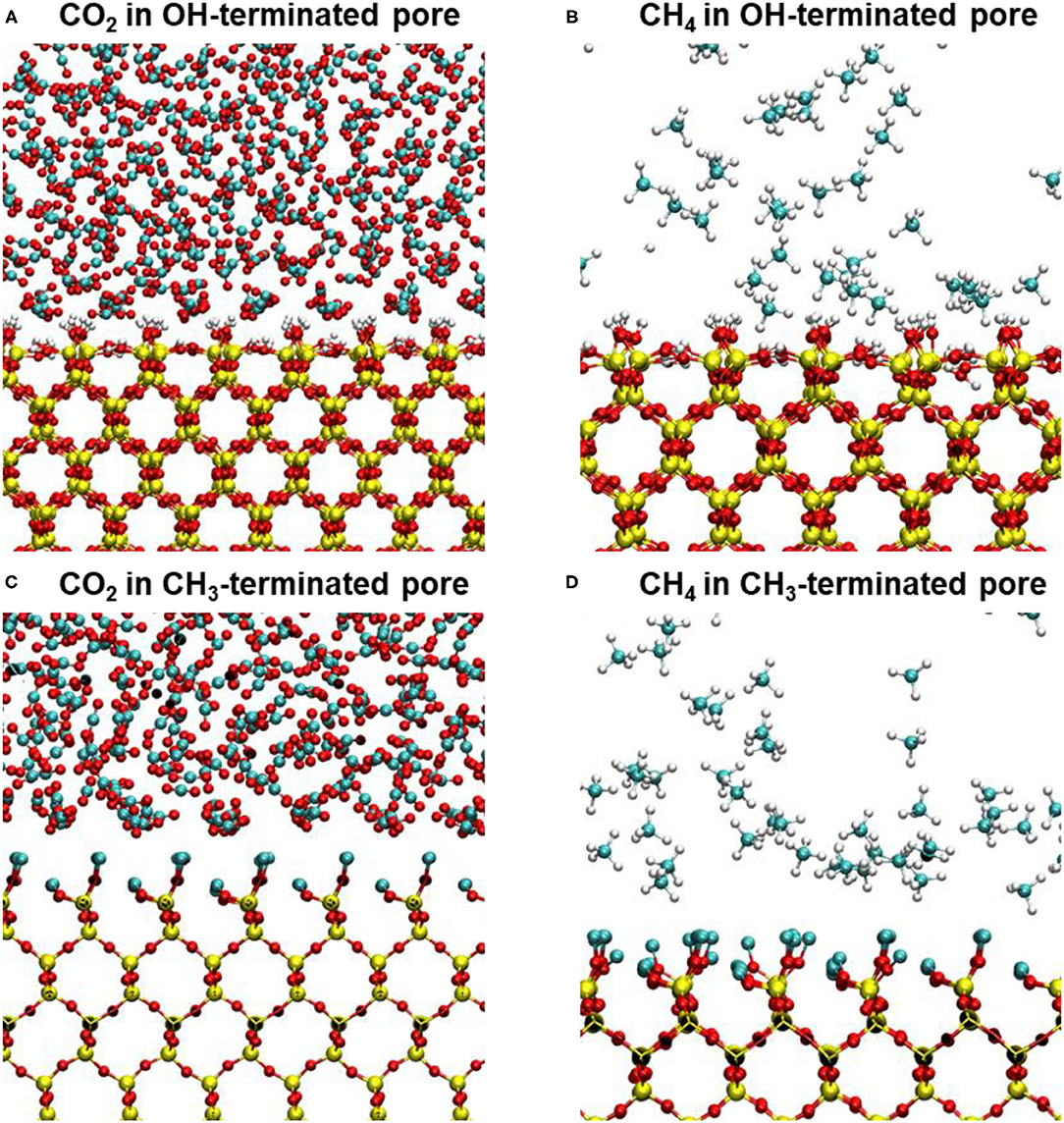
Figure 5. Snapshots showing the interfacial CO2 and CH4 molecules on (A,B) OH-terminated pore surfaces and (C,D) CH3-terminated pore surfaces. The snapshots represent the last trajectory frame of CO2 and CH4 molecules confined in 10 nm pores. The exclusion regions of interfacial CO2 molecules on CH3-terminated pores are evident in panels (C,D).
To contrast the adsorption behavior of CO2 and CH4, the adsorption extents (mmol/m2) of CO2 and CH4 on the pore surfaces are calculated from the number density profiles in OH-terminated and CH3-terminated pores and normalized by Avogadro number and the area of the pore surface (see Figure 6) as follows:
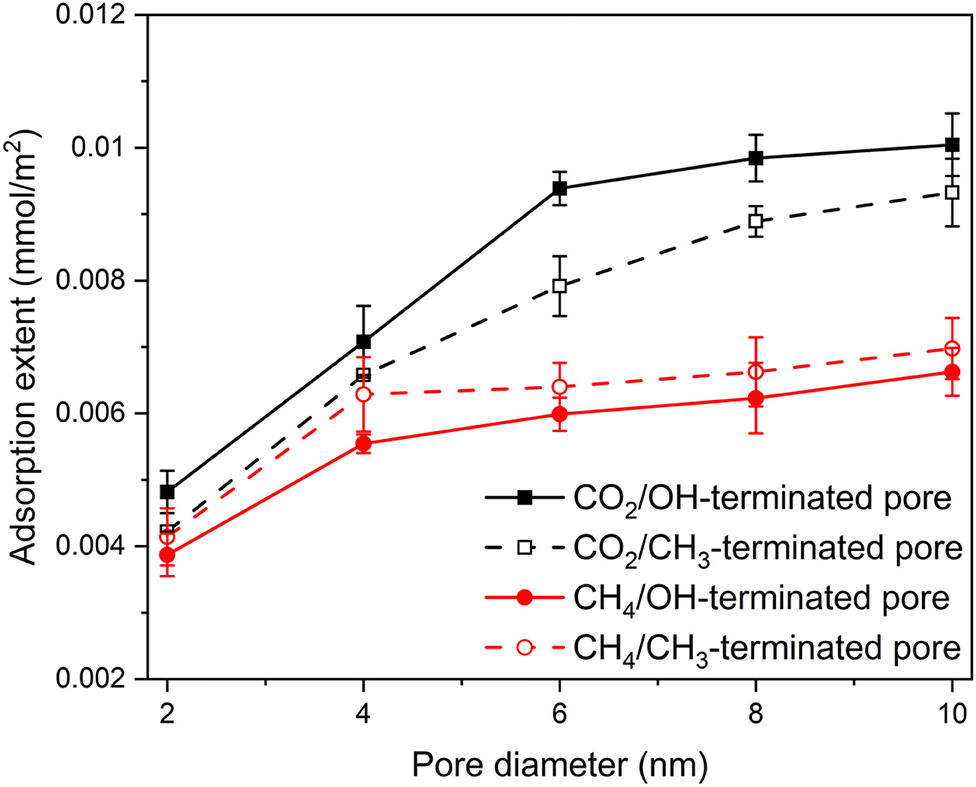
Figure 6. The adsorption extents of CO2 and CH4 on OH-terminated and CH3-terminated pore surfaces as a function of the pore diameter and pore surface chemistry. The adsorption extents are determined as the number of adsorbed molecules by Avogadro number and the pore surface area. The adsorption extents are averaged over the last 10 ns of the simulation time. Error bars represent the standard deviation from the mean values of three different simulations.
The adsorbed molecules were considered as the first peak in the density profile adjacent to the pore surface. This approach is consistent with prior studies (Mohammed and Gadikota, 2019a). The adsorption extent of CO2 and CH4 in OH-terminated and CH3-terminated pores increases systematically with the increase in the pore diameter. However, the adsorption extent of CO2 is higher than that of CH4 in OH-terminated and CH3-terminated pores. Further, the adsorption extent of CO2 in OH-terminated pores is higher than that in CH3-terminated pores. In contrast, the CH4 molecules adsorb preferentially on CH3-terminated pores compared to that in OH-terminated pores. As the pore diameter increases from 2 nm to 10 nm, the adsorption extent of CO2 in OH-terminated pores increases from 0.48 ± 0.03 to 1 ± 0.05 mmol/m2, while CO2 adsorption extent on CH3-terminated pores increases from 0.42 ± 0.01 to 0.92 ± 0.05 mmol/m2. Similarly, increasing the pore diameter from 2 to 10 nm causes the adsorption extent of CH4 to increase from 0.38 ± 0.03 to 0.66 ± 0.03 mmol/m2 in OH-terminated pores and from 0.41 ± 0.04 to 0.70 ± 0.04 mmol/m2 in CH3-terminated pores. The profiles of the adsorption extents are consistent with previous studies after accounting for the difference in the compositions of confined gases (Mohammed and Gadikota, 2019a).
The structural properties are further analyzed by calculating the radial distribution function (RDF) of the carbon atoms of CO2 and CH4 with respect to the -OH and -CH3 groups on the pore surface (Figure 7). RDFs are averaged over the last 10 ns of the simulations time to ensure equilibrium structure of the interfacial CO2 and CH4 molecules. RDF of C atoms of CO2 and CH4 showed two peaks with respect to the -OH and -CH3 groups on the silica pores surfaces. However, the first peak of OH-CCO2 is higher than that of CH3-CCO2 (see Figures 7A,C). Further, the first peak of OH-CCO2 is located at a radius of 0.34 nm while that of CH3-CCO2 is located at 0.41 nm. The lower first peak of CCH4 compared to CCO2 with respect to -OH and -CH3 functional groups (Figures 7B,D) on the pore surfaces agrees with the lower adsorption extent of CH4 on OH-terminated and CH3-terminated pores. The peak values of all RDFs decrease systematically as the pore diameter increases due to the larger number of gas molecules residing in the pore center compared the molecules adsorbed on the pore surface. Previous studies showed similar trends in the RDF of confined CO2 in OH-terminated and CH3-terminated pore surfaces (Qin et al., 2008).
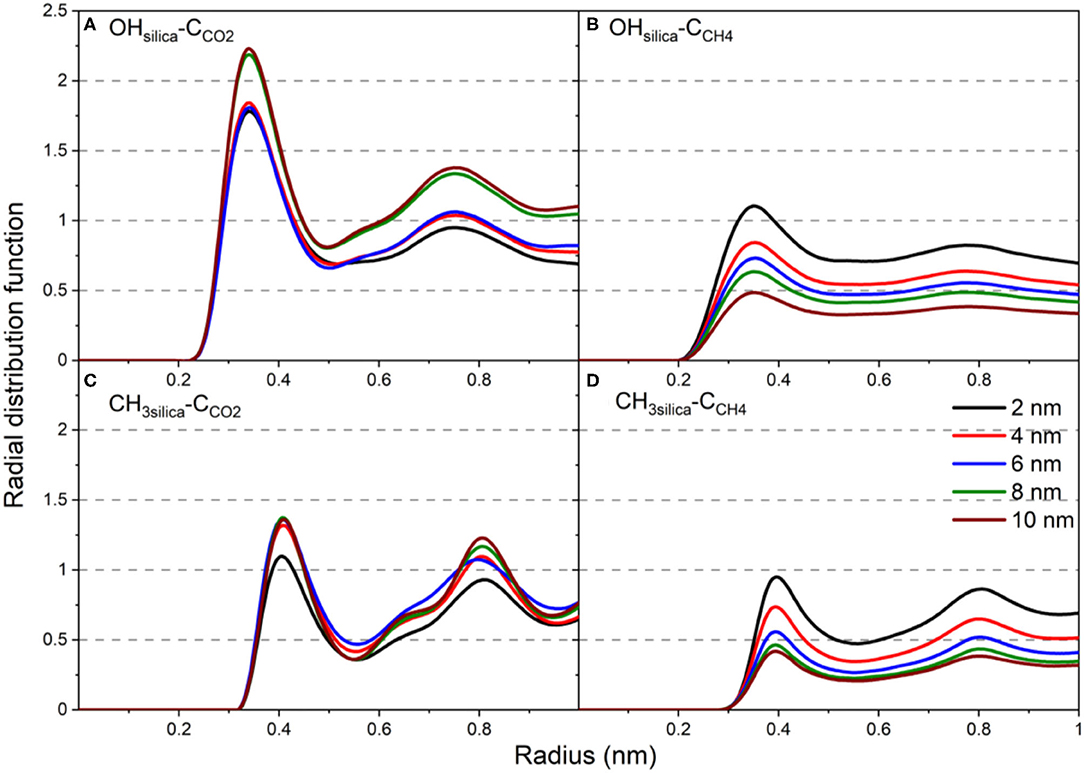
Figure 7. The radial distribution function of (A) OHsilica-CCO2, (B) OHsilica-CCH4, (C) CH3silica-CCO2, (D) CH3silica-CCH4 as a function of the pore diameter. Radial distribution functions are averaged over the last 10 ns of the simulation time.
The coordination numbers (n(r)) of CCO2 and CCH4 with respect to -OH and -CH3 groups on the pore surfaces (Figure 8) are calculated by integrating the radial distribution function as follows:
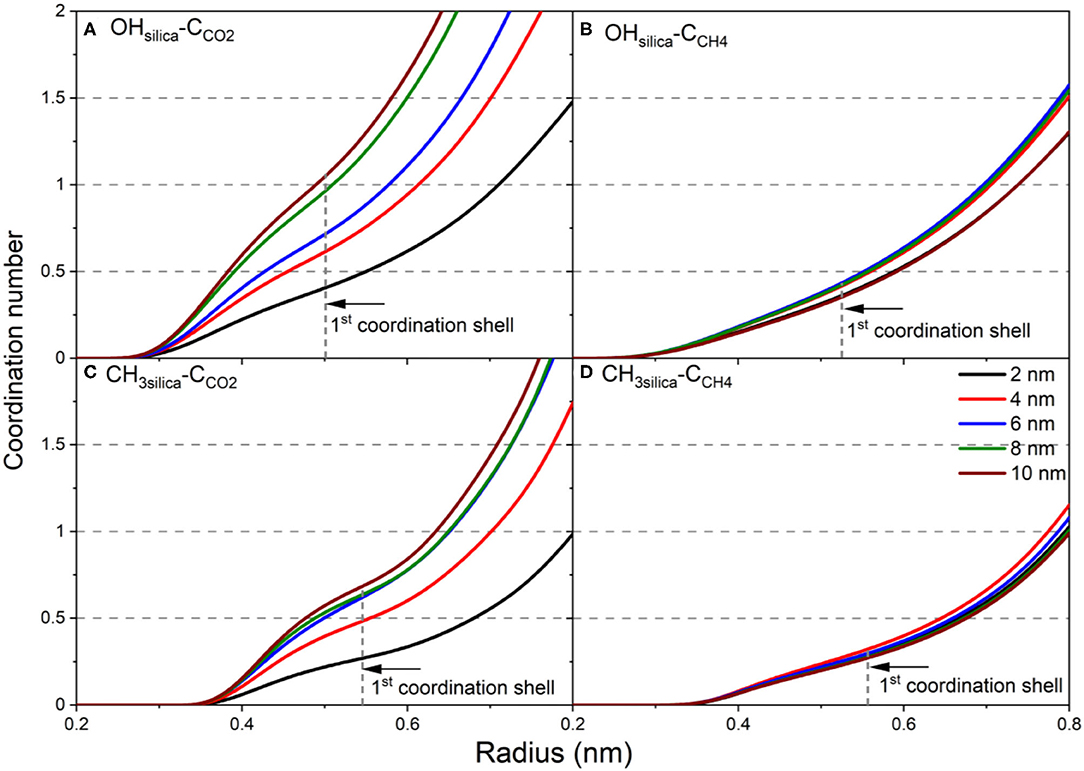
Figure 8. The coordination number of (A) CCO2 from OHsilica, (B) CCH4 from OHsilica, (C) CCO2 from CH3silica, and (D) CCH4 from CH3silica as a function of the pore diameter. The coordination number is calculated by integrating the radial distribution function and is averaged over the last 10 ns of the simulation time.
In the expression above, ρ is the density, r is the radius and g(r) is the radial distribution function. The number of CCO2 atoms in the first coordination shell of -OH and -CH3 increase significantly as the pore diameter increases from 2 nm to 10 nm (see Figures 8A,C). However, the number of CCO2 in the first coordination shell of OH is higher than that of CH3. In contrast, a slight increase is noted in the number of CCH4 in the first coordination shell of -OH and -CH3 groups on the silica surface (Figures 8B,D). In addition, the number of CCO2 in the first coordination shell of -OH and -CH3 is higher than CCH4 in all the pore sizes. The denser packing of the first coordination shell of CCO2 agrees with higher adsorption extent on OH-terminated and CH3-terminated pore surfaces.
The hypothesis that the structure of confined fluids influences their transport is investigated. The self-diffusion coefficient of confined CO2 and CH4 is calculated in the plane perpendicular to the pore diameter (xy plane) due to the negligible dimensionality of the pore diameter compared to xy plane (infinite) (see Figure 9). The self-diffusion coefficients are calculated based on the mean square displacement (l2) as follows (Gadikota et al., 2017):
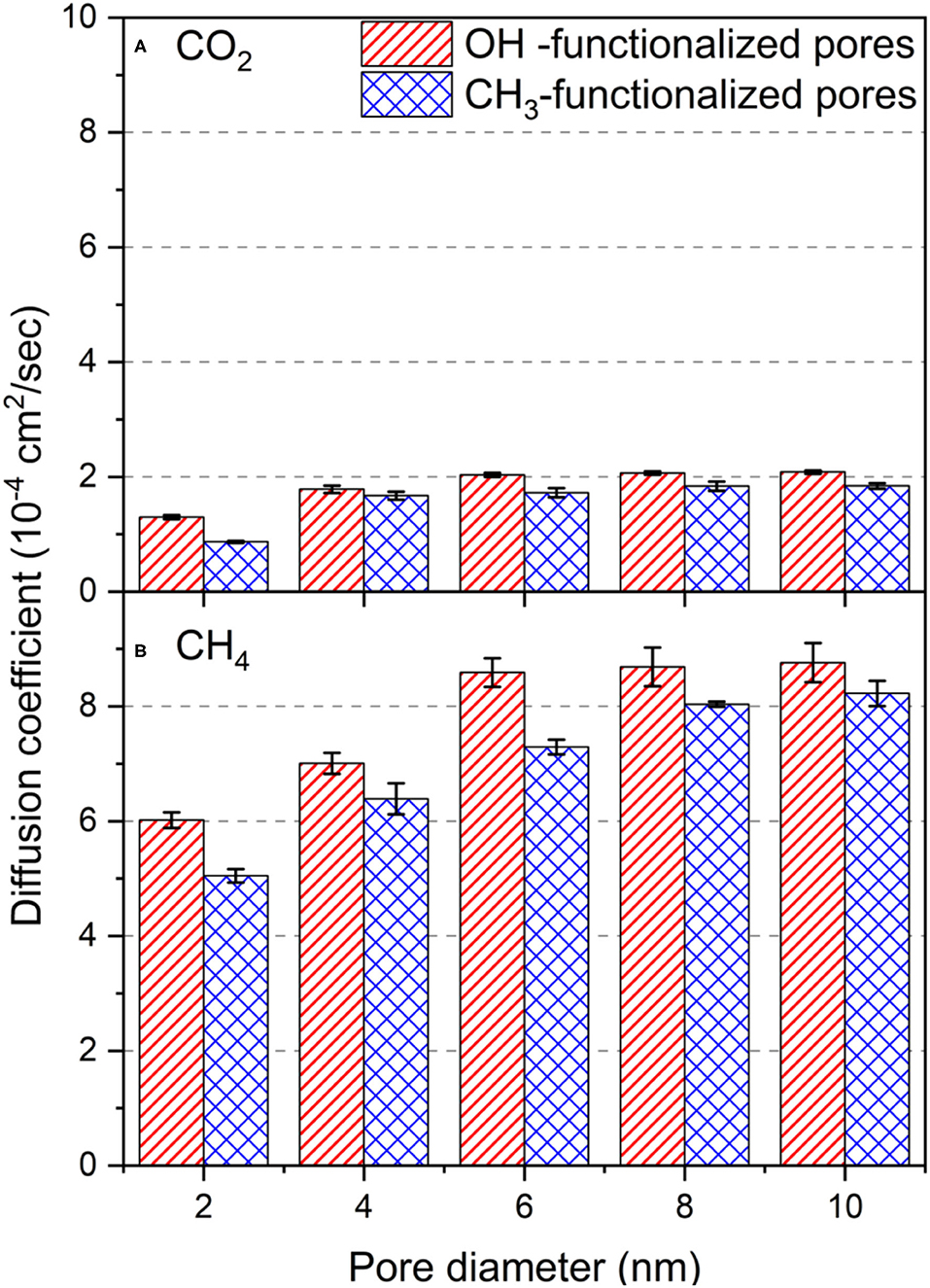
Figure 9. The self-diffusion coefficient of (A) CO2 and (B) CH4 molecules in OH-terminated and CH3-terminated pores as a function of the pore diameter. Diffusion coefficients are averaged over the last 10 ns of the simulation time. Error bars represent the standard deviation from the mean values of the three different simulations.
In the expression above, l2 was calculated in the xy plane (n = 2), and t is the time. The diffusion coefficients of confined CO2 and CH4 increase systematically with the pore diameter in OH-terminated and CH3-terminated pores. The diffusion coefficients of CO2 in OH-terminated pores increased from (1.29 ± 0.34) × 10−4 to (2.08 ± 0.29) × 10−4 cm2/sec as the pore diameter increases from 2 nm to 10 nm. Similarly, the diffusion coefficient of CO2 in CH3-terminated pores increased from (0.86 ± 0.16) × 10−4 to (1.84 ± 0.50) × 10−4 cm2/sec as the pore diameter increased from 2 to 10 nm. This increase in diffusivity with the pore diameter is attributed to the higher number of molecules confined in larger pores that occupy the adsorption sites on the pore surface and leave a large fraction of the confined molecules outside the attractive energy well near the pore surface. The non-adsorbed molecules on the pore surface can move freely along the xy direction due to the low molecular collisions and low steric hinderance in the pore center. The values of the self-diffusion coefficient of confined CO2 match those predicted by previous studies after accounting for the differences in the pore diameter, CO2 loading and the applied temperature (Qin et al., 2008; Le et al., 2015).
Interestingly, the diffusion coefficients in OH-terminated pores are higher than those confined in CH3-terminated pores. The higher diffusion coefficients in OH-terminated pores can be linked to the higher adsorption extent of CO2 molecules on the pore surface, such that it results in lower molecular density in the center of the pore. The lower molecular density in the center of the pore contributes to fewer molecular collisions in the OH-terminated pores compared to those in the CH3-terminated pores.
The lower adsorption extent of CH4 on OH-terminated and CH3-terminated silica pores, relative to CO2, result in higher in-plane self-diffusion coefficients in all the pore sizes. As the pore diameter increase from 2 nm to 10 nm, CH4 diffusion coefficients increase from (6.01 ± 0.13) × 10−4 to (8.76 ± 0.34) × 10−4 cm2/sec, respectively, in OH-terminated pores and from (5.04 ± 0.12) × 10−4 to (8.22 ± 0.21) × 10−4 cm2/sec, respectively, in CH3-terminated pores. The higher diffusion coefficient of CH4 in confinement is also attributed to the lower molecular weight and the lower intermolecular interactions with the pore walls that enable even the adsorbed atoms to move faster in the xy plane compared to the adsorbed CO2 molecules that bind strongly to the pore surfaces.
In addition, the diffusion coefficients of CO2 and CH4 in OH-terminated and CH3-terminated pores vary across the pore space such that the adsorbed molecules on the pore surface diffuse slower than the molecules in the center of the pore (see Table 2). The lower diffusion coefficients in the adsorbed layers are derived by the intermolecular interactions with the pore surface that results in the domination of surface diffusion. In contrast, the absence of surface interactions and lower density profiles in the pore center enable the gas molecules to diffuse faster relative to the adsorbed layers. The confined free gas molecules diffuse faster than the adsorbed molecules on the pore surfaces by an order of magnitude in the simulated pores.
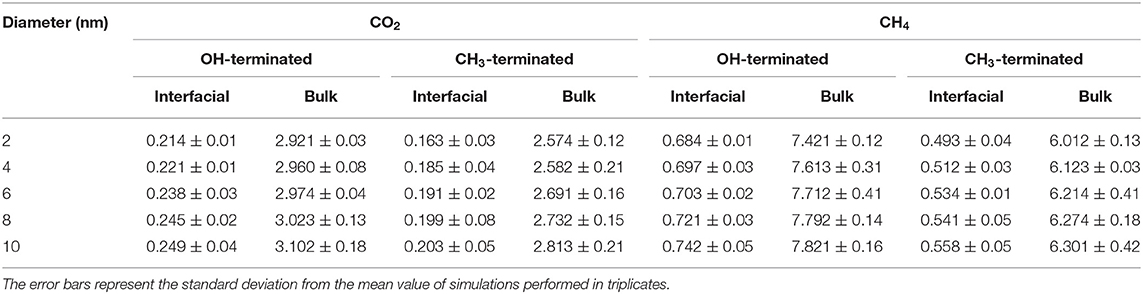
Table 2. Self-diffusion coefficients (10−4 cm−2/sec) of interfacial and confined free gas molecules (bulk) in OH-terminated and CH3-terminated pores as a function of the pore diameter.
The intermolecular interaction energies of CO2 and CH4 molecules with the silica surfaces are calculated to elucidate the energetic basis of the varying adsorption extents, organization of interfacial CO2 and CH4 molecules, and dynamics with changes in the functionalized surfaces and the pore diameter (see Figure 10). Van der Waals and electrostatic interactions are averaged over the last 10 ns of the simulations time as a function of the pore diameter. Error bars represent the standard deviation from the mean value of these three different simulations.
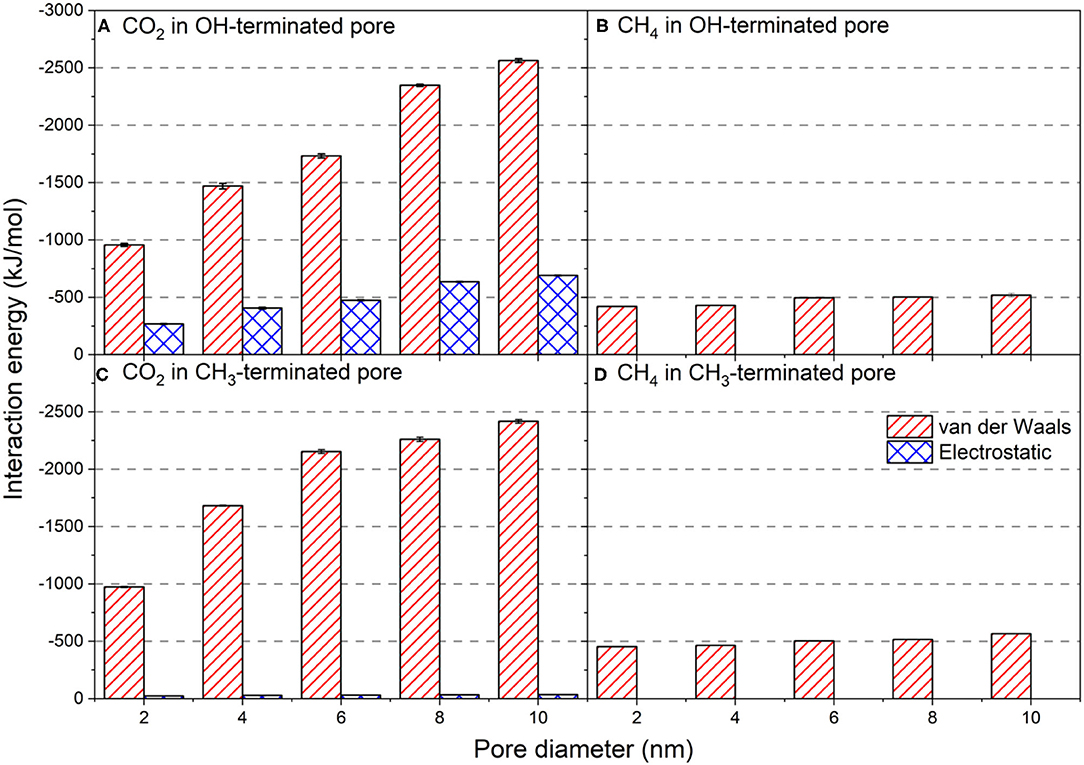
Figure 10. Van der Waals and electrostatic interaction energies of CO2 and CH4 with (A,C) OH-terminated and (B,D) CH3-terminated pores as a function of the pore diameter. The intermolecular interactions are averaged over the last 10 ns of the simulation time. Error bars represent the standard deviation from the means values of three different simulations.
Van der Waals interactions of CO2 with OH-terminated pores increase systematically with the pore diameter (Figure 10A). Van der Waals interactions in OH-terminated pores increased from −956.6 ± 13 to −2,563.5 ± 16 kJ/mol as the pore diameter increased from 2 to 10 nm. Similarly, van der Waals interactions between confined CO2 molecules and CH3-terminated pores have systematically increased from −972.6 ± 5 to −2,417.1 ± 16 kJ/mol as the pore diameter increase from 2 to 10 nm (Figure 10C). The increase in van der Waals interactions with the increase in pore diameter is attributed to the higher number of molecules in larger pores that are required to maintain the chemical potential. The larger number of molecules results in more intermolecular collisions between the interfacial CO2 molecules and functional groups (OH and CH3) on the pore surface. However, the difference in the magnitude of van der Waals interactions between the confined CO2 molecules with OH-terminated and CH3-terminated pores are not significant, suggesting that the intensity of intermolecular collisions between confined CO2 and pore surfaces is independent of the type of functional group on the pore surface.
Electrostatic interactions, on the other hand, showed a profound difference in OH-terminated pores compared to CH3-terminated pores. The electrostatic interactions between CO2 and OH-terminated pores are higher compared to those between CO2 and CH3-terminated pores by an order of magnitude. The electrostatic interactions between OH-terminated pores and the confined CO2 molecules increased from −267.5 ± 4 to −690.5 ± 4 kJ/mol as the pore diameter increased from 2 nm to 10 nm. The electrostatic interactions between CO2 molecules and CH3-terminated pores showed a slight increase from −23.0 ± 1 to −35.4 ± 1 kJ/mol as the pore diameter increased from 2 nm to 10 nm. Since the differences in the van der Waals interactions are insignificant, the profound difference in electrostatic interactions between CO2 and silica pores drives the preferential adsorption of confined CO2 on OH-terminated pores. The higher electrostatic interactions between CO2 and OH-terminated pores stems from the quadruple moment of CO2 molecules.
Further, CO2 molecules form hydrogen bonds with the OH-group on the silica surfaces that contribute positively to the overall intermolecular interactions. The hydrogen bonds between the confined CO2 molecules and OH-terminated surfaces increases from 33 ± 2 to 102 ± 4 as the pore diameter increases from 2 to 10 nm, respectively. The energy of a hydrogen bond can vary from 1 to 40 kcal/mol. The observed electrostatic interactions and hydrogen bonding between confined CO2 and OH-terminated silica surfaces are consistent with previous studies (Mohammed and Gadikota, 2019a).
The intermolecular interactions between the confined CH4 and pore walls are driven by van der Waals interactions with negligible contributions from the electrostatic interactions (see Figures 10B,D) due to the non-polar characteristics of CH4 molecules, and they slightly with the pore diameter. The intermolecular interactions of CH4 with the CH3-teminated pores are slightly lower than compared to the OH-terminated pores, which explains the higher adsorption extents of CH4 on CH3-terminated pores (see Figure 6). The higher van der Waals interactions combined with the contribution of the electrostatic interactions of CO2 with the OH-terminated and CH3-terminated pores results in a preferential adsorption of CO2 on the silica pores with different surface chemistries over non-polar hydrocarbons. This data suggests that the hydrophilic surfaces aid the preferential adsorption of CO2 and the displacement of hydrocarbons, including CH4, away from the silica surfaces.
The structure, dynamics and energetics of confined CO2 and CH4 in OH-terminated and CH3-terminated silica pores with diameters ranging from 2 nm to 10 nm are investigated using classical molecular dynamics simulations. In this study, anisotropic distribution of CO2 and CH4 molecules in confinement is noted with higher number densities observed adjacent to the pore surfaces. Preferential adsorption of CO2 on the surfaces of OH-terminated and CH3-terminated pores over CH4 is attributed to favorable intermolecular interactions with the hydroxyl functional groups. CO2 and CH4 molecules adsorb closer to the surfaces of OH-terminated pores than CH3-terminated pores. Higher adsorption extents of CO2 are noted compared to CH4, and the adsorption extent increases with the pore diameter. The diffusion coefficients of confined CO2 and CH4 showed higher values in larger pores, and confined CH4 diffuses faster than CO2 in all pore sizes. The higher adsorption extent of CO2 over CH4 on the surfaces of OH-terminated and CH3-terminated pores stems from significantly stronger van der Waals interactions with considerable contributions from electrostatic interactions. Thus, delineating the organization and transport behavior of CO2 and CH4 molecules in confinement in hydrophobic and hydrophilic pores provides the basis for tuning surface chemical interactions for storing gases in engineered materials and for predicting the fate of multicomponent fluids in porous environments in subsurface geologic formations.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
SM constructed the simulations and completed the first draft of the manuscript. AS and CW contributed to the simulations and data analyses. GG developed the concept and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported as part of the Multi-Scale Fluid-Solid Interactions in Architected and Natural Materials (MUSE), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award # DE-SC0019285.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors gratefully acknowledge the support from Akanksha Srivastava at Cornell University for assisting the effort.
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. doi: 10.1016/j.softx.2015.06.001
Aydin, G., Karakurt, I., and Aydiner, K. (2010). Evaluation of geologic storage options of CO2: applicability, cost, storage capacity, and safety. Energy Policy 38, 5072–5080. doi: 10.1016/j.enpol.2010.04.035
Bachu, S., Bonijoly, D., Bradshaw, J., Burruss, R., Holloway, S., Christensen, N. P., et al. (2007). CO2 storage capacity estimation: methodology and gaps. Int. J. Greenh. Gas Control 1, 430–443. doi: 10.1016/S1750-5836(07)00086-2
Baumgartner, B., Hayden, J., Loizillon, J., Steinbacher, S., Grosso, D., and Lendl, B. (2019). Pore size-dependent structure of confined water in mesoporous silica films from water adsorption/desorption using ATR–FTIR spectroscopy. Langmuir 35, 11986–11994. doi: 10.1021/acs.langmuir.9b01435
Berta, M., Dethlefsen, F., Ebert, M., SchäFer, D., and Dahmke, A. (2018). Geochemical effects of millimolar hydrogen concentrations in groundwater: an experimental study in the context of subsurface hydrogen storage. Environ. Sci. Technol. 52, 4937–4949. doi: 10.1021/acs.est.7b05467
Chaikittisilp, W., Khunsupat, R., Chen, T. T., and Jones, C. W. (2011). Poly (allylamine)–mesoporous silica composite materials for CO2 capture from simulated flue gas or ambient air. Ind. Eng. Chem. Res. 50, 14203–14210. doi: 10.1021/ie201584t
Chiang, W. S., Fratini, E., Baglioni, P., Chen, J. H., and Liu, Y. (2016a). Pore size effect on methane adsorption in mesoporous silica materials studied by small-angle neutron scattering. Langmuir 32, 8849–8857. doi: 10.1021/acs.langmuir.6b02291
Chiang, W. S., Fratini, E., Baglioni, P., Georgi, D., Chen, J. H., and Liu, Y. (2016b). Methane adsorption in model mesoporous material, SBA-15, studied by small-angle neutron scattering. J. Phys. Chem. C 120, 4354–4363. doi: 10.1021/acs.jpcc.5b10688
Cogswell, C. F., Jiang, H., Ramberger, J., Accetta, D., Willey, R. J., and Choi, S. (2015). Effect of pore structure on CO2 adsorption characteristics of aminopolymer impregnated MCM-36. Langmuir 31, 4534–4541. doi: 10.1021/la505037f
Cygan, R. T., Liang, J. J., and Kalinichev, A. G. (2004). Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 108, 1255–1266. doi: 10.1021/jp0363287
Dündar-Tekkaya, E., and Yürüm, Y. (2016). Mesoporous MCM-41 material for hydrogen storage: a short review. Int. J. Hydrogen Energ. 41, 9789–9795. doi: 10.1016/j.ijhydene.2016.03.050
Düren, T., Sarkisov, L., Yaghi, O. M., and Snurr, R. Q. (2004). Design of new materials for methane storage. Langmuir 20, 2683–2689. doi: 10.1021/la0355500
Elola, M. D., and Rodriguez, J. (2019). Preferential adsorption in ethane/carbon dioxide fluid mixtures confined within silica nanopores. J. Phys. Chem. C 123, 30937–30948. doi: 10.1021/acs.jpcc.9b07969
Evans, D. J., and Holian, B. L. (1985). The nose–hoover thermostat. J. Chem. Phys. 83, 4069–4074. doi: 10.1063/1.449071
Farajzadeh, R., Eftekhari, A. A., Dafnomilis, G., Lake, L. W., and Bruining, J. (2020). On the sustainability of CO2 storage through CO2-enhanced oil recovery. Appl. Energy 261:114467. doi: 10.1016/j.apenergy.2019.114467
Gadikota, G., Dazas, B., Rother, G., Cheshire, M. C., and Bourg, I. C. (2017). Hydrophobic solvation of gases (CO2, CH4, H2, noble gases) in clay interlayer nanopores. J. Phys. Chem. C 121, 26539–26550. doi: 10.1021/acs.jpcc.7b09768
Ghoufi, A., Hureau, I., Morineau, D., Renou, R., and Szymczyk, A. (2013). Confinement of tert-butanol nanoclusters in hydrophilic and hydrophobic silica nanopores. J. Phys. Chem. C 117, 15203–15212. doi: 10.1021/jp404702j
Giannozzi, P., Andreussi, O., Brumme, T., Bunau, O., Nardelli, M. B., Calandra, M., et al. (2017). Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Mat. 29:465901. doi: 10.1088/1361-648X/aa8f79
Han, Y., Liu, J., Huang, L., He, X., and Li, J. (2019). Predicting the phase diagram of solid carbon dioxide at high pressure from first principles. Npj Quantum Mater. 4, 1–7. doi: 10.1038/s41535-019-0149-0
Head, J. D., and Zerner, M. C. (1985). A broyden—fletcher—goldfarb—shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 122, 264–270. doi: 10.1016/0009-2614(85)80574-1
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Huo, P., Zhang, D., Yang, Z., Li, W., Zhang, J., and Jia, S. (2017). CO2 geological sequestration: displacement behavior of shale gas methane by carbon dioxide injection. Int. J. Greenh. Gas Control 66, 48–59. doi: 10.1016/j.ijggc.2017.09.001
Jiang, X. (2011). A review of physical modelling and numerical simulation of long-term geological storage of CO2. Appl. Energy 88, 3557–3566. doi: 10.1016/j.apenergy.2011.05.004
Jin, J., Asai, P., Wang, X., Miller, J. D., and Deo, M. (2021). Simulation and analysis of slip flow of water at hydrophobic silica surfaces of nanometer slit pores. Colloid. Surf. A 626:127032. doi: 10.1016/j.colsurfa.2021.127032
Jin, J., Wang, X., Wick, C. D., Dang, L. X., and Miller, J. D. (2019). Silica surface states and their wetting characteristics. Surf. Innov. 8, 145–157. doi: 10.1680/jsuin.19.00053
Jorgensen, W. L., Maxwell, D. S., and Tirado-Rives, J. (1996). Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236. doi: 10.1021/ja9621760
Klewiah, I., Berawala, D. S., Walker, H. C. A., Andersen, P. Ø., and Nadeau, P. H. (2020). Review of experimental sorption studies of CO2 and CH4 in shales. J. Nat. Gas Sci. Eng. 73:103045. doi: 10.1016/j.jngse.2019.103045
Knight, A. W., Kalugin, N. G., Coker, E., and Ilgen, A. G. (2019). Water properties under nano-scale confinement. Sci. Rep. 9, 1–12. doi: 10.1038/s41598-019-44651-z
Kuuskraa, V. A., Godec, M. L., and Dipietro, P. (2013). CO2 utilization from “next generation” CO2 enhanced oil recovery technology. Enrgy Procedia 37, 6854–6866. doi: 10.1016/j.egypro.2013.06.618
Le, T., Ogbe, S., Striolo, A., and Cole, D. R. (2016). N-octane diffusivity enhancement via carbon dioxide in silica slit-shaped nanopores–a molecular dynamics simulation. Mol. Simul. 42, 745–752. doi: 10.1080/08927022.2015.1089991
Le, T., Striolo, A., and Cole, D. R. (2015). CO2-C4H10 mixtures simulated in silica slit pores: relation between structure and dynamics. J. Phys. Chem. C 119, 15274–15284. doi: 10.1021/acs.jpcc.5b03160
Li, K. M., Jiang, J. G., Tian, S. C., Chen, X. J., and Yan, F. (2014). Influence of silica types on synthesis and performance of amine–silica hybrid materials used for CO2 capture. J. Phys. Chem. C 118, 2454–2462. doi: 10.1021/jp408354r
Liu, F. Q., Wang, L., Huang, Z. G., Li, C. Q., Li, W., Li, R. X., et al. (2014). Amine-tethered adsorbents based on three-dimensional macroporous silica for CO2 capture from simulated flue gas and air. ACS Appl. Mater. Interf. 6, 4371–4381. doi: 10.1021/am500089g
Mafra, L., Cendak, T., Schneider, S., Wiper, P. V., Pires, J., Gomes, J. R., et al. (2018). Amine functionalized porous silica for CO2/CH4 separation by adsorption: which amine and why. Chem. Eng. J. 336, 612–621. doi: 10.1016/j.cej.2017.12.061
Michael, K., Golab, A., Shulakova, V., Ennis-King, J., Allinson, G., Sharma, S., et al. (2010). Geological storage of CO2 in saline aquifers—a review of the experience from existing storage operations. Int. J. Greenh. Gas Control 4, 659–667. doi: 10.1016/j.ijggc.2009.12.011
Mohammed, S., Asgar, H., Kuzmenko, I., and Gadikota, G. (2020a). self-assembly of silica nanoparticles at water-hydrocarbon interfaces: insights from in-operando small-angle x-ray scattering measurements and molecular dynamics simulations. Energy Fuels 34, 12545–12555. doi: 10.1021/acs.energyfuels.0c02759
Mohammed, S., and Gadikota, G. (2018). The effect of hydration on the structure and transport properties of confined carbon dioxide and methane in calcite Nanopores. Front. Energy Res. 6:86. doi: 10.3389/fenrg.2018.00086
Mohammed, S., and Gadikota, G. (2019a). CO2-Induced displacement and diffusive transport of shale geofluids in silica nanopores of varying sizes. J. CO2 Utilizat. 32, 37–45. doi: 10.1016/j.jcou.2019.03.023
Mohammed, S., and Gadikota, G. (2019b). The role of calcite and silica interfaces on the aggregation and transport of asphaltenes in confinement. J. Mol. Liq. 274, 792–800. doi: 10.1016/j.molliq.2018.10.163
Mohammed, S., and Gadikota, G. (2020). Exploring the role of inorganic and organic interfaces on CO2 and CH4 partitioning: case study of silica, illite, calcite, and kerogen nanopores on gas adsorption and nanoscale transport behaviors. Energy Fuels 34, 3578–3590. doi: 10.1021/acs.energyfuels.0c00052
Mohammed, S., Liu, M., and Gadikota, G. (2021). Resolving the organization of CO2 molecules confined in silica nanopores using in situ small-angle neutron scattering and molecular dynamics simulations. Environ. Sci. Nano. 8, 2006–2018. doi: 10.1039/D0EN01282C
Mohammed, S., Liu, M., Liu, Y., and Gadikota, G. (2020b). Probing the core-shell organization of nano-confined methane in cylindrical silica pores using in-situ small-angle neutron scattering and molecular dynamics simulations. Energy Fuels 34, 15246–15256. doi: 10.1021/acs.energyfuels.0c02237
Neupane, H. K., and Adhikari, N. P. (2020). Structure, electronic and magnetic properties of 2D Graphene-Molybdenum diSulphide (G-MoS2) heterostructure (HS) with vacancy defects at Mo sites. Comput. Condens. Mater 24:e00489. doi: 10.1016/j.cocom.2020.e00489
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77:3865. doi: 10.1103/PhysRevLett.77.3865
Pfeiffer, W. T., and Bauer, S. (2015). Subsurface porous media hydrogen storage–scenario development and simulation. Energy Procedia 76, 565–572. doi: 10.1016/j.egypro.2015.07.872
Phan, A., Cole, D. R., and Striolo, A. (2014). Aqueous methane in slit-shaped silica nanopores: high solubility and traces of hydrates. J. Phys. Chem. C 118, 4860–4868. doi: 10.1021/jp500081t
Potoff, J. J., and Siepmann, J. I. (2001). Vapor–liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 47, 1676–1682. doi: 10.1002/aic.690470719
Qin, Y., Yang, X., Zhu, Y., and Ping, J. (2008). Molecular dynamics simulation of interaction between supercritical CO2 fluid and modified silica surfaces. J. Phys. Chem. C 112, 12815–12824. doi: 10.1021/jp711964e
Serna-Guerrero, R., Da'na, E., and Sayari, A. (2008). New insights into the interactions of CO2 with amine-functionalized silica. Ind. Eng. Chem. Res. 47, 9406–9412. doi: 10.1021/ie801186g
Sharma, V., Kagdada, H. L., Jha, P. K., Spiewak, P., and Kurzydłowski, K. J. (2019). Halogenation of SiGe monolayers: robust changes in electronic and thermal transport. Phys. Chem. Chem. Phys. 21, 19488–19498. doi: 10.1039/C9CP03822A
Shi, Z., Jessen, K., and Tsotsis, T. T. (2020). Impacts of the subsurface storage of natural gas and hydrogen mixtures. Int. J. Hydrogen Energy 45, 8757–8773. doi: 10.1016/j.ijhydene.2020.01.044
Siderius, D. W., Krekelberg, W. P., Chiang, W. S., Shen, V. K., and Liu, Y. (2017). Quasi-two-dimensional phase transition of methane adsorbed in cylindrical silica mesopores. Langmuir 33, 14252–14262. doi: 10.1021/acs.langmuir.7b03406
Wang, D., Wang, L., Zhang, L., Cai, C., Li, N., and Yang, M. (2020). A modified model for estimating excess adsorption of methane in moist nanoporous silica. Chem. Phys. 533:110740. doi: 10.1016/j.chemphys.2020.110740
Wiheeb, A. D., Kim, J., and Othman, M. R. (2015). Highly perm-selective micro-porous hydrotalcite-silica membrane for improved carbon dioxide-methane separation. Sep. Sci. Technol. 50, 1701–1708. doi: 10.1080/01496395.2014.987300
Wu, H., Chen, J., and Liu, H. (2015). Molecular dynamics simulations about adsorption and displacement of methane in carbon nanochannels. J. Phys. Chem. C 119, 13652–13657. doi: 10.1021/acs.jpcc.5b02436
Xiong, J., Liu, K., Liu, X., Liang, L., and Zeng, Q. (2016). Molecular simulation of methane adsorption in slit-like quartz pores. RSC Adv. 6, 110808–110819. doi: 10.1039/C6RA22803H
Xiong, J., Liu, X., Liang, L., and Zeng, Q. (2017). Adsorption of methane in organic-rich shale nanopores: an experimental and molecular simulation study. Fuel 200, 299–315. doi: 10.1016/j.fuel.2017.03.083
Yun, J. H., Düren, T., Keil, F. J., and Seaton, N. A. (2002). Adsorption of methane, ethane, and their binary mixtures on MCM-41: experimental evaluation of methods for the prediction of adsorption equilibrium. Langmuir 18, 2693–2701. doi: 10.1021/la0155855
Keywords: surface hydrophobicity, CO2, CH4, gas storage, structure, organization, dynamics, diffusivity
Citation: Mohammed S, Sunkara AK, Walike CE and Gadikota G (2021) The Role of Surface Hydrophobicity on the Structure and Dynamics of CO2 and CH4 Confined in Silica Nanopores. Front. Clim. 3:713708. doi: 10.3389/fclim.2021.713708
Received: 23 May 2021; Accepted: 05 July 2021;
Published: 02 August 2021.
Edited by:
Sarada Prasad Pradhan, Indian Institute of Technology Roorkee, IndiaReviewed by:
Tuli Bakshi, Indian Institute of Technology Bombay, IndiaCopyright © 2021 Mohammed, Sunkara, Walike and Gadikota. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Greeshma Gadikota, Z2c0NjRAY29ybmVsbC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.