 Atsushi Tahara
Atsushi Tahara Aska Mori3
Aska Mori3 Jun-ichiro Hayashi
Jun-ichiro Hayashi Shinji Kudo
Shinji Kudo- 1Creative Interdisciplinary Research Division, Frontier Research Institute for Interdisciplinary Sciences (FRIS), Tohoku University, Sendai, Japan
- 2Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan
- 3Division of Advanced Device Materials, Institute for Materials Chemistry and Engineering (IMCE), Kyushu University, Fukuoka, Japan
Introduction: With the growing global concern over CO2 emissions, reducing CO2 output has become an urgent requirement. The iron production industry is among those with the highest CO2 emissions, primarily due to the use of coke as a reductant and the use of a heat source at approximately 2,000°C. To address this issue, various alternative reductants, including CO, H2, and lignite, have been explored. Building on these efforts, we recently reported a novel ironmaking system using oxalic acid (HOOC–COOH) as the reductant. Formate salts, hydrogenated forms of CO2, are promising precursors for oxalate salts; however, their behavior during dimerization remains poorly understood. Herein, we investigate the influence of group 1 and 2 metal cations on the base-promoted dehydrogenative coupling of formate to form oxalate.
Methods: First, dehydrogenative coupling of sodium formate was executed by using various types of groups 1 and 2 metal carbonates. Second, the base was replaced from metal carbonates to metal hydroxides to check the reactivity. Finally, a countercation of sodium formate was replaced to various types of groups 1 and 2 metals. To elucidate the reaction mechanism, DFT calculation was executed.
Results and discussion: Treatment of sodium formate with various bases (group 1 and 2 metal carbonates or hydroxides) revealed that group 1 metal hydroxides are more effective than metal carbonates for oxalate formation, with cesium hydroxide (CsOH) exhibiting high reactivity. Density functional theory (DFT) calculations suggest that this kinetic advantage arises not only from increased basicity but also from intermediate destabilization in the Na/Cs mixed-cation system. Additionally, both experimental and theoretical investigations reveal that oxalate yield is influenced by the thermodynamic stability of intermediates and products (oxalate salts), highlighting the crucial role of cations in the reaction.
1 Introduction
Oxalic acid (HOOC–COOH) is the simplest dicarboxylic acid (Riemenschneider and Tanifuji, 2011; Schuler et al., 2021a). It undergoes thermal decomposition to form CO2 and formic acid, and formic acid further decomposes into water and CO in the presence of an acid. Its conjugate base, the oxalate dianion, is a reducing agent that acts as a two-electron donor, which subsequently decomposes into two CO2 molecules (Gibson et al., 2016). Oxalic acid is found in many plants, and its excessive ingestion and prolonged exposure to the skin are potential safety concerns (Zuo et al., 2016). Oxalic acid is widely used as a chemical feedstock in various industries including dyeing (Lee et al., 1999) and as an extractant in metallurgy of lanthanides from its ores (Abrahama et al., 2014; Zhang et al., 2022). In contrast to these conventional uses, our research team has proposed a novel ironmaking process with oxalic acid as the reductant (Santawaja et al., 2020; Santawaja et al., 2022). In iron and steel industries, high CO2 emission is one of the most pressing problems against sustainability (IEA. 2020). Traditionally, iron (0) is produced by the reaction of iron oxide with coke [C(0)] to produce CO2 as a byproduct. In the method proposed by our team, oxalic acid is used as a reductant instead of coke. The reaction of iron oxide with oxalic acid results in the formation of iron (III) oxalate, which was photochemically and thermally reduced to iron (0) powder concomitantly with the degradation of oxalate dianion to CO2 (Parker and Hatchard, 1959; Dudeney and Tarasova, 1998; Ogi et al., 2015). The regeneration of oxalic acid from CO2 could lead to the development of a sustainable carbon-neutral ironmaking system.
Oxalic acid has been traditionally produced from non-CO2 carbon sources (Schuler et al., 2021b; Fenton and Steinwand, 1974). Meanwhile, the reductive coupling of CO2 to form oxalate dianions has been examined in electrochemical studies (Savéant, 2008) and extended to the fields of metal-complex-catalyzed electrochemistry (Becker et al., 1985; Kushi et al., 1994; Kushi et al., 1995; Evans et al., 1998; Angamuthu et al., 2010) and biochemistry (Pastero et al., 2019) (Figure 1). Kanan et al. reported that thermal coupling of CO2 to form > C2 fragments, including oxalate dianions, using alkali metal carbonates is a non-electrochemical or non-biochemical approach (Banerjee and Kanan, 2018). In this approach, formate anions were produced first, followed by carbonite (CO22–) species. This approach not only produced oxalic acid but also promoted C2 (and >C2) chemistry via the transformation of oxalate to other organic compounds such as glycolic acid (Schuler et al., 2021a; Watanabe et al., 2015).
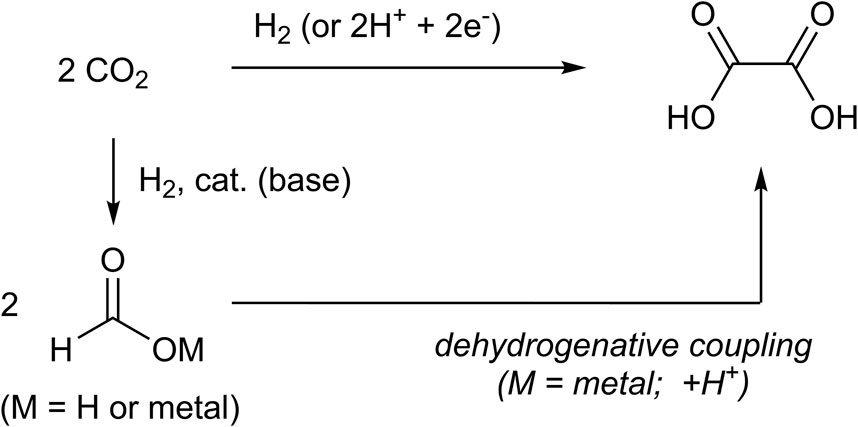
Figure 1. Syntheses of oxalic acid from CO2, formic acid, or metal formats.
There have been limited reports on the production of >C2 compounds, apart from oxalic acid, that use CO2 as a direct feedstock (Banerjee and Kanan, 2018; Prieto, 2017). In contrast, C1 compounds produced via CO2 reduction have been studied extensively, some of which find industrial applications (Álvarez al., 2018). Therefore, it is reasonable to synthesize oxalic acid by coupling formic acid or its salt that are potentially available via the reduction of CO2. To form a carbon–carbon bond from two formate anions, one formyl H atom, which normally acts as a δ–H atom or hydride (H−), must be abstracted as a proton (H+). The resulting carbonite species (CO22–) binds to the other formyl anion to form the oxalate species following hydrogen abstraction from the hydridic C–H bond. This reaction mechanism has been proposed in several reports (Andresen, 1977; Górski and Kraśnicka, 1987; Górski and Kraśnicka, 1987). The reaction is carried out in the presence of a base, typically an alkali carbonate. However, excess CO2 is generated when carbonate salts are used. Other reagents such as sodium amide or sodium borohydride were also used. As a breakthrough, in 2016, Lakkaraju and Batista et al. reported the dehydrogenative coupling of sodium formate to obtain the oxalate salt in the presence of a catalytic amount of sodium hydride (NaH) (Lakkaraju et al., 2016). Schuler et al. extended their approach to various types of bases, and it was found that potassium hydride (KH) and lithium hydride (LiH) also showed high catalytic activity at lower reaction temperatures, similar to NaH (Schuler et al., 2021a; Schuler et al., 2021b).
Given their report, we focused on the behavior of cations in the feedstock formate and bases during the coupling reaction. In the report by Lakkaraju and Batista et al., the catalytic activities of sodium hydroxide (NaOH) and potassium hydroxide (KOH) were investigated. Their activity was lower than that of NaH. It should be noted that not only the difference in anions (i.e., hydroxide and hydride) but also the difference in metal cations (i.e., Na+ and K+) affected the oxalate yield (Lakkaraju et al., 2016). In the report by Schuler, KH showed the highest catalytic activity compared with those of NaH or LiH (Schuler et al., 2021a; Schuler et al., 2021b). Although the reactivity of the other alkali metals (i.e., Rb+ and Cs+) seems to have generated interest, only cesium carbonate was studied in the report. Moreover, the effects of several types of metal cation combinations on the substrate (formate), product (oxalates), and bases have not been investigated in parallel, although self-thermolysis of formate salts bearing group 1 and 2 metal cations has been reported (Górski and Kraśnicka, 1987; Shishido and Masuda, 1971).
Herein, we report the base-promoted dehydrogenative coupling of the formate salts bearing different metal cations to oxalates. First, the dehydrogenative coupling of sodium formate was performed using various metal carbonates or hydroxides as bases. Based on the screening, it was observed that heavier alkali hydroxides showed higher reactivity toward the dimerization. Using the information obtained from density functional theory (DFT) calculations, the theoretical explanation for the reaction mechanism, in particular, the role of metal cations has been elucidated. The Na+ in the formate salts were also replaced with the other group 1 and 2 metal cations and evaluated in a similar manner.
2 Materials and methods
2.1 Experimental section
2.1.1 General procedure
Groups 1 and 2 metal formates, metal carbonates, metal hydroxides, and conc. HCl(aq) were purchased from Wako Pure Chemical Industries, Nacalai Tesque, Kanto Chemical, or Sigma-Aldrich. DSC measurements were performed using NETZSCH DSC 204 F1 Phoenix. Details of the reactor used for the coupling reaction are described in Supplementary Figure S1. The yield of oxalic acid was determined by HPLC analysis on a SHIMADZU HPLC unit (Prominent series), including the following instruments: a pump, a PDA detector, a column oven, and the Bio-Rad Aminex 87H column.
2.1.2 DSC measurement
Metal formate (5 mg) and metal carbonate or metal hydroxide (0.3 equivalents per mol) were set on a platinum pan. Under a flow of N2 gas (100 mL/min), DSC measurement for the mixture was performed at a temperature range of 30°C to 460 °C at a heating rate of 5 or 30°C/min. All the results are summarized in Supplementary Figures 2–5.
2.1.3 Synthesis of oxalic acid from metal formates
Metal formate (45.0 mmol) and the base (metal carbonate or metal hydroxide, 15.0 mmol) were added to a 15-mm ϕ glass insert and set to the reactor with a thermometer (drawn details are in Supplementary Figure S1). Under a flow of N2 gas (300 mL/min), the mixture was heated at 360°C for 1 h. After cooling, the resulting solid was dissolved in H2O, and conc. HCl(aq) was added until pH = 1. The water was removed under reduced pressure, and the white solid obtained was analyzed by HPLC to determine the yield of oxalic acid (mobile phase; 5 mM H2SO4, flow rate; 0.6 mL/min, Rt = 7.2 min, λ = 210 nm for oxalic acid.).
2.2 Computational methods
2.2.1 DFT calculations
All calculations were performed using the Gaussian 16 rev. C program to search for all intermediates and transition structures on potential energy surfaces (Frisch et al., 2016). For optimization, the B3LYP-D functional was selected (Frisch et al., 2016; Grimme, 2006). We also employed the SDD (Stuttgart/Dresden pseudopotentials) (Andrae et al., 1990) and 6-31G** basis sets (Gordon, 1980; Hariharan and Pople, 1974; Hariharan and Pople, 1973; Hehre et al., 1972; Ditchfield et al., 1971) for group 1 metal atoms (Li, Na, K, Rb, and Cs) and the other atoms, respectively [BS1]. All stationary-point structures were found to have an appropriate number of imaginary frequencies. An appropriate connection between a reactant and a product was confirmed by the intrinsic reaction coordinate (IRC) (Fukui, 1970; Fukui, 1981; Gonzalez and Schlegel, 1990) and quasi-IRC (qIRC) calculations. In the quasi-IRC calculation, the geometry of a transition state was first shifted by perturbing the geometries very slightly along the reaction coordinate and then released for equilibrium optimization. To determine the energy profile of the proposed reaction scheme, we performed single-point energy calculations at the optimized geometries using the SDD (Stuttgart/Dresden pseudopotentials) and 6-311++G** (Krishnan et al., 1980; McLean and Chandler, 1980; Clark et al., 1983) basis sets for group 1 metal atoms (Li, Na, K, Rb, and Cs) and the other atoms, respectively. The solvent effects of the molten salt (ε = 26.0) were evaluated using the polarizable continuum model (PCM) (Miertuš et al., 1981) [BS2]. Energy profiles of the calculated reaction pathways are presented as Gibbs free energy changes (ΔG) involving thermal corrections at 298.15 K. All the optimized structures (ball-and-stick models) and optimized geometries in the XYZ file format are summarized in Supplementary Tables 1–11.
3 Results
We report the formation of sodium oxalate via thermolysis of sodium formate at 360 °C in the presence of Na2CO3, followed by quenching with conc. HCl(aq). However, 2.0 equivalents of the base was required to quantitatively obtain oxalic acid (Andersen, 1977), and the base was decomposed into CO2 during quenching. Therefore, the relationship between the amount of Na2CO3 and oxalate yield was investigated before screening for the base. As shown in Table 1, quantitative formation of oxalic acid is observed when more than 0.5 equivalents of Na2CO3 (entries 1–4) is used, which are not helpful to compare the effects of the base. When the amount of the base was reduced to 0.3 equivalents, the yield decreased to 59% (entry 5), in which the moderate yield at the initial reaction condition enables both improved and less efficient outcomes to be observed by changing the bases. The oxalic acid could not be obtained in the absence of a base at 360 °C but at 410 °C (entries 6,7), which is in good correspondence with a previous report about self-dimerization of sodium formate without bases at the range 390°C–400 °C (Schuler et al., 2021a). The relatively low yield of oxalic acid (40%) is possibly due to the competitive reaction of its thermal degradation. No oxalic acid was produced in the absence of sodium formate (entry 8).
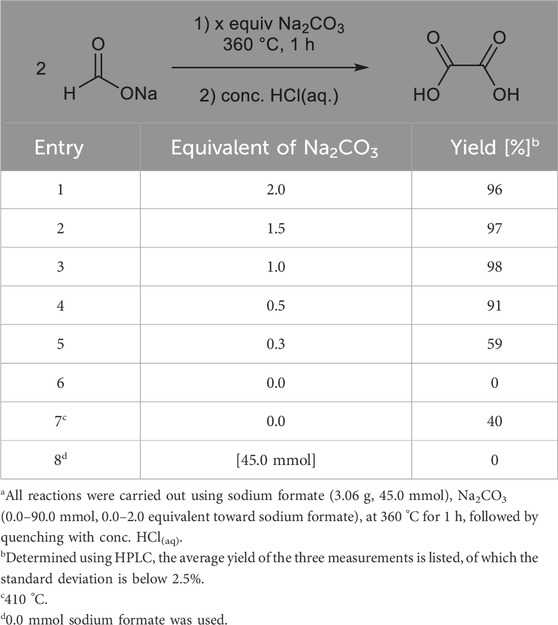
Table 1. Reaction of sodium formate with different equivalents of sodium carbonatea.
To evaluate the appropriate combination of base and metal formate, the amount of base for the coupling of metal formate was fixed at 0.3 equivalents. Before starting the experiments, differential scanning calorimetric (DSC) measurements of these mixtures were obtained. Some combinations showed large peaks at approximately 300°C–400 °C, which confirmed the exothermic coupling reaction (see Supplementary Figure S2). Table 2 shows the coupling of sodium formate with 0.3 equivalents of metal carbonates (group 1 and 2 metals). Li2CO3 weakly promoted the dehydrogenative coupling of sodium formate and gave a 14% yield of oxalic acid (entry 1). The other group 1 carbonate salts (i.e., K2CO3, Rb2CO3, and Cs2CO3) showed no reactivity (entries 3–5). Although MgCO3 and CaCO3 were inactive, some group 2 carbonate salts, such as SrCO3 and BaCO3, showed mild reactivity, resulting in a 40% and 50% yield of oxalic acid, respectively (entries 6–8, 11). An increase in the reaction temperature with use of SrCO3 and BaCO3 as bases reduced the yield, which was due to the thermal degradation of the generated oxalate salts (entries 8–13) (Górski and Kraśnicka, 1987; Shishido and Masuda, 1971). A unique reactivity was observed in the strontium and barium salts; however, the yields were less than those with Na2CO3.

Table 2. Reaction of sodium formate with different group 1 and 2 metal carbonates.a.
Next, the reactivities of group 1 and 2 metal hydroxide salts were investigated. The results are shown in Table 3. When NaOH was used as a base, 71% oxalic acid yield was obtained, which was higher than that obtained using Na2CO3 (entry 2). Not only NaOH but all the group 1 metal hydroxides also showed moderate or good reactivities (entries 3–5). Among group 2 metal hydroxides, however, only Mg(OH)2 showed good reactivity (entry 6), although it cannot be ignored that 0.6 equivalents of hydroxyl anions was generated from 0.3 equivalents of Mg(OH)2, in which additional –OH may affect the outcome.
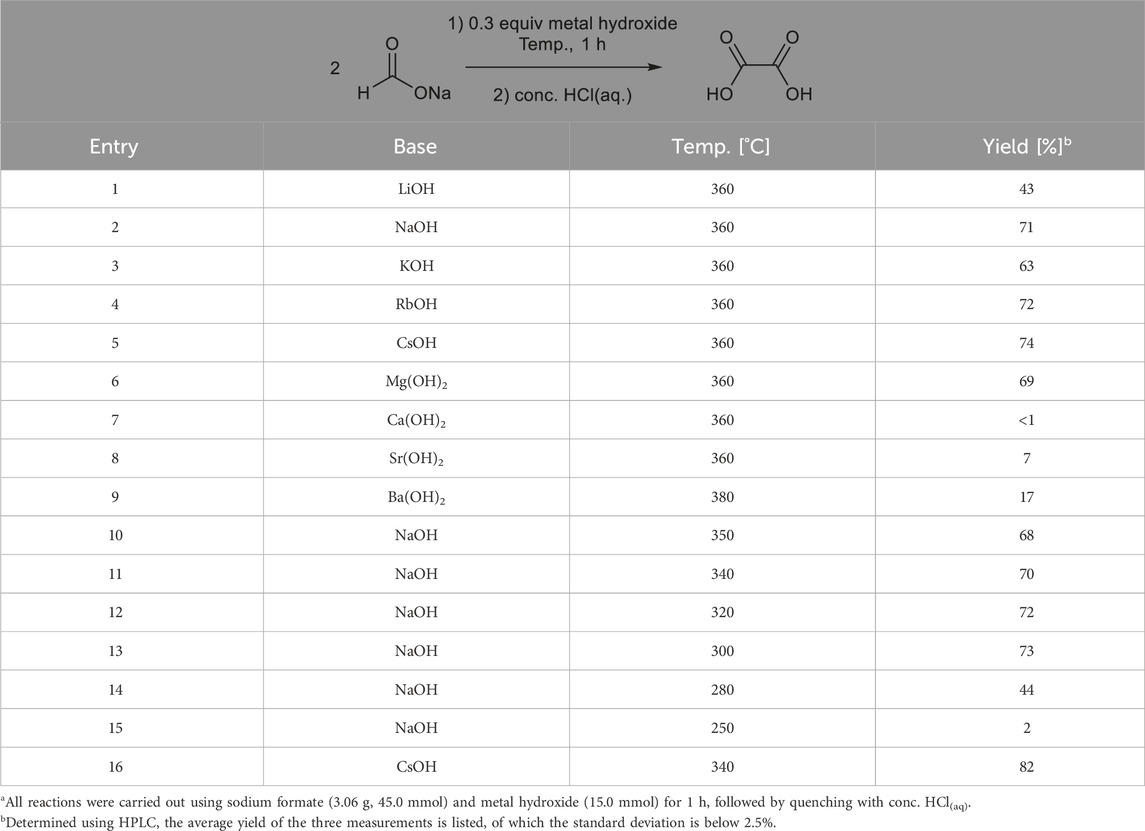
Table 3. Reaction of sodium formate with different group 1 and 2 metal hydroxides.a.
Further analysis revealed that lowering the reaction temperature improved the oxalic acid yield. In the present study, the reaction at 340 °C using CsOH gave the highest yield of oxalic acid (82%, entry 16). Using NaOH, oxalic acid was obtained at the lowest reaction temperature, 280 °C, without a decrease in the yield (entries 10–15). These trends could be rationalized by the thermal degradation of the resulting oxalate anions at higher temperatures (Górski and Kraśnicka, 1987; Shishido and Masuda, 1971).
Based on Tables 2 and 3, there is potential that the use of similar cations could affect the reactivity. A screening for formate salt cations was then performed. However, as summarized in Table 4, except for the combination of potassium formate and KOH, which had a 40% yield (entry 6), most metal formates could not be dimerized in the presence of carbonate or hydroxide salts having the same metal cations. These results indicate that similar cations do not always promote homocoupling. Entries 13 and 14 show possible cation exchange between formate salts and bases. When cesium formate was treated with NaOH, 65% oxalic acid was obtained, which was similar to that obtained from the combination of sodium formate and CsOH, as shown in Table 3.
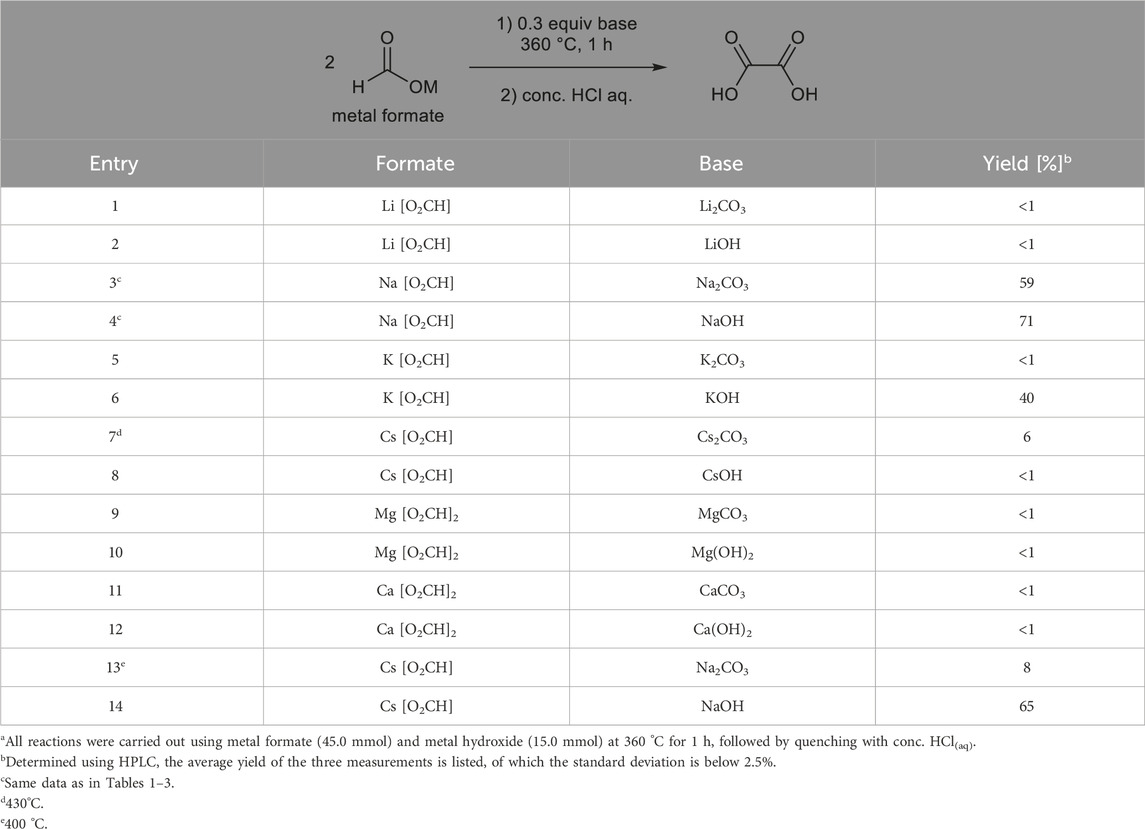
Table 4. Reaction of metal formates with metal hydroxides.a.
4 Discussion
The experimental results of base-promoted dehydrogenative couplings to form oxalate anions are summarized as follows: (1) sodium cations are appropriate when using carbonate salts as the base. Strontium and barium were relatively efficacious. (2) All group 1 metal hydroxides were better bases than metal carbonates, of which CsOH showed the highest reactivity. With the use of highly reactive bases such as NaOH and CsOH, the reaction temperature can be lowered down to 280 °C, enabling suppression of the thermal decomposition of the products. (3) The unification of metal cations between the formate salts and bases was not effective to improve the yield of oxalic acid. (4) A cation exchange between the formate salts and bases may occur during the reaction.
One of the biggest differences between metal carbonates/hydroxides/hydrides is the basicity. The strength of the basicity increases in the order of CO32– < OH− < H− (Lew, 2009), which explains the high yield of oxalic acid when using MOHs than when using carbonate salts. Although the result using CsOH could not reach those observed in the previous report using NaH in both catalytic amount (2 wt%, ca. 5.7 mol% toward sodium formate) and yield (99%) (Lakkaraju et al., 2016), or the combination of potassium formate and NaH or KH in the reaction temperature (below 200 °C) (Schuler et al., 2021b), metal hydroxides could be chosen as efficient bases for dimerization without further care of air (no need to use a glovebox). Notably, metal hydroxides also show a hygroscopic property, so usage of a glovebox may improve our results further.
It should be noted that not only the kind of anions but cations also affect the yield of oxalate salts. In addition to the results reported previously, with use of NaOH and KOH (Lakkaraju et al., 2016; Schuler et al., 2021a; Schuler et al., 2021b), the reactivities of LiOH, RbOH, and CsOH were reinvestigated, and it was revealed that heavier bases also worked as a good base for oxalate formation. It is well known that alkali metal hydroxides dissociate completely in solution to form M+ and OH−, and the basicity of group 1 metal hydroxides is nearly similar in a dilute solution. However, in a concentrated solution or a molten salt state, the strength of the basicity increases in the order of LiOH < NaOH < KOH < RbOH < CsOH due to the increase in the ionic radius of metal cations (Kennedy, 1938).
To acquire mechanistic insights and to compare the activation energy barriers between several bases, a computational study was performed. To simplify the discussion, the reactions using group 1 metal hydroxides were selected for the study. The group 1 metal carbonate salts or group 2 metal hydroxides/carbonates were not considered for the computational study because multivalent ions (CO32– or M2+) have the potential for multiple coordination styles to generate several reaction routes (see below).
Lakkaraju and Batista et al. proposed a reaction mechanism for the metal hydride-catalyzed dehydrogenative coupling of sodium formate (Lakkaraju et al., 2016), which consists of three main steps: (step 1) deprotonation of the formyl proton of the formate anion to form carbonite species, (step 2) C–C bond formation between (formally) dianionic carbonite and formate species stabilized by metal cations to form hydrooxalate species, and (step 3) dehydridation of hydrooxalate trianion to form metal hydride and oxalate species. In the previous study that used NaH as the catalyst, H2 was generated as a by-product in step 1 (Lakkaraju et al., 2016). In the present work, H2O is assumed to be obtained from metal hydroxide in step 1. Water then reacted with the metal hydride formed in step 3 to concomitantly regenerate the metal hydroxide and produce H2. In the absence of water, MOH may promote the reaction quantitatively, not catalytically, and the resulting MH could behave as a catalyst. In the gaseous outlet stream, detection of water was observed during the reaction by using GC, so there seems to be a low possibility of the regeneration of MOH species. On the other hand, if the 0.3 equivalent of MOH was fully consumed as the base, 60% of oxalate salt could be theoretically formed, while up to 82% of oxalic acid was experimentally obtained. Therefore, we cannot ignore three possibilities (case 1, MOH as stoichiometric; case 2, MOH as catalytic; and case 3, MOH as the first cycle and then MH after the second cycle). In this paper, the pathway using MOH was calculated to consider the complementarity with Lakkaraju and Batista’s report (Lakkaraju et al., 2016). The overall reaction scheme in the case of MOH is shown in Figure 2.

Figure 2. Proposed reaction scheme for the dehydrogenative dimerization of formate anions to oxalate (Lakkaraju et al., 2016).
The reactions shown in Table 3, i.e., dehydrogenative coupling of formate anions assisted by group 1 metal hydroxides (LiOH, NaOH, KOH, RbOH, and CsOH), were calculated, some of which have already been reported (Lakkaraju et al., 2016). All calculation data are given in Supplementary Tables 1–11.
The total Gibbs free energy of the free formate salt and metal hydroxide was set to 0 kcal/mol. The mixture of sodium formate and metal hydroxide was stabilized by aggregation to form A1NaM2. As shown in Table 5, there are three types of isomers for A1NaM2: κ2-formate anion on the Na atom (A1aNaM2), κ2-formate anion on the M2 atom (A1cNaM2), and µ-κ1:κ1-formate on both Na and M2 atoms (A1bNaM2) with bridged hydroxide anions. Each isomer was formed exergonically from the free formate salt and a metal hydroxide. When M2 = Na, A1aNaNa and A1cNaNa were same, and the Gibbs free energy (−4.1 kcal/mol) was higher than that of the bridging formate, A1bNaNa (−5.9 kcal/mol). However, the heavier cations, namely, K, Rb, and Cs, did not produce µ-κ1:κ1-formate species as optimized structures to converge into A1cNaM2, which was not energetically preferred compared to the normal adduct, A1aNaM2. This may be due to the instability that occurs because of the difference between two cations with different atomic radii. With sodium formate and LiOH, only A1bNaLi was formed, and it exhibited lower relative Gibbs free energy (−12.1 kcal/mol).

Table 5. Relative Gibbs free energy (ΔG [kcal/mol]) for the adduct of sodium formate with various metal hydroxidesa.
Next, the activation energy barrier for step 1 was estimated computationally. Since A1NaM2 was just a resting state, M2OH was needed to move next to the H atom of the formate salt. Deprotonation (note: not dehydridation) of the formic H atom by the hydroxyl group occurred via the transition state TSA1/A2NaM2 (see detailed structures in Supplementary Table 2), and the resulting carbonite species was stabilized with the assistance of M2 to form A2NaM2. Table 6 shows the activation energy barriers for step 1 (energy gap between TSA1/A2NaM2 and A1NaM2) for different metal hydroxides. Compared with NaOH (ΔG‡step1 = 36.6 kcal/mol), those for KOH, RbOH, and CsOH exhibited relatively small ΔG‡step1 (ca 30 kcal/mol). This behavior could be attributed to not only the stabilization of transition states due to the increase in the basicity but also the destabilization of the starting species, A1NaM2, as explained above. The thermodynamic stabilities of the resulting carbonite species are similar (ΔG = 33.3–34.5 kcal/mol, see Supplementary Table 1 in SI) regardless of the metal hydroxide used. For LiOH, the structure of the corresponding transition state TSA1/A2NaLi could not converge either due to its weak basicity or the strong affinity between Li and O (see below). The relative Gibbs energies of the carbonite species (M1 = Na, M2 = Li) were estimated to be 29.9 kcal/mol by the comparison of ΔG (sodium formate and LiOH) and ΔG (sodium lithium carbonite and water), as shown in Supplementary Table 2.

Table 6. Activation energy barriers (ΔG‡ [kcal/mol]) for steps 1–3 described in Figure 2.a
The resulting H2O molecule in A2NaM2 is replaced with the other sodium formate to form A3NaM2. In A3NaM2, a C–C bond is formed to generate A4NaM2 (step 2). Although the energy barrier for step 2 exhibits a marginal trend (Li > Na > K > Rb, Cs), as shown in Table 6, these differences are relatively small compared to those for step 1. A4NaM2 bears a hydrooxalate trianion that interacts with two Na cations and one M2 cation. The dehydridation of hydrooxalate H atom by the M2 cation produced an adduct, A5NaM2, composed of oxalate species and a metal hydride species (step 3). The remaining hydride is located between the M2 atom and the Na atom bearing one carboxylate group. Unlike for steps 1 and 2, ΔG‡ for step 3 increases in the order Li < Na < K < Rb < Cs due to the increase in the stability of A4NaM2 bearing heavier cations (right column in Table 6). The energy differences for step 3 for all M2 cations were lower.
There are two possible oxalate salt/metal hydride combinations. One is sodium oxalate, where both cations are Na atoms, and M2 hydride (A7NaM2). The other is an oxalate salt containing one Na and M2 atom and NaH (A6NaM2). According to the proposed mechanism, A6NaM2 is first produced and is then isomerized to A7NaM2. For K, Rb, and Cs, A7NaM2 is thermally stable compared to A6NaM2, which is reasonable for regenerating M2OH by hydration and completing the catalytic cycle (see details in the Supplementary Tables 1–11). Unlike the other four heavy metals of group 1, A6NaLi is energetically more favorable than A7NaLi, which results in its degradation into NaOH and [LiNa][oxalate] salt. Although the generation of NaOH seems stoichiometrically unreasonable for the proposed LiOH reaction scheme, these calculations, including a high activation energy barrier for step 1, imply that it is NaOH and not LiOH that is the active species, which does not contradict the moderate yield of oxalic acid (entry 1, Table 3).
Furthermore, a computational study was carried out to investigate the effects of alkali cations in formate salts on the coupling reaction. Here, the thermal stability of the resulting metal oxalates depended on the cation of the metal formates (M1), whereas the same product, sodium oxalate, was obtained using sodium formate regardless of the base (except for LiOH). A comparison of the Gibbs free energies, as shown in Table 7, reveals that the coupling reactions for K, Rb, and Cs formates are highly endergonic (ΔG ≈ 10 kcal/mol) compared to that for sodium (ΔG = 5.5 kcal/mol). In addition, most of the intermediates formed via the reaction of K, Rb, and Cs formates were less stable than those formed via Na formate by 5–10 kcal/mol (Table 7). Although the activation energy barriers for step 1 showed the same tendency as that in Table 3 (exact values are in the Supplementary Tables 1–11), sodium formate gave the best yields due to thermodynamic reasons rather than kinetics.
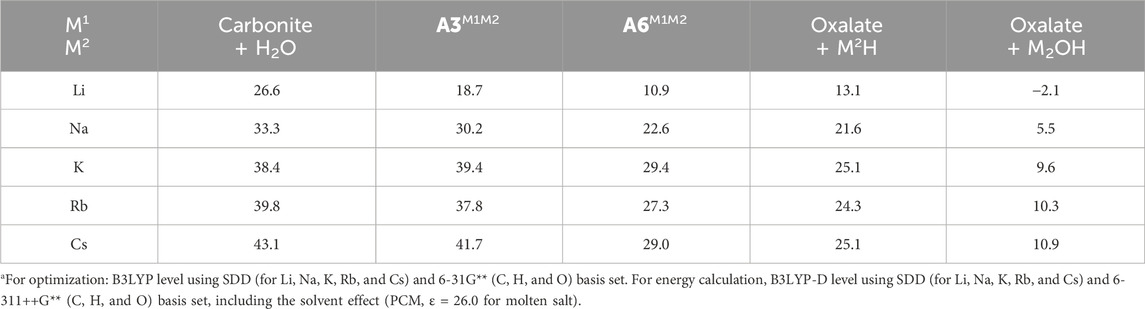
Table 7. Relative Gibbs free energy (ΔG [kcal/mol]) for intermediate species described in Figure 2.a
Except Li, these results clearly support several points and trends that are listed below: (1) independent of the type of the metal cation, step 1, i.e., the generation of the carbonite species, is the rate-determining step, and its activation energy barrier decreases in the order NaOH > KOH > RbOH > CsOH. This supports the experimental results, which indicate that CsOH and RbOH improved oxalic acid yield compared to NaOH. (2) Although steps 2 and 3 show some trend (ΔG‡ decreases for step 2 and increases for step 3 from LiOH to CsOH, respectively), the differences are too small to affect the reaction rate. (3) The relative Gibbs free energies for the dimerization of formate salts bearing group 1 metal cations to form their corresponding metal oxalates increased in the order of Na < K < Rb < Cs, which agrees with the experimental results presented in Table 4.
Finally, the discussion will be with regard to the exceptions in the computational trends. For the LiOH/sodium formate combination, the transition state estimation for step 1 failed. For the NaOH/cesium formate system, all prospective intermediates, including transition states, could not be computationally converged. These failures imply that the proposed configuration has extremely high energy potential to proceed, resulting in the convergence of the other stable structures. However, both mixtures produced oxalic acid in moderate yields (41% and 65%, respectively). These discrepancies between experimental and theoretical results imply that the reaction mechanism depicted in Figure 2 may not describe these two reaction systems and that cation exchange between metal formates and metal hydroxides may occur during the reaction, which easily proceeds via the “bridged” A1bM1M2 resting state. The NaOH/cesium formate system behaves as a CsOH/sodium formate system, generating similar amounts of the desired product (74%, as shown in Table 3, entry 5). There are several reports on the cation exchange of Na and Cs in the presence of formate anion to form a complicated crystal structure (Alcock et al., 2006). Although these configurations might be very fluid in molten salt, which does not always form a complicated structure like the crystal, these reports support our proposed hypothesis. We recalculated the reaction route for the NaOH/lithium formate system, assuming cation exchange, and found an activation energy barrier. However, the activation barrier was as high as 40.5 kcal/mol, which implied that reactions without lithium salts (i.e., only the NaOH/sodium formate system worked after Li/Na cation exchange) could not be ignored (Table 6).
As described above, computational studies for only a combination of group 1 metal salts were executed, while those for group 2 were not. Tables 2, 3 suggest that SrCO3, BaCO3, and Mg(OH)2 are efficient bases for the synthesis of oxalate salts. Therefore, it is worth considering their reaction mechanisms. If Mg(OH)2 is applied as base to the reaction scheme drawn in Figure 2; thus, the role of an additional hydroxy group in Mg(OH)2 needs to be investigated. There are many possibilities of interaction of this hydroxy group, such as whether it activates the same formyl anion, another formyl anion, or stabilizes some intermediates, or remains inert. Furthermore, versatile discussions will be needed when these pathways are extended to SrCO3 or BaCO3, which consist of multivalent ions (M2+ and CO32–).
In addition to group 1 and 2 metals, more optimized cation combinations can be employed, including organocations, lanthanides, actinides, and transition metals such as Cu, Zn, Fe, and Al. In particular, one of the ideal goals is to replace group 1 and 2 metal cations in formate salts with iron atoms. If iron formate is dehydrogenatively coupled under similar reaction conditions, the resulting iron oxalate can be utilized directly for the ironmaking system described in the Introduction. For such a system, the need to add acid (conc. HCl(aq)) for the conversion of oxalate salt into oxalic acid will be eliminated, leading to zero emission of metal waste (MCl, etc.). Concomitant with the development of procedures for the formation of iron formate from CO2, H2, and iron oxides, a new carbon-cycling ironmaking system could be realized. Studies investigating the thermal degradation of iron formate to iron oxides have been reported (Morando et al., 1987; Viertelhaus et al., 2005). Therefore, a multifaceted discussion is required to determine the most suitable cation for the reaction in terms of stability, reactivity, price, abundance, and safety.
5 Conclusion
To summarize, the effect of metal cations during the base-promoted dehydrogenative coupling of formate salts to oxalates was discussed both experimentally and theoretically. Experimentally, it was revealed that metal hydroxides were effective bases for the coupling reaction, compared to carbonate salts, and CsOH showed high activity. Theoretically, DFT calculations suggested that heavier metal hydroxides such as CsOH caused not only stabilization of the transition state (due to stronger basicity) but also destabilization of the initial structure (due to different cations) in the rate-determining step (i.e., the deprotonation of formyl H atom for the formation of carbonite species), leading to the decrease in activation energy barriers. Further attempts to study various cation combinations to realize a carbon-cycling ironmaking system are ongoing.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
AT: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, visualization, writing – original draft, and writing – review and editing. AM: formal analysis, investigation, and writing – review and editing. J-iH: conceptualization, project administration, supervision, and writing – review and editing. SK: conceptualization, funding acquisition, investigation, project administration, supervision, and writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was financially supported by the Feasibility Study Program on Uncharted Territory Challenge 2050 (No. 18101678-0) and a subsidized project (No. JPNP16002) by the New Energy and Industrial Technology Development Organization (NEDO). This work was also supported by JSPS KAKENHI grant numbers JP22H02089 (AT), JP22H05554 (AT), and JP24H01304 (AT) and MEXT Leading Initiative for Excellent Young Researchers grant number JPMXS0320200558 (AT).
Acknowledgments
The authors are grateful to the Integrated Research Consortium on Chemical Sciences (IRCCS), the Cooperative Research Program of “Network Joint Research Center for Materials and Devices”, the Cooperative Research Program of Network Joint Research Center for Materials and Devices that has been supported by MEXT, and The Iron and Steel Institute of Japan for 2020–2024.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1588773/full#supplementary-material
References
Abrahama, F., Arab-Chapeletb, B., Riveneta, M., Tamainb, C., and Grandjeanb, S. (2014). Actinide oxalates, solid state structures and applications. Coord. Chem. Rev. 266-267, 28–68. doi:10.1016/j.ccr.2013.08.036
Alcock, N. W., Wilson, M. P., and Rodger, P. M. (2006). Caesium sodium bis(formate). Acta Cryst. E 62, m388–m390. doi:10.1107/S1600536806003047
Álvarez, A., Bansode, A., Urakawa, A., Bavykina, A. V., Wezendonk, T. A., Makkee, M., et al. (2017). Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 117, 9804–9838. doi:10.1021/acs.chemrev.6b00816
Andrae, D., Häußermann, U., Dolg, M., Stoll, H., and Preuß, H. (1990). Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 77, 123–141. doi:10.1007/BF01114537
Andresen, B. D. (1977). Synthesis of sodium formate-13C and oxalic acid-13C2. J. Org. Chem. 42, 2790. doi:10.1021/jo00436a035
Angamuthu, R., Byers, P., Lutz, M., Spek, A. L., and Bouwman, E. (2010). Electrocatalytic CO 2 conversion to oxalate by a copper complex. Sci. (1979). 327, 313–315. doi:10.1126/science.1177981
Banerjee, A., and Kanan, M. W. (2018). Carbonate-promoted hydrogenation of carbon dioxide to multicarbon carboxylates. ACS Cent. Sci. 4, 606–613. doi:10.1021/acscentsci.8b00108
Becker, J. Y., Vainas, B., Eger (née Levin), R., and Kaufman (Née Orenstein), L. (1985). Electrocatalytic reduction of CO to oxalate by Ag and Pd porphyrins. J. Chem. Soc. Chem. Commun., 1471–1472. doi:10.1039/C39850001471
Clark, T., Chandrasekhar, J., Spitznagel, G. W., and Schleyer, P. v. R. (1983). Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements. Li–F. J. Comput. Chem. 4, 294–301. doi:10.1002/jcc.540040303
Ditchfield, R., Hehre, W. J., and Pople, J. A. (1971). Self-Consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 54, 724–728. doi:10.1063/1.1674902
Dudeney, A. W. L., and Tarasova, I. I. (1998). Photochemical decomposition of trisoxalatoiron(III):A hydrometallurgical application of daylight. Hydrometallurgy 47, 243–257. doi:10.1016/S0304-386X(97)00049-2
Evans, W. J., Seibel, C. A., and Ziller, J. W. (1998). Organosamarium-mediated transformations of CO2 and COS: monoinsertion and disproportionation reactions and the reductive coupling of CO2 to [O2CCO2]2-. Inorg. Chem. 37, 770–776. doi:10.1021/ic971381t
Fenton, D. M., and Steinwand, P. J. (1974). Noble metal catalysis. III. Preparation of dialkyl oxalates by oxidative carbonylation. J. Org. Chem. 39, 701–704. doi:10.1021/jo00919a026
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16, revision C.01. Wallingford CT: Gaussian, Inc.
Fukui, K. (1970). Formulation of the reaction coordinate. J. Phys. Chem. 74, 4161–4163. doi:10.1021/j100717a029
Fukui, K. (1981). The path of chemical reactions - the IRC approach. Acc. Chem. Res. 14, 363–368. doi:10.1021/ar00072a001
Gibson, M. I., Chen, P. Y., Johnson, A. C., Pierce, E., Can, M., Ragsdale, S. W., et al. (2016). One-carbon chemistry of oxalate oxidoreductase captured by X-ray crystallography. Proc. Nat. Acad. Sci. 113, 320–325. doi:10.1073/pnas.1518537113
Gonzalez, C., and Schlegel, H. B. (1990). Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 94, 5523–5527. doi:10.1021/j100377a021
Gordon, M. S. (1980). The isomers of silacyclopropane. Chem. Phys. Lett. 76, 163–168. doi:10.1016/0009-2614(80)80628-2
Górski, A., and Kraśnicka, A. D. (1987). Formation of oxalates and carbonates in the thermal decompositions of alkali metal formates. J. Therm. Anal. 32, 1243–1251. doi:10.1007/BF01913982
Gorski, A., and Krasnicka, A. D. (1987). Influence of the cation on the formation of free hydrogen and formaldehyde in the thermal decomposition of formates. J. Therm. Anal. 32, 1345–1354. doi:10.1007/BF01913334
Grimme, S. (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799. doi:10.1002/jcc.20495
Hariharan, Pc., and Pople, J. A. (1973). The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chem. Acc. 28, 213–222. doi:10.1007/BF00533485
Hariharan, Pc., and Pople, J. A. (1974). Accuracy of AHn equilibrium geometries by single determinant molecular orbital theory. Mol. Phys. 27, 209–214. doi:10.1080/00268977400100171
Hehre, W. J., Ditchfield, R., and Pople, J. A. (1972). Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261. doi:10.1063/1.1677527
IEA (2020). Industry direct CO2 emissions in the sustainable development scenario. Available online at: https://www.iea.org/data-and-statistics/charts/industry-direct-co2-emissions-in-the-sustainable-development-scenario-2000-2030 (Accessed February 28, 2025).
Kennedy, J. J. (1938). The alkali metal cesium and some of its salts. Chem. Rev. 23, 157–163. doi:10.1021/cr60074a008
Krishnan, R., Binkley, J., Seeger, R., and Pople, J. A. (1980). Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654. doi:10.1063/1.438955
Kushi, Y., Nagao, H., Nishioka, T., Isobe, K., and Tanaka, K. (1994). Oxalate Formation in electrochemical CO2 reduction catalyzed by rhodium-sulfur cluster. Chem. Lett. 23, 2175–2178. doi:10.1246/cl.1994.2175
Kushi, Y., Nagao, H., Nishioka, T., Isobe, K., and Tanaka, K. (1995). Remarkable decrease in overpotential of oxalate formation in electrochemical CO2 reduction by a metal–sulfide cluster J. Chem. Soc. Chem. Commun. J. Chem. Soc. Chem. Commun. 12, 1223–1224. doi:10.1039/C39950001223
Lakkaraju, P. S., Askerka, M., Beyer, H., Ryan, C. T., Dobbins, T., Bennett, C., et al. (2016). Formate to oxalate: a crucial step for the conversion of carbon dioxide into multi-carbon compounds. ChemCatChem 8, 3453–3457. doi:10.1002/cctc.201600765
Lee, S., Oh, J., and Shin, B., (1999). Dissolution of iron oxide rust materials using oxalic acid, 115, 815–819. doi:10.2473/shigentosozai.115.815
Lew, K. (2009). Acids and bases. Infobase Publishing. Available online at: https://www.amazon.com/Acids-Bases-Essential-Chemistry-Kristi/dp/0791097838.
McLean, A. D., and Chandler, G. S. (1980). Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 72, 5639–5648. doi:10.1063/1.438980
Miertuš, S., Scrocco, E., and Tomasi, J. (1981). Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 55, 117–129. doi:10.1016/0301-0104(81)85090-2
Morando, P. J., Piacquadio N, H., Blesa, M. A., and Vedova, C. O. D. (1987). The thermal decomposition of iron(III) formate. Thermochim. Acta 117, 325–330. doi:10.1016/0040-6031(87)88126-1
Ogi, Y., Obara, Y., Katayama, T., Suzuki, Y.-I., Liu, S. Y., Bartlett, N. C.-M., et al. (2015). Ultraviolet photochemical reaction of [Fe(III)(C2O4)3]3- in aqueous solutions studied by femtosecond time-resolved X-ray absorption spectroscopy using an X-ray free electron laser. Struct. Dyn. 2, 034901. doi:10.1063/1.4918803
Parker, C. A., and Hatchard, C. G. (1959). Photodecomposition of complex oxalates–some preliminary experiments by flash photolysis. J. Phys. Chem. 63, 22–26. doi:10.1021/j150571a009
Pastero, A., Curetti, N., Ortenzi, M. A., M, S., Destefanis, E., and Pavese, A. (2019). CO2 capture and sequestration in stable Ca-oxalate, via Ca-ascorbate promoted green reaction. Sci. Total Environ. 666, 1232–1244. doi:10.1016/j.scitotenv.2019.02.114
Prieto, G. (2017). Carbon dioxide hydrogenation into higher hydrocarbons and oxygenates: thermodynamic and kinetic bounds and progress with heterogeneous and homogeneous catalysis. ChemSusChem 10, 1056–1070. doi:10.1002/cssc.201601591
Riemenschneider, W., and Tanifuji, M. (2011). “Oxalic acid,” in Ullmann's encyclopedia of industrial chemistry (Weinheim: Wiley-VCH Verl.).
Santawaja, P., Kudo, S., Mori, A., Tahara, A., Asano, S., and Hayashi, J. (2020). Sustainable iron-making using oxalic acid: the concept, A brief review of key reactions, and an experimental demonstration of the iron-making process. ACS Sus. Chem. Eng. 8, 13292–13301. doi:10.1021/acssuschemeng.0c03593
Santawaja, P., Kudo, S., Tahara, A., Asano, S., and Hayashi, J. (2022). Dissolution of iron oxides highly loaded in oxalic acid aqueous solution for a potential application in iron-making. ISIJ Int. 62, 2466–2475. doi:10.2355/isijinternational.isijint-2020-726
Savéant, J. M. (2008). Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem. Rev. 108, 2348–2378. doi:10.1021/cr068079z
Schuler, E., Demetriou, M., Shiju, N. R., and Gruter, G. J. M. (2021). Towards sustainable oxalic acid from CO2 and biomass. ChemSusChem 14, 3636–3664, and references therein. doi:10.1002/cssc.202101272
Schuler, E., Ermolich, P. A., Shiju, N. R., and Gruter, G. J. M. (2021a). Monomers from CO2: superbases as catalysts for formate-to-oxalate coupling. ChemSusChem 14, 1517–1523. doi:10.1002/cssc.202002725
Schuler, E., Stoop, M., Shiju, N. R., and Gruter, G. J. M. (2021b). Stepping stones in CO2 utilization: optimizing the formate to oxalate coupling reaction using response surface modeling. ACS Sus. Chem. Eng. 9, 14777–14788. doi:10.1021/acssuschemeng.1c04539
Shishido, S., and Masuda, Y. (1971). Thermogravimetric analysis of various formates. Nikkashi 92, 309–312. doi:10.1246/nikkashi1948.92.309
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J. (1994). Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627. doi:10.1021/j100096a001
Viertelhaus, M., Adler, P., Cle´rac, R., Anson, C. E., and Powell, A. K. (2005). Iron(II) formate [Fe(O2CH)2]·1/3HCO2H: a mesoporous magnet–solvothermal syntheses and crystal structures of the isomorphous framework metal(II) formates [M(O2CH)2]·n(Solvent) (M = Fe, Co, Ni, Zn, Mg). Eur. J. Inorg. Chem. 2005, 692–703. doi:10.1002/ejic.200400395
Watanabe, R., Yamauchi, M., Sadakiyo, M., Abe, R., and Takeguchi, T. (2015). CO2-free electric power circulation via direct charge and discharge using the glycolic acid/oxalic acid redox couple. Energy Environ. Sci. 8, 1456–1462. doi:10.1039/c5ee00192g
Zhang, E., Noble, A., Ji, B., and Li, Q. (2022). Effects of contaminant metal ions on precipitation recovery of rare earth elements using oxalic acid. J. Rare Earths. 40, 482–490. doi:10.1016/j.jre.2020.11.008
Keywords: formate anion, oxalate dianion, CO2, alkali metal cations, DFT calculation
Citation: Tahara A, Mori A, Hayashi J-i and Kudo S (2025) Effect of alkali metal cations on dehydrogenative coupling of formate anions to oxalate. Front. Chem. 13:1588773. doi: 10.3389/fchem.2025.1588773
Received: 06 March 2025; Accepted: 25 March 2025;
Published: 23 April 2025.
Edited by:
Alessandro Pellis, University of Genoa, ItalyReviewed by:
Cameron Weber, The University of Auckland, New ZealandTommaso Tabanelli, University of Bologna, Italy
Copyright © 2025 Tahara, Mori, Hayashi and Kudo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsushi Tahara, dGFoYXJhLmEuYWFAdG9ob2t1LmFjLmpw