 Vera S. Dorokhova1
Vera S. Dorokhova1 Bozhena S. Komarova1
Bozhena S. Komarova1 José O. Previato2
José O. Previato2 Lúcia Mendonça Previato2
Lúcia Mendonça Previato2 Vadim B. Krylov1,3
Vadim B. Krylov1,3 Nikolay E. Nifantiev1*
Nikolay E. Nifantiev1*- 1Laboratory of Glycoconjugate Chemistry, N.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Moscow, Russia
- 2Laboratório de Glicobiologia, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 3Laboratory of Synthetic Glycovaccines, N.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Moscow, Russia
This study focuses on the synthesis of a series of oligo-α-(1→6)-D-galactopyranosides bearing β-D-galactofuranosyl residues at O-2 and/or O-3, which relate structurally to fragments of glucuronoxylomannogalactan (GXMGal) from the fungal pathogen Cryptococcus neoformans that causes severe diseases in immunocompromised patients. The preparation of target compounds is based on the use of a selectively O-protected N-phenyltrifluoroacetimidoyl galactopyranoside donor with an allyl group at O-2, levulinoyl group (Lev) at O-3, pentafluorobenzoyl (PFB) group at O-4, and fluorenylmethoxycarbonyl (Fmoc) group at O-6. The choice of protecting groups for this donor ensures the stereospecific formation of α-(1→6)-glycosidic bonds due to the stereodirecting effect of acyls at O-3, O-4, and O-6. At the same time, this combination of O-substituents permits the selective recovery of free OH groups at O-2, O-3, and O-6 for chain elongation via the introduction of β-D-galactofuranosyl and α-D-galactopyranosyl residues. The reported compounds are obtained as aminopropyl glycosides, which are transformed into biotinylated conjugates for further use as coating antigens in immunological studies. The obtained oligosaccharides were subjected to detailed 13C NMR analysis to show the spatial similarity of the obtained hexasaccharide with the corresponding fragment in the GXMGal chain, making this compound suitable for further immunological studies of C. neoformans.
Introduction
Cryptococcus neoformans is a human fungal pathogen capable of causing severe diseases in patients with a weakened immune system (especially in patients with HIV/AIDS) (Bermas and Geddes-McAlister, 2020; Zhao et al., 2023). This fungus can attack the central nervous system, thus causing cryptococcal meningitis, a fatal disease if untreated (Chen et al., 2022). In recent years, serious concerns have arisen about the increasing cases of cryptococcal meningitis in HIV-seronegative individuals (Paccoud et al., 2023). This fungus can also attack the lungs, skin, and other organs, which also leads to serious complications (Rivera et al., 1998). This pathogen spreads through bird droppings and enters the human body through inhaled dust (Maziarz and Perfect, 2016). C. neoformans is most commonly found in territories of Africa and Southern and Southeastern Asia, but the affected area is expanding every year (Rajasingham et al., 2022).
One of the main factors contributing to the virulence of C. neoformans is its bulk polysaccharide capsule (Vecchiarelli, 2000; Doering, 2009). It is composed mainly of glucuronoxylomannan (GXM), with minor components—glucuronoxylomannogalactan (GXMGal) and mannoprotein. The structure and immunological properties of GXM were studied in detail (Cherniak et al., 1998; McFadden and Casadevall, 2004; Oscarson et al., 2005; Nakouzi et al., 2009; Hargett et al., 2024), and their heterogeneity was shown for different serotypes. In contrast, the minor polysaccharide GXMGal of the C. neoformans capsule, which has not attracted significant attention until recently, is of great interest from an immunological point of view due to its immunomodulatory effect (Villena et al., 2008; Vecchiarelli et al., 2011; Decote-Ricardo et al., 2019). Unlike GXM (Cherniak et al., 1980; Skelton et al., 1991a; 1991b; James and Cherniak, 1992), GXMGal is a conserved polysaccharide that seems to be structurally similar in all C. neoformans serotypes studied to date (Cherniak et al., 1982; James and Cherniak, 1992; Vaishnav et al., 1998; Heiss et al., 2009).
Generally, GXMGal consists of a poly-α-(1→6)-D-galactopyranan backbone bearing β-Xylp-(1→3)-α-Manp-(1→3)[β-Xylp-(1→2)-]-α-Manp-1→4)[β-GlcpA-1→3)]-β-Galp and β-D-galactofuranosyl residues (Figure 1) (Heiss et al., 2013; Previato et al., 2017). However, C. neoformans GXMGal does not contain a regular and defined repeating unit due to the variable addition of β-GlcpA, β-Xylp, and O-acetyl groups on the β-Galp side chains and a variable number of β-Galf branches on the polysaccharide backbone. Given the high immunological activity of galactofuranosyl-bearing epitopes demonstrated on a number of other polysaccharide antigens (Turco and Pedersen, 2003; Peltier et al., 2008; Richards and Lowary, 2009; Tefsen et al., 2012; Krylov et al., 2021; Argunov et al., 2024), we started the systematic synthesis of spacer-armed oligosaccharides related to GXMGal fragments bearing galactofuranosyl residues for their immunological studies toward the development of potential immunomodulators, diagnostic kits, and vaccines.
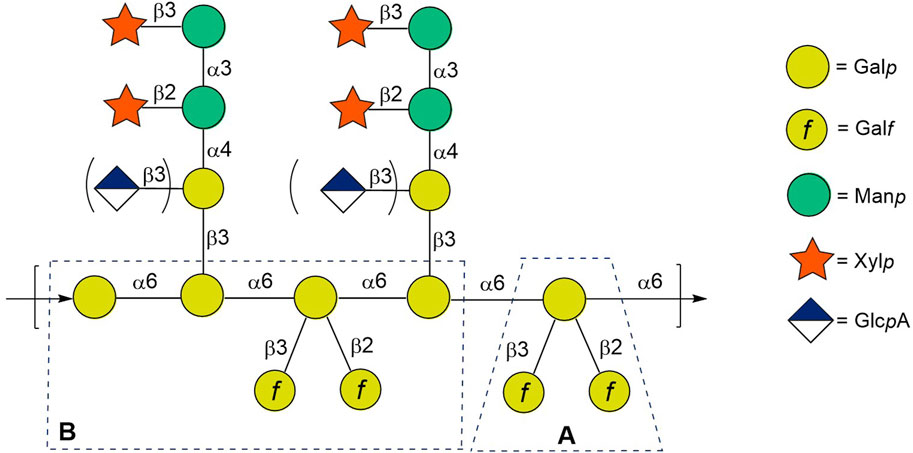
Figure 1. Structure of glucuronoxylomannogalactan of Cryptococcus neoformans. Presumed immunodominant vicinal branched fragment is framed.
Previously (Dorokhova et al., 2021), we described the preparation, nuclear magnetic resonance (NMR), and conformational studies of the model trisaccharide with two β-D-galactofuranosyl residues at O-2 and O-3, which relates to branch point A (Figure 1), as well as of its constituent monofuranosylated disaccharides. In this study, we report on the synthesis and NMR studies of spacered hexasaccharide 5a related to fragment B (Figure 1) of the GXMGal chain (Figure 1), which includes not only 2,3-vicinal branching but also 1,2-cis-pseudo-branching. These elements may influence the 3D structure of oligo and polysaccharides and, therefore, should be taken into account during the selection of the oligosaccharide, which is spatially equivalent to the target antigenic polysaccharide GXMGal. In addition to 5a, the synthesis of a series of its constituting oligosaccharide derivatives 1a–4a is also described, along with the preparation of corresponding biotinylated glycoconjugates 1b–5b required for use as molecular probes and coating antigens in a variety of immunological investigations (Figure 2).
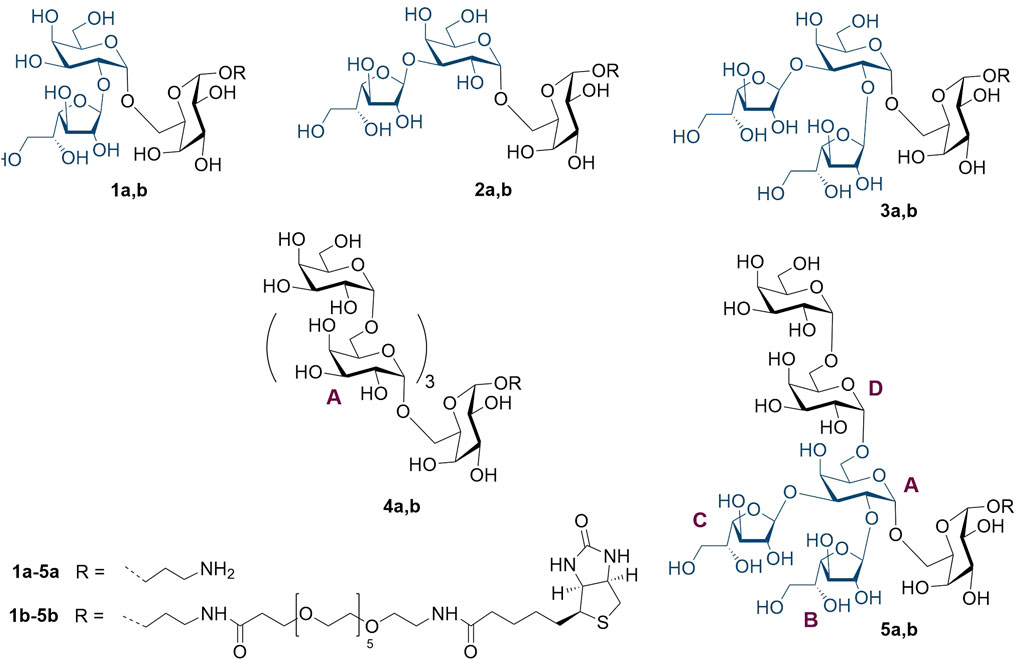
Figure 2. Studied set of oligosaccharides related to the vicinally branched galactoside fragment of GXMGal.
Results and discussion
The galactopyranosyl units in target compounds have an α-anomeric configuration and, thus, are connected to other parts of the molecules through 1,2-cis-glycosidic bonds. Their stereoselective construction can be accomplished by the remote anchimeric assistance of remote acyl groups at O-3, O-4, and O-6 of the glycosyl donor. The stereocontrolling participation of a single acyl group, as well as the combined effect of two or three acyls, was previously applied for stereoselective 1,2-cis-glycosylation by gluco- and galacto-glycosyl donors [for reviews, see Nigudkar and Demchenko (2015); Komarova et al. (2016); Hettikankanamalage et al. (2020); Tokatly et al. (2021)] toward the synthesis of biologically relevant oligosaccharides (Gerbst et al., 2001; Calin et al., 2013; Komarova et al., 2014; 2015; 2018a; 2018b; Vinnitskiy et al., 2015; Zou et al., 2018; Zhang et al., 2022). In the present work, we explored this approach in the case of galactosylation and used a stereodirecting acyl group at O-6 as the temporary substituent needed for the selective liberation of OH to be glycosylated.
It was also shown that the introduction of fluoro-substituted benzoates at O-6 in glucosyl donors also favors the selectivity of 1,2-cis-glycosylation (Cato et al., 2005; Vohra et al., 2009; Komarova et al., 2018a; 2023). In this work, we report an integrated approach of both participating and withdrawing acceptor groups to achieve high α-selectivity in the synthesis of target structures.
Synthesis of compounds 1–5
In order to obtain target compounds 1–5, universal synthetic blocks 13, 21, and 33 were designed. They, on one hand, would ensure the stereoselective building of 1,2-cis-glycosidic bonds between galactopyranose residues and, on the other hand, would allow the regioselective deprotection of hydroxyl groups at C-2, C-3, and C-6 for the efficient synthesis of branched fragments and chain extension. Both goals were achieved by the rational selection of protecting groups in the galactopyranosyl donors. Thus, donor 13, which is the precursor of the branched unit, carries a non-participating allyl (All)-protecting group at O-2 and participating acyl groups at O-3 (levulinoyl, Lev) (Komarova et al., 2014; 2015; 2023), O-4 (pentafluorobenzoyl, PFB) (Komarova et al., 2018a), and O-6 atoms (fluorenylmethyloxycarbonyl, Fmoc), which favored the α-stereocontrol of the glycosylation reaction. According to the literature data, each of the above groups can be selectively removed under orthogonal conditions without affecting the other protecting groups (Prabhu et al., 2003; Ágoston et al., 2016).
Galactosyl donor 13 was synthesized from the well-known monosaccharide precursor 6 (Thijssen et al., 1998) (Scheme 1). Its primary hydroxyl group at C-6 was regioselectively protected by Fmoc to form 3,4-diol 8 (Argunov et al., 2016). The introduction of the levulinoyl group using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) hydrochloride and dimethylaminopyridine (DMAP) at −18оС proceeded exclusively at O-3 (Hirose et al., 2015), and the subsequent treatment of product 10 with pentafluorobenzoyl chloride in the presence of pyridine (Py) allowed obtaining a fully protected monosaccharide 11. The p-methoxyphenyl-protecting group was removed from the anomeric center by ceric ammonium nitrate (CAN) in a mixture of acetonitrile, benzene, and water to form the corresponding hemiacetal 12, which was then converted to N-phenyltrifluoroacetimidate 13. It is important to note that glycosyl donors of this type are usually purified on silica gel with the addition of triethylamine to the eluent in order to neutralize the silica gel and reduce the cleavage of the acid-labile leaving group. In the case of Fmoc-containing donors, the presence of triethylamine led to a loss of yield as a result of partial Fmoc removal (Oberli et al., 2008). Chromatography of donor 13 on neutral aluminum oxide without triethylamine has made it possible to mitigate side reactions and isolate the product with a sufficiently high yield of 82%.
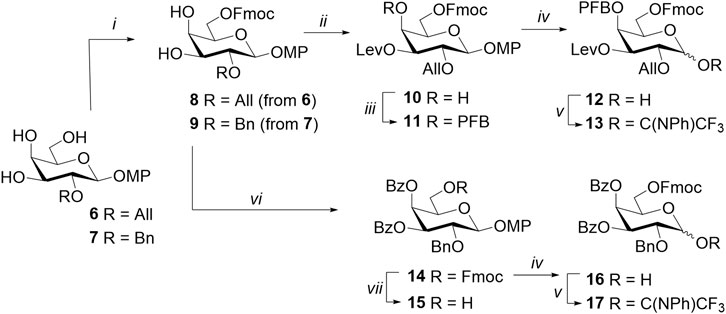
Scheme 1. Synthesis of the monosaccharide donors 13 and 17 and acceptor 15. Reagents and conditions: (i) FmocCl, 2,6-lutidine, MeCN, 2–3 days, 65% for 8 and 64% for 9; (ii) LevOH, CMPI, DMAP, CH2Cl2, -18°C, 20 h, 87%; (iii) pentafluorobenzoyl chloride, Py, DMAP, 12 h, 82%; (iv) CAN, MeCN, benzene, H2O, 0 °C, 10–12 min, 88% for 12 and 74% for 16; (v) ClC(NPh)CF3, K2CO3, acetone, 12 h, 82% for 13 and 84% for 17; (vi) BzCl, Py, DMAP, CH2Cl2, 12 h, 99%; and (vii) piperidine, CH2Cl2, 0 °C, 15 min, 69%.
Monosaccharide blocks 15 and 17 were obtained based on 2-O-benzylated triol 7 (Zhu and Yang, 2012) (Scheme 1). Protecting groups (6-O-Fmoc and two benzoate groups at C-3 and C-4) were introduced sequentially with considerably high yields at each step. The resulting monosaccharide 14 was partially converted to acceptor 15 after Fmoc removal under the action of piperidine in tetrahydrofuran (THF). Hemiacetal 16 was also obtained from monosaccharide 14 and then treated with N-phenyltrifluoroacetimidoyl chloride in acetone. As in the case of donor 13, chromatographic purification of the resulting 6-O-Fmoc-bearing donor was performed on neutral Al2O3, yielding compound 17 with a considerably high yield of 84%.
The conditions for the selective removal of the chosen protecting groups (Fmoc, Lev, and All) were optimized using the model monosaccharide 11 (Scheme 2). The use of the standard Fmoc-removal procedure in the piperidine/THF system led to a rapid cleavage of the Fmoc group (Pennington and Dunn, 1994; Werz, 2012). However, under these conditions, a side reaction was observed, which consisted of the substitution of a fluorine atom in 4-O-pentafluorobenzoate by piperidine. This process was confirmed by HRMS data and the emergence of piperidine ring signals in 1H NMR spectra at 3.31 ppm 1.65 ppm and 13C NMR at 52.1 ppm and 23.9 ppm, respectively. Thus, these conditions can be used only for compounds without a pentafluorobenzoyl group or only at the last synthetic steps of the complete deprotection. Nevertheless, the Fmoc group in the presence of pentafluorobenzoate was successfully removed under milder conditions under the action of N-methylmorpholine in methylene chloride for 2 days. The levulinoyl group was efficiently and selectively removed by hydrazine acetate in pyridine, resulting in compound 19 with a 96% yield. The allyl substituent at O-2 was selectively removed using (1,5-cyclooctadiene)bis (methyldiphenylphosphine)iridium(I) hexafluorophosphate ([Ir(COD) (PMePh2)2]PF6), which was pre-reduced with hydrogen (Laroussarie et al., 2015). This resulted in compound 20 with an almost quantitative yield.
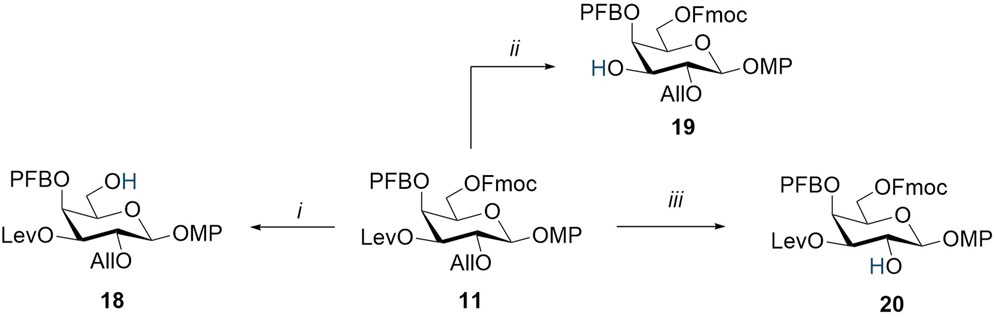
Scheme 2. Regioselective removal of orthogonal-protecting groups in monosaccharide 11. Reagents and conditions: (i) N-methylmorpholine, CH2Cl2, 2 days, 65%; (ii) NH2NH2∙H2O, AcOH, Py, 20 min, 96%; and (iii) [Ir(COD) (PMePh2)2]PF6, H2, I2, THF, 2 h, 99%.
In order to increase the efficiency of the α-(1→6)-glycoside bond formation, the conditions for the glycosylation of the spacer-containing acceptor 21 by donor 13 were optimized. Originally, the coupling was carried out in the presence of trimethylsilyl trifluoromethane sulfonate (TMSOTf) at −35°C (Table 1, entry 1). The desired disaccharide 23 was obtained with an insufficient yield of 28%; however, full α-stereospecificity was achieved that can be explained by the presence of three α-directing protecting groups in donor 13. The low yield may be attributed to the presence of three electron-withdrawing groups in the donor, which lower its activity; hence, the side processes of its destruction occur before the glycosylation reaction is completed. An increase in the reaction yield to 47% was achieved by replacing the promoter with triflic acid, along with a gradual increase in temperature to −15°C (Table 1, entry 2). One of the reasons for the low yield of disaccharide 23, in this case, is the removal of Fmoc from the O-6 product by triethylamine, which is used to neutralize unreacted acid after the reaction is completed (Oberli et al., 2008). This is confirmed by an increase in the yield of disaccharide 23 up to 64% when the addition of triethylamine was omitted, and the reaction mixture was immediately filtered and washed with a saturated NaHCO3 solution (Table 1, entry 3).
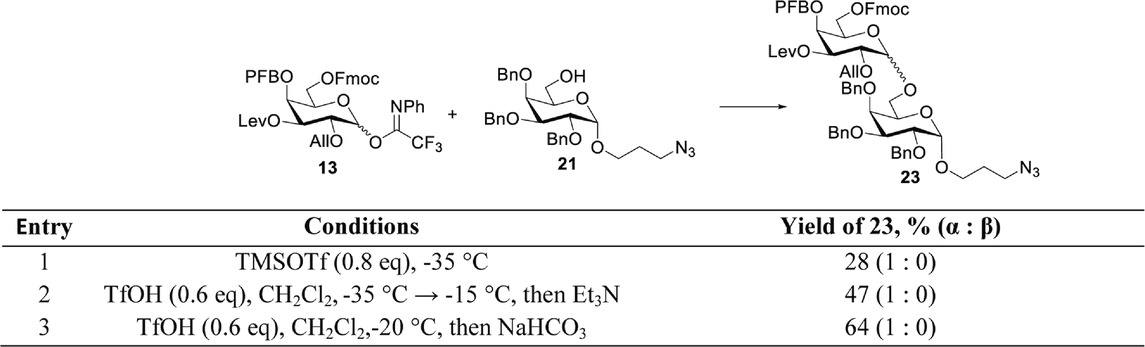
Table 1. Optimization of acceptor 21 glycosylation by donor 13.
The disaccharide acceptor 25 was obtained from 23 by the removal of the 2-O-allyl group with the iridium complex [Ir(COD) (PMePh2)2]PF6 pre-reduced with hydrogen (Scheme 3). Glycosylation of acceptor 25 by galactofuranosyl donor 22, previously obtained by us (B. Krylov et al., 2018), in the presence of TMSOTf, resulted in trisaccharide 28 with a high yield as a pure β-isomer. In the next step, the hydroxyl group at the C-3 atom of the non-reducing residue was recovered by hydrazine acetate in pyridine. However, in addition to the expected disaccharide 29, we observed the formation of a migration product of pentafluorobenzoate from O-4 to O-3 (compound 29i). Both regioisomers 29 and 29i were successfully separated by column chromatography and found applications in the synthetic scheme.
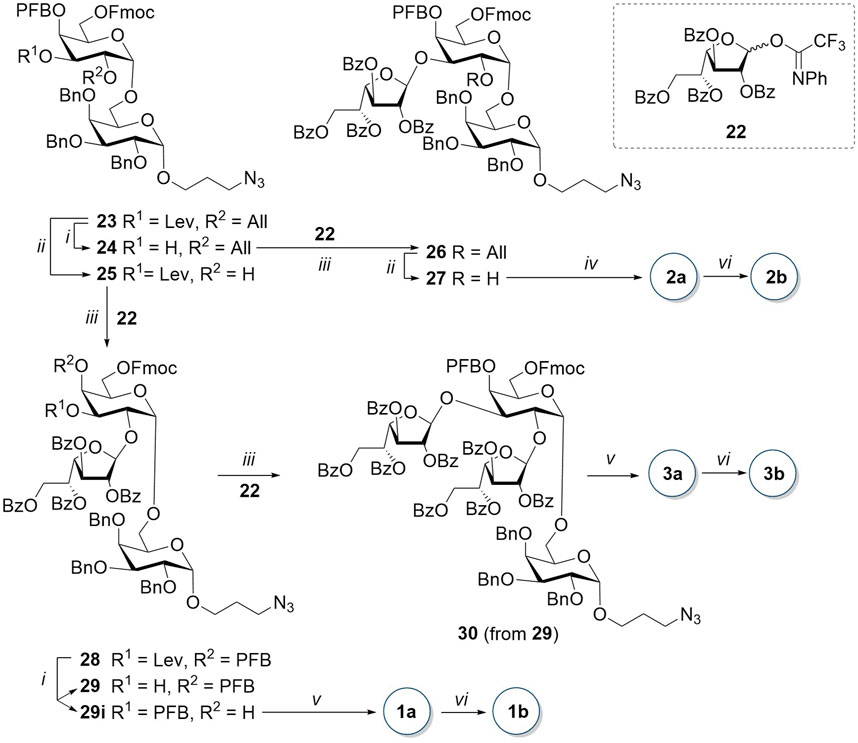
Scheme 3. Synthesis of tri- and tetrasaccharides 1, 2, and 3. Reagents and conditions: (i) NH2NH2∙H2O, AcOH, Py, 10–11 min, 92% for 24, 27% for 29, and 29% for 29i; (ii) [Ir(COD) (PMePh2)2]PF6, H2, I2, THF, 2 h, 83% for 25 and 78% for 27; (iii) TMSOTf, AW300, CH2Cl2, -20°C, only β, 1.5 h, 86% for 26, 3 h, 87% for 28, and 17 min, 70% for 30; (iv) 1) piperidine, THF, 0 C, 40 min; 2) NaOCH3, MeOH, 12 h; 3) H2, Pd(OH)2/C, HCl, MeOH, EtOAc, 25 min, 79% in three steps; (v) 1) piperidine, THF, 0 C, 25–60 min; 2) NaOCH3, MeOH, 3.5–12 h; and 3) Na, NH3, THF, 50–60 min, 56% for 1a, 35% for 3a in three steps; (vi) AEB, Et3N, DMF, 30 min, 50% for 1b, 85% for 2b, and 2.5 h, 44% for 3b.
The target trisaccharide 1a was synthesized from the pentafluorobenzoyl migration product 29i in three steps. First, the Fmoc-protecting group was removed with piperidine in THF. Then, without intermediate purification, benzoyl substituents were removed in the presence of sodium methylate in methanol. The following reduction of the azide group in the spacer to the amino group and the simultaneous removal of benzyl groups by treatment with sodium in liquid ammonia yielded the unprotected (1→2)-trisaccharide 1a with a high yield of 98%. Tetrasaccharide 30 was obtained by coupling galactofuranosyl donor 22 and trisaccharide acceptor 29 with a fairly high yield of 87% and exclusively as a pure β-isomer. The sequential removal of protecting groups using a scheme similar to that described above for trisaccharide 29i resulted in unprotected tetrasaccharide 3a.
Unlike in trisaccharide 28, the removal of the 3-O-levulinoyl group in disaccharide 23 did not result in the migration of the pentafluorobenzoyl group from O-4 to O-3. The furanosyl residue was introduced by glycosylation with donor 22, resulting in the formation of β-(1→3)-trisaccharide 26 with an 86% yield. The removal of all protecting groups in compound 26 included (1) 2-O-deallylation (→27); (2) removal of 6-O-Fmoc with piperidine in THF; (3) removal of 4-O-pentafluorobenzoyl and benzoyl groups with sodium methylate in methanol; and (4) hydrogenolysis on Pd(OH)2/C in the presence of a small amount of hydrochloric acid, which prevents the methylation of the amino group of the target (1→3)-trisaccharide 2a.
The α-(1→6)-linked galactopyranosyl chain in the synthesis of target compounds 4a and 5a was elongated with disaccharide donor 33 (Scheme 4). The glycosylation reaction of p-methoxygalactoside 15 by donor 17 in the presence of TfOH proceeded with the exclusive formation of α-isomer 33 due to the concerted action of three α-stereodirecting acyl groups at O-3, O-4, and O-6 (Baek et al., 2015). The absence of the β-isomer among the reaction products was confirmed by the NMR spectra of the untreated reaction mixture. The removal of the p-methoxyphenyl-protecting group of the anomeric center, followed by the addition of the N-phenyltrifluoroacetimidoyl-leaving group to the hemiacetal 32, yielded disaccharide donor 33.
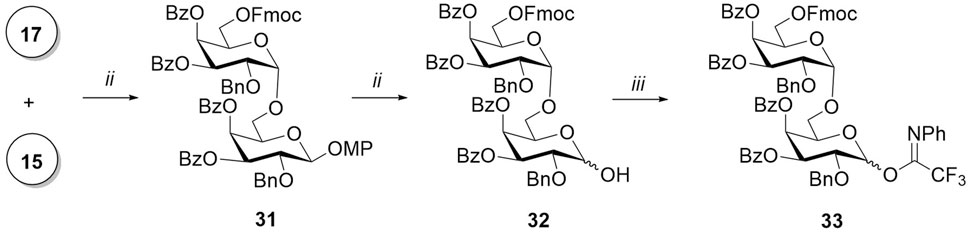
Scheme 4. Synthesis of disaccharide donors 42 and 44. Reagents and conditions: (i) TfOH, MS AW300, CH2Cl2, -20°C, 7 min, 79%, only α; (ii) CAN, MeCN, benzene, H2O, 0 °C, 10 min, 71%; and (iii) ClC(NPh)CF3, K2CO3, acetone, 12 h, 60%.
Glycosylation of prespacer-containing monosaccharide 21 by donor 33 in the presence of TfOH (Scheme 5) resulted in a mixture of α- and β-isomeric trisaccharides in the ratio of 20:1. Their ratio was determined by the integration of the 1H NMR spectrum of the reaction mixture. After the successful separation of the two isomers by column chromatography, the desired α-product 34 was isolated with a yield of 75%. The trisaccharide acceptor 35 was obtained after the removal of 6-O-Fmoc with piperidine in THF. An attempt of a TfOH-assisted glycosylation of acceptor 35 with a disaccharide donor 33 failed. After an optimization of conditions, it was found that in the presence of TMSOTf and with an increase in temperature from −20°C to −5°C, α-pentasaccharide 36 is formed with a sufficient yield of 55% without any β-isomer admixture.
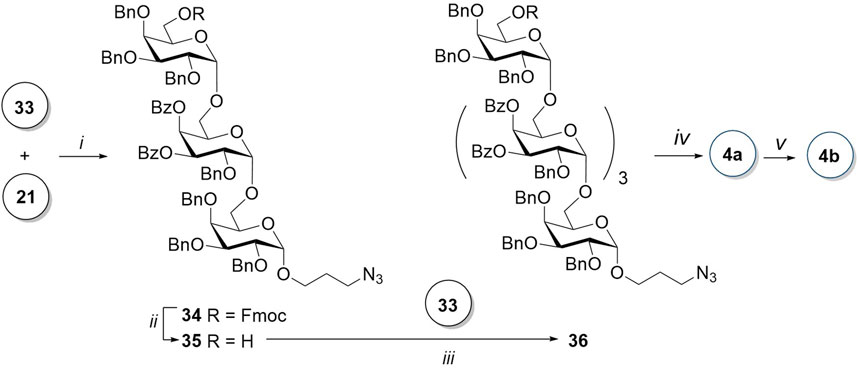
Scheme 5. Synthesis of the linear pentasaccharides 4a and 4b. Reagents and conditions: (i) TfOH, MS AW300, CH2Cl2, -20°C, 6 min, α:β = 20:1, 75%; (ii) piperidine, THF, 0 C, 20 min, 95%; (iii) TMSOTf, MS AW300, CH2Cl2, -5°C, 2.5 h, only α, 55%; (iv) 1) piperidine, THF, 0 C, 35 min; 2) NaOMe, MeOH, 12 h; and 3) H2, Pd(OH)2/C, HCl, MeOH, EtOAc, 5 h, 59% for the three steps; and (v) AEB, Et3N, DMF, 12 h, 92%.
Protecting groups in pentasaccharide 36 were removed according to a standardized algorithm: first, Fmoc was removed with piperidine; then, benzoate groups were removed in the presence of sodium methylate in methanol; and, in the last step, the azide group was reduced and benzyl groups removed in the course of catalytic hydrogenolysis. Unprotected pentasaccharide 4a was isolated by gel permeation chromatography with a 70% yield after all stages of deprotection. The 1H NMR spectrum of the product contains five anomeric proton signals. For each of the monosaccharide residues, α-configuration of the C-1 atom is confirmed both by spin–spin coupling constants (less than 4 Hz) and chemical shifts of the related carbon atoms (signals at 99.4, 98.9, and 98.8 ppm and two more signals at 98.7 ppm in 13C NMR).
The synthesis of hexasaccharide 5a began with the removal of 6-O-Fmoc in disaccharide 23 by N-methylmorpholine in a mixture of dichloromethane and THF (Scheme 6). The 19F NMR spectrum, as well as the absence of piperidine signals in the 1H spectrum, confirmed that the pentafluorobenzoyl group was not affected in this transformation. The TfOH-promoted glycosylation of the resulting acceptor 38 by disaccharide donor 33 was very slow and required a gradual increase in temperature from −20°C to −8°C. The low reaction rate and, accordingly, the accumulation of a large number of by-products of the destruction of the donor may be attributed to the presence of a strong electron-withdrawing PFB group in the immediate vicinity of the nucleophilic center in the acceptor. Attempts to vary the temperature regime of this reaction, as well as to change the promoter from TfOH to TMSOTf, tert-butyldimethylsilyl trifluoromethane sulfonate (TBDMSOTf), and C4F9SO3H, were not successful. After the separation of α- and β-isomers, obtained in a ratio of 13:1 in the reaction promoted with TfOH, tetrasaccharide 39a was isolated by HPLC with a yield of 51%.
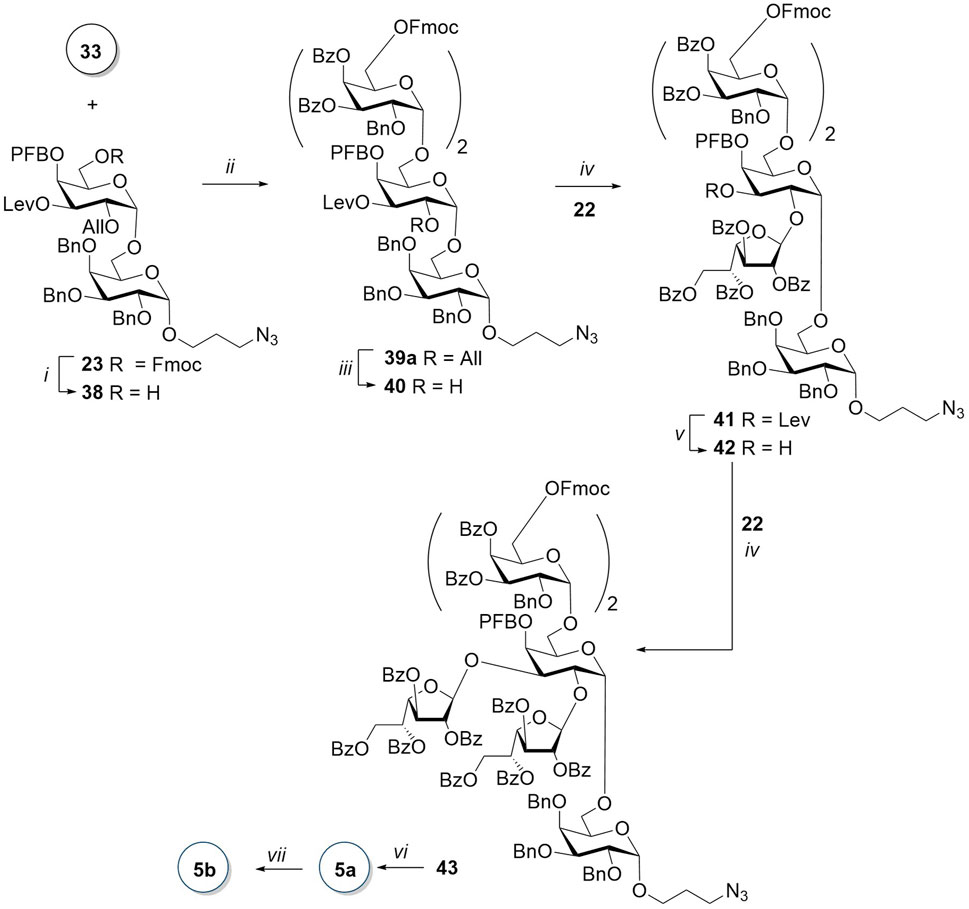
Scheme 6. Synthesis of the branched hexasaccharide 5. Reagents and conditions: (i) N-methylmorpholine, THF/CH2Cl2, 19 h, 78%; (ii) TfOH, MS AW300, CH2Cl2, -20 C → −8°C, 2 h, 55%, α:β = 13:1; (iii) [Ir(COD) (PMePh2)2]PF6, H2, I2, THF, 2 h, 86%; (iv) TMSOTf, AW300, CH2Cl2, -20°C, 84% for 41 and 87% for 43; (v) NH2NH2∙H2O, AcOH, Py, 40 min, 89%; (vi) 1) N-methylmorpholine, THF/CH2Cl2, 48 h; 2) NaOMe MeOH, 48 h; and 3) H2, Pd(OH)2/C, HCl, MeOH, EtOAc, 7 h, 46% for the three steps; and (vii) AEB, Et3N, DMF, 1 h, 65%.
For the synthesis of the branched hexasaccharide, we first obtained acceptor 40, which was then reacted with donor 22. Monofuranosylated pentasaccharide 41 was isolated with a fairly high yield of 84% and only as a β-isomer. The introduction of a second galactofuranosyl residue after the removal of the levulinoyl-protecting group from O-3 resulted in a protected hexasaccharide 43 with an 87% yield and absolute β-stereoselectivity. The target hexasaccharide 5a was obtained after the sequential removal of Fmoc and benzoate groups and hydrogenolysis, with a total yield of 46% for the three steps. Two singlets (δ 5.19 and 5.17 ppm) corresponding to H-1 of furanose rings are observed in the 1H NMR spectrum. Further notable are four doublets (δ 5.07–4.96 ppm) with J in the range 2.6–3.3 Hz, attributable to H-1 in pyranose residues of the main chain. Chemical shifts of the corresponding carbon atoms in the 1H–13C HSQC spectra also confirm the configurations of the anomeric centers of all six carbohydrate residues of compound 5a (for 13C-NMR data of 1a–5a, see Table 2).
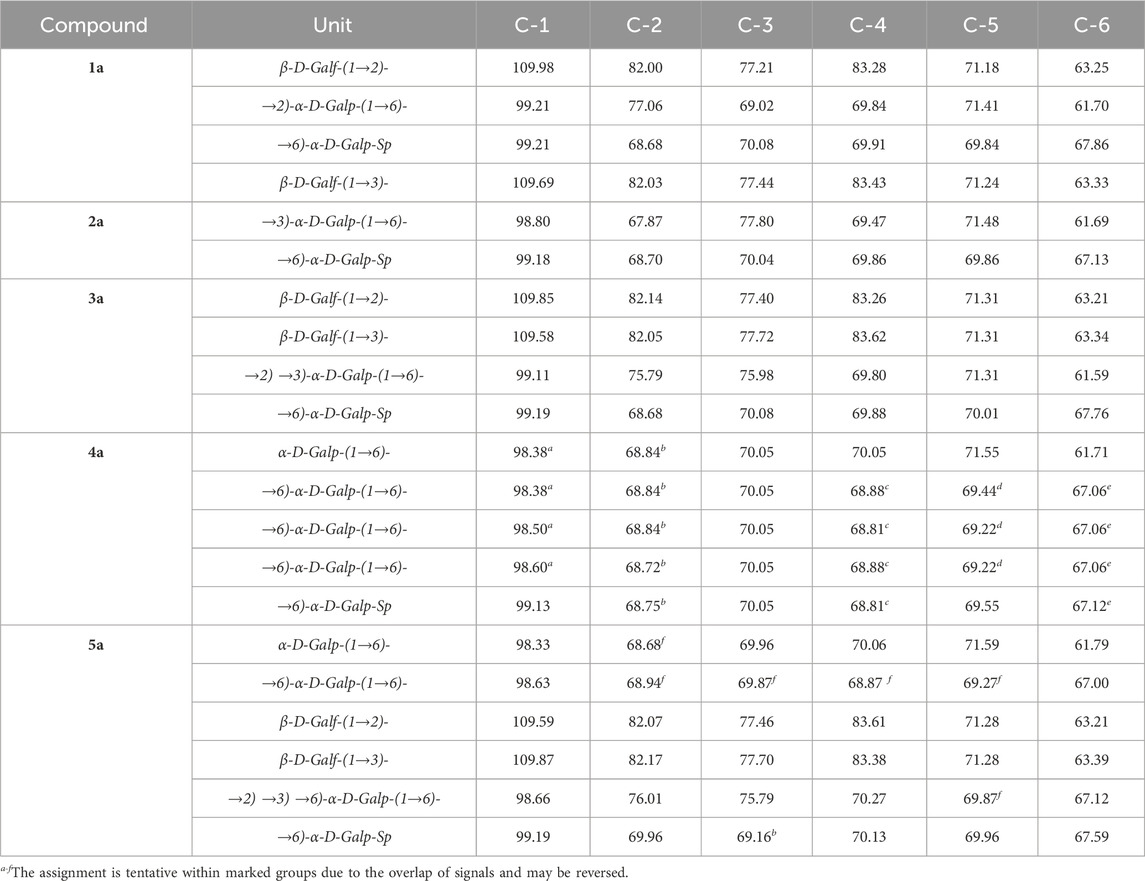
Table 2. 13C-NMR chemical shifts (δ, ppm, D2O, 303 K) for oligosaccharides 1a–5a.
Biotinylated oligosaccharides 1b–5b were obtained by the treatment of aminopropyl glycosides 1a–5a with an activated biotin ester and triethylamine in DMF according to the described procedure (Tsvetkov et al., 2012). Following purification on gel TSK-40 afforded 1b–5b in good to excellent yields. The attachment of the biotin entity was confirmed by the presence of characteristic signals in the 1H NMR spectra (for NMR data of corresponding biotin conjugates, see the Supplementary Material) and by HRMS data.
NMR analysis of obtained oligosaccharides 1a–5a
The NMR spectra of oligosaccharides 1a–5a were totally assigned by applying 2D NMR experiments (Table 2. For 1H NMR shifts, see Supplementary Table S1 in Supplementary Material). The effects of glycosylation (Table 3, units are labeled A–D as in Figure 2) were calculated as the difference in the 13C chemical shifts between two structures, one with and one without a particular type of glycosylation, as described before (Dorokhova et al., 2021). Upon the introduction of a glycosylating residue, the most pronounced spectral effect was observed on the glycosylated carbons, which underwent a down-field shift by 5–8 ppm (α-effect), while the resonances of the adjacent carbon atoms moved up-field to a smaller extent (β-effect) (Lipkind et al., 1988; Shashkov et al., 1988; Kochetkov et al., 1991; Gerbst et al., 2015). The C-1 of the glycosylating residue also underwent a significant down-field shift. Other carbon resonances were much less affected and were excluded from consideration. The α- and β-glycosylation effects for β-(1→2)- and β-(1→3)-galactofuranosylation measured using trisaccharides 1a and 2a agreed well with previously reported data (Dorokhova et al., 2021). The deviations from additivity in the vicinally branched fragment of hexasaccharide 5a (Table 4) were calculated as the difference between the experimental (δexp) and calculated (δcalc) 13C chemical shifts, where δcalc was calculated by the summation of all glycosylation effects.
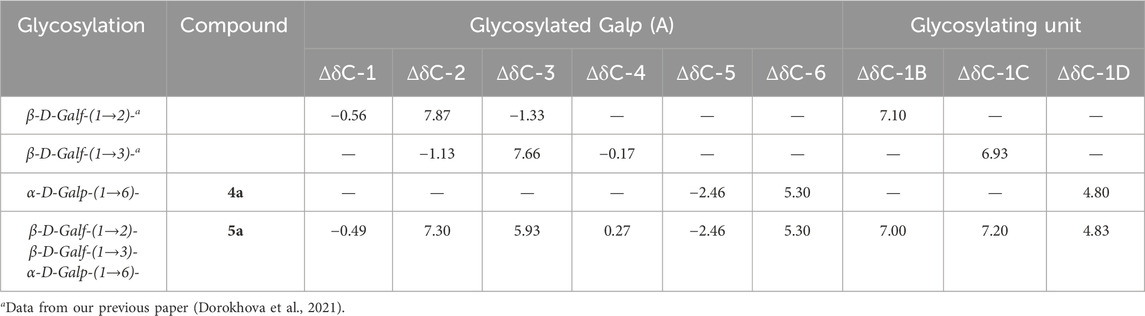
Table 3. 13C NMR α- and β-glycosylation effects (Δδ, ppm) of a branched galactose fragment in hexasaccharide 5a and related monoglycosylated oligosaccharides.
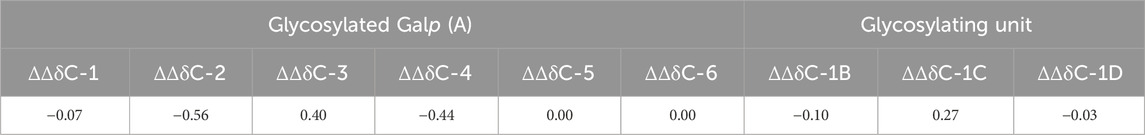
Table 4. Deviations from additivity (ΔΔδ, ppm) in the 13C-NMR spectra of a branched fragment in hexasaccharide 5a.
In spite of the presence of 2,3-vicinal branching and 1,2-cis-pseudobranching in hexasaccharide 5a, the good agreement between theoretical and experimental 13C chemical shifts was determined (deviation from additivity did not exceed 0.56 ppm; Table 4). It suggests the independence of conformational flexibility around corresponding interunit linkages connected with the branched Galp-residue of 5a. These suggest that the spatial similarity of 5a to the corresponding fragment within the chain of GXMGal makes 5a a the reliable model for future immunological studies of C. neoformans.
Conclusion
In conclusion, the oligosaccharides 1a–5a and their biotinylated derivatives 1b–5b were synthesized according to the convergent scheme, achieving good to excellent yields at each step. The sequential introduction of β-galactofuranosyl residues at O-2 and/or O-3, along with the elongation of the α-(1→6)-galactopyranoside core chain, was achieved using a galactosyl donor bearing orthogonal groups: 2-O-allyl, 3-O-levulinoyl, and Fmoc group at O-6. High, nearly absolute α-stereoselectivity in each glycosylation step was achieved due to the joint stereoredirecting effects of O-protecting acyl groups in galactosyl donors, including the 4-O-pentafluorobenzoyl group. The analysis of 13C NMR shifts and corresponding glycosylation effects for oligosaccharides 1a–5a confirms the spatial equivalence of synthetic hexasaccharide 5a to the corresponding branched fragment of the polysaccharide GXMGal, supporting the use of 5a as a reliable model hapten for further immunological studies of C. neoformans.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
VD: investigation and writing–original draft. BK: data curation and writing–original draft. JP: conceptualization and writing–review and editing. LM: conceptualization and writing–review and editing. VK: data curation, formal analysis, methodology, and writing–review and editing. NN: conceptualization, data curation, funding acquisition, project administration, resources, supervision, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Russian Science Foundation (Grant 19-73-30017-P) and Fundação de Apoio à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Acknowledgments
The authors thank their colleagues A.S. Dmitrenok and A.S. Shashkov for recording NMR spectra and A.O. Chizhov for recording of HRMS spectra of the synthesized compounds.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1501766/full#supplementary-material
References
Ágoston, K., Streicher, H., and Fügedi, P. (2016). Orthogonal protecting group strategies in carbohydrate chemistry. Tetrahedron Asymmetry 27, 707–728. doi:10.1016/j.tetasy.2016.06.010
Argunov, D. A., Aladysheva, U. S., Krylov, V. B., and Nifantiev, N. E. (2024). Acid-catalyzed transformation of pyranosides into furanosides as a tool for preparation of furanoside synthetic blocks. Org. Lett. 26, 8090–8094. doi:10.1021/acs.orglett.4c02984
Argunov, D. A., Krylov, V. B., and Nifantiev, N. E. (2016). The use of pyranoside-into-furanoside rearrangement and controlled O(5) → O(6) benzoyl migration as the basis of a synthetic strategy to assemble (1→5)- and (1→6)-linked galactofuranosyl chains. Org. Lett. 18, 5504–5507. doi:10.1021/acs.orglett.6b02735
Baek, J. Y., Kwon, H.-W., Myung, S. J., Park, J. J., Kim, M. Y., Rathwell, D. C. K., et al. (2015). Directing effect by remote electron-withdrawing protecting groups at O-3 or O-4 position of donors in glucosylations and galactosylations. Tetrahedron 71, 5315–5320. doi:10.1016/j.tet.2015.06.014
Bermas, A., and Geddes-McAlister, J. (2020). Combatting the evolution of antifungal resistance in Cryptococcus neoformans. Mol. Microbiol. 114, 721–734. doi:10.1111/mmi.14565
Calin, O., Eller, S., Hahm, H. S., and Seeberger, P. H. (2013). Total synthesis of the Escherichia coli O111 O-specific polysaccharide repeating unit. Chem. – Eur. J. 19, 3995–4002. doi:10.1002/chem.201204394
Cato, D., Buskas, T., and Boons, G. (2005). Highly efficient stereospecific preparation of tn and TF building blocks using thioglycosyl donors and the Ph2SO/Tf2O promotor system. J. Carbohydr. Chem. 24, 503–516. doi:10.1081/CAR-200067091
Chen, Y., Shi, Z. W., Strickland, A. B., and Shi, M. (2022). Cryptococcus neoformans infection in the central nervous system: the battle between host and pathogen. J. Fungi 8, 1069. doi:10.3390/jof8101069
Cherniak, R., Reiss, E., Slodki, M. E., Plattner, R. D., and Blumer, S. O. (1980). Structure and antigenic activity of the capsular polysaccharide of Cryptococcus neoformans serotype A. Mol. Immunol. 17, 1025–1032. doi:10.1016/0161-5890(80)90096-6
Cherniak, R., Reiss, E., and Turner, S. H. (1982). A galactoxylomannan antigen of Cryptococcus neoformans serotype A. Carbohydr. Res. 103, 239–250. doi:10.1016/S0008-6215(00)80686-2
Cherniak, R., Valafar, H., Morris, L. C., and Valafar, F. (1998). Cryptococcus neoformans chemotyping by quantitative analysis of 1H nuclear magnetic resonance spectra of glucuronoxylomannans with a computer-simulated artificial neural network. Clin. Diagn. Lab. Immunol. 5, 146–159. doi:10.1128/CDLI.5.2.146-159.1998
Decote-Ricardo, D., LaRocque-de-Freitas, I. F., Rocha, J. D. B., Nascimento, D. O., Nunes, M. P., Morrot, A., et al. (2019). Immunomodulatory role of capsular polysaccharides constituents of Cryptococcus neoformans. Front. Med. 6, 129. doi:10.3389/fmed.2019.00129
Doering, T. L. (2009). How sweet it is! Cell wall biogenesis and polysaccharide capsule formation in Cryptococcus neoformans. Annu. Rev. Microbiol. 63, 223–247. doi:10.1146/annurev.micro.62.081307.162753
Dorokhova, V. S., Gerbst, A. G., Komarova, B. S., Previato, J. O., Previato, L. M., Dmitrenok, A. S., et al. (2021). Synthesis and conformational analysis of vicinally branched trisaccharide β-d-Galf-(1 → 2)-[β-d-Galf-(1 → 3)-]-α-Galp from Cryptococcus neoformans galactoxylomannan. Org. Biomol. Chem. 19, 2923–2931. doi:10.1039/D0OB02071K
Gerbst, A. G., Krylov, V. B., Vinnitskiy, D. Z., Dmitrenok, A. S., Shashkov, A. S., and Nifantiev, N. E. (2015). 13C-NMR glycosylation effects in (1→3)-linked furanosyl-pyranosides. Carbohydr. Res. 417, 1–10. doi:10.1016/j.carres.2015.08.014
Gerbst, A. G., Ustuzhanina, N. E., Grachev, A. A., Khatuntseva, E. A., Tsvetkov, D. E., Whitfield, D. M., et al. (2001). Synthesis, nmr, and conformational studies of fucoidan fragments. III. Effect of benzoyl group at O-3 on stereoselectivity of glycosylation by 3-O- and 3,4-di-O-benzoylated 2-O-benzylfucosyl bromides. J. Carbohydr. Chem. 20, 821–831. doi:10.1081/CAR-100108659
Hargett, A. A., Azurmendi, H. F., Crawford, C. J., Wear, M. P., Oscarson, S., Casadevall, A., et al. (2024). The structure of a C. neoformans polysaccharide motif recognized by protective antibodies: a combined NMR and MD study. Proc. Natl. Acad. Sci. 121, e2315733121. doi:10.1073/pnas.2315733121
Heiss, C., Skowyra, M. L., Liu, H., Klutts, J. S., Wang, Z., Williams, M., et al. (2013). Unusual galactofuranose modification of a capsule polysaccharide in the pathogenic yeast Cryptococcus neoformans. J. Biol. Chem. 288, 10994–11003. doi:10.1074/jbc.M112.441998
Heiss, C., Stacey Klutts, J., Wang, Z., Doering, T. L., and Azadi, P. (2009). The structure of Cryptococcus neoformans galactoxylomannan contains β-d-glucuronic acid. Carbohydr. Res. 344, 915–920. doi:10.1016/j.carres.2009.03.003
Hettikankanamalage, A. A., Lassfolk, R., Ekholm, F. S., Leino, R., and Crich, D. (2020). Mechanisms of stereodirecting participation and ester migration from near and far in glycosylation and related reactions. Chem. Rev. 120, 7104–7151. doi:10.1021/acs.chemrev.0c00243
Hirose, H., Tamai, H., Gao, C., Imamura, A., Ando, H., Ishida, H., et al. (2015). Total syntheses of disulphated glycosphingolipid SB1a and the related monosulphated SM1a. Org. Biomol. Chem. 13, 11105–11117. doi:10.1039/C5OB01744K
James, P. G., and Cherniak, R. (1992). Galactoxylomannans of Cryptococcus neoformans. Infect. Immun. 60, 1084–1088. doi:10.1128/iai.60.3.1084-1088.1992
Kochetkov, N. K., Lipkind, G. M., Shashkov, A. S., and Nifantiev, N. E. (1991). N.m.r. and conformational analysis of some 2,3-disubstituted methyl α-L-rhamnopyranosides. Carbohydr. Res. 221, 145–168. doi:10.1016/0008-6215(91)80053-P
Komarova, B. S., Dorokhova, V. S., Tsvetkov, Y. E., and Nifantiev, N. E. (2018a). Synthesis of a biotinylated penta-α-(1→6)-D-glucoside based on the rational design of an α-stereoselective glucosyl donor. Org. Chem. Front. 5, 909–928. doi:10.1039/C7QO01007A
Komarova, B. S., Novikova, N. S., Gerbst, A. G., Sinitsyna, O. A., Rubtsova, E. A., Kondratyeva, E. G., et al. (2023). Combination of 3-O-levulinoyl and 6-O-trifluorobenzoyl groups ensures α-selectivity in glucosylations: synthesis of the oligosaccharides related to Aspergillus fumigatus α-(1 → 3)-D-glucan. J. Org. Chem. 88, 12542–12564. doi:10.1021/acs.joc.3c01283
Komarova, B. S., Orekhova, M. V., Tsvetkov, Y. E., Beau, R., Aimanianda, V., Latgé, J., et al. (2015). Synthesis of a pentasaccharide and neoglycoconjugates related to fungal α-(1→3)-glucan and their use in the generation of antibodies to trace Aspergillus fumigatus cell wall. Chem. – Eur. J. 21, 1029–1035. doi:10.1002/chem.201404770
Komarova, B. S., Orekhova, M. V., Tsvetkov, Y. E., and Nifantiev, N. E. (2014). Is an acyl group at O-3 in glucosyl donors able to control α-stereoselectivity of glycosylation? The role of conformational mobility and the protecting group at O-6. Carbohydr. Res. 384, 70–86. doi:10.1016/j.carres.2013.11.016
Komarova, B. S., Tsvetkov, Y. E., and Nifantiev, N. E. (2016). Design of α-selective glycopyranosyl donors relying on remote anchimeric assistance. Chem. Rec. 16, 488–506. doi:10.1002/tcr.201500245
Komarova, B. S., Wong, S. S. W., Orekhova, M. V., Tsvetkov, Y. E., Krylov, V. B., Beauvais, A., et al. (2018b). Chemical synthesis and application of biotinylated oligo-α-(1→3)-D-glucosides to study the antibody and cytokine response against the cell wall α-(1→3)-D-glucan of Aspergillus fumigatus. J. Org. Chem. 83, 12965–12976. doi:10.1021/acs.joc.8b01142
Krylov, V. B., Argunov, D. A., Solovev, A. S., Petruk, M. I., Gerbst, A. G., Dmitrenok, A. S., et al. (2018). Synthesis of oligosaccharides related to galactomannans from Aspergillus fumigatus and their NMR spectral data. Org. Biomol. Chem. 16, 1188–1199. doi:10.1039/C7OB02734F
Krylov, V. B., Solovev, A. S., Puchkin, I. A., Yashunsky, D. V., Antonets, A. V., Kutsevalova, O. Y., et al. (2021). Reinvestigation of carbohydrate specificity of EBCA-1 monoclonal antibody used for the detection of Candida mannan. J. Fungi 7, 504. doi:10.3390/jof7070504
Laroussarie, A., Barycza, B., Andriamboavonjy, H., Tamigney Kenfack, M., Blériot, Y., and Gauthier, C. (2015). Synthesis of the tetrasaccharide repeating unit of the β-kdo-containing exopolysaccharide from Burkholderia pseudomallei and B. cepacia complex. J. Org. Chem. 80, 10386–10396. doi:10.1021/acs.joc.5b01823
Lipkind, G. M., Shashkov, A. S., Knirel, Y. A., Vinogradov, E. V., and Kochetkov, N. K. (1988). A computer-assisted structural analysis of regular polysaccharides on the basis of 13C-n.m.r. data. Carbohydr. Res. 175, 59–75. doi:10.1016/0008-6215(88)80156-3
Maziarz, E. K., and Perfect, J. R. (2016). Cryptococcosis. Infect. Dis. Clin. 30, 179–206. doi:10.1016/j.idc.2015.10.006
McFadden, D. C., and Casadevall, A. (2004). Unexpected diversity in the fine specificity of monoclonal antibodies that use the same V region gene to glucuronoxylomannan of Cryptococcus neoformans. J. Immunol. 172, 3670–3677. doi:10.4049/jimmunol.172.6.3670
Nakouzi, A., Zhang, T., Oscarson, S., and Casadevall, A. (2009). The common Cryptococcus neoformans glucuronoxylomannan M2 motif elicits non-protective antibodies. Vaccine 27, 3513–3518. doi:10.1016/j.vaccine.2009.03.089
Nigudkar, S., and Demchenko, V. (2015). Stereocontrolled 1,2-cis-glycosylation as the driving force of progress in synthetic carbohydrate chemistry. Chem. Sci. 6, 2687–2704. doi:10.1039/C5SC00280J
Oberli, M. A., Bindschädler, P., Werz, D. B., and Seeberger, P. H. (2008). Synthesis of a hexasaccharide repeating unit from Bacillus anthracis vegetative cell walls. Org. Lett. 10, 905–908. doi:10.1021/ol7030262
Oscarson, S., Alpe, M., Svahnberg, P., Nakouzi, A., and Casadevall, A. (2005). Synthesis and immunological studies of glycoconjugates of Cryptococcus neoformans capsular glucuronoxylomannan oligosaccharide structures. Vaccine 23, 3961–3972. doi:10.1016/j.vaccine.2005.02.029
Paccoud, O., Desnos-Ollivier, M., Cassaing, S., Boukris-Sitbon, K., Alanio, A., Bellanger, A.-P., et al. (2023). Cryptococcus neoformans infections differ among human immunodeficiency virus (HIV)–Seropositive and HIV-seronegative individuals: results from a nationwide surveillance program in France. Open Forum Infect. Dis. 11, ofad658. doi:10.1093/ofid/ofad658
Peltier, P., Euzen, R., Daniellou, R., Nugier-Chauvin, C., and Ferrières, V. (2008). Recent knowledge and innovations related to hexofuranosides: structure, synthesis and applications. Carbohydr. Res. 343, 1897–1923. doi:10.1016/j.carres.2008.02.010
Prabhu, A., Venot, A., and Boons, G.-J. (2003). New set of orthogonal protecting groups for the modular synthesis of heparan sulfate fragments. Org. Lett. 5, 4975–4978. doi:10.1021/ol0359261
Previato, J. O., Vinogradov, E., Maes, E., Fonseca, L. M., Guerardel, Y., Oliveira, P. A. V., et al. (2017). Distribution of the O-acetyl groups and β-galactofuranose units in galactoxylomannans of the opportunistic fungus Cryptococcus neoformans. Glycobiology 27, 582–592. doi:10.1093/glycob/cww127
Rajasingham, R., Govender, N. P., Jordan, A., Loyse, A., Shroufi, A., Denning, D. W., et al. (2022). The global burden of HIV-associated cryptococcal infection in adults in 2020: a modelling analysis. Lancet Infect. Dis. 22, 1748–1755. doi:10.1016/S1473-3099(22)00499-6
Richards, M. R., and Lowary, T. L. (2009). Chemistry and biology of galactofuranose-containing polysaccharides. ChemBioChem 10, 1920–1938. doi:10.1002/cbic.200900208
Rivera, J., Feldmesser, M., Cammer, M., and Casadevall, A. (1998). Organ-dependent variation of capsule thickness in Cryptococcus neoformans during experimental murine infection. Infect. Immun. 66, 5027–5030. doi:10.1128/iai.66.10.5027-5030.1998
Shashkov, A. S., Lipkind, G. M., Knirel, Y. A., and Kochetkov, N. K. (1988). Stereochemical factors determining the effects of glycosylation on the 13C chemical shifts in carbohydrates. Magn. Reson. Chem. 26, 735–747. doi:10.1002/mrc.1260260904
Skelton, M. A., Cherniak, R., Poppe, L., and van Halbeek, H. (1991a). Structure of the de-O-acetylated glucuronoxylomannan from Cryptococcus neoformans serotype D, as determined by 2D NMR spectroscopy. Magn. Reson. Chem. 29, 786–793. doi:10.1002/mrc.1260290808
Skelton, M. A., van Halbeek, H., and Cherniak, R. (1991b). Complete assignment of the 1H- and 13C-n.m.r. spectra of the O-deacetylated glucuronoxylomannan from Cryptococcus neoformans serotype B. Carbohydr. Res. 221, 259–268. doi:10.1016/0008-6215(91)80062-R
Tefsen, B., Ram, A. F., van Die, I., and Routier, F. H. (2012). Galactofuranose in eukaryotes: aspects of biosynthesis and functional impact. Glycobiology 22, 456–469. doi:10.1093/glycob/cwr144
Tokatly, A. I., Vinnitskiy, D. Z., Ustuzhanina, N. E., and Nifantiev, N. E. (2021). Protecting groups as a factor of stereocontrol in glycosylation reactions. Russ. J. Bioorg. Chem. 47, 53–70. doi:10.1134/S1068162021010258
Tsvetkov, Y. E., Burg-Roderfeld, M., Loers, G., Ardá, A., Sukhova, E. V., Khatuntseva, E. A., et al. (2012). Synthesis and molecular recognition studies of the HNK-1 trisaccharide and related oligosaccharides. The specificity of monoclonal anti-HNK-1 antibodies as assessed by surface plasmon resonance and STD NMR. J. Am. Chem. Soc. 134, 426–435. doi:10.1021/ja2083015
Turco, S. J., and Pedersen, L. L. (2003). Galactofuranose metabolism: a potential target for antimicrobial chemotherapy. Cell. Mol. Life Sci. CMLS 60, 259–266. doi:10.1007/s000180300021
Thijssen, M. J., van Rijswijk, M. N., Kamerling, J. P., and Vliegenthart, J. F. (1998). Synthesis of spacer-containing di-and tri-saccharides that represent parts of the capsular polysaccharide of Streptococcus pneumoniae type 6B. Carbohydrate. Research. 306, 93–109. doi:10.1016/S0008-6215(97)00271-1
Vaishnav, V. V., Bacon, B. E., O’Neill, M., and Cherniak, R. (1998). Structural characterization of the galactoxylomannan of Cryptococcus neoformans Cap67. Carbohydr. Res. 306, 315–330. doi:10.1016/S0008-6215(97)10058-1
Vecchiarelli, A. (2000). Immunoregulation by capsular components of Cryptococcus neoformans. Med. Mycol. 38, 407–417. doi:10.1080/mmy.38.6.407.417
Vecchiarelli, A., Pericolini, E., Gabrielli, E., Chow, S.-K., Bistoni, F., Cenci, E., et al. (2011). Cryptococcus neoformans galactoxylomannan is a potent negative immunomodulator, inspiring new approaches in anti-inflammatory immunotherapy. Immunotherapy 3, 997–1005. doi:10.2217/imt.11.86
Villena, S. N., Pinheiro, R. O., Pinheiro, C. S., Nunes, M. P., Takiya, C. M., DosReis, G. A., et al. (2008). Capsular polysaccharides galactoxylomannan and glucuronoxylomannan from Cryptococcus neoformans induce macrophage apoptosis mediated by Fas ligand. Cell. Microbiol. 10, 1274–1285. doi:10.1111/j.1462-5822.2008.01125.x
Vinnitskiy, D. Z., Krylov, V. B., Ustyuzhanina, N. E., Dmitrenok, A. S., and Nifantiev, N. E. (2015). The synthesis of heterosaccharides related to the fucoidan from Chordaria flagelliformis bearing an α-L-fucofuranosyl unit. Org. Biomol. Chem. 14, 598–611. doi:10.1039/C5OB02040A
Vohra, Y., Buskas, T., and Boons, G.-J. (2009). Rapid assembly of oligosaccharides: a highly convergent strategy for the assembly of a glycosylated amino acid derived from PSGL-1. J. Org. Chem. 74, 6064–6071. doi:10.1021/jo901135k
Werz, D. B. (2012). in Chemical synthesis of carbohydrates and their surface immobilization: a brief introduction. Editors C. Microarrays, and Y. Chevolot (Totowa, NJ: Humana Press), 13–29. doi:10.1007/978-1-61779-373-8_2
Zhang, Y., Hu, Y., Liu, S., He, H., Sun, R., Lu, G., et al. (2022). Total synthesis of Lentinus giganteus glycans with antitumor activities via stereoselective α-glycosylation and orthogonal one-pot glycosylation strategies. Chem. Sci. 13, 7755–7764. doi:10.1039/D2SC02176E
Zhao, Y., Ye, L., Zhao, F., Zhang, L., Lu, Z., Chu, T., et al. (2023). Cryptococcus neoformans, a global threat to human health. Infect. Dis. Poverty 12, 20. doi:10.1186/s40249-023-01073-4
Zhu, S.-Y., and Yang, J.-S. (2012). Synthesis of tetra- and hexasaccharide fragments corresponding to the O-antigenic polysaccharide of Klebsiella pneumoniae. Tetrahedron 68, 3795–3802. doi:10.1016/j.tet.2012.03.074
Keywords: Cryptococcus neoformans, oligosaccharides, glucuronoxylomannogalactan, stereoselective glycosylation, orthogonal protecting groups
Citation: Dorokhova VS, Komarova BS, Previato JO, Mendonça Previato L, Krylov VB and Nifantiev NE (2024) Synthesis of branched and linear galactooligosaccharides related to glucuronoxylomannogalactan of Cryptococcus neoformans. Front. Chem. 12:1501766. doi: 10.3389/fchem.2024.1501766
Received: 25 September 2024; Accepted: 23 October 2024;
Published: 14 November 2024.
Edited by:
Teresa M. V. D. Pinho e Melo, University of Coimbra, PortugalReviewed by:
Roman Bielski, Wilkes University, United StatesRajendra Rohokale, University of Florida, United States
Copyright © 2024 Dorokhova, Komarova, Previato, Mendonça Previato, Krylov and Nifantiev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolay E. Nifantiev, bmVuQGlvYy5hYy5ydQ==