 Lamya H. Al-Wahaibi1
Lamya H. Al-Wahaibi1 Ali M. Elshamsy2
Ali M. Elshamsy2 Taha F. S. Ali3
Taha F. S. Ali3 Bahaa G. M. Youssif4*
Bahaa G. M. Youssif4* Stefan Bräse5*Mohamed Abdel-Aziz3*
Stefan Bräse5*Mohamed Abdel-Aziz3* Nawal A. El-Koussi2,6
Nawal A. El-Koussi2,6- 1Department of Chemistry, College of Sciences, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
- 2Pharmceutical Chemistry Department, Faculty of Pharmacy, Deraya University, Minia, Egypt
- 3Medicinal Chemistry Department, Faculty of Pharmacy, Minia University, Minia, Egypt
- 4Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Assiut University, Assiut, Egypt
- 5Institute of Biological and Chemical Systems, IBCS-FMS, Karlsruhe Institute of Technology, Karlsruhe, Germany
- 6Department of Pharmaceutical Medicinal Chemistry, Faculty of Pharmacy, Assiut University, Assiut, Egypt
Introduction: Tubulin polymerization inhibitors have emerged as interesting anticancer therapies. We present the design, synthesis, and structural elucidation of novel thiazole-based derivatives to identify novel tubulin inhibitors with potent antiproliferative efficacy and strong inhibition of tubulin polymerization.
Methods: The novel compounds consist of two scaffolds. Scaffold A compounds 10a-e and scaffold B compounds 13a-e. the structures of the newly synthesized compounds 10a-e and 13a-e were validated using 1H NMR, 13C NMR, and elemental analysis.
Results and Discussion: The most effective antitubulin derivative was 10a, exhibiting an IC50 value of 2.69 μM. Subsequently, 10o and 13d exhibited IC50 values of 3.62 μM and 3.68 μM, respectively. These compounds exhibited more potency than the reference combretastatin A-4, which displayed an IC50 value of 8.33 μM. These compounds had no cytotoxic effects on normal cells, preserving over 85% cell viability at 50 μM. The antiproliferative experiment demonstrated that compounds 10a, 10o, and 13d displayed significant activity against four cancer cell lines, with average GI50 values of 6, 7, and 8 μM, equivalent to the reference’s doxorubicin and sorafenib. Compounds 10a, 10o, and 13d were demonstrated to activate caspases 3, 9, and Bax, while down-regulating the anti-apoptotic protein Bcl2. Molecular docking studies demonstrated superior binding affinities for 10a (-7.3 kcal/mol) at the colchicine binding site of tubulin, forming key hydrophobic and hydrogen bonding interactions that enhance its activity. ADMET analysis confirmed favorable drug-like properties, establishing these compounds as promising candidates for further development as anticancer agents targeting tubulin polymerization.
1 Introduction
Cancers are a group of disorders characterized by unregulated and disorganized cellular proliferation. These disorders can spread to nearby tissues through a process called metastasis, which is the main cause of cancer-related death (El-Sherief et al., 2018; Hagar et al., 2023; Chu and Mehrzad, 2023). Factors associated with an elevated cancer risk include tobacco consumption, insufficient physical exercise, alcohol consumption, inadequate intake of fruits and vegetables, and obesity. These factors are believed to contribute to roughly one-third of cancer deaths (Seiler et al., 2018; Sun et al., 2020; Gregory et al., 2024). Cancer development entails many genetic changes in aberrant cell proliferation (Madhurya et al., 2024; Hanahan and Weinberg, 2011). The rising global cancer incidence has resulted in significant advancements in the identification of novel, safer, and curative chemotherapeutic drugs (Anand et al., 2023). In prominently developing new antitumor drugs, we focus on the current study of thiazoles, which are bioactive heterocyclic compounds known for their numerous biological activities and have prominent anticancer properties (Sahil et al., 2022; Kassem et al., 2024; Rana et al., 2023; Sabry et al., 2022; Wang et al., 2025).
Thiazole derivatives featuring either one or more thiazole rings constitute a significant class of heterocyclic compounds recognized for their anticancer properties, attributed to their notable affinity for various biological targets implicated in cancer development, including tiazofurin (Tricot et al., 1990; Franchetti et al., 1995) and bleomycin (Hecht, 2000). Efforts have been made to improve the antitumor activity of the 2-aminothiazole core in anticancer therapies, such as dasatinib (I, Figure 1; Saha et al., 2016), thia-netropsin (II, Figure 1; Plouvier et al., 1989), and alpelisib (III, Figure 1; Juric et al., 2019), which received medical approval in 2019.
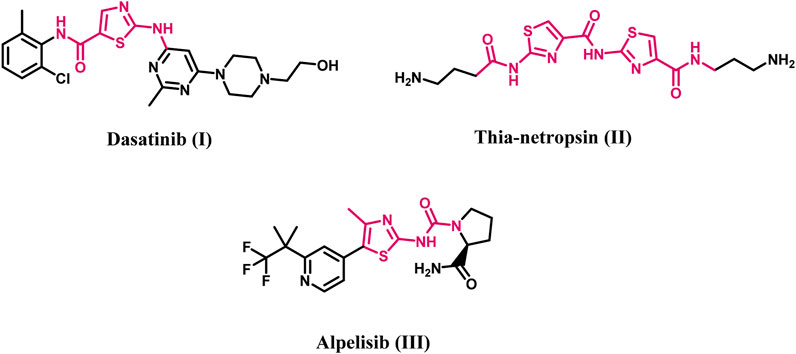
Figure 1. Structures of Dasatinib, thia-netropsin, and Alpelisib.
Additionally, studies have shown that several 2-amino-4-phenylthiazol derivatives hinder tubulin polymerization, interfere with microtubule assembly, and impair cellular division (Lee et al., 2010; Sun et al., 2017). A novel series of 2,4-disubstituted thiazole derivatives was developed and assessed for their potential anticancer action as tubulin polymerization inhibitors (El-Abd et al., 2022). All synthesized compounds were evaluated for their cytotoxic activities against four human cancer cell lines. The results indicated that compound IV (Figure 2) was the most effective inhibitor of tubulin polymerization, with an IC50 value of 2.00 ± 0.12 μM, surpassing that of the reference compound combretastatin A-4 (CA-4) (IC50 = 2.96 ± 0.18 μM). The docking analysis of compound IV into the colchicine binding site indicated that both the sulfur atom of the thiazole ring and the amidic NH established hydrogen bonds with the residue ThrB353, which is crucial for receptor site interaction. Additionally, the amidic NH formed a hydrogen bond with the GlnB247 amino acid residue, demonstrating the importance of this group (amidic NH) for activity.
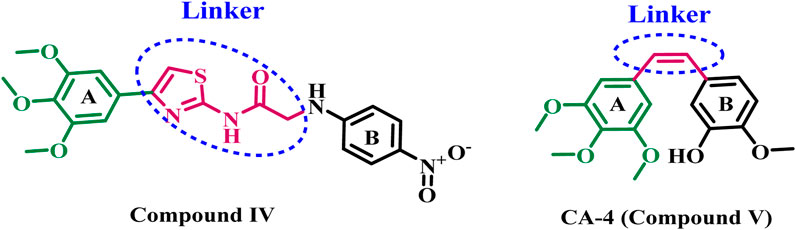
Figure 2. Structures of compound IV and CA-4 (V).
Microtubules are long, filamentous, tubular protein polymers that help cells form and maintain their structure. They help with tasks like transporting vesicles and protein complexes, maintaining cell shape, and directing cell migration and division (Gudimchuk and McIntosh, 2021; Logan and Menko, 2019). Microtubules comprising tubulin heterodimers are very important in mitosis, marked by increased dynamic instability during spindle formation and chromosome segregation (Vicente and Wordeman, 2015). Disruption of microtubules can induce cell cycle arrest in the G2-M phase and produce abnormal mitotic spindles. Because of their role in mitosis and cell division, microtubules are an attractive target for anticancer drug development.
The South African tree Combretum caffrum naturally generates Combretastatin A4 (CA-4, compound V, Figure 2), a stilbene that inhibits tubulin polymerization and exhibits significant cytotoxicity to a variety of human cancer cell lines. Furthermore, investigations have shown that CA-4 inhibits the blood flow to cancer cells. Nonetheless, the isomerization of the cis-double bond to a more stable and inactive trans-form impeded its potential therapeutic application (Tron et al., 2006; Seddigi et al., 2017). As a result, researchers are increasingly interested in developing novel tubulin inhibitors for cancer treatment.
1.1 Rational design
El-Abd et al. (2022) have reported the development of a new series of thiazole-based tubulin inhibitors. Compound IV, depicted in Figure 2, was the most potent tubulin inhibitor. In this series, the authors maintain the trimethoxy phenyl moiety (ring A) in CA-4 while substituting the cis-alkene linker in CA-4 with a thiazole-2-acetamide moiety as a rigid heterocyclic linker. Diversity in structure was achieved by including various bioactive side arms at the 2-position of the chosen backbone (Ring B), specifically amide derivatives, urea derivatives, or other heterocyclic moieties, including thiazole.
Gaspari et al. (2017) present a high-resolution crystal structure of the tubulin-cis-CA-4 complex. Researchers discovered that CA-4 interacts with the tubulin site and is distinct from the standard microtubule-destabilizing drug colchicine. The 3,4,5-trimethoxy-substituted phenyl (ring A) is deeply embedded in the tubulin-cis-CA-4 complex via a network of hydrophobic interactions with the amino acid residues that define the receptor site’s hydrophobic pocket. Hydrophobic interactions with various amino acid residues that define the pocket help to stabilize the 3-hydroxy-4-methoxy-substituted phenyl moiety (ring B) of cis-CA-4. The hydroxyl group in ring B also establishes two hydrogen bonds with the Val181 and Thr179 residues.
Inspired by the previously mentioned data, we continue our initiative (Abdelbaset et al., 2021; Abdelbaset et al., 2018) to develop specific anticancer agents as tubulin inhibitors by designing and synthesizing a novel series of thiazole-based inhibitors. The newly synthesized compounds comprise three major components: In ring A, we replace the tri-methoxy phenyl group in compound IV with a chalcone moiety, seeking to improve binding to the colchicine binding site through an array of hydrophobic interactions. The second component is the linker, the thiazole-2-acetamide moiety, as seen in compound IV. The final component is ring B, which is the thiazole or 4-phenyl thiazole moiety responsible for both hydrophobic and hydrophilic interactions. As shown in Figure 3.
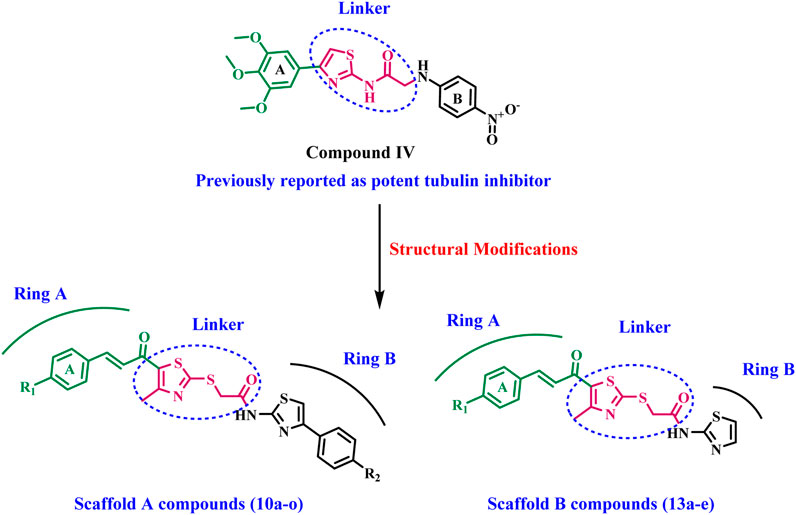
Figure 3. Structures of previously reported compound IV and new compounds 10a-o and 13a-e.
The newly synthesized compounds are of two scaffolds: Scaffold A, which contains compounds 10a–o, and Scaffold B, which comprises compounds 13a–e. All newly synthesized compounds were tested for their tubulin inhibitory activity using CA-4 as a reference compound. Next, the most potent tubulin inhibitors were tested for their antiproliferative activity against a panel of four cancer cell lines. Finally, a docking study was performed to explore the binding method and interactions within the colchicine binding site.
2 Results and discussion
2.1 Chemistry
The synthesis of chalcone intermediates 5a-e employs a multi-step pathways, as illustrated in Scheme 1, which begins with the synthesis of 3-chloroacetylacetone (2) from acetylacetone (1) via chlorination using sulfuryl chloride in toluene at 0°C. This reaction chlorinates the methylene group, generating the reactive electrophilic intermediate (Abo-Ashour et al., 2018). Compound 2 is then cyclized into 1-(2-mercapto-4-methylthiazol-5-yl)ethan-1-one (3) reacting with ammonia and carbon disulfide in absolute ethanol. The acetyl group of the thiazole ring then undergoes a Claisen-Schmidt condensation reaction with different substituted benzaldehydes (4a-e) in ethanol under basic conditions, resulting in chalcone derivatives (5a-e) with the development of a new carbon-carbon double bond (Hashem et al., 2024).
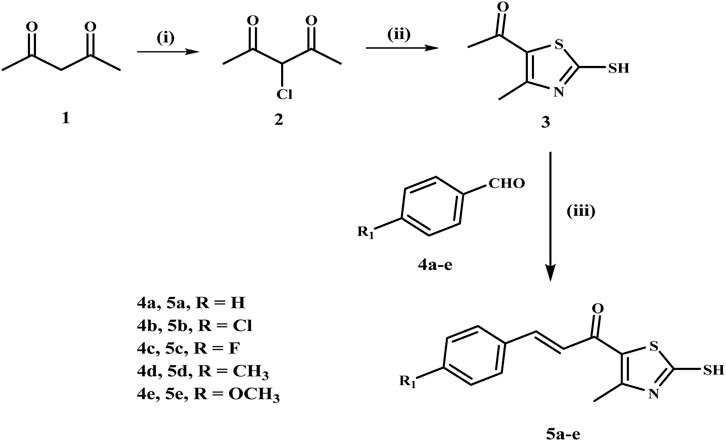
Scheme 1. Synthesis of the chalcone intermediates 5a–e. Reagents and Reaction conditions: (i) SO2Cl2, toluene, 0°C, 12 h; (ii) NH3, CS2, EtOH, RT, 6 h; (iii) appropriate aromatic aldehyde, 60% NaOH, EtOH, 0°C, 18 h.
Simultaneously, phenacyl bromides (7a-c) are synthesized from acetophenone derivatives (6a-c) via α-bromination using N-bromo succinimide (NBS) with p-toluene sulfonic acid as a catalyst in refluxing acetonitrile, as illustrated in Scheme 2. Phenacyl bromides (7a-c) react with thiourea in ethanol under reflux to yield 2-amino-4-aryl-thiazoles (8a-c), which are subsequently acylated with chloroacetyl chloride in dichloromethane using sodium carbonate as a base, resulting in N-acylated thiazoles (9a-c) containing an electrophilic chloroacetyl moiety (Valiveti et al., 2015).
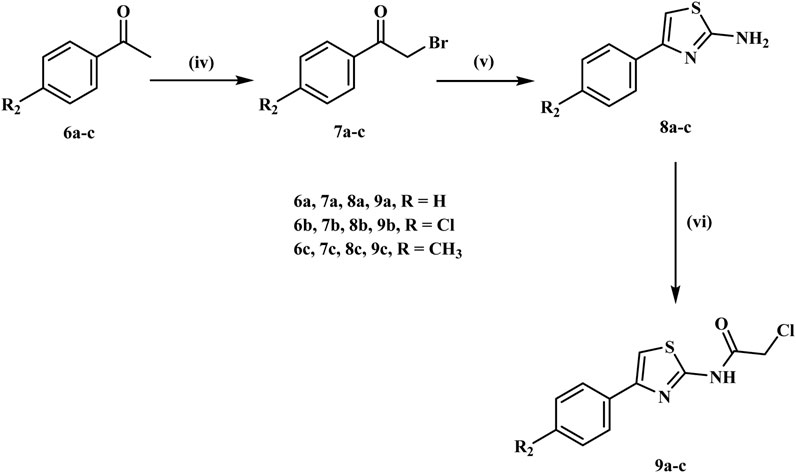
Scheme 2. Synthesis of the key intermediates 9a-c. Reagents and Reaction conditions: (iv) NBS, PTSA. H2O, acetonitrile, reflux, 10 h; (v) thiourea, Na2CO3, EtOH, reflux, 5 h; (vi) Chloroacetyl chloride, DCM, Na2CO3, H2O, 0°C, 12 h.
The final step comprises alkylation of the chalcone derivatives 5a-e with the acylated thiazoles 9a-c in acetone using anhydrous sodium carbonate and sodium iodide to afford the target compounds 10a-o, which were isolated in good yields after recrystallization from ethanol, Scheme 3. The structures of the final compounds 10a-o were elucidated by 1H NMR, 13C NMR, and elemental analysis. The 1H NMR spectra of the target compounds 10a-o showed distinct peaks, including a singlet peak at δ 4.40–4.44 ppm, which indicates the methylene group. The methyl group on the thiazole ring displays a singlet signal at δ 2.64–2.66 ppm, whereas the chalcone protons provide two doublets at around δ 7.30 and 7.65 ppm. In addition, compounds having methoxy groups show a characteristic peak at around δ 3.81–3.82 ppm. The NH of the amide group displayed a singlet at δ 12.67–12.74 ppm, thereby corroborating the structure of these compounds. Compound 10j is a good example of this group because it had characteristic peaks in 1H NMR, like two singlets at δ 2.65 and 4.41 ppm, which are the methyl and methylene groups. The methoxy group appeared as a singlet peak at δ 3.81 ppm, whereas the typical two doublets of the chalcone protons appeared at δ 7.23 and 7.65 ppm, respectively. Additionally, the NH of the amide group exhibited a singlet signal at δ 12.74 ppm. The 13C NMR spectrum of 10j detected the methyl group on the thiazole ring at δ 18.72 ppm and the methylene linker at δ 37.37 ppm. In addition, the carbonyl carbon signal was observed at δ 181.85 ppm and the methoxy group signal at 55.89 ppm.
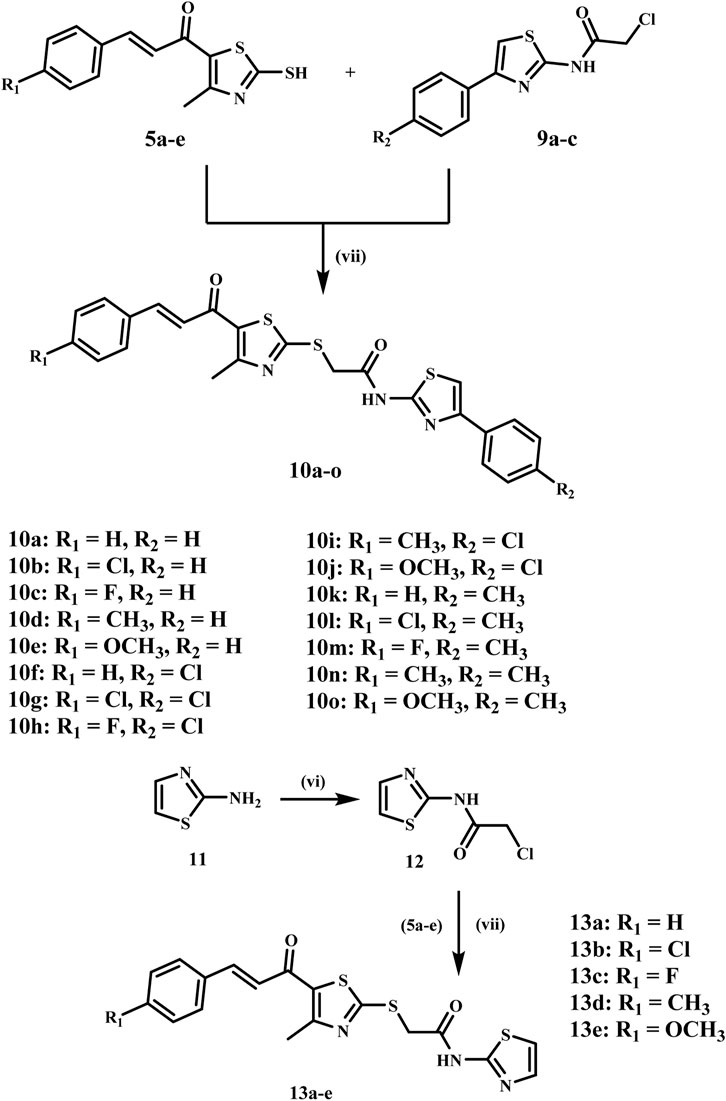
Scheme 3. Synthesis of the target compounds 10a-o and 13a-e. Reagents and Reaction conditions: (vi) Chloroacetyl chloride, DCM, Na2CO3, H2O, 0°C, 12 h; (vii) Na2CO3, NaI, acetone, RT, 6 h.
Similarly, we synthesized scaffold B compounds 13a-e by reacting 2-aminothiazole 11 with chloroacetyl chloride, which produced an intermediate 12, Scheme 3 (Tilekar et al., 2020). The intermediate 12 then reacts with thiazole chalcone derivatives 5a-e to form the target compound 13a-e in high yields. 1H NMR exhibited a comparable pattern, with the methylene protons appearing as a singlet signal at δ 4.40–4.41 ppm and the methyl protons displaying a singlet signal at δ 2.64–2.66 ppm. Chalcone protons exhibited two doublets at δ 7.23–7.39 and 7.65–7.68, and the NH of the amide group appeared as a singlet at δ 12.54–12.55 ppm. Compound 13d, as a representative example, exhibited three singlets at δ 2.35, 2.65, and 4.40 ppm, corresponding to the two methyl groups and the methylene group, respectively. The chalcone protons exhibit doublets at δ 7.31 and 7.65 ppm, while the NH of the amidic group showed a singlet signal at δ 12.54 ppm.
2.2 Biology
2.2.1 Cell viability assay
The impact of novel compounds 10a-o and 13a-e on cell viability was examined using the MCF-10A (human mammary gland epithelial) normal cell line. The MTT test was employed to assess the cell viability of the novel compounds after a four-day incubation with MCF-10A cells (El-Sherief et al., 2019; Ramadan et al., 2020). Table 1 indicates that none of the analyzed compounds exhibited cytotoxic effects on normal cells; all compounds preserved cell viability over 85% at a concentration of 50 µM.

Table 1. Results of cell viability assay and anti-tubulin (IC50) for compounds 10a-o and 13a-e.
2.2.2 Tubulin inhibitory assay
The effects of all novel synthetic compounds 10a-o and 13a-e on tubulin polymerization, using CA-4 as a reference compound (Abdelbaset et al., 2021; Abdelbaset et al., 2018), are detailed in Table 1. Compounds 10a-o and 13a-e displayed strong anti-tubulin activity, with IC50 values ranging from 2.69 to 59.37 µM, compared to the reference CA-4 (IC50 = 8.33 µM). Compounds 10a, 10d, 10f, 10h, 10m, 10o, and 13b-d had the most significant anti-tubulin activity, with IC50 values between 2.69 and 7.95 µM, surpassing the reference compound CA-4 (IC50 = 8.33 µM).
Compound 10a (R1 = R2 = H, Scaffold A) outperformed all other evaluated compounds, demonstrating an IC50 of 2.69 µM, making it three times more effective than CA-4’s IC50 of 8.33 µM, as indicated in Table 1. The anti-tubulin efficacy of compounds 10a-o is significantly affected by the substitution pattern on the chalcone moiety’s phenyl groups and at the thiazole moiety’s fourth position. For example, compounds 10b (R1 = Cl; R2 = H), 10c (R1 = F; R2 = H), 10d (R1 = CH3; R2 = H), and 10e (R1 = OCH3; R2 = H), which possess a phenyl group at the four positions of the thiazole moiety, demonstrated diminished efficacy as antitubulin agents relative to 10a (R1 = R2 = H).
Compounds 10b, 10c, 10d, and 10e exhibit IC50 values of 8.75, 22.50, 5.26, and 15.30 µM, respectively, demonstrating that the unsubstituted phenyl group of the chalcone moiety is more favorable for anti-tubulin activity than the substituted phenyl group, with efficacy increasing in the sequence: H > CH3 > Cl > OCH3 > F. Compound 10d exhibited superior potency compared to the standard CA-4.
Furthermore, substituting the hydrogen atom of the phenyl group at the four positions of the thiazole moiety in compound 10a with a 4-chloro atom in compound 10f (R1 = H, R2 = 4-Cl) or with a methyl group in compound 10k (R1 = H, R2 = 4-CH3) resulted in a significant decrease in anti-tubulin activity. The IC50 values for 10f and 10k were 5.62 µM and 47.75 µM, respectively, indicating a potency decrease of 2-fold and 18-fold relative to 10a (IC50 = 2.69 µM). These findings indicated that hydrogen and chlorine atoms at the phenyl group of the fourth position of the thiazole moiety are more tolerated than the methyl group for anti-tubulin activity.
Compound 10o (R1 = OCH3, R2 = CH3) exhibited the second highest anti-tubulin activity, with an IC50 value of 3.62 µM, which is 1.4-fold less effective than 10a (IC50 = 2.69 µM). Yet, 10o demonstrated superior activity (2.4-fold) compared to CA-4 (IC50 = 8.33 µM), as indicated in Table 1. Ultimately, compound 10g (R1 = R2 = Cl) exhibited the lowest potency among the anti-tubulin derivatives, with an IC50 value of 59.37 µM, rendering it 22-fold less potent than 10a and 7-fold less potent than the reference CA-4, indicating that the incorporation of chlorine atoms on both phenyl groups is not conducive to activity. This supports the notion that the unsubstituted phenyl rings of the chalcone and thiazole moieties exhibited greater tolerance for activity.
The anti-tubulin activity of Scaffold B compounds, 13a-e, was moderate to high relative to Scaffold A compounds, 10a-o. Compounds 13a-e exhibited enhanced IC50 values between 3.68 and 6.05 µM, with the exceptions of 13a and 13e, which displayed IC50 values of 15.82 and 39.93 µM, respectively. Compounds 13b, 13c, and 13d exhibited the highest efficacy among scaffold B derivatives, with IC50 values of 3.73, 6.05, and 3.68 µM, respectively, rendering them more effective than the reference CA-4 (IC50 = 8.33 µM), but less potent than compound 10a (IC50 = 2.69 µM). Compound 13a (R1 = H) has the same structure as 10a, but it does not have the phenyl group at the fourth position of the thiazole moiety. Its IC50 value is 15.82 µM, 6 times lower than 10a′s. The observations underscore the phenyl group’s importance at the thiazole moiety’s fourth position for anti-tubulin action.
2.2.3 Antiproliferative assay
The antiproliferative efficacy of novel compounds 10a, 10d, 10f, 10h, 10m, 10o (Scaffold A) and 13b-d (Scaffold B) against four human cancer cell lines (colon - HCT-116, prostatic - PC-3, breast - MCF-7, and pancreatic - MDAMB-231) was assessed utilizing the MTT test (Mahmoud et al., 2022; Hisham et al., 2022; Al-Wahaibi et al., 2023). Doxorubicin and Sorafenib were employed as controls in this investigation. Table 2 displays the median inhibitory concentration (IC50) and GI50 (mean IC50) values for the four cancer cell lines.
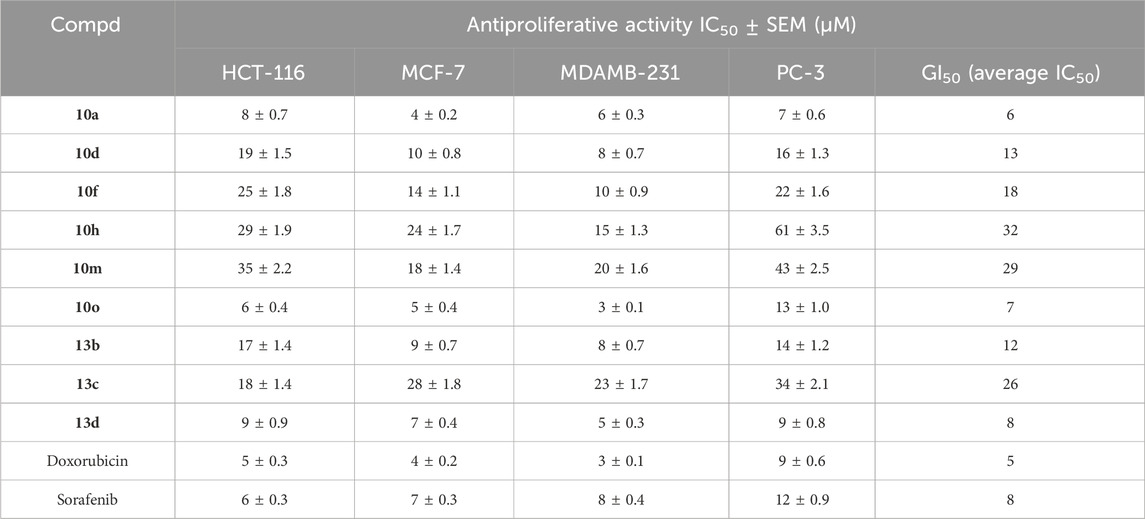
Table 2. Antiproliferative activity (IC50) for compounds 10a-o and 13a-e.
The outcomes of this antiproliferative assay are congruent with the in vitro antitubulin assay findings. Compounds 10a (R1 = R2 = H), 10o (R1 = OCH3, R2 = CH3), and 13d (R1 = CH3), the most potent anti-tubulin derivatives, exhibited the highest antiproliferative efficacy with GI50 values of 6, 7, and 8 μM, respectively, rendering them comparably potent to the reference drugs doxorubicin and sorafenib, which have GI50 values of 5 and 8 μM, respectively.
Compound 10a (R1 = R2 = H) was the most effective among the derivatives tested against the prostate PC-3 cancer cell line, with an IC50 value of 7 ± 0.6 µM. It had more potency than doxorubicin and sorafenib. Compound 10a demonstrated 1.7-fold higher efficacy than sorafenib against the PC-3 prostate cancer cell line, as shown in Table 2. Also, compound 10a showed strong antiproliferative activity against the MCF-7 breast cancer cell line, with an IC50 value of 4 ± 0.2 µM, which was higher than other derivatives and equal to doxorubicin (IC50 = 4 ± 0.2 µM) and twice as strong as sorafenib (IC50 = 7 ± 0.3 µM).
Compound 10o (R1 = OCH3, R2 = CH3), the second most efficient derivative in all assays, had the highest potency among the derivatives evaluated against the pancreatic - MDAMB-231 cancer cell line, with an IC50 value of 3 ± 0.2 µM. It exhibited equivalent potency to doxorubicin (IC50 = 3 ± 0.2 µM) but showed more potency than sorafenib. Compound 10o demonstrated 2.7-fold more potency than sorafenib against the pancreatic MDAMB-231 cancer cell line, Table 2. Compound 10o exhibited equal efficacy to doxorubicin against both colon (HCT-116) and breast (MCF-7) cancer cell lines. However, it was marginally less efficient than doxorubicin against the prostatic-PC-3 cancer cell line.
The third most potent compound, 13d (R1 = CH3), had significant antiproliferative activity with IC50 values equivalent to sorafenib across all four cancer cell lines analyzed, but it was often somewhat less effective than doxorubicin, as illustrated in Table 2. Ultimately, all other evaluated derivatives 10d, 10f, 10h, 10m, 13b, and 13c exhibited weak to moderate antiproliferative activity, with GI50 values between 12 and 29 μM, demonstrating at least 2.5-fold and 1.5-fold reduced potency compared to doxorubicin and sorafenib, respectively.
2.2.4 Apoptotic markers assay
Apoptosis, or anticipated cell death, encompasses various biochemical and morphological processes (Cavalcante et al., 2019). Antiapoptotic proteins, such as Bcl-2, coexist with proapoptotic proteins like Bax (Qian et al., 2022). Pro-apoptotic proteins facilitate the release of cytochrome c, whereas anti-apoptotic proteins modulate apoptosis by inhibiting cytochrome c release. The outer mitochondrial membrane becomes permeable when proapoptotic protein concentrations exceed those of antiapoptotic proteins, triggering a cascade of events. The release of cytochrome c initiates caspase-3 and caspase-9 activation. Caspase-3 causes apoptosis by targeting many critical proteins necessary for cellular function (Ponder and Boise, 2019).
Compounds 10a, 10o, and 13d, the most efficient in all in vitro experiments, were further investigated as activators of apoptotic markers.
2.2.4.1 Caspase 3/9 activation assay
Compounds 10a, 10o, and 13d were assessed as activators of caspase-3/9 in the MDAMB-231 pancreatic cancer cell line (Youssif et al., 2019), with results detailed in Table 3. The results indicated that cells treated with derivatives 10a, 10o, and 13d exhibited markedly elevated levels of caspase-3 protein (625 ± 5, 605 ± 5, and 690 ± 5 pg/mL, respectively), compared to the cells treated with standard compound staurosporine (510 ± 4 pg/mL). Compounds 10a, 10o, and 13g induced an increase of the overall amount of caspase-3 protein in the MDAMB-231 cancer cell line, reaching levels almost 10-fold greater than the untreated control cells and surpassing those of the reference compound staurosporine.
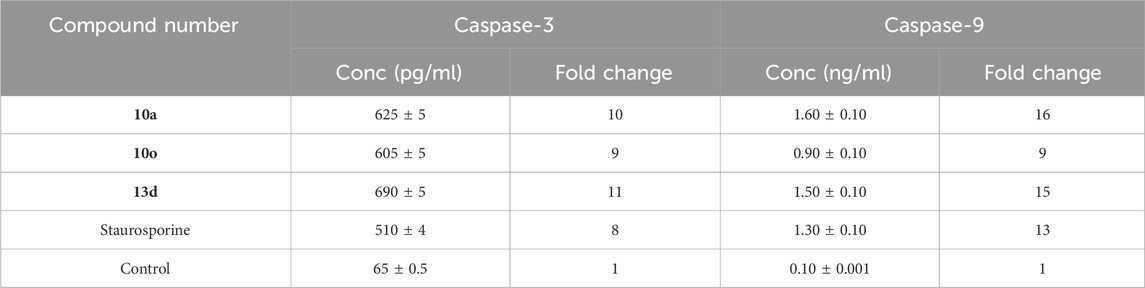
Table 3. Caspase-3/9 activation of compounds 10a, 10o, and 13d against MDAMB-231 pancreatic cancer cell line.
Additionally, caspase-9 activation testing results showed that test compounds 10a, 10o, and 13g significantly increased caspase-9 levels compared to staurosporine. Compound 10a exhibited the highest overexpression of caspase-9 (1.60 ng/mL), proceeded by compound 13d (1.50 ng/mL) and staurosporine as a control (1.30 ng/mL). Compound 10o was the least efficient derivative as caspase-9 activators (0.90 ng/mL). These data suggest that apoptosis may contribute to the antiproliferative actions of the examined compounds, possibly due to the activation of caspase-3 and 9.
2.2.4.2 Assay for levels of bax and Bcl2
We additionally investigated the impact of compounds 10a, 10o, and 13d on the apoptotic marker Bax and the anti-apoptotic Bcl2 levels in the MDAMB-231 cancer cell line, applying staurosporine as a reference (Youssif et al., 2019). The results are presented in Table 4.
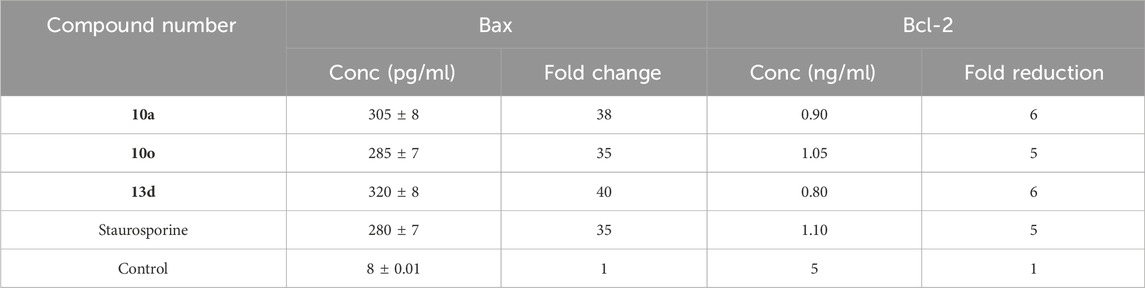
Table 4. Bax and Bcl-2 levels of 10a, 10o and 13d in pancreatic MDAMB-231 cell line.
Compounds 10a, 10o, and 13d exhibited superior Bax induction (305, 285, and 320 pg/mL, respectively) compared to staurosporine (280 pg/mL), reflecting a 38-fold, 35-fold, and 40-fold improvement relative to untreated control MDAMB-213 carcinoma cells. Ultimately, compound 13d induced a significant reduction in the anti-apoptotic Bcl-2 protein concentration (0.80 ng/mL), got by compound 10a (0.90 ng/mL) in the MDAMB-231 cell line, in contrast to staurosporine (1.10 ng/mL). These apoptosis experiments demonstrated that compounds 10a and 13d exhibited a significant apoptotic antiproliferative activity.
2.3 Computational studies
2.3.1 Molecular docking study
The most potent compounds from Scaffold A (10a) and Scaffold B (13d) were selected for molecular docking studies to explore their interactions with the colchicine binding site of tubulin. The crystal structure of tubulin (PDB code: 5LYJ) was used for docking (Wang et al., 2024; Afzal et al., 2023), and the simulations were performed using Auto-Dock Vina (Trott and Olson, 2010). Visualization and analysis of the docking results were carried out using Discovery Studio Visualizer (Podila et al., 2024; Fatima et al., 2023), enabling a detailed understanding of the molecular interactions at the target site. To validate the docking protocol and ensure its reliability for subsequent docking studies, the co-crystallized ligand combretastatin A4 (CA-4) was redocked into the colchicine binding site of tubulin. The redocking yielded a low root-mean-square deviation (RMSD) of 0.4994 Å, indicating a high degree of alignment between the redocked and the original co-crystallized ligand. Additionally, the redocked ligand demonstrated a favorable binding affinity of −6.6 kcal/mol, consistent with its known interaction profile. The superimposition of the redocked and co-crystallized CA-4 is illustrated in Figure 4.
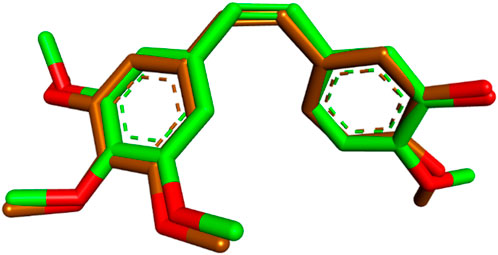
Figure 4. Superimposition of redocked (brown) and co-crystallized (green) poses of CA-4 in the colchicine binding site (RMSD = 0.4994 Å).
After validating the docking protocol with CA-4, the most potent compound in Scaffold A, compound 10a, was docked into the colchicine binding site of tubulin. Docking results showed that 10a exhibited an improved binding affinity of −7.3 kcal/mol, surpassing the native ligand CA-4. Compared with the CA-4 binding mode (Figure 5), 10a retained most of the key binding interactions demonstrated by CA-4 while introducing additional interactions that potentially account for its superior potency.
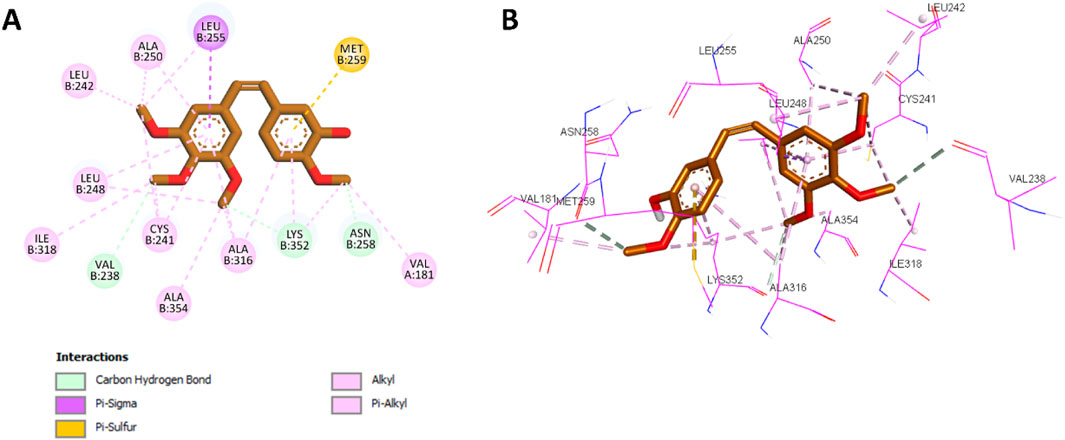
Figure 5. (A) 2D interaction diagram and (B) 3D binding mode of CA-4 within the colchicine binding site.
Docking analysis revealed that 10a was well-positioned within the hydrophobic pocket of the colchicine binding site. It preserved key hydrophobic interactions with residues Leu248, Ala354, Ala316, Cys241, Ala250, Leu255, and Leu242, which were also observed with CA-4. Furthermore, 10a formed new hydrophobic interactions with Lys352 and additional hydrogen bonds with Tyr202, absent in the CA-4 complex. The amide group of 10a further contributed a novel hydrogen bond with Val238, another interaction not observed with CA-4. The phenyl ring of the chalcone scaffold in 10a participated in hydrophobic interactions, including π-sulfur interactions with Met259 and π-alkyl interactions with Val181 and Lys352, which were also observed in the CA-4 binding profile. Additionally, the phenyl ring attached to the thiazole moiety engaged in π-sigma interactions with Leu242 (a conserved interaction with CA-4) while introducing unique contacts such as π-alkyl interactions with Leu252 and π–π T-shaped interactions with Tyr202. These distinct interactions are illustrated in Figure 6, showing the 2D and 3D interaction profiles of 10a within the colchicine binding site.
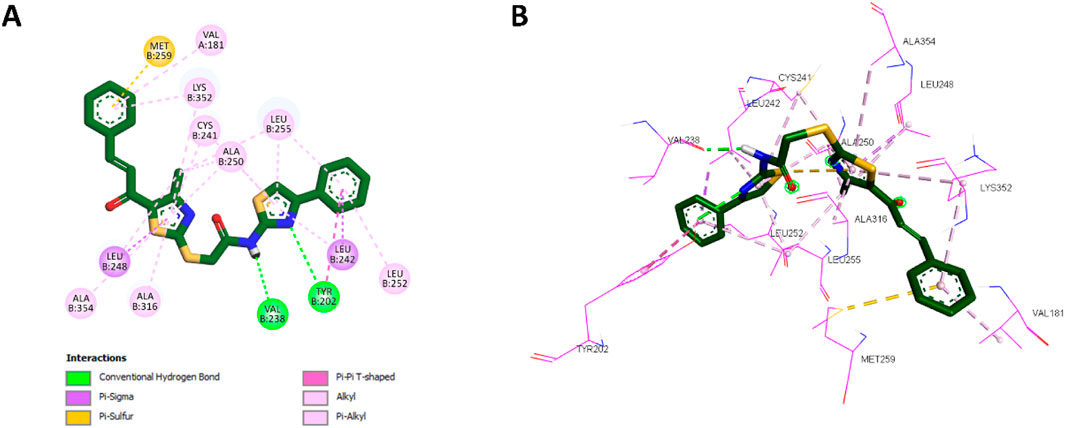
Figure 6. (A) 2D interaction diagram and (B) 3D binding mode of 10a within the colchicine binding site.
These docking results correlate strongly with in vitro findings, where 10a exhibited greater inhibitory potency than CA-4. Its higher binding affinity, retention of key interactions, and novel hydrophobic and hydrogen bonding interactions collectively explain its enhanced activity.
Following the docking analysis of 10a, the most potent compound in Scaffold B, 13d, was also docked into the colchicine binding site of tubulin. Compound 13d exhibited a binding affinity of −7.2 kcal/mol, slightly lower than 10a. Its interaction profile revealed similar hydrophobic interactions with the same amino acids as 10a, including Leu248, Ala316, Cys241, Ala250, Leu242, Met259, Val181, and Lys352. These conserved interactions indicate that 13d retains a significant portion of the binding mode observed for 10a. However, the absence of the phenyl ring attached to the thiazole moiety in 13d limited its ability to form certain hydrophobic interactions. Specifically, 13d could not establish π–π T-shaped interactions with Tyr202 or π-alkyl interactions with Leu252, key contributors to 10a’s enhanced binding and potency. This structural difference likely accounts for the slightly lower potency of 13d as a tubulin polymerization inhibitor compared to 10a. The detailed 2D and 3D interaction profiles of 13d within the colchicine binding site are presented in Figure 7.
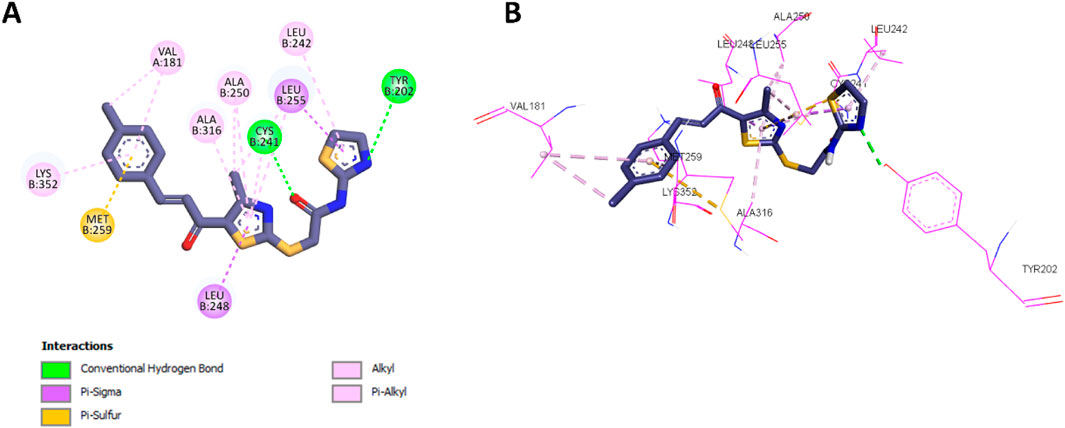
Figure 7. (A) 2D interaction diagram and (B) 3D binding mode of 13d within the colchicine binding site.
The comparative docking analysis of CA-4, 10a, and 13d underscores the importance of specific structural features in enhancing binding affinity and inhibitory potency at the colchicine binding site of tubulin. These results emphasize the critical role of molecular interactions in determining potency and offer insights for the rational design of more effective tubulin polymerization inhibitors.
2.3.2 ADME prediction
The SwissADME approach reveals various helpful features that distinguish compound 10a as a potential lead chemotherapeutic agent targeting tubulin polymerization (Daina et al., 2017; Afridi et al., 2024). The molecular weight of 477.62 g/mol, although on the higher end of the permitted range for drug-like compounds, adheres to Lipinski’s rule of five without any violations. This adherence to Lipinski’s guidelines strongly indicates the compound’s drug-like properties and potential for future development. The large topological polar surface area (TPSA) of 153.73 Å2 indicates that it may interact with tubulin’s polar areas, making it more selective for the target. Additionally, the compound’s moderate molar refractivity (133.55) supports its compatibility with biological membranes and receptor sites.
The lipophilicity profile is notably promising, including a consensus Log P of 5.10, which balances hydrophobicity and hydrophilicity. This degree of lipophilicity is advantageous for traversing cell membranes and engaging with intracellular targets such as tubulin. Higher Log P values may be associated with solubility issues. Still, they also make it simpler for the compound to concentrate in lipid-rich cancer cell membranes, potentially increasing its efficacy and selectivity. Moreover, 10a does not serve as a substrate for P-glycoprotein (P-gp), which is a notable advantage. P-glycoprotein-mediated efflux affects numerous anticancer agents, leading to drug resistance; 10a’s resistance to this mechanism enhances its potential effectiveness in cancer treatment.
The compound’s pharmacokinetic characteristics enhance its attractiveness. Despite the anticipated inhibition of CYP3A4 and CYP2C9, clinical environments may alleviate these interactions with careful dose modifications and monitoring. The lack of BBB permeability is advantageous in this situation, as it diminishes the likelihood of central nervous system side effects, enhancing its suitability for systemic anticancer uses. The low skin permeability (Log Kp = −4.79 cm/s) suggests a reduced risk of unintentional cutaneous absorption, which is advantageous for handling and formulation.
Regarding drug-likeness and medicinal chemistry, 10a is notable for its bioavailability score of 0.55, which is commendable for an orally delivered medication candidate. The bioavailability radar (Figure 8) visibly depicts the compound’s strengths. Although certain qualities, such as solubility and flexibility, are undesirable, they pose reasonable challenges in pharmaceutical development. The significant number of rotatable bonds (9) indicates that its structure is flexible, making it more likely to attach to tubulin. Also, using nanocarriers or co-crystals in the formulation process can help with its low solubility, making it easier to distribute while maintaining its natural activity.
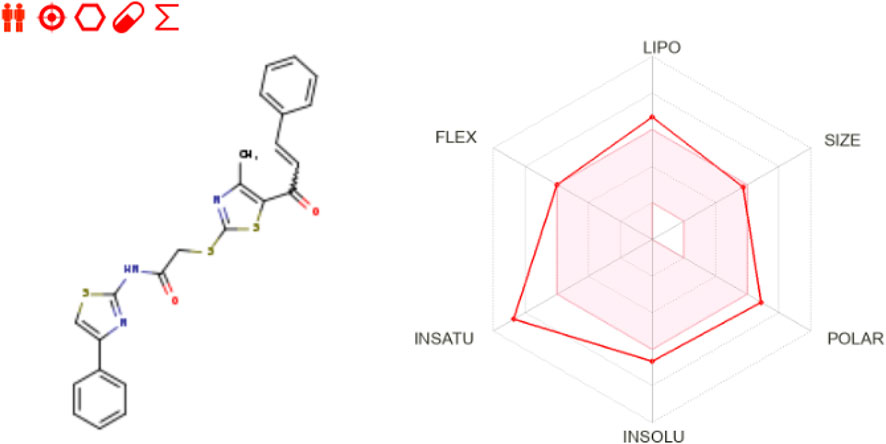
Figure 8. Bioavailability radar of 10a as retrieved from SwissADME server.
The findings for compound 10a indicate a robust basis for continued advancement as a tubulin polymerization inhibitor. Its drug-likeness, advantageous pharmacokinetics, and potential anticancer efficacy beyond tolerable constraints establish it as a highly useful lead compound for anticancer therapy.
3 Conclusion
This work describes a new series of thiazole-based compounds that inhibit tubulin polymerization, indicating their potential use in cancer therapy. All target compounds were first examined for cell viability and tubulin inhibition efficacy, and the most effective derivatives were studied as antiproliferative agents. The results show that compound 10a is the most potent and selectively active compound at both the enzyme and cellular levels with potential apoptotic effect. Molecular docking and dynamic testing revealed that compound 10a strongly binds to the colchicine binding site and interacts with enzymes in various ways. The findings of this investigation suggested that 10a could be a promising candidate for additional biological testing against various cancer cell lines. This may also lead to more research into how it works, in vivo carcinogenic animal models, and lead optimization.
4 Experimental
4.1 Chemistry
General Details: Refer to Supplementary Appendix A.
3-Chloroacetylacetone (2) (Abo-Ashour et al., 2018), 1-(2-mercapto-4-methylthiazol-5-yl)ethan-1-one (3) and (5a-e) (Hashem et al., 2024), phenacyl bromides (7a-c), 2-amino-4-aryl-thiazoles (8a-c), and (9a-c) (Valiveti et al., 2015) were prepared according to reported procedures.
A mixture of chalcone derivatives 5a-e (1 mmol), the corresponding acylated thiazole 9a-c or 12 (1 mmol), anhydrous sodium carbonate (1.5 mmol), and sodium iodide (2 mmol) in acetone was stirred at ambient temperature for 6 h. The solvent was subsequently evaporated under reduced pressure, and the residue was thoroughly washed with a 10% sodium thiosulfate solution and distilled water. The crude product underwent recrystallization from ethanol.
4.1.1 (E)-2-((5-cinnamoyl-4-methylthiazol-2-yl)thio)-N-(4-phenylthiazol-2-yl)acetamide (10a)
Yellow powder; 0.440 g, 92% yield; mp 203-207 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 7.91 (d, J = 7.2 Hz, 2H, Ar-H), 7.82–7.77 (m, 2H, Ar-H), 7.68 (d, J = 14.6 Hz, 2H, Ar & CH=CH), 7.46–7.43 (m, 5H, Ar-H), 7.38 (d, J = 15.6 Hz, 1H, CH=CH), 7.34 (t, J = 7.4 Hz, 1H, Ar-H), 4.43 (s, 2H, CH2), 2.66 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 182.01, 168.69, 166.36, 158.38, 158.33, 149.43, 144.19, 134.67, 134.62, 132.66, 131.38, 129.50, 129.30, 129.25, 128.34, 126.15, 124.79, 108.85, 37.27, 18.78; Anal. Calcd. For C24H19N3O2S3: C, 60.35%; H, 4.01%; N, 8.80%. Found: C, 60.57%; H, 4.20%; N, 8.68%.
4.1.2 (E)-2-((5-(3-(4-chlorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(4-phenylthiazol-2-yl)acetamide (10b)
Yellow powder; 0.356 g, 70% yield; mp 245-249 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.73 (s, 1H, NH), 7.91 (d, J = 7.3 Hz, 2H, Ar-H), 7.83 (d, J = 8.5 Hz, 2H, Ar-H), 7.66 (d, J = 13.4 Hz, 2H, Ar-H & CH=CH), 7.50 (d, J = 8.5 Hz, 2H, Ar-H), 7.45 (t, J = 7.7 Hz, 2H, Ar-H), 7.39 (d, J = 15.6 Hz, 1H, CH=CH), 7.34 (t, J = 7.4 Hz, 1H, Ar-H), 4.44 (s, 2H, CH2), 2.66 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 181.87, 168.76, 166.28, 158.49, 158.17, 149.45, 142.71, 135.85, 134.63, 133.59, 132.56, 130.99, 129.52, 129.20, 128.36, 126.15, 125.48, 108.91, 37.19, 18.78; Anal. Calcd. For C24H18ClN3O2S3: C, 56.30%; H, 3.54%; N, 8.21%. Found: C, 56.16%; H, 3.62%; N, 8.11%.
4.1.3 (E)-2-((5-(3-(4-fluorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(4-phenylthiazol-2-yl)acetamide (10c)
Yellow powder; 0.427 g, 86% yield; mp 211-213 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 7.94–7.86 (m, 4H, Ar-H), 7.71–7.66 (m, 2H, Ar-H & CH=CH), 7.44 (t, J = 7.7 Hz, 2H, Ar-H & CH=CH), 7.36 (s, 1H, Ar-H), 7.33 (d, J = 7.2 Hz, 1H, Ar-H), 7.29 (t, J = 8.8 Hz, 2H, Ar-H), 4.43 (s, 2H, CH2), 2.66 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 181.94, 168.64, 166.31, 158.33, 149.44, 143.00, 134.65, 132.63, 131.77 (d, J = 8.67), 131.70, 131.30 (d, J = 3.07), 129.25, 128.35, 126.15, 124.68, 116.62, 116.44 (d, J = 21.83), 108.89, 37.21, 18.77; Anal. Calcd. For C24H18FN3O2S3: C, 58.16%; H, 3.66%; N, 8.48%. Found: C, 58.37%; H, 3.88%; N, 8.34%.
4.1.4 (E)-2-((4-methyl-5-(3-(p-tolyl)acryloyl)thiazol-2-yl)thio)-N-(4-phenylthiazol-2-yl)acetamide (10d)
Yellow powder; 0.441 g, 90% yield; mp 227-230 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 7.92 (dd, J = 8.3, 1.1 Hz, 2H, Ar-H), 7.68 (d, J = 3.1 Hz, 2H), 7.65 (d, J = 17.0 Hz, 2H), 7.45 (t, J = 7.7 Hz, 2H), 7.35 (d, J = 8.5 Hz, 1H), 7.30 (d, J = 15.5 Hz, 1H), 7.25 (d, J = 8.1 Hz, 2H), 4.43 (s, 2H), 2.65 (s, 3H), 2.33 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.92, 168.41, 166.31, 158.19, 149.45, 144.30, 141.56, 134.64, 132.67, 131.87, 130.14, 130.11, 129.33, 129.25, 128.35, 126.16, 123.71, 108.90, 37.19, 21.58, 18.74; Anal. Calcd. For C25H21N3O2S3: C, 61.08%; H, 4.31%; N, 8.55%. Found: C, 60.87%; H, 4.22%; N, 8.69%.
4.1.5 (E)-2-((5-(3-(4-methoxyphenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(4-phenylthiazol-2-yl)acetamide (10e)
Yellow powder; 0.334 g, 66% yield; mp 220-223 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.73 (s, 1H, NH), 7.91 (d, J = 7.3 Hz, 2H), 7.76 (d, J = 8.7 Hz, 2H), 7.66 (d, J = 17.2 Hz, 2H), 7.44 (t, J = 7.7 Hz, 2H), 7.34 (t, J = 7.4 Hz, 1H), 7.23 (d, J = 15.4 Hz, 1H), 7.00 (d, J = 8.7 Hz, 2H), 4.42 (s, 2H), 3.81 (s, 3H), 2.65 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.86, 168.14, 166.35, 162.09, 158.29, 157.90, 149.43, 144.31, 134.66, 132.81, 131.29, 129.25, 128.35, 127.21, 126.15, 122.21, 114.99, 108.87, 55.89, 37.22, 18.72; Anal. Calcd. For C25H21N3O3S3: C, 59.15%; H, 4.17%; N, 8.28%. Found: C, 58.94%; H, 4.11%; N, 8.46%.
4.1.6 (E)-N-(4-(4-chlorophenyl)thiazol-2-yl)-2-((5-cinnamoyl-4-methylthiazol-2-yl)thio) acetamide (10f)
Yellow powder; 0.360 g, 70% yield; mp 251-253 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 7.93 (d, J = 8.5 Hz, 2H), 7.82–7.78 (m, 2H), 7.74 (s, 1H), 7.68 (d, J = 15.5 Hz, 1H), 7.51 (d, J = 8.6 Hz, 2H), 7.47–7.43 (m, 3H), 7.38 (d, J = 15.5 Hz, 1H), 4.44 (s, 2H), 2.66 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 182.00, 168.57, 166.38, 158.38, 158.30, 148.21, 144.19, 134.61, 133.50, 132.81, 132.68, 131.38, 129.49, 129.29, 129.28, 127.87, 124.76, 109.65, 37.19, 18.78; Anal. Calcd. For C24H18ClN3O2S3: C, 56.30%; H, 3.54%; N, 8.21%. Found: C, 56.25%; H, 3.72%; N, 8.12%.
4.1.7 (E)-2-((5-(3-(4-chlorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(4-(4-chlorophenyl) thiazol-2-yl)acetamide (10g)
Yellow powder; 0.467 g, 85.45% yield; mp 260-263 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.73 (s, 1H, NH), 7.92 (d, J = 8.3 Hz, 2H), 7.84 (d, J = 8.3 Hz, 2H), 7.74 (s, 1H), 7.67 (d, J = 15.5 Hz, 1H), 7.51 (d, J = 8.3 Hz, 4H), 7.39 (d, J = 15.6 Hz, 1H), 4.43 (s, 2H), 2.65 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.90, 168.77, 166.39, 158.49, 148.20, 143.42, 142.72, 135.85, 133.60, 133.52, 132.80, 132.55, 131.01, 129.52, 129.28, 127.87, 125.51, 109.63, 37.23, 18.78; Anal. Calcd. For C24H17Cl2N3O2S3: C, 52.75%; H, 3.14%; N, 7.69%. Found: C, 52.86%; H, 3.20%; N, 7.60%.
4.1.8 (E)-N-(4-(4-chlorophenyl)thiazol-2-yl)-2-((5-(3-(4-fluorophenyl)acryloyl)-4-methyl thiazol-2-yl)thio)acetamide (10h)
Yellow powder; 0.482 g, 90.93% yield; mp 217-219 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 7.93 (d, J = 8.6 Hz, 2H), 7.89 (dd, J = 8.7, 5.6 Hz, 2H), 7.74 (s, 1H), 7.68 (d, J = 15.6 Hz, 1H), 7.51 (d, J = 8.6 Hz, 2H), 7.34 (d, J = 15.6 Hz, 1H), 7.29 (t, J = 8.8 Hz, 2H), 4.43 (s, 2H), 2.65 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.94, 168.61, 166.40, 165.01, 163.03, 158.32, 148.20, 143.00, 133.52, 132.80, 132.63, 131.70 (d, J = 8.68), 131.32 (d, J = 2.94), 129.28, 127.87, 124.66, 116.44 (d, J = 21.86), 109.63, 37.22, 18.77; Anal. Calcd. For C24H17ClFN3O2S3: C, 54.38%; H, 3.23%; N, 7.93%. Found: C, 54.31%; H, 3.11%; N, 7.98%.
4.1.9 (E)-N-(4-(4-chlorophenyl)thiazol-2-yl)-2-((4-methyl-5-(3-(p-tolyl)acryloyl)thiazol-2-yl)thio)acetamide (10i)
Yellow powder; 0.445 g, 84.59% yield; mp 240-241 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 7.93 (d, J = 8.6 Hz, 2H), 7.71 (d, J = 18.6 Hz, 2H), 7.68–7.62 (m, 2H), 7.50 (d, J = 8.6 Hz, 2H), 7.31 (d, J = 15.5 Hz, 1H), 7.25 (d, J = 8.0 Hz, 2H), 4.42 (s, 2H), 2.65 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.93, 168.51, 166.49, 158.21, 148.17, 144.30, 141.56, 133.58, 132.83, 132.76, 132.62, 131.88, 130.12, 129.34, 129.27, 127.87, 123.73, 109.55, 37.35, 21.58, 18.74; Anal. Calcd. For C25H20ClN3O2S3: C, 57.08%; H, 3.83%; N, 7.99%. Found: C, 57.30%; H, 3.67%; N, 7.88%.
4.1.10 (E)-N-(4-(4-chlorophenyl)thiazol-2-yl)-2-((5-(3-(4-methoxyphenyl)acryloyl)-4-methylthiazol-2-yl)thio)acetamide (10j)
Yellow powder; 0.361 g, 67% yield; mp 223-226 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H, NH), 7.93 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.72 (s, 1H), 7.65 (d, J = 15.4 Hz, 1H), 7.50 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 15.4 Hz, 1H), 7.00 (d, J = 8.8 Hz, 2H), 4.41 (s, 2H), 3.81 (s, 3H), 2.65 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.85, 168.23, 166.53, 162.09, 157.91, 148.15, 144.30, 133.59, 132.77, 132.74, 131.33, 131.30, 129.27, 127.86, 127.21, 122.22, 114.99, 109.53, 55.89, 37.37, 18.72; Anal. Calcd. For C25H20ClN3O3S3: C, 55.39%; H, 3.72%; N, 7.75%. Found: C, 55.59%; H, 3.8%; N, 7.53%.
4.1.11 (E)-2-((5-cinnamoyl-4-methylthiazol-2-yl)thio)-N-(4-(p-tolyl)thiazol-2-yl)acetamide (10k)
Yellow powder; 0.364 g, 74% yield; mp 255-257 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H, NH), 7.80 (d, J = 7.8 Hz, 4H), 7.68 (d, J = 15.5 Hz, 1H), 7.59 (s, 1H), 7.47–7.44 (m, 3H), 7.38 (d, J = 15.6 Hz, 1H), 7.25 (d, J = 8.0 Hz, 2H), 4.43 (s, 2H), 2.66 (s, 3H), 2.33 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 182.01, 168.59, 166.21, 158.31, 157.97, 149.55, 144.19, 137.69, 134.60, 132.69, 131.97, 131.38, 129.81, 129.50, 129.29, 126.10, 124.76, 108.04, 37.14, 21.29, 18.78; Anal. Calcd. For C25H21N3O2S3: C, 61.08%; H, 4.31%; N, 8.55%. Found: C, 60.96%; H, 4.18%; N, 8.72%.
4.1.12 (E)-2-((5-(3-(4-chlorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(4-(p-tolyl)thiazol-2-yl)acetamide (10l)
Pale yellow crystal; 0.358 g, 68% yield; mp 229-232 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H, NH), 7.83 (d, J = 8.6 Hz, 2H), 7.80 (d, J = 8.1 Hz, 2H), 7.67 (d, J = 15.6 Hz, 1H), 7.60 (s, 1H), 7.50 (d, J = 8.5 Hz, 2H), 7.39 (d, J = 15.6 Hz, 1H), 7.25 (d, J = 8.0 Hz, 2H), 4.42 (s, 2H), 2.66 (s, 3H), 2.33 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.90, 170.25, 166.21, 165.49, 158.50, 151.75, 142.73, 137.70, 137.45, 135.85, 133.60, 132.56, 131.01, 129.82, 129.53, 126.10, 125.51, 108.04, 37.42, 21.29, 18.78; Anal. Calcd. For C25H20ClN3O2S3: C, 57.08%; H, 3.83%; N, 7.99%. Found: C, 57.27%; H, 4.05%; N, 7.91%.
4.1.13 (E)-2-((5-(3-(4-fluorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(4-(p-tolyl)thiazol-2-yl)acetamide (10m)
Yellow powder; 0.399 g, 78% yield; mp 237-238 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H, NH), 7.89 (dd, J = 8.6, 5.7 Hz, 2H), 7.80 (d, J = 8.1 Hz, 2H), 7.68 (d, J = 15.5 Hz, 1H), 7.59 (s, 1H), 7.34 (d, J = 15.5 Hz, 1H), 7.29 (t, J = 8.8 Hz, 2H), 7.25 (d, J = 8.2 Hz, 2H), 4.42 (s, 2H), 2.66 (s, 3H), 2.33 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.94, 168.64, 166.24, 165.01, 163.03, 158.34, 149.52, 143.00, 137.67, 132.62, 132.00, 131.76 (d, J = 8.71), 131.30 (d, J = 3.11), 129.80, 126.10, 124.66, 116.44 (d, J = 21.83), 108.00, 37.21, 21.28, 18.77; Anal. Calcd. For C25H20FN3O2S3: C, 58.92%; H, 3.96%; N, 8.25%. Found: C, 7.91%; H, 3.79%; N, 8.06%.
4.1.14 (E)-2-((4-methyl-5-(3-(p-tolyl)acryloyl)thiazol-2-yl)thio)-N-(4-(p-tolyl)thiazol-2-yl)acetamide (10n)
Yellow powder; 0.420 g, 83% yield; mp 248-250 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.68 (s, 1H, NH), 7.80 (d, J = 8.1 Hz, 2H), 7.68 (d, J = 8.1 Hz, 2H), 7.65 (d, J = 15.6 Hz, 1H), 7.59 (s, 1H), 7.31 (d, J = 15.5 Hz, 1H), 7.25 (d, J = 7.9 Hz, 4H), 4.42 (s, 2H), 2.65 (s, 3H), 2.33 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 181.95, 168.45, 166.25, 158.20, 144.32, 141.57, 137.67, 132.66, 132.01, 131.88, 130.13, 129.81, 129.34, 126.36, 126.30, 126.11, 123.73, 108.02, 37.19, 21.58, 21.29, 18.74; Anal. Calcd. For C26H23N3O2S3: C, 61.76%; H, 4.58%; N, 8.31%. Found: C, 61.61%; H, 4.63%; N, 8.10%.
4.1.15 (E)-2-((5-(3-(4-methoxyphenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(4-(p-tolyl) thiazol-2-yl)acetamide (10o)
Yellow powder; 0.378 g, 72% yield; mp 240-242 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H, NH), 7.80 (d, J = 8.1 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.65 (d, J = 15.4 Hz, 1H), 7.59 (s, 1H), 7.26 (s, 1H), 7.22 (d, J = 15.6 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 4.42 (s, 2H), 3.81 (s, 3H), 2.65 (s, 3H), 2.33 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.86, 168.11, 166.25, 162.09, 158.04, 157.90, 149.54, 144.31, 137.68, 132.82, 131.99, 131.29, 129.81, 127.20, 126.10, 122.21, 114.99, 108.03, 55.88, 37.16, 21.29, 18.71; Anal. Calcd. For C26H23N3O3S3: C, 59.86%; H, 4.44%; N, 8.06%. Found: C, 59.64%; H, 4.50%; N, 8.16%.
4.1.16 (E)-2-((5-cinnamoyl-4-methylthiazol-2-yl)thio)-N-(thiazol-2-yl)acetamide (13a)
Yellow powder; 0.272 g, 67% yield; mp 215-219 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.55 (s, 1H, NH), 7.80 (dd, J = 6.7, 2.9 Hz, 2H), 7.68 (d, J = 15.5 Hz, 1H), 7.51 (d, J = 3.6 Hz, 1H), 7.46 (dd, J = 5.0, 1.7 Hz, 3H), 7.37 (d, J = 15.5 Hz, 1H), 7.26 (d, J = 3.6 Hz, 1H), 4.41 (s, 2H), 2.66 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.99, 168.62, 165.97, 158.30, 158.26, 144.19, 138.26, 134.61, 132.65, 131.39, 129.50, 129.30, 124.77, 114.38, 37.17, 18.76; Anal. Calcd. For C18H15N3O2S3: C, 53.85%; H, 3.77%; N, 10.47%. Found: C, 53.68%; H, 3.56%; N, 10.67%.
4.1.17 (E)-2-((5-(3-(4-chlorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(thiazol-2-yl)acetamide (13b)
Yellow powder; 0.335 g, 77% yield; mp 221-224 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.55 (s, 1H, NH), 7.85 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 15.6 Hz, 1H), 7.52 (d, J = 8.5 Hz, 2H), 7.51 (d, J = 3.6 Hz, 1H), 7.39 (d, J = 15.6 Hz, 1H), 7.26 (d, J = 3.6 Hz, 1H), 4.41 (s, 2H), 2.65 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.89, 168.77, 165.95, 158.46, 158.24, 142.72, 138.27, 135.85, 133.60, 132.57, 131.02, 129.53, 125.48, 114.39, 37.16, 18.78; Anal. Calcd. For C18H14ClN3O2S3: C, 49.59%; H, 3.24%; N, 9.64%. Found: C, 49.77%; H, 3.09%; N, 9.73%.
4.1.18 (E)-2-((5-(3-(4-fluorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(thiazol-2-yl)acetamide (13c)
Yellow powder; 0.344 g, 82% yield; mp 218-222 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H, NH), 7.89 (dd, J = 8.7, 5.6 Hz, 2H), 7.68 (d, J = 15.5 Hz, 1H), 7.51 (d, J = 3.6 Hz, 1H), 7.36–7.28 (m, 3H), 7.26 (d, J = 3.6 Hz, 1H), 4.40 (s, 2H), 2.65 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.94, 168.64, 165.97, 165.02, 163.04, 158.31, 143.00, 138.26, 132.62, 131.71 (d, J = 8.72), 131.30 (d, J = 3.13), 124.67, 116.63 (d, J = 21.80), 114.37, 37.18, 18.76; Anal. Calcd. For C18H14FN3O2S3: C, 51.54%; H, 3.36%; N, 10.02%. Found: C, 51.37%; H, 3.30%; N, 10.18%.
4.1.19 (E)-2-((4-methyl-5-(3-(p-tolyl)acryloyl)thiazol-2-yl)thio)-N-(thiazol-2-yl)acetamide (13d)
Yellow powder; 0.356 g, 86% yield; mp 212-215 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H, NH), 7.69 (d, J = 8.1 Hz, 2H), 7.65 (d, J = 15.5 Hz, 1H), 7.51 (d, J = 3.5 Hz, 1H), 7.31 (d, J = 15.5 Hz, 1H), 7.28 (s, 1H), 7.26 (d, J = 3.6 Hz, 2H), 4.40 (s, 2H), 2.65 (s, 3H), 2.35 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.95, 168.42, 165.98, 158.27, 158.14, 144.32, 141.59, 138.26, 132.70, 131.89, 130.13, 129.36, 123.70, 114.37, 37.17, 21.59, 18.73; Anal. Calcd. For C19H17N3O2S3: C, 54.92%; H, 4.12%; N, 10.11%. Found: C, 55.06%; H, 4.01%; N, 10.04%.
4.1.20 (E)-2-((5-(3-(4-methoxyphenyl)acryloyl)-4-methylthiazol-2-yl)thio)-N-(thiazol-2-yl)acetamide (13e)
Yellow powder; 0.332 g, 77% yield; mp 225-227 ̊C; 1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H, NH), 7.77 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 15.4 Hz, 1H), 7.50 (d, J = 3.5 Hz, 1H), 7.26 (d, J = 3.6 Hz, 1H), 7.23 (d, J = 15.4 Hz, 1H), 7.02 (d, J = 8.7 Hz, 2H), 4.40 (s, 2H), 3.82 (s, 3H), 2.64 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 181.86, 168.17, 166.03, 162.10, 158.40, 157.87, 144.31, 138.25, 132.81, 131.31, 127.21, 122.19, 115.01, 114.33, 55.90, 37.22, 18.70; Anal. Calcd. For C19H17N3O3S3: C, 52.88%; H, 3.97%; N, 9.74%. Found: C, 52.75%; H, 3.89%; N, 9.96%.
4.2 Biology
4.2.1 Cell viability assay
The effects of compounds 10a-o and 13a-e on cell viability were assessed using the human mammary gland epithelial normal cell line (MCF-10A). The MTT assay assessed the cell viability of compounds 10a-o and 13a-e following a four-day incubation with MCF-10A cells (El-Sherief et al., 2019; Ramadan et al., 2020). Check Supplementary Appendix A for additional information.
4.2.2 Tubulin polymerization inhibition assay
Compounds 10a-o and 13a-e effects on tubulin polymerization were investigated using the Tubulin Polymerization Assay Kit (Cytoskeleton Inc., Denver, CO, USA) according to the supplier’s instructions (Abdelbaset et al., 2021; Abdelbaset et al., 2018). Details are presented in Supplementary Appendix A.
4.2.3 Antiproliferative assay
The antiproliferative efficacy of novel compounds 10a, 10d, 10f, 10h, 10m, 10o (Scaffold A) and 13b-d (Scaffold B) against four human cancer cell lines (colon - HCT-116, prostatic - PC-3, breast -MCF-7, and pancreatic-MDAMB-231) was evaluated utilizing the MTT test (Mahmoud et al., 2022; Hisham et al., 2022; Al-Wahaibi et al., 2023). Doxorubicin and Sorafenib were used as controls in this study.
4.2.4 Apoptotic markers assays
Compounds 10a, 10o, and 13d were assessed for their ability to activate caspase-3, caspase-9, and Bax, as well as to downregulate the anti-apoptotic protein Bcl2 in the MDAMB-231 pancreatic cancer cell line (Youssif et al., 2019). Supplementary Appendix A provides more information.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
LA-W: Funding acquisition, Project administration, Writing – review and editing. AE: Data curation, Formal Analysis, Methodology, Software, Writing – original draft, Writing – review and editing. TA: Conceptualization, Supervision, Writing – original draft. BY: Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review and editing. SB: Formal Analysis, Writing – original draft, Writing – review and editing. MA-A: Conceptualization, Formal Analysis, Investigation, Resources, Supervision, Visualization, Writing – original draft, Writing – review and editing. NE-K: Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2025R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Acknowledgments
The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2025R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1565699/full#supplementary-material
References
Abdelbaset, M. S., Abdelrahman, M. H., Bukhari, S. N. A., Gouda, A. M., Youssif, B. G., Abdel-Aziz, M., et al. (2021). Design, synthesis, and biological evaluation of new series of pyrrol-2 (3H)-one and pyridazin-3 (2H)-one derivatives as tubulin polymerization inhibitors. Bioorg. Chem. 107, 104522. doi:10.1016/j.bioorg.2020.104522
Abdelbaset, M. S., Abuo-Rahma, G.E.-D. A., Abdelrahman, M. H., Ramadan, M., Youssif, B. G., Bukhari, S. N. A., et al. (2018). Novel pyrrol-2 (3H)-ones and pyridazin-3 (2H)-ones carrying quinoline scaffold as anti-proliferative tubulin polymerization inhibitors. Bioorg. Chem. 80, 151–163. doi:10.1016/j.bioorg.2018.06.003
Abo-Ashour, M. F., Eldehna, W. M., George, R. F., Abdel-Aziz, M. M., Elaasser, M. M., Gawad, N. M. A., et al. (2018). Novel indole-thiazolidinone conjugates: design, synthesis and whole-cell phenotypic evaluation as a novel class of antimicrobial agents. Eur. J. Med. Chem. 160, 49–60. doi:10.1016/j.ejmech.2018.10.008
Afridi, M. B., Sardar, H., Serdaroğlu, G., Shah, S. W. A., Alsharif, K. F., and Khan, H. (2024). SwissADME studies and Density Functional Theory (DFT) approaches of methyl substituted curcumin derivatives. Comput. Biol. Chem. 112, 108153. doi:10.1016/j.compbiolchem.2024.108153
Afzal, O., Ali, A., Ali, A., Altamimi, A. S. A., Alossaimi, M. A., Bakht, M. A., et al. (2023). Synthesis and anticancer evaluation of 4-Chloro-2-((5-aryl-1, 3, 4-oxadiazol-2-yl) amino) phenol analogues: an insight into experimental and theoretical studies. Molecules 28 (16), 6086. doi:10.3390/molecules28166086
Al-Wahaibi, L. H., Mahmoud, M. A., Mostafa, Y. A., Raslan, A. E., and Youssif, B. G. (2023). Novel piperine-carboximidamide hybrids: design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzyme Inhibition Med. Chem. 38 (1), 376–386. doi:10.1080/14756366.2022.2151593
Anand, U., Dey, A., Chandel, A. K. S., Sanyal, R., Mishra, A., Pandey, D. K., et al. (2023). Cancer chemotherapy and beyond: current status, drug candidates, associated risks and progress in targeted therapeutics. Genes & Dis. 10 (4), 1367–1401. doi:10.1016/j.gendis.2022.02.007
Cavalcante, G. C., Schaan, A. P., Cabral, G. F., Santana-da-Silva, M. N., Pinto, P., Vidal, A. F., et al. (2019). A cell’s fate: an overview of the molecular biology and genetics of apoptosis. Int. J. Mol. Sci. 20 (17), 4133. doi:10.3390/ijms20174133
Chu, J. J., and Mehrzad, R. (2023). The biology of cancer, the link between obesity and cancer. Elsevier, 35–45.
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7 (1), 42717. doi:10.1038/srep42717
El-Abd, A. O., Bayomi, S. M., El-Damasy, A. K., Mansour, B., Abdel-Aziz, N. I., and El-Sherbeny, M. A. (2022). Synthesis and molecular docking study of new thiazole derivatives as potential tubulin polymerization inhibitors. ACS omega 7 (37), 33599–33613. doi:10.1021/acsomega.2c05077
El-Sherief, H. A., Youssif, B. G., Abdelazeem, A. H., Abdel-Aziz, M., and Abdel-Rahman, H. M. (2019). Design, synthesis and antiproliferative evaluation of novel 1, 2, 4-triazole/schiff base hybrids with EGFR and B-RAF inhibitory activities. Anti-Cancer Agents Med. Chem. Former. Curr. Med. Chemistry-Anti-Cancer Agents 19 (5), 697–706. doi:10.2174/1871520619666181224115346
El-Sherief, H. A., Youssif, B. G., Bukhari, S. N. A., Abdelazeem, A. H., Abdel-Aziz, M., and Abdel-Rahman, H. M. (2018). Synthesis, anticancer activity and molecular modeling studies of 1, 2, 4-triazole derivatives as EGFR inhibitors. Eur. J. Med. Chem. 156, 774–789. doi:10.1016/j.ejmech.2018.07.024
Fatima, S., Zaki, A., Madhav, H., Khatoon, B. S., Rahman, A., Manhas, M. W., et al. (2023). Design, synthesis, and biological evaluation of morpholinopyrimidine derivatives as anti-inflammatory agents. RSC Adv. 13 (28), 19119–19129. doi:10.1039/d3ra01893h
Franchetti, P., Cappellacci, L., Grifantini, M., Barzi, A., Nocentini, G., Yang, O, H., et al. (1995). Furanfurin and thiophenfurin: two novel tiazofurin analogs. Synthesis, structure, antitumor activity, and interactions with inosine monophosphate dehydrogenase. Med. Chem. 38, 3829–3837. doi:10.1021/jm00019a013
Gaspari, R., Prota, A. E., Bargsten, K., Cavalli, A., and Steinmetz, M. O. (2017). Structural basis of cis-and trans-combretastatin binding to tubulin. Chem 2 (1), 102–113. doi:10.1016/j.chempr.2016.12.005
Gregory, K., Zhao, L., Felder, T. M., Clay-Gilmour, A., Eberth, J. M., Murphy, E. A., et al. (2024). Prevalence of health behaviors among cancer survivors in the United States. J. Cancer Surviv. 18 (3), 1042–1050. doi:10.1007/s11764-023-01347-8
Gudimchuk, N. B., and McIntosh, J. R. (2021). Regulation of microtubule dynamics, mechanics and function through the growing tip. Nat. Rev. Mol. cell Biol. 22 (12), 777–795. doi:10.1038/s41580-021-00399-x
Hagar, F. F., Abbas, S. H., Abdelhamid, D., Gomaa, H. A., Youssif, B. G., and Abdel-Aziz, M. (2023). New 1, 3, 4-oxadiazole-chalcone/benzimidazole hybrids as potent antiproliferative agents. Arch. Pharm. 356 (2), 2200357. doi:10.1002/ardp.202200357
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Hashem, H., Hassan, A., Abdelmagid, W. M., Habib, A. G., Abdel-Aal, M. A., Elshamsy, A. M., et al. (2024). Synthesis of new thiazole-privileged chalcones as tubulin polymerization inhibitors with potential anticancer activities. Pharmaceuticals 17 (9), 1154. doi:10.3390/ph17091154
Hecht, S. M. (2000). Bleomycin: new perspectives on the mechanism of action. J. Nat. Prod. 63 (1), 158–168. doi:10.1021/np990549f
Hisham, M., Hassan, H. A., Gomaa, H. A., Youssif, B. G., Hayallah, A. M., and Abdel-Aziz, M. (2022). Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as EGFR inhibitors. J. Mol. Struct. 1254, 132422. doi:10.1016/j.molstruc.2022.132422
Juric, D., Janku, F., Rodón, J., Burris, H. A., Mayer, I. A., Schuler, M., et al. (2019). Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor–positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol. 5 (2), e184475. doi:10.1001/jamaoncol.2018.4475
Kassem, A. F., Althomali, R. H., Anwar, M. M., and El-Sofany, W. I. (2024). Thiazole moiety: a promising scaffold for anticancer drug discovery. J. Mol. Struct. 1303, 137510. doi:10.1016/j.molstruc.2024.137510
Lee, J., Kim, S. J., Choi, H., Kim, Y. H., Lim, I. T., Yang, H.-m., et al. (2010). Identification of CKD-516: a potent tubulin polymerization inhibitor with marked antitumor activity against murine and human solid tumors. J. Med. Chem. 53 (17), 6337–6354. doi:10.1021/jm1002414
Logan, C. M., and Menko, A. S. (2019). Microtubules: evolving roles and critical cellular interactions. Exp. Biol. Med. 244 (15), 1240–1254. doi:10.1177/1535370219867296
Madhurya, M. S., Thakur, V., Dastari, S., and Shankaraiah, N. (2024). Pyrrolo [2, 3-d] pyrimidines as potential kinase inhibitors in cancer drug discovery: a critical review. Bioorg. Chem. 153, 107867. doi:10.1016/j.bioorg.2024.107867
Mahmoud, M. A., Mohammed, A. F., Salem, O. I., Gomaa, H. A., and Youssif, B. G. (2022). New 1, 3, 4-oxadiazoles linked with the 1, 2, 3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch. Pharm. 355 (6), 2200009. doi:10.1002/ardp.202200009
Plouvier, B., Houssin, R., Bailly, C., and Hénichart, J. P. (1989). Synthesis and DNA-binding study of A thiazole-containing analog of netropsin. J. Heterocycl. Chem. 26 (6), 1643–1647. doi:10.1002/jhet.5570260625
Podila, N., Penddinti, N. K., Rudrapal, M., Rakshit, G., Konidala, S. K., Pulusu, V. S., et al. (2024). Design, synthesis, biological and computational screening of novel pyridine-based thiadiazole derivatives as prospective anti-inflammatory agents. Heliyon 10 (8), e29390. doi:10.1016/j.heliyon.2024.e29390
Ponder, K. G., and Boise, L. H. (2019). The prodomain of caspase-3 regulates its own removal and caspase activation. Cell death Discov. 5 (1), 56. doi:10.1038/s41420-019-0142-1
Qian, S., Wei, Z., Yang, W., Huang, J., Yang, Y., and Wang, J. (2022). The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 12, 985363. doi:10.3389/fonc.2022.985363
Ramadan, M., Abd El-Aziz, M., Elshaier, Y. A., Youssif, B. G., Brown, A. B., Fathy, H. M., et al. (2020). Design and synthesis of new pyranoquinolinone heteroannulated to triazolopyrimidine of potential apoptotic antiproliferative activity. Bioorg. Chem. 105, 104392. doi:10.1016/j.bioorg.2020.104392
Rana, R., Kumar, N., Gulati, H. K., Sharma, A., Khanna, A., Badhwar, R., et al. (2023). A comprehensive review on thiazole based conjugates as anti-cancer agents. J. Mol. Struct. 1292, 136194. doi:10.1016/j.molstruc.2023.136194
Sabry, M. A., Ghaly, M. A., Maarouf, A. R., and El-Subbagh, H. I. (2022). New thiazole-based derivatives as EGFR/HER2 and DHFR inhibitors: synthesis, molecular modeling simulations and anticancer activity. Eur. J. Med. Chem. 241, 114661. doi:10.1016/j.ejmech.2022.114661
Saha, S. K., Gordan, J. D., Kleinstiver, B. P., Vu, P., Najem, M. S., Yeo, J.-C., et al. (2016). Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. 6 (7), 727–739. doi:10.1158/2159-8290.cd-15-1442
Sahil, , Kaur, K., and Jaitak, V. (2022). Thiazole and related heterocyclic systems as anticancer agents: a review on synthetic strategies, mechanisms of action and SAR Studies. Curr. Med. Chem. 29 (29), 4958–5009. doi:10.2174/0929867329666220318100019
Seddigi, Z. S., Malik, M. S., Saraswati, A. P., Ahmed, S. A., Babalghith, A. O., Lamfon, H. A., et al. (2017). Recent advances in combretastatin based derivatives and prodrugs as antimitotic agents. MedChemComm 8 (8), 1592–1603. doi:10.1039/c7md00227k
Seiler, A., Chen, M. A., Brown, R. L., and Fagundes, C. P. (2018). Obesity, dietary factors, nutrition, and breast cancer risk. Curr. breast cancer Rep. 10, 14–27. doi:10.1007/s12609-018-0264-0
Sun, D., Li, H., Cao, M., He, S., Lei, L., Peng, J., et al. (2020). Cancer burden in China: trends, risk factors and prevention. Cancer Biol. & Med. 17 (4), 879–895. doi:10.20892/j.issn.2095-3941.2020.0387
Sun, M., Xu, Q., Xu, J., Wu, Y., Wang, Y., Zuo, D., et al. (2017). Synthesis and bioevaluation of N, 4-diaryl-1, 3-thiazole-2-amines as tubulin inhibitors with potent antiproliferative activity. PLoS One 12 (3), e0174006. doi:10.1371/journal.pone.0174006
Tilekar, K., Upadhyay, N., Hess, J. D., Macias, L. H., Mrowka, P., Aguilera, R. J., et al. (2020). Structure guided design and synthesis of furyl thiazolidinedione derivatives as inhibitors of GLUT 1 and GLUT 4, and evaluation of their anti-leukemic potential. Eur. J. Med. Chem. 202, 112603. doi:10.1016/j.ejmech.2020.112603
Tricot, G., Jayaram, H. N., Weber, G., and Hoffman, R. (1990). Tiazofurin: biological effects and clinical uses. Int. J. Cell Cloning 8 (3), 161–170. doi:10.1002/stem.5530080303
Tron, G. C., Pirali, T., Sorba, G., Pagliai, F., Busacca, S., and Genazzani, A. A. (2006). Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 49 (11), 3033–3044. doi:10.1021/jm0512903
Trott, O., and Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31 (2), 455–461. doi:10.1002/jcc.21334
Valiveti, A. K., Bhalerao, U. M., Acharya, J., Karade, H. N., Acharya, B. N., Raviraju, G., et al. (2015). Synthesis and in vitro kinetic evaluation of N-thiazolylacetamido monoquaternary pyridinium oximes as reactivators of sarin, O-ethylsarin and VX inhibited human acetylcholinesterase (hAChE). Bioorg. & Med. Chem. 23 (15), 4899–4910. doi:10.1016/j.bmc.2015.05.027
Vicente, J. J., and Wordeman, L. (2015). Mitosis, microtubule dynamics and the evolution of kinesins. Exp. cell Res. 334 (1), 61–69. doi:10.1016/j.yexcr.2015.02.010
Wang, B., Hao, S., Han, F., Wu, T., Jia, S., Ruan, X., et al. (2025). Discovery of 5-Phenylthiazol-2-amine derivatives as novel PI4KIIIβ inhibitors with efficacious antitumor activity by inhibiting the PI3K/AKT Axis. J. Med. Chem. 68, 6270–6291. doi:10.1021/acs.jmedchem.4c02588
Wang, C., Zhang, Y., Yang, S., Shi, L., Xiu, Y., Wu, Y., et al. (2024). 3-aryl-4-(3, 4, 5-trimethoxyphenyl) pyridines inhibit tubulin polymerisation and act as anticancer agents. J. Enzyme Inhibition Med. Chem. 39 (1), 2286939. doi:10.1080/14756366.2023.2286939
Keywords: tubulin, colchicine, CA-4, antiproliferative, cell viability, docking
Citation: Al-Wahaibi LH, Elshamsy AM, Ali TFS, Youssif BGM, Bräse S, Abdel-Aziz M and El-Koussi NA (2025) Design, synthesis, in silico studies, and apoptotic antiproliferative activity of novel thiazole-2-acetamide derivatives as tubulin polymerization inhibitors. Front. Chem. 13:1565699. doi: 10.3389/fchem.2025.1565699
Received: 23 January 2025; Accepted: 01 April 2025;
Published: 16 April 2025.
Edited by:
Geoff Wells, University College London, United KingdomCopyright © 2025 Al-Wahaibi, Elshamsy, Ali, Youssif, Bräse, Abdel-Aziz and El-Koussi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamed Abdel-Aziz, YWJ1bG5pbEBob3RtYWlsLmNvbQ==; Bahaa G. M. Youssif, Ymd5b3Vzc2lmMkBnbWFpbC5jb20=; Stefan Bräse, YnJhZXNlQGtpdC5lZHU=