 Justice Akwensi
Justice Akwensi Robert T. Kumah
Robert T. Kumah Dorcas Osei-Safo
Dorcas Osei-Safo Richard K. Amewu
Richard K. Amewu- 1Department of Chemistry, School of Physical and Mathematical Sciences, College of Basic and Applied Sciences, University of Ghana, Legon, Ghana
- 2Department of Chemical and Petrochemical Engineering, School of Petroleum Studies, University of Mines and Technology, Tarkwa, Ghana
The Hosomi–Sakurai allylation reaction has been widely applied in the total synthesis of biologically active natural products, especially in synthesising complex polycyclic compounds containing multi-stereogenic centres since its discovery in 1976. The Hosomi-Sakurai allylation is the allylation of ketones and aldehyde with nucleophilic allylsilanes catalyzed with Lewis acid mainly used to extend the C-C bond in a molecule and also create a new site for manipulation due to the facile transformation of the pi (π) bond at the end of its chain. This review highlights only portions of natural product synthetic works that feature the Hosomi-Sakurai allylation reaction or its modification as a key transformation in the synthetic route.
1 Introduction
1.1 Catalytic allylation reactions
Homoallylic alcohols are a significant class of compounds with extensive applications in the synthesis of biologically active substances. Their unique structural properties make them valuable in various chemical processes, contributing to advancements in medicinal chemistry (Farina et al., 2006; Li et al., 2021; Pellissier, 2016; Shen et al., 2013). Homoallylic alcohols are products of catalytic allylation reactions that employ the use of allylic reagents under mild reaction conditions. Allylation reactions are particularly advantageous due to their use of cost-effective, nontoxic, and stable nucleophiles such as allyltrialkylsilanes, allyltrialkoxysilanes among others. Although significant advancements have been made in allylation reactions, only a limited number of catalytic asymmetric allylation reactions have been reported. This is particularly interesting due to the importance of chiral tertiary homoallylic alcohols in organic synthesis. (Fleming et al., 1989; 1997; Langkopf and Schinzer, 1995; Lee, 2020).
Allylation of carbonyl compounds is one of the most used methods of forming C-C bonds in the most versatile manner (Lee, 2020). In 1964, Mikhailov and co-workers reported the first reaction between an allyl-metal and a carbonyl catalysed by triallylborane (Stepanyan et al., 2001). A number of allyl-(semi)-metals such as Si, Mn, Sn, Sb, Mg, Bi, Al, B, Cr, Li, Zn, Ti, Zr and In have been applied in carbonyl allylation in the total synthesis of simple and complex compounds (Bols and Skrydstrup, 1995; Denmark and Fu, 2003; Dilman and Ioffe, 2003; Fleming et al., 1989; 1997; Langkopf and Schinzer, 1995; Lee, 2020; Masse and Panek, 1995; Stankevich and Cook, 2023; Yamamoto and Asao, 1993). Allylation of carbonyl compounds to produce corresponding alcohols and enols is a well-established synthetic method that employs allylic reagents in the presence of Lewis acids catalysts to form C-C bonds and alcohol derivatives in high-yields (Yamamoto and Asao, 1993). Several other allylation reactions including the Tsuji-Trost allylic reaction, Keck radical allylation, Hosomi-Sakurai allylation reactions among others have been reported in the past 2 decades. For example, the Tsuji-Trost allylic reaction is a versatile and high-yielding reaction in this category. The reaction involves the use of Pd (0) to catalyze the allylation of several nucleophilic species, including methylenes, enolates, amines, and phenols (Li, 1999; Song et al., 2007). These nucleophiles are combined with allylic compounds, such as allyl acetates and allyl bromides, (Supplementary Scheme S1a) (Hegedus et al., 1978; Keck and Yates, 1982; Trost and Van Vranken, 1996; Tsuji, 1969). Tsuji-Trost allylic reaction is applicable in the total synthesis of indole alkaloids such as Desethylibogamine, Isoquinuclidine and (±)-Catharanthine (Trost, 2015; Trost et al., 1978; Tsuji, 1986a; 1986b; Tsuji and Takahashi, 1965). Keck radical allylation is also frequently used in C-C bond formation (Supplementary Scheme S1b) (Aiqin et al., 2011; Keck and Yates, 1982). Keck radical allylation proceeds through the generation of radicals from alkyl bromides, chlorides, phenyl-selenides, and thioacylimidazoles in the presence of a radical initiator such as azobisisobutyronitrile (AIBN) and benzoyl peroxide (BPO) (Sumino et al., 2018). The application of Keck allylation is limited by the challenges associated with the purification of the corresponding products and the inherent toxic nature of organotin reagents used in the reaction (Supplementary Scheme S1b) (Fent, 1996).
The Hosomi-Sakurai allylation reaction (HSR) is the type of allylation reaction that efficiently creates C-C bonds in the total synthesis of optically active natural products (Scheme 1) (Majetich et al., 1990). HSR involves the allylation of carbonyl compounds using allylic reagents in the presence of Lewis acid catalysts to give the corresponding alcohols in good yields. The HSR usually gives quantitative yields and hence produces fewer by-products. The substituents of the allylsilane are useful in controlling the stereochemistry of the products although the reaction is not centered on the silicon atom in most cases except for chiral allysilanes which usually yield the chiral product (Chan and Wang, 1992; Kira et al., 1990).
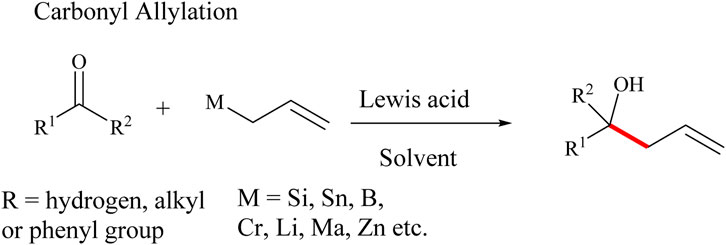
Scheme 1. A typical example of an HSR (allylation of carbonyl compounds) reaction catalyzed by Lewis acid to give the corresponding alcohols.
HSR is also applicable in the synthesis of polymers by repetitively allylating dialdehydes with bisallylsilanes to give a hydroxyl and olefinic polymer with repeating units (Supplementary Scheme S4) (Itsuno, 2005). A typical example is the preparation of Si-phenyl linkage polymers using HSR in the presence of TBAF/THF overnight (Supplementary Scheme S4). However, low molecular weight monomer units of homoallylic alcohols with high enantiomeric purity were reported (Kumagai and Itsuno, 2000).
Bauer and co-workers discovered a reductive HSR of acetals (Bauer and Maulide, 2018) through the 1,5-hydride transfer transformations (Scheme 2a). The reaction exploits in situ generation of homoallylic ether after the HSR with the acetal substrate. Subsequent protonation to generate a carbocation at the γ-position to the ether group leads to an intramolecular 1,5-hydride transfer rearrangement to form an oxocarbenium ion. The addition of water leads to the formation of the corresponding alcohol product. The reaction mechanism yields a similar product to a direct Grignard/Alkyl-Li reagent addition to an aldehyde (Scheme 2b). Air- and water-sensitive Grignard reagents bearing functional groups such as α,β-unsaturated esters, and alkyl bromides are tolerated under these reaction conditions (Bauer and Maulide, 2018). However, the reaction is not applicable when carbonyl substrates bearing aromatic groups, unprotected alcohols, and thioethers are used, as illustrated in Supplementary Scheme S2. In addition, the homo-ether intermediate formed in the HSR could subsequently yield the desired homoallylic products with phenyl-allylsilane if the amount of catalyst was reduced to 2.7 mol%, (dr = 4.5:1) (Bauer and Maulide, 2018). It is interesting to note that this reaction was not only chemoselective but also stereoselective when pro-chiral carbonyl substrates were used (Scheme 2).
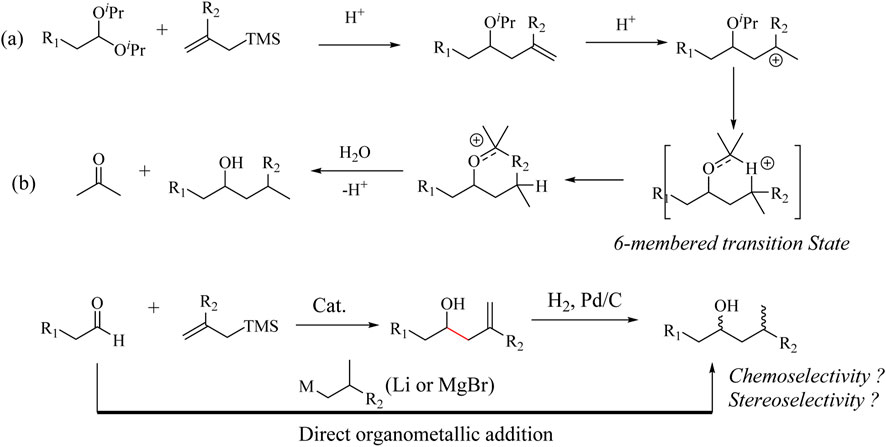
Scheme 2. Reductive Hosomi-Sakurai allylation reaction (a) Intramolecular 1,5-hydride transfer rearrangement and (b) direct Grignard reaction.
1.2 History of Hosomi-Sakurai allylation reactions
The HSR, commonly referred to as the Sakurai allylation or Sakurai reaction, was discovered in the 1970s as an alternative to the classical allylation method of C-C bond formation and has been extensively studied over the years (Bauer and Maulide, 2018). The HSR of carbonyl compounds and their derivatives with the allyl trialkylsilane as the allylating agent in the presence of a Lewis acid as a catalyst, proceeds rapidly even at an extremely low temperature of −78°C (Hosomi and Sakurai, 1976). The H-SR is applicable in the allylation of aliphatic, alicyclic, and aromatic carbonyl compounds using allylmethylsilane and TiCl4 as a catalyst. Allylsilanes are reported to be weakly nucleophilic hence the Lewis acid catalyst is used to activate and increase the reactivity of the carbon electrophile in the solution. This results in a C-C bond formation that occurs only at the γ-position to the silicon moiety (Yamamoto and Sasaki, 1989).
Allylsilanes are very useful in organic synthesis because they are easy to handle, can be used at room temperatures, and do not require special storage conditions. Their reactions are usually smooth (homogeneous) with different electrophiles under Lewis acid conditions. On the contrary, other allylating agents such as allyl-magnesium halides, -Li, -Cu, and -Ti compounds require specific temperatures and moisture-free working atmosphere and reaction conditions (Yamamoto and Sasaki, 1989). The major advantage of the HSR is that allylsilanes are readily available inexpensive starting materials. They are stable, have low toxicity, compatible with multiple functional groups, and chemically inert to atmospheric conditions (Fleming, 1991; Hosomi, 1988; Yamamoto and Asao, 1993). However, the reaction depends on the presence of Lewis acids such as TiCl4, BF3⋅OEt2, SnCl4, and EtAlCl2 as activators because allylsilanes do not readily react with non-activated electrophiles (Lade et al., 2017).
1.3 Mechanism, scope and features
A typical reaction pathway of the HSR is illustrated in Scheme 3. An oxocarbenium ion is first generated from the reaction of the carbonyl compound with the Lewis acid which makes the carbonyl carbon more electrophilic. It begins with a nucleophilic attack of the γ-carbon of the allylsilane, 1 on the carbonyl to generate a carbocation which is stabilized by the silyl group (Schweig et al., 1974). The α-Si-C bond at this stage is weak and can be easily cleaved to neutralize the carbocation, hence forming the alkene. Halide from the catalyst facilitates the facile cleavage to form the corresponding intermediate (Ponomarenko & Mironov, 1954). The reaction is usually completed by the addition of H2O to give the desired allyl-alcohol products (Scheme 3). The propylene unit created through a reaction between an aldehyde and allylic reagents is very useful in the synthesis of optically active molecules. One of the key features of silyl groups is the ability to stabilize charges on their immediate. Specifically, this includes stabilizing geminal anions (α-effect) and vicinal cations (β-effect). The α-effect is often observed through the metalation of silanes in the α-position using strong bases (Britten et al., 2021). However, the stabilizing influence of the silyl group is significantly more pronounced for the β-silicon effect compared to the α-effect (Roberts and McLaughlin, 2022).
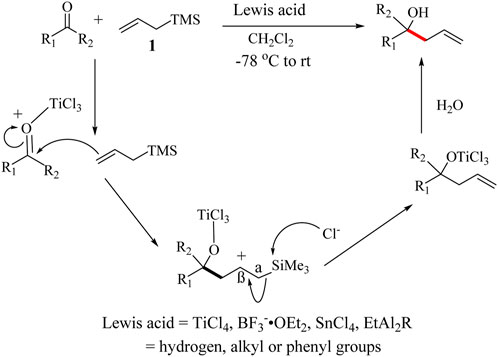
Scheme 3. General reaction and mechanism of the HSR (Kira et al., 1990).
Under Lewis acid conditions, it was observed that the reaction proceeds through a less open cyclic transition state, and more asymmetric products could be achieved (Kira et al., 1990). The HSR is regioselective due to the formation of a stable silylcarbocation intermediate (Hosomi et al., 1978a). In 1978, tetra-n-butylammonium fluoride (TBAF) was introduced as an alternative catalyst to Lewis acids. TBAF reacts with allylsilanes to give a stable Si-F intermediate which attacks carbonyl leading to the formation of silyl ethers (Hosomi et al., 1978b). TBAF is also effective in catalysing allylation of synthetic intermediates such as acetals, aldimines/imines/iminium ions (Shingo et al., 2002), aldehydes, acids chlorides (Yadav et al., 2003), epoxides (Yamasaki et al., 2002), ketals, keto-imines, ketones, oxocarbenium ions (Hosomi et al., 1978a) and α,β-unsaturated carbonyls (Lade et al., 2017; Roberts and McLaughlin, 2022). Allylation of α,β-unsaturated carbonyl compounds, however, generates the ketone-enol tautomers instead of the alcohol which continues the allylation cycle as illustrated in Supplementary Scheme S2 (Fürstner and Voigtländer, 2000). A number of Lewis acids have been reported as pre-catalysts in HSR in the past 3–5 decades. Lewis acid reagents such as SnCl4, AlCl3, BF3⋅OEt2, Me3SiOTf, Me3SiI, and Me3O+BF4− or their combinations have also been used (Birch et al., 1981; Colvin, 1978; Hosomi et al., 1984; Nishiyama et al., 1982; Noyori et al., 1981; Yotsu-Yamashita et al., 1995).
Despite the numerous applications, the use of Lewis acid catalysts in HSR is limited by the inability to recover and reuse them in the subsequent reaction cycles (Supplementary Scheme S3). In addition, Lewis acids are susceptible to degenerate to allylic metal oxides and are therefore added in stoichiometric amounts to ensure the completion of the reaction (Mahlau and List, 2013). The HSR typically yields syn-addition products, making it highly diastereoselective in the allylation of carbonyl compounds. The Newman projection (Scheme 4) of the allylation of aldehyde 3 with (2-bromoallyl) trimethylsilane 4 to form 5, a key intermediate in the synthesis of amphidinol 3, (2) demonstrates the diastereospecificity of the HSR. In the Newman projection, the intermediate coordinated to the titanium tetrachloride with the two oxygen atoms tethered together, fixing the conformation of the substrate. This is known as the Cram chelation control mechanism (Cram and Elhafez, 1952; Stanton et al., 2010). The (2-bromoallyl) trimethylsilane nucleophile then approaches along the Bürgi-Dunitz angle, but only one side of the approach is possible as the other is hindered by the R groups. This ensures the formation of only one diastereomer in high yields. Amphidinol 3 (2) is a potent antifungal compound produced by the dinoflagellate Amphidinium klebsii (Grisin and Evans, 2015; Huckins et al., 2007; Oishi, 2024; Wakamiya et al., 2020).
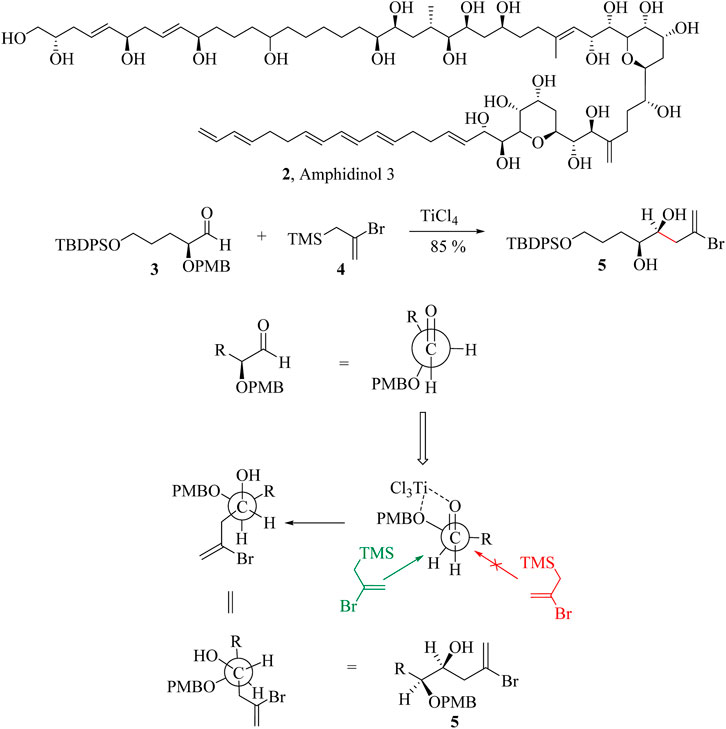
Scheme 4. An illustration of the stereospecificity of HSR.
1.4 Drawbacks of the HSR
The HSR typically proceeds through an open-chain transition state mechanism, which inherently limits the scope of the method. Since carbonyl compounds are not highly reactive, there is a need to increase their reactivity by using strong Lewis acids as catalysts or by activating the allylsilane, especially those with electron-withdrawing groups (Mahlau and List, 2013). However, this approach is not favourable for late-stage functionalization, as it can be intolerant of functional groups and lead to loss of regioselectivity in some cases, resulting in low yields (Chemler and Roush, 2000). Efforts to increase the electrophilicity of carbonyl compounds often lead to a loss of regioselectivity when the allylsilane is activated by a fluoride source (Denmark and Fu, 2003). While the open anti-periplanar transition state leads to the formation of the syn-product in most cases, it also impedes the enantioselectivity of the mechanism (Denmark and Almstead, 1994).
1.5 Modifications to the HSR
Attempts to address the challenges of HSR have led to the discovery and use of a chiral base catalyst (S44), transition metal fluoride (S45), chiral Lewis acids (S47) Supplementary Figure S1, and tartrate-modified allylsilanes to improve the selectivity and versatility of the HSR (Denmark and Fu, 2001; Gauthier and Carreira, 1996; Ishihara et al., 1993; Malkov et al., 2002; 2008; Schäfers et al., 2022; Zhang et al., 1997). Due to the inherent challenges associated with the HSR, Schäfers et al. developed a dual catalytic system that uses less basic allyl chromium instead of allylsilanes (Schäfers et al., 2022). This reaction proceeds through a ring-closed Zimmerman−Traxler transition state, initiated using the sterically hindered acridinium photocatalyst and blue light-emitting diode (LED) irradiation (Supplementary Scheme S13). These reaction conditions often yield undesirable homoallylic products with different chemo- and diastereoselectivity. Mechanistic analysis showed that the reaction proceeds through a cyclic mechanism, leading to the formation of the anti-product.
This alternative approach using allyl chromium and photocatalytic activation represents a valuable strategy to overcome the inherent limitations of the HSR, providing access to complementary stereochemical outcomes (Schäfers et al., 2022). The HSR has seen numerous modifications to its reaction conditions since its discovery. For example, new chiral catalysts were introduced with allyltrialkylsilanes, allyltrialkoxysilanes, or allyl trichlorosilane to attain enantiomerically pure allylated products (Kaib et al., 2016; Kampen et al., 2008; Lee, 2020; Sai and Yamamoto, 2015; Schreyer et al., 2019).
The allylation of ketones to generate homoallylic alcohols through the HSR has been a challenge and has proven unsuccessful over the years. Despite these challenges, several asymmetric allylations of ketone have been reported (Niwa et al., 2019; Pellissier, 2006; Tietze et al., 1995; 2004; Wadamoto et al., 2003). The most efficient one is the use of fluoroallylsilane derivatives and Pd-based catalysts instead of the usual Lewis acids (Hayashi et al., 2010). Bismuth bromide has also been reported as a useful catalyst in the allylation of carbonyls by Komatsu and co-workers. Allylation with bismuth bromide is compatible with trimethylsilyl, trifluoromethane sulfonate (TMSOTf) (Noyori et al., 1981), and trimethylsilyl iodide (TMSI) (Komatsu et al., 1997).
Alternatively, Sakurai-Hosomi-Yamamoto allylation has been an effective approach for the stereoselective synthesis of a number of chiral homoallylic alcohols in high yields by employing Chiral (R)- and (S)-BINAP.AgF catalysts (Supplementary Scheme S8) (Makita et al., 2003; Mirabdolbaghi and Dudding, 2012). Although the reaction mechanism for the asymmetric (R)- or (S)-BINAP.AgF Sakurai-Hosomi-Yamamoto allylation reaction remains unknown, Mirabdolbaghi et al., proposed a transition state (TS1) that rationalized the enantioselectivity of the mechanism as shown in Supplementary Scheme S8 (Mirabdolbaghi and Dudding, 2012).
Yamamoto and his coworkers further disclosed the synthesis of the γ- and anti-products upon using AgF and a chiral bisphosphine catalyst such as (R)-DIFLUOROPHOS (Nokami et al., 2003; Wadamoto et al., 2003). This reaction is also assumed to progress through the cyclic transition state that is generated in situ from the transmetalation of allylsilanes to AgF (Supplementary Scheme S5). This modified Ag-catalyzed asymmetric allylation reaction is amenable to ketone as well as aldehydes (Komiyama et al., 2017; Wadamoto et al., 2003).
The aza-Hosomi-Sakurai (aHS) reaction, developed by Veenstra and Schmid (1997), is an additional variation of the HSR. This method allows for the one-step synthesis of homoallylamines which involves reacting an allylsilane with an aldehyde, a carbamate, or a sulfonamide, in the presence of a Lewis acid catalyst (Scheme 5). This reaction mechanism permits the synthesis of N-protected homoallylamines, which can be utilised to make N-heteroatomic rings in a few steps (Ella-Menye et al., 2005) leading to the synthesis of pyrrolidine and piperidine rings and eventually (+)-Allo-sedamine (9) (Girard, 2015).
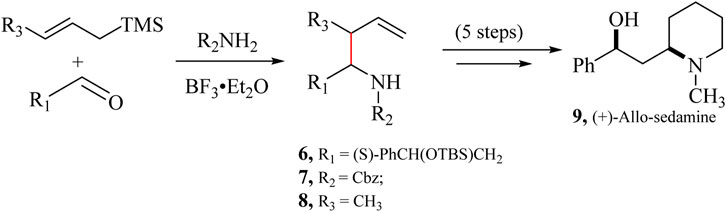
Scheme 5. Modified Aza-Hosomi-Sakurai (aHS) allylation reaction for synthesizing (+)-Allo-sedamine (9) (Girard, 2015).
Onishi et al. (2002), developed a highly effective Lewis acid by combining InCl3 with Me3SiCl. The hybrid catalytic system is effective for HSR and Friedel-Crafts allylation and hydrosilylation reactions. Hexafluoroisopropanol (HFIP) has also proven to be an efficient source of a hydrogen bond donor and as a catalyst in the form HFIP–DCM (4:1 (v/v)) solution (Motiwala et al., 2022). To form the desired targeted allylated aldehyde and acetal products product, HFIP (2–5 eq.) was required (Supplementary Scheme S6) (Berkessel et al., 2006; Motiwala et al., 2022). HSR of hydroxy-aldehydes with allytrifluorosilanes was discovered to be stereospecific in the presence of triethylamine and chiral Lewis acid catalysts (Kira et al., 1990). Asymmetric Hosomi-Sakurai reactions (AHSR) could be effectively achieved with ee up to 92% using chiral Lewis (Denmark et al., 1994; Iseki et al., 1999; Wang et al., 1999).
Furthermore, a new trityl tetrakis (pentafluophenyl) borate [(Ph3C)[BPh(F)4] has been discovered as a catalyst to mediate HSR of β,γ-unsaturated α-ketoesters to yield γ,γ-disubstituted α-ketoesters in excellent yields using allylsilane reagent (Chien et al., 1991; Hollis and Bosnich, 1995; Kosugi et al., 1985; Mukaiyama et al., 1984). HSR of crotyl geminal bis(silane) with aldehydes (Supplementary Scheme S7) for instance can be carried out more efficiently by using the (Ph3C)[BPh(F)4] catalyst (Chu et al., 2017; Santos and Silva, 2018).
2 Applications of HSR in total synthesis of biologically active natural products
A plethora of synthetic methods have been studied and advanced in the synthesis of complex compounds including natural products over the past decades. Among them is HSR of carbonyl intermediates which provides a straightforward and versatile method for preparing these compounds. The HSR is characterised by the formation of a new carbon-carbon bond with high functional group compatibility. The stereoselectivity of the reaction is largely regulated by substituents on substrates under mild reaction conditions. The use of allylsilanes makes this allylation a useful tool for synthesizing complex natural products. Recently, Lee (2020) reported the application of Hosomi-Sakurai in the total synthesis of a number of natural products. In this review, we will focus only on selected examples of total synthesis of biologically active natural products that have utilized HSR as a key transformation method in the past 2 decades and were not included in Lee work.
2.1 Total synthesis of aburatubolactam A
Aburatubolactam A (10) is a natural product isolated from the culture broth of marine mollusc bacterium Streptomyces sp., SCRC-A20 in Japan (Bai et al., 2016; Henderson et al., 2008). Uemura and co-workers determined the complete structure and absolute stereochemistry of the natural product using X-ray crystallographic analysis (Bae et al., 1996). Biological screening of Aburatubolactam A (10) exhibited cytotoxicity properties against human cancer cells by inhibiting cell proliferation, anti-microbial activity, and the inhibition of superoxide generation (Israr et al., 2024). Aburatubolactam A (10) has been observed to be an inhibitor of protein kinase C in both rat brain tissue samples and rat liver cell membrane preparations (Israr et al., 2024). Additionally, Aburatubolactam A (10) has been found to suppress the growth of Mycobacterium tuberculosis bacteria by disrupting the synthesis of DNA, RNA, and proteins within the bacterial cells (Henderson et al., 2008).
A total of 23 steps were required to synthesise Aburatubolactam A (10) starting from commercially available ketone 11, followed by five steps to form bicyclo [3.3.0] octene 12 with a 90% yield (Scheme 6) (Henderson et al., 2008). The fluoride-mediated Sakurai allylation using TBAF in a solvent mixture of dimethylformamide and N,N′-Dimethylpropyleneurea (DMF-DMPU) resulted in the formation of intermediate 13 (Majetich et al., 1986). This intermediate was obtained as a 4:1 mixture of inseparable diastereomers, achieving a yield of 78%. This ratio was then improved in favour of the desired isomer to 2:1 by the protonation of the silylketene acetyl derived from 13. The resulting product was taken through 11 other sequential steps to achieve Aburatubolactam A (14) in 46% total yield (Bai et al., 2016; Henderson et al., 2008) (Scheme 6).
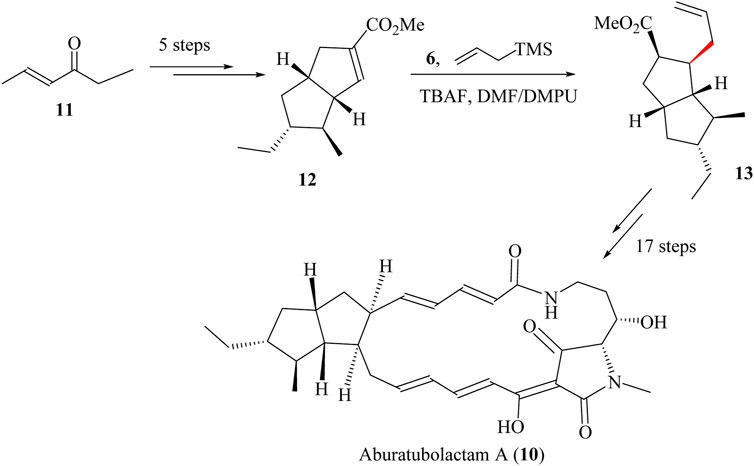
Scheme 6. Total synthesis of Aburatubolactam A (10) (Bai et al., 2016).
2.2 Total syntheses of (−)-acutumine
(−)-Acutumine 14, is an alkaloid isolated first from the roots of Sinomenium acutum in 1929 by Goto and co-workers (Li et al., 2009). It has also been discovered in Hypserpa nitida and Apis cerana. The family of (−)-Acutumine has structural similarities to the Hasubanan family of alkaloids such as (−)-Dechloroatunine (15), and (−)-Hasubanonine (16). (−)-Acutumine (14) inhibits the proliferation of human T-cells and memory-enhancing properties in the Wistar rat model (Yu et al., 2002). The structure complexity with embedded chirality in the spirocyclopentenone rings 1 and 2 of (−)-Acutumines poses difficulty in its total synthesis (King et al., 2013).
In 2009, Castle and co-workers reported the total synthesis of (−)-Acutumine (14) (Li et al., 2009). In the build-up to the synthesis of (−)-Acutumine spirocyclopentenone ring derived from acetonide 17 was prepared in 5 steps from the starting D-ribose, 18 (Scheme 7) (Kim et al., 2020; Smith et al., 2005). Palladium was employed to catalyze the converting of 1,4-disilylation, (Ogoshi et al., 2002), and cleavage of the enoxysilane generated the β-dimethylsilyl ketone 19 as a single diastereomer which was taken through seven linear steps to obtain intermediate 20. HSR was then carried out on 20 using tetrabutylammonium fluoride (TBAF) to induce an intramolecular cyclisation to obtain the key tetracycle 21 as a single diastereomer in 37% yield. The tetracycle 21 was taken through eight other linear steps to yield (−)-Acutumine, 14 (King et al., 2013).
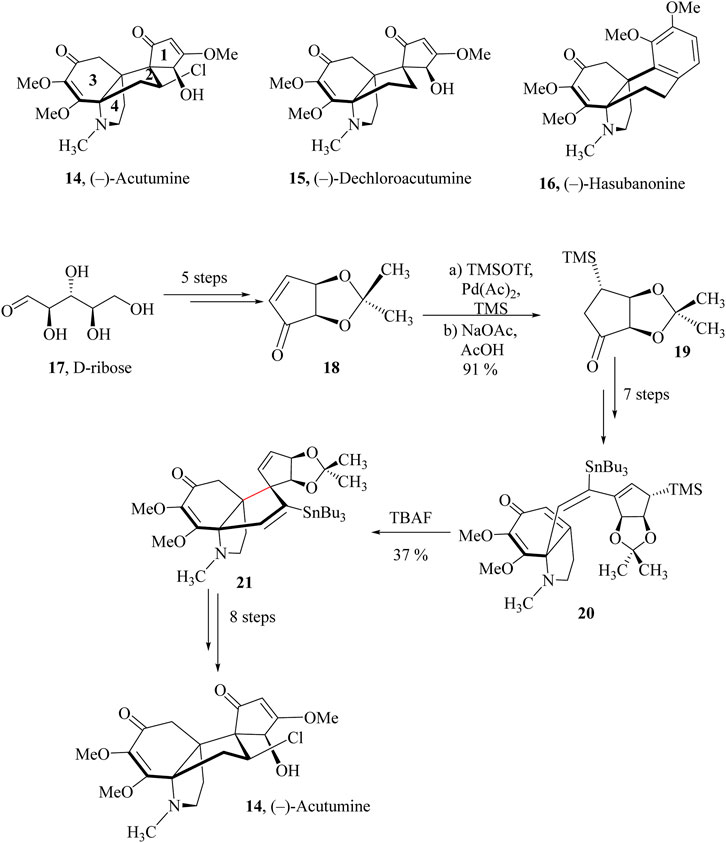
Scheme 7. Total synthesis of (−)-Acutumine, 45 (King et al., 2013).
2.3 Total synthesis of alotaketals and phorbaketals
Alotaketals and Phorbaketals are sesterterpenoid natural products that have been discovered in extracts of marine sponge species (Hubert et al., 2015; Joung et al., 2017; Shirley et al., 2018; Yao et al., 2020). Alotaketal A (22) (Scheme 8) was first isolated from a marine sponge Hamigera sp in Milne Bay, Papua New Guinea by Kieffer and Andersen in 2009 (Forestieri et al., 2009). To date, five Alotaketals (A- E) have been isolated from different other species. Alotaketals share the same tricyclic spiroketal backbone with Phorbaketals (A- N) which have had fourteen members isolated and structurally characterised using NMR spectroscopic techniques. Despite their structural similarity, they have different biological activity profiles. Alotaketal A (22) was found to increase protein kinase activity and also activate the cyclic adenosine monophosphate (cAMP) cell (cAMP is a derivative of adenosine triphosphate, i.e., ATP) signalling pathway in human embryonic kidney cells (HEK293T) (Beavo and Brunton, 2002; Forestieri et al., 2009). Phorbaketal (23) on the other hand is cytotoxic against human colorectal, hepatoma, and lung cancer cell lines (Daoust et al., 2013; Lee et al., 2014; Wang et al., 2016; Yao et al., 2020).
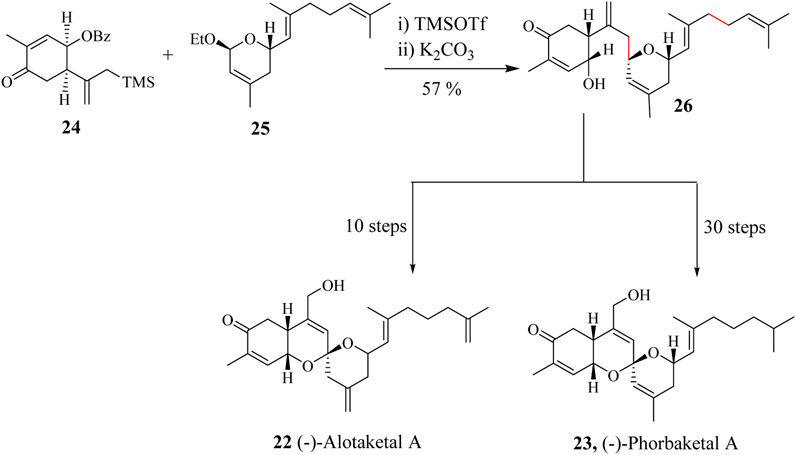
Scheme 8. Total synthesis of (−)-Alotaketal A (22) and (−)-Phorbaketal A (23) involving HSR protocol (Yao et al., 2020).
Yao et al., used TMSOTf as Lewis acid in catalysing the reaction between moiety 24 and 25 through an HSR to prepare a single diastereomer 26 in 75% yield (Scheme 8) (Yao et al., 2020). Yang completed the synthesis of (−)-Alotaketal A (22) in 30 steps with an overall yield of 2.6%. Alotaketals and Phorbaketal, i.e., Spiroketals of this nature have been very challenging to synthesise (Yao et al., 2020). Lee et al., accomplished the synthesis of (−)-Phorbaketal A (23) in 10 steps with a yield of 1.04% being the shortest linear sequence steps (Lee et al., 2014).
2.4 Synthesis of (+)-Amphidinolide P
Amphidinolides are bioactive natural products isolated from marine dinoflagellates. Kobayashi and co-workers isolated the first amphidinolide A in 1986 (Ishibashi and Kobayashi, 1997; Kobayashi et al., 1986) and other amphidinolides including amphidinolides P 27 from a cultured diniflagellate Amphidinium sp. Amphidinolides are characterized by their unique 26-membered macrolide structure have shown antineoplastic activity against cancer cell lines and possess IC50 values in the low micromolar range (Heravi and Mohammadkhani, 2018).
Total synthesis of (−)-amphidinolide P, 27 was reported for the first time in 2000 by Williams and co-workers (Williams and Meyer, 2001). (+)-Amphidinolide P 27 was also prepared from a mixture of chiral precursors 29 and 30 starting from commercially available 28 (Williams et al., 2013). Aldehyde allylation with allylsilane 31 to achieve key intermediate 32 was achieved by adopting the HSR using boron trifluoride etherate as a catalyst Scheme 9 (Carreira and Joliton, 2015; Williams et al., 2013; Williams and Meyer, 2001). Traeatment of intermediate 62 through 7 steps reactions resulted in the formation of the Amphidinolides P (27).
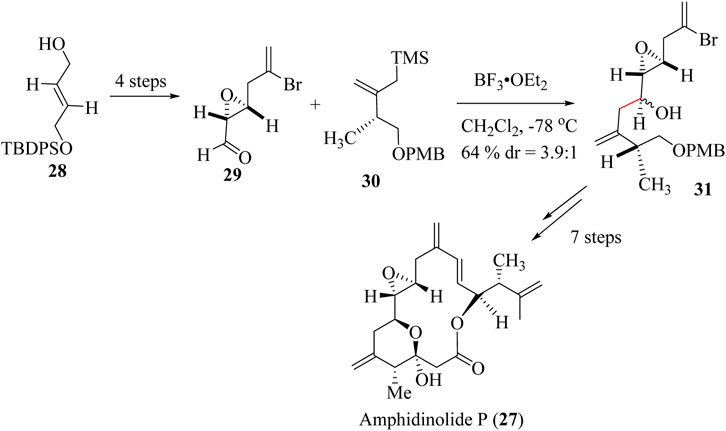
Scheme 9. Total synthesis of Amphidinolides P (27) (Carreira and Joliton, 2015).
2.5 Total synthesis of (±)-Aspidospermidine
(±)-Aspidospermidine (32) belongs to the Aspidosperma family of alkaloids (Du et al., 2017) which are made up of a pentacyclic ring system and five chiral centres. Members of this family of alkaloids possess antibiotic, anticancer, antimalarial and anti-inflammatory activities. However, the structural complexity of this class of compounds presents a challenge to successful asymmetric synthesis (Du et al., 2017; Ramirez and Garcia-Rubio, 2005).
The key intermediate in the synthetic route of (−)-Aspidospermidine 32 was first prepared by Stork and Dolfini (Stork and Dolfini, 1963). Sabot and co-workers used an alternative approach known as the “aromatic ring umpolung” which starts with polysubstituted phenol 33 which was converted to dienone 34 through HSR using allyltrimethylsilane and trifluoroethanol (TFE) or Hexafluoroisopropanol (HFIP) in the presence of PhI(OAc)2 (Scheme 10). The entire total synthesis was completed in 10 steps starting from phenol 33 to give the overall yield of 5.4% (Sabot et al., 2009).
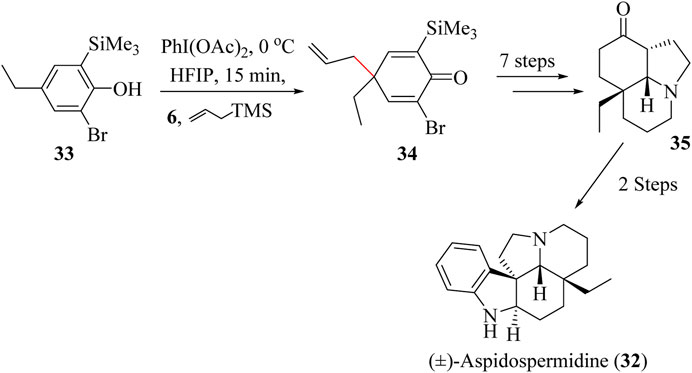
Scheme 10. Total synthesis of (±)-Aspidospermidine (32) (Stork and Dolfini, 1963).
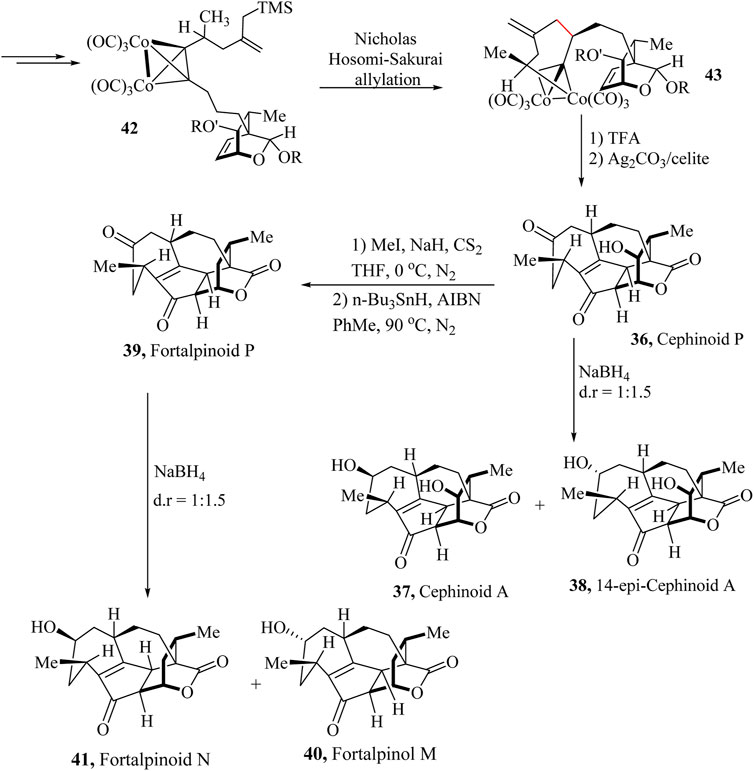
Scheme 11. Total asymmetric total synthesis of Cephalotaxus diterpenoids: Cephinoid P, Cephafortoid A, 14-epi-cephafortoid A and Fortalpinoids M, N, (Wang et al., 2023).
2.6 Total synthesis of Cephalotaxus diterpenoids: Cephinoid P, Cephafortoid A, 14-epi-Cephafortoid A and Fortalpinoids M-N, P
Cephalotaxus diterpenoids are structurally diverse diterpenoids. In 2016, Yue and co-workers isolated this family of compounds from Cephalotaxus species (Fan et al., 2017; Shao et al., 2024). This family of natural products showed potential anticancer properties. The intriguing and challenging structure of these compounds was established using X-ray crystallography techniques. Gao and coworkers have recently reported the first asymmetric total synthesis of these natural products and subgroups such as Cephinoid P (36), Cephafortoid A (37), 14-Epi-cephafortoid A (38) and Fortalpinoids M (39), N (40) and P (41) from the precursor (S)-trimethyl (4-methyl-2-methylenehex-5-yn-1-yl) silane (Wang et al., 2023).
A universal strategy for the total synthesis of Cephalotaxus diterpenoids bearing unique cycloheptene A rings with chiral methyl groups was adopted. The 7-5-6 tricycle rings of the Cephalotaxus diterpenoids were obtained from the dicobalt hexacarbonyl-propargylic alcohol complex (Scheme 20). An intramolecular Nicholas/Hosomi-Sakurai cascade reaction was developed to form the cycloheptene A-ring bearing a chiral methyl group (Scheme 11). The Pauson-Khand reaction was then used to complete the skeleton of the target molecules. The asymmetric total syntheses of Cephinoid P (36), Cephafortoid A (37), 14-Epi-cephafortoid A (38) and Fortalpinoids M (39), N (40) and P (41) were successfully achieved over 15–18 steps from compound 42. The proposed strategy presents a novel approach for the total synthesis of Cephalotaxus diterpenoids and structurally related akin polycyclic natural products (Wang et al., 2023).
2.7 Total Synthesis of Dehaloperophoramidine and (+)-Perophoramidine
Naturally occurring (+)-Perophoramidine 44 was isolated from marine ascidian Perophora namei by Ireland and co-workers in the Philippine ascidian organism, P. namei (Verbitski et al., 2002). Dehalogenated product Dehaloperophoramidine 44 was obtained from the hydrogenation of (+)-Perophoramidine 45. The two compounds, 44 and 45 bear a structural resemblance to a complex family of natural products called communesin alkaloids (Communesin F, 46) (Trost and Osipov, 2015; Wilkie et al., 2016; Zuo and Ma, 2011). They both possess complex chemistry of quaternary chiral centres that makes them synthetically challenging. (+)-Perophoramidine 45 has been reported to possess anticancer activity and this has drawn the synthetic chemist’s attention to them.
Synthesis of Dehaloperophoramidine 44 started with the reaction of commercially available 47 and 48 to make intermediate 49, which was taken through seven steps to yield 50. Key intermediate 51 was treated with allyltrimethylsilane in the presence of TiCl4 to give 52 which was taken through 12 extra steps to achieve Dehaloperophoramidine 44 (Scheme 12) (Wilkie et al., 2016).
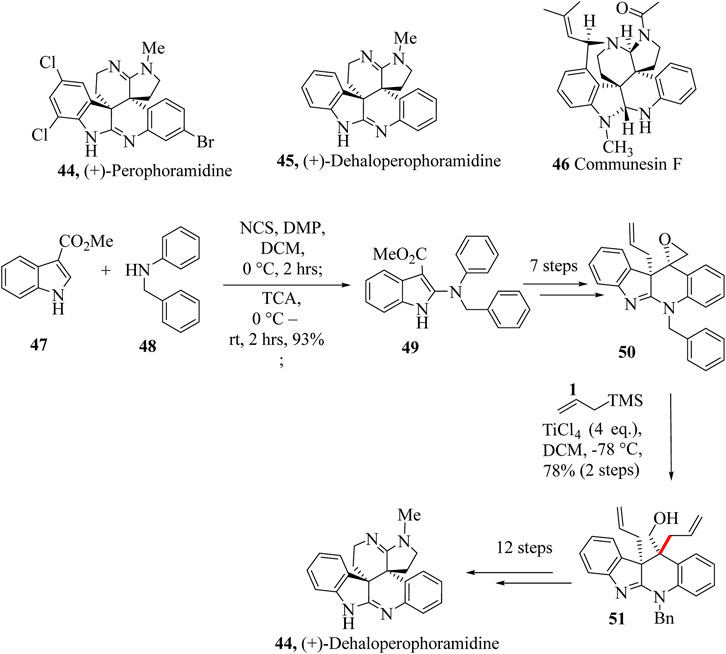
Scheme 12. Total synthesis of (+)-Dehaloperophoramidine 51 (Wilkie et al., 2016).
2.8 Total synthesis of deoxopinguisone
Deoxopinguisone (52) is a type of Pinguisanes (sesquiterpenes) that occurs naturally in liverworts (Asakawa et al., 2013). Pinguisanes was first isolated and characterised successfully using spectroscopic techniques by Krutov and co-workers in 1972 (Krutov et al., 1973; Uyehara et al., 1985). The distinctive feature of this compound is its spatial configuration of the four methyl group substituents. These methyl groups are specifically oriented in a β-configuration in the molecular framework.
Formation of the C4–C9 bond was facilitated by the HSR to give a cis stereochemistry between C8 and C9 since the cis hindrance system is more stable in solution compared to the trans-derivative. The synthesis started with commercially available ketone 53 which was taken through seven sequential steps to obtain intermediate 54 (Supplementary Scheme S9). This intermediate 54 was oxidized with pyridinium dichromate and tert-butylhydroperoxide to obtain the enone intermediate 55 which was subjected to Hosomi-Sakurai cyclisation (HSC) in dry TBAF that led to the concomitant cleavage of the C-Si bond to form intermediate 55. The HSC of 55 favoured only the formation of the desired cis isomer 56 as shown in Supplementary Scheme S9 below (Tori et al., 2001).
From Supplementary Scheme S10, Dthe conformation of the cis-isomer shows that the β-position from the enone is near the allylic proton that is in the γ-position from the TMS group. Also, cis -isomer of cyclohexanone is favoured due to steric hindrance (Supplementary Scheme S10) (Tori et al., 2001). The intramolecular cyclisation was accomplished by the HSR in dry TBAF as a catalyst in THF as a solvent to yield 99% compound 56, but 56 was not formed through the trans conformation due to steric hindrance as shown (Supplementary Scheme S10) in the intermediate (Tori et al., 2001). Conversion of intermediate 55 to the final product Deoxopinguisone, 52 was accomplished by Uyehara and coworkers (Uyehara et al., 1985; 1986).
2.9 Total synthesis of Ent-Callilongisin B
Ent-Callilogisin B (57) was isolated from the leaves of Callicarpa longissima, along with related compounds, Callilongisin A (58), C (59), and D (60). Ent-Callilongisin B (59) is an analogue of 3,4-seco-abietane-type diterpenoid whose structure was established using NMR spectroscopic techniques (Liu et al., 2012). The natural product 57 demonstrated cytotoxic activities against a human prostate cancer cell line. An anti-inflammatory activity of 57 was also observed when tested for superoxide anion production from human neutrophil cells (Kamiya et al., 2021).
In 2021, the first asymmetric total synthesis of tricyclic diterpenoid Ent-callilongisin B (57) was reported by Kamiya et al. (2021). The synthesis started with the Sharpless oxidation of enantiomerically pure (S)-perillyl alcohol 61, to form acetoxy ketone 62 (Indictor and Brill, 1965; Sharpless and Michaelson, 1973; Tanaka et al., 1974). The synthesis was accomplished following stereo-controlled Michael 1,4-addition and Hosomi−Sakurai allylation of enone 63, followed by Wacker oxidation, and intramolecular aldol reaction of diketone to construct the six-membered ring and oxidative dearomatisation of phenol intermediate accompanied by diastereoselective δ-lactonisation (Scheme 13). The same research group is also developing enantioselective synthetic methods for other Callilongisin analogues A (58), C (59) and D (60) (Kamiya et al., 2022).
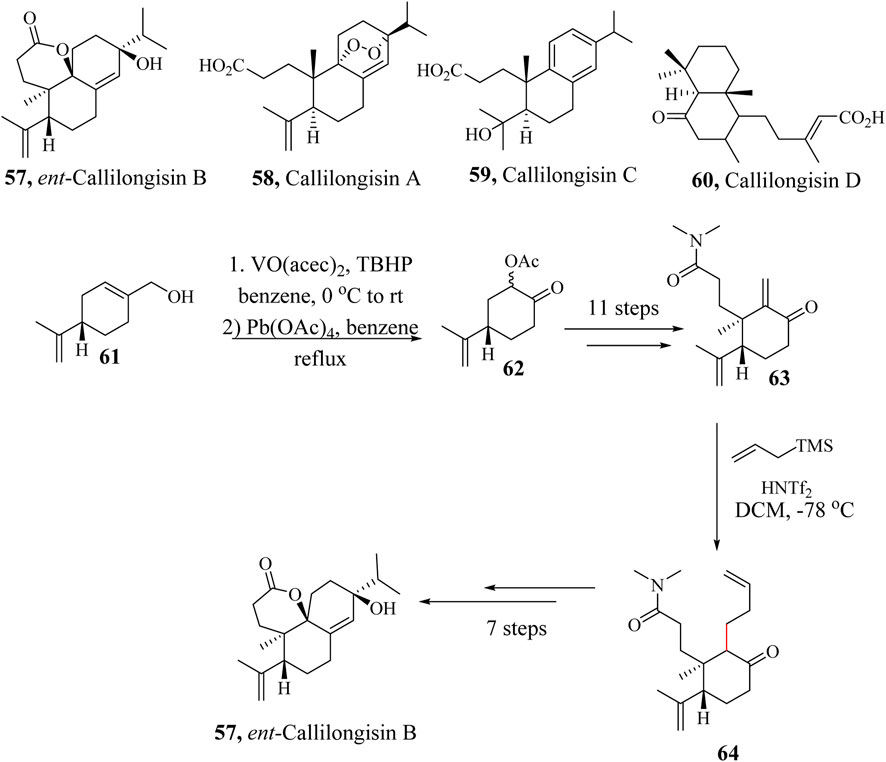
Scheme 13. Total synthesis of Ent-callilongisin B (57) (Kamiya et al., 2021).
2.10 Total synthesis of eribulin mesylate
Eribulin mesylate (65) is an anticancer drug made from halichondrin B (66), a natural marine polyether macrolide product (Ortega and Cortés, 2012; Paterson et al., 2011). In 1986, halichondrin B was first isolated from the marine sponge Halichondrin okadai by Uemura and co-workers (Kulkarni et al., 2016). Further studies on the structure of halichondrin B led to the discovery of the analogue known as Eribulin mesylate (65) with improved anticancer activity against metastatic cancer cell lines. In 1997, Eisai Pharmaceutical Company in collaboration with Kishi et al., reported the total synthesis of the drug in an industrial viable way (Fang et al., 1992; Lee et al., 2016; Li et al., 2018; Stamos et al., 1997).
Currently, Pabbaraja et al., reported a more economical, and industrially scalable, approach toward the easy production of the drug (Nasam and Pabbaraja, 2024). The modified stereoselective synthetic approach to the preparation of Eribulin mesylate (65) involved the use of commercially available 1,4-butanediol (67) to generate key intermediate 68 which was coupled with intermediate 69 in HSR to achieve the key intermediate 70. The total synthesis of Eribulin mesylate was accomplished by taking the intermediate 70 through several steps (Scheme 14) (Nasam and Pabbaraja, 2024).
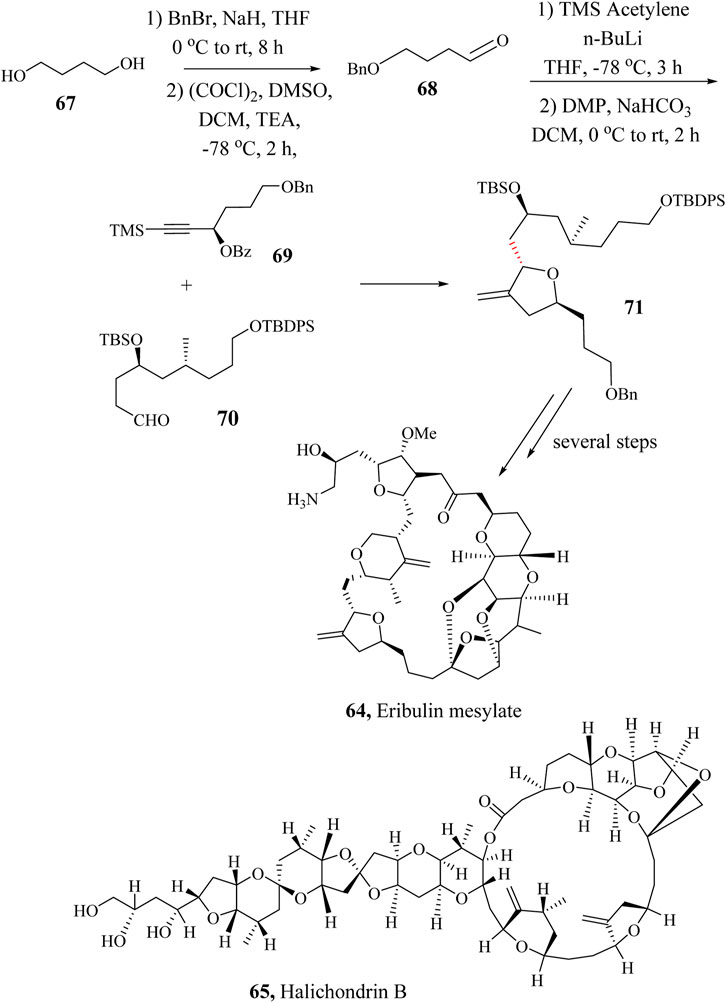
Scheme 14. Total synthesis of Eribulin mesylate, 64 (Nasam and Pabbaraja, 2024).
2.11 Total synthesis of (−)-Galiellalactone
(−)-Galiellalactone 72, (Scheme 15), a fungal metabolite was originally isolated from ascomycete Galiella rufa in 1990 (Johansson and Sterner, 2001; Kim et al., 2015). It has been found to possess inhibitory properties against STAT3 (signal transducer and activator of transcription 3) that block the DNA binding of phosphorylated STAT3 without inhibiting phosphorylation and dimerization (Don-Doncow et al., 2014). (−)-Galiellalactone also induces the apoptosis and growth inhibition of human prostate cancer cells. Sterner and co-workers were the first to report the synthesis of the (−)-Galiellalactone and confirmed its stereochemistry unambiguously (Johansson and Sterner, 2001).
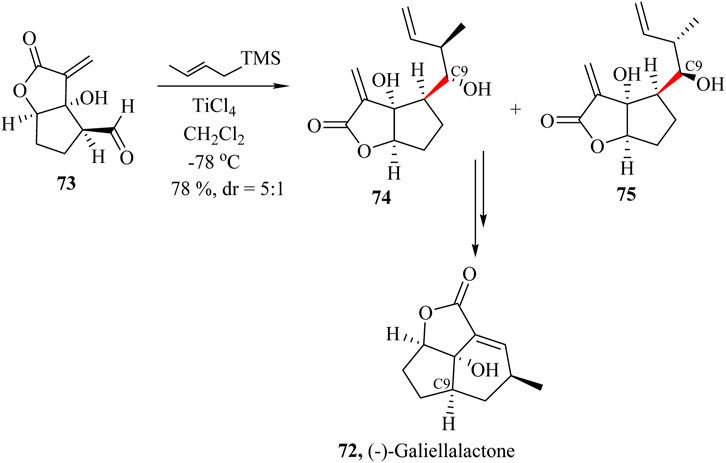
Scheme 15. Total synthesis of (−)-Galiellalactone (72) (Kim et al., 2015).
Kim et al also used a modified Hosomi−Sakurai crotylation method (Hosomi and Sakurai, 1977; Shing and Li, 1997) to diastereoselectively introduce the C9 side chain with the required terminal alkene for ring closure metathesis. The intermediate 73 was reacted with (E)-crotyltrimethylsilane and BF3⋅Et2O as the promoter to yield compounds 74 and 75 in 88% yield in favour of the undesired 75. However, when the catalyst was changed to TiCl4, the yields dropped to 78% but favoured compound 74 to give a dr of 5:1 (Scheme 15). Further, intermolecular cyclisation of the 75 resulted in the formation of the product with the exact configuration (Kim et al., 2015).
2.12 Total syntheses of grayanane diterpenoids: (−)-grayanotoxin III, (+)-Principinol E, and (−)-Rhodomollein XX
Grayanane diterpenoids are naturally occurring secondary metabolites exclusively found in flowering plants of the Ericaceae family (Li et al., 2019) with over 4,000 species (Liu et al., 2024). In addition to grayanane diterpenoids (Cai et al., 2018; Ho and Lin, 1995; Lv et al., 2017; Zhou et al., 2012), Ericaceae plants contain other types of terpenoids, including triterpenoids (Teng et al., 2018; Zhang et al., 2018; Zhao et al., 2018) and meroterpenoids (Huang et al., 2018; Liao et al., 2015; Liao et al., 2017). These compounds feature a distinctive tetracyclic backbone system with an intricate 5/7/6/5 configuration, contributing to their complexity and diversity (Huang et al., 2005; Wang et al., 1998). This family of compounds exhibits various properties, including analgesic, antinociceptive, anticancer, antiviral, antifeedant, insecticidal effects, as well as toxicity and inhibition of protein tyrosine phosphatase 1B (PTP1B) (Li et al., 2019; Liu et al., 2024).
Grayanotoxins were isolated from the leaves of Leucothoe grayana, a poisonous shrub native to Japan, between 1930 and 1960 (Kakisawa et al., 1961; Kong et al., 2023) but the stereochemistry at the ring junction was only ambiguously determined in 1970 using X-ray crystallography by Narayanan and coworkers (Narayanan et al., 1970). Grayanotoxins are known to be toxic to both humans and animals, acting by modulating voltage-gated sodium channels. This mechanism accounts for their poisoning effects as well as their analgesic properties. Grayanotoxin III, 76 is the alkaline hydrolysed derivative of Grayanotoxin I, which naturally occurs in L. grayana (Kakisawa et al., 1961). Principinol E, 77 was isolated from the aerial parts of Rhododendron principis and exhibited significant in vitro inhibitory activity against PTP1B with an IC50 value of 3.14 ± 0.12 µM (Liu et al., 2014; Z. R; Zhang et al., 2012). Rhodomollein XX, 78 was isolated from fruits of Rhododendron mole (Li et al., 2000; Zhou et al., 2014) and exhibited moderate antinociceptive activity (Li et al., 2019).
The total synthesis of 76, 77, and 78 began with the synthesis of the key intermediate 80 from 79. The intermediate 81 was prepared by the asymmetric 1,4-conjugated addition developed by May and co-workers (May et al., 2011) and was treated with TMSOTf in the presence of 83 to foster an intramolecular Mukaiyama aldol reaction followed by the addition of EtAlCl2 which mediated the HSR to produce 82 in 62% yield (1.7:1 dr) in a one-pot. A total of 18, 19 or 20 and 18 synthetic steps were followed to obtain (−)-Grayanotoxin III, 76, (+)-Principinol E, 77 and (−)-Rhodomollein XX, 78 (Scheme 16) (Kong et al., 2022; 2023).
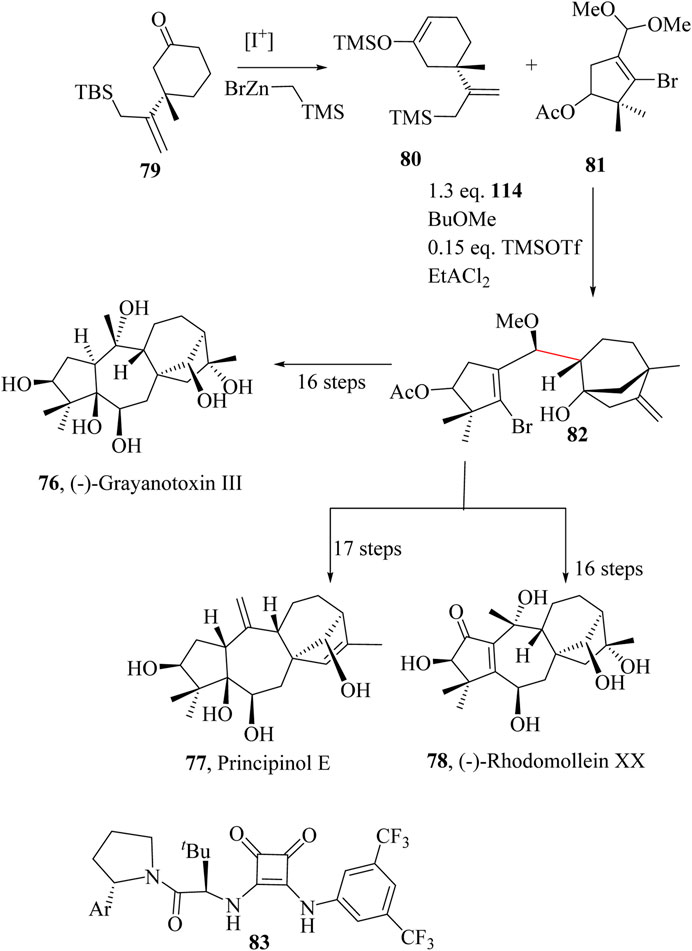
Scheme 16. Total syntheses of Grayanane Diterpenoids: (−)-Grayanotoxin III (76), (+)-Principinol E (77), and (−)-Rhodomollein (78) (Kong et al., 2022; 2023).
2.13 Total synthesis of Heliespirone A and C
Heliespirone A (84) is a member of a sesquiterpene group of alkaloids that was isolated from cultivar sunflowers, i.e., Helianthus annuus L. by Huang et al. (2011). Heliespirone C (85) was isolated in the later part of 2006 and reported with six- and five-membered oxaspirocyclic skeletons, respectively (Huang and Liu, 2010). Heliespirone A (84) is anticipated to have a significant impact on the allelopathic action of cultivar sunflowers. Heliespirone C (85) demonstrated an inhibitory activity in coleoptile bioassay. Due to their intriguing structural features, biological profiles, and limited availability, these natural products present appealing targets for total synthesis (Huang and Liu, 2010).
Miyawaki et al., in 2012 reported the total synthesis of (−)-Heliespirone A (84) and (+)-Heliespirone C (85) (Miyawaki et al., 2012). A combination of several synthetic approaches including the HSR was carried out to introduce two stereogenic centers at C-8 and C-10 (Scheme 17). Intramolecular allylation of substrate 86 which had an allylsilane and p-benzoquinone substituents was accomplished using tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) as a catalyst in isobutyronitrile (Me2CHCN) to give a mixture of 87 and 88 in 68% with 8:1 dr. The excellent stereoselectivity in this transformation could be attributed to the preference for transition state T1 over T2. This is due to the stereoelectronically favourable antiperiplanar conformation of T1, as well as the steric repulsion between the allylsilane and the benzoquinone groups in T2 as shown in Scheme 17. Intermediate 88 was taken through 4 linear steps to achieve the two diastereoisomers (±)-Heliespirones A, 84 and C, 85 (Miyawaki et al., 2012).
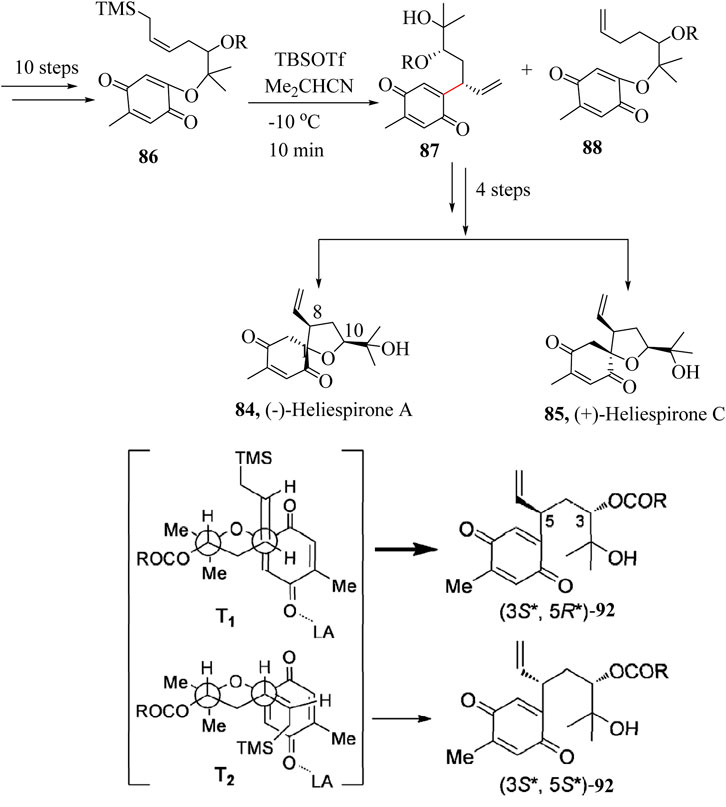
Scheme 17. Total synthesis of (−)-Heliespirone A and C (Miyawaki et al., 2012).
2.14 Total synthesis of Herboxidiene/GEX1A
Herboxidiene GEX1A (89) a potent phytotoxic polyketide was isolated from Streptomyces chromofuscus A7847 by Isaac and co-workers in 1992 (Isaac et al., 1992; Thirupathi and Mohapatra, 2016). Yoshida et al. also isolated six polyketide analogues [series (GEX1)] from Streptomyces sp, in 2002 (Miyawaki et al., 2012; Sakai et al., 2002; Yun and Panek, 2007). The compound exhibited selective phytotoxicity against a range of broadleaf annual weeds but harmless to wheat (Blakemore, 2002; Sakai et al., 2002). Herboxidiene/GEX1A (89) is also able to induce GAP1 and GAP2/mitosis (G1 and G2/M) cell cycle arrest in human tumour cell line WI-38 (Ghosh and Li, 2011). It also binds to SAP155, the protein responsible for the pre-mRNA splicing thereby acting as a novel slicing inhibitor (Thirupathi and Mohapatra, 2016).
The first total synthesis attempts of herboxidiene/GEX1A, 89 was in 1999 by Kocienski and co-workers using direct aldol reaction, Ireland-Claisen rearrangement, and hydroxy-directed epoxidation. The structural complexity of herboxidiene/GEX1A (89) comprising the presence of nine chiral centres, trisubstituted tetrahydropyran core, and conjugated diene side chain has made its synthesis particularly challenging (Smith et al., 1996). Mohapatra and Thirupathi reported a 22 linear sequence steps toward the synthesis of herboxidiene/GEX1A (89) (Thirupathi and Mohapatra, 2016). HSR was used to couple lactol 90 which was achieved in 7 steps from 2-butyne-1,4-diol (91), with allyltrimethylsilane and AuCl3 as a diastereoselective allylation catalyst previously developed for cyclic hemiacetals as shown in Scheme 18. Compound 92 was the key intermediate needed to synthesize Herboxidiene 89 (Thirupathi and Mohapatra, 2016).
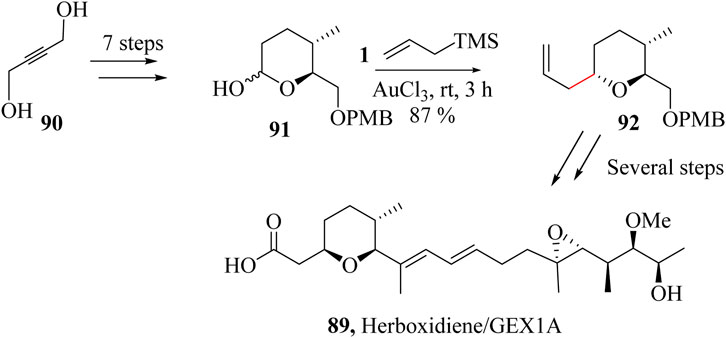
Scheme 18. Total synthesis of Herboxidiene GEX1A, 89 (Thirupathi and Mohapatra, 2016).
2.15 Total synthesis of Huperserratines A and B
Lycopodium alkaloids are a large group of natural products with unique heterocyclic frameworks found in the Lycopodiaceae family. Following the isolation of lycopodine by Bödeker (1881), Harrison et al. (1961), Wada et al. (2019), more than 300 Lycopodium alkaloids have been isolated, and their structures established using various spectroscopic techniques including X-ray crystallography (Ma and Gang, 2004; Siengalewicz et al., 2013). These compounds exhibit distinguishable polyfused-bridged structures and demonstrate remarkable biological activities. For instance, Huperzine A, a lycodine-type lycopodium alkaloid isolated from Huperzia serrata, is a highly specific and potent inhibitor of AChE (Xing et al., 2014). It is also found to be effective in the treatment of Alzheimer’s disease (AD) in China, and used as a dietary supplement in the United States (Liu et al., 1986; Xu et al., 1999). In 2020, ZhaO and co-workers isolated two Lycopodium alkaloids Huperserratines A (93) and B (94), from H. serrata (Wu et al., 2020). The absolute configurations of these compounds were determined using single-crystal X-ray diffraction. The compounds were the first reported macrocyclic lycopodium alkaloids with a 5-azabicyclo [10.4.0]hexadecane structure. They were also the second and third examples of lycopodium alkaloids containing an oxime group in the molecule. In addition, Huperserratine A (93) showed moderate anti-HIV-1 activity with an EC50 value of 52.91 μg mL−1 (Deng et al., 2022).
ZhaO and co-workers reported the first total synthesis of Huperserratines A (93) and B (94) through a 12-step sequence including Suzuki-Miyaura coupling, the HSR, the ring-closing metathesis, the dihydroxylation, and Swern oxidation (Scheme 19) (Zhao et al., 2021). The synthesis of Huperserratines A (93) and B (94) started with (5R)-2-iodo-5-methyl-2-cyclohexen-1-one (95), which was transformed into the intermediate, 96 by a Pd-catalysed Suzuki–Miyaura reaction followed by modification through N-alkylation to obtain compound 97. The 1,4-addition was completed using a TiCl4-catalyzed HSR of enone 98 with allyltrimethylsilane, yielding 3,5-trans-cyclohexanone 99. Epimerization at the α-position of the ketone occurred under these conditions, resulting in a 1:1 mixture of diastereomers. After three steps, diastereomers 101 and 102 were produced, respectively. After the preparation of the oxime followed by deprotection to give Huperserratines A (93) and B (94) (Deng et al., 2022; Wu et al., 2020).
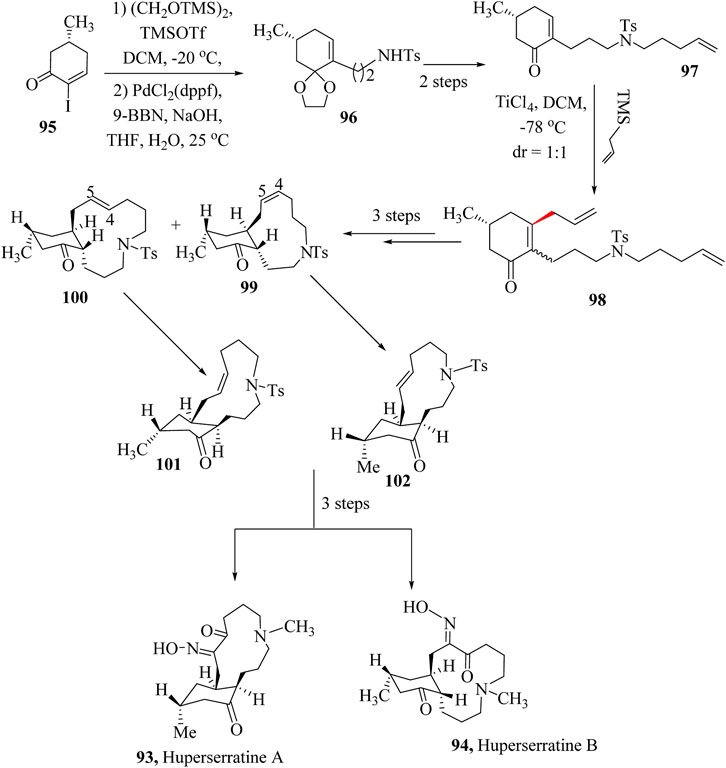
Scheme 19. Total synthesis of Huperserratine A (93) and B (94) (Deng et al., 2022).
2.16 Total syntheses of (±)-Isonitramine, (−)-Sibirine, and (+)-Nitramine
(±)-Isonitramine (103), (−)-Sibirine (104), and (+)-Nitramine (105), are spiropiperidine class of alkaloids possessing chiral quaternary carbon centres on their 2-azaspiro [5,5]undecane-7-ol core and were all isolated from Nitraria sibrica and N. Schoberi (Manske, 1960; Pandey et al., 2011). The three compounds demonstrated good biological activities against the proliferation of cancer cell lines (Boubaker et al., 2011).
The total synthesis of these alkaloids has been reported although targeted products are obtained as racemic mixtures (Westermann et al., 1993). These molecules appear simple but are challenging to synthesise due to their contiguous chiral centres. The total synthesis of (−)-Isonitramine (103) and (−)-Sibirine (104) started with the pure enantiomer 106 (Scheme 20). The ester group on 107 was reduced to an alcohol using LiAlH4 followed by protection of the amine group using benzyl chloroformate in dioxane/water and subsequent oxidation to yield intermediate 108. An allylation of intermediate 108 with allyltrimethylsilane in the presence of BF3⋅Et2O was accomplished via the HSR to give the key intermediate 109 in 92% with 78:22 diastereoselectivity. Synthesis of (+)-Nitramine (105) started with the conversion of the ester group of enantiopure 106 to aldehyde 109 which was treated with allyltrimethylsilane in the presence of SnCl4 to yield intermediate 110 in 87% with diastereoselectivity of 96:4. The intermediates 108 and 110 were each taken through 2 and 3 steps independently to yield 42%, 38%, and 25% for (−)-Isonitramine (103), (−)-Sibirine (104), and (+)-Nitramine (105), respectively (Pandey et al., 2011; Yang et al., 2021).

Scheme 20. Application of HSR total synthesis of (−)-Isonitramine (103), (−)-Sibirine (104), and (+)-Nitramine (105) (Pandey et al., 2011).
2.17 Total synthesis of (−)-Kumausallene
(−)-Kumausallene, 111 is sesquiterpene secondary metabolite belonging to a family of non-isoprenoids with a unique bromoallene or enyne moiety and also containing a dioxa-bicyclo [3.3.0] octane core (Das and Ramana, 2015; Hoffmann-Röder and Krause, 2004). Kurosawa et al first reported the isolation of (−)-Kumausallene, 111 from red algae Laurencia Nipponica Yamada collected at Kumausu, near Otaru, Hokkaido, Japan (Evans et al., 1999; Suzuki et al., 1983). Other members of the enyne containing compounds with dioxa-bicyclo [3.3.0] octane core shown in Supplementary Figure S2 include (+)-Panacene, 112 (Boukouvalas et al., 2006; Feldman, 1982; Feldman et al., 1982) and (−)-Aplysiallene, 113 (Wang and Pagenkopf, 2007); the eight- and nine-membered cyclic ethers (+)-Laurallene, 114 (Crimmins et al., 2010; Saitoh et al., 2003), (+)-Isolaurallene, 115 (Crimmins et al., 2002; Crimmins and Emmitte, 2001) (+)-Itomanallene A, 116 (Jeong et al., 2010) and (+)-Microcladallene B, 117 (Park et al., 2007).
The first total synthesis of (±)-Kumausallene (111) was reported by Overman and his group using a ring annulation strategy (Brown et al., 1991; Grese et al., 1993). It has also been enantioselectively synthesized by Evans et al., using an acyl radical cyclization to construct the tetrahydrofuran ring followed by an efficient biomimetic strategy (Evans et al., 1999). Several other successful syntheses of (±)-Kumausallene (111) has been reported in literature (Grese et al., 1993; Werness and Tang, 2011). In their 20 steps synthetic attempt, Werness et al oxidized hydroxyl ester intermediate 119 which was synthesized through 5 steps starting from compound 118 to the corresponding aldehyde and subsequently installed the pentenyl side chain through (Werness et al., 2013; Werness and Tang, 2011). The final product (−)-Kumausallene was achieved after 14 sequential steps as shown in Scheme 21a. Tang et al., used BF3⋅OEt2 as a Lewis acid in a HSR after the ozonolysis of compound 122 which was synthesized in four steps from acetylacetone 151 to generate pentene linker with the right stereochemistry (Scheme 21b). The HSR proceeded with a diastereoselectivity of a 4:1 ratio favoring the desired stereoisomer. Compound 123 was taken through 8 extra steps to give the title compound in 5.4% yield (Werness and Tang, 2011).
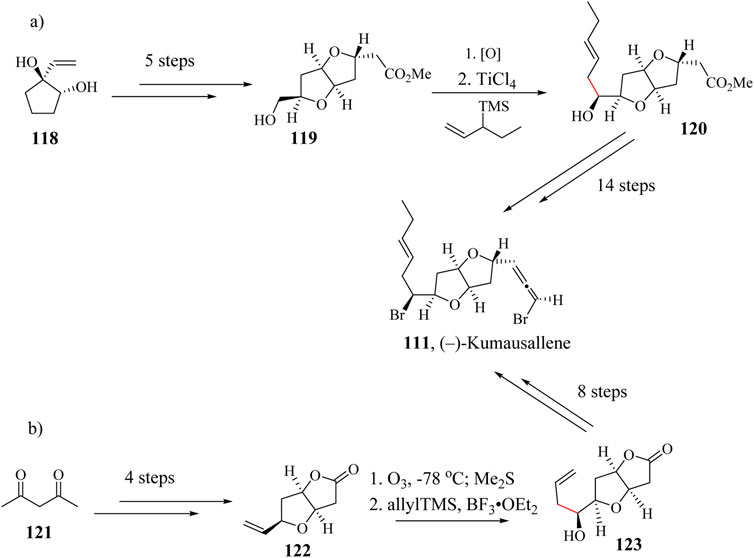
Scheme 21. Total synthesis of Kumausallene reported by (a) Werness et al. (2013) and (b) Werness and Tang (2011).
2.18 Total synthesis of Leiodermatolide A
Leiodermatolide A, 124 is a 16-membered polyketide macrolide skeleton, featuring an unsaturated side chain terminating in a δ-lactone. It was isolated in 2008 from marine invertebrate sponge lithistid Leiodermatium Sp. in Florida (Paterson et al., 2011). It possessed antimitotic selective cytotoxicity towards the pancreatic cancer cell lines AsPC-1, PANC-1, BxPC-3, and MIA PaCa-2, and potent cytotoxicity against skin, breast and colon cancer cell lines (Guzmán et al., 2016; Siu et al., 2021).
Krische and coworkers reported a 13-step total synthesis of leiodermatolide A, by reacting together fragments 124, 125 and 126 synthesised independently from commercially available starting materials (Scheme 22). Intermediates 127 and 128 were synthesized from starting materials 125 and 126 independently in 5 and 6 steps respectively. Aldehyde 127 and allyl silane 158 were reacted in a chelation-controlled procedure using AlEtCl2 followed by TBAF mediated HSR and exhaustive deprotection of the product to yield fragment 129. Fragments 129, 130 and 131 were then combined sequentially to yield Leiodermatolide A, 124 (Siu et al., 2021).
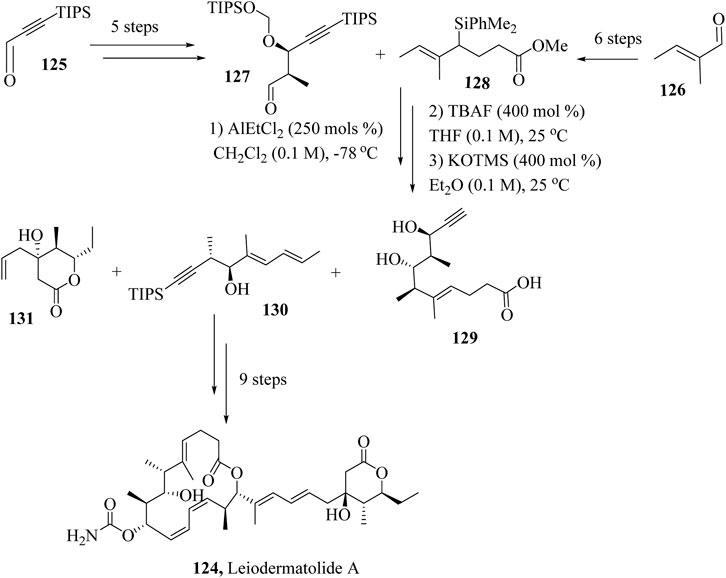
Scheme 22. Total synthesis of Leiodermatolide A, 124 (Siu et al., 2021).
2.19 Total synthesis of Lycopodine
Lycopodine, 132 belongs to a family of over 300 naturally occurring Lycopodium alkaloids with a diverse structural backbone (Harrison et al., 1961). It was first isolated in 1881 by Ayer et al (Harrison et al., 1961; Yang and Carter, 2010) but its structure was not elucidated until 1960 by Harrison and coworkers (Ayer and Cruz, 1993). Some of the members of this family of compounds possess medicinal properties that can reversibly inhibit acetylcholinesterase and increase learning and memory efficiency (Liu et al., 1986). Lycopodine is made up of four haxacyclic rings fused with five asymmetric centres like lycodine (Liu et al., 1986).
Several total synthetic techniques have been reported for the preparation of natural 132. Asymmetric synthesis was first reported by Yang and Carter (2010), Ma et al. (2016). Wada and co-workers also reported a concise asymmetric total synthesis of 162 by applying the HSR in the first step (Scheme 23a). Seven synthetic pathways were followed in the total synthesis of Lycopodine which started with the conversion of the commercially available crotonamide 133 to diastereoselectively make 134 (22.3: 1 dr) as shown in Scheme 23a (Wada et al., 2019).
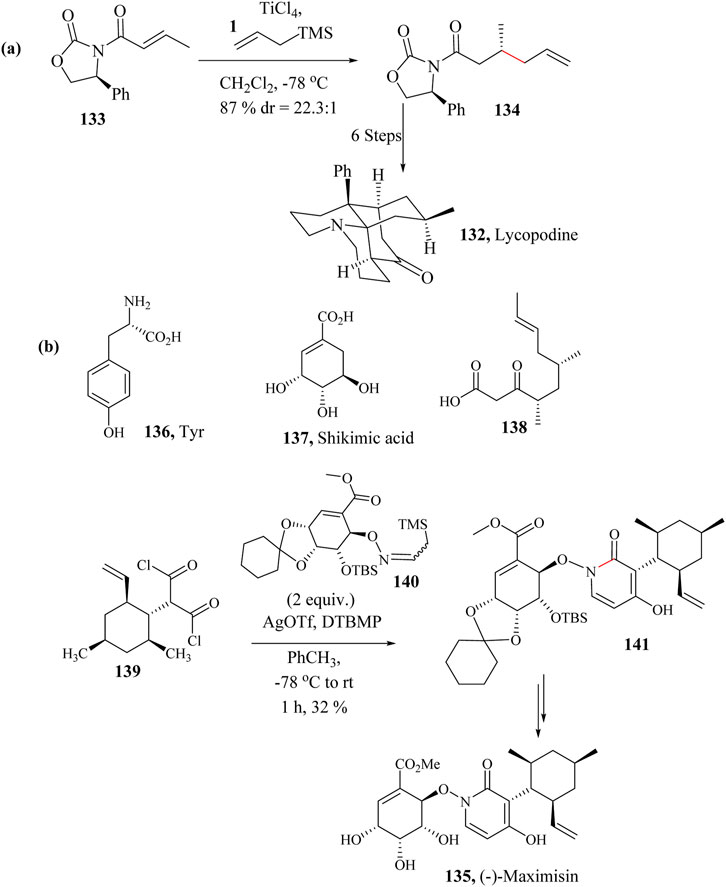
Scheme 23. Total syntheses of (a) Lycopodine (132) and (b) (−)-Maximiscin (135) involving HSR step (Mcclymont et al., 2020; Wada et al., 2019).
2.20 Total synthesis of (−)-Maximiscin
(−)-Maximiscin 135 is an alkaloid made from three different metabolic pathways that incorporates a central 1,4-dihydroxy-2-pyridone derived from tyrosine 137 (Scheme 23b), attached to the ester of shikimic acid 137 and cyclohexyl of a polyketide 141 (Schmidt et al., 2003). 4-Hydroxy-2-pyridone family of alkaloids (Du et al., 2014) to which (−)-Maximiscin belongs are known to be unstable and usually fragment in isolation but possess interesting biological activities (Schmidt et al., 2003). (−)-Maximiscin 135 is a fungal metabolite isolated from the fungus Tolypocladium which demonstrated tumor suppression activity in animal models (Du et al., 2013; Du, et al., 2014).
The total synthesis of (−)-Maximiscin 135 faced challenges due to the instability of its structure. Specifically, shikimate and pyridone residues tend to fragment when synthesised. Mcclymont and co-workers completed the total synthesis of Maximiscin 135 by following a modified enantioselective HSR using AgOTf as a promoter to activate the diacid chloride 139 as an electrophile in the presence of 140 and use the β-silicon effect of the Si−C to enhance the nucleophilicity of the nitrogen (Déléris et al., 1980; Mcclymont et al., 2020) (Scheme 23b).
2.21 Total synthesis of (+)-Ophiobolin A
Ophiobolins A-K (142–147) are terpenoids with 5,8,5-membered carbocyclic ring systems (Scheme 24). The complexity of the structure and intriguing biological activities of Ophiobolins A-K have attracted the attention of the organic synthetic community. (+)-Ophiobolin A, 142 has a unique 5,8,5,5 tetracyclic ring system and eight chiral centres isolated from Ophiobolus miyabeanus in 1958 by Ishibashi and co-workers. Its structure was fully established in 1965 by Nozoe et al. (1965), Rowley et al. (1989). Biological screening of 172 has revealed that it induces apoptotic cell death in the L1210 cell-lines, (Fujiwara et al., 2000), inhibits calmodulin-activated cyclic nucleotide phosphodiesterase, and also shows cytotoxicity to cancer cell lines A-549, Mel-20, and P-335 (IC50 values that range from 62.5 to 125 µM) (Li et al., 2013).
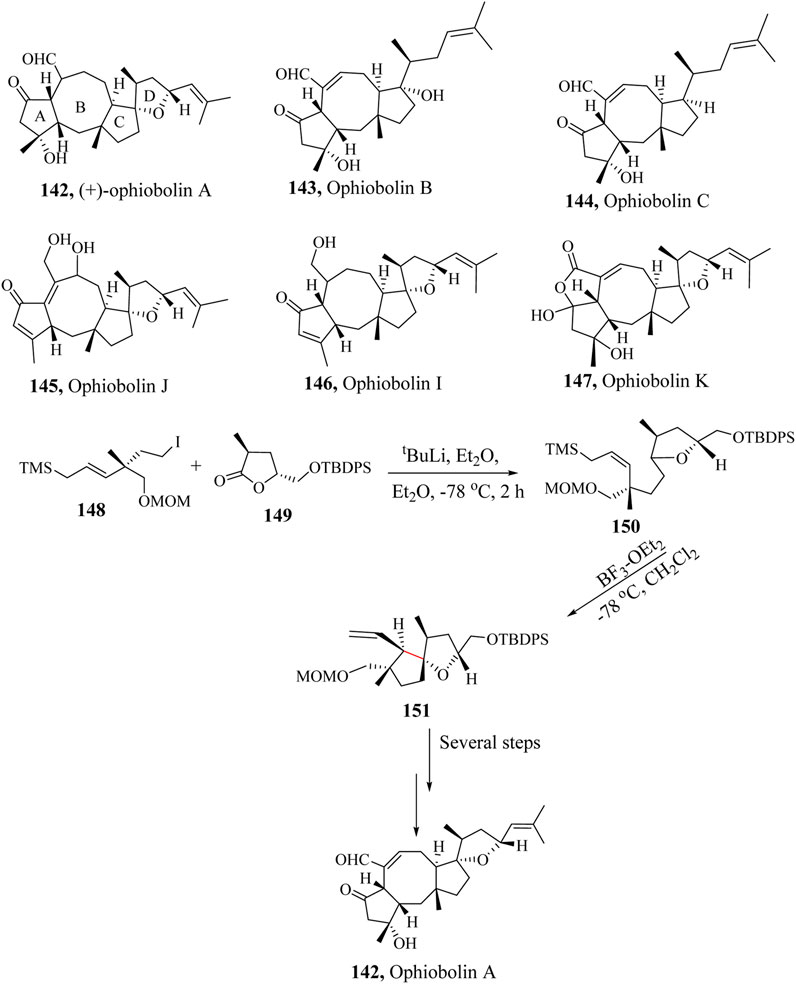
Scheme 24. Application of HSR in total synthesis of Ophiobolin A, (142) (Tsuna et al., 2013).
A number of synthetic attempts have been made toward the total synthesis of Ophiobolin by Kishi and co-workers (Li et al., 2013). Tsuna et al., reported the first total synthesis of (+)-ophiobolin A (142) in 2013. A protected tert-butyldiphenylsilyl (TBDPS) intermediate 148 was reacted with the methoxymethyl (MOM) intermediate 149 to generate the complex intermediate 150 which was taken through an intramolecular HSR to form the key intermediate spirocyclic compound 151 in 45% yield using BF3⋅OEt2 catalytic system. Further transformation of the key intermediate result in the production of the targeted product 152 as illustrated in Scheme 24 (Tsuna et al., 2013).
2.22 Total synthesis of (+)-Paniculatine
(+)-Paniculatine 152 belongs to the Lycopodium family of alkaloids and was first isolated by Castillo and his group in 1975 (Castillo et al., 1975; 2011). Other members of the Lycopodium family such as (−)-Magellanine 153 (Loyola et al., 1979) and (+)-Magellaninone 154 (Maclean, 1985) were also isolated from Lycopodium paniculatum by Casillo and co-workers. (+)-Paniculatine 152 has a complicated 6-5-5-6-fused tetracyclic diquinane core structure made up of seven chiral centres woven in a tetracyclic framework (Ma and Gang, 2004). Lycopodium family possesses anti-inflammatory activity and acetylcholinesterase inhibitory properties and are being studied as potential drugs for the treatment of Alzheimer’s and other neurodegenerative diseases (Ma and Gang, 2004; Saito et al., 2023).
The first total synthesis of (+)-Paniculatine (152) was reported by Sha and co-workers in 1999 using tandem cyclisation reactions that involve HSR (Sha et al., 1999). Mukai and co-workers, stereospecifically synthesized (+)-Paniculatine 152, (−)-Magellanine 153, (+)-Magellaninone 154 (Supplementary Scheme S12) in 43–45 unprecedented synthetic steps in 2007 (Kozaka et al., 2007). Yan et al. proposed a concise palladium-catalyzed reaction in 2014, leading to the synthesis of (+)-Paniculatine, (−)-Magellanine, and (+)-Magellaninone in 12 steps (Chen et al., 2018; Liu et al., 2019). Liu et al., developed a more concise synthetic route for the synthesis of (+)-Paniculatine, achieving the synthesis in 10 steps. This involved the HSR of compound 155 to yield the desired intermediate 156 with a high diastereoselective ratio (dr > 20:1) at −78°C using TiCl4 as depicted in Supplementary Scheme S12. The overall yeild was 12%. The authors proposed that the same reaction protocol could be used to synthesise (−)-Magellanine 155 and (+)-Magellaninone 154 as well (Liu et al., 2019).
2.23 Total synthesis of (+)-Penostatin E
Penostatin E 157 was isolated from a marine-based alga Enteromorpha intestinalis, a strain of Penicillium sp. (Jansma and Hoye, 2012). The biological activity of this compound attracts much interest from the organic synthetic community. When screened against cancer cell lines (P388 leukaemia cell lines), an ED50 value of 0.9 μg/mL was obtained, making it a potential anticancer agent.
Fujioka et al reported the first successful synthesis of (+)-Penostatin E, 157 following the HSR method to prepare enyne 150 from aldehyde 158 and allysilane 159 which were both synthesized from (R)-glycidyl isobutyrate and (E)-4-(trimethylsilyl)but-2-en-1-ol, respectively (Scheme 25a). Enyne 160 was diastereoselectively obtained in 96% yield after reacting 158 and 159 in the presence of SnCl4 without loss of enantiopurity. The synthetic pathway was simple and could be employed in the synthesis of other Penostatin derivatives. From compound 160 to the title compound 157 took several steps (Fujioka et al., 2014; Jansma and Hoye, 2012).
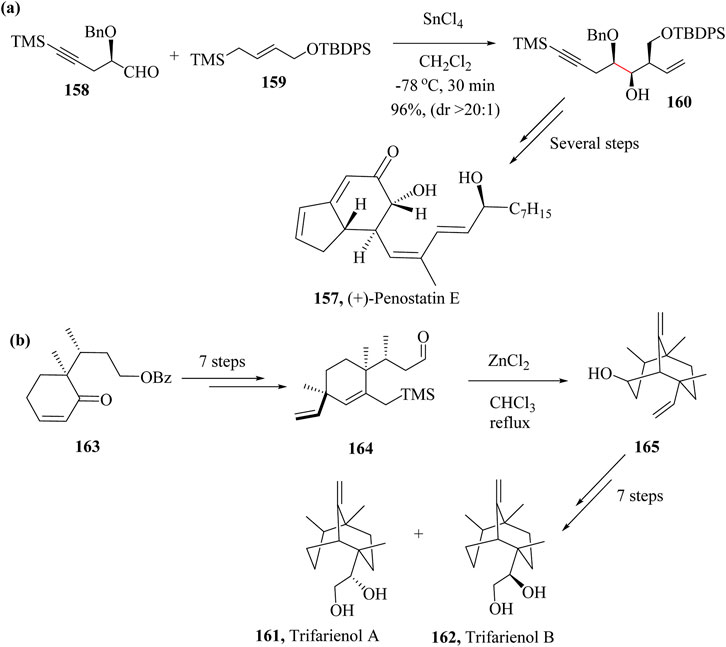
Scheme 25. Total syntheses of (a) (+)-Penostatin E, (157) and (b) Trifarienols A (161) and B (162) showing applications of HSR step (Fujioka et al., 2014; Takahashi et al., 2009).
2.24 Total synthesis of Trifarienols A and B
Trifarienols A (161), and B (162) are sesquiterpenes that were isolated from Cheilolejeunea trifaria, a liverwort of Malaysian origin with a trifarane carbon backbone (Hashimoto et al., 1994; Huang and Forsyth, 1995). These compounds 161 and 162 are structurally complicated biologically active sesquiterpenes that exhibit anticancer, antifungal, and insect antifeedant properties. The complexity of this class of compounds is hidden in the highly substituted bicyclo [3.3.1] nonane moiety and the exo-methylene group. The bicyclo [3.3.1] nonane backbone of Trifarienols A (161) and B (162) is also found in some naturally occurring compounds such as hyperforin, aristophenones, guttiferones, garsubellin A, papuaforin A, and upial.
The first successful Trifarienols A (161) and B (162) synthesis was by Huang and co-workers (Huang and Forsyth, 1995) in 16 steps with an overall enantiomeric yield of 3%. Tori and co-workers attempted enantioselective synthesis of Trifarienols A (161) and B (162) in 1999 where they used (2RS,3R)-2,3-dimethylcyclohexanone as starting material and had an overall yield of 1.8% and 1.4% respectively (Tori et al., 1999). In 2009, Takahashi et al., synthesised Trifarienols A (161) and B (162) from compound 165 (Scheme 25b) which was previously prepared by the group and attempted construction of the bicyclo [3.3.1]nonane ring system. HSR was employed to achieve product 165 as the sole product in 93% yield by treating intermediate 164 with ZnCl2 in chloroform. The entire synthetic process took 15 steps to accomplish 2% and 9% yields of Trifarienols A (161) and B (162), respectively (Scheme 25b) (Takahashi et al., 2009).
3 Conclusion
In summary, the HSR and its applications in the total synthesis of complex and contiguous bioactive organic molecules from natural sources, have been reviewed. The HSR is applied in the formation of C-C bonds and complex transformations via activated Lewis acid-promoted allylation of various electrophiles. HSR is a facile reaction that has enabled organic chemists to undertake key transformations for the formation of complex carbocyclic compounds. Examples of such transformation include hetero-Diels–Alder (HDA), allylation of aldehydes and ketones, allylation of cyclic oxonium cation, β,γ-unsaturated α-ketoesters, carbocyclization, allylation of imines, epoxide opening/allylsilylation, cyanation of carbonyls amongst others.
The versatility of the HSR has been enhanced following the various modifications to the original method to broaden the scope of its application and molecular transformations of both intermolecularly and intramolecularly. Modifications such as the use of homoallylic allowed the preparation of a wide range of compounds including acetals, aldimines, aldehydes, carboxylic acid chlorides, epoxides, ketals, ketoimines, ketones, and α,β-unsaturated carbonyl compounds. Furthermore, HSRs occur under mild conditions and have low turnover time, are typically high yielding as well as are diastereoselective, stereoselective and installing multiple stereogenic centres concurrently.
In this review, we describe the applications of HSR in the synthesis of key intermediates and final products from several natural and synthetic products.
Author contributions
JA: Writing – original draft, Writing – review and editing. RK: Conceptualization, Writing – original draft, Writing – review and editing. DO-S: Conceptualization, Supervision, Writing – original draft, Writing – review and editing. RA: Conceptualization, Resources, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1527387/full#supplementary-material
References
Aiqin, W., Jingpei, X., Wenyan, W., and Jiwen, L. (2011). Microstructural morphology and formation mechanism of rapidly solidified Al-21Si multi-alloys. Adv. Mater. Res. 146–147, 1597–1600. doi:10.4028/www.scientific.net/AMR.146-147.1597
Asakawa, Y., Ludwiczuk, A., and Nagashima, F. (2013). Chemical diversity of bryophytes. Prog. Chem. Org. Nat. Prod., 95, pp. 21–24. doi:10.1007/978-3-7091-1084-3_3
Ayer, W. A., and Cruz, E. R. (1993). The tremulanes, a new group of sesquiterpenes from the aspen rotting fungus phellinus tremulae. J. Org. Chem. 58 (26), 7529–7534. doi:10.1021/jo00078a035
Bae, M. A., Yamada, K., Ijuin, Y., Tsuji, T., Yazawa, K., Tomono, Y., et al. (1996). Aburatubolactam A, a novel inhibitor of superoxide anion generation from a marine microorganism. Heterocycl. Commun. 2 (4), 315–318. doi:10.1515/HC.1996.2.4.315
Bai, W. J., Lu, C., and Wang, X. (2016). Recent advances in the total synthesis of tetramic acid-containing natural products. J. Chem. 2016 (1), 1–13. doi:10.1155/2016/8510278
Bauer, A., and Maulide, N. (2018). A stereoselective reductive hosomi-sakurai reaction. Org. Lett. 20 (5), 1461–1464. doi:10.1021/acs.orglett.8b00276
Beavo, J. A., and Brunton, L. L. (2002). Cyclic nucleotide research — still expanding after half a century. Nat. Rev. Mol. Cell Biol. 3 (9), 710–717. doi:10.1038/nrm911
Berkessel, A., Adrio, J. A., Hüttenhain, D., and Neudörfl, J. M. (2006). Unveiling the “booster effect” of fluorinated alcohol solvents: aggregation-induced conformational changes and cooperatively enhanced H-bonding. J. Am. Chem. Soc. 128 (26), 8421–8426. doi:10.1021/ja0545463
Birch, A. J., Kelly, L. F., and Thompson, D. J. (1981). Organometallic compounds in organic synthesis. Part 10. Preparations and some reactions of tricarbonyl-1,3- and -1,4-dimethoxycyclohexa-1,3-dieneiron and related compounds: the preparation of the tricarbonyl-3- methoxycyclohexadienyliumiron cation. J. Chem. Soc. Perkin Trans. 1, 1006–1012. doi:10.1039/P19810001006
Blakemore, P. R. (2002). The modified Julia olefination: alkene synthesis via the condensation of metallated heteroarylalkylsulfones with carbonyl compounds. J. Chem. Soc. Perkin Trans. 1, 2563–2585. doi:10.1039/b208078h
Bödeker, K. (1881). Lycopodin, das erste Alkaloïd der Gefässkryptogamen. Justus Liebigs Ann. Der Chem. 208 (1), 363–367. doi:10.1002/jlac.18812080308
Bols, M., and Skrydstrup, T. (1995). Silicon-tethered reactions. Chem. Rev. 95 (5), 1253–1277. doi:10.1021/cr00037a006
Boubaker, J., Bhouri, W., Sghaier, M. B., Bouhlel, I., Skandrani, I., Ghedira, K., et al. (2011). Leaf extracts from Nitraria retusa promote cell population growth of human cancer cells by inducing apoptosis. Cancer Cell Int. 11 (37), 37–2468. doi:10.1186/1475-2867-11-37
Boukouvalas, J., Pouliot, M., Robichaud, J., MacNeil, S., and Snieckus, V. (2006). Asymmetric total synthesis of (-)-panacene and correction of its relative configuration. Org. Lett. 8 (16), 3597–3599. doi:10.1021/ol061385e
Britten, T. K., Basson, A. J., Roberts, D. D., and McLaughlin, M. G. (2021). Aza-peterson olefinations: rapid synthesis of (E)-Alkenes. Synthesis 53 (19), 3535–3544. doi:10.1016/j.tet.2018.06.015
Brown, M. J., Harrison, T., and Overman, L. E. (1991). General approach to halogenated tetrahydrofuran natural products from red algae of the genus Laurencia. Total synthesis of (.+-.)-trans-kumausyne and demonstration of an asymmetric synthesis strategy. J. Am. Chem. Soc. 113 (14), 5378–5384. doi:10.1021/ja00014a032
Cai, Y. Q., Hu, J. H., Qin, J., Sun, T., and Li, X. L. (2018). Rhododendron Molle (Ericaceae): phytochemistry, pharmacology, and toxicology. Chin. J. Nat. Med. 16 (6), 401–410. doi:10.1016/S1875-5364(18)30073-6
Carreira, E. M., and Joliton, A. (2015). Total synthesis of (–)-Amphidinolide P. Synfacts 11 (09), 0903. doi:10.1055/s-0034-1378759
Castillo, M., Morales, G., Loyola’, L. A., Calvo, C., Holland, H. L., Maclean, D. B., et al. (2011). The alkaloids of L. paniculatum and the structure of paniculatine. Can. J. Chem. 54 (18), 2900–2908. doi:10.1139/v76-410
Castillo, M., Morales, G., Loyola, L. A., Singh, I., Calvo, C., Holland, H. L., et al. (1975). Paniculatine. A new alkaloid from L. paniculatum desvaux. Can. J. Chem. 53 (16), 2513–2514. doi:10.1139/v75-357
Chan, T. H., and Wang, D. (1992). Chiral organosilicon compounds in asymmetric synthesis. Chem. Rev. 92 (5), 995–1006. doi:10.1021/cr00013a012
Chemler, S. R., and Roush, W. R. (2000). Recent applications of the allylation reaction to the synthesis of natural products 1, pp. 403–490. John Wiley and Sons, Ltd. doi:10.1002/9783527613267.ch11
Chen, S., Wang, J., and Qiu, F. G. (2018). A divergent and concise total synthesis of (-)-lycoposerramine R and (+)-lycopladine A. Chem. Commun. 54 (29), 3598–3600. doi:10.1039/c8cc01626g
Chien, J. C. W., Tsai, W. M., Rausch, M. D., and Rausch, M. D. (1991). Isospecific polymerization of propylene catalyzed by rac-Ethylenebis(indenyl)methylzirconium “cation.”. J. Am. Chem. Soc. 113 (22), 8570–8571. doi:10.1021/ja00022a081
Chu, Z., Wang, K., Gao, L., and Song, Z. (2017). Chiral crotyl geminal bis(silane): a useful reagent for asymmetric Sakurai allylation by selective desilylation-enabled chirality transfer. Chem. Commun. 53 (21), 3078–3081. doi:10.1039/C7CC00632B
Colvin, E. W. (1978). Silicon in organic synthesis. Chem. Soc. Rev. 7 (1), 15–64. doi:10.1039/cs9780700015
Cram, D. J., and Elhafez, F. A. A. (1952). Studies in stereochemistry. X. The rule of “steric control of asymmetric induction” in the syntheses of acyclic systems. J. Am. Chem. Soc. 74 (23), 5828–5835. doi:10.1021/ja01143a007
Crimmins, M. T., and Emmitte, K. A. (2001). Asymmetric total synthesis of (−)-Isolaurallene. J. Am. Chem. Soc. 123 (7), 1533–1534. doi:10.1021/ja005892h
Crimmins, M. T., Emmitte, K. A., and Choy, A. L. (2002). Ring closing metathesis for the formation of medium ring ethers: the total synthesis of (-)-isolaurallene. Tetrahedron 58 (10), 1817–1834. doi:10.1016/S0040-4020(02)00040-6
Crimmins, M. T., Mans, M. C., and Rodríguez, A. D. (2010). Total synthesis of the proposed structure of briarellin J. Org. Lett. 12 (21), 5028–5031. doi:10.1021/ol102169w
Daoust, J., Chen, M., Wang, M., Williams, D. E., Chavez, M. A. G., Wang, Y. A., et al. (2013). Sesterterpenoids isolated from a northeastern pacific phorbas sp. J. Org. Chem. 78 (17), 8267–8273. doi:10.1021/jo4014589
Das, S., and Ramana, C. V. (2015). A formal total synthesis of (−)-kumausallene. Tetrahedron 71 (45), 8577–8584. doi:10.1016/j.tet.2015.09.027
Déléris, G., Pillot, J. P., and Rayez, J. C. (1980). Influence of a silyl group on an allylic position. A theoretical approach. Tetrahedron 36 (15), 2215–2218. doi:10.1016/0040-4020(80)80114-1
Deng, Z. T., Wu, X.De, Yuan, Z. F., Yu, N. R., Ou, Y. F., and Zhao, Q. S. (2022). Total synthesis of huperserratines A and B. Org. Chem. Front. 9 (14), 3664–3668. doi:10.1039/D2QO00608A
Denmark, S. E., and Almstead, N. G. (1994). Stereochemical studies on the addition of allylsilanes to aldehydes. The SE′ component. J. Org. Chem. 59 (18), 5130–5132. doi:10.1021/jo00097a008
Denmark, S. E., Coe, D. M., Pratt, N. E., and Griedel, B. D. (1994). Asymmetric allylation of aldehydes with chiral lewis bases. J. Org. Chem. 59 (21), 6161–6163. doi:10.1021/jo00100a013
Denmark, S. E., and Fu, J. (2001). Catalytic, enantioselective addition of substituted allylic trichlorosilanes using a rationally-designed 2,2‘-bispyrrolidine-based bisphosphoramide. J. Am. Chem. Soc. 123 (38), 9488–9489. doi:10.1021/ja016552e
Denmark, S. E., and Fu, J. (2003). Catalytic enantioselective addition of allylic organometallic reagents to aldehydes and ketones. Chem. Rev. 103 (8), 2763–2794. doi:10.1021/cr020050h
Dilman, A. D., and Ioffe, S. L. (2003). Carbon-carbon bond forming reactions mediated by Silicon Lewis acids. Chem. Rev. 103 (3), 733–772. doi:10.1021/cr020003p
Don-Doncow, N., Escobar, Z., Johansson, M., Kjellström, S., Garcia, V., Munoz, E., et al. (2014). Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells. J. Biol. Chem. 289 (23), 15969–15978. doi:10.1074/jbc.M114.564252
Du, K., Yang, H., Guo, P., Feng, L., Xu, G., Zhou, Q., et al. (2017). Efficient syntheses of (−)-crinine and (−)-aspidospermidine, and the formal synthesis of (−)-minfiensine by enantioselective intramolecular dearomative cyclization. Chem. Sci. 8 (9), 6247–6256. doi:10.1039/C7SC01859B
Du, L., Robles, A., King, J., Powell, D., Miller, A., Mooberry, S., et al. (2013). Maximiscin, a novel shikimate-polyketide-NRPS hybrid metabolite obtained from Tolypocladium sp. with potent antitumor activities. Planta Medica 79 (10), CL3. doi:10.1055/s-0033-1348528
Du, L., Robles, A. J., King, J. B., Powell, D. R., Miller, A. N., Mooberry, S. L., et al. (2014). Crowdsourcing natural products discovery to access uncharted dimensions of fungal metabolite diversity. Angew. Chem. 126 (3), 823–828. doi:10.1002/ange.201306549
Ella-Menye, J. R., Dobbs, W., Billet, M., Klotz, P., and Mann, A. (2005). Unexpected 1,2 syn diastereoselectivity in the three-component “aza Sakurai-Hosomi” reaction. Tetrahedron Lett. 46 (11), 1897–1900. doi:10.1016/j.tetlet.2005.01.077
Evans, P., Murthy, V., Roseman, J., and Rheingold, A. L. (1999). Enantioselective total synthesis of the nonisoprenoid sesquiterpene (−)-Kumausallene. Angew. Chem. Int. Ed. 38 (21), 3175–3177. doi:10.1002/(SICI)1521-3773(19991102)38:21<3175::AID-ANIE3175>3.0.CO;2-M
Fan, Y. Y., Xu, J. B., Liu, H. C., Gan, L. S., Ding, J., and Yue, J. M. (2017). Cephanolides A-J, cephalotane-type diterpenoids from Cephalotaxus sinensis. J. Nat. Prod. 80 (12), 3159–3166. doi:10.1021/acs.jnatprod.7b00412
Fang, F. G., Kishi, Y., Matclich, M. C., and Scola, P. M. (1992). Synthetic studies towards halichondrins synthesis of the left halves of norhalichondrins and homohalichondrins. Tetrahedron Lett. 33 (12), 1557–1560. doi:10.1016/S0040-4039(00)91673-3
Farina, V., Reeves, J. T., Senanayake, C. H., and Song, J. J. (2006). Asymmetric synthesis of active pharmaceutical ingredients. Chem. Rev. 106 (7), 2734–2793. doi:10.1021/cr040700c
Feldman, K. S. (1982). Biomimetic synthesis of (±) panacene. Tetrahedron Lett. 23 (30), 3031–3034. doi:10.1016/s0040-4039(00)87525-5
Feldman, K. S., Mechem, C. C., and Nader, L. (1982). Total synthesis of (±)-Panacene. J. Am. Chem. Soc. 104 (14), 4011–4012. doi:10.1021/cr040700c
Fent, K. (1996). Ecotoxicology of organotin compounds. In critical reviews in toxicology. 26, 3–117. doi:10.3109/10408449609089891
Fleming, I. (1991). “Allylsilanes, allylstannanes and related systems,” in Comprehensive organic synthesis (Elsevier), 563–593. doi:10.1016/b978-0-08-052349-1.00041-x
Fleming, I., Barbero, A., and Walter, D. (1997). Stereochemical control in organic synthesis using silicon-containing compounds. Chem. Rev. 97 (6), 2063–2192. doi:10.1021/cr941074u
Fleming, I., Dunoguès, J., and Smithers, R. (1989). The electrophilic substitution of allylsilanes and vinylsilanes. Org. React. 37 (57), 57–575. doi:10.1002/0471264180.or037.02
Forestieri, R., Merchant, C. E., De Voogd, N. J., Matainaho, T., Kieffer, T. J., and Andersen, R. J. (2009). Alotaketals A and B, sesterterpenoids from the marine sponge hamigera species that activate the camp cell signaling pathway. Org. Lett. 11 (22), 5166–5169. doi:10.1021/ol902066e
Fujioka, K., Yokoe, H., Inoue, A., Soga, K., Tsubuki, M., and Shishido, K. (2014). Enantioselective synthesis of (+)-Penostatin E. J. Org. Chem. 79 (16), 7512–7519. doi:10.1021/jo501225y
Fujiwara, H., Matsunaga, K., Kumagai, H., Ishizuka, M., and Ohizumi, Y. (2000). Ophiobolin A, a novel apoptosis-inducing agent from fungus strain f-7438. Pharm. Pharmacol. Commun. 6 (9), 427–431. doi:10.1211/146080800128736312
Fürstner, A., and Voigtländer, D. (2000). “Halophilic activation” of chlorosilanes: allylation of aldehydes catalyzed by platinum dichloride or silver triflate. Synthesis 12 (7), 959–969. doi:10.1055/s-2000-6290
Gauthier, D. R., and Carreira, E. M. (1996). Catalytic, enantioselective addition of allylsilanes to aldehydes: generation of a novel, reactive TiIV complex from TiF4. Angewandte Chemie Int. Ed. Engl. 35 (20), 2363–2365. doi:10.1002/anie.199623631
Ghosh, A. K., and Li, J. (2011). A stereoselective synthesis of (+)-herboxidiene/GEX1A. Org. Lett. 13 (1), 66–69. doi:10.1021/ol102549a
Girard, N. (2015). “Synthesis of alkaloids containing a quinolizidine core by means of strategies based on a hydroformylation reaction,” in Strategies and tactics in organic synthesis. 1st ed., Vol. 11 (Elsevier Ltd). doi:10.1016/B978-0-08-100023-6.00008-7
Grese, T. A., Hutchinson, K. D., and Overman, L. E. (1993). General approach to halogenated tetrahydrofuran natural products from red algae of the genus Laurencia. Total synthesis of (.+-.)-kumausallene and (.+-.)-1-epi-kumausallene. J. Org. Chem. 58 (9), 2468–2477. doi:10.1021/jo00061a021
Grisin, A., and Evans, P. A. (2015). A highly convergent synthesis of the C1–C31 polyol domain of amphidinol 3 featuring a TST-RCM reaction: confirmation of the revised relative stereochemistry. Chem. Sci. 6 (11), 6407–6412. doi:10.1039/C5SC00814J
Guzmán, E. A., Xu, Q., Pitts, T. P., Mitsuhashi, K. O., Baker, C., Linley, P. A., et al. (2016). Leiodermatolide, a novel marine natural product, has potent cytotoxic and antimitotic activity against cancer cells, appears to affect microtubule dynamics, and exhibits antitumor activity. Int. J. Cancer 139 (9), 2116–2126. doi:10.1002/IJC.30253
Harrison, W. A., Curcumelli-Rodostamo, M., Carson, D. F., Barclay, I. R. C., and MacLean, D. B. (1961). Lycopodium alkaloids x. The structure of Lycopodine. Can. J. Chem., 39(1), 2086–2099. doi:10.1139/v61-277
Hashimoto, T., Koyama, H., Takaoka, S., Tori, M., and Asakawa, Y. (1994). Trifarienols A and B, Isolated from the liverwort Cheilolejeunea trifaria. Sesquiterpenes having a new carbon skeleton, trifarane. Tetrahedron Lett. 35 (27), 4787–4788. doi:10.1016/S0040-4039(00)76968-1
Hayashi, T., Usuki, Y., and Iio, H. (2010). 2-Fluoro-3-phenyl-allyltrimethylsilane: a new fluorinated reagent for Hosomi-Sakurai reaction. J. Fluor. Chem. 131 (6), 709–713. doi:10.1016/j.jfluchem.2010.03.005
Hegedus, L. S., Allen, G. F., Bozell, J. J., and Waterman, E. L. (1978). Palladium-Assisted intramolecular amination of olefins. Synthesis of nitrogen heterocycles. J. Am. Chem. Soc. 100 (18), 5800–5807. doi:10.1021/ja00486a035
Henderson, J. A., Phillips, A. J., Henderson, J. A., and Phillips, A. J. (2008). Total synthesis of Aburatubolactam A. Angew. Chem. Int. Ed. 47 (44), 8499–8501. doi:10.1002/anie.200803593
Heravi, M. M., and Mohammadkhani, L. (2018). Recent applications of Stille reaction in total synthesis of natural products: an update. J. Organomet. Chem. 869 (15), 106–200. doi:10.1016/j.jorganchem.2018.05.018
Ho, L.-K., and Lin, W.-N. (1995). Quercetin 5,4′-dimethyl ether from Rhododendron ellipticum. Phytochemistry 39 (2), 463–464. doi:10.1016/0031-9422(94)00905-9
Hoffmann-Röder, A., and Krause, N. (2004). Synthesis and properties of allenic natural products and pharmaceuticals. Angew. Chem. - Int. Ed. 43 (10), 1196–1216. doi:10.1002/ANIE.200300628
Hollis, T. K., and Bosnich, B. (1995). Homogeneous catalysis. Mechanisms of the catalytic Mukaiyama aldol and Sakurai allylation reactions. J. Am. Chem. Soc. 117 (16), 4570–4581. doi:10.1021/ja00121a015
Hosomi, A. (1988). Characteristics in the reactions of allylsilanes and their applications to versatile synthetic equivalents. Accounts Chem. Res. 21 (5), 200–206. doi:10.1021/ar00149a004
Hosomi, A., Endo, M., and Sakurai, H. (1978a). Selective allylations of α,β-unsaturated acetals with allylsilanes in the presence of lewis acid. Chem. Lett. 7 (5), 499–502. doi:10.1246/cl.1978.499
Hosomi, A., Sakata, Y., and Sakurai, H. (1984). N-(trimethylsilylmethyl)aminomethyl ethers as azomethine ylide synthons. A new and convenient access to pyrrolidine derivatives. Chem. Lett. 13 (7), 1117–1120. doi:10.1246/cl.1984.1117
Hosomi, A., and Sakurai, H. (1976). Syntheses of γ,δ-unsaturated alcohols from allylsilanes and carbonyl compounds in the presence of titanium tetrachloride. Tetrahedron Lett. 17 (16), 1295–1298. doi:10.1016/S0040-4039(00)78044-0
Hosomi, A., and Sakurai, H. (1977). Chemistry of organosilicon compounds. 99. Conjugate addition of allylsilanes to.alpha., beta.-enones. A New method of stereoselective introduction of the angular allyl group in fused cyclic.alpha., beta.-enones. J. Am. Chem. Soc. 99 (5), 1673–1675. doi:10.1021/ja00447a080
Hosomi, A., Shirahata, A., and Sakurai, H. (1978b). Chemistry of organosilicon compounds 113 chemoselective allylation of carbonyl compounds with allylsilanes promoted by tetra-butylammonium fluoride. A new synthesis of homoallyl alcohols. Tetrahedron Lett. 19 (33), 3043–3046. doi:10.1016/S0040-4039(01)94934-2
Huang, C., and Liu, B. (2010). Asymmetric total synthesis of ent-heliespirones A and C. Chem. Commun. 46 (29), 5280–5282. doi:10.1039/C0CC00481B
Huang, C., Zhang, W., and Liu, B. (2011). Racemic and enantioselective total synthesis of heliespirones A and C. Sci. China Chem. 54 (1), 43–55. doi:10.1007/s11426-010-4173-y
Huang, G. H., Hu, Z., Lei, C., Wang, P. P., Yang, J., Li, J. Y., et al. (2018). Enantiomeric pairs of meroterpenoids with diverse heterocyclic systems from Rhododendron nyingchiense. J. Nat. Prod. 81 (8), 1810–1818. doi:10.1021/acs.jnatprod.8b00273
Huang, H., and Forsyth, C. J. (1995). Anti selective spirocarbomercuration: synthesis and stereochemistry of the spirobicyclic sesquiterpenes spirojatamol and erythrodiene. J. Org. Chem. 60 (9), 2773–2779. doi:10.1021/jo00114a027
Huang, X. Z., Wang, Y. H., Yu, S. S., Fu, G. M., Hu, Y. C., Liu, Y., et al. (2005). Iridoid glycosides and grayanane diterpenoids from the roots of Craibiodendron henryi. J. Nat. Prod. 68 (11), 1646–1650. doi:10.1021/np050261s
Hubert, J. G., Furkert, D. P., and Brimble, M. A. (2015). Synthesis of the spirocyclic framework of sesterterpenoid natural products. J. Org. Chem. 80 (5), 2715–2723. doi:10.1021/jo502897u
Huckins, J. R., De Vicente, J., and Rychnovsky, S. D. (2007). Synthesis of the C1-C52 fragment of amphidinol 3, featuring a β-alkoxy alkyllithium addition reaction. Org. Lett. 9 (23), 4757–4760. doi:10.1021/ol7020934
Indictor, N., and Brill, W. F. (1965). Metal acetylacetonate catalyzed epoxidation of olefins with t-butyl hydroperoxide. J. Org. Chem. 30 (6), 2074–2075. doi:10.1021/jo01017a520
Isaac, B. G., Ayer, S. W., Elliott, R. C., and Stonard, R. J. (1992). Herboxidiene: a potent phytotoxic polyketide from Streptomyces sp. A7847. J. Org. Chem. 57 (26), 7220–7226. doi:10.1021/jo00052a042
Iseki, K., Mizuno, S., Kuroki, Y., and Kobayashi, Y. (1999). Asymmetric allylation with chiral formamide catalysts. Tetrahedron 55 (4), 977–988. doi:10.1016/S0040-4020(98)01097-7
Ishibashi, M., and Kobayashi, J. (1997). Amphidinolides: unique macrolides from marine dinoflagellates. Heterocycles 44 (1), 543–572. doi:10.3987/rev-96-sr1
Ishihara, K., Mouri, M., Gao, Q., Maruyama, T., Furuta, K., and Yamamoto, H. (1993). Catalytic asymmetric allylation using a chiral (Acyloxy)borane complex as a versatile lewis acid catalyst. J. Am. Chem. Soc. 115 (24), 11490–11495. doi:10.1021/ja00077a054
Israr, M., Kim, W. H., Kim, D. H., and Bae, H. Y. (2024). Trityl tetrakis(pentafluorophenyl)borate: a sustainable precursor for multifunctional silylium Lewis acid organocatalysis. Chem. Catal. 4 (4). doi:10.1016/J.CHECAT.2023.100866/ASSET/C6DF1B90-8A65-4DB5-ABA8-FFE0B53E3589/MAIN.ASSETS/SC12.SML
Itsuno, S. (2005). Chiral polymer synthesis by means of repeated asymmetric reaction. Prog. Polym. Sci. Oxf. 30 (5), 540–558. doi:10.1016/j.progpolymsci.2005.01.008
Jansma, M. J., and Hoye, T. R. (2012). Oxyanionic sigmatropic rearrangements relevant to cyclooctadienone formation in penostatins I and F. Org. Lett. 14 (18), 4738–4741. doi:10.1021/ol3019488
Jeong, W., Kim, M. J., Kim, H., Kim, S., Kim, D., and Shin, K. J. (2010). Substrate-controlled asymmetric total synthesis and structure revision of (+)-itomanallene a. Angew. Chem. - Int. Ed. 49 (4), 752–756. doi:10.1002/anie.200905826
Johansson, M., and Sterner, O. (2001). Synthesis of (+)-Galiellalactone. Absolute configuration of galiellalactone. Org. Lett. 3 (18), 2843–2845. doi:10.1021/ol016286+
Joung, S., Kim, R., and Lee, H. Y. (2017). Total synthesis of (−)-Phorbaketal A. Org. Lett. 19 (14), 3903–3906. doi:10.1021/ACS.ORGLETT.7B01797
Kaib, P. S. J., Schreyer, L., Lee, S., Properzi, R., and List, B. (2016). Extremely active organocatalysts enable a highly enantioselective addition of allyltrimethylsilane to aldehydes. Angew. Chem. Int. Ed. 55 (42), 13200–13203. doi:10.1002/anie.201607828
Kakisawa, H., Kurono, M., Takahashi, S., and Hirata, Y. (1961). Structure of grayanotoxin-I and -III. Tetrahedron Lett. 2 (2), 59–67. doi:10.1016/S0040-4039(01)99208-1
Kamiya, A., Kawamoto, Y., Kobayashi, T., and Ito, H. (2021). Total synthesis of ent-callilongisin B. Org. Lett. 23 (17), 6916–6918. doi:10.1021/acs.orglett.1c02473
Kamiya, A., Kawamoto, Y., Kobayashi, T., and Ito, H. (2022). Asymmetric syntheses of ent-callilongisins B and C. Tetrahedron 111 (12), 132712–141337. doi:10.1016/j.tet.2022.132712
Kampen, D., Ladépêche, A., Claßen, G., and Lista, B. (2008). Brønsted acid-catalyzed three-component hosomi–sakurai reactions. Adv. Synthesis and Catal. 350 (7–8), 962–966. doi:10.1002/adsc.200800036
Keck, G. E., and Yates, J. B. (1982). Carbon-carbon bond formation via the reaction of trialkylallylstannanes with organic halides. J. Am. Chem. Soc. 104 (21), 5829–5831. doi:10.1021/ja00385a066
Kim, C. Y., Mitchell, A. J., Glinkerman, C. M., Li, F. S., Pluskal, T., and Weng, J. K. (2020). The chloroalkaloid (−)-acutumine is biosynthesized via a Fe(II)- and 2-oxoglutarate-dependent halogenase in Menispermaceae plants. Nat. Commun. 11 (1), 1867–7. doi:10.1038/s41467-020-15777-w
Kim, T., Han, Y. T., An, H., Kim, K., Lee, J., and Suh, Y. G. (2015). Diastereoselective total synthesis of (-)-Galiellalactone. J. Org. Chem. 80 (24), 12193–12200. doi:10.1021/acs.joc.5b02121
King, S. M., Calandra, N. A., and Herzon, S. B. (2013). Total syntheses of (−)-Acutumine and (−)-Dechloroacutumine. Angew. Chem. 125 (13), 3642–3645. doi:10.1002/anie.201210076
Kira, M., Sato, K., and Sakurai, H. (1990). Highly stereoselective allylation of aldehydes with pentacoordinate allylsilicates in hydroxylic media. Discrimination between linear and.alpha.-branched alkanals. J. Am. Chem. Soc. 112 (1), 257–260. doi:10.1021/ja00157a040
Kobayashi, J., Ishibashi, M., Nakamura, H., Ohizumi, Y., Yamasu, T., Sasaki, T., et al. (1986). Amphidinolide-A, a novel antineoplastic macrolide from the marine dinoflagellate Amphidinium sp. Tetrahedron Lett. 27 (47), 5755–5758. doi:10.1016/S0040-4039(00)85318-6
Komatsu, N., Uda, M., Suzuki, H., Takahashi, T., Domae, T., and Wada, M. (1997). Bismuth bromide as an efficient and versatile catalyst for the cyanation and allylation of carbonyl compounds and acetals with organosilicon reagents. Tetrahedron Lett. 38 (41), 7215–7218. doi:10.1016/S0040-4039(97)01719-X
Komiyama, T., Minami, Y., and Hiyama, T. (2017). Recent advances in transition-metal-catalyzed synthetic transformations of organosilicon reagents. ACS Catalysis,, 7 (1), 631–651. doi:10.1021/acscatal.6b02374
Kong, L., Yu, H., Deng, M., Wu, F., Chen, S. C., and Luo, T. (2023). Enantioselective total syntheses of grayanane diterpenoids and (+)-Kalmanol: evolution of the bridgehead carbocation-based cyclization and late-stage functional group manipulation strategies. J. Org. Chem. 88 (9), 6017–6038. doi:10.1021/acs.joc.3c00365
Kong, L., Yu, H., Deng, M., Wu, F., Jiang, Z., and Luo, T. (2022). Enantioselective total syntheses of grayanane diterpenoids: (-)-Grayanotoxin III, (+)-Principinol E, and (-)-Rhodomollein XX. J. Am. Chem. Soc. 144 (12), 5268–5273. doi:10.1021/jacs.2c01692
Kosugi, M., Sumiya, T., Ohhashi, K., Sano, H., and Migita, T. (1985). Novel hydroxymethylation of aryl bromides by means of organotin reagents. Chem. Lett. 14 (7), 997–998. doi:10.1246/cl.1985.997
Kozaka, T., Miyakoshi, N., and Mukai, C. (2007). Stereoselective total syntheses of three Lycopodium alkaloids, (-)-magellanine, (+)-magellaninone, and (+)-paniculatine, based on two Pauson-Khand reactions. J. Org. Chem. 72 (26), 10147–10154. doi:10.1021/jo702136b
Krutov, S. M., Samek, Z., Benešova, V., and Herout, V. (1973). Isolation and the structure of deoxopinguisone from the liverwort Ptilidium ciliare. Phytochemistry 12 (6), 1405–1407. doi:10.1016/0031-9422(73)80574-6
Kulkarni, Y. A., Garud, M. S., Gaud, R. S., and Gaikwad, A. B. (2016). “Recent developments in using plant-derived natural products as tubulin inhibitors for the management of cancer,” in Recent developments in using plant-derived natural products as tubulin inhibitors for the management of cancer, 1. Academic Press, 507–524. doi:10.1016/B978-0-12-802972-5.00024-X
Kumagai, T., and Itsuno, S. (2000). Asymmetric allylation polymerization: novel polyaddition of bis(allylsilane) and dialdehyde using chiral (acyloxy)borane catalyst. Macromolecules 33 (14), 4995–4996. doi:10.1021/ma000502x
Lade, J. J., Pardeshi, S. D., Vadagaonkar, K. S., Murugan, K., and Chaskar, A. C. (2017). The remarkable journey of catalysts from stoichiometric to catalytic quantity for allyltrimethylsilane inspired allylation of acetals, ketals, aldehydes and ketones. RSC Adv. 7 (13), 8011–8033. doi:10.1039/C6RA27813B
Langkopf, E., and Schinzer, D. (1995). Uses of silicon-containing compounds in the synthesis of natural products. Chem. Rev. 95 (5), 1375–1408. doi:10.1021/cr00037a011
Lee, J. H. (2020). Use of the Hosomi-Sakurai allylation in natural product total synthesis. Tetrahedron 76 (33), 131351–131367. doi:10.1016/j.tet.2020.131351
Lee, J. H., Li, Z., Osawa, A., and Kishi, Y. (2016). Extension of Pd-mediated one-pot ketone synthesis to macrocyclization: application to a new convergent synthesis of Eribulin. J. Am. Chem. Soc. 138 (50), 16248–16251. doi:10.1021/jacs.6b11663
Lee, Y., Wang, W., Kim, H., Giri, A. G., Won, D. H., Hahn, D., et al. (2014). Phorbaketals L-N, cytotoxic sesterterpenoids isolated from the marine sponge of the genus Phorbas. Bioorg. Med. Chem. Lett. 24 (17), 4095–4098. doi:10.1016/j.bmcl.2014.07.066
Li, C. H., Zhang, J. Y., Zhang, X. Y., Li, S. H., and Gao, J. M. (2019). An overview of grayanane diterpenoids and their biological activities from the Ericaceae family in the last seven years. Eur. J. Med. Chem. 166, 400–416. doi:10.1016/J.EJMECH.2019.01.079
Li, C. J., Wang, L. Q., Chen, S. N., and Qin, G. W. (2000). Diterpenoids from the fruits of Rhododendron molle. J. Nat. Prod. 63 (9), 1214–1217. doi:10.1021/NP000009E
Li, F., Tartakoff, S. S., and Castle, S. L. (2009). Enantioselective total synthesis of (-)-acutumine. J. Org. Chem. 74 (23), 9082–9093. doi:10.1021/jo902006q
Li, H., Zhang, J., and She, X. (2021). The total synthesis of diquinane-containing natural products. Chem. – A Eur. J. 27 (15), 4839–4858. doi:10.1002/chem.202003741
Li, J. J. (1999). Applications of palladium chemistry to the total syntheses of naturally occurring indole alkaloids. Alkaloids Chem. Biol. Perspect. 14 (C), 437–503. doi:10.1016/S0735-8210(99)80006-5
Li, K., Wang, C., Yin, G., and Gao, S. (2013). Construction of the basic skeleton of ophiobolin A and variecolin. Org. Biomol. Chem. 11 (43), 7550–7558. doi:10.1039/C3OB41693C
Li, X., Zhang, W., and Gao, S. (2018). Total synthesis of complex natural products: combination of chemical synthesis and biosynthesis strategies. Chin. J. Org. Chem. 38 (9), 2185–2198. doi:10.6023/cjoc201806019
Liao, H. B., Huang, G. H., Yu, M. H., Lei, C., and Hou, A. J. (2017). Five pairs of meroterpenoid enantiomers from Rhododendron capitatum. J. Org. Chem. 82 (3), 1632–1637. doi:10.1021/ACS.JOC.6B02800
Liao, H. B., Lei, C., Gao, L. X., Li, J. Y., Li, J., and Hou, A. J. (2015). Two enantiomeric pairs of meroterpenoids from Rhododendron capitatum. Org. Lett. 17 (20), 5040–5043. doi:10.1021/ACS.ORGLETT.5B02515
Liu, C. C., Lei, C., Zhong, Y., Gao, L. X., Li, J. Y., Yu, M. H., et al. (2014). Novel grayanane diterpenoids from Rhododendron principis. Tetrahedron 70 (29), 4317–4322. doi:10.1016/J.TET.2014.05.019
Liu, J., Chen, S., Li, N., and Qiu, F. G. (2019). A concise total synthesis of (+)-Paniculatine. Adv. Synthesis Catal. 361 (15), 3514–3517. doi:10.1002/adsc.201900376
Liu, J. S., Zhu, Y. L., Yu, C. M., Zhou, Y. Z., Han, Y. Y., Wu, F. W., et al. (1986). The structures of huperzine A and B,’ two new alkaloids exhibiting marked anticholinesterase activity. Can. J. Chem. 64 (4), 837–839. doi:10.1139/v86-137@cjc-csc.issue01
Liu, S., Sun, L., Zhang, P., and Niu, C. (2024). Recent advances in grayanane diterpenes: isolation, structural diversity, and bioactivities from Ericaceae family (2018–2024). Molecules 29 (7), 1649. doi:10.3390/molecules29071649
Liu, Y. W., Cheng, Y. B., Liaw, C. C., Chen, C. H., Guh, J. H., Hwang, T. L., et al. (2012). Bioactive diterpenes from Callicarpa longissima. J. Nat. Prod. 75 (4), 689–693. doi:10.1021/NP200932K
Loyola, L. A., Morales, G., and Castillo, M. (1979). Alkaloids of lycopodium magellanicum. Phytochemistry 18 (10), 1721–1723. doi:10.1016/0031-9422(79)80193-4
Lv, X. J., Li, Y., Ma, S. G., Qu, J., Liu, Y. B., Li, L., et al. (2017). Isopimarane and nor-diterpene glucosides from the twigs and leaves of Lyonia ovalifolia. Tetrahedron 73 (6), 776–784. doi:10.1016/J.TET.2016.12.060
Ma, D., Zhong, Z., Liu, Z., Zhang, M., Xu, S., Xu, D., et al. (2016). Protecting-group-free total synthesis of (-)-Lycopodine via phosphoric acid promoted alkyne aza-prins cyclization. Org. Lett. 18 (17), 4328–4331. doi:10.1021/acs.orglett.6b02072
Ma, X., and Gang, D. R. (2004). The Lycopodium alkaloids. Nat. Product. Rep. 21 (6), 752–772. doi:10.1039/B409720N
Maclean, D. B. (1985). Chapter 5 lycopodium alkaloids. Alkaloids Chem. Pharmacol. 26 (C), 241–298. doi:10.1016/S0099-9598(08)60196-0
Mahlau, M., and List, B. (2013). Asymmetric counteranion-directed catalysis: concept, definition, and applications. Angew. Chem. Int. Ed. 52 (2), 518–533. doi:10.1002/anie.201205343
Majetich, G., Casares, A., Chapman, D., and Behnke, M. (1986). A general allylation procedure using trimethylallylsilane and fluoride catalysis. J. Org. Chem. 51 (10), 1745–1753. doi:10.1021/jo00360a021
Majetich, G., Lowery, D., and Khetani, V. (1990). A direct synthesis of (±)-neolemnanyl acetates. Tetrahedron Lett. 31 (1), 51–54. doi:10.1016/S0040-4039(00)94331-4
Makita, N., Hoshino, Y., and Yamamoto, H. (2003). Asymmetric epoxidation of homoallylic alcohols and application in a concise total synthesis of (-)-α-bisabolol and (-)-8-epi-α-bisabolol. Angew. Chem. - Int. Ed. 42 (8), 941–943. doi:10.1002/anie.200390250
Malkov, A. V., Orsini, M., Pernazza, D., Muir, K. W., Langer, V., Meghani, P., et al. (2002). Chiral 2,2′-bipyridine-type N-monoxides as organocatalysts in the enantioselective allylation of aldehydes with allyltrichlorosilane. Org. Lett. 4 (6), 1047–1049. doi:10.1021/ol025654m
Malkov, A. V., Ramírez-López, P., Biedermannová, L., Rulíšek, L., Dufková, L., Kotora, M., et al. (2008). On the mechanism of asymmetric allylation of aldehydes with allyltrichlorosilanes catalyzed by QUINOX, a chiral isoquinoline N-oxide. J. Am. Chem. Soc. 130 (15), 5341–5348. doi:10.1021/ja711338q
Manske, R. H. F. (1960). The alkaloids. Chemistry and physiology. Academic Press. doi:10.1016/S1876-0813(08)60018-4
Masse, C. E., and Panek, J. S. (1995). Diastereoselective reactions of chiral allyl and allenyl silanes with activated C:X.pi.-Bonds. Chem. Rev. 95 (5), 1293–1316. doi:10.1021/cr00037a008
May, T. L., Dabrowski, J. A., and Hoveyda, A. H. (2011). Formation of vinyl-vinylhalide-or acyl-substituted quaternary carbon stereogenic centers through NHC-Cu-catalyzed enantioselective conjugate additions of Si-containing vinylaluminums to β-substituted cyclic enones. J. Am. Chem. Soc. 133 (4), 736–739. doi:10.1021/ja110054q
Mcclymont, K. S., Wang, F., Minakar, A., and Baran, P. S. (2020). Total synthesis of (−)-Maximiscin. J. Am. Chem. Soc. 142 (19), 8608–8613. doi:10.1021/jacs.0c03202
Mirabdolbaghi, R., and Dudding, T. (2012). A catalytic asymmetric approach to C 1-chiral 3-methylene-indan-1-ols. Tetrahedron 68 (7), 1988–1991. doi:10.1016/j.tet.2011.12.040
Miyawaki, A., Kikuchi, D., Niki, M., Manabe, Y., Kanematsu, M., Yokoe, H., et al. (2012). Total synthesis of natural enantiomers of heliespirones A and C via the diastereoselective intramolecular Hosomi-Sakurai reaction. J. Org. Chem. 77 (18), 8231–8243. doi:10.1021/jo3016055
Motiwala, H. F., Armaly, A. M., Cacioppo, J. G., Coombs, T. C., Koehn, K. R. K., Norwood, V. M., et al. (2022). HFIP in organic synthesis. Chem. Rev. 122 (15), 12544–12747. doi:10.1021/acs.chemrev.1c00749
Mukaiyama, T., Kobayashi, S., and Murakami, M. (1984). Trityl perchlorate as an efficient catalyst in the aldol-type reaction. Chem. Lett. 13 (10), 1759–1762. doi:10.1246/cl.1984.1759
Narayanan, P., Röhrl, M., Zechmeister, K., and Hoppe, W. (1970). Crystal and molecular structure of grayanotoxin - I, C22H36O7. Tetrahedron Lett. 11 (45), 3943–3944. doi:10.1016/S0040-4039(01)98631-9
Nasam, R., and Pabbaraja, S. (2024). A convergent approach for the synthesis of C14–C26 fragment of anticancer drug Eribulin mesylate. Synthesis 56 (03), 462–468. doi:10.1055/a-2202-5597
Nishiyama, H., Narimatsu, S., Sakuta, K., and Itoh, K. (1982). Reaction of allylsilanes and monothioacetals in the presence of Lewis acids: regioselectivity in the cleavage of the acetals. J. Chem. Soc. Chem. Commun. 2 (8), 459–460. doi:10.1039/C9CC01159E
Niwa, Y., Miyake, M., Hayakawa, I., and Sakakura, A. (2019). Catalytic enantioselective Hosomi-Sakurai reaction of α-ketoesters promoted by chiral copper(ii) complexes. Chem. Commun. 55 (27), 3923–3926. doi:10.1039/C9CC01159E
Nokami, J., Nomiyama, K., Matsuda, S., Imai, N., and Kataoka, K. (2003). Highly enantioselective alk-2-enylation of aldehydes through an allyl-transfer reaction. Angew. Chem. Int. Ed. 42 (11), 1273–1276. doi:10.1002/anie.200390327
Noyori, R., Murata, S., and Suzuki, M. (1981). Trimethysilyl triflate in organic synthesis11Part 11 of this series. Part 10: S. Murata and R. Noyori, Tetrahedron Letters 2107 (1981). Tetrahedron 37 (23), 3899–3910. doi:10.1016/S0040-4020(01)93263-6
Nozoe, S., Morisaki, M., Tsuda, K., Iitaka, Y., Takahashi, N., Tamura, S., et al. (1965). The structure of ophiobolin, a C25 terpenoid having a novel skeleton. J. Am. Chem. Soc. 87 (21), 4968–4970. doi:10.1021/ja00949a061
Ogoshi, S., Tomiyasu, S., Morita, M., and Kurosawa, H. (2002). Palladium/Me3SiOTf-catalyzed bis-silylation of α,β-unsaturated carbonyl compounds without involving oxidative addition of disilane. J. Am. Chem. Soc. 124 (39), 11598–11599. doi:10.1021/ja0272944
Oishi, T. (2024). A long journey toward structure revision and total synthesis of amphidinol 3. Mod. Nat. Product. Synth. 23 (11), 55–81. doi:10.1007/978-981-97-1619-7_3
Onishi, Y., Ito, T., Yasuda, M., and Baba, A. (2002). Remarkable enhancement of Lewis acidity of chlorosilane by the combined use of indium(III) chloride. Tetrahedron 58 (41), 8227–8235. doi:10.1016/S0040-4020(02)00972-9
Ortega, V., and Cortés, J. (2012). Potential clinical applications of halichondrins in breast cancer and other neoplasms. Breast Cancer Targets Ther. 4 (2), 9–19. doi:10.2147/BCTT.S12423
Pandey, G., Prasanna Kumara, C., Kumar Burugu, S., and Puranik, V. G. (2011). Enantioselective total syntheses of (-)-Isonitramine, (-)-Sibirine, and (+)-Nitramine by ring-closing metathesis. Eur. J. Org. Chem. 2011 (36), 7372–7377. doi:10.1002/ejoc.201101256
Park, J., Kim, B., Kim, H., Kim, S., and Kim, D. (2007). Substrate-controlled asymmetric total synthesis of (+)-microcladallene B with a bromination strategy based on a nucleophile-assisting leaving group. Angew. Chem. - Int. Ed. 46 (25), 4726–4728. doi:10.1002/anie.200700854
Paterson, I., Dalby, S. M., Roberts, J. C., Naylor, G. J., Guzmán, E. A., Isbrucker, R., et al. (2011). Leiodermatolide, a potent antimitotic macrolide from the marine sponge Leiodermatium sp. Angewandte Chemie Int. Ed. Engl. 50 (14), 3219–3223. doi:10.1002/ANIE.201007719
Pellissier, H. (2006). Asymmetric domino reactions. Part A: reactions based on the use of chiral auxiliaries. Tetrahedron 62 (8), 1619–1665. doi:10.1016/j.tet.2016.04.053
Pellissier, H. (2016). Recent developments in organocatalytic dynamic kinetic resolution. Tetrahedron 72 (23), 3133–3150. doi:10.1016/j.tet.2016.04.053
Ponomarenko, V. A., and Mironov, V. F. (1954). Synthesis of some alkenyl- and alkyl-silanes and their derivatives. Bull. Acad. Sci. USSR Div. Chem. Sci. 3 (3), 423–427. doi:10.1007/BF01167820
Ramirez, A., and Garcia-Rubio, S. (2005). Current progress in the chemistry and pharmacology of akuammiline alkaloids. Curr. Med. Chem. 10 (18), 1891–1915. doi:10.2174/0929867033457016
Roberts, D. D., and McLaughlin, M. G. (2022). Strategic applications of the β-silicon effect. Adv. Synthesis and Catal. 364 (14), 2307–2332. doi:10.1002/adsc.202200237
Rowley, M., Tsukamoto, M., and Kishi, Y. (1989). Total synthesis of (+)-Ophiobolin C. J. Am. Chem. Soc. 111 (7), 2735–2737. doi:10.1021/ja00189a069
Sabot, C., Guérard, K. C., and Canesi, S. (2009). Concise total synthesis of (±)-aspidospermidine via an oxidative Hosomi-Sakurai process. Chem. Commun. 20, 2941–2943. doi:10.1039/B903530C
Sai, M., and Yamamoto, H. (2015). Chiral brønsted acid as a true catalyst: asymmetric Mukaiyama aldol and hosomi–sakurai allylation reactions. J. Am. Chem. Soc. 137 (22), 7091–7094. doi:10.1021/jacs.5b04168
Saito, T., Awad, J. M., and Zhang, W. (2023). Synthetic studies on tetracyclic diquinane lycopodium alkaloids magellanine, magellaninone and paniculatine. Molecules 28 (3), 1501–1357. doi:10.3390/molecules28031501
Saitoh, T., Suzuki, T., Sugimoto, M., Hagiwara, H., and Hoshi, T. (2003). Total synthesis of (+)-laurallene. Tetrahedron Lett. 44 (15), 3175–3178. doi:10.1016/S0040-4039(03)00432-5
Sakai, Y., Tsujita, T., Akiyama, T., Yoshida, T., Mizukami, T., Akinaga, S., et al. (2002). GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. II. The effects on cell cycle progression and gene expression. J. Antibiotics 55 (10), 863–872. doi:10.7164/antibiotics.55.863
Santos, C. M. M., and Silva, A. M. S. (2018). Six-membered ring systems: with O and/or S atoms. Prog. Heterocycl. Chem. 30 (22), 473–532. doi:10.1016/B978-0-323-98410-2.00015-1
Schäfers, F., Dutta, S., Kleinmans, R., Mück-Lichtenfeld, C., and Glorius, F. (2022). Asymmetric addition of allylsilanes to aldehydes: a Cr/photoredox dual catalytic approach complementing the hosomi-sakurai reaction. ACS Catal. 12 (19), 12281–12290. doi:10.1021/acscatal.2c03960
Schmidt, K., Riese, U., Li, Z., and Hamburger, M. (2003). Novel tetramic acids and pyridone alkaloids, militarinones B, C, and D, from the insect pathogenic fungus Paecilomyces militaris. J. Nat. Prod. 66 (3), 378–383. doi:10.1021/np020430y
Schreyer, L., Properzi, R., and List, B. (2019). IDPi catalysis. Angew. Chem. Int. Ed. 58 (37), 12761–12777. doi:10.1002/anie.201900932
Schweig, A., Weidner, U., and Manuel, G. (1974). Theory and application of photoelectron spectroscopy. J. Organomet. Chem. 67 (1), C4–C6. doi:10.1016/S0022-328X(00)93659-3
Sha, C. K., Lee, F. K., and Chang, C. J. (1999). Tandem radical cyclizations initiated with α-carbonyl Radicals: first total synthesis of (+)-Paniculatine. J. Am. Chem. Soc. 121 (42), 9875–9876. doi:10.1021/ja992315o
Shao, H., Ma, Z. H., Cheng, Y. Y., Guo, X. F., Sun, Y. K., Liu, W. J., et al. (2024). Bioinspired total synthesis of Cephalotaxus diterpenoids and their structural analogues. Angew. Chem. Int. Ed. 63 (22), 2931–2948. doi:10.1002/ange.202402931
Sharpless, K. B., and Michaelson, R. C. (1973). High stereo- and regioselectivities in the transition metal catalyzed epoxidations of olefinic alcohols by tert-butyl hydroperoxide. J. Am. Chem. Soc. 95 (18), 6136–6137. doi:10.1021/ja00799a061
Shen, Q., Wang, L., Zhou, H., Jiang, H. D., Yu, L. S., and Zeng, S. (2013). Stereoselective binding of chiral drugs to plasma proteins. Acta Pharmacol. Sin. 34 (8), 998–1006. doi:10.1038/aps.2013.78
Shing, T. K. M., and Li, L. H. (1997). Asymmetric Hosomi - Sakurai reaction of allylsilanes containing arabinose-derived alcohols as chiral auxiliaries. J. Org. Chem. 62 (5), 1230–1233. doi:10.1021/jo962240w
Shingo, Y., Kunihiko, F., Reiko, W., Motomu, K., and Masakatsu, S. (2002). A general catalytic allylation using allyltrimethoxysilane. J. Am. Chem. Soc. 124 (23), 6536–6537. doi:10.1021/ja0262582
Shirley, H. J., Jamieson, M. L., Brimble, M. A., and Bray, C. D. (2018). A new family of sesterterpenoids isolated around the Pacific Rim. Nat. Product. Rep. 35 (3), 210–219. doi:10.1039/C7NP00049A
Siengalewicz, P., Mulzer, J., and Rinner, U. (2013). Lycopodium alkaloids – synthetic highlights and recent developments. Alkaloids Chem. Biol. 72 (22), 1–151. doi:10.1016/B978-0-12-407774-4.00001-7
Siu, Y. M., Roane, J., and Krische, M. J. (2021). Total synthesis of leiodermatolide A via transfer hydrogenative allylation, crotylation, and propargylation: polyketide construction beyond discrete allyl- or allenylmetal reagents. J. Am. Chem. Soc. 143 (28), 10590–10595. doi:10.1021/JACS.1C06062
Smith, A. B., Han, Q., Breslin, P. A. S., and Beauchamp, G. K. (2005). Synthesis and assignment of absolute configuration of (-)-oleocanthal: a potent, naturally occurring non-steroidal anti-inflammatory and anti-oxidant agent derived from extra virgin olive oils. Org. Lett. 7 (22), 5075–5078. doi:10.1021/ol052106a
Smith, N. D., Kocienski, P. J., and Street, S. D. A. (1996). A synthesis of (+)-Herboxidiene A. Synthesis 5, 652–666. doi:10.1055/s-1996-4254
Song, F., Garner, A. L., and Koide, K. (2007). A highly sensitive fluorescent sensor for palladium based on the allylic oxidative insertion mechanism. J. Am. Chem. Soc. 129 (41), 12354–12355. doi:10.1021/ja073910q
Stamos, D. P., Chen, S. S., and Kishi, Y. (1997). New synthetic route to the C.14-C.38 segment of halichondrins. J. Org. Chem. 62 (22), 7552–7553. doi:10.1021/jo971713b
Stankevich, K. S., and Cook, M. J. (2023). The Hosomi-Sakurai allylation in carbocyclization reactions. Tetrahedron Lett. 121 (2), 154415–154422. doi:10.1016/j.tetlet.2023.154415
Stanton, G. R., Johnson, C. N., and Walsh, P. J. (2010). Overriding felkin control: a general method for highly diastereoselective chelation-controlled additions to α-silyloxy aldehydes. J. Am. Chem. Soc. 132 (12), 4399–4408. doi:10.1021/ja910717p
Stepanyan, S., Burkert, V. D., Elouadrhiri, L., Adams, G. S., Anciant, E., Anghinolfi, M., et al. (2001). Observation of exclusive deeply virtual compton scattering in polarized electron beam asymmetry measurements. APS 87 (18), 182002. doi:10.1103/PhysRevLett.87.182002
Stork, G., and Dolfini, J. E. (1963). The Total Synthesis of dl-Aspidospermine and of dl-Quebrachamine. J. Am. Chem. Soc. 85 (18), 2872–2873. doi:10.1021/ja00901a061
Sumino, S., Uno, M., Huang, H. J., Wu, Y. K., and Ryu, I. (2018). Palladium/Light induced radical alkenylation and allylation of alkyl iodides using alkenyl and allylic sulfones. Org. Lett. 20 (4), 1078–1081. doi:10.1021/acs.orglett.7b04050
Suzuki, T., Koizumi, K., Suzuki, M., and Kurosawa, E. (1983). Kumausallene, A new bromoallene from the marine red alga Laurencia Nipponica Yamada. Chem. Lett. 12 (10), 1639–1642. doi:10.1246/CL.1983.1639
Takahashi, K., Akao, R., and Honda, T. (2009). Efficient diastereoselective synthesis of trifarane-type sesquiterpenes, trifarienols A and B. J. Org. Chem. 74 (9), 3424–3429. doi:10.1021/jo900369t
Tanaka, S., Yamamoto, H., Nozaki, H., Sharpless, K. B., Michaelson, R. C., and Cutting, J. D. (1974). Stereoselective epoxidations of acyclic allylic alcohols by transition metal-hydroperoxide reagents. Synthesis of dl-C18 cecropia juvenile hormone from farnesol. J. Am. Chem. Soc. 96 (16), 5254–5255. doi:10.1021/ja00823a042
Teng, Y., Zhang, H., Zhou, J., Zhan, G., and Yao, G. (2018). Hebecarposides A-K, antiproliferative lanostane-type triterpene glycosides from the leaves of Lyonia ovalifolia var. hebecarpa. Phytochemistry 151, 32–41. doi:10.1016/J.PHYTOCHEM.2018.03.012
Thirupathi, B., and Mohapatra, D. K. (2016). Gold-catalyzed Hosomi-Sakurai type reaction for the total synthesis of herboxidiene. Org. Biomol. Chem. 14 (26), 6212–6224. doi:10.1039/C6OB00321D
Tietze, L. F., Hölsken, S., Adrio, J., Kinzel, T., and Wegner, C. (2004). Facial-selective allylation of methyl ketones for the asymmetric synthesis of tertiary homoallylic ethers. Synthesis, 23, 2236–2239. doi:10.1002/1521-3765(20010316)7:63.0
Tietze, L. F., Schiemann, K., and Wegner, C. (1995). Enantioselective synthesis of tertiary homoallylic alcohols via diastereoselective addition of allylsilanes to ketones. J. Am. Chem. Soc. 117 (21), 5851–5852. doi:10.1021/ja00126a025
Tori, M., Hisazumi, K., Wada, T., Sono, M., and Nakashima, K. (1999). Total synthesis of optically active liverwort sesquiterpenes, trifarienols a and B, using phenylethylamine as a chiral auxiliary. Tetrahedron Asymmetry 10 (5), 961–971. doi:10.1016/S0957-4166(99)00067-1
Tori, M., Makino, C., Hisazumi, K., Sono, M., and Nakashima, K. (2001). Synthesis of a homochiral ketone having a pinguisane skeleton using phenylethylamine as a chiral auxiliary: a formal total synthesis of deoxopinguisone. Tetrahedron Asymmetry 12 (2), 301–307. doi:10.1016/S0957-4166(01)00041-6
Trost, B. M. (2015). Metal catalyzed allylic alkylation: its development in the Trost laboratories. Tetrahedron 71 (35), 5708–5733. doi:10.1016/j.tet.2015.06.044
Trost, B. M., Godleski, S. A., and Genet, J. P. (1978). A total synthesis of racemic and optically active ibogamine. Utilization and mechanism of a new silver ion assisted palladium catalyzed cyclization. Chem. Inf. 9 (38), 3930–3931. doi:10.1021/ja00480a047
Trost, B. M., and Osipov, M. (2015). Recent advances on the total syntheses of communesin alkaloids and perophoramidine. Chem. - A Eur. J. 21 (46), 16318–16343. doi:10.1002/chem.201501735
Trost, B. M., and Van Vranken, D. L. (1996). Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 96 (1), 395–422. doi:10.1021/cr9409804
Tsuji, J. (1969). Carbon—carbon bond formation via palladium complexes. Accounts Chem. Res. 2 (5), 144–152. doi:10.1021/ar50017a003
Tsuji, J. (1986a). 25 Years in the organic chemistry of palladium. J. Organomet. Chem. 300 (1–2), 281–305. doi:10.1016/0022-328X(86)84066-9
Tsuji, J. (1986b). New general synthetic methods involving π-allylpalladium complexes as intermediates and neutral reaction conditions. Tetrahedron 42 (16), 4361–4401. doi:10.1016/S0040-4020(01)87277-X
Tsuji, J., and Takahashi, H. (1965). Organic syntheses by means of noble metal compounds. XII.1 reaction of the cyclooctadiene-palladium chloride complex with ethyl malonate. J. Am. Chem. Soc. 87 (14), 3275–3276. doi:10.1021/ja01092a067
Tsuna, K., Noguchi, N., and Nakada, M. (2013). Enantioselective total synthesis of (+)-ophiobolin A. Chem. - A Eur. J. 19 (17), 5476–5486. doi:10.1002/chem.201204119
Uyehara, T., Kabasawa, Y., and Kato, T. (1986). Rearrangement approaches to cyclic skeletons. IV. The total synthesis of (±)-Pinguisone and (±)-Deoxopinguisone based on photochemical [1,3] acyl migration of a bicyclo[3.2.2]non-6-en-2-one. Bull. Chem. Soc. Jpn. 59 (8), 2521–2528. doi:10.1246/bcsj.59.2521
Uyehara, T., Kabasawa, Y., Kato, T., and Furuta, T. (1985). Photochemical rearrangement approach to the total synthesis of (±)-pinguisone and (±)-deoxopinguisone. Tetrahedron Lett. 26 (19), 2343–2346. doi:10.1016/S0040-4039(00)95094-9
Veenstra, S. J., and Schmid, P. (1997). One-pot synthesis of protected homoallyl amines. Tetrahedron Lett. 38 (6), 997–1000. doi:10.1016/S0040-4039(96)02458-6
Verbitski, S. M., Mayne, C. L., Davis, R. A., Concepcion, G. P., and Ireland, C. M. (2002). Isolation, structure determination, and biological activity of a novel alkaloid, perophoramidine, from the Philippine ascidian Perophora namei. J. Org. Chem. 67 (20), 7124–7126. doi:10.1021/jo026012f
Wada, K., Kogure, N., Kitajima, M., and Takayama, H. (2019). Concise asymmetric total synthesis of lycopodine and flabelliformine via cascade cyclization reaction. Tetrahedron Lett. 60 (2), 187–190. doi:10.1016/j.tetlet.2018.12.007
Wadamoto, M., Ozasa, N., Yanagisawa, A., and Yamamoto, H. (2003). BINAP/AgOTf/KF/18-crown-6 as new bifunctional catalysts for asymmetric Sakurai-Hosomi allylation and Mukaiyama aldol reaction. J. Org. Chem. 68 (14), 5593–5601. doi:10.1021/jo020691c
Wakamiya, Y., Ebine, M., Matsumori, N., and Oishi, T. (2020). Total synthesis of amphidinol 3: a general strategy for synthesizing amphidinol analogues and structure-activity relationship study. J. Am. Chem. Soc. 142 (7), 3472–3478. doi:10.1021/jacs.9b11789
Wang, D., Wang, Z. G., Wang, M. W., Chen, Y. J., Liu, L., and Zhu, Y. (1999). Asymmetric allylation of aldehydes and glyoxylates through `C-centered' chiral pentacoordinate allylsilicates or promoted by Lewis acid. Tetrahedron Asymmetry 10 (2), 327–338. doi:10.1016/S0957-4166(99)00004-X
Wang, H., Liu, Y., Zhang, H., Yang, B., He, H., and Gao, S. (2023). Asymmetric total synthesis of Cephalotaxus diterpenoids: cephinoid P, cephafortoid A, 14-epi-Cephafortoid A and Fortalpinoids M-N, P. J. Am. Chem. Soc. 145 (31), 16988–16994. doi:10.1021/jacs.3c05455
Wang, J., and Pagenkopf, B. L. (2007). First total synthesis and structural reassignment of (-)-aplysiallene. Org. Lett. 9 (18), 3703–3706. doi:10.1021/ol701797e
Wang, L. Q., Ding, B. Y., Qin, G. W., Lin, G., and Cheng, K. F. (1998). Grayanoids from pieris formosa1Part 2 in the series: “chemical studies on Ericaceae Plants”.1. Phytochemistry 49 (7), 2045–2048. doi:10.1016/S0031-9422(98)00410-5
Wang, M., Tietjen, I., Chen, M., Williams, D. E., Daoust, J., Brockman, M. A., et al. (2016). Sesterterpenoids isolated from the sponge phorbas sp. activate latent HIV-1 provirus expression. J. Org. Chem. 81 (22), 11324–11334. doi:10.1021/acs.joc.6b02312
Werness, J. B., and Tang, W. (2011). Stereoselective total synthesis of (-)-kumausallene. Org. Lett. 13 (14), 3664–3666. doi:10.1021/ol201477u
Werness, J. B., Zhang, W., and Tang, W. (2013). Stereoselective addition of halogen to conjugated enynes and its application in the total synthesis of (−)-Kumausallene. Strategies Tactics Org. Synthesis 9 (6), 275–291. doi:10.1016/B978-0-08-099362-1.00010-2
Westermann, B., Scharmann, H. G., and Kortmann, I. (1993). Ple-catalyzed resolution of α-substituted β-ketoesters application to the synthesis of (+)-nitraniine and (-)-Isonitramine. Tetrahedron Asymmetry 4 (10), 2119–2122. doi:10.1016/S0957-4166(00)80054-3
Wilkie, R. P., Neal, A. R., Johnston, C. A., Voute, N., Lancefield, C. S., Stell, M. D., et al. (2016). Total synthesis of dehaloperophoramidine using a highly diastereoselective Hosomi-Sakurai reaction. Chem. Commun. 52 (71), 10747–10750. doi:10.1039/C6CC05747K
Williams, D. R., and Meyer, K. G. (2001). Total synthesis of (+)-Amphidinolide K. J. Am. Chem. Soc. 123 (4), 765–766. doi:10.1021/ja005644l
Williams, D. R., Myers, B. J., Mi, L., and Binder, R. J. (2013). Studies for the total synthesis of amphidinolide P. J. Org. Chem. 78 (10), 4762–4778. doi:10.1021/jo4002382
Wu, X.De, Li, X. N., Peng, L. Y., and Zhao, Q. S. (2020). Huperserratines A and B, two macrocyclic lycopodium alkaloids with an unusual skeleton from Huperzia serrata. J. Org. Chem. 85 (10), 6803–6807. doi:10.1021/acs.joc.0c00623
Xing, S. H., Zhu, C. X., Zhang, R., and An, L. (2014). Huperzine A in the treatment of Alzheimer’s disease and vascular dementia: a meta-analysis. Evid. Based Complement. Altern. Med. 2014 (2), 363985–363997. doi:10.1155/2014/363985
Xu, S. S., Cai, Z. Y., Qu, Z. W., Yang, R. M., Cai, Y. L., Wang, G. Q., et al. (1999). Huperzine-A in capsules and tablets for treating patients with Alzheimer disease. Clin. Trial 20 (6), 486–490.
Yadav, J. S., Reddy, B. V. S., Reddy, M. S., and Parimala, G. (2003). InBr3-Catalyzed alkynylation and allylation of acid chlorides: a facile synthesis of alkynyl and allyl ketones. Synthesis 23 (15), 2390–2394. doi:10.1055/s-2003-42438
Yamamoto, Y., and Asao, N. (1993). Selective reactions using allylic metals. Chem. Rev. 93 (6), 2207–2293. doi:10.1021/cr00022a010
Yamamoto, Y., and Sasaki, N. (1989). “The stereochemistry of the Sakurai reaction,” in Chemical bonds–better ways to make them and break them (Elsevier Science Publishers B.V), 363–441.
Yamasaki, S., Fujii, K., Wada, R., Kanai, M., and Shibasaki, M. (2002). A general catalytic allylation using allyltrimethoxysilane. J. Am. Chem. Soc. 124 (23), 6536–6537. doi:10.1021/ja0262582
Yang, H., and Carter, R. G. (2010). Development of an enantioselective route toward the lycopodium alkaloids: total synthesis of lycopodine. J. Org. Chem. 75 (15), 4929–4938. doi:10.1021/jo100916x
Yang, J., Park, Y., Yang, S., Lee, G., Ha, M. W., Kim, M. H., et al. (2021). Enantioselective total synthesis of Nitraria alkaloids: (+)-Nitramine, (+)-Isonitramine, (-)-Isonitramine, and (-)-Sibirine via asymmetric phase-transfer catalytic α-allylations of α-carboxylactams. J. Org. Chem. 86 (6), 4375–4390. doi:10.1021/acs.joc.0c02573
Yao, H., Zhou, N., Zhang, Z., Guan, W., Wang, H., and Cheng, H. (2020). Recent developments in syntheses of alotaketals and phorbaketals. Tetrahedron Lett. 61 (8), 151480–151510. doi:10.1016/j.tetlet.2019.151480
Yotsu-Yamashita, M., Yamagishi, Y., and Yasumoto, T. (1995). 5,6,11-trideoxytetrodotoxin from the puffer fish, fugu poecilonotus. Tetrahedron Lett. 36 (51), 9329–9332. doi:10.1016/0040-4039(95)02020-P
Yu, B. W., Chen, J. Y., Wang, Y. P., Cheng, K. F., Li, X. Y., and Qin, G. W. (2002). Alkaloids from menispermum dauricum. Phytochemistry 61 (4), 439–442. doi:10.1016/S0031-9422(02)00162-0
Yun, Z., and Panek, J. S. (2007). Total synthesis of herboxidiene/GEX 1A. Org. Lett. 9 (16), 3141–3143. doi:10.1021/ol701427k
Zhang, L. C., Sakurai, H., and Kira, M. (1997). Enantioselective allylation of aldehydes using tartrate ester modified allylsilanes. Chem. Lett. 26 (2), 129–130. doi:10.1246/cl.1997.129
Zhang, R., Tang, C., Li, Y., Ke, C. Q., Lin, G., Xie, H., et al. (2018). The first phytochemical investigation of Rhododendron websterianum: triterpenoids and their cytotoxic activity. Phytochem. Lett. 25, 43–46. doi:10.1016/J.PHYTOL.2018.03.015
Zhang, Z. R., Zhong, J. D., Li, H. M., Li, H. Z., Li, R. T., and Deng, X. L. (2012). Two new grayanane diterpenoids from the flowers of Rhododendron molle. J. Asian Nat. Prod. Res. 14 (8), 764–768. doi:10.1080/10286020.2012.691095
Zhao, D. R., Su, L. H., Li, R. T., Chen, X. Q., and Li, H. M. (2018). Chemical constituents from the twigs and leaves of Lyonia ovalifolia. Biochem. Syst. Ecol. 78, 1–4. doi:10.1016/J.BSE.2018.02.006
Zhao, S., Sirasani, G., and Andrade, R. B. (2021). Aspidosperma and strychnos alkaloids: chemistry and biology. Alkaloids Chem. Biol. 86 (12), 1–143. doi:10.1016/bs.alkal.2021.05.001
Zhou, C., Li, X., Li, H., and Li, R. (2012). Chemical constituents from the leaves of Craibiodendron yunnanense. Biochem. Syst. Ecol. 45, 179–182. doi:10.1016/J.BSE.2012.07.004
Zhou, S. Z., Yao, S., Tang, C., Ke, C., Li, L., Lin, G., et al. (2014). Diterpenoids from the flowers of Rhododendron molle. J. Nat. Prod. 77 (5), 1185–1192. doi:10.1021/NP500074Q
Zuo, Z., and Ma, D. (2011). Enantioselective total syntheses of communesins A and B. Angew. Chem. - Int. Ed. 50 (50), 12008–12011. doi:10.1002/anie.201106205
Glossary
AHSR Asymmetric Hosomi-Sakurai reaction
AIBN Azobisisobutyronitrile
AD Alzheimer’s disease
aSH aza-Hosomi-Sakurai
ATP adenosine triphosphate
BINAP 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
BPO Benzoyl peroxide
CAB Chiral (acyloxy)borane
cAMP cyclic adenosine monophosphate
DCM Dichloromethane
DIFLUOROPHOS (2,2,2′,2′-Tetrafluoro-4,4′-bi-1,3-benzodioxole-5,5′-diyl)bis(diphenylphosphine)
DMF Dimethylformamide
DMPU N,N′-Dimethylpropyleneurea
DNA Deoxyribonucleic Acid
EC50 half maximal effective concentration
ED50 median effective dose
GAP GTPase-accelerating proteins
GETFund Ghana Education Trust Fund
GNPC Ghana National Petroleum Corporation
HDA Hetero-Diels–Alder
HEK human embryonic kidney cells
HFIP Hexafluoroisopropanol
HIV Human immunodeficiency virus
HSC Hosomi-Sakurai cyclisation
HSR Hosomi-Sakurai reaction
IC50 Half-maximal inhibitory concentration
LED light-emitting diode
MOM Methoxymethyl ether
NMR Nuclear Magnetic Resonance spectroscopy
NOE Nuclear Overhauser effect
RNA Ribonucleic acid
STAT3 signal transducer and activator of transcription 3
TBAF tetrabutylammonium fluoride
TBDPS tert-butyldiphenylsilyl
TFE trifluoroethanol
THF tetrahydrofuran
TMS trimethylsilyl
TMSI trimethylsilyl iodide
TMSO Trimethylsulfoxonium
TMSOT Trimethylsilyl trifluoromethanesulfonate
Keywords: Hosomi-Sakurai allylation, carbonylation, total synthesis, stereoselectivity, natural products, lewis acid-promotors
Citation: Akwensi J, Kumah RT, Osei-Safo D and Amewu RK (2025) Application of Hosomi-Sakurai allylation reaction in total synthesis of biologically active natural products. Front. Chem. 13:1527387. doi: 10.3389/fchem.2025.1527387
Received: 13 November 2024; Accepted: 28 February 2025;
Published: 28 March 2025.
Edited by:
Jian-Wei Han, East China University of Science and Technology, ChinaReviewed by:
Lucia Chiummiento, University of Basilicata, ItalyN. Vijaya Ganesh, LGC Standards, United States
Copyright © 2025 Akwensi, Kumah, Osei-Safo and Amewu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard K. Amewu, cmFtZXd1QHVnLmVkdS5naA==; Dorcas Osei-Safo, ZG9zZWktc2Fmb0B1Zy5lZHUuZ2g=