 Lamya H. Al-Wahaibi1Mohamed A. Mahmoud2Hayat Ali Alzahrani3
Lamya H. Al-Wahaibi1Mohamed A. Mahmoud2Hayat Ali Alzahrani3 Hesham A. Abou-Zied4
Hesham A. Abou-Zied4 Alshaimaa Abdelmoez2
Alshaimaa Abdelmoez2 Bahaa G. M. Youssif2*
Bahaa G. M. Youssif2* Stefan Bräse5*Safwat M. Rabea6,7
Stefan Bräse5*Safwat M. Rabea6,7- 1Department of Chemistry, College of Sciences, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
- 2Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut, Egypt
- 3Applied Medical Science College, Medical Laboratory Technology Department, Northern Border University, Arar, Saudi Arabia
- 4Medicinal Chemistry Department, Faculty of Pharmacy, Deraya University, Minia, Egypt
- 5Institute of Biological and Chemical Systems - Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology, Karlsruhe, Germany
- 6Medicinal Chemistry Department, Faculty of Pharmacy, Minia University, Minia, Egypt
- 7Apogee Pharmaceuticals, Burnaby, BC, Canada
Introduction: Bacteria have acquired resistance to almost all antibiotics currently in use due to their extensive, broad, and improper utilization over a prolonged period. DNA gyrase and DHFR exhibit significant promise as targets for antibacterial therapeutics.
Methods: We have developed a series of disalicylic acid methylene/Schiff bases hybrids (6a-l) that function as antibacterial agents by targeting DNA gyrase and DHFR.
Results and discussion: The findings showed that 6a-l have significant antibacterial activity against both Gram-positive and Gram-negative bacteria, with inhibition zones (IZ) comparable to or even higher than the reference Ciprofloxacin. MIC testing revealed that 6h and 6l were 1.5 times as effective than ciprofloxacin against S. aureus. Compounds 6h and 6l had MBC values of 28 and 33 nM for S. aureus, compared to Ciprofloxacin’s 45 nM, indicating that they are more potent bactericidal agents. The MIC values for compounds 6c, 6e, 6h, 6j, and 6l against A. flavus were between 14.50 and 19.50 µM, while the MIC value for fluconazole was 11.50 µM. Also, the studied compounds had MIC values between 18.20 and 22.90 µM against C. albicans, while Fluconazole had a MIC value of 17.50 µM. Compound 6h showed a MIC value of 1.70 µM against the clinical strain S. aureus (ATCC 43300) (MRSA), making it an effective antibacterial agent. Compounds 6h, 6j, and 6l inhibited E. coli DNA gyrase with IC50 values of 79, 117, and 87 nM, respectively, compared to the reference novobiocin (IC50 = 170 nM). Additionally, compounds 6h and 6l, the most potent E. coli gyrase inhibitors, showed encouraging results on DHFR. Compounds 6h and 6l exhibit IC50 values of 3.80 µM and 4.25 µM, respectively. These values are significantly lower and hence more effective than Trimethoprim’s IC50 of 5.20 µM.
1 Introduction
Bacterial infections, caused by either Gram-positive or Gram-negative pathogens, are the most common type of infections acquired in hospitals or by the general public (Chu et al., 2019; Abdel-Aziz et al., 2023). Moreover, bacteria have developed resistance to nearly all currently used antibiotics as a result of their long-term, widespread, and incorrect use, complicating the situation (Gao C. et al., 2018; Gao F. et al., 2018; Kumar et al., 2023a). Annually, some 0.7 million deaths occur worldwide due to drug-resistant infections, and this figure might rise to 10 million by the year 2050 if current patterns persist (Agrawal and Patel, 2024; Kumar et al., 2023b). Therefore, it is imperative to expedite the development of novel antibacterials that demonstrate exceptional efficacy against both susceptible and resistant infections (Kumar et al., 2022).
The enzyme dihydrofolate reductase (DHFR) is a crucial target for numerous anticancer and antibacterial medications. It holds significant value in the field of medicinal chemistry due to its role as a cofactor in the production of nucleic acids and amino acids (Roth, 1986; Abdolmaleki and Maddah, 2023). The mechanism of action for DHFR involves the inhibition of DNA, RNA, and protein synthesis, which leads to the stop of cell growth (Alrohily et al., 2019; Chawla et al., 2021). On the other hand, DNA gyrase is a type II topoisomerase enzyme that catalyzes modifications in the topology of DNA (Abdel-Aziz et al., 2023; Hirsch and Klostermeier, 2021). Furthermore, it consists of two chains, GyrA and GyrB subunits, which are accountable for the temporary disruption of two DNA strands and the induction of negative supercoiling in DNA during replication. Antibacterial drugs that specifically target DNA gyrase exert their activity through two mechanisms: gyrase poisoning, as seen in ciprofloxacin, or by blocking the ATP binding site, as observed in novobiocin (Tiz et al., 2019). Due to its crucial role, DNA gyrase has become an appealing target for the development of antibacterial drugs (Elbastawesy et al., 2023). As a result, DHFR and DNA topoisomerases have a demonstrated track record of supporting their function in microbial disorders (Hassan et al., 2020; Alqurashi et al., 2023).
The development of biofilms is another contributing component to bacterial resistance’s pathogenicity. Biofilms consist of an intricate community of bacteria enclosed within a polysaccharide matrix (Singh et al., 2021; Shree et al., 2023). A significant portion of bacterial pathogens generate biofilms as a pathogenic mechanism to adhere to surfaces and protect themselves from antimicrobial agents. More than 80% of persistent microbial infections are associated with biofilms, which pose a significant health concern (Vestby et al., 2020). As a result, given the significant role biofilms play in infection transmission and resistance evolution, there is an urgent need for the discovery of new chemotherapeutic drugs that interfere with biofilm formation and/or break pre-formed ones.
The investigation of medically advantageous heterocyclic frameworks is an essential arena in the field of drug discovery. The compound is referred to as isatin (1H-indole-2,3-dione, Figure 1) moiety is widely present in nature, and its derivatives exhibit a wide range of pharmacological effects, with anti-bacterial activity (Zhang et al., 2018; Liu et al., 2022) being the most prominent.
Figure 1. Structures of isatin, semaxanib, and indirubin.
The isatin moiety offers a wide range of modification possibilities, with the N-1, C-3, and C-5 positions being the most important ones for chemical modification (Wang et al., 2018). Additionally, a number of isatin-based medications, such as Semaxanib and Indirubin, are currently in clinics or undergoing clinical trials for the treatment of a variety of disorders (Xu et al., 2017a; Xu et al., 2017b). The extensive variety of biological activities, along with the ability to make various structural alterations, and the effective use in clinical practice, have motivated researchers to investigate isatins and develop many derivatives with different structures.
Recently, researchers investigated various isatin compounds to establish their efficacy against bacteria. Some of these compounds (I and II, Figure 2) have demonstrated promise in vitro assays as DNA gyrase inhibitors (Alzahrani et al., 2022).
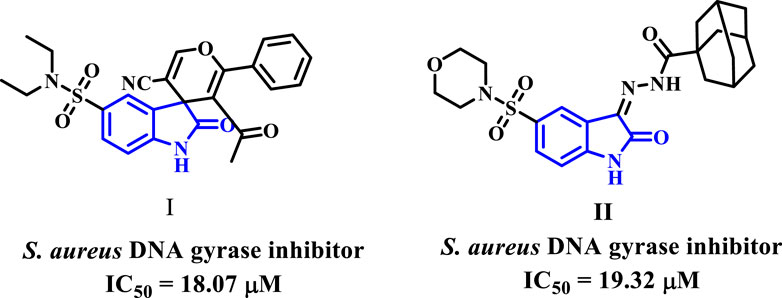
Figure 2. Isatin-based DNA gyrase inhibitors I and II.
Schiff base structures are becoming increasingly popular among researchers due to their ease of synthesis, versatility, and diverse range of activities, including antibacterial properties (Bayeh et al., 2020). In addition, the imine bond found in Schiff base offers the potential to form interactions with various nucleophiles and electrophiles, thereby impeding the activity of enzymes or the replication of DNA (Nastasă et al., 2018).
The combination of multiple pharmacophores into a single hybrid molecule is a promising approach to the development of innovative drugs. This approach has the potential to overcome cross-resistance and increase the effectiveness of the original medications (Hisham et al., 2023; Frejat et al., 2022). In addition, many hybrids, such as Ro 23-9424 and TD-1792, are currently undergoing clinical studies to help battle a variety of diseases. So, combining isatin with other antibacterial pharmacophores could result in very potent options that function against both drug-sensitive and drug-resistant Gram-positive and Gram-negative bacteria.
1.1 Rational design
In a recent work from our lab (Al-Wahaibi et al., 2024), we reported on the design, synthesis, and antibacterial activity of a new series of Schiff bases of disalicylic acid methylene hybrid with various aldehydes as DNA gyrase and Topoisomerase IV inhibitors. The novel hybrids 5a-k, Figure 3, were tested for antibacterial efficacy against Gram-positive pathogens S. aureus (S. aureus) and B. subtilis (B. subtilis), as well as Gram-negative organisms E. coli (E. coli) and P. aeruginosa (P. aeruginosa). Ciprofloxacin was utilized as the reference medication.

Figure 3. Structures of compounds 5a-k and new targets 6a-l.
The in vitro assay test showed that compound 5h (X = 2-OMe) had the highest potency among the compounds examined, with MIC values of 0.030, 0.065, and 0.060 μg/mL against S. aureus, E. coli, and P. aeruginosa. It was as effective as ciprofloxacin against the studied species but had a MIC value of 0.050 μg/mL against B. subtilis, making it five times less potent than ciprofloxacin. Additionally, compound 5i (X = 4-dimethylamino) exhibited the second greatest activity. At the MIC level, it was equally effective against S. aureus, E. coli, and P. aeruginosa as compound 5h and ciprofloxacin were. However, against B. subtilis, it was 7 times less effective than ciprofloxacin. Additionally, the inhibitory efficacy of 5h and 5i against E. coli DNA gyrase was assessed using the E. coli DNA gyrase test. Compounds 5h and 5i exhibited greater inhibitory activity against E. coli DNA gyrase, with IC50 values of 92 ± 5 nM and 97 ± 6 nM, respectively, compared to the positive control novobiocin (IC50 = 170 nM). Compounds 5h and 5i also demonstrated promising effects on Topoisomerase IV. Compounds 5h and 5i exhibit IC50 values of 3.50 µM and 5.80 µM, respectively. These results are much lower and more potent than Novobiocin’s IC50 value of 11 µM.
In the present study, the newly synthesized compounds (6a-l) were developed by making two modifications to the previously disclosed 5a-k compounds. Our first modification involves replacing aldehydes with isatin or isatin derivatives during the synthesis of our new hybrids. Second, in compounds 6j-l, we use a dichloro-disalicylic acid methylene derivative rather than disalicylic acid methylene to increase the antibacterial activity.
Based on the data above and our ongoing research into developing medicinally active antimicrobials (Abdel-Aziz et al., 2023; Elbastawesy et al., 2023; Frejat et al., 2022; Al-Wahaibi et al., 2024; Al-Wahaibi et al., 2021a; Al-Wahaibi et al., 2021b; Aly et al., 2024; Abdu-Allah et al., 2017; Alkhaldi et al., 2018; Alkhaldi et al., 2019; Youssif, 2013; Hofny et al., 2021; Youssif et al., 2016), we report the design and synthesis of new isatin-based Schiff bases 6a–l (Figure 3), obtained from methylene disalicylic acid hydrazide. The novel compounds 6a-l were tested for antibacterial activity against Gram-positive pathogens S. aureus and B. subtilis, as well as Gram-negative organisms E. coli and P. aeruginosa. Ciprofloxacin was utilized as the reference medication. Furthermore, the synthetic compounds 6a-l were tested for antifungal activity against Aspergillus flavus (A. flavus) and Candida albicans (C. albicans), using fluconazole as a reference medication. The minimum inhibitory concentrations (MICs), bactericidal concentrations (MBCs), and fungicidal concentrations (MFCs) of the most active derivatives against the tested microorganisms were compared to ciprofloxacin and/or fluconazole. This study looked at the antibacterial efficacy of the most potent Schiff bases against a panel of multi-drug resistant bacteria (MDRB), which included one clinical strain of S. aureus (ATCC 43300) (MRSA), two standard strains of E. coli (ATCC BAA-196), and P. aeruginosa (ATCC BAA-2111). Norfloxacin, a broad-spectrum antibiotic, was used as the positive control. Additionally, the evaluation focused on assessing the inhibitory potency of the most active compounds against E. coli DNA gyrase and DHFR, which were identified as potential targets. Finally, the cell viability and antibiofilm assays for the most potent compounds were evaluated.
2 Results and discussion
2.1 Chemistry
Scheme 1 depicts the synthesis steps for the key intermediates 4a and 4b, as well as the target compounds 6a-l. Compounds 2, 3, and 4b were previously synthesized and well described (Al-Wahaibi et al., 2024). The procedures used in the current investigation differ slightly from those used in Cushman and Suseela’s study (Cushman and Kanamathareddy, 1990). The synthesis commenced by preparing the methylene bridged 3-chlorosalicylic acid dimer 2 (DSA-Cl2) with a yield of 98%. This was achieved through the condensation of two molecules of 3-chlorosalicylic acid 1 with paraformaldehyde in concentrated sulfuric acid. The chlorine atom was used as a protective group to control the regio-chemistry in the dimerization reaction and prevent further interaction of DSA-Cl2 with formaldehyde in the presence of acid. This interaction would lead to the formation of phenol-formaldehyde polymers.
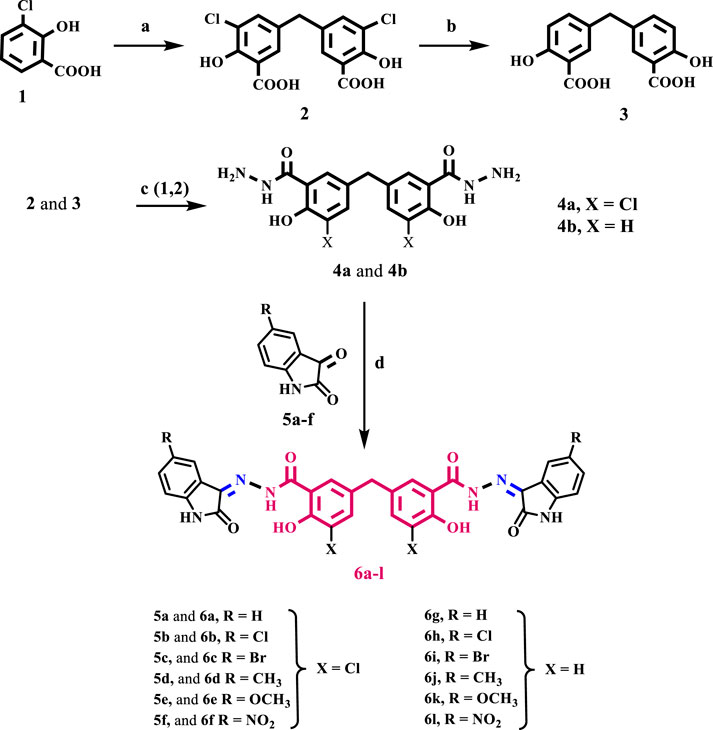
Scheme 1. Reagents and reaction conditions: (A) HCHO, H2SO4, 35°C, 18 h, 98%; (B) KHCOO, Pd/C, KOH, 2- PrOH/H2O, 70°C, 4 h, 92%; (C) 1-EtOH/Conc. H2SO4, 2- Hydrazine hydrate, reflux 6 h; (D) isatin derivative, EtOH, reflux overnight.
The subsequent step involved the reduction of compound 2 through reductive dehalogenation using potassium formate as a source of hydride and Pd/C as a catalyst, resulting in the formation of disalicylic acid dimer 3 with a yield of 92%. Initially, the hydrodehalogenation of the chlorine atom in compound 2 to produce disalicylic acid dimer 3 was attempted using molecular hydrogen (Youssif, 2013). However, this reaction proceeded slowly and was incomplete even after 24 h. To overcome this, catalytic transfer hydrogenation (CTH) was employed, utilizing organic hydrogen donors such as potassium formate (HCOOK) as a reducing agent. CTH demonstrated a significantly higher reaction rate with potassium formate compared to molecular hydrogen. Additionally, potassium formate is easily manageable, less toxic, and less flammable, making it more advantageous for large-scale synthesis.
Compound 4a was prepared from compound 2 using a two-step process. Firstly, compound 2 was heated in absolute ethanol with a catalytic amount of concentrated sulfuric acid. Then, the resulting ester product was further refluxed with an excess of hydrazine hydrate. Compound 4a was obtained in a 65% yield as white crystals after purification. The structure of compound 4a was confirmed using IR spectroscopy, which showed a strong absorption peak at υ 2,616–3,370 cm−1, of phenolic OH groups. In addition, the spectrum exhibited prominent absorption peaks at υ 3,370 and 3,286 cm−1, corresponding to the NH2 group. Additionally, the spectrum revealed a characteristic peak at υ 1,654 cm−1, corresponding to the amidic C=O bond. Unfortunately, due to solubility issues, we were unable to obtain a distinct 1H NMR or 13C NMR spectrum for 4a using commonly used NMR solvents. We use LC-MS spectroscopy, which revealed the molecular ion peak of 4a in the anticipated region of the mass spectrum (See Supplementary Material).
Schiff base derivatives, 6a-l, were prepared by refluxing compounds 4a and 4b with proper (un) substituted isatin in ethanol for 14–18 h, giving 6a-l in good yields. The validity of the structures of 6a-l was verified by employing 1H NMR, 13C NMR, and elemental microanalysis experiments. The 1H NMR spectrum of compound 6k, as a representative example, displayed five distinct singlet signals, which are characteristic of this compound. There is a singlet at δ 14.38 ppm which corresponds to two phenolic OH groups. There is another singlet at δ 11.52 ppm which corresponds to amidic NH groups. Additionally, there is a singlet at δ 10.93 ppm which corresponds to isatin NH groups. There is also a singlet at δ 3.95 ppm which corresponds to an aryl methylene group (Ar-CH2). Lastly, there is a singlet at δ 3.76 ppm which corresponds to six protons of two methoxy groups. Additionally, aromatic protons have distinct signals.
2.2 Biology
2.2.1 In vitro antimicrobial activities
The antimicrobial effects of the currently synthesized Schiff bases (6a-l) were tested against two Gram-positive bacteria (S. aureus and B. subtilis), two Gram-negative bacteria (E. coli and P. aeruginosa), and two strains of fungus (A. flavus and C. albicans). The modified disk diffusion method (Alkhaldi et al., 2019; Manso et al., 2021) was used to determine the inhibition zones (IZ, mm/mL) and the minimal inhibitory concentration (MIC, nM). Ciprofloxacin and Fluconazole was used as positive controls. Tables 1–3 display the findings.
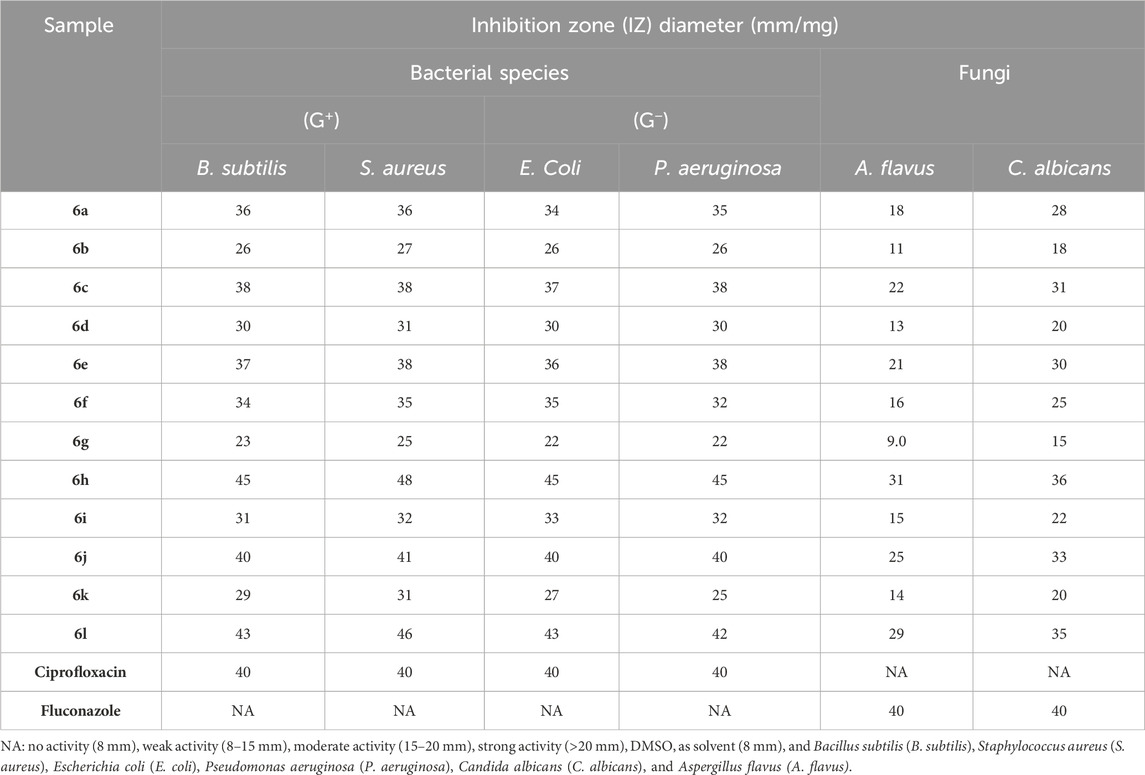
Table 1. Inhibition zone diameter (mm/mg) of compounds 6a-l and reference drugs.

Table 2. MICs and MBCs of compounds 6c, 6e, 6h, 6j, and 6l.
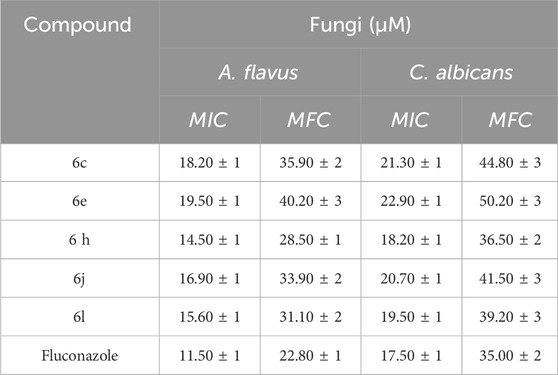
Table 3. MICs and MFCs of compounds 6c, 6e, 6h, 6j, and 6l.
Based on the inhibition zones (IZ) measurements in Table 1, we can conclude that five Schiff bases (6c, 6e, 6h, 6j, and 6l) had inhibition zones that were either greater than or comparable to the inhibition zones of the reference medications (ciprofloxacin and fluconazole) against pathogenic organisms. Schiff bases 6a-l were found to have strong antibacterial activity against both Gram-positive and Gram-negative bacteria, with inhibition zones (IZ) ranging from 22 to 48 mm for all pathogens tested, compared to Ciprofloxacin’s IZ of 40 mm. Moreover, Table 1 demonstrated that compounds 6h and 6l had superior efficacy compared to the reference ciprofloxacin against both gram-positive and gram-negative bacteria. In addition, most of the newly synthesized Schiff bases 6a-l had IZ values between 11 and 36 mm against A. flavus and C. albicans, whereas fluconazole, a broad spectrum antifungal medication, had a value of 40 mm. We proceeded with the investigation to ascertain the minimal inhibitory concentrations (MIC, nM) of the potent Schiff bases (6c, 6e, 6h, 6j, and 6l). The results are displayed in Tables 2, 3.
2.2.2 Minimum inhibitory concentration (MIC) assay
The most potent components 6c, 6e, 6h, 6j, and 6l were evaluated for antibacterial activity using a twofold serial dilution method on a 96-well microtiter plate (Wiegand et al., 2008). Table 2 showed the MICs (nM) of these compounds against the tested bacteria, with ciprofloxacin as the reference medicine. The results of this in vitro assay test are consistent with the findings of the antimicrobial sensitivity test. Compound 6h (R = Cl, X = H) was the most potent of the compounds examined, having MIC values of 19, 54, and 55 nM against S. aureus, E. coli, and P. aeruginosa. It displayed greater potency to ciprofloxacin against the investigated species but had a MIC value of 24 nM against B. subtilis, which is 2.5 times less effective than ciprofloxacin (MIC = 10 nM). Compound 6l (R = NO2, X = H) exhibited the second highest activity. Its MIC values were comparable to those of ciprofloxacin against E. coli and P. aeruginosa, Table 2. However, it was 2.5 times less effective than ciprofloxacin against B. subtilis. Compounds 6h and 6l were 1.5 times more effective than ciprofloxacin against S. aureus, with MIC values of 19 and 21 nM, respectively, whereas ciprofloxacin had a MIC value of 30 nM. Compounds 6c (R = Br, X = Cl) and 6j (R = CH3, X = H) exhibited significant activity against the tested species, especially against S. aureus, with MICs values of 29 and 25 nM, respectively. These values were comparable to that of ciprofloxacin (MIC = 30 nM). Finally, compound 6e (with R = OCH3 and X = Cl) demonstrated the lowest level of efficacy. It has lower antimicrobial activity than ciprofloxacin against all pathogens tested.
2.2.3 Minimum bactericidal concentration (MBC) assay
The MBC differs from the MIC. The MIC test finds the lowest concentration of an antimicrobial agent that significantly inhibits growth, whereas the MBC identifies the lowest concentration that causes microbiological organisms to die. In contrast to the MBC, which does result in mortality, the MIC just inhibits, meaning that the antibacterial action does not cause death (Abdullahi et al., 2023). MBC is typically reported as MBC50, indicating that the antibiotic concentration kills 50% of the initial bacterial population (Sun et al., 2019).
In general, components 6c, 6e, 6h, 6j, and 6l had strong bactericidal activity. The MBC values for Gram-positive bacteria ranged from 28 to 58 nM, whereas ciprofloxacin MBC values were 19 and 45 nM, Table 2. Compounds 6h (R = Cl, X = H) and 6l (R = NO2, X = H), the most potent antibacterial agents, displayed bactericidal activity of 28 and 33 nM, respectively, for S. aureus, compared to ciprofloxacin’s MBC value of 45 nM, making 6h and 6l more effective as bactericidal agents. Compound 6j (R = CH3, X = H) scored third in bactericidal activity with MBC value of 40 nM against S. aureus, which was comparable to the reference ciprofloxacin. Regrettably, compounds 6h, 6j, and 6l exhibited lower effectiveness as bactericidal agents compared to ciprofloxacin against B. subtilis. The MBC values for compounds 6h, 6j, and 6l were 38, 47, and 43 nM, respectively, while ciprofloxacin had an MBC value of 19 nM.
In the case of Gram-negative bacteria, compounds 6h, 6j, and 6l had higher MBC values than the other compounds examined. These compounds have MBC values of 74, 80, and 78 nM, making them more effective than ciprofloxacin (MBC = 90 nM) against both E. Coli and P. aeruginosa species. Again, compound 6e (with R = OCH3 and X = Cl) was the least effective as bactericidal agents against all of the bacterial species tested.
2.2.4 Antifungal assay
Compounds 6c, 6e, 6h, 6j, and 6l were tested for antifungal activity with a twofold serial dilution method (Wiegand et al., 2008). Table 3 displays the MICs (µM) and MFCs (minimum fungicidal concentration, µM) of these derivatives against A. flavus and C. albicans fungus, using fluconazole as the reference medication. Overall, the investigated compounds displayed robust antifungal efficacy against the selected fungal species when compared to ciprofloxacin. Compounds 6c, 6e, 6h, 6j, and 6l exhibited MIC values ranging from 14.50 to 19.50 µM against A. flavus, while fluconazole had a MIC value of 11.50 µM. Moreover, the tested compounds demonstrated MIC values ranging from 18.20 to 22.90 µM against C. albicans, while the reference fluconazole had a MIC of 17.50 µM.
Compound 6h (R = Cl, X = H), the most potent antibacterial agent, was also the most potent antifungal agent, with MIC values of 14.50 µM against A. flavus and 18.20 µM against C. albicans, being comparable to the reference fluconazole, which had MIC values of 11.50 and 17.50 µM, respectively. Compound 6l (R = NO2, X = H) demonstrated the second highest antifungal activity. It exhibited MIC values of 15.60 µM against A. flavus and 19.50 µM against C. albicans. Finally, Table 3 shows that all investigated compounds had fungicidal activity, with MFC/MIC ratios ranging around two.
2.2.5 Antibacterial assay against multi-drug resistant strains
The most potent Schiff bases 6c, 6e, 6h, 6j, and 6l were examined for their antibacterial efficacy against a panel of MDRB (multi-drug resistant bacteria), including one clinical strain S. aureus (ATCC 43300) (MRSA), two standard strain E. coli (ATCC BAA-196) and P. aeruginosa (ATCC BAA-2111). Norfloxacin, a broad-spectrum antibiotic, worked as positive control. Results are displayed in Table 4.
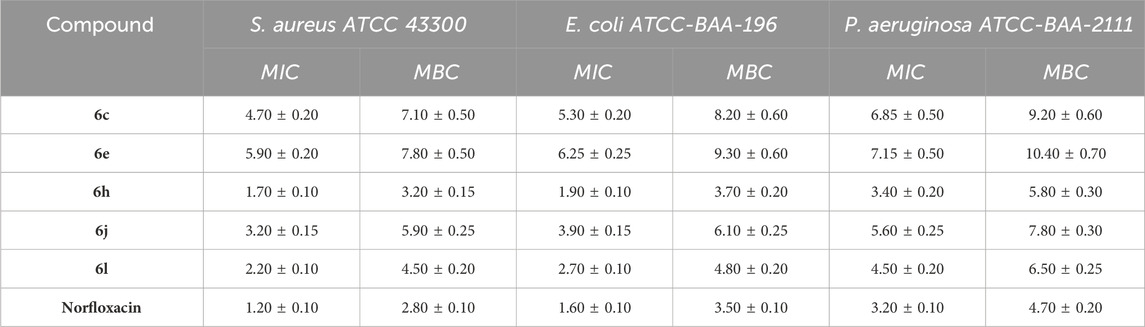
Table 4. MICs and MBCs of compounds 6c, 6e, 6h, 6j, and 6l against MDRB strains.
The MIC values for the tested compounds vary from 1.70 to 7.15 µM, while the MBC values range from 3.50 to 10.40 µM against MDRB bacteria. These values are compared to the MIC values of norfloxacin, which range from 1.20 to 3.20 µM, and the MBC values, which range from 2.80 to 4.70 µM. The majority of the investigated Schiff bases, compounds 6c, 6e, 6h, 6j, and 6l, had substantial broad-spectrum effects on both Gram-positive and Gram-negative bacteria, either by inhibiting their growth or by killing them (bactericidal effect). Notably, compounds 6h and 6l show the most potent actions compared to other derivatives. Compound 6h (R = Cl, X = H) showed a MIC value of 1.70 µM against the clinical strain S. aureus (ATCC 43300) (MRSA), making it an effective antibacterial agent. It was found to be approximately as potent as the reference drug norfloxacin, which had a MIC of 1.20 µM. Compound 6h, on the other hand, exhibited an MBC of 3.90 µM, making it around 1.2 times more potent against the MRSA strain than the reference norfloxacin. Furthermore, compound 6h demonstrated similar efficacy to norfloxacin against E. coli (ATCC BAA-196) and P. aeruginosa (ATCC BAA-2111), with MICs of 1.9 and 3.4 µM, respectively. Norfloxacin, on the other hand, showed MIC values of 1.6 and 3.2 µM. Additionally, compound 6l (R = NO2, X = H) showed significant antibacterial activity against MDRB strains, with MIC values ranging from 2.20 to 4.50 µM and MBC values ranging from 4.50 to 6.50 µM. Based on the MBC/MIC ratio, we identified that all of the compounds tested had values less than two, indicating bactericidal activity.
2.2.6 DNA gyrase and DHFR inhibitory assay
The inhibitory potency of derivatives 6h, 6j, and 6l, the most potent antibacterial agents, against E. coli DNA gyrase and DHFR was determined using the E. coli DNA gyrase and DHFR assay (Durcik et al., 2018). The findings are shown as IC50 values for the investigated compounds and reference drugs (Table 5). The results of this assay complement those of the antibacterial activity investigation. Compounds 6h, 6j, and 6l inhibited E. coli DNA gyrase at IC50 values of 79, 117, and 87 nM, respectively, compared to the reference novobiocin (IC50 = 170 nM). Compounds 6h, 6j, and 6l exhibited greater potency compared to the reference compound novobiocin, with compounds 6h and 6l being twice as potent as novobiocin against DNA gyrase.
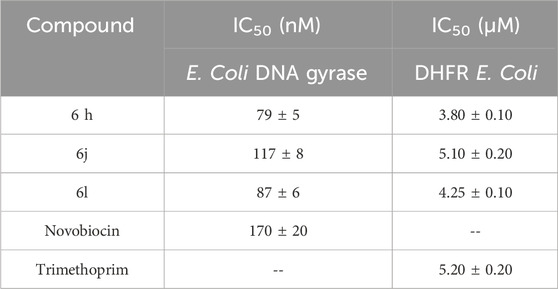
Table 5. IC50 values of 6h, 6j, and 6l against E. coli DNA gyrase and DHFR.
Compounds 6h, 6j, and 6l were further evaluated against the DHFR enzyme as indicated in Table 5. Compounds 6h and 6l, the most potent E. coli gyrase inhibitors, also demonstrated promising effects on DHFR. Compounds 6h and 6l have IC50 values of 3.80 µM and 4.25 µM, respectively. These values are much lower and more potent than trimethoprim’s IC50 value of 5.20 µM. Based on these observations, we may infer that both compounds 6h and 6j show promise as dual-target inhibitors against DNA gyrase and DHFR, especially after optimization.
2.2.7 Antibiofilm assay
Bacterial biofilms provide health risks in hospitals, the food industry, and drinking water systems. In the current work, we investigated the antibiofilm activity of compound 6h, the most potent derivative, against S. aureus using Microtiter plate assay for biofilm quantification (Antunes et al., 2010; Niu and Gilbert, 2004). The assay was performed with three different concentrations, the first of which was equivalent to the MIC of 6h against S. aureus ATCC 43300 (Table 4), the second of which was equivalent to 1/2 MIC, and finally 1/4 MIC. The results were presented in Table 6 as Biofilm inhibition%.
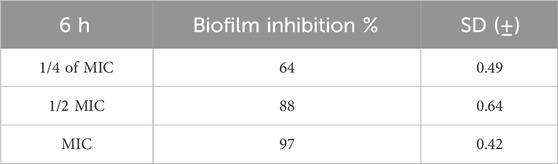
Table 6. Antibiofilm assay of compound 6h.
The results showed that 6h has significant antibiofilm action, with biofilm inhibition percentage equal to 97 at the MIC dose. Compound 6h inhibited biofilms by 88% and 64% at ½ and ¼ MIC levels, respectively.
Blank represented absorbance of media only.
Control represented absorbance of test organism without any treatment.
2.2.8 Cell viability assay
This test examines the impact of compounds 6c, 6e, 6h, 6j, and 6l, which are the most potent derivatives, on normal cell lines in order to assess the safety level of these compounds. The vitality of the investigated compounds was assessed using the MCF-10A cell line, which is a normal human mammary gland epithelial cell line. Following a 4-day incubation period on MCF-10A cells with a concentration of 50 µM for each compound being studied, the viability of the cells was assessed using the MTT test (Hisham et al., 2022; Mahmoud et al., 2022). The results from Table 7 indicate that none of the compounds tested exhibited cytotoxicity, and all compounds show a cell viability of more than 89% at a concentration of 50 µM.
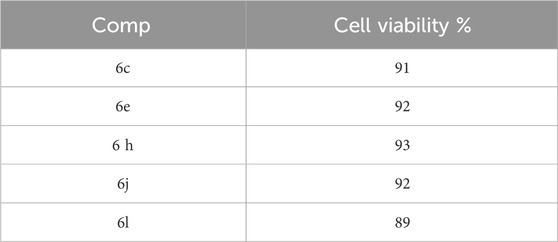
Table 7. Cell viability assay of compounds 6c, 6e, 6h, 6j, and 6l
2.3 Docking studies
2.3.1 Docking study of E. Coli DNA gyrase B
This investigation performed an extensive computational docking analysis to determine the binding affinities of compounds 6b, 6h, and 6l with E. coli DNA gyrase B and E. coli DHFR. Novobiocin served as the reference drug for E. coli DNA gyrase B, while trimethoprim was used as the reference for E. coli DHFR. Utilizing Discovery Studio software (Baroroh et al., 2023), the study provided an in-depth examination of the interaction mechanisms between these compounds and their respective target proteins. To ensure the accuracy and relevance of our investigation, we incorporated the crystallographic structure of the E. coli DNA gyrase B ligand complex (PDB ID: 4DUH) from the Protein Data Bank (Marinho et al., 2023). The OPLS-AA (Optimized Potentials for Liquid Simulations - All Atom) force field was utilized during the energy minimization process for the molecular systems under examination. Implementing this force field was crucial for achieving conformational stability of the molecular structures, thereby enhancing the precision and reliability of our computational analyses (Hazarika et al.; Kumar H. et al., 2023). Prior to initiating the docking procedure, the protein structure underwent comprehensive preparation to ensure its accuracy. This preparation included protonation, a critical step that significantly contributed to the robustness and reliability of the subsequent docking analysis. A comparative analysis between docking scores and in vitro activity levels of E. coli DNA gyrase B for the compounds studied revealed a direct correlation. Compound 6h, which demonstrated the highest in vitro activity against E. coli DNA gyrase B, achieved a docking score of −7.88 kcal/mol. In contrast, compound 6b, with lower in vitro antibacterial activity and a smaller inhibition zone (IZ) measurement compared to the ciprofloxacin, had a docking score of −5.78 kcal/mol. Compound 6l recorded a docking score of −7.51 kcal/mol, reflecting its relatively strong binding affinity and correlating with its moderate in vitro activity. Moreover, Novobiocin, used as a reference drug in the study, displayed a docking score of −7.26 kcal/mol, indicating its considerable binding affinity. In analyzing interactions between the tested compounds and the E. coli DNA gyrase B protein, compound 6h has exhibited notable binding characteristics. An important hydrogen bond interaction between the carbonyl oxygen of the salicylate moiety and Arg136 stabilizes 6h within the active site. This hydrogen bond is crucial as it anchors the compound, preventing its dissociation from the active site and enhancing its inhibitory efficacy (Figure 4). Moreover, the absence of a bulky atom, such as the 3-chloro group on the di-salicylate nucleus of 6h, facilitates the free rotation of the two salicylate moieties around the methylene axis. This rotational freedom is crucial as it enables the compound to adopt a conformation that effectively blocks the entrance to the active site. By fitting more snugly into the binding pocket, 6h can comprehensively obstruct substrate access, thereby enhancing its ability to inhibit E. coli gyrase activity. The isatin nucleus within 6h significantly enhances its binding affinity. The docking interactions reveal that the isatin moiety forms essential interactions with several amino acid residues within the active site. Notably, the π-π T-shaped interactions with residues such as His55 and Lys103 underscore the crucial role of the isatin nucleus in stabilizing the enzyme-inhibitor complex. The aromatic nature of isatin allows for these non-covalent interactions, which strengthen the overall binding and stability of 6h within the active site (Figure 4). Furthermore, the presence of substituents like the 5-chloro group on the isatin nucleus of 6h further amplifies its binding affinity. The 5-chloro substituent increases hydrophobic interactions with the active site residues and contributes to the electronic distribution of the molecule. The electron-withdrawing nature of the 5-chloro group enhances interactions with residues like His99. Additionally, this substituent helps position the isatin moiety in an optimal orientation for interaction with the enzyme, thereby boosting the inhibitory potential of 6h (Figure 4).
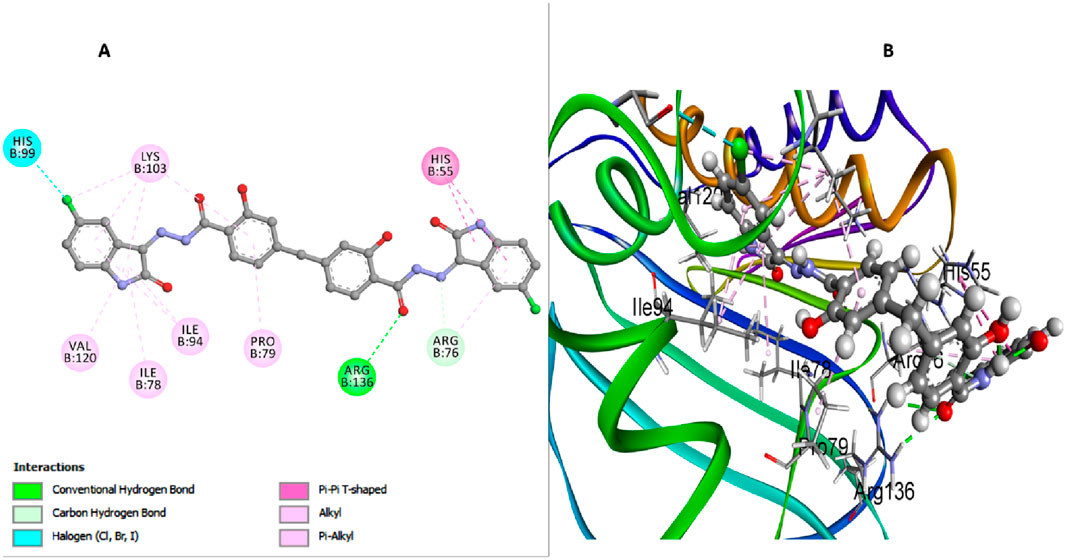
Figure 4. Docking representation models of compound 6h within the binding site of E. coli DNA gyrase B protein; (A) 2D-docked model of compound 6h; (B) 3D-docked model of compound 6h.
In contrast, compound 6l, which also lacks the bulky 3-chloro group on the di-salicylate nucleus like 6 h, exhibits less binding affinity due to fewer interactions with the active site. The absence of the 3-chloro group allows 6l to have a certain degree of rotational freedom, but not as effectively as 6h. This results in a less snug fit within the binding pocket, making it less efficient in blocking substrate access (Figure 5). Arg136 forms a Pi-donor hydrogen bond with the salicylate moiety, while Gly77 and Arg76 interact with the isatin moiety. Thr165 and Asp73 contribute to stabilizing the binding conformation through additional hydrogen bonds.
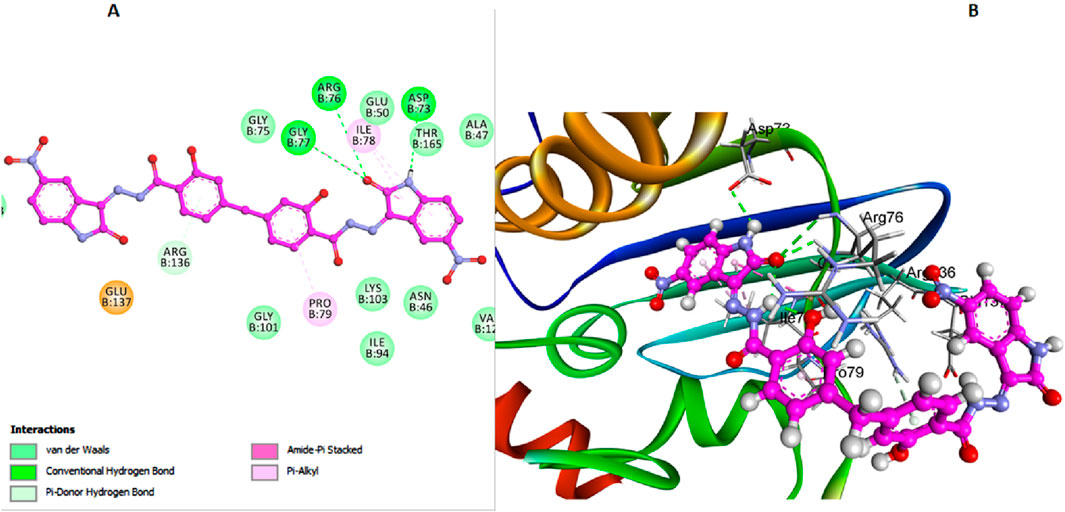
Figure 5. Docking representation models of compound 6l within the binding site of E. coli DNA gyrase B protein; (A) 2D-docked model of compound 6l; (B) 3D-docked model of compound 6l.
In addition, Novobiocin exhibited notable interactions with the E. coli DNA gyrase B protein, with its amide nitrogen functioning as a hydrogen bond acceptor in conjunction with Gly 101, as depicted in Figure 6. However, this significant interaction results in Novobiocin having a comparatively lower binding affinity to the active site than compounds 6h and 6l. This reduced affinity can be attributed to the lack of additional stabilizing interactions that compounds 6h and 6l possess, such as the π-π interactions with His55 and Lys103 in compound 6h and the hydrophobic interactions facilitated by the 5-chloro group in compound 6h (Figure 4). Moreover, the rigid structure of Novobiocin, which might limit its flexibility and ability to adopt an optimal conformation within the active site. This structural rigidity can result in fewer contacts with key residues within the binding pocket, leading to weaker overall binding interactions (Figure 6).
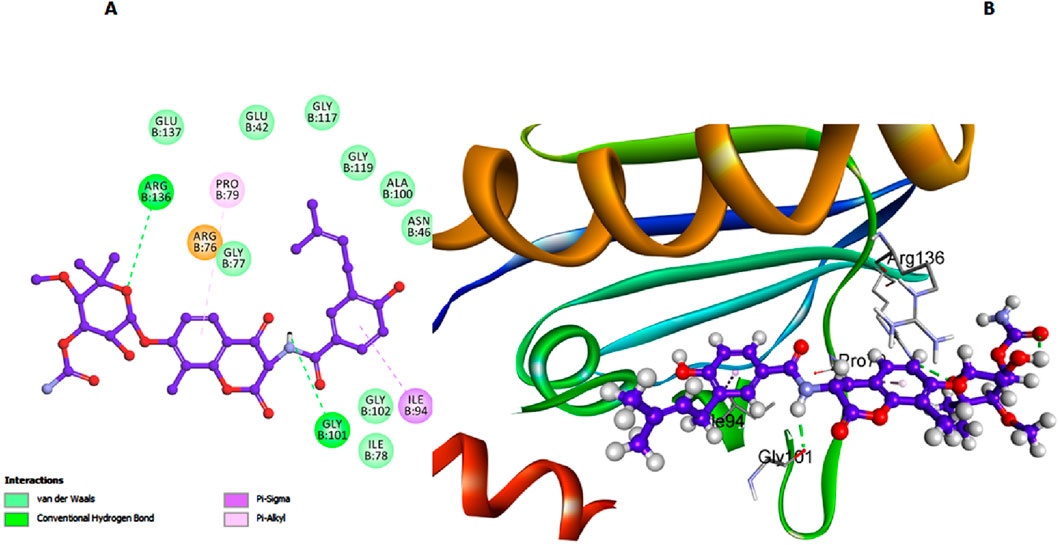
Figure 6. Docking representation models of Novobiocin within the binding site of E. coli DNA gyrase B protein; (A) 2D-docked model of Novobiocin; (B) 3D-docked model of Novobiocin.
On the other hand, compound 6b features a bulky atom, the 3-chloro group on the di-salicylate nucleus, which significantly influences its interaction with E. coli gyrase. The presence of the 3-chloro group restricts the free rotation of the salicylate moieties around the methylene axis. This restriction limits the conformational flexibility of 6b, potentially reducing its ability to snugly fit into the binding pocket and block substrate access as effectively as 6h (Figure 7).
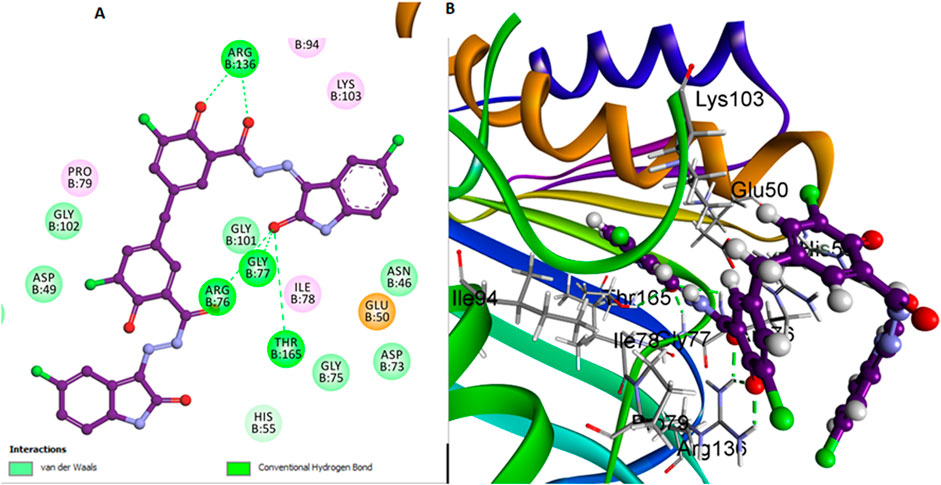
Figure 7. Docking representation models of compound 6b within the binding site of E. coli DNA gyrase B protein; (A) 2D-docked model of compound 6b; (B) 3D-docked model of compound 6b.
Comparative analysis between compounds 6h, 6l, and 6b reveals insights into their flexibility and stability. Compound 6h benefits from the absence of bulky substituents, allowing for greater conformational flexibility and a snugger fit within the active site, leading to potentially higher inhibitory efficacy. Compound 6l, while also lacking the bulky 3-chloro group on salicylate, forms fewer interactions within the active site, resulting in reduced binding affinity and inhibitory potency compared to 6h. On the other hand, compound 6b, with its bulky 3-chloro group on salicylate, faces a steric hindrance that reduces its conformational adaptability.
2.3.2 Docking study of E. Coli DHFR
In our study on E. coli DHFR, we utilized the crystallographic structure of its ligand complex (PDB ID: 6CXK) from the Protein Data Bank as a foundational framework for computational modeling (Bhati, 2024). Compound 6h, which demonstrated the highest in vitro activity against E. coli DHFR, achieved a docking score of −7.42 kcal/mol. Similarly, compound 6l, with nearly comparable in vitro activity, recorded a docking score of −7.28 kcal/mol. Trimethoprim, used as the reference compound in this study, exhibited a docking score of −6.94 kcal/mol. The significance of the docking score of trimethoprim lies in its role as a benchmark, highlighting that compounds 6h and 6l exhibit stronger binding affinities, as evidenced by their lower docking scores, which correlates with their higher in vitro activities against E. coli DHFR. In the comprehensive analysis focusing on the interaction profiles between the investigated compounds and E. coli DHFR, compound 6h exhibited significant binding characteristics. The docking interactions of compound 6h with the DHFR E. coli enzyme provide detailed insights into its inhibitory potential. The carbonyl and phenolic oxygens of the salicylate moiety form two crucial hydrogen bonds with Lys32, anchoring 6h within the active site (Figure 8). Additionally, π-stacked interactions with Leu28 and Leu54 further enhance the binding affinity of 6h, while hydrogen bonds with Ile5 and Asn18 help stabilize its overall conformation within the active site. These interactions highlight the importance of specific residues in stabilizing the binding of compound 6h, allowing it to fit snugly and effectively block substrate access, thereby enhancing its inhibitory efficacy against the DHFR E. coli enzyme.
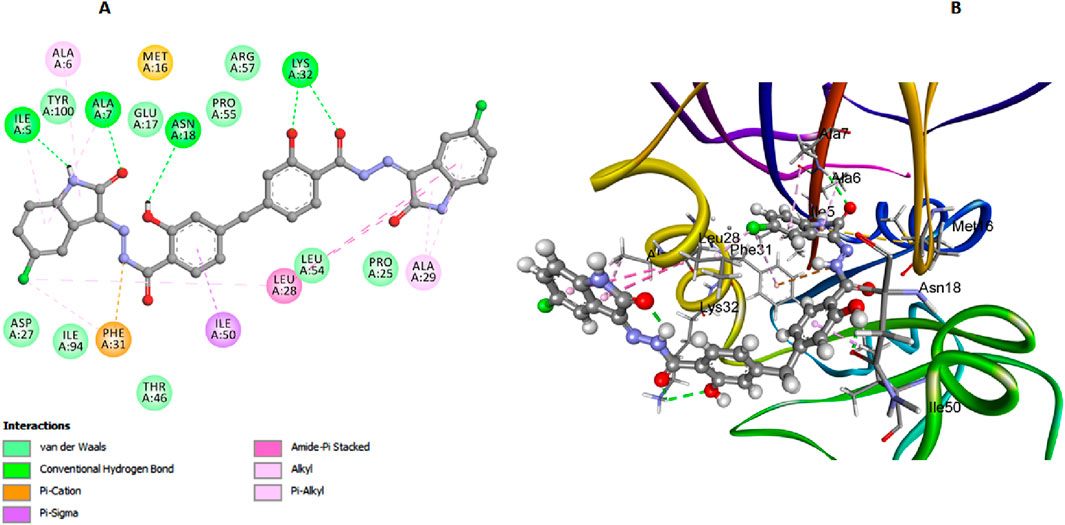
Figure 8. Docking representation models of compound 6h within the binding site of E. coli DHFR enzyme; (A) 2D-docked model of compound 6h; (B) 3D-docked model of compound 6h.
Similarly, compound 6l exhibits a binding affinity to DHFR E. coli enzyme that is comparable to 6h. The docking interactions of 6l reveal several key interactions contributing to its stability and inhibitory potential. The carbonyl oxygen of the salicylate moiety in 6l forms a crucial hydrogen bond with Lys32, mirroring the interaction seen with 6h (Figure 9). Pi-cation interactions between the aromatic ring of the salicylate moiety and Ala29 further stabilize the enzyme-inhibitor complex. Additionally, isatin-pi stacked interactions with Leu54, and Pro25 enhance the binding affinity of 6l, while hydrogen bonds with Asn23 and Ile50 contribute to the overall stability of the compound within the active site. These interactions allow 6l to occupy the DHFR active site effectively, forming stable bonds with key residues and blocking substrate access, thus inhibiting enzyme activity.

Figure 9. Docking representation models of compound 6l within the binding site of E. coli DHFR enzyme; (A) 2D-docked model of compound 6l; (B) 3D-docked model of compound 6L.
Trimethoprim exhibited notable interactions with the E. coli DHFR enzyme, with its amino nitrogen functioning as a hydrogen bond acceptor in conjunction with Asp27, as depicted in Figure 10. However, this significant interaction results in Trimethoprim having a comparatively lower binding affinity to the active site than compounds 6h and 6l. This reduced affinity can be attributed to the lack of additional stabilizing interactions that compounds 6h and 6l possess, such as the π-π stacking interactions with Ala29 and Lys32 of di-salicylate and also, the network interactions facilitated by the isatin nucleus.
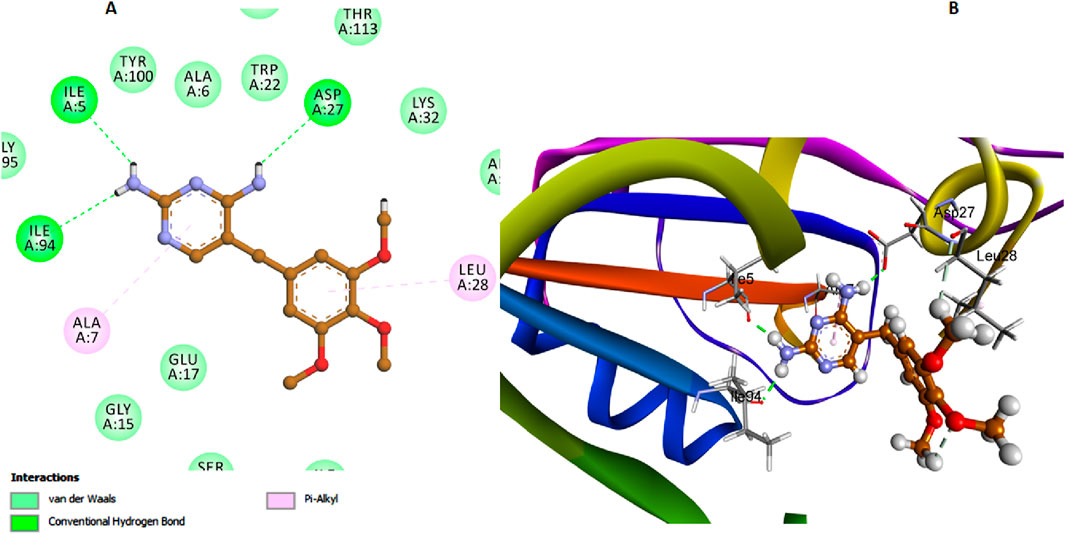
Figure 10. Docking representation models of Trimethoprim within the binding site of E. coli DHFR enzyme; (A) 2D-docked model of Trimethoprim; (B) 3D-docked model of Trimethoprim.
This dual modeling against E. coli DNA gyrase B and DHFR enzymes has crucial implications for antibacterial drug development. Particularly, the favorable docking poses of 6h suggest its potential as dual inhibitor against E. coli DNA gyrase B and DHFR enzymes.
In conclusion, The molecular docking results provided crucial insights into the binding affinities and interaction mechanisms of the compounds with E. coli DNA gyrase B and DHFR enzymes. As summarized in Table 8, compound 6h demonstrated the strongest binding affinity due to key hydrogen bonds and π-π interactions with active site residues, making it particularly effective in blocking substrate access. Comparatively, compound 6l and reference drugs like Novobiocin and Trimethoprim exhibited fewer stabilizing interactions, resulting in relatively lower binding affinities.
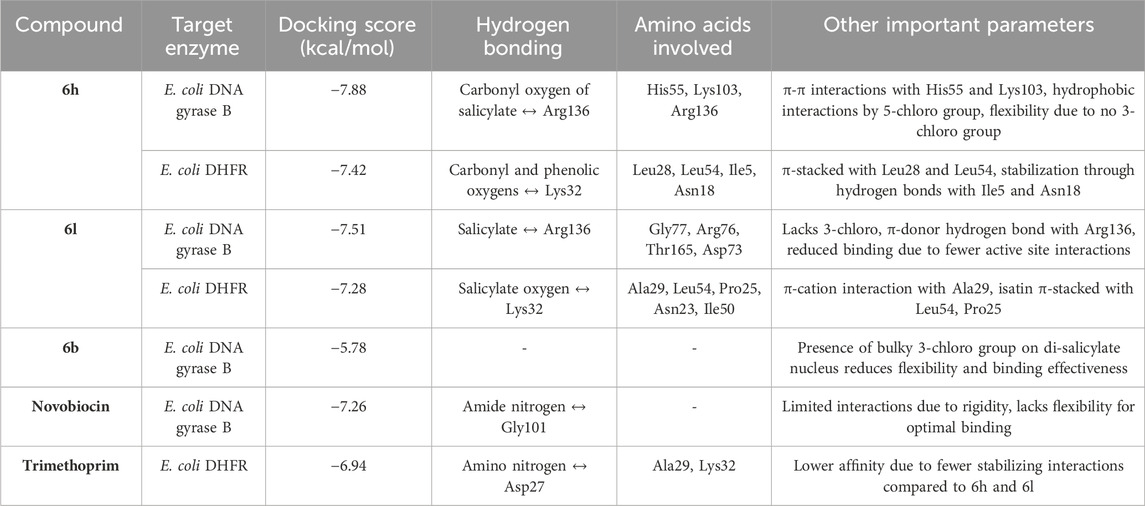
Table 8. Binding affinity and molecular interactions of compounds 6b, 6h and 6l with E. coli DNA gyrase B and DHFR enzymes in molecular docking studies.
2.4 Molecular dynamics (MD) simulation against E. coli DNA gyrase B
To validate the docking study results, a molecular dynamics (MD) simulation was performed using GROMACS 2023 (Vieira et al., 2023). The MD simulations for compound 6h, the top hit against E. coli DNA gyrase B enzyme, provided comprehensive insights into the stability and binding interactions of the compound within the active site over 180 ns simulation period. The key parameters analyzed include RMSD, RMSF, radius of gyration, hydrogen bonds, and potential energy. The Root Mean Square Deviation (RMSD) plot (Figure 11) indicates the stability of the compound-enzyme complex over the 180 ns simulation period. The RMSD of the backbone atoms of E. coli DNA gyrase B (red line) remained stable around 0.5 nm, signifying minimal fluctuations and indicating that the protein structure was well-maintained throughout the simulation. The RMSD of compound 6h (blue line) showed initial fluctuations within the first 20 ns, stabilizing thereafter around 2.5 nm. This initial fluctuation is typical as the compound adjusts within the binding pocket, after which the stable RMSD suggests a consistent binding conformation.
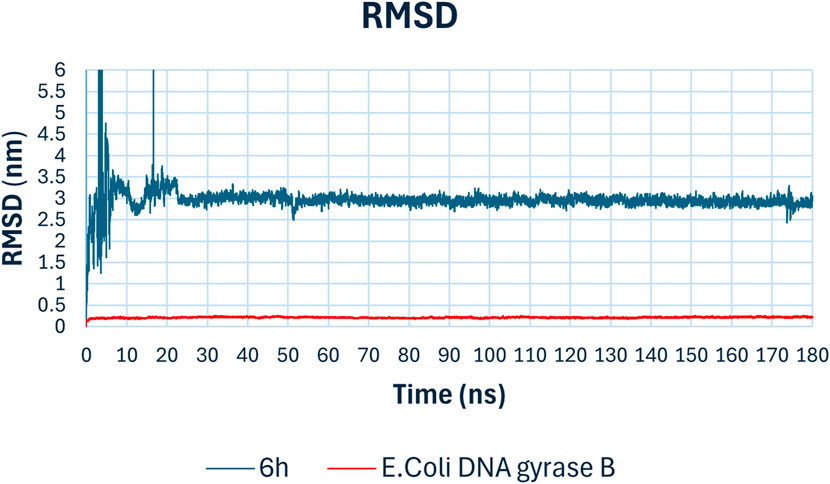
Figure 11. Rmsd analysis of compound 6h with E. coli DNA gyrase B.
The Root Mean Square Fluctuation (RMSF) chart (Figure 12) provides insights into the flexibility of individual amino acid residues. Peaks in the RMSF plot indicate regions of higher flexibility. Most residues showed low fluctuations, suggesting a rigid binding environment. However, certain residues around the binding site exhibited slightly higher fluctuations, indicating their involvement in dynamic interactions with compound 6h.
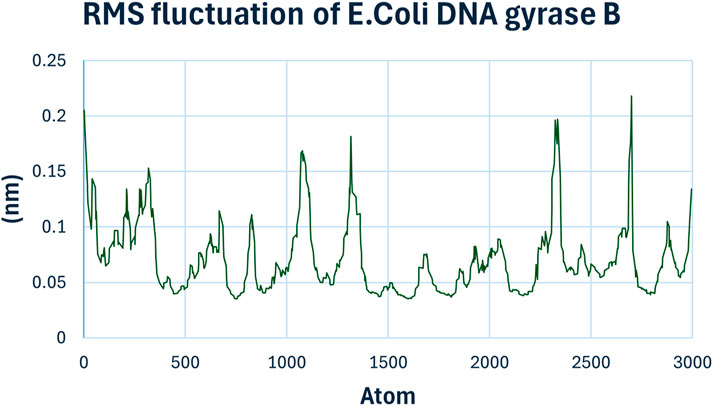
Figure 12. RMSF Analysis of compound 6h with E. coli DNA Gyrase B.
The radius of gyration (Rg), (Figure 13), measures the compactness of the protein structure over time. The Rg values for the complex remained consistently around 1.59 nm, with minor fluctuations, indicating that the overall compactness and tertiary structure of the protein were maintained during the simulation. This suggests that the binding compound 6h does not induce significant conformational changes in the protein structure.
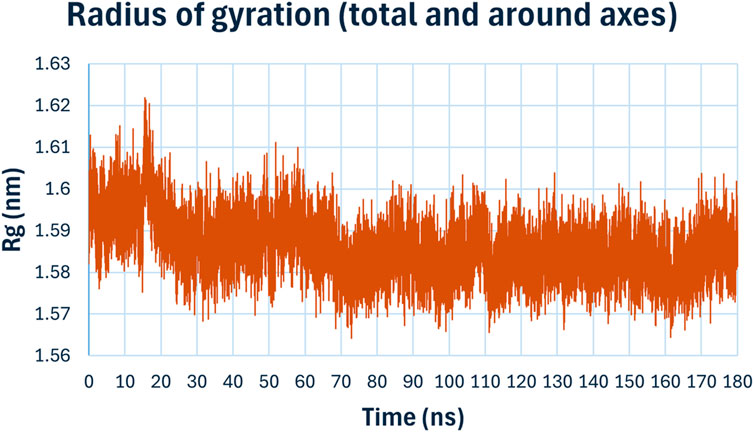
Figure 13. Radius of gyration (Rg) of compound 6h with E. coli DNA Gyrase B.
Hydrogen bonding analysis (Figure 14) is crucial for understanding the stability and specificity of ligand binding. The number of hydrogen bonds between compound 6h and E. coli DNA gyrase B fluctuated between 1 and 4 throughout the simulation, stabilizing mostly around 2 hydrogen bonds. These persistent hydrogen bonds are indicative of strong and stable interactions between the ligand and the protein, contributing to the high binding affinity observed in docking studies.
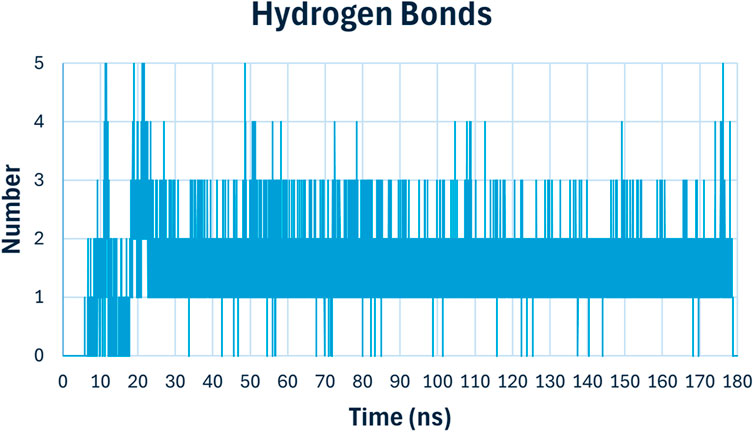
Figure 14. Hydrogen bonding analysis of compound 6h with E. coli DNA Gyrase B.
The potential energy (Figure 15) of the system remained relatively stable throughout the simulation, fluctuating around −438000 kJ/mol. The stability in potential energy further confirms the stability of the protein-ligand complex during the MD simulation, indicating that no significant energetic disturbances occurred, and the system was equilibrated.
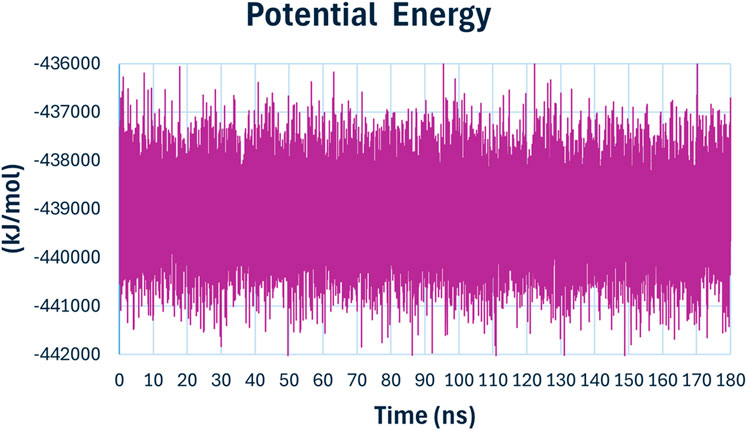
Figure 15. The potential energy of compound 6h with E. coli DNA Gyrase B.
The MD simulation results demonstrate that compound 6h forms a stable complex with E. coli DNA gyrase B. The consistent RMSD, stable radius of gyration, persistent hydrogen bonding, and stable potential energy all point towards a robust binding interaction. These findings support the potential of compound 6h as a promising lead compound for further development as an antibacterial agent targeting replication mechanisms. The observed molecular interactions and stability underscore its efficacy and provide a strong foundation for subsequent experimental validation and optimization studies.
3 Conclusion
Twelve compounds (6a-l) were developed by combining disalicylic acid methylene hydrazide with various isatin derivatives. The antibacterial activity of newly synthesized compounds 6a-l was assessed against a variety of gram-negative and gram-positive bacterial strains, as well as fungal species. These novel targets were tested for DNA gyrase and DHFR inhibitory activities. The results showed that Compounds 6h and 6l were the most potent antibacterial agents, with MIC and MBC values comparable to or even lower than the reference Ciprofloxacin. Compound 6h had a promising MIC value against the clinical strain S. aureus (ATCC 43300) (MRSA), indicating that it is an efficient antibacterial agent. It was shown to be about as potent as the reference medication, Norfloxacin. DNA gyrase and DHFR inhibitory experiments revealed that compounds 6h and 6l were twice as potent as novobiocin against DNA gyrase and more potent as DHFR inhibitors than the standard trimethoprim. Based on these data, we may conclude that both compounds 6h and 6j show promise as dual-target inhibitors of DNA gyrase and DHFR, especially following optimization. Additionally, compound 6 h demonstrated substantial antibiofilm effect, with a biofilm inhibition percentage of 97 at the MIC level. Docking experiments demonstrated that compound 6h has a high affinity for both the DNA gyrase and the DHFR enzyme. MD simulations lasting 180 nanoseconds demonstrated compound 6h′s stability and binding interactions in the active region of DNA gyrase. The computational results, together with the promising antibacterial activity exhibited in laboratory tests for compound 6h, indicate that it has the potential to be developed as a lead compound in combating bacterial infections that are resistant to many treatments. Subsequent study will focus on improving the molecular structure of 6h to optimize its pharmacokinetic characteristics and efficacy in biological systems.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
LA-W: Funding acquisition, Methodology, Resources, Software, Writing–review and editing. MM: Methodology, Writing–original draft, Writing–review and editing. HA: Formal Analysis, Methodology, Writing–review and editing. HA-Z: Methodology, Software, Writing–original draft, Writing–review and editing. AA: Data curation, Formal Analysis, Software, Writing–review and editing. BY: Conceptualization, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. SB: Supervision, Visualization, Writing–review and editing. SR: Methodology, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2023R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Conflict of interest
Author SR was employed by Apogee Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1493906/full#supplementary-material
References
Abdel-Aziz, S. A., Cirnski, K., Herrmann, J., Abdel-Aal, M. A., Youssif, B. G., and Salem, O. I. (2023). Novel fluoroquinolone hybrids as dual DNA gyrase and urease inhibitors with potential antibacterial activity: design, synthesis, and biological evaluation. J. Mol. Struct. 1271, 134049. doi:10.1016/j.molstruc.2022.134049
Abdolmaleki, B., and Maddah, M. (2023). Screening of indole derivatives as the potent anticancer agents on dihydrofolate reductase: pharmaco-informatics and molecular dynamics simulation. J. Biomol. Struct. Dyn. 41 (8), 3667–3679. doi:10.1080/07391102.2022.2053745
Abdu-Allah, H. H., Youssif, B. G., Abdelrahman, M. H., Abdel-Hamid, M. K., Reshma, R. S., Yogeeswari, P., et al. (2017). Synthesis and anti-mycobacterial activity of 4-(4-phenyl-1 H-1, 2, 3-triazol-1-yl) salicylhydrazones: revitalizing an old drug. Archives pharmacal Res. 40, 168–179. doi:10.1007/s12272-016-0882-x
Abdullahi, Z. U., Musa, S. S., Abu-Odah, H., Ahmed, A., Lawan, A. A., and Bello, U. M. (2023). Bactericidal effects of snake venom phospholipases A2: a systematic review and analysis of minimum inhibitory concentration. Physiologia 3 (1), 30–42. doi:10.3390/physiologia3010003
Agrawal, A., and Patel, A. K. (2024). Antibiotic resistance profile and detection in ESKAPE pathogens, ESKAPE pathogens: detection, mechanisms and treatment strategies, 33–77. Springer2024.
Alkhaldi, A. A., Abdelgawad, M. A., and Youssif, B. G. (2018). Synthesis, antimicrobial evaluation and docking studies of new pyrazolone derivatives. Trop. J. Pharm. Res. 17 (11), 2235–2241. doi:10.4314/tjpr.v17i11.18
Alkhaldi, A. A., Abdelgawad, M. A., Youssif, B. G., El-Gendy, A. O., and De Koning, H. P. (2019). Synthesis, antimicrobial activities and GAPDH docking of novel 1, 2, 3-triazole derivatives. Trop. J. Pharm. Res. 18 (5), 1101–1108. doi:10.4314/tjpr.v18i5.27
Alqurashi, R. M., Farghaly, T. A., Sabour, R., and Shaabana, M. R. (2023). Design, Synthesis, antimicrobial screening and molecular modeling of novel 6, 7 dimethylquinoxalin-2 (1H)-one and thiazole derivatives targeting DNA gyrase enzyme. Bioorg. Chem. 134, 106433. doi:10.1016/j.bioorg.2023.106433
Alrohily, W. D., Habib, M. E., El-Messery, S. M., Alqurshi, A., El-Subbagh, H., and Habib, E.-S. E. (2019). Antibacterial, antibiofilm and molecular modeling study of some antitumor thiazole based chalcones as a new class of DHFR inhibitors. Microb. Pathog. 136, 103674. doi:10.1016/j.micpath.2019.103674
Al-Wahaibi, L. H., Amer, A. A., Marzouk, A. A., Gomaa, H. A., Youssif, B. G., and Abdelhamid, A. A. (2021a). Design, synthesis, and antibacterial screening of some novel heteroaryl-based ciprofloxacin derivatives as DNA gyrase and topoisomerase IV inhibitors. Pharmaceuticals 14 (5), 399. doi:10.3390/ph14050399
Al-Wahaibi, L. H., Mahmoud, M. A., Alzahrani, H. A., Abou-Zied, H. A., Gomaa, H. A., Youssif, B. G., et al. (2024). Discovery of new Schiff bases of the disalicylic acid scaffold as DNA gyrase and Topoisomerase IV inhibitors endowed with antibacterial properties. Front. Chem. 12, 1419242. doi:10.3389/fchem.2024.1419242
Al-Wahaibi, L. H., Mostafa, A., Mostafa, Y. A., Abou-Ghadir, O. F., Abdelazeem, A. H., Gouda, A. M., et al. (2021b). Discovery of novel oxazole-based macrocycles as anti-coronaviral agents targeting SARS-CoV-2 main protease. Bioorg. Chem. 116, 105363. doi:10.1016/j.bioorg.2021.105363
Aly, A. A., Abdallah, E. M., Ahmed, S. A., Rabee, M. M., Fuhr, O., Ibrahim, M. A., et al. (2024). Synthesis and characterization of new palladium (II) and silver (I) thiosemicarbazone derived by acenaphthenequinone complexes and their antimicrobial activity. Polyhedron 251, 116851. doi:10.1016/j.poly.2024.116851
Alzahrani, A. Y., Ammar, Y. A., Salem, M. A., Abu-Elghait, M., and Ragab, A. (2022). Design, synthesis, molecular modeling, and antimicrobial potential of novel 3-[(1H-pyrazol-3-yl) imino] indolin-2-one derivatives as DNA gyrase inhibitors. Arch. Pharm. 355 (1), 2100266. doi:10.1002/ardp.202100266
Antunes, A. L. S., Trentin, D. S., Bonfanti, J. W., Pinto, C. C. F., Perez, L. R. R., Macedo, A. J., et al. (2010). Application of a feasible method for determination of biofilm antimicrobial susceptibility in staphylococci. Apmis 118 (11), 873–877. doi:10.1111/j.1600-0463.2010.02681.x
Baroroh, U., Biotek, M., Muscifa, Z. S., Destiarani, W., Rohmatullah, F. G., and Yusuf, M. (2023). Molecular interaction analysis and visualization of protein-ligand docking using Biovia Discovery Studio Visualizer. Indonesian J. Comput. Biol. (IJCB) 2 (1), 22–30. doi:10.24198/ijcb.v2i1.46322
Bayeh, Y., Mohammed, F., Gebrezgiabher, M., Elemo, F., Getachew, M., and Thomas, M. (2020). Synthesis, characterization and antibacterial activities of polydentate Schiff bases, based on salicylaldehyde. Adv. Biol. Chem. 10 (05), 127–139. doi:10.4236/abc.2020.105010
Beu, T. A., and Farcaş, A. (2017). CHARMM force field and molecular dynamics simulations of protonated polyethylenimine. J. Comput. Chem. 38 (27), 2335–2348. doi:10.1002/jcc.24890
Bhati, S. (2024). Synthesis, characterization, antimicrobial screening and in-silico studies of thiadiazole derivatives. J. Mol. Struct. 1316, 138793. doi:10.1016/j.molstruc.2024.138793
Chawla, P., Teli, G., Gill, R. K., and Narang, R. K. (2021). An insight into synthetic strategies and recent developments of dihydrofolate reductase inhibitors. ChemistrySelect 6 (43), 12101–12145. doi:10.1002/slct.202102555
Chu, X.-M., Wang, C., Liu, W., Liang, L.-L., Gong, K.-K., Zhao, C.-Y., et al. (2019). Quinoline and quinolone dimers and their biological activities: an overview. Eur. J. Med. Chem. 161, 101–117. doi:10.1016/j.ejmech.2018.10.035
Cushman, M., and Kanamathareddy, S. (1990). Synthesis of the covalent hydrate of the incorrectly assumed structure of aurintricarboxylic acid (ATA). Tetrahedron 46 (5), 1491–1498. doi:10.1016/s0040-4020(01)81957-8
Durcik, M., Tammela, P., Barančoková, M., Tomašič, T., Ilaš, J., Kikelj, D., et al. (2018). Synthesis and evaluation of N-phenylpyrrolamides as DNA gyrase B Inhibitors. ChemMedChem 13 (2), 186–198. doi:10.1002/cmdc.201700549
Elbastawesy, M. A., Mohamed, F. A., Zaki, I., Alahmdi, M. I., Alzahrani, S. S., Alzahrani, H. A., et al. (2023). Design, synthesis and antimicrobial activity of novel quinoline-2-one hybrids as promising DNA gyrase and topoisomerase IV inhibitors. J. Mol. Struct. 1278, 134902. doi:10.1016/j.molstruc.2023.134902
Frejat, F. O., Cao, Y., Wang, L., Zhai, H., Abdelazeem, A. H., Gomaa, H. A., et al. (2022). New 1, 2, 4-oxadiazole/pyrrolidine hybrids as topoisomerase IV and DNA gyrase inhibitors with promising antibacterial activity. Arch. Pharm. 355 (7), 2100516. doi:10.1002/ardp.202100516
Gao, C., Fan, Y.-L., Zhao, F., Ren, Q.-C., Wu, X., Chang, L., et al. (2018a). Quinolone derivatives and their activities against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem. 157, 1081–1095. doi:10.1016/j.ejmech.2018.08.061
Gao, F., Wang, P., Yang, H., Miao, Q., Ma, L., and Lu, G. (2018b). Recent developments of quinolone-based derivatives and their activities against Escherichia coli. Eur. J. Med. Chem. 157, 1223–1248. doi:10.1016/j.ejmech.2018.08.095
Hassan, A. S., Askar, A. A., Naglah, A. M., Almehizia, A. A., and Ragab, A. (2020). Discovery of new Schiff bases tethered pyrazole moiety: design, synthesis, biological evaluation, and molecular docking study as dual targeting DHFR/DNA gyrase inhibitors with immunomodulatory activity. Molecules 25 (11), 2593. doi:10.3390/molecules25112593
Hazarika, Z., Rajkhowa, S., and Jha, A. N. “Role of force fields in protein function prediction,” in Homol. Mol. modeling-perspectives Appl.
Hirsch, J., and Klostermeier, D. (2021). What makes a type IIA topoisomerase a gyrase or a Topo IV? Nucleic Acids Res. 49 (11), 6027–6042. doi:10.1093/nar/gkab270
Hisham, M., Hassan, H. A., Gomaa, H. A., Youssif, B. G., Hayalah, A. M., and Abdel-Aziz, M. (2023). Design, synthesis, and antiproliferative activity of quinazolin-4-one/chalcone hybrids via the EGFR inhibition pathway. Anti-Cancer Agents Med. Chem. Former. Curr. Med. Chemistry-Anti-Cancer Agents 23 (17), 1932–1943. doi:10.2174/1871520623666230727104933
Hisham, M., Hassan, H. A., Gomaa, H. A., Youssif, B. G., Hayallah, A. M., and Abdel-Aziz, M. (2022). Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as EGFR inhibitors. J. Mol. Struct. 1254, 132422. doi:10.1016/j.molstruc.2022.132422
Hofny, H. A., Mohamed, M. F., Gomaa, H. A., Abdel-Aziz, S. A., Youssif, B. G., El-Koussi, N. A., et al. (2021). Design, synthesis, and antibacterial evaluation of new quinoline-1, 3, 4-oxadiazole and quinoline-1, 2, 4-triazole hybrids as potential inhibitors of DNA gyrase and topoisomerase IV. Bioorg. Chem. 112, 104920. doi:10.1016/j.bioorg.2021.104920
Kumar, D., Aggarwal, N., Kumar, H., Kapoor, G., Deep, A., Bibi, S., et al. (2023a). 2-Substituted-3-(5-Substituted-1,3,4-oxadiazol/thiadiazol-2-yl) thiazolidin-4-one derivatives: synthesis, anticancer, antimicrobial, and antioxidant potential. Pharmaceuticals 16 (6), 805. doi:10.3390/ph16060805
Kumar, D., Aggarwal, N., Kumar, V., Kumar, H., Deep, A., Bibi, S., et al. (2023b). Synthesis, anticancer, antimicrobial and antioxidant potential of novel 4-(substituted phenyl-1,3,4-oxadiazol/thiadiazol-2-yl)-4-(4-substituted phenyl) azetidin-2-one derivatives. Pharmaceuticals 16 (4), 517. doi:10.3390/ph16040517
Kumar, H., Aggarwal, N., Marwaha, M. G., Deep, A., Chopra, H., Matin, M. M., et al. (2022). Thiazolidin-2,4-Dione scaffold: an insight into recent advances as antimicrobial, antioxidant, and hypoglycemic agents. Molecules 27 (19), 6763. doi:10.3390/molecules27196763
Kumar, H., Sharma, A., Kumar, D., Marwaha, M. G., Dhanawat, M., Aggarwal, N., et al. (2023c). Synthesis, biological evaluation and in silico studies of some new analogues of 3, 5-disubstituted thiazolidin-2, 4-dione. Future Med. Chem. 15 (24), 2257–2268. doi:10.4155/fmc-2023-0237
Liu, B., Jiang, D., and Hu, G. (2022). The antibacterial activity of isatin hybrids. Curr. Top. Med. Chem. 22 (1), 25–40. doi:10.2174/1568026621666211116090456
Mahmoud, M. A., Mohammed, A. F., Salem, O. I., Gomaa, H. A., and Youssif, B. G. (2022). New 1, 3, 4-oxadiazoles linked with the 1, 2, 3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch. Pharm. 355 (6), 2200009. doi:10.1002/ardp.202200009
Manso, T., Lores, M., and de Miguel, T. (2021). Antimicrobial activity of polyphenols and natural polyphenolic extracts on clinical isolates. Antibiotics 11 (1), 46. doi:10.3390/antibiotics11010046
Marinho, A. M. d.R., de Oliveira, C. M. S., Silva-Silva, J. V., de Jesus, S. C. A., Siqueira, J. E. S., de Oliveira, L. C., et al. (2023). Antimicrobial activity and molecular docking studies of the biotransformation of diterpene acanthotic acid using the fungus Xylaria sp. Antibiotics 12 (8), 1331. doi:10.3390/antibiotics12081331
Nastasă, C., Vodnar, D. C., Ionuţ, I., Stana, A., Benedec, D., Tamaian, R., et al. (2018). Antibacterial evaluation and virtual screening of new thiazolyl-triazole Schiff bases as potential DNA-gyrase inhibitors. Int. J. Mol. Sci. 19 (1), 222. doi:10.3390/ijms19010222
Niu, C., and Gilbert, E. (2004). Colorimetric method for identifying plant essential oil components that affect biofilm formation and structure. Appl. Environ. Microbiol. 70 (12), 6951–6956. doi:10.1128/aem.70.12.6951-6956.2004
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30 (1), 70–82. doi:10.1002/pro.3943
Roth, B. (1986). Design of dihydrofolate reductase inhibitors from X-ray crystal structures. Fed. Proc. 45, 2765–2772.
Shree, P., Singh, C. K., Sodhi, K. K., Surya, J. N., and Singh, D. K., Biofilms: understanding the structure and contribution towards bacterial resistance in antibiotics, Medicine in Microecology (2023). 100084.
Singh, S., Datta, S., Narayanan, K. B., and Rajnish, K. N. (2021). Bacterial exo-polysaccharides in biofilms: role in antimicrobial resistance and treatments. J. Genet. Eng. Biotechnol. 19, 1–19. doi:10.1186/s43141-021-00242-y
Sun, N., Li, M., Cai, S., Li, Y., Chen, C., Zheng, Y., et al. (2019). Antibacterial evaluation and mode of action study of BIMQ, a novel bacterial cell division inhibitor. Biochem. biophysical Res. Commun. 514 (4), 1224–1230. doi:10.1016/j.bbrc.2019.05.086
Tiz, D. B., Skok, Ž., Durcik, M., Tomašič, T., Mašič, L. P., Ilaš, J., et al. (2019). An optimised series of substituted N-phenylpyrrolamides as DNA gyrase B inhibitors. Eur. J. Med. Chem. 167, 269–290. doi:10.1016/j.ejmech.2019.02.004
Vestby, L. K., Grønseth, T., Simm, R., and Nesse, L. L. (2020). Bacterial biofilm and its role in the pathogenesis of disease. Antibiotics 9 (2), 59. doi:10.3390/antibiotics9020059
Vieira, I. H. P., Botelho, E. B., de Souza Gomes, T. J., Kist, R., Caceres, R. A., and Zanchi, F. B. (2023). Visual dynamics: a web application for molecular dynamics simulation using GROMACS. BMC Bioinforma. 24 (1), 107. doi:10.1186/s12859-023-05234-y
Wang, G., Chen, M., Qiu, J., Xie, Z., and Cao, A. (2018). Synthesis, in vitro α-glucosidase inhibitory activity and docking studies of novel chromone-isatin derivatives. Bioorg. and Med. Chem. Lett. 28 (2), 113–116. doi:10.1016/j.bmcl.2017.11.047
Wiegand, I., Hilpert, K., and Hancock, R. E. (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3 (2), 163–175. doi:10.1038/nprot.2007.521
Xu, Z., Song, X.-F., Hu, Y.-Q., Qiang, M., and Lv, Z.-S. (2017b). Azide-alkyne cycloaddition towards 1H-1, 2, 3-triazole-tethered gatifloxacin and isatin conjugates: design, synthesis and in vitro anti-mycobacterial evaluation. Eur. J. Med. Chem. 138, 66–71. doi:10.1016/j.ejmech.2017.05.057
Xu, Z., Zhang, S., Song, X., Qiang, M., and Lv, Z. (2017a). Design, synthesis and in vitro anti-mycobacterial evaluation of gatifloxacin-1H-1, 2, 3-triazole-isatin hybrids. Bioorg. and Med. Chem. Lett. 27 (16), 3643–3646. doi:10.1016/j.bmcl.2017.07.023
Youssif, B. G., Mohamed, Y. A., Salim, M. T., Inagaki, F., Mukai, C., and Abdu-Allah, H. H. (2016). Synthesis of some benzimidazole derivatives endowed with 1, 2, 3-triazole as potential inhibitors of hepatitis C virus. Acta Pharm. 66 (2), 219–231. doi:10.1515/acph-2016-0014
Youssif, B. G. M. (2013). Synthesis and biological evaluation of some new coumarin derivatives as antimicrobial agents. Bull. Pharm. Sci. Assiut Univ. 36 (2), 105–116. doi:10.21608/bfsa.2013.63201
Keywords: bacterial resistance, biofilm, DNA, isatin, salicylic acid, bactericidal
Citation: Al-Wahaibi LH, Mahmoud MA, Alzahrani HA, Abou-Zied HA, Abdelmoez A, Youssif BGM, Bräse S and Rabea SM (2024) Synthesis, enzyme inhibition, and docking studies of new schiff bases of disalicylic acid methylene-based derivatives as dual-target antibacterial agents. Front. Chem. 12:1493906. doi: 10.3389/fchem.2024.1493906
Received: 09 September 2024; Accepted: 30 October 2024;
Published: 12 November 2024.
Edited by:
Diego Brancaccio, University of Naples Federico II, ItalyReviewed by:
Penke Vijaya Babu, Tikvah Pharma Solutions Pvt Ltd., IndiaNavidha Aggarwal, Maharishi Markandeshwar University, India
Copyright © 2024 Al-Wahaibi, Mahmoud, Alzahrani, Abou-Zied, Abdelmoez, Youssif, Bräse and Rabea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bahaa G. M. Youssif, YmFoYWEueW91c3NpZkBwaGFybS5hdW4uZWR1LmVn, Ymd5b3Vzc2lmMkBnbWFpbC5jb20=; Stefan Bräse, YnJhZXNlQGtpdC5lZHU=