 Dongxiao Yue
Dongxiao Yue Hajime Hirao*
Hajime Hirao*- Warshel Institute for Computational Biology, School of Medicine, The Chinese University of Hong Kong, Shenzhen, China
Previous theoretical studies have revealed that high-spin states possess flatter potential energy surfaces than low-spin states in reactions involving iron(IV)-oxo species of cytochrome P450 enzymes (P450s), nonheme enzymes, or biomimetic complexes. Therefore, actively utilizing high-spin states to enhance challenging chemical transformations, such as C–H bond activation, represents an intriguing research avenue. However, the inherent instability of high-spin states relative to low-spin states in pre-reaction complexes often hinders their accessibility around the transition state, especially in heme systems with strong ligand fields. Counterintuitively, our investigation of the metabolic hydroxylation of paclitaxel by human CYP2C8 using a hybrid quantum mechanics and molecular mechanics (QM/MM) approach showed that the high-spin sextet state exhibits unusually high stability, when the reaction follows a secondary reaction pathway leading to 6β-hydroxypaclitaxel. We thoroughly analyzed the factors contributing to the enhanced stabilization of the high-spin state, and the knowledge obtained could be instrumental in designing competent biomimetic catalysts and biocatalysts for C–H bond activation.
1 Introduction
The efficient activation of inert C–H bonds is a paramount goal with far-reaching implications across chemical and materials science (Gandeepan et al., 2019; Yamaguchi et al., 2012; Bergman, 2007; Balcells et al., 2010; Anastas and Eghbali, 2010). This capability directly translates into enhanced molecular diversity and streamlined synthesis of complex molecules, such as in drug discovery processes, thereby promoting more sustainable practices. Cytochrome P450 enzymes (P450s) constitute a superfamily of heme-containing proteins and stand out as a remarkable class of biological systems adept at catalyzing such reactions (Ortiz de Montellano, 2015; Werck-Reichhart and Feyereisen, 2000). P450s play a central role in metabolizing a broad range of endogenous or exogenous substrates across various organisms through activating C–H bonds and facilitating other types of reactions. Their ability to activate C–H bonds also makes them invaluable platforms for engineered biocatalyst development (Kumar, 2010; Li et al., 2020; Hu et al., 2023). A precise understanding of the catalytic machinery in P450s could significantly aid in the rational design of biomimetic catalysts.
The ability of P450s to activate C–H bonds originates from the formation of a high-valent iron(IV)-oxo porphyrin π-cation radical intermediate, known as Compound I (Cpd I). This intermediate is formed through the catalytic cycle that requires the supply of two electrons, two protons, and one O2 molecule (Denisov et al., 2005; Sono et al., 1996; Rittle and Green, 2010). Inspired by the remarkable reactivity of Cpd I, biomimetic iron(IV)-oxo complexes have been synthesized (Que, 2007; Nam, 2007). Additionally, our understanding of the electronic structure of Cpd I has advanced through computational studies. In particular, density functional theory (DFT) and hybrid quantum mechanics and molecular mechanics (QM/MM) calculations have highlighted the active involvement of triradicaloid doublet and quartet states in substrate reactions of P450 Cpd I (Shaik et al., 2005; Shaik et al., 2007; Shaik et al., 2010; de Visser et al., 2001). In these states, the π*xz, π*yz, and a2u orbitals are singly occupied (Scheme 1A), and one of these orbitals, typically the a2u orbital, receives one electron from the substrate during the initial H-abstraction step, yielding a substrate radical (Scheme 1B). While the sextet state at the pre-reaction complex stage containing Cpd I is less stable than the doublet and quartet states, it displays a relatively flat potential energy surface for H-abstraction. This is attributed to additional stabilization from exchange enhancement resulting from the electron shift from the substrate toward the σ*z2 orbital (Scheme 1C) (Hirao et al., 2005).
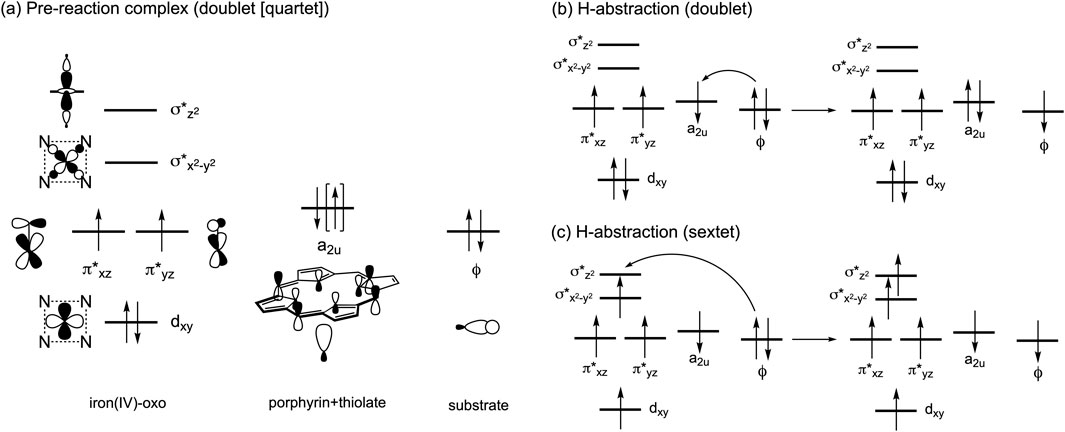
Scheme 1. (A) Electron configurations in the low-lying doublet and quartet spin states. (B) Typical electron shift pattern during H-abstraction in the doublet state. (C) Typical electron shift pattern in the sextet state.
Consequently, a common observation in computationally derived energy profiles for P450-catalyzed reactions, regardless of the reaction type, is a reduced energy gap between the sextet and lower-spin states at the bond-activation transition state (TS), compared to the pre-reaction state. For instance, a DFT study on C–H hydroxylation and olefin epoxidation reactions of P450 Cpd I demonstrated that the relative stability of the sextet TS increases as the reactions progress (Hirao et al., 2005). In our recent QM/MM study on the CYP3A4-catalyzed aromatic hydroxylation reaction of paclitaxel (PTX, or Taxol), the energy gap was initially 6.4 kcal/mol. However, at the TS for C–O bond formation between an aromatic ring and Cpd I, the gap significantly narrowed to just 0.3 kcal/mol, with the sextet state exhibiting slightly higher stability than the doublet state (Yue and Hirao, 2023).
Given the relatively high energy of P450 Cpd I or its pre-reaction complexes in high-spin states, stabilizing them is considered a promising strategy for enhancing the stability of the subsequent TSs. This can be achieved by substituting the heme ligand with a nonheme ligand to reduce the iron(IV)-oxo ligand field strength (Kumar et al., 2005; Hirao et al., 2006; Sastri et al., 2007; Hirao et al., 2008a; Hirao et al., 2015). Nonheme iron enzymes, such as taurine dioxygenase (TauD), are known to produce iron(IV)-oxo species with a high-spin (S = 2) ground state (Krebs et al., 2007). Furthermore, various high-spin iron(IV)-oxo complexes have been successfully synthesized through meticulous nonheme ligand design (Puri and Que, 2015; England et al., 2009; England et al., 2010; England et al., 2011; Lacy et al., 2010; Bigi et al., 2012; Bae et al., 2016; Hou et al., 2023). Theoretical studies have also suggested that external electric fields could further stabilize high-spin TSs in the reactions of nonheme iron (IV)-oxo complexes (Hirao et al., 2008b).
Thus, nonheme ligands clearly offer better support for stabilizing high-spin states of the pre-reaction complex. Nevertheless, this study explores a less examined area by investigating the high-spin reactivity of P450 Cpd I. Specifically, we investigate the CYP2C8-catalyzed hydroxylation reaction of PTX, a renowned anticancer compound (Stage et al., 2018; Singla et al., 2002; Weaver, 2014). Experimental evidence suggests that PTX undergoes hepatic metabolic transformations catalyzed by CYP2C8 and CYP3A4 following administration (Harris et al., 1994b; Rahman et al., 1994). As illustrated in Scheme 2A, these enzymes target different sites, resulting in distinct products. Our recent study focused on the CYP3A4-catalyzed aromatic hydroxylation of PTX at the 3′-phenyl ring (Yue and Hirao, 2023). We have also recently investigated the mechanism of the CYP2C8-catalyzed hydroxylation of PTX into 6α-hydroxypaclitaxel using QM/MM calculations (Yue and Hirao, submitted). While further exploring CYP2C8-catalyzed hydroxylation of PTX, we unexpectedly discovered that the high-spin sextet state could exhibit remarkably enhanced stability compared to other spin states at the H-abstraction TS when PTX is converted to 6β-hydroxypaclitaxel (Scheme 2B). Although 6β-hydroxypaclitaxel formation has not been experimentally observed, our computational findings reveal an intriguing aspect of high-spin iron (IV)-oxo reactivity. The unexpected stability of the high-spin state in this reaction prompted us to investigate the contributing factors beyond pre-reaction complex stabilization, which could aid in the rational design of high-spin catalysts.
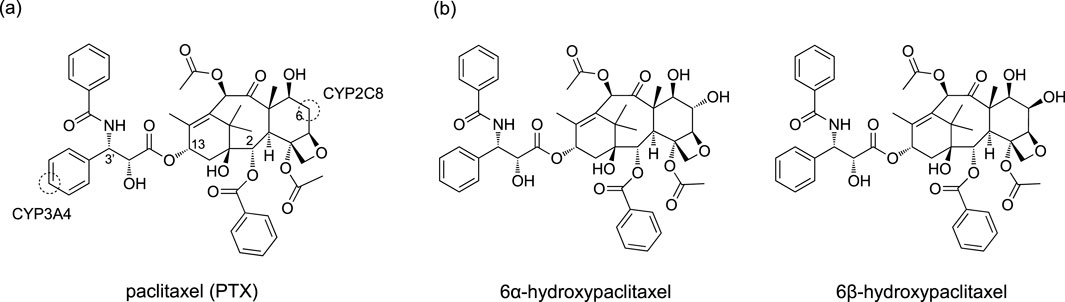
Scheme 2. (A) Chemical structure of PTX and its metabolic sites targeted by CYP2C8 and CYP3A4. (B) 6α-hydroxypaclitaxel and 6β-hydroxypaclitaxel.
2 Computational methods
2.1 Molecular docking and molecular dynamics simulations
To identify a plausible binding structure of PTX in CYP2C8, we initially performed molecular docking simulations. We utilized MOE 2020 software (Molecular Operating Environment (MOE), 2022) and docked PTX into several crystal structures of CYP2C8 (PDB codes 2NNH, 2NNI, 2VN0, and 2NNJ). Unlike CYP3A4, these crystal structures exhibit minimal structural variations, leading to comparable docking results. Consequently, we proceeded with the structure from 2NNJ. To further refine the CYP2C8–PTX complex structure, we performed molecular dynamics (MD) simulations using AMBER (Case et al., 2020). The MD simulations essentially followed the same protocol employed in our prior study of CYP3A4 (Yue and Hirao, 2023), including clustering to select a representative structure.
2.2 QM/MM calculations
To investigate the reaction mechanism, we employed the ONIOM(QM:MM) method, a subtractive QM/MM scheme (Chung et al., 2012; Chung et al., 2015). Gaussian 16 software was used for QM/MM computations (Frisch et al., 2016). The CYP2C8–PTX complex geometry, derived from MD simulations, served as the starting point. To simplify the system, a significant portion of the solvent water molecules outside the enzyme was removed. The QM region encompassed the porphine and iron-oxo units, the CβH3S−segment of the cysteine axial ligand, and the PTX molecule. DFT methods were applied to describe the QM atoms. Geometry optimization and vibrational frequency calculations were performed using the ONIOM mechanical-embedding scheme and the B3LYP/6-31G(d) QM method. The ONIOM energy obtained with this basis set is referred to as E1. For single-point energy calculations, the B3LYP/def2-TZVP method and the electronic-embedding scheme were utilized (Becke, 1993; Lee et al., 1988; Vosko et al., 1980; Hehre et al., 1972; Weigend and Ahlrichs, 2005). MM calculations within the ONIOM framework employed AMBER and TIP3P parameters (Cornell et al., 1995; Jorgensen et al., 1983). Force field parameters for PTX and the heme group were consistent with those in the MD simulations. Free energies were determined by summing the ONIOM single-point energy with a larger basis set (E2), the DFT-D3BJ dispersion correction to the QM energy (Edisp) (Grimme et al., 2011; Becke and Johnson, 2005), and the free energy correction obtained from ONIOM vibrational frequency analysis (Gcorr). The sum of these energy values (G) was subsequently used to construct reaction energy diagrams.
3 Results and discussion
3.1 Active-site feature and possible binding pose
Human P450s generally possess spacious active sites, enabling them to accommodate a wide range of substrates. CYP2C8 (PDB code 2NNJ), with a 746 Å3 active site, readily binds PTX (Figure 1A). However, an α-helix overlying the heme restricts ligand access to this region. This structural feature is also observed in CYP2C9, which shares 78% sequence identity with CYP2C8 (Williams et al., 2003). As a result, molecular docking identified a binding mode (Pose A) avoiding the α-helix region and positioning the metabolic site’s Hβ closer to the heme iron compared to Hα (Figure 1B). At the C6 position of PTX in Scheme 2, Hα and Hβ are potential sites for hydroxylation. While C–Hα hydroxylation would produce the experimentally observed 6α-hydroxypaclitaxel metabolite (Harris et al., 1994a; Kumar et al., 1994), C–Hβ hydroxylation would yield the unobserved 6β-hydroxypaclitaxel. Thus, Pose A is inconsistent with experimental findings. Further analysis revealed that the Pose A conformation of PTX is not very stable in its isolated form, which could lead to an overestimation of its binding affinity in docking simulations. Consequently, a lower-ranked binding mode, involving a substrate with an intrinsically more stable conformation, should be a more probable candidate for C–Hα hydroxylation (Yue and Hirao, submitted). Nevertheless, in this study, we delved deeper into the reactivity of Pose A, which unexpectedly led us to uncover intriguing insights into high-spin reactivity.
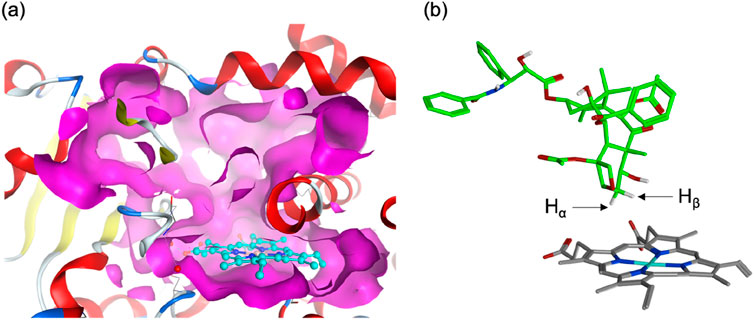
Figure 1. (A) Surface representation of the active site of human CYP2C8 (PDB code 2NNJ). (B) A top-ranked binding structure obtained from docking simulations.
3.2 QM/MM mechanistic study of PTX hydroxylation
Following MD refinement of the Pose A structure, we conducted QM/MM mechanistic studies. P450-catalyzed alkane hydroxylation typically initiates with H-abstraction from a C–H bond (Schöneboom et al., 2004). As illustrated in Figure 2A, the geometry-optimized pre-reaction complex between Cpd I and PTX (1) in the doublet ground state (21) exhibits an O–H distance of 2.28 Å between the Hβ atom and the Cpd I oxo group. This proximity suggests a favorable conformation for subsequent H-abstraction. Figure 2B highlights key interactions between PTX and surrounding amino-acid residues. PTX is securely anchored within the CYP2C8 active site via non-bonded interactions including hydrogen bonds and π-π interactions. Specifically, Asn99, Ser103, Asn209, and Gly365 form hydrogen bonds with PTX’s polar groups. Moreover, a hydrogen-bond network involving Asn99, Ser100, Gln214, Ser103, Leu208, and Asn209 stabilizes the Cpd I–PTX complex. Phe201 and Phe205 engage in hydrophobic interactions with the phenyl moiety of the benzoyloxy group in PTX. Figure 2C depicts the available space around PTX using a surface model, clearly demonstrating that PTX’s excellent fit within the active site.
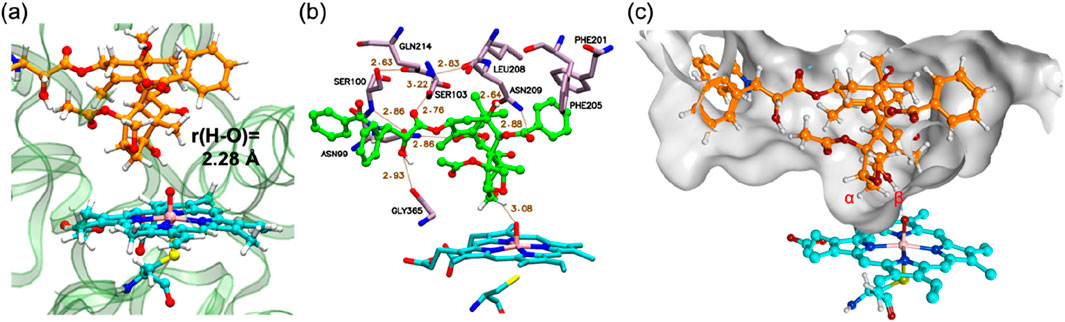
Figure 2. (A) QM/MM-optimized structure of 21. (B) 3D ligand interaction diagram of 21. (C) A surface representation of the active site in 21. Key distances are presented in Å.
Through detailed QM/MM calculations on 6β-hydroxypaclitaxel formation pathways, we identified four distinct electron-shift patterns for the H-abstraction step. One doublet-state pathway (Path A) involves an electron shift from the substrate orbital (ϕ) to the a2u-type orbital of Cpd I, yielding intermediate 23a with a negative spin density (ρ) value (∼–1.0) on the PTX moiety (Scheme 1B; Supplementary Table S2). Another doublet-state pathway (Path B) features an electron shift from ϕ to the π* orbital of Cpd I, forming intermediate 23b with a positive ρ value (∼1.0) on PTX (Supplementary Table S2). In the quartet spin state, an electron migrates from the substrate to the a2u-type orbital of Cpd I, producing intermediate 43 with a positive ρ value (∼1.0) on PTX (Supplementary Table S2). Finally, the sextet spin state involves electron migration from ϕ to the σ*z2 orbital of Cpd I, resulting in intermediate 63 with a negative ρ value (∼–1.0) on PTX (Supplementary Table S2).
Figure 3 presents the free energy profile for PTX hydroxylation. As described, two electron-shift patterns can occur during the doublet-state H-abstraction step, resulting in two TSs (22a* and 22b*) for Paths A and B, respectively. TSs were also obtained in the quartet (42*) and sextet (62*) states. Comparing relative energies reveals 22b* as slightly more stable (25.6 kcal/mol) than 22a* (26.7 kcal/mol). Without corrections, the uncorrected E1 values produce barriers exceeding 30 kcal/mol for both doublet states (Supplementary Table S1), significantly surpassing typically observed values of around 20 kcal/mol (Schöneboom et al., 2004). These results suggest that the substrate experiences significant strain around the TS in the protein environment. Intermediates 23a and 23b exhibit reversed stability (5.3 and 9.5 kal/mol, respectively). The stability of the quartet-state TS (42*, 25.4 kcal/mol) is comparable to that of 22b*, with subsequent intermediate 43 at 5.6 kcal/mol.
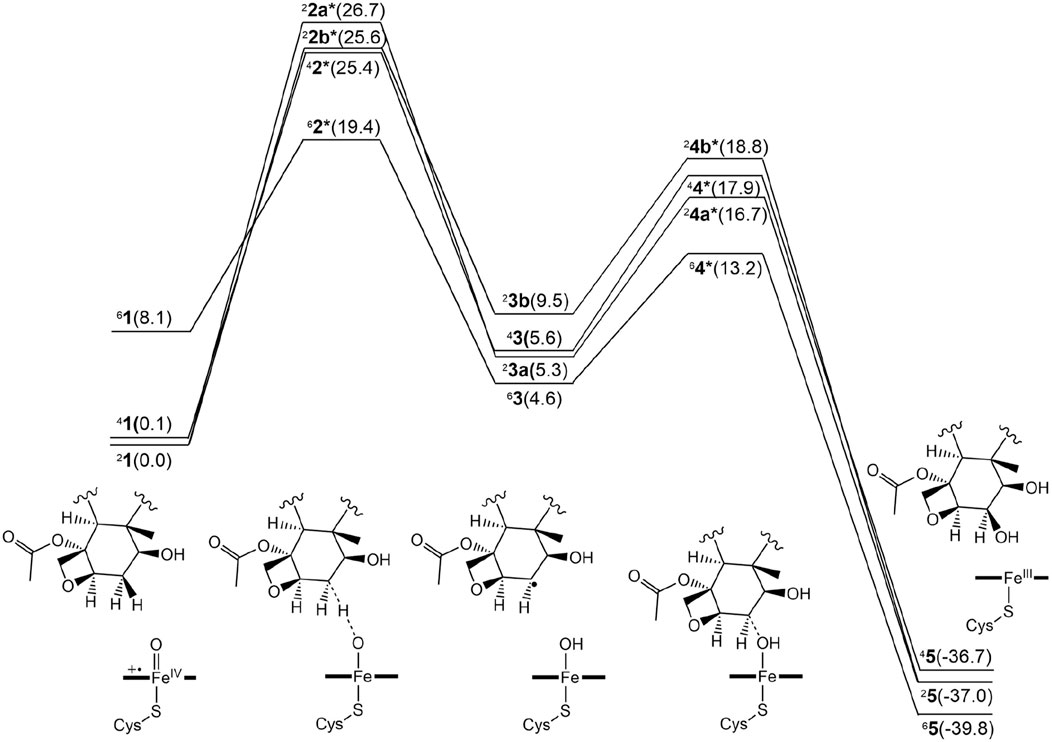
Figure 3. Free energy profile for the β-hydroxylated PTX metabolite formation in the doublet, quartet, and sextet spin states. The numerical values within parentheses indicate relative free energy values in kcal/mol.
Surprisingly, the sextet-state TS (62*, 19.4 kcal/mol) is significantly more stable than the others, with energy differences of at least 6 kcal/mol. This exceptional stability arises from a remarkably low barrier of 11.3 kcal/mol on the sextet-state free energy surface, contrasting sharply with the higher barriers observed for the other spin states. While the sextet state typically benefits from transition-state stabilization due to exchange enhancement (Hirao et al., 2005), the substantially lower energy of the sextet TS in the current system is an unusual observation. Normally, the sextet state’s stability at the TS is comparable to or slightly inferior to that of lower spin states. In contrast, the CYP3A4-catalyzed aromatic hydroxylation reaction exhibited a slight energetic preference for the sextet TS, with a small energy gap of 0.3 kcal/mol (Yue and Hirao, 2023). The significantly greater stability of the sextet TS in the current CYP2C8-catalyzed reaction is expected to increase the likelihood of this reaction channel being accessed.
Figure 4 displays the optimized geometries of key TSs and intermediates. The larger O–H distance observed in 62* (1.25 Å) than those in the other corresponding species (1.16–1.24 Å) indicates an earlier TS in the sextet pathway, consistent with the lower energy barrier observed in the sextet-state energy profile (Figure 3). A closer examination of Fe–O–H angles across different spin states reveals a larger angle for 62* (135.1°) compared to the others (around 127.0°). This larger Fe–O–H angle in the sextet state is characteristic of the high-spin H-abstraction mechanism, involving an electron shift from the substrate’s ϕ orbital to the axial σ*z2 orbital of iron(IV)-oxo. Optimal orbital overlap is achieved when the substrate approaches the iron (IV)-oxo unit from above, leading to a larger Fe–O–H angle (Hirao et al., 2005).
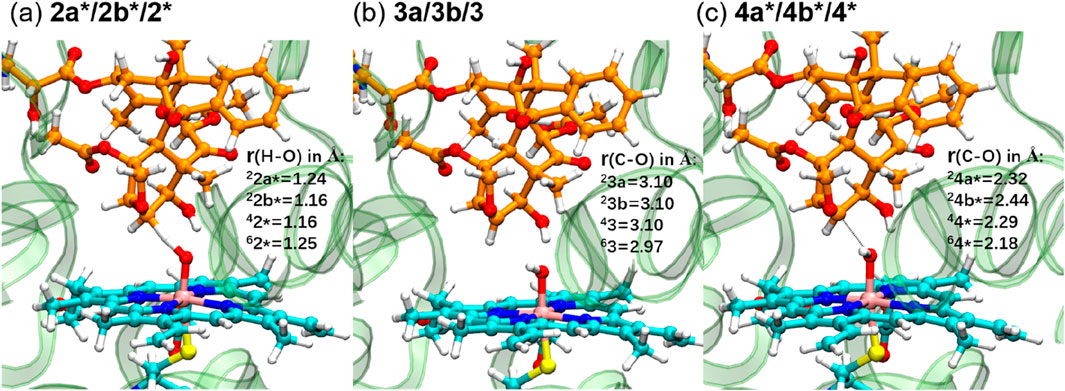
Figure 4. QM/MM-optimized geometries of key TSs ((A) and (C)) and intermediates (B).
We further investigated the rebound step leading to cycloalkanol product formation (Figures 3, 4) (Groves, 1985). Starting from intermediates 23a and 33b in the doublet spin state, we obtained product 25 (−37.0 kcal/mol) via TSs 24a* (16.7 kcal/mol) and 24b* (18.8 kcal/mol), respectively. While Path A exhibits a lower overall energy, its rebound barrier of 11.3 kcal/mol is unusually high for a P450 reaction in the doublet state compared to other reported values, likely due to steric constraints within the enzyme’s active site. The quartet TS 44* (17.9 kcal/mol) lies energetically between 24a* and 24b*, leading to product 45 with a relative free energy of −36.7 kcal/mol, which is less stable than the doublet product (25, −37.0 kcal/mol). From the most stable intermediate 63 (4.6 kcal/mol), we obtained the most stable product 65 (−39.8 kcal/mol) via TS 64* (13.2 kcal/mol). Surprisingly, the sextet state exhibits the lowest rebound barrier in the sextet state (8.6 kcal/mol), contrary to the typical trend of higher rebound barriers in the sextet state.
3.3 Energy decomposition analysis
Our QM/MM mechanistic study on CYP2C8-catalyzed PTX hydroxylation unexpectedly revealed a dominant role for the high-spin sextet state from an early stage of the reaction. To understand the origins of this pronounced sextet state stability, we conducted further theoretical analyses. The ONIOM-based G value can be expressed as follows (Equation 1):
where EQM is the gas-phase energy of the QM atoms at the QM/MM geometry, and EMM and Epol are defined by Equations 2, 3, respectively:
E2(EE) and E2(ME) are ONIOM-EE and ONIOM-ME energies obtained from single-point energy calculations with the def2-TZVP basis set (Hirao, 2011a; Hirao, 2011b). The relative free energy (ΔG) of the H-abstraction TS with respect to 21 is given by Equation 4:
While ∆EQM can be obtained from DFT calculations on the QM atoms, the remaining four terms also contribute to the overall ΔG value. Figure 5 displays the latter four terms for the H-abstraction TSs in different spin states. The consistently negative ∆Edisp values indicate stronger dispersion stabilization in TSs compared to 21, with the largest stabilization observed for the sextet state. Therefore, although modest, dispersion contributes to the sextet state’s stability. The relatively large ∆Edisp for the sextet TS is attributed to appropriate positioning of the QM atoms, which enhances interatomic dispersion stabilization. The MM energy term (∆EMM) is also consistently negative, with the largest stabilization for 62*. Therefore, the MM effect partly contributes to the pronounced stability of 62*. While the polarization effect (∆Epol) does not enhance the sextet TS’s stability, the ∆Gcorr term stabilizes all TSs, with the greatest stabilization observed for the sextet state. This likely results from the sextet TS’s flexible and entropically favorable structure. Among the four terms examined, the free energy correction term (∆Gcorr) provides the most significant stabilization for 62*.
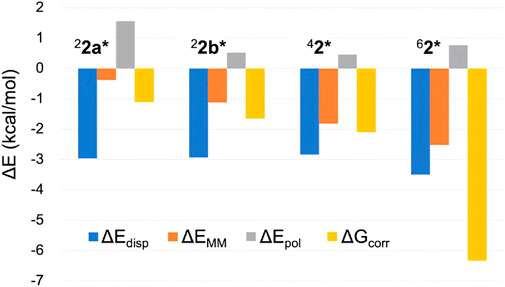
Figure 5. Comparison of several energy terms constituting ΔG (in kcal/mol) of H-abstraction TSs in different spin states.
Despite the insights gained from the above analysis, the substantial relative stability of 62* remained only partially explained. Therefore, we further investigated the ΔEQM values across different spin states. In addition to the QM/MM approach, DFT calculations were performed using a P450 Cpd I model and cyclohexane to represent the reaction. Figure 6 compares the ΔEQM values for different spin states obtained from both methods. Interestingly, significant discrepancies were observed between the QM/MM and DFT results. QM/MM calculations consistently yielded higher ΔEQM by 9.1–11.9 kcal/mol compared to DFT. In other words, the TS geometries are highly strained in the protein environment. However, the smallest destabilization was observed for the sextet state (9.1 kcal/mol). Thus, the protein environment destabilizes TSs more in the other spin states, contributing to the enhanced relative stability of 62*. The smaller destabilization in the sextet state is likely due to the protein-imposed geometric constraints that orient the substrate more favorably for the sextet-state TS geometry.
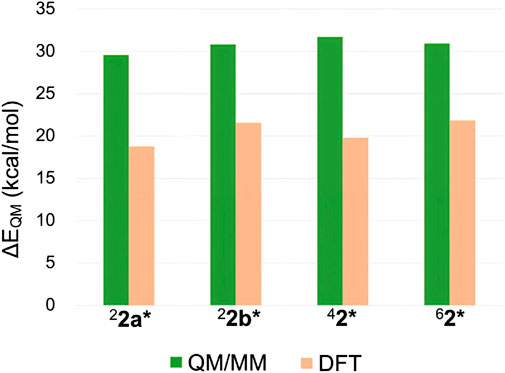
Figure 6. ΔEQM values obtained by QM/MM and DFT calculations.
Based on the present theoretical analysis and previous studies, several possible strategies for promoting high-spin reactivity of iron(IV)-oxo complexes can be summarized (Scheme 3). The most fundamental approach involves stabilizing the high-spin pre-reaction complex, which lowers the energy of the entire high-spin energy surface (Scheme 3A). This can be achieved through the use of nonheme ligands that reduce ligand field strength. This study highlights additional factors beyond this approach (Scheme 3B-E), with entropy and QM effects being particularly influential (Scheme 3D,E). We propose that these effects are key to enhancing high-spin reactivity. While pronounced entropy benefits for high-spin TSs have been observed in previous studies (Yue and Hirao, 2023), further enhancing the high-spin reactivity requires increasing the quantum mechanical stability of the high-spin TS relative to others. The surrounding environment plays a crucial role in influencing this effect, as the QM effect is largely related to differing degrees of geometric strain of the substrate. If the environment imposes steric constraints on the substrate, enforcing a linear approach toward the iron(IV)-oxo unit from above, a lower degree of destabilization in the high-spin TS compared to others can be expected, as the high-spin state can undergo an efficient electron shift in an exchange enhanced fashion within this configuration. Therefore, even in nonenzymatic environments, ligand design should consider such steric effects.

Scheme 3. Possible strategies for increasing the relative stability of high-spin TSs in reactions of iron(IV)-oxo species.
4 Conclusion
Actively harnessing the high-spin reactivity of iron(IV)-oxo species presents a promising strategy for activating inert C–H bonds of organic substrates. Traditionally, nonheme ligands have been employed to reduce ligand field strength and stabilize the pre-reaction complex, while high-spin reactivity of P450s has not been widely explored. Our current QM/MM study on CYP2C8-catalyzed PTX hydroxylation revealed that the high-spin TS can be remarkably stabilized even within a heme ligand environment, particularly when forming 6β-hydroxypaclitaxel. Detailed energy decomposition analysis identified the critical roles of entropy and the substrate’s quantum mechanical (strain) effects in stabilizing the high-spin TS. To enhance the latter effect, it is essential to impose steric constraints on the substrate using surrounding atoms, thereby reducing the relative destabilization of the high-spin state. These insights could be strategically applied to the rational design of high-spin iron(IV)-oxo catalysts for C–H bond activation.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
DY: Data curation, Formal Analysis, Investigation, Visualization, Writing–review and editing. HH: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Ganghong Young Scholar Development Fund, the Changjiang Scholarship, the Guangdong Pearl River Talent Program, the Shenzhen Natural Science Foundation 2022 fund, and the funding for the Warshel Institute for Computational Biology from Shenzhen City and Longgang District (LGKCSDPT2024001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1471741/full#supplementary-material
References
Anastas, P., and Eghbali, N. (2010). Green chemistry: principles and practice. Chem. Soc. Rev. 39, 301–312. doi:10.1039/B918763B
Bae, S. H., Seo, M. S., Lee, Y. M., Cho, K. B., Kim, W. S., and Nam, W. (2016). Mononuclear nonheme high-spin (S=2) versus intermediate-spin (S=1) iron (IV)–Oxo complexes in Oxidation reactions. Angew. Chem. Int. Ed. 55, 8027–8031. doi:10.1002/anie.201603978
Balcells, D., Clot, E., and Eisenstein, O. (2010). C-H bond activation in transition metal species from a computational perspective. Chem. Rev. 110, 749–823. doi:10.1021/cr900315k
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98 (7), 5648–5652. doi:10.1063/1.464913
Becke, A. D., and Johnson, E. R. (2005). A density-functional model of the dispersion interaction. J. Chem. Phys. 123, 154101. doi:10.1063/1.2065267
Bigi, J. P., Harman, W. H., Lassalle-Kaiser, B., Robles, D. M., Stich, T. A., Yano, J., et al. (2012). A high-spin iron (IV)–oxo complex supported by a trigonal nonheme pyrrolide platform. J. Am. Chem. Soc. 134, 1536–1542. doi:10.1021/ja207048h
Case, D. A., Belfon, K., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., Cheatham, T. E., et al. (2020). Amber 2020. San Francisco, CA: University of California.
Chung, L. W., Hirao, H., Xin, L., and Morokuma, K. (2012). The ONIOM method: its foundation and applications to metalloenzymes and photobiology. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 327–350. doi:10.1002/wcms.85
Chung, L. W., Sameera, W. M. C., Ramozzi, R., Page, A. J., Hatanaka, M., Petrova, G. P., et al. (2015). The ONIOM method and its applications. Chem. Rev. 115, 5678–5796. doi:10.1021/cr5004419
Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M., et al. (1995). A Second generation Force field for the simulation of proteins, Nuncleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197. doi:10.1021/ja00124a002
Denisov, I. G., Makris, T. M., Sligar, S. G., and Schlichting, I. (2005). Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2278. doi:10.1021/cr0307143
de Visser, S. P., Ogliaro, F., Harris, N., and Shaik, S. (2001). Multi-state epoxidation of ethene by cytochrome P450: a quantum chemical study. J. Am. Chem. Soc. 123, 3037–3047. doi:10.1021/ja003544+
England, J., Guo, Y., Farquhar, E. R., Young, V. G., Munck, E., and Que, L. (2010). The crystal structure of a high-spin oxoiron (IV) complex and characterization of its self-decay pathway. J. Am. Chem. Soc. 132, 8635–8644. doi:10.1021/ja100366c
England, J., Guo, Y., Van Heuvelen, K. M., Cranswick, M. A., Rohde, G. T., Bominaar, E. L., et al. (2011). A more reactive trigonal-bipyramidal high-spin oxoiron (IV) complex with a cis-labile site. J. Am. Chem. Soc. 133, 11880–11883. doi:10.1021/ja2040909
England, J., Martinho, M., Farquhar, E. R., Frisch, J. R., Bominaar, E. L., Münck, E., et al. (2009). A synthetic high-spin oxoiron (IV) complex: generation, spectroscopic characterization, and reactivity. Angew. 121, 3622–3626. doi:10.1002/anie.200900863
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16, revision C.01. Wallingford CT: Gaussian, Inc.
Gandeepan, P., Muller, T., Zell, D., Cera, G., Warratz, S., and Ackermann, L. (2019). 3d Transition metals for C–H activation. Chem. Rev. 119, 2192–2452. doi:10.1021/acs.chemrev.8b00507
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the Daping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465. doi:10.1002/jcc.21759
Groves, J. T. (1985). Key elements of the chemistry of cytochrome P-450: the oxygen rebound mechanism. J. Chem. Educ. 62, 928. doi:10.1021/ed062p928
Harris, J. W., Katki, A., Anderson, L. W., Chmurny, G. N., Paukstelis, J. V., and Collins, J. M. (1994a). Isolation, structural determination, and biological activity of 6. alpha.-Hydroxytaxol, the principal human metabolite of taxol. J. Med. Chem. 37, 706–709. doi:10.1021/jm00031a022
Harris, J. W., Rahman, A., Kim, B. R., Guengerich, F. P., and Collins, J. M. (1994b). Metabolism of taxol by human hepatic microsomes and liver slices: participation of cytochrome P450 3A4 and an unknown P450 enzyme. Cancer Res. 54, 4026–4035.
Hehre, W. J., Ditchfield, R., and Pople, J. A. (1972). Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56 (5), 2257–2261. doi:10.1063/1.1677527
Hirao, H. (2011a). The effects of protein environment and dispersion on the formation of ferric-superoxide species in myo-inositol oxygenase (MIOX): a combined ONIOM(DFT:MM) and energy decomposition analysis. J. Phys. Chem. B 115, 11278–11285. doi:10.1021/jp2057173
Hirao, H. (2011b). Energy decomposition analysis of the protein environmental effect: the case of cytochrome P450cam compound I. Chem. Lett. 40, 1179–1181. doi:10.1246/cl.2011.1179
Hirao, H., Chen, H., Carvajal, M. A., Wang, Y., and Shaik, S. (2008b). Effect of external electric fields on the C−H bond activation reactivity of nonheme iron−oxo reagents. J. Am. Chem. Soc. 130, 3319–3327. doi:10.1021/ja070903t
Hirao, H., Kumar, D., Que Jr, L., and Shaik, S. (2006). Two-state reactivity in alkane hydroxylation by non-heme Iron−Oxo complexes. J. Am. Chem. Soc. 128, 8590–8606. doi:10.1021/ja061609o
Hirao, H., Kumar, D., Thiel, W., and Shaik, S. (2005). Two states and two more in the mechanisms of hydroxylation and epoxidation by cytochrome P450. J. Am. Chem. Soc. 127, 13007–13018. doi:10.1021/ja053847+
Hirao, H., Ng, W. K. H., Moeljadi, A. M. P., and Bureekaew, S. (2015). Multiscale model for a metal–organic framework: high-spin rebound mechanism in the reaction of the oxoiron (IV) species of Fe-MOF-74. ACS Catal. 5, 3287–3291. doi:10.1021/acscatal.5b00475
Hirao, H., Que Jr, L., Nam, W., and Shaik, S. (2008a). A two-state reactivity Rationale for Counterintuitive axial ligand effects on the CøH activation reactivity of nonheme FeIVO Oxidants. Chem. Eur. J. 14, 1740–1756. doi:10.1002/chem.200701739
Hou, K., Börgel, J., Jiang, H. Z. H., SantaLucia, D. J., Kwon, H., Zhuang, H., et al. (2023). Reactive high-spin iron(IV)-Oxo sites through dioxygen activation in a metal-organic framework. Science 382, 547–553. doi:10.1126/science.add7417
Hu, B., Yu, H., Zhou, J., Li, J., Chen, J., Du, G., et al. (2023). Whole-cell P450 Biocatalysis using engineered Escherichia coli with Fine-Tuned heme Biosynthesis. Adv. Sci. 10, 2205580. doi:10.1002/advs.202205580
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of Simple potential Functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi:10.1063/1.445869
Krebs, C., Fujimori, D. G., Walsh, C. T., and Bollinger, J. M. (2007). Non-heme Fe(IV)–Oxo intermediates. Acc. Chem. Res. 40, 484–492. doi:10.1021/ar700066p
Kumar, D., Hirao, H., Que, L., and Shaik, S. (2005). Theoretical investigation of C-H hydroxylation by (N4Py) FeIVO2+: an Oxidant more Powerful than P450? J. Am. Chem. Soc. 127, 8026–8027. doi:10.1021/ja0512428
Kumar, G. N., Oatis, J. E., Thornburg, K. R., Heldrich, F. J., Hazard, E. S., and Walle, T. (1994). 6 alpha-Hydroxytaxol: isolation and identification of the major metabolite of taxol in human liver microsomes. Drug Metab. Dispos. 22, 177–179.
Kumar, S. (2010). Engineering cytochrome P450 biocatalysts for biotechnology, medicine and bioremediation. Expert Opin. Drug Metab. Toxicol. 6, 115–131. doi:10.1517/17425250903431040
Lacy, D. C., Gupta, R., Stone, K. L., Greaves, J., Ziller, J. W., Hendrich, M. P., et al. (2010). Formation, structure, and EPR Detection of a high spin FeIV-oxo species derived from Either an FeIII-oxo or FeIII-OH complex. J. Am. Chem. Soc. 132, 12188–12190. doi:10.1021/ja1047818
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37 (2), 785–789. doi:10.1103/physrevb.37.785
Li, Z., Jiang, Y., Guengerich, F. P., Ma, L., Li, S., and Zhang, W. (2020). Engineering cytochrome P450 enzyme systems for biomedical and biotechnological applications. J. Biol. Chem. 295, 833–849. doi:10.1074/jbc.REV119.008758
Molecular Operating Environment (MOE) (2022). 02 chemical computing group ULC, 1010 Sherbooke St. West, suite #910. Montreal, QC, Canada. H3A 2R7, 2023.
Nam, W. (2007). High-valent iron (IV)–Oxo complexes of heme and non-heme ligands in bxygenation reactions. Acc. Chem. Res. 40, 522–531. doi:10.1021/ar700027f
Ortiz de Montellano, P. R. (2015). Cytochrome P450: structure, mechanism, and Biochemistry. 4th ed. Switzerland: Springer.
Puri, M., and Que, L. (2015). Toward the synthesis of more reactive S=2 non-heme oxoiron (IV) complexes. Acc. Chem. Res. 48, 2443–2452. doi:10.1021/acs.accounts.5b00244
Que, L. (2007). The Road to non-heme fxoferryls and beyond. Acc. Chem. Res. 40, 493–500. doi:10.1021/ar700024g
Rahman, A., Korzekwa, K. R., Grogan, J., Gonzalez, F. J., and Harris, J. W. (1994). Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2C8. Cancer Res. 54, 5543–5546.
Rittle, J., and Green, M. T. (2010). Cytochrome P450 compound I: capture, characterization, and C-H bond activation Kinetics. Science 330, 933–937. doi:10.1126/science.1193478
Sastri, C. V., Lee, J., Oh, K., Lee, Y. J., Lee, J., Jackson, T. A., et al. (2007). Axial ligand tuning of a nonheme iron (IV)–oxo unit for hydrogen atom abstraction. Proc. Nat. Acad. Sci. 104, 19181–19186. doi:10.1073/pnas.0709471104
Schöneboom, J. C., Cohen, S., Lin, H., Shaik, S., and Thiel, W. (2004). Quantum mechanical/molecular mechanical investigation of the mechanism of C−H hydroxylation of camphor by cytochrome P450cam: theory supports a two-state rebound mechanism. J. Am. Chem. Soc. 126, 4017–4034. doi:10.1021/ja039847w
Shaik, S., Cohen, S., Wang, Y., Chen, H., Kumar, D., and Thiel, W. (2010). P450 enzymes: their structure, reactivity, and belectiveity modeled by QM/MM calculations. Chem. Rev. 110, 949–1017. doi:10.1021/cr900121s
Shaik, S., Hirao, H., and Kumar, D. (2007). Reactivity of high-valent iron–oxo species in enzymes and synthetic reagents: a tale of many states. Acc. Chem. Res. 40, 532–542. doi:10.1021/ar600042c
Shaik, S., Kumar, D., de Visser, S. P., Altun, A., and Thiel, W. (2005). Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 105, 2279–2328. doi:10.1021/cr030722j
Singla, A. K., Garg, A., and Aggarwal, D. (2002). Paclitaxel and its formulations. Int. J. Pharm. 235, 179–192. doi:10.1016/s0378-5173(01)00986-3
Sono, M., Roach, M. P., Coulter, E. D., and Dawson, J. H. (1996). Heme-containing oxygenases. Chem. Rev. 96, 2841–2888. doi:10.1021/cr9500500
Stage, T. B., Bergmann, T. K., and Kroetz, D. L. (2018). Clinical pharmacokinetics of paclitaxel monotherapy: an updated literature review. Clin. Pharmacokinet. 57, 7–19. doi:10.1007/s40262-017-0563-z
Vosko, S. H., Wilk, L., and Nusair, M. (1980). Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 58 (8), 1200–1211. doi:10.1139/p80-159
Weaver, B. A. (2014). How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell. 25, 2677–2681. doi:10.1091/mbc.E14-04-0916
Weigend, F., and Ahlrichs, R. (2005). Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence qurality for H to Rn: design and assessment of acuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/B508541A
Werck-Reichhart, D., and Feyereisen, R. (2000). Cytochromes P450: a success story. Genome Biol. 1, REVIEWS3003. doi:10.1186/gb-2000-1-6-reviews3003
Williams, P. A., Cosme, J., Ward, A., Angove, H. C., Matak Vinković, D., and Jhoti, H. (2003). Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 424, 464–468. doi:10.1038/nature01862
Yamaguchi, J., Yamaguchi, A. D., and Itami, K. (2012). C-H bond functionalization: emerging synthetic tobols for natural products and pharmaceuticals. Angew. Int. Ed. Engl. 51, 8960–9009. doi:10.1002/anie.201201666
Keywords: cytochrome P450, high-spin reactivity, C-H bond activation, QM/MM, CYP2C8, paclitaxel
Citation: Yue D and Hirao H (2024) Enhancing the high-spin reactivity in C–H bond activation by Iron (IV)-Oxo species: insights from paclitaxel hydroxylation by CYP2C8. Front. Chem. 12:1471741. doi: 10.3389/fchem.2024.1471741
Received: 28 July 2024; Accepted: 26 August 2024;
Published: 05 September 2024.
Edited by:
Haibo Ge, Texas Tech University, United StatesReviewed by:
Hao Su, Chinese Academy of Sciences (CAS), ChinaXuesong Wu, Huazhong University of Science and Technology, China
Copyright © 2024 Yue and Hirao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hajime Hirao, aGlyYW9AY3Voay5lZHUuY24=