
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Child Adolesc. Psychiatry, 07 December 2022
Sec. Autism and Other Neurodevelopmental Disorders
Volume 1 - 2022 | https://doi.org/10.3389/frcha.2022.987339
This article is part of the Research TopicAdvancements and Challenges in Autism and Other Neurodevelopmental DisordersView all 8 articles
 Dandan Wu
Dandan Wu Rong Li*
Rong Li*Purpose: To explore the genetic cause of children with unidentified etiology of neurodevelopmental disorders, thus providing references for the diagnosis, treatment and genetic counseling.
Design and methods: Children with neurodevelopmental disorders but unidentified etiology in the Child Healthcare Department, Children's Hospital of Nanjing Medical University from November 2018 to December 2021 were retrospectively analyzed. A total of 2 ml of peripheral venous blood was collected from the child and their parents for the whole exome sequencing (WES) and copy number variation (CNV) detection. Male children were subjected to fragile X syndrome testing to determine the genetic mutations. For those with positive results, Sanger sequencing was performed to explore the mutations in the gene sites and pedigrees.
Results: A total of 488 (33.5%) pathogenic variations were detected among 1,457 global developmental/intellectual disabilities (GDD/ID) children, including 362 (24.9%) cases of monogenic mutations, and 111 (7.6%) cases of chromosomal microdeletions or microduplications. There were 15/780 (1.92%) male children with fragile X syndrome. Single point mutations were detected in 277/362 (76.5%) and 85/362 (23.5%) male and female GDD/ID children, respectively, including 295 (81.5%) cases of missense mutations, 32 (8.8%) cases of frameshift mutations, 5 (2.2%) cases of non-sense mutations and 30 (8.3%) cases of splice site mutations. In addition, there were 166 (45.8%) cases of autosomal inheritance and 196 (54.2%) cases of X-linked inheritance. The X chromosome abnormalities were mostly observed in 362 GDD/ID children with monogenic mutations, including 15 cases of the AFF2 gene mutation, 13 cases of the MECP2 gene mutation and 12 cases of the HUWEI gene mutation. The CREBBP gene mutation was the most common autosome abnormality in GDD/ID children with monogenic mutations, which was detected in five cases. There were 74 cases of chromosomal microdeletions, 31 cases of chromosomal microduplications and six cases of both. A total of 114 novel pathogenic mutations responsible for GDD/ID were found, including four novel mutations in MECP2 and TRAPPC9 genes.
Conclusion: The incidence of genetic abnormalities remains high in NDD children. Abundant novel mutations are responsible for GDD/ID in children, and can be used as references in the diagnosis of neurogenetic diseases.
Neurodevelopmental disorders (NDD), an array of multifactorial chronic diseases, seriously impair children's physical and psychological health, even cross their lifespan. NDD include at least global developmental/intellectual disabilities (GDD/ID), autism spectrum disorders (ASD), both as main contributors to pediatric disability worldwide. GDD/ID exhibit highly heterogeneous clinical and genetic features, often complicated with ASD and attention deficit and hyperactivity disorder (ADHD) [1]. ID has a global prevalence of 1%, and serious ID of 0.6%. ASD was first reported in 1943, with a global incidence ever-growing to 1% in 2012 according to the WHO data [2–4].
NDD are caused by environmental and genetic factors. In an era with updating livelihood and enriching medical resources, exogenous factors, like infection, poisoning, trauma, hypoxia, malnutrition, illiteracy, psychological impairment, have been well-uncovered and controlled. In contrast, genetic factors remain enigmatic by large. About 2/3 of ID are caused by genetic factors, including chromosome abnormality, single or multiple-gene mutation, or inborn errors of metabolism. With plural manifestations, ID meets with no effective treatments, leading to a high rate of disability. Special education or rehabilitation training may alleviate ID symptoms, but their application across the lifespan is restricted by a huge cost [5–8].
Genetic diagnosis and counseling can benefit the treatment of NDD children, even prevent secondary adverse pregnancy outcomes. In the present study, a total of 1,457 GDD/ID and ASD children with unidentified etiology were collected and analyzed with the whole exome sequencing (WES). For those with positive results, Sanger sequencing was performed to explore the mutations in the gene sites and pedigrees. Male children were subjected to fragile X syndrome testing to determine the genetic mutations. Our findings are expected to provide references for the diagnosis and treatment of children with GDD/ID or ASD.
We recruited 1,457 children treated for GDD/ID and ASD of unclear etiology from November 2018 to December 2021 at the Department of Children Health Care, Children's Hospital of Nanjing Medical University (Nanjing, China), including 1,152 males (79.1%) and 305 females (20.9%). Inclusion criteria were as follows: (1) Assessment of GDD/ID. Gesell Developmental Scales (Gesell) [9], Griffiths Development Scales-Chinese Edition (GDS-C) [10], Wechsler Preschool and Primary Scale of Intelligence (WPPSI-IV) [11], Wechsler Intelligence Scale for Children (WISC-IV) [12], and Infants-Junior Middle School Student's Social-Life Abilities Scale (S-M scale) [13] were adopted to assess GDD/ID in children. Children with two or more domains of Gesell and GDS-C that were significantly lower than those in children of the same age (>2 standard deviations) were assessed as GDD. WPPSI-IV and WISC-IV were used to assess GDD/ID in children with 2.5–6 and 6–16 years, respectively. According to the intelligence quotient (IQ) range and social adaptability, ID was classified into mild (IQ: 50–69), moderate (IQ: 35–49), severe (IQ: 20–34), and profound (IQ <20). Corresponding developmental assessment scales were used according to the developmental level and age of children (Table 1). (2) Assessment of ASD. The Autism Behavior Checklist (ABC) [14] and the Childhood Autism Rating Scale (CARS) [15] were adopted to assess autism symptoms in children. ASD was diagnosed in children with the total ABC score ≥67 points and CARS score ≥30 points.

Table 1. Corresponding developmental assessment scales for GDD/ID children at different ages.
Exclusion criteria: (1) children with specific genetic diseases and genetic metabolic diseases diagnosed by clinical symptoms and signs, and laboratory testing; (2) children with GDD/ID and ASD caused by perinatal ischemia, hypoxic brain injury, bilirubin encephalopathy, and central nervous system infection, poisoning and trauma.
After obtaining the written informed consent of the guardians, 2 ml of peripheral venous blood was collected from each child and his/her parents for WES and CNV detection. Male children were subjected to fragile X syndrome testing to determine the genetic mutations. For those with positive results, Sanger sequencing was performed to explore the mutations in the gene sites and pedigrees.
DNA fragments of the target region were enriched by microarray hybridization, then subjected to the next-generation high-throughput sequencing. In the present study, the exon regions of 23,000 genes were captured using the Gen Cap custom exome enrichment kit (My Genostics, Beijing, China). Briefly, genomic DNA fragments were ligated to the Illumina-supplied adapter sequences, then subjected to ligation-mediated polymerase chain reaction (LM-PCR). After amplification and purification, the quality of DNA library was checked. PCR products were hybridized with the target capture microarray, and the captured sequence was analyzed using the Illumina Nova 6000 sequencer (Illumina, San Diego, CA, USA). Raw data were processed for image recognition and differentiation.
After trimming adapter sequences from short read data, the Illumina sequencing reads were mapped to the human reference genome (GRCh37/hg19, 2009 Assembly) using the BWA-MEM alignment algorithm of Burrows-Wheeler Aligner (BWA, http://bio-bwa.sourceforge.net) [16, 17]. Single nucleotide variation (SNV) and inserts and deletions (INDEL) were analyzed using the Genome Analysis Toolkit (GATK, http://software.broadinstitute.org) [18], and annotated by ANNOVAR (http://annovar.openbioinformatics.org/en/latest/) [19]. Mutation sites with a frequency of <0.05 were screened out of the following databases, including the 1,000 Genomes Project (http://www.1000genomes.org), Exome Variant Server (http://evs.gs.washington.edu) and EXAC (http://exac.broadinstitute.org/). Pathogenicity and conservative prediction of missense mutations were carried out using SIFT (http://sift.jcvi.org/) [20–22], PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) [23–25], MutationTaster (http://www.mutationtaster.org/) [26], and GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html) [27]. Changes in splice sites were analyzed for pathogenicity using SPIDEX (http://www.openbioinformatics.org/annovar/spidex_download_form.php).
Mutations of interest were subjected to PCR and Sanger sequencing. PCR primers were synthesized using Primer 3.0 (http://primer3.ut.ee/) [28–30]. In addition, PCR products were subjected to Sanger sequencing and analyzed using the ABI 3130 Genetic Analyzer (Applied Biosystems), followed by the family cosegregation analysis.
DNA extracted from peripheral blood was analyzed by PCR, and the PCR fragment was designed to flank the area of repeats and effectively adjust the size of the PCR product correspondingly to the repeat size. After allele-specific methylation PCR, capillary electrophoresis was performed to measure the number of CTG repeats.
Data were expressed as medians and interquartile ranges (IQRs), and processed using SPSS 22.0. P < 0.05 was considered as statistically significant.
There were 1,253/1,457 (85.9%) children with GDD/ID and 204/1,457 (14.1%) with GDD/ID and autism. A total of 488 (33.5%) pathogenic variations were detected among 1,457 GDD/ID children, including 362 (24.9%) monogenic mutations, and 111 (7.6%) chromosomal microdeletions or microduplications. Fifteen male children (1.92%, 15/780) aged 2–13 years presented with fragile X syndrome, and four of their mothers were determined as premutation carriers, while FMR1 premutation was not detected in their fathers. Notably, full mutation in FMR1 gene was detected in 9 male children of 4 pedigrees (Table 2).
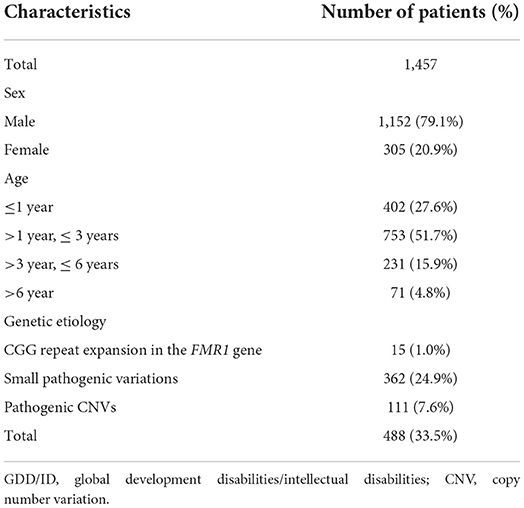
Table 2. Baseline characteristics of GDD/ID children.
Most of the X chromosome abnormalities were observed in 362 GDD/ID children with monogenic mutations, including 15 with AFF2 gene mutation, 13 with MECP2 gene mutation and 12 with HUWEI gene mutation. The CREBBP gene mutation, detected in five cases, was the most common autosome abnormality in GDD/ID children with monogenic mutations (Table 3).
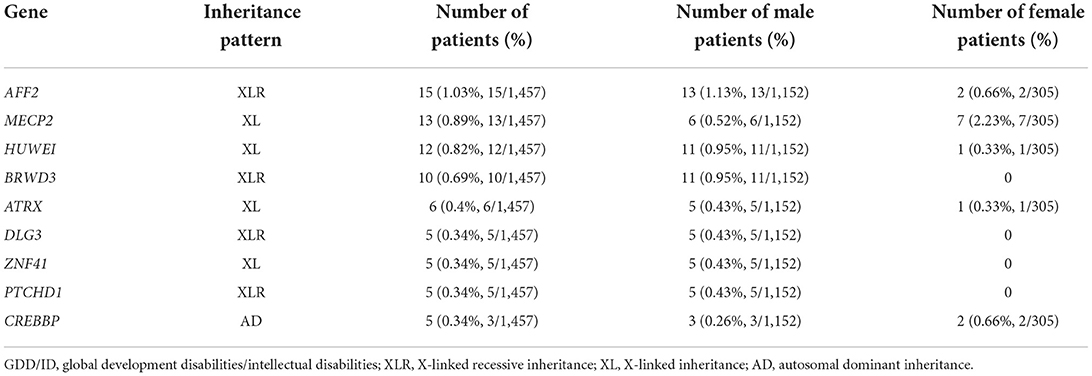
Table 3. Pathogenic variations in GDD/ID children.
Monogenic mutations were detected in 362 GDD/ID children, involving 277 males and 85 females. Among them, 295 carried missense mutations, 32 carried frameshift mutations, five carried non-sense mutations, and 30 carried splice site mutations (Table 4). In particular, non-sense mutations were inherited in 3 GDD/ID children with an autosomal recessive pattern, involving 1 combined with splice site mutation and 2 with frameshift mutations.
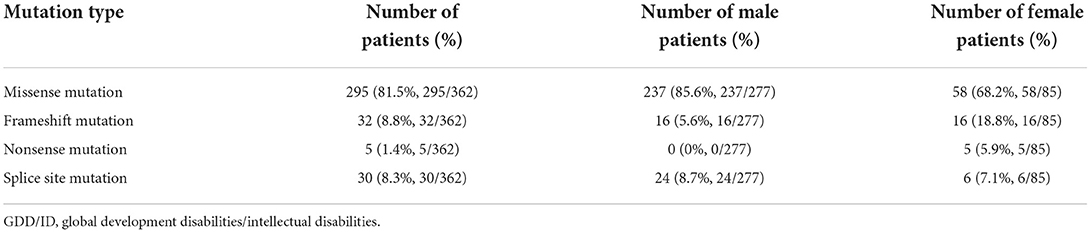
Table 4. Single site mutations in GDD/ID children.
Newly discovered single point mutations were abundant in 362 cases of GDD/ID children, all never reported before. The mutations were considered pathogenic (P) based on protein damage due to mutation-induced premature termination codon and clinical manifestations of affected children. Moreover, their pathogenicity (p) was considered as positive results from more than four online tools in predicting the pathogenicity of rare missense variants (e.g., SIFT, PolyPhen_2MutationTaster, GERP++, and REVEL). At last, 114 novel pathogenic mutations responsible for GDD/ID were found (Table 5).
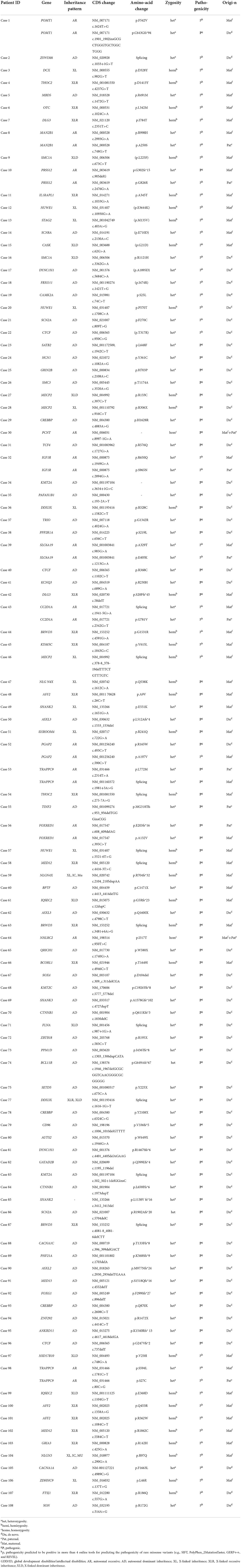
Table 5. Novel single point mutations in GDD/ID children.
Among the 362 GDD/ID children with single point mutations, 166 (45.8%) presented with autosomal inheritance, and 196 (54.2%) presented with X-linked inheritance (Table 6).

Table 6. Inheritance patterns of the single site mutations in GD/IDD children.
We identified 74 children with chromosomal microdeletions, 31 with chromosomal microduplications and 6 with both. Among the 111 (7.6%) cases of chromosomal microdeletions or microduplications, we detected 16 cases of Williams syndrome (WS), four cases of Phelan-McDermid syndrome (22q13 deletion syndrome), four cases of Prader-Willi syndrome or Angelman syndrome with deletions or duplications at the 15q11.2q13.1 locus (Table 7).
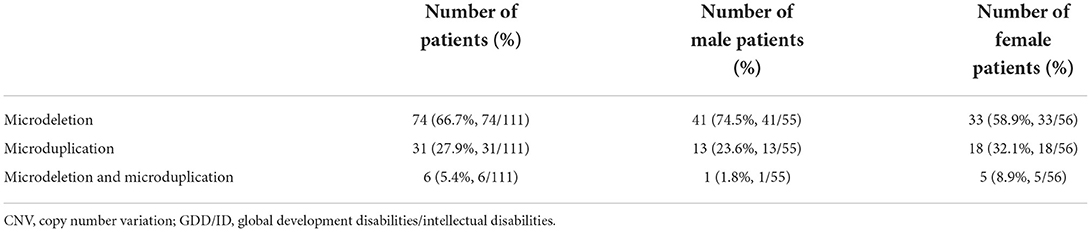
Table 7. Pathogenic CNVs in GD/IDD children.
Clinical manifestations of NDD in children are diverse, the disability rate of which remains high. However, effective treatment for NDD lacks. Genetic factors are predominantly responsible for NDD, including chromosomal abnormalities, single or multiple gene mutations and congenital metabolic defects. In the present study, 362 (24.9%) pathogenic mutations were found in 1,457 GDD/ID children, dominated by missense mutation (295/362, 81.5%).
We found abundant single site mutations in GDD/ID children that have not been previously reported, and 114 of them were confirmed as pathogenic, including 51 (44.7%) of autosomal dominant, 21 (18.4%) of autosomal recessive, 8 (7.0%) of X-linked dominant, 18 (15.8%) of X-linked recessive and 16 (14.0%) of X-linked. Among them, 54 (47.4%) were inherited from parents, and 60 (52.6%) were spontaneous mutations. The latter was considered to pose a stronger destructive effect on biological functions than genetic mutations. Our findings provide valuable clinical evidence supporting the genetic causes of sporadic GDD/ID in children.
The newly discovered mutations in GDD/ID children have expanded the genetic spectrum of this disease. Notably, four novel mutations were found in MECP2 and TRAPPC9 genes, respectively. A total of seven female GDD/ID children carrying MECP2 gene presented obvious intellectual disability and stereotyped behavior. MECP2 gene is also the second most common gene for sex-linked genetic diseases. With the discovery of novel mutation sites, the incidence of neurodevelopmental disorders caused by MECP2 gene is estimated to increase [31]. Novel mutations in the TRAPPC9 gene were detected in 3 GDD/ID children. Notably, a GDD/ID child carried two novel mutations in the TRAPPC9 gene with the clinical manifestations of severe intellectual disability and autism. NIBP/TRAPPC9 mutations are associated with non-syndromic autosomal recessive intellectual disability (NS-ARID), which is known as NIBP syndrome, and assigned as the intellectual disability-obesity-brain malformations facial dysmorphism syndrome in the human disease database Mala Cards [32].
All included GDD/ID children received WES and CNV detection. Chromosomal abnormalities and genomic microdeletions and microduplications act as critical genetic causes for NDD in children. In the present study, a total of 111 (7.6%) children with pathogenic CNVs were detected, involving 74 (66.7%) with microdeletions, 16 (27.9%) with microduplications and 6 (5.4%) with both. The incidence of microdeletions was much higher than that of microduplications in GDD/ID children. Theoretically, the incidence of genomic microdeletions is close to that of microduplications. In the early stage of gamete formation, however, neither duplication nor deletion is prone to occur. It is reported that the phenotype of microduplication is often milder (no clinical phenotype occurs) than that of microdeletion. Even at the chromosomal level, triploids are more likely to survive than haploids [33–35]. WS, a 7q11.23 duplication syndrome, was the most common disorder in GDD/ID children. Our findings differed from the previous studies of microdeletion or microduplication, which may be linked with the bigger data in our institution.
Sex-linked genetic diseases are characterized by an obvious between-sex difference. Healthy females can carry pathogenic genes of X-linked dominant and recessive disorders, bringing with a high risk of genetic diseases inherited through the pedigree. Among 1,457 GDD/ID children, we detected 196 (54.1%), 51 (14.1%), and 145 (40.1%) children X-linked inheritance, X-linked dominant inheritance, and X-linked recessive inheritance, respectively. The incidence of X-linked inheritance was similar to that of autosomal inheritance. However, X-linked recessive inheritance is more recessive and harmful. Chromosome positioning of 473 GDD/ID children carrying definite pathogenic genes showed that the X chromosome was the most common position of gene abnormalities. Consistently, the top 3 pathogenic genes (the AFF2, MECP2, and HUWEI gene) in GDD/ID children were also located in the X chromosome.
AFF2 gene (OMIM 300806) maps to Xq28 and is highly expressed in brain regions involved in learning, cognition, and memory, including the hippocampus and amygdala. Murine AFF2 has an 88% amino acid similarity with human AFF2. In situ hybridization of adult mouse brains showed labeling in the cingulate gyrus, hippocampus, piriform cortex, and Purkinge layers [36, 37].
The expansion of CCG trinucleotide in the AFF2 gene causes a form of X-linked intellectual disability (ID) related to Fragile site E (FRAXE) at Xq28. The clinical presentations of FRAXE fragility vary a lot, including mild intellectual disability, learning disabilities, communication deficits, attention deficit hyperactivity disorder (ADHD), and autistic spectrum. Translocations disrupting AFF2, partial or entire deletions and partial duplication have been described in ID patients. Partial AFF2 deletions, especially smaller deletions, are usually associated with a milder phenotype or autism, whereas complete loss of gene function causes FRAXE. A partial AFF2 duplication has been reported associated with mild ID [38–40].
In addition, Fragile X syndrome is inherited with an X-linked incomplete dominance, manifested as hereditary intellectual disability and autism. Mainly caused by the dynamic (CGG)n mutation, it develops with the silence or severe downregulation of Fragile X intellectual disability protein (FMRP). Fragile X syndrome is considered as the most common cause of intellectual disability in men in Europe and North America, which is observed in 10–20%. The prevalence of a full mutation of FMR1 in the general population and females is 1/4,000–1/5,000 and 1/4,000–1/8,000, respectively. A large-scale screening of Fragile X syndrome in 51,000 newborns of mainland China in 2021 has shown that the frequencies of CGG repeats >100 in males and females are 1/9,371 and 1/5,887, respectively. In addition, the premutation rate of Fragile X syndrome in the Chinese population is lower than that in Caucasians [41]. In the present study, 15/780 (1.9%) of male GDD/ID children were diagnosed as Fragile X syndrome. Notably, the full mutation in the FMR1 gene was detected in nine male children in four pedigrees.
Neurodevelopmental and psychiatric disorders are a highly heterogeneous group of developmental and mental disorders, resulting from the complex interaction between genetic and environmental factors and leading to various disabilities. The diagnosis of neurodevelopmental disorders is challenging, due to its multifactor, comorbidity and polygenicity. Its genetic mechanism has been firmly established, but the its causative variants remain to be discovered. At present, new tools, such as WES, have unearthed an increasing number of genetic variants responsible for human diseases.
In the present study, we performed the genetic analysis of neurodevelopmental disorders in 1,457 children from Jiangsu and Anhui Provinces, China, and provided etiological diagnosis and genetic consultation to 488 (33.5%) cases with identified pathogenic mutations. Our study provides references for prenatal diagnosis, thus preventing adverse reproductive outcomes. In addition, we found 114 novel pathogenic mutations responsible for GDD/ID, which expanded the genetic spectrum of GDD/ID. The TRAPPC9 gene was not familiar with neurodevelopmental disorders, but showed 4 mutations in GDD/ID children. More neurodevelopment-associated loci in the TRAPPC9 gene may be discovered in the future. In addition, the incidence of Fragile X syndrome in 488 children with identified pathogenic mutations was lower than those of AFF2 (3.6%) and HUWEI (3.0%) in male GDD/ID children. Our findings are different from the previous that Fragile X syndrome is the second leading cause of male ID, which may be attributed to the genetic variations in different races. AFF2 gene is closely linked with FRAXE, which was also the top pathogenic gene in the present study. Here, a total of 30 (2.1%) GDD/ID children presented both AFF2 gene mutation and Fragile X syndrome. Sex-linked genetic diseases are the main cause of neurodevelopmental disorders in children, especially in males. Newborn screening of AFF2 gene and Fragile X syndrome is recommended for early diagnosis and intervention of potential genetic diseases, especially for those high-risk families.
The genetic background of one disease varies much across China with a large population and 56 ethnicities. Rich dada about various genetic diseases have accumulated in medical institutions around China. However, a large-scale or state-level biological sample database has not been built up. Current research on genetic diseases is usually limited in Jiangsu and Anhui Provinces, China. Multi-center cohort studies of genetic diseases should be conducted in the future to further improve the diagnosis and treatment of neurogenetic diseases.
Some limitations in this study should be noted. First, all GDD/ID children were from Nanjing and surrounding regions. Second, they were all Han Chinese and we did not recruited subjects an ethnic minority. Our findings should be further validated in the large-scale study involving multiple ethnic minorities.
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA003395) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
The studies involving human participants were reviewed and approved by the Ethics Committee of Children's Hospital of Nanjing Medical University (202110083-1). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
RL designed the study and helped write the manuscript. DW performed the interpreted data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors gratefully acknowledge the contributions of the doctors and nurses in the Nanjing Children's Hospital and the participation of the child and his parents.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lecomte J, Mercier C. The WHO atlas on global resources for persons with intellectual disabilities: a right to health perspective. Salud Publica Mex. (2008) 50(Suppl.2):s160–6. doi: 10.1590/S0036-36342008000800009
2. Friedman A. Outdated language: use of “Mental Retardation” in medicaid HCBS waivers post-Rosa's Law. Intellect Dev Disabil. (2016) 54:342–53. doi: 10.1352/1934-9556-54.5.342
3. Maenner MJ, Blumberg SJ, Kogan MD, Christensen D, Yeargin-Allsopp M, Schieve LA. Prevalence of cerebral palsy and intellectual disability among children identified in two U.S. National Surveys, 2011-2013. Ann Epidemiol. (2016) 26:222–6. doi: 10.1016/j.annepidem.2016.01.001
4. Beadle-Brown J, Mansell J, García-Ibañez J, Magallanes T, Novell R, Poole M, et al. Intellectual disability in Europe: overview. In: European Intellectual Disability Research Network, editor, Intellectual Disability in Europe. Working Papers. (2003). p. 1–11.
5. Ma Y, Chen C, Wang Y, Wu L, He F, Zhang C, et al. Analysis copy number variation of Chinese children in early-onset epileptic encephalopathies with unknown cause. Clin Genet. (2016) 90:428–36. doi: 10.1111/cge.12768
6. Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. (2016) 17:9–18. doi: 10.1038/nrg3999
7. Chiurazzi P, Pirozzi F. Advances in understanding - genetic basis of intellectual disability. F1000Res. (2016) 5:1. doi: 10.12688/f1000research.7134.1
8. O'Byrne JJ, Lynch SA, Treacy EP, King MD, Betts DR, Mayne PD, et al. Unexplained developmental delay/learning disability: guidelines for best practice protocol for first line assessment and genetic/metabolic/radiological investigations. Ir J Med Sci. (2016) 185:241–8. doi: 10.1007/s11845-015-1284-7
9. Jin X, Sun Y, Jiang F, Ma J, Morgan C, Shen X. “Care for Development” intervention in rural China: a prospective follow-up study. J Dev Behav Pediatr. (2007) 28:213–8. doi: 10.1097/dbp.0b013e31802d410b
10. Cirelli M, Bickle Graz A, Tolsa JF. Comparison of Griffiths-II and Bayley-II tests for the developmental assessment of high-risk infants. Infant Behav Dev. (2015) 41:17–25. doi: 10.1016/j.infbeh.2015.06.004
11. Watkins MW, Beaujean AA. Bifactor structure of the Wechsler Preschool and Primary Scale of Intelligence–Fourth Edition. Sch Psychol Q. (2014) 29:52–63. doi: 10.1037/spq0000038
12. Baron IS. Test review: Wechsler Intelligence Scale for Children-Fourth Edition (WISC-IV). Child Neuropsychol. (2005) 11:471–5. doi: 10.1080/09297040590951587
13. Ji C, Yao D, Chen W, Li M, Zhao Z. Adaptive behavior in Chinese children with Williams syndrome. BMC Pediatr. (2014) 14:90. doi: 10.1186/1471-2431-14-90
14. Kaat AJ, Lecavalier L, Aman MG. Validity of the aberrant behavior checklist in children with autism spectrum disorder. J Autism Dev Disord. (2014) 44:1103–16. doi: 10.1007/s10803-013-1970-0
15. Rellini E, Tortolani D, Trillo S, Carbone S, Montecchi F. Childhood Autism Rating Scale (CARS) and Autism Behavior Checklist (ABC) correspondence and conflicts with DSM-IV criteria in diagnosis of autism. J Autism Dev Disord. (2004) 34:703–8. doi: 10.1007/s10803-004-5290-2
16. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
17. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
18. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
19. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
20. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. (2012) 7:e46688. doi: 10.1371/journal.pone.0046688
21. Choi Y. A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein. Bioinformat Comput Biol. (2012) 2012:2382989. doi: 10.1145/2382936.2382989
22. Choi Y, Chan AP. PROVEAN. web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. (2015) 31:2745–7. doi: 10.1093/bioinformatics/btv195
23. Adzhubei A, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. (2013) 7:20. doi: 10.1002/0471142905.hg0720s76
24. Ramensky, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. (2002) 30:3894–900. doi: 10.1093/nar/gkf493
25. Sunyaev SR, Eisenhaber F, Rodchenkov IV, Eisenhaber B, Tumanyan VG, Kuznetsov EN, et al. Profile extraction from sequence alignments with position-specific counts of independent observations. Protein Eng. (1999) 12:387–94. doi: 10.1093/protein/12.5.387
26. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
27. Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. (2012) 40:e115. doi: 10.1093/nar/gks596
28. Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. (2007) 23:1289–91. doi: 10.1093/bioinformatics/btm091
29. Koressaar T, Lepamets M, Kaplinski L, Raime K, Andreson R, Remm M. Primer3_masker: integrating masking of template sequence with primer design software. Bioinformatics. (2018) 34:1937–8. doi: 10.1093/bioinformatics/bty036
30. Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. (2005) 15:901–13. doi: 10.1101/gr.3577405
32. Amberger JS, Bocchini CA, Scott AF, Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. (2019) 47:D1038–43. doi: 10.1093/nar/gky1151
33. Panigrahi, Jain P, Didel S, Agarwal S, Muthuswamy S, Saha A, et al. Identification of microdeletion and microduplication syndromes by chromosomal microarray in patients with intellectual disability with dysmorphism. Neurol India. (2018) 66:1370–6. doi: 10.4103/0028-3886.241346
34. Mostovoy Y, Yilmaz F, Chow SK, Chu C, Lin C, Geiger EA, et al. Genomic regions associated with microdeletion/microduplication syndromes exhibit extreme diversity of structural variation. Genetics. (2021) 217:iyaa038. doi: 10.1093/genetics/iyaa038
35. Mohammadzadeh A, Akbaroghli S, Aghaei-Moghadam E, Mahdieh N, Badv RS, Jamali P, et al. Investigation of chromosomal abnormalities and microdeletion/ microduplication(s) in fifty iranian patients with multiple congenital anomalies. Cell J. (2019) 21:337–49. doi: 10.22074/cellj.2019.6053
36. Takata A. Estimating contribution of rare non-coding variants to neuropsychiatric disorders. Psychiatry Clin Neurosci. (2019) 73:2. doi: 10.1111/pcn.12774
37. Servetti M, Pisciotta L, Tassano E, Cerminara M, Nobili L, Boeri S, et al. Neurodevelopmental disorders in patients with complex phenotypes and potential complex genetic basis involving non-coding genes, and double CNVs. Front Genet. (2021) 12:732002. doi: 10.3389/fgene.2021.732002
38. Mondal K, Ramachandran D, Patel VC, Hagen KR, Bose P, Cutler DJ, et al. Excess variants in AFF2 detected by massively parallel sequencing of males with autism spectrum disorder. Hum Mol Genet. (2012) 21:4356–64. doi: 10.1093/hmg/dds267
39. RK CY, Merico D, Bookman M, Thiruvahindrapuram B, Patel RV. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. (2017) 20:602–11. doi: 10.1038/nn.4524
40. Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Reports. (2018) 11:1211–25. doi: 10.1016/j.stemcr.2018.10.003
Keywords: neurodevelopmental disorders, global developmental/intellectual disabilities, autism spectrum disorders, children, gene
Citation: Wu D and Li R (2022) Genetic analysis of neurodevelopmental disorders in children. Front. Child Adolesc. Psychiatry 1:987339. doi: 10.3389/frcha.2022.987339
Received: 06 July 2022; Accepted: 21 September 2022;
Published: 07 December 2022.
Edited by:
Sara Calderoni, Stella Maris Foundation (IRCCS), ItalyReviewed by:
Maria Pia Pia Riccio, Federico II University Hospital, ItalyCopyright © 2022 Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong Li, MTU4NTA3ODAwNjJAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.