 Nick Gerrits
Nick Gerrits- Gorlaeus Laboratories, Leiden Institute of Chemistry, Leiden University, Leiden, Netherlands
The dissociation of methane is not only an important reaction step in catalytic processes, but also of fundamental interest. Dynamical effects during the dissociative chemisorption of methane on metal surfaces cause significant differences in computed reaction rates, compared to what is predicted by typical transition state theory (TST) models. It is clear that for a good understanding of the catalytic activation of methane dynamical simulations are required. In this paper, a general blueprint is provided for performing dynamical simulations of the dissociative chemisorption of methane on metal surfaces, by employing either the quasi-classical trajectory or ring polymer molecular dynamics approach. If the computational setup is constructed with great care–since results can be affected considerably by the setup – chemically accurate predictions are achievable. Although this paper concerns methane dissociation, the provided blueprint is, so far, applicable to the dissociative chemisorption of most molecules.
1 Introduction
Methane steam reforming is an important industrial process to produce syngas, where the dissociative chemisorption (DC) of methane (i.e., breaking the first CH bond) is typically the rate controlling step (Wei and Iglesia, 2004; Zhang et al., 2021). Unfortunately, methane dissociation is a highly activated catalytic reaction, requiring a large amount of energy in the form of high temperature and pressure. In order to meet future sustainability goals, the energy consumption of methane activation needs to be reduced. Therefore, simulations of the DC of the methane molecule on a metal surface are not just of fundamental interest, but also practical.
In surface science, single crystal surface facets are investigated instead of real catalytic surfaces that exhibit many different facets. The reduced complexity helps gaining a clear understanding of the surface properties and how they affect molecule-surface interactions, while still being valuable for understanding heterogeneous catalysis (Ertl, 1983; Ertl, 1990). Such investigations are performed experimentally using, e.g., supersonic molecular beams, with very accurate Miller indexed cuts through the metal that ensure a defect rate lower than 0.1% (Kroes, 2021). For molecular beam studies of DC, defects that are extremely more reactive or are highly accessible through mobile trapping of the molecule rarely affect the results significantly. Additionally, so-called stepped instead of flat single crystal surfaces can often yield good understanding of defects. Although, sometimes it is necessary to simulate considerably large unit cell sizes to accurately represent catalytic materials (Imbihl et al., 2007; Gerrits, 2021a).
Unfortunately, the success of atomistic theoretical simulations hinges on many factors, e.g., the electronic structure theory, the (dynamical) model, and the tractability. For the electronic structure, density functional theory (DFT) is the workhorse method of choice, but which density functional (DF) to employ is not straightforward (Kroes, 2021; Díaz et al., 2009; Nattino et al., 2016a; Nattino et al., 2016b; Migliorini et al., 2017; Gerrits et al., 2020a). For example, if the difference between the surface’ work function and molecule’s electron affinity is smaller than 7 eV, all generalized gradient approximation (GGA) DFs are expected to underestimate the barrier height (Gerrits et al., 2020a). Fortunately, for methane, this difference is typically much larger than the threshold of 7 eV, generally allowing the use of affordable GGA DFs. But even then, not any GGA DF can be employed (Nattino et al., 2016a; Nattino et al., 2016b; Migliorini et al., 2017; Chadwick et al., 2018a; Tchakoua et al., 2023).
The employed (dynamical) model is also very important as it can have large consequences for the determination of reaction rates. For example, molecular dynamics (MD) simulations of the DC of
Finally, AIMD simulations are expensive, because they often require 500–2000 on-the-fly DFT calculations per trajectory (one DFT calculation per time step) under the conditions typically simulated. Moreover, if one compares to
From above, it is clear that, at present, some form of MD simulations is required to accurately compute reaction rates for the DC of methane and to analyse the reaction mechanism. Unfortunately, many non-trivial considerations go into setting up such calculations, which can influence the computed reactivity considerably. Therefore, in this paper, I will discuss what choices need to be made and why (with a focus on the quasi-classical trajectory (QCT) approach), as well as provide a blueprint for future dynamical simulations of methane. The key aspects of setting up these dynamical calculations are shown in Figure 1. This blueprint is, so far, largely applicable to the activated DC of any molecule.
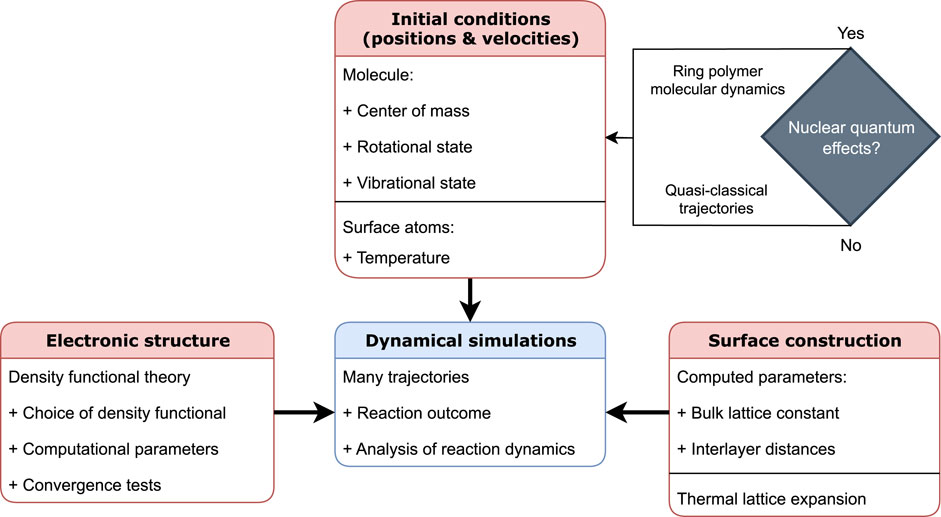
Figure 1. Key aspects of setting up and performing dynamical simulations of DC of molecules on surfaces. The three main categories that are needed as “inputs” are the electronic structure, construction of the surface, and initial conditions (i.e., atomic positions and velocities) of both the molecule and the metal surface.
2 Choice of density functional
The choice of the DF is important, because it underpins the entire simulation and conclusions drawn from it. As mentioned above, GGA DFs should be suitable for the DC of methane. So far, the semi-empirical SRP32-vdW-DF1 DF is the only chemically accurate DF for
Interestingly, PBE (Perdew et al., 1996) yields similar errors across the methane subset of SBH17 as SRP32-vdW-DF1,14 but it is likely that the PESs yielded by PBE are too reactive for methane (Chadwick et al., 2018a). The disagreement in
It should also be noted that SRP32-vdW-DF1 performed poorly for the DC of methane on Pt(210) and reconstructed Pt(110)-
In short, when simulating the DC of methane on close-packed surfaces, where the reactive site is reasonably coordinated, I advice to use SRP32-vdW-DF1, because it will usually yield chemically accurate results. When the reactive site involves a metal atom that is considerably undercoordinated, a more advanced DF is required. Although more expensive than GGA (roughly a factor 3 Mejia-Rodriguez and Trickey, 2008), mGGA DFs of the MS-PBEl family seem to offer a good balance between performance and computational cost (Gerrits et al., 2020a; Tchakoua et al., 2023; Smeets et al., 2019; Smeets and Kroes, 2021; Wei et al., 2021; Cai et al., 2024; Gerrits et al., 2020b; Gerrits et al., 2021). Future efforts should establish which specific mGGA DF is a more general-purpose DF for the DC of methane. Moreover, technical advancements can bring the computational cost down, e.g., regularization of the iso-orbital indicator (Cai et al., 2024; Furness and Sun, 2019; Furness et al., 2020) and de-orbitalization to remove the expensive dependence on the kinetic energy density (Mejia-Rodriguez and Trickey, 2018; Mejia-Rodriguez and Trickey, 2017; Tran et al., 2018; Mejía-Rodríguez and Trickey, 2020). Hopefully, developments will also make the use of exact exchange tractable.
3 Geometries
Although dynamical simulations are a necessity in order to compute (chemically) accurate reaction probabilities, properties extracted from the PES with static calculations can still provide valuable insights. When computing static PES properties, one needs to be aware of the precise reaction mechanism that is at play. Since the DC of methane is a highly activated process, the reaction proceeds typically directly from the gas phase towards the TS at the surface, without prior physisorption or (thermal) equilibration. Although it should be noted that precursor mediated reaction of methane has also been observed (Moiraghi et al., 2020; Seets et al., 1997a; Seets et al., 1997b). This means that the asymptotic value used to compute the barrier height (since the barrier height is a relative energy) corresponds to gaseous
Moreover, the surface needs to be treated with care, as it affects results considerably (Tchakoua et al., 2023; Mondal et al., 2013). The bulk lattice constant should be obtained with a similar computational setup to the rest of the calculations. With the computed bulk lattice constant, a slab with specific Miller indices can be constructed. Subsequently, the interlayer distances are optimized, where the bottom interlayer distances are often fixed to their bulk values, in order to retain a bulk-like behaviour, even if the slab is rather thin. The rule of thumb is to leave at least the top three layers mobile. If the simulated surface temperature is non-zero, as it should be in dynamical simulations of methane, the lattice is expanded in all directions with the experimental thermal expansion coefficient (Mondal et al., 2013).
Obviously, the convergence of, e.g., the number of layers, supercell size, and vacuum distance needs to be validated. A typical benchmark is to compute the TS geometry with a reasonable computational setup and the dimer method (Henkelman and Jónsson, 1999). The resulting geometry is then used in single point calculations using different computational setups to gauge the convergence. Of course, other parameters than the aforementioned ones can be checked this way as well. From personal experience, the following parameters are generally the bare minimum if chemical accuracy (i.e., an error lower than 4.2 kJ/mol) is desired: 4 surface layers, supercell size in the
It should be emphasized that a vacuum distance of 13Å is not converged for methane when employing a non-local correlation DF (as one should, vide supra). Considerably larger vacuum gaps are required, but are computationally more expensive: Even though the vacuum is empty, a larger distance between the slabs still yields a larger computational cost, because it scales with the real-space system size. Typically, the error is about 2–5 kJ/mol for a vacuum distance of 13Å and only dependents on the maximum distance between the periodic slabs and molecule. Thus, a common trick in MD simulations of reactive scattering is to compute the error in the minimum barrier height due to the vacuum distance not being converged. Then, to compensate, the error in the barrier height is added to the initial incidence energy of the molecule (Nattino et al., 2016b).
4 Dynamical simulations
The dynamical simulations can be performed with the QCT approach. In this approach, microcanonical (
4.1 Center of mass
For the center of mass, the most straightforward approach is to simulate only a single incidence energy
where
where
Many experiments assume normal energy scaling (i.e.,
4.2 Rotational state
So far, rotational excitation of methane is observed to have a very limited effect on the rotational state-specific
Here, I discuss how to set up the rotational state of the symmetric top rotor
Additionally, the quantum number
and
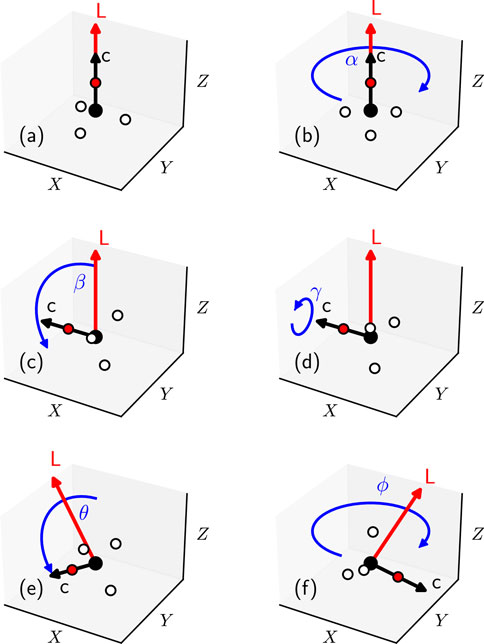
Figure 2. (A) The initial orientation of a symmetric top molecule (black arrow), here
Finally, both the figure axis and the angular momentum vector are rotated by the spherical
The azimuthal angle
4.3 Vibrational state
In QCT, the vibrational initial conditions of a molecule are obtained by micro-canonical sampling of each of its vibrational modes (Karplus et al., 1965). A 1D MD simulation is performed along each mode (i.e., the vibrational modes are not coupled), from which the initial displacement and concomitant velocity is selected by randomly sampling the phase of the vibration. Subsequently, the sum of the mode-specific displacements and velocities are added to the atomic positions and velocities, while also taking into account the orientation of the molecule given by its rotational state. The 1D potentials are computed along the normal mode Cartesian vectors extracted from the Hessian, which is obtained through finite differences. The vibrational quantum mechanical energies are determined through 1D quantum dynamics (QD) calculations on the same potential [see, e.g., Ref (Colbert and Miller, 1992)].
In principle, more accurate (semi-)classical methods can be employed to obtain the vibrational distributions, as long as it is done on the same PES as the rest of the calculations (Nguyen and Barker, 2010). But it is likely that the accuracy would mainly increase when multiple modes are simultaneously excited. Unfortunately, in this case, the QCT approach is considerably less accurate due to artificial intramolecular vibrational energy redistribution (IVR), causing overestimation of the reactivity (Gerrits et al., 2024). Multi-mode excitation of a molecule is also difficult to model, due to the mixing of modes (Hundt et al., 2017). Ring polymer molecular dynamics (RPMD) has been found to be a suitable alternative to QCT for the DC of methane on Pt(111), especially if the translational energy is low or the vibrational energy is high (Gerrits et al., 2024). Due to the approximate inclusion of nuclear quantum effects in RPMD, artificial IVR is reduced, zero-point energy (ZPE) is conserved, and tunneling effects are included. For RPMD, a large portion of the initial conditions are obtained in the same manner as for QCT, because the translational and rotational motion are only applied to the “classical” centroid, ignoring related quantum effects in the ring polymer normal modes. For the vibrational initial conditions, a different approach is required (Gerrits et al., 2024). At present, it is only possible to simulate a thermal Boltzmann distribution of the vibrational modes, instead of state-specific initial conditions. Specifically, a canonical (
So far, I have only discussed how to obtain a rovibrational state, but not its thermostatistical weight. The rovibrational state population
where
4.4 Surface atom motion
To simulate the effect of surface temperature
The frequency
The simulated surface temperature should be above the Debye temperature, which also reduces the issue of trapped trajectories (Nattino et al., 2016b; Migliorini et al., 2017; Manson, 1991; Manson, 1994). Otherwise, classical MD yields incorrect phonon distributions. At present, it is unclear whether RPMD would correctly describe the phonon distribution at low surface temperatures. Regardless, if you use RPMD, it is advised to use
4.5 Reaction outcome
For dynamical simulations of the DC of methane, typically three different reaction outcomes are defined: scattering, dissociation, and trapping. Methane is often considered to be scattered when the distance between the surface macroscopic plane and methane’s center of mass is larger than half of the vacuum distance (i.e., larger than the initial condition) and its momentum is pointing away from the surface. It is possible to reduce the distance criterium to save computational cost, but it should be checked whether this affects results (e.g., how bouncing trajectories are counted). Furthermore, methane is considered to be reacted if one of the intramolecular bonds is considerably extended beyond the TS value, or a smaller length for a certain amount of time. Typically safe parameters are
The reaction probability
The energy transfer
where
Finally, if RPMD is employed, observables are generally computed in the same fashion as with QCT, by simply using the centroid.
5 Conclusion
The DC of methane on metal surfaces is an important reaction step in catalytic processes. Dynamical effects cause significant deviation in reaction rates and mechanisms, compared to what is predicted by TST models. Therefore, for an accurate description and understanding of the DC of methane, dynamical simulations are required. Performing such calculations is not trivial and many choices have to be made. In this paper, I have described how an accurate dynamical simulation might be set up within the QCT approach, or alternatively using RPMD. Perhaps the most important points are the choice of DF, the way the surface geometry is obtained, the dynamical model, and the construction of the initial conditions. If the dynamical calculations are carefully constructed, chemically accurate predictions are possible. Moreover, most of the choices made here are the same or similar for simulations of the DC of molecules other than methane. Therefore, this work also serves as a blueprint for simulating DC in general.
Author contributions
NG: Conceptualization, Investigation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author declares that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Beck, R. D., Maroni, P., Papageorgopoulos, D. C., Dang, T. T., Schmid, M. P., and Rizzo, T. R. (2003). Vibrational mode-specific reaction of methane on a nickel surface. Science 302, 98–100. doi:10.1126/science.1088996
Behler, J., and Parrinello, M. (2007). Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401. doi:10.1103/physrevlett.98.146401
Bernard, M. E., and Harrison, I. (2024). Microcanonical treatment of HCl dissociative chemisorption on Au(111): reactive dampening through inefficient translational energy coupling and an active surface. J. Chem. Phys. 160, 084702. doi:10.1063/5.0193675
Bisson, R., Sacchi, M., and Beck, R. D. (2010). Mode-specific reactivity of CH4 on Pt(110)-(1×2): the concerted role of stretch and bend excitation. Phys. Rev. B 82, 121404. doi:10.1103/physrevb.82.121404
Bisson, R., Sacchi, M., Dang, T. T., Yoder, B., Maroni, P., and Beck, R. D. (2007). State-resolved reactivity of CH4(2nu3) on Pt(111) and Ni(111): effects of barrier height and transition state location. J. Phys. Chem. A 111, 12679–12683. doi:10.1021/jp076082w
Bukoski, A., Blumling, D., and Harrison, I. (2003). Microcanonical unimolecular rate theory at surfaces. I. Dissociative chemisorption of methane on Pt(111). J. Chem. Phys. 118, 843–871. doi:10.1063/1.1525803
Cai, Y., Michiels, R., De Luca, F., Neyts, E., Tu, X., Bogaerts, A., et al. (2024). Improving molecule–metal surface reaction networks using the meta-generalized gradient approximation: CO2 hydrogenation. J. Phys. Chem. C 128, 8611–8620. doi:10.1021/acs.jpcc.4c01110
Campbell, V. L., Chen, N., Guo, H., Jackson, B., and Utz, A. L. (2015). Substrate vibrations as promoters of chemical reactivity on metal surfaces. J. Phys. Chem. A 119, 12434–12441. doi:10.1021/acs.jpca.5b07873
Ceriotti, M., Bussi, G., and Parrinello, M. (2009). Nuclear quantum effects in solids using a colored-noise thermostat. Phys. Rev. Lett. 103, 030603. doi:10.1103/physrevlett.103.030603
Ceriotti, M., and Manolopoulos, D. E. (2012). Efficient first-principles calculation of the quantum kinetic energy and momentum distribution of nuclei. Phys. Rev. Lett. 109, 100604. doi:10.1103/physrevlett.109.100604
Ceriotti, M., Parrinello, M., Markland, T. E., and Manolopoulos, D. E. (2010). Efficient stochastic thermostatting of path integral molecular dynamics. J. Chem. Phys. 133, 124104. doi:10.1063/1.3489925
Chadwick, H., Gutiérrez-González, A., Beck, R. D., and Kroes, G.-J. (2019a). CHD3 dissociation on the kinked Pt(210) surface: a comparison of experiment and theory. J. Phys. Chem. C 123, 14530–14539. doi:10.1021/acs.jpcc.9b03051
Chadwick, H., Gutiérrez-González, A., Beck, R. D., and Kroes, G.-J. (2019b). Transferability of the SRP32-vdW specific reaction parameter functional to CHD3 dissociation on Pt(110)-(2x1). J. Chem. Phys. 150, 124702. doi:10.1063/1.5081005
Chadwick, H., Gutiérrez-González, A., Migliorini, D., Beck, R. D., and Kroes, G.-J. (2018b). Incident angle dependence of CHD3 dissociation on the stepped Pt(211) surface. J. Phys. Chem. C 122, 19652–19660. doi:10.1021/acs.jpcc.8b05887
Chadwick, H., Migliorini, D., and Kroes, G. J. (2018a). CHD3 dissociation on Pt(111): a comparison of the reaction dynamics based on the PBE functional and on a specific reaction parameter functional. J. Chem. Phys. 149, 044701. doi:10.1063/1.5039458
Colbert, D. T., and Miller, W. H. (1992). A novel discrete variable representation for quantum mechanical reactive scattering via the S-matrix kohn method. J. Chem. Phys. 96, 1982–1991. doi:10.1063/1.462100
Díaz, C., Pijper, E., Olsen, R. A., Busnengo, H. F., Auerbach, D. J., and Kroes, G. J. (2009). Chemically accurate simulation of a prototypical surface reaction: H2 dissociation on Cu(111). Science 326, 832–834. doi:10.1126/science.1178722
Dion, M., Rydberg, H., Schröder, E., Langreth, D. C., and Lundqvist, B. I. (2004). Van der waals density functional for general geometries. Phys. Rev. Lett. 92, 246401. doi:10.1103/physrevlett.92.246401
Ertl, G. (1983). Primary steps in catalytic synthesis of ammonia. J. Vac. Sci. Technol. A 1, 1247–1253. doi:10.1116/1.572299
Ertl, G. (1990). Elementary steps in heterogeneous catalysis. Angew. Chem. Int. Ed. 29, 1219–1227. doi:10.1002/anie.199012191
Furness, J. W., Kaplan, A. D., Ning, J., Perdew, J. P., and Sun, J. (2020). Accurate and numerically efficient r2SCAN meta-generalized gradient approximation. J. Phys. Chem. Lett. 11, 8208–8215. doi:10.1021/acs.jpclett.0c02405
Furness, J. W., and Sun, J. (2019). Enhancing the efficiency of density functionals with an improved iso-orbital indicator. Phys. Rev. B 99, 041119. doi:10.1103/physrevb.99.041119
Gee, A. T., Hayden, B. E., Mormiche, C., Kleyn, A. W., and Riedmüller, B. (2003). The dynamics of the dissociative adsorption of methane on Pt(533). J. Chem. Phys. 118, 3334–3341. doi:10.1063/1.1538184
Gerrits, N. (2021a). Accurate simulations of the reaction of H2 on a curved Pt crystal through machine learning. J. Phys. Chem. Lett. 12, 12157–12164. doi:10.1021/acs.jpclett.1c03395
Gerrits, N. (2021b). Accurate modeling of the dynamics of dissociative chemisorption on metal surfaces. Leiden, Netherlands: Leiden University. Ph.D. thesis.
Gerrits, N., Chadwick, H., and Kroes, G.-J. (2019b). Dynamical study of the dissociative chemisorption of CHD3 on Pd(111). J. Phys. Chem. C 123, 24013–24023. doi:10.1021/acs.jpcc.9b05757
Gerrits, N., Geweke, J., Auerbach, D. J., Beck, R. D., and Kroes, G.-J. (2021). Highly efficient activation of HCl dissociation on Au(111) via rotational preexcitation. J. Phys. Chem. Lett. 12, 7252–7260. doi:10.1021/acs.jpclett.1c02093
Gerrits, N., Geweke, J., Smeets, E. W. F., Voss, J., Wodtke, A. M., and Kroes, G.-J. (2020b). Closing the gap between experiment and theory: reactive scattering of HCl from Au(111). J. Phys. Chem. C 124, 15944–15960. doi:10.1021/acs.jpcc.0c03756
Gerrits, N., Jackson, B., and Bogaerts, A. (2024). Accurate reaction probabilities for translational energies on both sides of the barrier of dissociative chemisorption on metal surfaces. J. Phys. Chem. Lett. 15, 2566–2572. doi:10.1021/acs.jpclett.3c03408
Gerrits, N., Migliorini, D., and Kroes, G.-J. (2018). Dissociation of CHD3 on Cu(111), Cu(211), and single atom alloys of Cu(111). J. Chem. Phys. 149, 224701. doi:10.1063/1.5053990
Gerrits, N., Shakouri, K., Behler, J., and Kroes, G.-J. (2019a). Accurate probabilities for highly activated reaction of polyatomic molecules on surfaces using a high-dimensional neural network potential: CHD3 + Cu(111). J. Phys. Chem. Lett. 10, 1763–1768. doi:10.1021/acs.jpclett.9b00560
Gerrits, N., Smeets, E. W. F., Vuckovic, S., Powell, A. D., Doblhoff-Dier, K., and Kroes, G.-J. (2020a). Density functional theory for molecule–metal surface reactions: when does the generalized gradient approximation get it right, and what to do if it does not. J. Phys. Chem. Lett. 11, 10552–10560. doi:10.1021/acs.jpclett.0c02452
Guo, H., Farjamnia, A., and Jackson, B. (2016). Effects of lattice motion on dissociative chemisorption: toward a rigorous comparison of theory with molecular beam experiments. J. Phys. Chem. Lett. 7, 4576–4584. doi:10.1021/acs.jpclett.6b01948
Hammer, B., Hansen, L. B., and Nørskov, J. K. (1999). Improved adsorption energetics within density-functional theory using revised perdew-burke-ernzerhof functionals. Phys. Rev. B 59, 7413–7421. doi:10.1103/physrevb.59.7413
Henkelman, G., and Jónsson, H. (1999). A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 111, 7010–7022. doi:10.1063/1.480097
Higgins, J., Conjusteau, A., Scoles, G., and Bernasek, S. L. (2001). State selective vibrational (2v3) activation of the chemisorption of methane on Pt(111). J. Chem. Phys. 114, 5277–5283. doi:10.1063/1.1349895
Holmblad, P. M., Wambach, J., and Chorkendorff, I. (1995). Molecular beam study of dissociative sticking of methane on Ni(100). J. Chem. Phys. 102, 8255–8263. doi:10.1063/1.468955
Hundt, P. M., Ueta, H., van Reijzen, M. E., Jiang, B., Guo, H., and Beck, R. D. (2015). Bond-selective and mode-specific dissociation of CH3D and CH2D2 on Pt(111). J. Phys. Chem. A 119, 12442–12448. doi:10.1021/acs.jpca.5b07949
Hundt, P. M., van Reijzen, M. E., Beck, R. D., Guo, H., and Jackson, B. (2017). Quantum-state-resolved reactivity of overtone excited CH4 on Ni(111): comparing experiment and theory. J. Chem. Phys. 146, 054701. doi:10.1063/1.4975025
Imbihl, R., Behm, R., and Schlögl, R. (2007). Bridging the pressure and material gap in heterogeneous catalysis. Phys. Chem. Chem. Phys. 9, 3459. doi:10.1039/b706675a
Jackson, B. (2022). Quantum studies of methane-metal inelastic diffraction and trapping: the variation with molecular orientation and phonon coupling. Chem. Phys. 559, 111516. doi:10.1016/j.chemphys.2022.111516
Jackson, B., Nattino, F., and Kroes, G.-J. (2014). Dissociative chemisorption of methane on metal surfaces: tests of dynamical assumptions using quantum models and ab initio molecular dynamics. J. Chem. Phys. 141, 054102. doi:10.1063/1.4891327
Jackson, B., and Nave, S. (2013). The dissociative chemisorption of methane on Ni(111): the effects of molecular vibration and lattice motion. J. Chem. Phys. 138, 174705. doi:10.1063/1.4802008
Juurlink, L. B., Smith, R. R., and Utz, A. L. (2000). The role of rotational excitation in the activated dissociative chemisorption of vibrationally excited methane on Ni(100). Faraday Discuss. 117, 147–160. doi:10.1039/b003708g
Juurlink, L. B. F., Killelea, D. R., and Utz, A. L. (2009). State-resolved probes of methane dissociation dynamics. Prog. Surf. Sci. 84, 69–134. doi:10.1016/j.progsurf.2009.01.001
Karplus, M., Porter, R. N., and Sharma, R. D. (1965). Exchange reactions with activation energy. I. Simple barrier potential for (H, H2). J. Chem. Phys. 43, 3259–3287. doi:10.1063/1.1697301
King, D. A., and Wells, M. G. (1972). Molecular beam investigation of adsorption kinetics on bulk metal targets: nitrogen on tungsten. Surf. Sci. 29, 454–482. doi:10.1016/0039-6028(72)90232-4
Kresse, G., and Joubert, D. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775. doi:10.1103/physrevb.59.1758
Kroes, G.-J. (2021). Computational approaches to dissociative chemisorption on metals: towards chemical accuracy. Phys. Chem. Chem. Phys. 23, 8962–9048. doi:10.1039/d1cp00044f
Larsen, J. H., Holmblad, P. M., and Chorkendorff, I. (1999). Dissociative sticking of CH4 on Ru(0001). J. Chem. Phys. 110, 2637–2642. doi:10.1063/1.477985
Manson, J. R. (1991). Inelastic scattering from surfaces. Phys. Rev. B 43, 6924–6937. doi:10.1103/physrevb.43.6924
Manson, J. R. (1994). Multiphonon atom-surface scattering. Comput. Phys. Commun. 80, 145–167. doi:10.1016/0010-4655(94)90101-5
Marcus, R. A. (1966). On the analytical mechanics of chemical reactions. Quantum mechanics of linear collisions. J. Chem. Phys. 45, 4493–4499. doi:10.1063/1.1727528
McCullough, E. A., and Wyatt, R. E. (1969). Quantum dynamics of the collinear (H, H2) reaction. J. Chem. Phys. 51, 1253–1254. doi:10.1063/1.1672133
McMaster, M. C., and Madix, R. J. (1993). Alkane dissociation dynamics on Pt(110)–(1×2). J. Chem. Phys. 98, 9963–9976. doi:10.1063/1.464322
Mejia-Rodriguez, D., and Trickey, S. B. (2017). Deorbitalization strategies for meta-generalized-gradient-approximation exchange-correlation functionals. Phys. Rev. A 96, 052512. doi:10.1103/physreva.96.052512
Mejia-Rodriguez, D., and Trickey, S. B. (2018). Deorbitalized meta-GGA exchange-correlation functionals in solids. Phys. Rev. B 98, 115161. doi:10.1103/physrevb.98.115161
Mejía-Rodríguez, D., and Trickey, S. B. (2020). Meta-GGA performance in solids at almost GGA cost. Phys. Rev. B 102, 121109. doi:10.1103/physrevb.102.121109
Michelsen, H. A., and Auerbach, D. J. (1991). A critical examination of data on the dissociative adsorption and associative desorption of hydrogen at copper surfaces. J. Chem. Phys. 94, 7502–7520. doi:10.1063/1.460182
Migliorini, D., Chadwick, H., Nattino, F., Gutiérrez-González, A., Dombrowski, E., High, E. A., et al. (2017). Surface reaction barriometry: methane dissociation on flat and stepped transition-metal surfaces. J. Phys. Chem. Lett. 8, 4177–4182. doi:10.1021/acs.jpclett.7b01905
Moiraghi, R., Lozano, A., Peterson, E., Utz, A., Dong, W., and Busnengo, H. F. (2020). Nonthermalized precursor-mediated dissociative chemisorption at high catalysis temperatures. J. Phys. Chem. Lett. 11, 2211–2218. doi:10.1021/acs.jpclett.0c00260
Mondal, A., Wijzenbroek, M., Bonfanti, M., Díaz, C., and Kroes, G.-J. (2013). Thermal lattice expansion effect on reactive scattering of H2 from Cu(111) at ts = 925 K. J. Phys. Chem. A 117, 8770–8781. doi:10.1021/jp4042183
Nattino, F., Díaz, C., Jackson, B., and Kroes, G.-J. (2012). Effect of surface motion on the rotational quadrupole alignment parameter of D2 reacting on Cu(111). Phys. Rev. Lett. 108, 236104. doi:10.1103/physrevlett.108.236104
Nattino, F., Migliorini, D., Bonfanti, M., and Kroes, G.-J. (2016a). Methane dissociation on Pt(111): searching for a specific reaction parameter density functional. J. Chem. Phys. 144, 044702. doi:10.1063/1.4939520
Nattino, F., Migliorini, D., Kroes, G.-J., Dombrowski, E., High, E. A., Killelea, D. R., et al. (2016b). Chemically accurate simulation of a polyatomic molecule-metal surface reaction. J. Phys. Chem. Lett. 7, 2402–2406. doi:10.1021/acs.jpclett.6b01022
Nattino, F., Ueta, H., Chadwick, H., van Reijzen, M. E., Beck, R. D., Jackson, B., et al. (2014). Ab initio molecular dynamics calculations versus quantum-state-resolved experiments on CHD3 + Pt(111): new insights into a prototypical gas–surface reaction. J. Phys. Chem. Lett. 5, 1294–1299. doi:10.1021/jz500233n
Nave, S., Tiwari, A. K., and Jackson, B. (2014). Dissociative chemisorption of methane on Ni and Pt surfaces: mode-specific chemistry and the effects of lattice motion. J. Phys. Chem. A 118, 9615–9631. doi:10.1021/jp5063644
Nguyen, T. L., and Barker, J. R. (2010). Sums and densities of fully coupled anharmonic vibrational states: a comparison of three practical methods. J. Phys. Chem. A 114, 3718–3730. doi:10.1021/jp100132s
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Peverati, R., and Truhlar, D. G. (2014). Quest for a universal density functional: the accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. R. Soc. A 372, 20120476. doi:10.1098/rsta.2012.0476
Polanyi, J. C. (1972). Concepts in reaction dynamics. Acc. Chem. Res. 5, 161–168. doi:10.1021/ar50053a001
Salmeron, M., Gale, R. J., and Somorjai, G. A. (1977). Molecular beam study of the H2–D2 exchange reaction on stepped platinum crystal surfaces: dependence on reactant angle of incidence. J. Chem. Phys. 67, 5324–5334. doi:10.1063/1.434711
Seets, D. C., Reeves, C. T., Ferguson, B. A., Wheeler, M. C., and Mullins, C. B. (1997b). Dissociative chemisorption of methane on Ir(111): evidence for direct and trapping-mediated mechanisms. J. Chem. Phys. 107, 10229–10241. doi:10.1063/1.475306
Seets, D. C., Wheeler, M. C., and Mullins, C. B. (1997a). Trapping-mediated and direct dissociative chemisorption of methane on Ir(110): a comparison of molecular beam and bulb experiments. J. Chem. Phys. 107, 3986–3998. doi:10.1063/1.474754
Smeets, E. W., Voss, J., and Kroes, G.-J. (2019). Specific reaction parameter density functional based on the meta-generalized gradient approximation: application to H2 + Cu(111) and H2 + Ag(111). J. Phys. Chem. A 123, 5395–5406. doi:10.1021/acs.jpca.9b02914
Smeets, E. W. F., and Kroes, G.-J. (2021). Performance of made simple meta-GGA functionals with rVV10 nonlocal correlation for H2 + Cu(111), D2 + Ag(111), H2 + Au(111), and D2 + Pt(111). J. Phys. Chem. C 125, 8993–9010. doi:10.1021/acs.jpcc.0c11034
Tchakoua, T., Gerrits, N., Smeets, E. W. F., and Kroes, G.-J. (2023). SBH17: benchmark database of barrier heights for dissociative chemisorption on transition metal surfaces. J. Chem. Theory Comput. 19, 245–270. doi:10.1021/acs.jctc.2c00824
Tran, F., Kovács, P., Kalantari, L., Madsen, G. K. H., and Blaha, P. (2018). Orbital-free approximations to the kinetic-energy density in exchange-correlation MGGA functionals: tests on solids. J. Chem. Phys. 149, 144105. doi:10.1063/1.5048907
Verhoef, R. W., Kelly, D., Mullins, C. B., and Weinberg, W. H. (1993). Isotope effects for the direct dissociative chemisorption of methane and ethane on Ir(110) and vibrationally assisted chemisorption. Surf. Sci. 287–288, A370–A398. doi:10.1016/0167-2584(93)90398-3
Wei, F., Smeets, E. W., Voss, J., Kroes, G.-J., Lin, S., and Guo, H. (2021). Assessing density functionals for describing methane dissociative chemisorption on Pt(110)-(2×1) surface. Chin. J. Chem. Phys. 34, 883–895. doi:10.1063/1674-0068/cjcp2110207
Wei, J., and Iglesia, E. (2004). Mechanism and site requirements for activation and chemical conversion of methane on supported Pt clusters and turnover rate comparisons among noble metals. J. Phys. Chem. B 108, 4094–4103. doi:10.1021/jp036985z
Yoder, B. L., Bisson, R., and Beck, R. D. (2010). Steric effects in the chemisorption of vibrationally excited methane on Ni(100). Science 329, 553–556. doi:10.1126/science.1191751
Zhang, H., Sun, Z., and Hu, Y. H. (2021). Steam reforming of methane: current states of catalyst design and process upgrading. Renew. Sustain. Energy Rev. 149, 111330. doi:10.1016/j.rser.2021.111330
Zhou, X., and Jiang, B. (2019). A modified generalized Langevin oscillator model for activated gas-surface reactions. J. Chem. Phys. 150, 024704. doi:10.1063/1.5078541
Keywords: heterogeneous catalysis, density functional theory, surface science, dissociative chemisorption, methane, metal surfaces, theoretical chemistry, chemical reactivity
Citation: Gerrits N (2024) How to simulate dissociative chemisorption of methane on metal surfaces. Front. Chem. 12:1481235. doi: 10.3389/fchem.2024.1481235
Received: 15 August 2024; Accepted: 23 September 2024;
Published: 09 October 2024.
Edited by:
Shaodong Zhou, Zhejiang University, ChinaReviewed by:
Uttam Pal, S.N. Bose National Centre for Basic Sciences, IndiaCopyright © 2024 Gerrits. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nick Gerrits, bi5nZXJyaXRzQGxpYy5sZWlkZW51bml2Lm5s