 N. Sai Kumar
N. Sai Kumar Anubendu Adhikary
Anubendu Adhikary- School of Advanced Sciences, VIT-AP University, Amaravati, Andhra Pradesh, India
The storage and transportation of hydrogen gas, a non-polluting alternative to carbon-based fuels, have always been challenging due to its extreme flammability. In this regard, formic acid (FA) is a promising liquid organic hydrogen carrier (LOHC), and over the past decades, significant progress has been made in dehydrogenating FA through transition metal catalysis. In this review, our goal is to provide a detailed insight into the existing processes to expose various mechanistic challenges associated with FA dehydrogenation (FAD). Specifically, methodologies catalyzed by pincer-ligated metal complexes were chosen. Pincer ligands are preferred as they provide structural rigidity to the complexes, making the isolation and analysis of reaction intermediates less challenging and consequently providing a better mechanistic understanding. In this perspective, the catalytic activity of the reported pincer complexes in FAD was overviewed, and more importantly, the catalytic cycles were examined in detail. Further attention was given to the structural modifications, role of additives, reaction medium, and their crucial effects on the outcome.
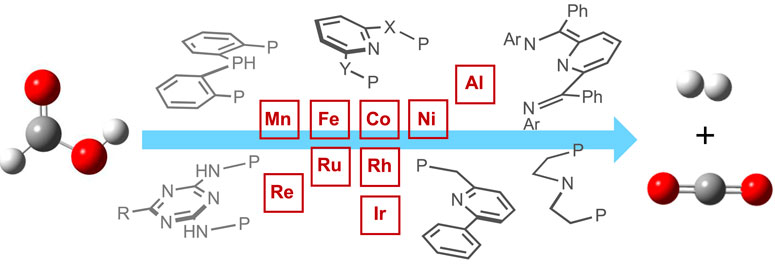
1 Introduction
The demand for dihydrogen (H2) as an alternative to carbon-based fossil fuels is growing enormously as it is renewable and produces clean energy. However, because of its gaseous state and high flammability, the storage and transportation of H2 have always been challenging (Schlapbach and Züttel, 2001; Turner, 2004; Felderhoff et al., 2007; Schüth, 2009; Myers and Berben, 2014; Zhu and Xu, 2015; He et al., 2016; Lu et al., 2016). Compared to current storage techniques such as compression, liquefaction, or adsorption, storing H2 chemically through liquid organic hydrogen carriers (LOHCs) is far safer and more convenient. In this regard, formic acid (FA) is a promising candidate due to its high volumetric capacity of H2 (53.4 g/L at STP), non-flammability, and non-toxicity (Grasemann and Laurenczy, 2012). For utilizing and implementing formic acid to store and transport H2, two chemical processes must be efficient and economically viable: a) formic acid production and b) H2 generation from FA. Current industrial processes for synthesize FA through its long-known straightforward procedures using carbon monoxide are inexpensive. Most importantly, convenient FA production methods from the hydrogenation of CO2 have been achieved through transition metal catalysis (Wang et al., 2015; Álvarez et al., 2017; Yin et al., 2019). Therefore, combining H2 with CO2 to obtain formic acid, followed by its decomposition to release the H2, provides a viable, carbon-neutral solution for using formic acid as an equivalent to liquid hydrogen (Scheme 1).
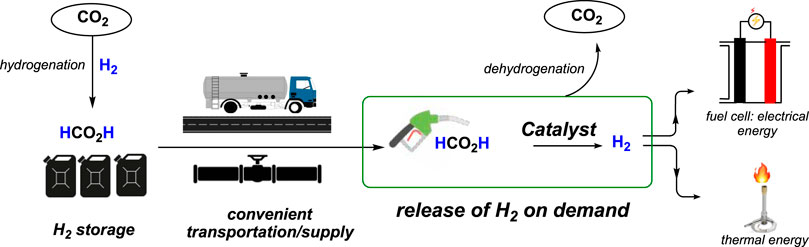
Scheme 1. Formic acid as a hydrogen carrier for CO2 hydrogenation/dehydrogenation.
Studying the convenience and applicability of various LOHCs has been a burgeoning area of research, particularly in the automobile industry. Along this line, at the Eindhoven University of Technology in the Netherlands, the world’s first bus powered by formic acid has been developed. This breakthrough prototype by the startup “Team Fast” (Formic Acid Sustainable Transportation) has shown how safe, more efficient, and a carbon-neutral fuel formic acid can be (http://teamfast.nl, http://www.dens.one). In the past decades, several catalytic systems, under both homogeneous and heterogeneous conditions, have been studied and applied for the dehydrogenation of formic acid (Grasemann and Laurenczy, 2012; Andérez-Fernández et al., 2016; Mellmann et al., 2016; Singh et al., 2016; Bernskoetter and Hazari, 2017; Li and Xu, 2017; Algi et al., 2018; Katerina et al., 2018; Onishi et al., 2022; Wei et al., 2022). Although these catalysts have significantly contributed to the understanding and advancement of the formic acid dehydrogenation (FAD) process, most reported methodologies are associated with one or more limitations: a) generation of CO-contaminated H2; b) short-lived and non-reusable catalysts; c) costly and complicated synthetic procedures of the catalyst; and most commonly, d) use of additives and e) costly or organic reaction medium. To overcome these challenges, it is essential not only to develop a new generation of catalysts but also to study and understand the details of existing procedures. In this review, our primary focus is to understand and analyze various mechanistic pathways involved in metal-catalyzed FAD.
In general, the formic acid decomposition pathway can be divided into two major parts: (a) formation of H2 and (b) release of CO2. Among most of the existing catalytic cycles, it is noted that two intermediates play the major role: metal-bound hydride (M-H) and metal-bound formate (M-OCHO) (Mellmann et al., 2016; Bernskoetter and Hazari, 2017; Algi et al., 2018; Katerina et al., 2018; Onishi et al., 2022; Wei et al., 2022). In the dihydrogen formation step, which can also be broadly termed “protonation” or “FA activation,” the M-H reacts with formic acid, producing H2 and metal formate (Figure 1). In another step, the formate complex undergoes CO2 elimination, regenerating the M-H. In principle, it appears as a straightforward cycle; however, a number of factors may critically be involved in both steps, which brings up numerous possibilities in the course of the reaction (Scheme 2). Based on the catalyst structure, properties, and reaction conditions, various options in FA activation are possible, which include traditional coordination, acid–base-type interaction, ligand participation, or oxidative addition (less likely). Similarly, the decarboxylation pathways may follow classical β-hydride elimination, isomerization followed by CO2 release, or through metal–ligand cooperation. Furthermore, these processes were found to be influenced by different additives, such as salts, bases, or Lewis acids. Unfortunately, elucidation of the operating mechanism of a particular process is often challenging due to the high reactivities of transient intermediates. Hence, for a comprehensive understanding of these mechanisms, those catalytic cycles will be preferred where intermediates are isolable or can be characterized in situ. In this context, the pincer ligands are well known for providing stability to plenty of metal complexes through stronger binding (Koten and Milstein, 2012; Szabó and Wendt, 2014; Morales-Morales, 2018; Piccirilli et al., 2020). Thus, we became particularly interested in examining catalysts with rigid pincer backbones, expecting to acquire a finer mechanistic insight. In addition, these ligands allow us to tune the reactivity of the metal center by accommodating different substituents with flexible electronic and steric properties, which will provide additional findings about the processes. In the last 15 years, metal complexes ligated with tridentate pincers have shown a tremendous contribution in dehydrogenating formic acid (Table 1, 1A–24A). In this report, we presented these catalysts collectively and compared their activity in FAD. More importantly, a thorough inspection of the reaction mechanisms was conducted, and the effects of essential factors such as backbone structure, ancillary ligands, various substituents, and additives were discussed in detail. The connection between strategic modifications and their consequences was also illustrated.
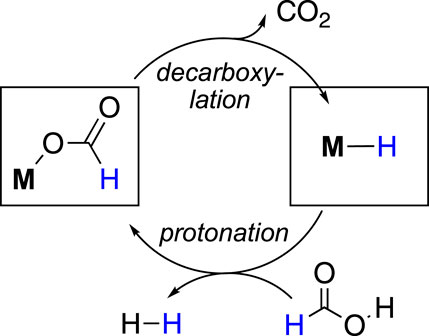
Figure 1. Formic acid dehydrogenation cycle involving metal–hydride and metal–formate complexes.
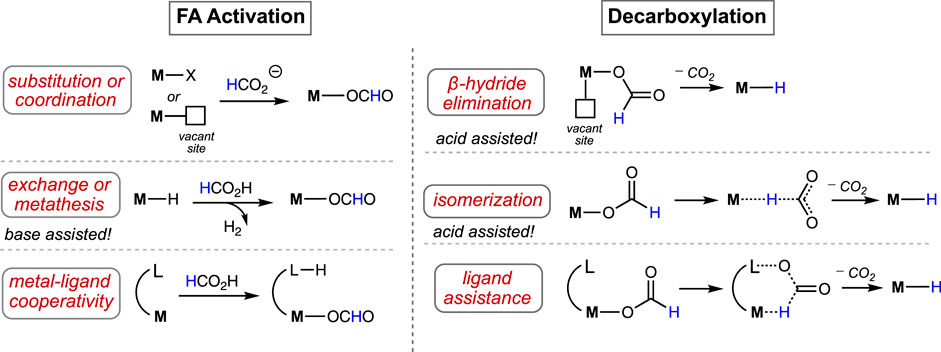
Scheme 2. Various pathways to activate formic acid (left) and decarboxylate the metal–formate complex (right).
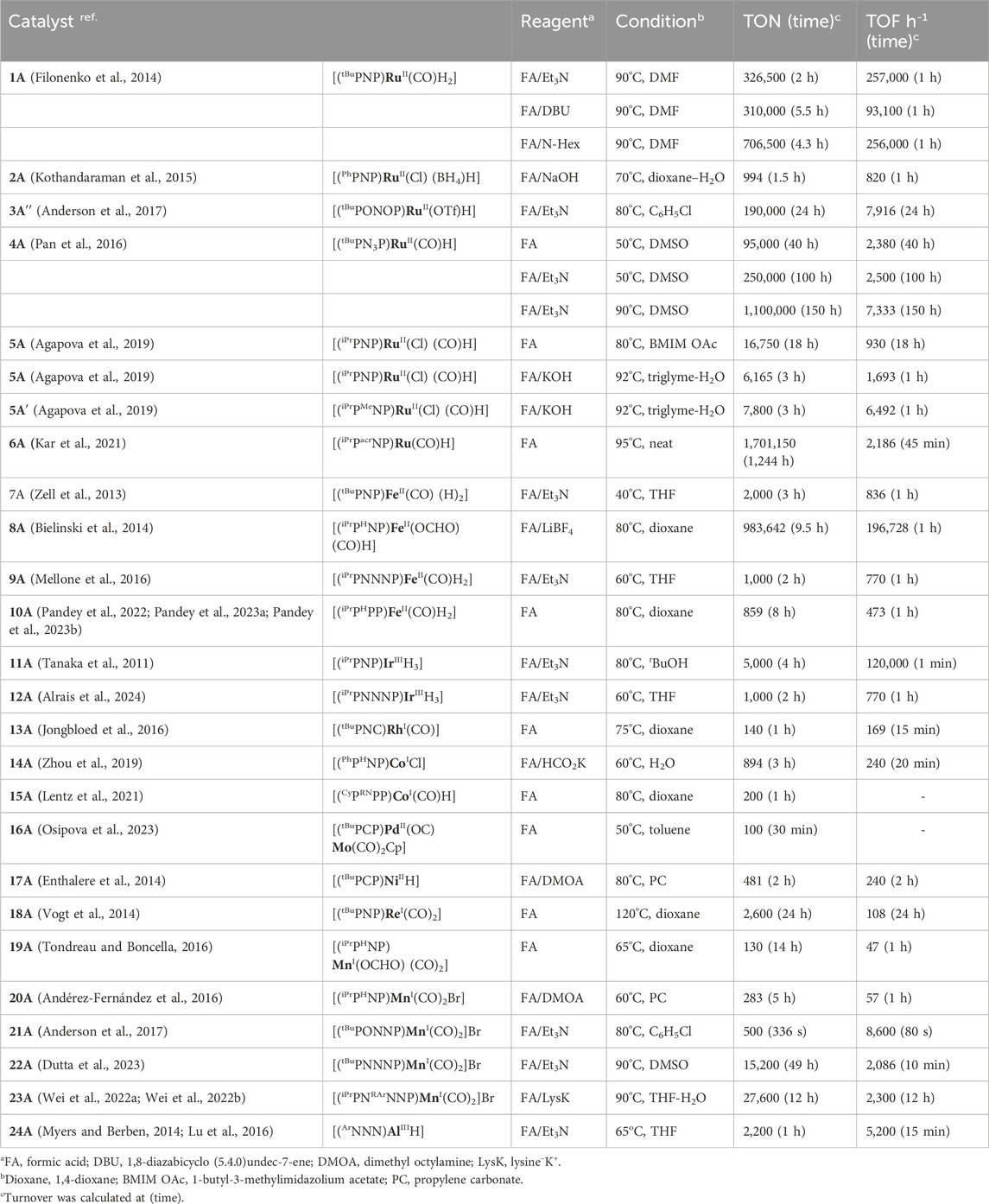
Table 1. Pincer catalysts for formic acid dehydrogenation.
2 Group VIII (Ru and Fe) metal complexes
2.1 Ruthenium
In 2014, a ruthenium complex with a PNP-ligand [PNP = 2,6-bis(di-tertbutylphosphinomethyl) pyridine; Figure 2, 1A] was applied in formic acid dehydrogenation by Filonenko et al. (2014). The catalytic reactions were set up in a batch reactor and studied under continuous FA addition. Over a range of temperatures (65–90°C), high turnovers by 1A were obtained in the presence of a base such as triethylamine, trihexylamine, and 1,8-diazabicyclo (5.4.0)undec-7-ene (DBU). Among different polar solvents, DMF was found to be most effective considering the catalytic activity and their stability. However, no conversion was observed in the polar protic solvent, e.g., ethanol, possibly due to the insolubility of the catalyst. After a thorough experimental and computational study, a comprehensive mechanistic cycle was proposed, as shown in Scheme 3. In the first step, protonation of the metal-bound hydride resulted in the formation of H2. Afterward, its rapid dissociation generated a cationic Ru intermediate. The coordination of the formate ion to this intermediate produced Ru–formate, which was successfully isolated and characterized in a separate experiment. The release of CO2 from Ru–formate completed the cycle, regenerating 1A. As noted, the base additives played an important role in the process, and interestingly, their proportion with respect to FA was found to be critical. It was observed that Et3N and DBU influenced the rate in an opposite way. In the case of triethylamine, the rate decreased with an increase in the acid-to-amine ratio, whereas, with DBU, the rate varied proportionally with the acid/amine concentration. This behavior was believed to be controlled by the strength of basicity and the reverse nature of the protonation step (Scheme 3, 1A→1B). In the protonation step, DBU being a stronger base (pKa = ∼11.5 in water), its conjugate acid is reluctant to protonate the ruthenium-bound hydride, making H2 formation a rate-limiting step. On the other hand, for Et3NH+ (pKa = ∼10.5 in water), protonation of Ru-H is more energetically favorable, ensuing decarboxylation as the slower step (Scheme 3, 1D→1A). This base-dependent switching of rds was further supported by the kinetic isotope effect study. The activation energy of the decarboxylation step, which is rate-limiting for the Et3N-assisted cycle, was calculated as 74 kJ mol-1.
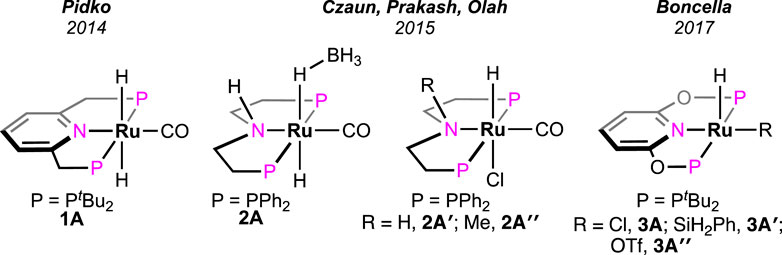
Figure 2. Ruthenium pincer catalysts for the dehydrogenation of FA with base additives.
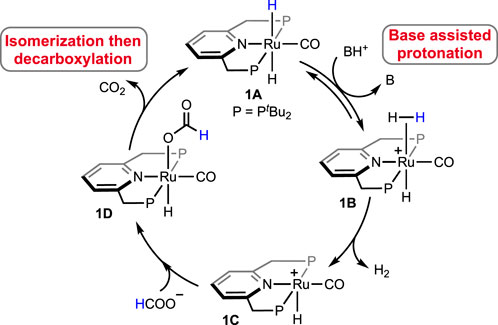
Scheme 3. Proposed mechanism of (PNP)Ru-catalyzed FA dehydrogenation.
Olah et al. investigated the catalytic activity of another ruthenium–pincer complex with a non-aromatic PHNP backbone (PHNP = HN{CH2CH2(PR2)}2; R = Ph; Figure 2, 2A) (Kothandaraman et al., 2015). Using the complex, different formate salts were successfully dehydrogenated in a dioxane–water medium under convenient conditions. Lithium and sodium formates were more reactive in producing H2 than their potassium and cesium salts. A catalytic cycle similar to that shown in Scheme 3, involving Ru–formate and Ru-hydride as intermediates, was proposed. To uncover the presence of any ligand cooperativity, complexes having a methyl substituent on the central nitrogen were examined (complex 2A″). Although pre-catalysts 2A′ and 2A″ were equally efficient in FAD, the possibility of ligand participation was not ruled out. Reaction kinetics and isotopic studies suggested the CO2 elimination step as rate-determining, whose activation energy was also calculated (80.7 kJ mol-1). In another report, Boncella et al. explored the reactivity of the (PONOP)-ruthenium catalyst, which was structurally similar to 1A (PONOP = 2,6-bis(diisopropylphosphinito)pyridine; Figure 2, 3A) (Anderson et al., 2017). These catalysts were found to be highly efficient in dehydrogenating an equimolar mixture of FA and Et3N, even under high pressure. For instance, at 80°C, a pressure of 10 atm was built up in 2 min, starting from 1.58 mL of reagents. A fast reaction with TOF of 36,000 h-1 was achieved; however, the catalyst was found to be ineffective without the amine.
All the aforementioned Ru catalysts were air-sensitive and required external bases for effective dehydrogenation. Zheng and Huang et al. prepared an air- and moisture-stable (PN3P)-ligated ruthenium pincer (Scheme 4, 4A) that displayed comparable reactivity but at a lower temperature (50°C) (Pan et al., 2016). Interestingly, the catalyst worked under base-free conditions at a high speed (TOF of 2,380 h-1). Furthermore, in the presence of a stoichiometric quantity of triethylamine, the TOF increased up to 2,500 h-1. An increase in efficiency was also attained at elevated temperatures (90°C; TOF of 7,333 h-1 and TON up to 1,100,000). This catalyst was successful in bypassing the requirement of an amine, suggesting internal assistance from the ligand. In an observation, the reaction between 4A and one equivalent of formic acid instantly generated complex 4B bearing a protonated N-center, which strongly supported the capability of imine nitrogen to perform as an internal base. Accordingly, in the base-free catalytic cycle, the activation of formic acid via ligand protonation was well established as a critical step (Scheme 4). Nevertheless, the deinsertion of CO2 from Ru-OCHO was determined as the slower step. In another study, complexes 4A′ and 4A″, having similar ligand frameworks, were examined; however, they displayed slower catalysis than 4A, illustrating the influence of phosphine donors.
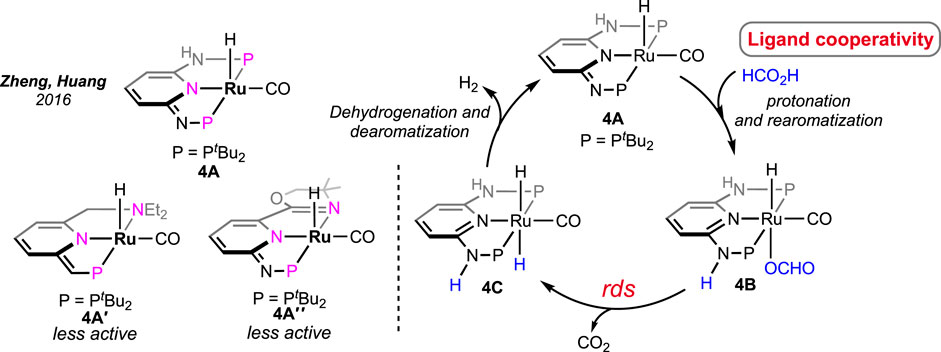
Scheme 4. Ruthenium pincer catalysts for base-free FAD (left). Proposed mechanistic cycle involving ligand cooperation (right).
In 2021, Beller et al. performed a comparative study between (PHNP)-Ru pincer complex 5A and its N-methylated version 5A′ (PMeNP-Ru; PMeNP = MeN{CH2CH2(PR2)}2; R = iPr, Figure 3) (Agapova et al., 2019). Surprisingly, the complex with a methylated backbone exhibited a faster FA dehydrogenation irrespective of the reaction condition. In the case of (PHNP)-Ru pincer 5A, the involvement of Ru-N in the catalytic cycle was expected, whereas for 5A′, ligand participation is not possible, as shown in Scheme 5. The enhanced reactivity of 5A′ implied that ligand cooperativity was not advantageous. When the effect of pH on the reaction rate was tested, H2 generation was found to be faster under acidic conditions (pH 4.5). Although the rate was moderate at a pH value of 13, a more satisfactory conversion and rate were obtained when the reaction was carried out in a buffer solution (pH range: 4.5–8.5). Displaying a low-pH preference indicates the crucial role of the protonation step in the mechanism (cycle 5a, 5A′→5D′; cycle 5b, 5A″→5E). During the catalysis by the (PMeNP)Ru pincer, both the intermediates 5A′ and 5B′ were detected in the reaction mixture (cycle 5a). Possibly, the protonation step is highly influential in these cycles, and in the case of N-methylated ruthenium hydride, it is energetically more favorable. Activation energies for 5A and 5A′-catalyzed FAD were experimentally calculated as 85.9 kJ mol-1 and 66.9 kJ mol-1, respectively. A difference of ∼20 kJ mol-1 between their activation energies supports the no-benefit of ligand cooperativity on the overall rate. However, in an ionic liquid medium, complex 5A and its derivatives were proven to be excellent catalysts for CO2 hydrogenation–dehydrogenation. Das and Nielsen et al. successfully applied complexes 2A, 2A′, 5A, 5A″, and 5A‴ in producing H2 from formic acid under additive-free conditions (Figure 3) (Piccirilli et al., 2023). Although similar turnovers were shown by all these catalysts, those having phenyl groups on P-donors (2A and 2A′) exhibited higher stability over the P-isopropyl analogs (5A, 5A″, and 5A‴). The ionic liquid, 1-butyl-3-methylimidazolium acetate (BMIM OAc), acted not only as an effective medium but also played a crucial role in activating the catalyst. Spectroscopic investigation disclosed that irrespective of the applied (PNP)Ru complexes, the resting state during catalysis was the acetate complex 5A‴. This observation confirmed that the acetate ion from the solvent was directly involved in triggering the catalytic activity.
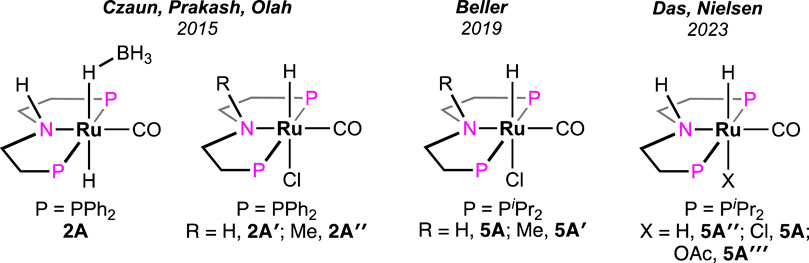
Figure 3. Bis(phosphine)-based non-aromatic (PNP) Ru pincer complexes applied in FAD.
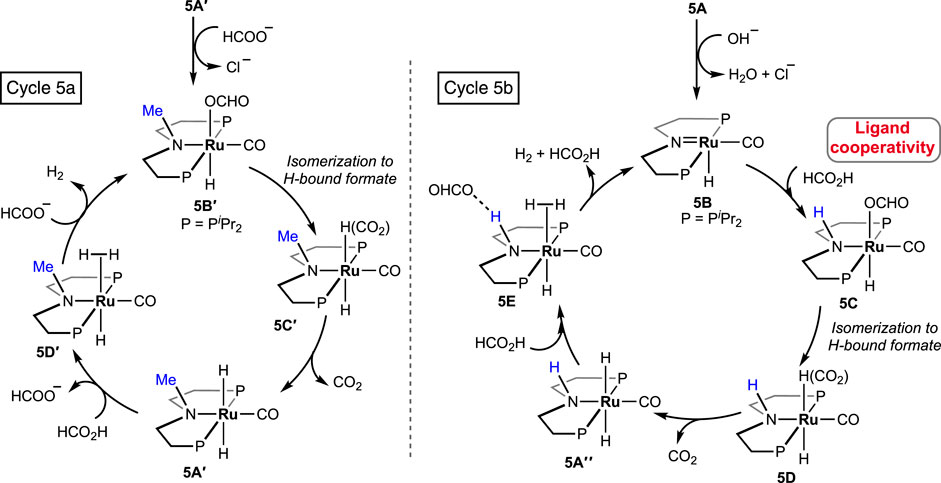
Scheme 5. FAD cycles by catalysts 5Aʹ (left) and 5A (right).
Without using any solvent or additive, the catalytic dehydrogenation of formic acid was achieved by Kar et al. (2021). The ruthenium hydride complex supported on the 9H-acridine-based bis(phosphine) ligand has shown remarkable turnovers in producing H2 at 95°C (Figure 4, 6A). More importantly, a pressure of 100 bar was attained starting from 5 mL of 85% formic acid. Experimental investigation revealed an initial binding of formic acid to the ruthenium center (Scheme 6, 6B). The presence of the FA-coordinated ruthenium complex in the reaction mixture was confirmed using NMR spectroscopy. A detailed theoretical calculation showed a stabilizing hydrogen-bond interaction between the FA proton and Ru-H, which favored dihydrogen generation. Through a six-membered cyclic transition state, eventually, the formation of the ruthenium–formate complex was proposed (Scheme 6, 6B → 6C). Although the formate complex was not observed during catalysis, it was isolated from a separate experiment by reacting 6A with excess CO2 (complex 6C, κ2-coordination mode of HCO2−). As anticipated, under a CO2-free environment, decarboxylation from 6C was observed, resulting in the regeneration of the hydride 6A. Kinetic isotope effects and DFT calculation demonstrated the CO2 release as a slower process than the elimination of H2. Furthermore, the activation parameters were experimentally calculated as 17.01 kcal mol-1 (ΔH≠) and −20.36 cal mol-1 K−1 (ΔS≠). On an interesting note, even though the formation of CO was below 20 ppm, complex 6A′ was detected during the reaction, indicating an interference from the byproduct CO. However, complexes 6A′ and 6A″ were observed to be catalytically slower than 6A.
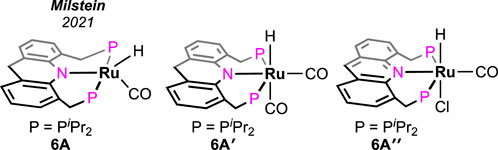
Figure 4. Acridine-based (PNP) ruthenium catalysts for solvent-less, additive-less FA decomposition.
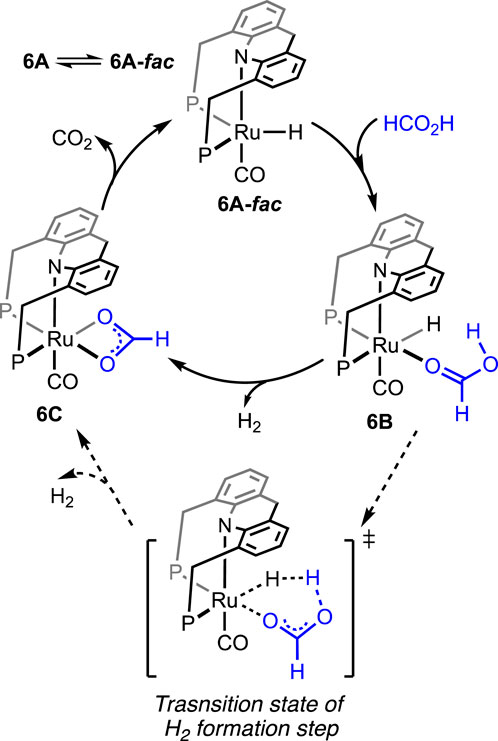
Scheme 6. Proposed mechanistic cycle of 6A-catalyzed dehydrogenation of FA.
2.2 Iron
Supported by ligand frameworks similar to those used in Ru systems, a few iron complexes were synthesized and explored for formic acid decomposition. The (bis)phosphine-ligated iron hydride complex was first reported by Zell et al. (2013) (Figure 5, 7A). Compared to the ruthenium analog (see 1A in Figure 2), the iron hydride performed under a milder condition (40°C) but at a slower rate. In the presence of one equivalent of triethylamine, TON up to 2,000 with an initial TOF of 836 h-1 was obtained. The mechanistic cycle involved the usual protonation of the iron-bound hydride to produce H2 and its dissociation to generate a reactive cationic intermediate. Then, smooth coordination of formate to the iron center, followed by the release of CO2, completed the cycle (Scheme 7, cycle 7a). Although the Fe intermediates involved in this cycle are similar, as shown in Scheme 3, it is important to note the unusual role of the base additive. The amine has shown no effect on the rate of protonation (cycle 7a, 7A→7B); rather, it was compelling to find experimentally that Et3NH+ facilitated the CO2 elimination process. As hypothesized by the group, a stabilizing interaction between the metal formate and the Bronsted acid (Et3NH+) sped up the decarboxylation. In addition, a classical β-hydride elimination of Fe-OCHO cannot be followed due to the unavailability of a vacant coordination site around the Fe(II) center; therefore, switching in the coordination mode of formate prior to CO2 release was necessary. As expected, DFT calculations strongly supported an intramolecular isomerization of iron-bound formate to carboxylate (Fe--H--CO2) in order to release the CO2.
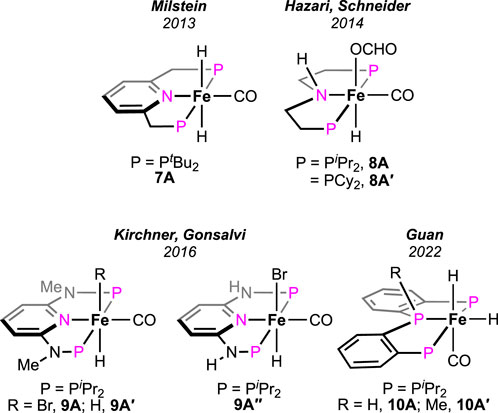
Figure 5. Different iron pincer complexes for FA decomposition.
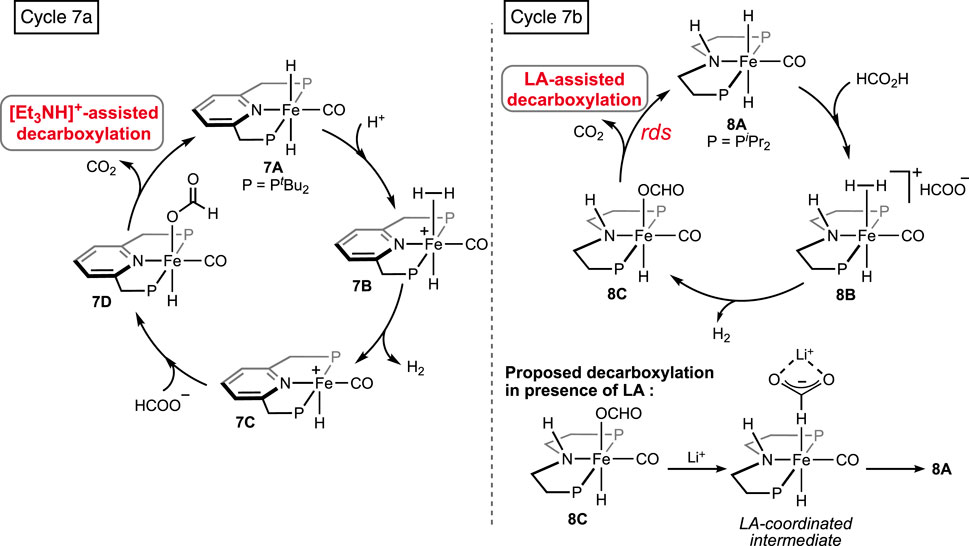
Scheme 7. FAD catalytic cycles by iron pincers involving additive-assisted decarboxylation.
Later, additive-promoted decarboxylation of iron–formate was well proven by Schneider and Hazari et al. (Bielinski et al., 2014; Zhang et al., 2015; Curley et al., 2018; Curley et al., 2021). The specialty of their finding was the role of the Lewis acid (LA) as a co-catalyst in FA dehydrogenation. Applying a (PHNP)–iron formate 8A-A′ as catalyst with 10 mol% Lewis acid, high conversion and turnover were accomplished (Figure 5; Scheme 7, cycle 7b). As evidenced, in the presence of Lewis acids, the decarboxylation from iron–formate was accelerated, which also suggested an energetically favorable interaction between the two. Among various LA additives, LiBF4 was found to be most effective in facilitating the deinsertion of CO2. As shown in Scheme 7, coordination of the formate with the lithium ion was proposed to be the key factor in lowering the energy barrier. In a comparison of potency between the Lewis acid and Bronsted acid (LiBF4 vs [Et3NH]+), a faster CO2 formation was observed for the triethylammonium ion under 0.1 mol% catalyst loading. However, in the case of LiBF4, interestingly, a higher rate and conversion were achieved with a lower catalyst concentration (0.001%). Irrespective of the additives used, during catalysis, iron–formate (8c) was the only observed species in the reaction mixture, which suggested its decarboxylation as the rate-determining step.
In addition to FA dehydrogenation, the group extensively studied its reverse process, i.e., CO2 hydrogenation. By substituting the hydrogen atom on the N-donor with a methyl group (Scheme 8, 8A-1), a huge improvement in the catalytic efficiency for CO2 hydrogenation was observed (Zhang et al., 2015). However, the corresponding effect on FA dehydrogenation was not reported. The group further studied the influences of the ancillary ligands by replacing the iron-bound CO with an isonitrile moiety. Prepared iron complexes 8A-2 and 8A-3 were found to be less efficient in catalytic FAD (Scheme 8). The presence of isonitrile functionality was proposed to destabilize the iron intermediates involved in catalysis, leading to catalyst decomposition. Later, it was discovered that the presence of small-size phosphines, such as PMe3 was useful in suppressing catalyst degradation (Curley et al., 2018; Curley et al., 2021).
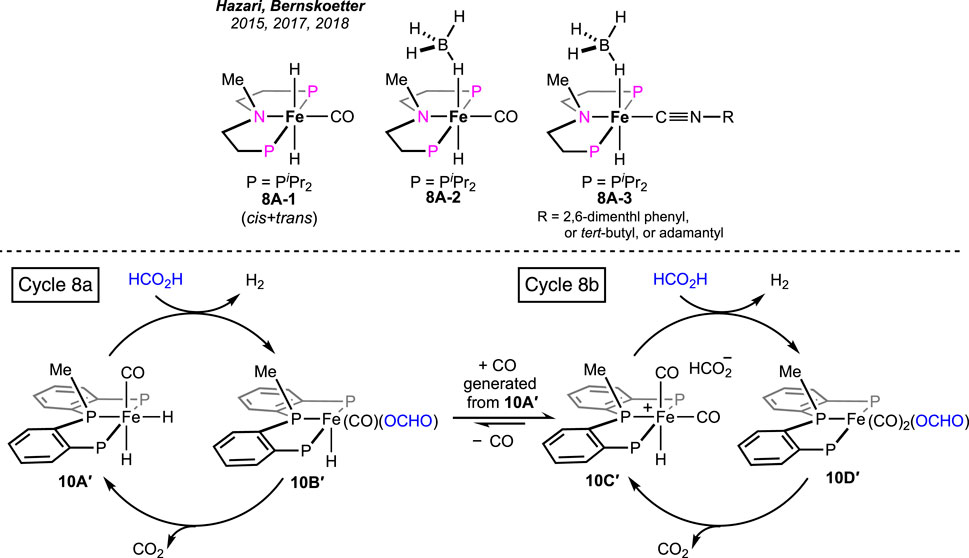
Scheme 8. Tertiary amine-based (PNP)iron hydrides (above). Parallel catalytic cycles in FAD by (PPP)iron pincer complexes (stereochemistry is not confirmed for all the intermediates) (below).
Pyridine-based (PNNNP)-iron pincer catalysts showed a comparable efficiency with structurally similar 7A, as reported by Gonsalvi and Kirchner (Figure 5, 9A-A′) (Mellone et al., 2016). Amines were an optimum need to carry out the transformation. A similar catalytic pathway as in cycle 7a was proposed; however, the role of amine was found to be dissimilar. Experimentally, it was noted that the amine was involved in promoting the protonation step in contrast to favoring the decarboxylation, as observed in the case of the 7A-catalyzed process. The presence of other Lewis acids, such as LiBF4, did not show any acceleration on the turnovers, implying no favor in the CO2 elimination step. In line with these experimental observations, computational calculations revealed the initial coordination of FA to the catalyst, followed by amine-led proton abstraction, resulting in the corresponding iron–formate, which further supported the positive impact of the amine on the protonation step. However, a point to note is that DBU, a stronger base, was less effective than triethylamine during catalysis. On another interesting note, poor activity was observed with complex 9A″, despite being structurally eligible for metal–ligand cooperativity (Figure 5). Then, in 2022, novel (PPP)–iron dihydride complexes were investigated by Guan and Pandey [(o-iPr2PC6H4)2PR (R = H or Me); Figure 5, 10A-A′] (Pandey et al., 2022; Pandey et al., 2023a; Pandey et al., 2023b). Additive-less FAD was accomplished under convenient conditions using these catalysts. However, slow ligand dissociation, followed by decomposition of the catalyst, was observed, along with carbon monoxide formation. An extensive investigation led to a complicated reaction mechanism involving two parallel catalytic cycles (Scheme 8). Initial protonation of Fe-cis-(H)2, 10A′, resulted in a trans-hydridoformate complex (cycle 8a, 10B′), which was found to be in equilibrium with the iron–dicarbonyl complex (10C′), the proposed entry point to another cycle (cycle 8b). Nevertheless, as revealed experimentally, the rate-limiting step was the elimination of CO2 from iron–formate. No significant difference in the catalytic turnovers between 10A and 10A′ was noted; thus, the possibility of ligand assistance during catalysis can be ruled out.
3 Group IX (Ir, Rh, and Co) metal complexes
3.1 Iridium
Morokuma and Nozaki et al. studied the catalytic dehydrogenation of formic acid using a (iPrPNP)-ligated iridium(III) trihydride pincer (Figure 6, 11A) (Tanaka et al., 2011). Without an amine additive, the catalyst showed a low conversion rate (18%) at 60°C. However, the catalytic performance significantly increased in the presence of triethylamine. At 80°C, quantitative conversion to H2 and CO2 was achieved within 4 h, displaying a high turnover frequency (120,000 h-1). Although no mechanistic investigation was performed for this FA dehydrogenation, the group looked into the details of CO2 hydrogenation, the reverse process of FAD. The participation of the ligand in the catalytic cycle was successfully demonstrated by experimental observations and DFT calculations. Following the principle of microscopic reversibility, these lines of evidence recommend ligand involvement in the FA dehydrogenation process. However, no further experiments were conducted to support the metal–ligand cooperativity. A recent advancement in iridium catalysis was made by Huang et al., where the formic acid dehydrogenation took place without any solvent. From a mixture of formic acid and cesium formate, dihydrogen was successfully produced by the (tBuPHNNHNP)iridium trihydride 12A (Figure 6) (Alrais et al., 2024). Under the reaction condition (80–90°C), the cesium salt not only played the role of a base but also served as a potent medium. Fascinatingly, the group immobilized this highly active catalyst on an aluminum-bound fibrous silica nanosphere, which exhibited a remarkable upgrade in turnovers. As noted, the immobilized catalyst was two times more efficient in generating dihydrogen (TON 260,000 vs 540,000) (Figure 6). The catalytic pathway involved the formation of a metal formate from the reaction between FA and the catalyst, followed by its decarboxylation. Interestingly, after carrying out the transformation in an aqueous medium, an important role of water was found in stabilizing the reaction intermediates through hydrogen bond interactions. The detailed theoretical study further revealed the participation of ligand N-H and H2O molecules in the formic acid activation step. In an explanation of the higher reactivity of the immobilized catalyst, DFT calculations showed a greater negative charge density on the iridium of immobilized 12A, which stimulated its protonation by FA.
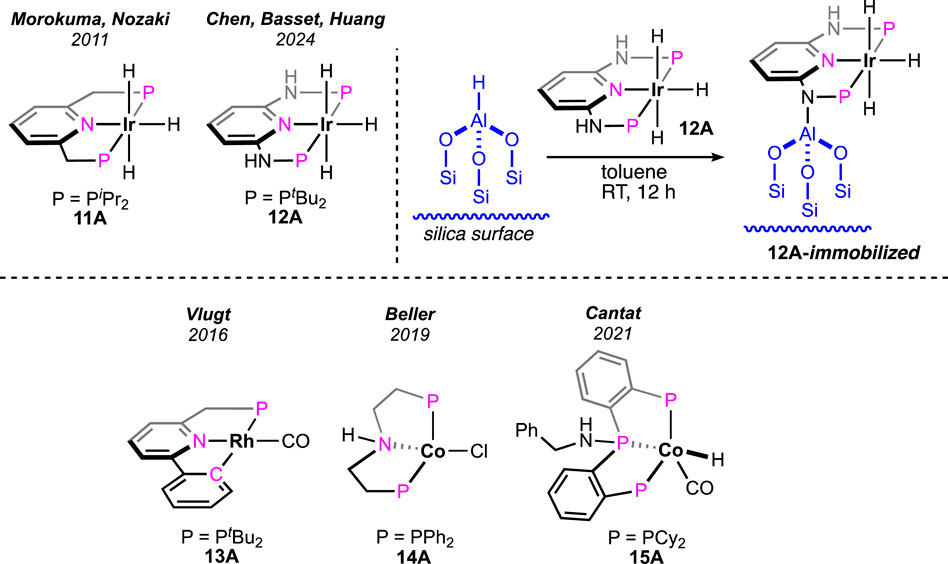
Figure 6. Iridium pincer complexes studied in FAD and the (tBuPHNNHNP)Ir trihydride immobilized on silica (above). Rhodium and cobalt pincer complexes studied in FAD (below).
3.2 Rhodium
A rare type of metal–ligand cooperativity in activating formic acid was reported by van der Vlugt et al. (Figure 6) (Jongbloed et al., 2016). The rhodium(I) complex (13A), bearing a phenyl group as the pincer arm, satisfactorily decomposed formic acid under base-free conditions. Although a lower turnover than Ru and Fe catalysts was displayed, unlike previous cycles, the catalysis took an interesting route, which made it mechanistically valuable. The cycle started with the expected reaction between 13A and formic acid following a different path. The attached phenyl arm of 13A abstracted H+, resulting in the opening of the metallacycle and the formation of a rhodium–formate bond (Scheme 9, 13B). Then, the intermediate 13B underwent decarboxylation, generating Rh(I)-hydride (13C). Oxidative addition of sp2C-H to Rh(I) led to Rh(III)-dihydride, and finally, the reductive elimination of H2 completed the cycle. The use of HCO2D as the starting reagent resulted in deuteriation at the ortho-positions of the phenyl ring, which strongly supported the reversible cyclometallation pathway, as shown in Scheme 9. The computational study confirmed that Rh-OCHO, 13B, was the most stable intermediate and the CO2 release was the slowest step. Evidently, the phenyl arm of the pincer ligand acted as an internal base, and as expected, no effect of external bases was noticed on the reaction rate. Furthermore, related complexes where carbanionic ligands were absent, such as (tBuPCP)- and (tBuPNN)-Rh(CO), were found completely inactive (Scheme 9). Another complex having a methyl arm, 13A′, appeared as an active catalyst in the presence of a strong proton acceptor, tert-butoxide, emphasizing the crucial participation of the carbanionic ligand in the process. Despite these convincing lines of evidence, although less likely, mechanisms involving oxidative addition of FA to the Rh(I) center cannot be excluded.
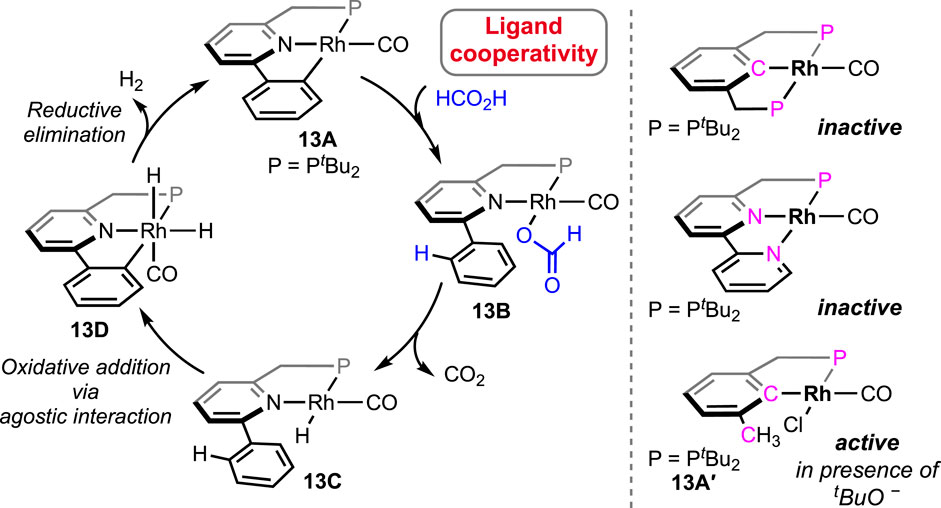
Scheme 9. Mechanistic cycle of (tBuPNC)Rh(CO)-catalyzed FAD (left). Analogous rhodium pincer complexes attempted in FAD (right).
3.3 Cobalt
(PNP)Pincer-bound cobalt complexes were investigated as pre-catalysts by Beller et al. (Figure 6, 14A; Scheme 10, 14A′-A′′) (Zhou et al., 2019). Looking at various pincer catalysts discussed earlier, the presence of a metal-bound strongly anionic group such as the hydride or aryl (in 13A) was found in common. As obvious from the proposed catalytic cycles, these ancillary anionic ligands are critically involved in initializing the cycle by abstracting the proton from FA. On the other hand, the metal–formate can also be a starting point for these catalytic cycles. In this particular case, the cobalt halides were the catalyst precursor, and their activation required the formate salts. Nucleophilic displacement of the cobalt-bound chloride by HCO2−-generated cobalt–formate, which was found to initiate the dehydrogenation cycle (Scheme 10, cycle 10a). Isomerization of 14B to 14C occurred, followed by the CO2 deinsertion. Ultimately, proton transfer in the next step, generating H2, completed the mechanistic cycle. Interestingly, the critical involvement of pincer N-H to stabilize the intermediates through hydrogen bonding was unveiled by a detailed computational study. It is to be noted that possessing the same ligand, ruthenium, and iron pincers (see Figures 2, 5, 2A and 8A-A′, respectively) did not show any such N-H assistance or involvement in the catalysis. We believe that the tetrahedral configuration of the cobalt allowed an optimum fit to attain a geometrically stable transition state, which might be energy-demanding for octahedral pincers (for Ru and Fe). The results from kinetic isotopic studies strongly suggested the decarboxylation step as rate-limiting. In the improvement of aqueous-medium, air-stable cobalt dihalides, 14A′-A″, showed satisfactory conversions. However, to activate these precursors, sodium triethyl borohydride was applied. Then, in 2021, an intelligent ligand design was reported by Cantat et al., accomplishing a co-catalyzed base-free FAD method (Lentz et al., 2021). The closer proximity of N-H to the metal center was attained after adding an amine functionality to the central P-donor of the cobalt (PPP)-pincer (Figure 6, 15A). This structural refinement, as a consequence, successfully ruled out the need for an external base, as shown in cycle 10b (Scheme 10). A DFT study showed distinct N-H participation in the rate-limiting step, and hydrogen bond interactions were firmly proposed to stabilize the intermediates. In general, the FAD pathway involves polar intermediates and transition states, which makes the polar medium more suitable for carrying out the transformation. However, in this case, the efficiency of catalysts was found to be independent of the solvent polarity. This is an uncommon observation, and possibly, the stabilizing H-bond interactions present in the intermediates were strong enough to overcome the necessity of a polar environment.
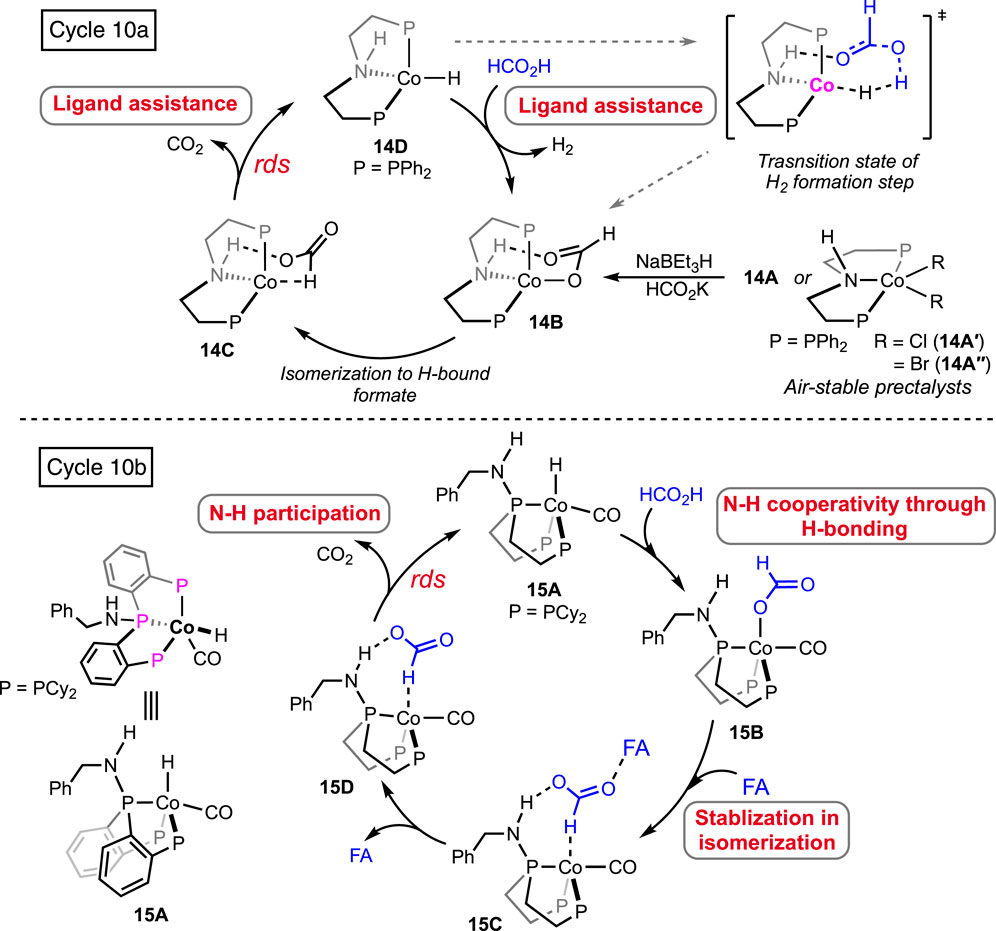
Scheme 10. Proposed catalytic cycle of FA dehydrogenation by cobalt pincer 14A (above, cycle 10a). DFT calculated catalytic pathway by cobalt pincer 15A (below, cycle 10b).
4 Group X (Pd and Ni) metal complexes
4.1 Palladium
Shubina and Belkova et al. developed base-free FAD under milder temperatures (25–50°C) using palladium-based heterobimetallic catalysts (Figure 7; Scheme 11) (Osipova et al., 2023). These complexes behaved as a “transition metal analog of frustrated Lewis pairs.” Among the series of bimetallic complexes, 16A was found to be the most efficient (Figure 7). Collaborative action from both metals was observed, where palladium acted as the acidic half and molybdenum as the basic counterpart. Systematic experimental investigation and evidence from infrared spectroscopy have led to a comprehensive mechanistic cycle (Scheme 11). At first, the proton was abstracted by the anionic counterpart [Mo(CO)3Cp]−, and the simultaneous coordination of HCO2− ended up forming Pd-OCHO (16A–16B), resulting in the dissociation of the metal pair. Next, with the help of the Bronsted acid H-Mo or another molecule of HCO2H, the Pd–formate underwent decarboxylation, generating the hydride 16D. Subsequently, in the last step, Pd-H and H-Mo combined, releasing dihydrogen. A spectral study, along with independent experiments, demonstrated a Pd-OCHO⋅⋅⋅⋅Hδ+ hydrogen bond interaction, boosting the deinsertion of CO2. As shown in the catalytic cycle, the molybdenum unit plays a dual role: (a) the anion, i.e., [Mo(CO)3Cp]−, acts as the proton acceptor and (b) the conjugate acid, [Mo(CO)3Cp]H, actively participates in decarboxylation. Under the additive-free conditions, the use of bi(phosphinite)palladium-only complex 16D, unfortunately, showed poor reactivity (Scheme 11). Moreover, slow decomposition of 16D was noted, forming Pd(0) particles. These findings indicated the importance of metal–metal cooperativity not only in the catalysis but also in balancing the catalyst stability. Interestingly, toluene was used to perform all the studies, and no other solvent was tried, which might suggest the inessentiality of a polar medium to conduct dehydrogenation.
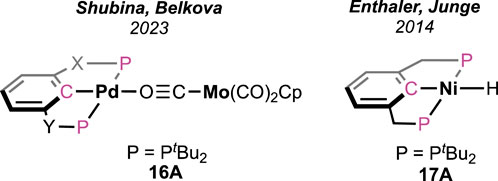
Figure 7. Palladium and nickel pincer complexes for FAD.
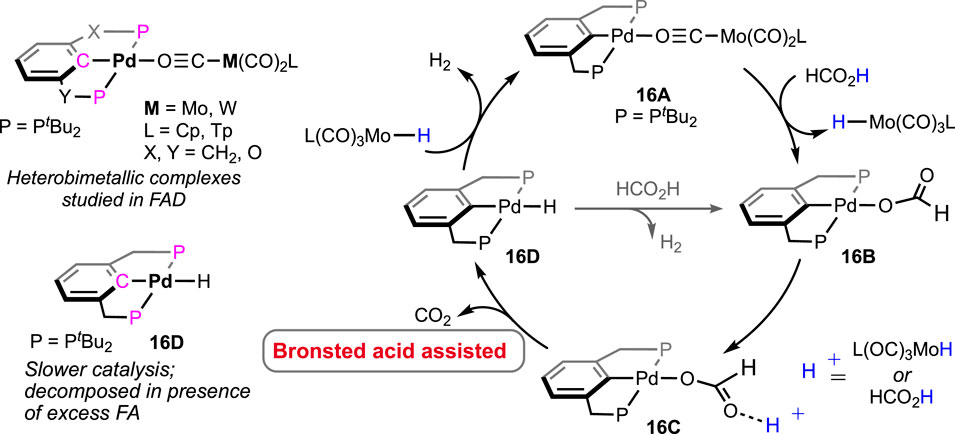
Scheme 11. Various heterobimetallic complexes of palladium and molybdenum/tungsten in FAD (left) and the proposed catalytic cycle (right).
4.2 Nickel
Similar to complex 16D, a bis(phosphonite)-ligated nickel hydride complex (Figure 7, 17A) was investigated by Enthaler and Junge but in the presence of triethylamine (Enthalere et al., 2014). As expected, without amine, no reaction was observed, which verifies the role of an internal or external base in initializing the process. The best catalytic performance was observed in the propylene carbonate medium with dimethyl-n-octylamine as an additive. The formation of the intermediate Ni-OCHO and its decarboxylation are proposed to be the two major steps.
5 Group VI (Re and Mn) metal complexes
5.1 Rhenium
Milstein et al. intensely studied the pyridine-based (PNP)-rhenium complex in base-free FAD (Scheme 12) (Vogt et al., 2014). A low loading (0.03%) of the rhenium pincer successfully generated CO-free H2 at 180°C. When the reaction was carried out in a closed vessel, a maximum TON of 2,600 was measured after 24 h. During the catalysis, rhenium hydride and formate were observed as the two crucial intermediates. On the basis of experimental results, a straightforward two-step catalytic path was proposed; Re-H reacted with FA in the first step to produce the formate complex 18B, which underwent decarboxylation to regenerate the hydride. The high-temperature stability of the catalyst and the production of CO-free H2 are the two major advantages of this method.
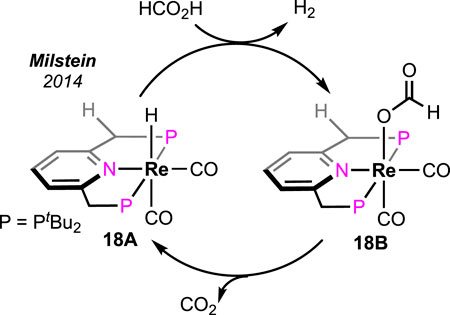
Scheme 12. Base-free formic acid dehydrogenation by the rhenium hydride complex.
5.2 Manganese
In the literature, among diverse homogeneous catalysts, the application of manganese complexes has been extensively studied in recent times. Boncella et al. examined a manganese pincer complex 19A (Figure 8), which is isoelectronic to catalyst 8A (see Figure 5) (Tondreau and Boncella, 2016). Although positive activity in FA dehydrogenation was displayed by the catalyst, formation of CO and water were noticed from simultaneous FA dehydration. As noted during the process, these side products became responsible for catalyst decomposition. In contrast to this observation, only a negligible quantity of CO was formed in the case of iron catalyst 8A. Additionally, for 8A, an accelerating effect by Lewis acid was observed on the decarboxylation rate (Scheme 7, cycle 7b). In the case of 19A, surprisingly, the conversion rate significantly slowed down after the use of LiBF4. In the same year, an analogous (PNP)-based manganese pincer with P-isopropyl substituents was reported by Beller et al. for methanol dehydrogenation (Figure 8, 20A) (Anderez-Fernandez et al., 2016). The same catalyst, 20A, was applied to FA decomposition, and it afforded carbon monoxide-free H2. Moderate turnovers were obtained in a propylene carbonate medium and in the presence of dimethyl octylamine. The proposed mechanism supported the participation of the manganese–nitrogen bond during catalysis, which differs from the (PNP)iron-catalyzed pathway (Scheme 7, cycle 7b). In a separate investigation, the group performed a reactivity comparison between 20A and its N-methylated analog 20A′. Poor catalytic activity was observed for 20A′, suggesting an acceleration in the reaction rate through the cooperation of the central nitrogen in 20A. A point to note is that in the case of similar ruthenium catalysts 5A and 5A′, surprisingly, the outcome was the opposite (see Figure 3; Scheme 5).
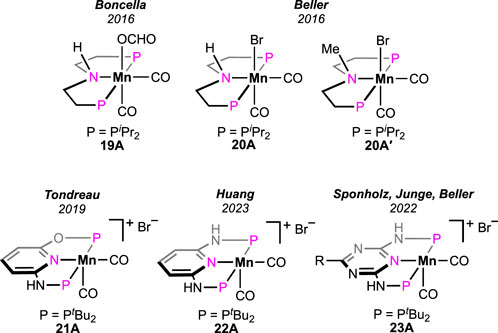
Figure 8. Manganese pincer catalysts for dehydrogenating formic acid.
The catalytic efficiency of the manganese was hugely improved by applying the alternative (PNNOP)-ligand (Figure 8) (Anderson et al., 2019). Complex 21A displayed a high TOF of 8,500 h-1 at 80°C. In the presence of bases such as triethyl and tributyl amine, a mixture of H2 and CO2 was produced in a closed vessel, building the pressure up to 18 atm. Furthermore, the catalyst maintained remarkable stability under the reaction condition even after 10 consecutive runs, projecting its rigidity and recyclability. Although mechanistic details were not described, complex 21B was revealed as the resting state (Scheme 13), which strongly supported metal–ligand cooperativity. However, from the DFT study on 21A-catalyzed FAD, Marino and Prejanò did not find a low-energy pathway involving ligand participation (Marino and Prejanò, 2021). Rather, a typical amine-assisted formation of Mn-OCHO, followed by the CO2 release without any backbone engagement, was found to be energetically favorable. Nevertheless, decarboxylation appeared as the rds, with the energy barrier determined to be 73.0 kJ mol-1. By comparison, structurally similar (tBuPONOP)- and (tBuPNNNP)-ligated complexes exhibited lower efficiency than 21A (Scheme 13; Table 2).
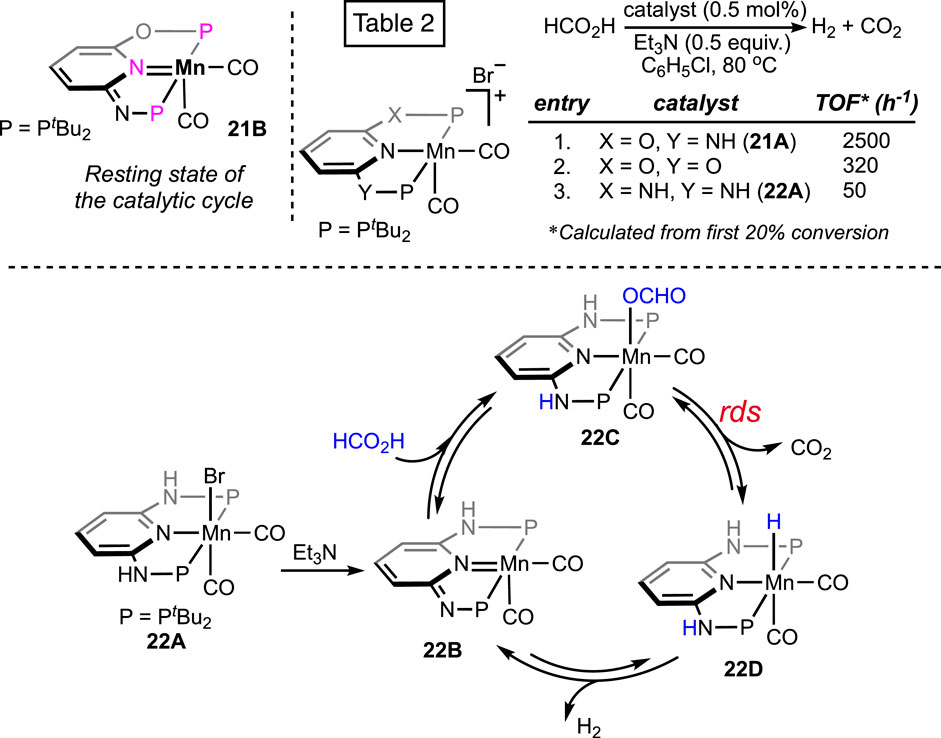
Scheme 13. Proposed resting state of 21A-catalyzed FAD and efficiency comparison of various manganese pincer complexes (above). Plausible base-free formic acid dehydrogenation pathway by the (PNNNP)Mn complex (below).
Recently, the reactivity of the (PNNNP)-Mn pincer (22A) was explored by Huang et al. (Dutta et al., 2023). Although 22A showed poor activity in chlorobenzene (Scheme 13; Table 2, entry 3), interestingly, when switching the solvent to DMSO, the TON increased to 274 at 80°C. With further escalation in temperature to 90°C, the turnover increased up to 15,200. A catalytic cycle involving ligand cooperativity was well established based on a series of experimental and spectroscopic data (Scheme 13). Complex 22B was verified to be the entry point, which was generated through amine-assisted HBr elimination from the manganese bromide 22A. Proposed intermediates 22B–D were also detected in 1H NMR spectroscopy. The presence of reversibility throughout the whole catalytic cycle was further demonstrated. CO2 release was established to be the rate-determining step by the kinetic isotope effect studies.
In 2022, extensive research on CO2 hydrogenation, along with FA dehydrogenation, was reported by Sponholz, Junge, and Beller using a new class of manganese pincer complexes (23A, see Figure 8) (Wei et al., 2022a; Wei et al., 2022b). The unique component in their method was the amino acid lysine, which was found to be essential for catalysis. At 90°C, under aqueous conditions (H2O:THF = 1:1), notably high turnovers were achieved in the presence of an equimolar amount of lysine. In a parallel study, after screening 14 amino acids, lysine and its salt were proven to be highly effective in trapping CO2 in an aqueous solution. Under the FA dehydrogenation condition, interestingly, the presence of a stoichiometric quantity of lysine successfully captured the produced CO2, which resulted in CO2-less H2 as the outcome. The ability to supply pure H2 (up to >99%) directly from the reaction mixture is another advantage of this procedure. Although both lysine (LysH) and its potassium salt (LysK) were found to be equally effective in facilitating catalysis, the latter is more potent in trapping CO2 and, hence, in generating pure H2 (>99%). A detailed mechanistic analysis demonstrated a lysine-mediated dehydrogenation pathway (Scheme 14). Mixing the pre-catalyst 23A with LysK generated the active catalyst (23B). An NMR study supported the deprotonation of N-H on ligand arms by LysK. Using another molecule of LysK, the reaction between 23B and FA produced the Mn–formate (23C), followed by its decarboxylation. In the next step, manganese hydride complex 23D was protonated by lysine (LysH), forming H2 and regenerating the active catalyst. The substantial role of lysine and its salt in proton abstraction-donation throughout the cycle is quite explanatory for its essentiality in this process. In addition, an advantage of these catalysts was their reusability in H2 production from FA. In a follow-up study, under the same reaction condition, a reactivity comparison among various non-aromatic (PNP)-Mn pincers with varying P-substituents was carried out (Scheme 14; Table 3, entries 1–3), along with the effect of a distant ring substituent in catalysts 23A-Aʹʹ (Scheme 14; Table 3, entries 4–6). The group also examined the H2 production from different formate salts (HCO2M; M+ = Li+, Na+, Cs+, NH4+, Mg2+, and Ca2+). All of them were found to behave similarly, and interestingly, glutamic acid was noted to be an effective CO2-trapping agent in addition to lysine.
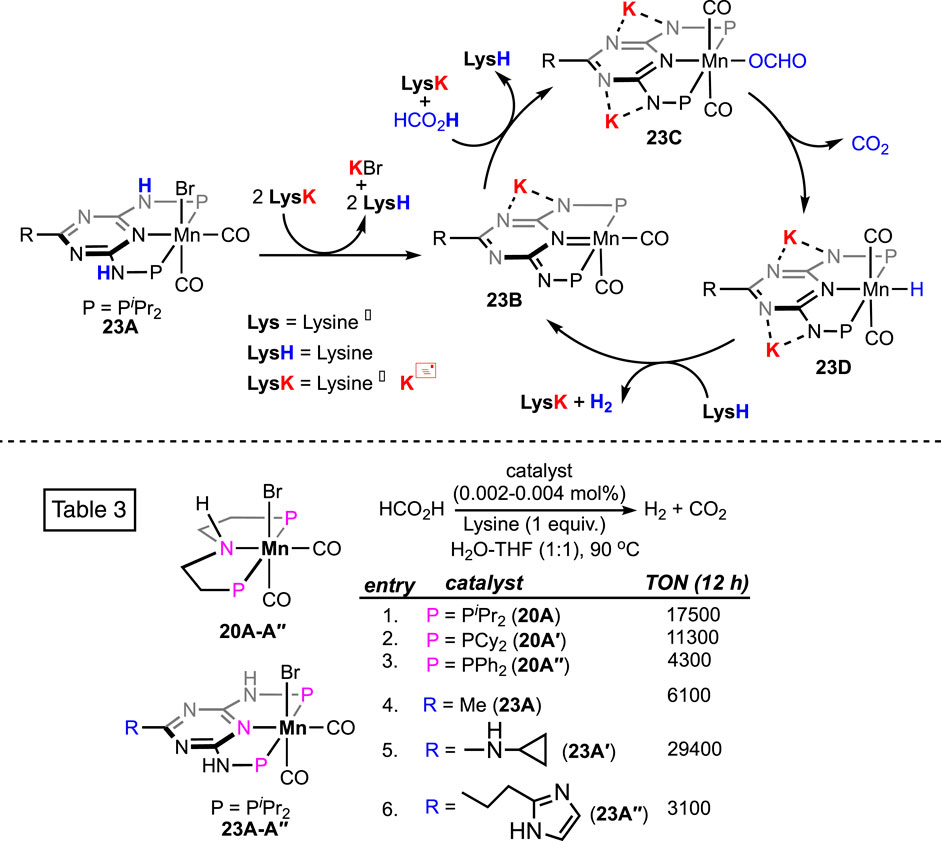
Scheme 14. Proposed FA dehydrogenation pathway by (PNArNNP)Mn complexes in the presence of lysine as additive (above). Manganese pincer catalysts and their reactivity in formic acid dehydrogenation in the presence of lysine (below).
6 Aluminum
Main group metal-based pincer complexes are rare in formic acid dehydrogenation. Berben and Myers prepared an aluminum hydride, 24A, ligated with a pincer having three nitrogen donors (Myers and Berben, 2014; Lu et al., 2016). The complex offered decent reactivity in H2 production. In a stoichiometric reaction between the complex and formic acid, participation of the amide arm was observed (Scheme 15, 24A→24B→24C). Moreover, double protonation of the ligand backbone was noted in the presence of three equivalent FAs, which was not observed for any other pincer complex. The diformate 24C was believed to be the active catalyst, and a β-hydride elimination through a ligand-stabilized transition state was proposed by the group (Scheme 15, 24C→24D). Fu et al. further studied this mechanism using DFT calculations. A non-hydrogen-bonded transition state was found to be associated with a lower energy barrier, suggesting no metal–ligand cooperativity during catalysis.
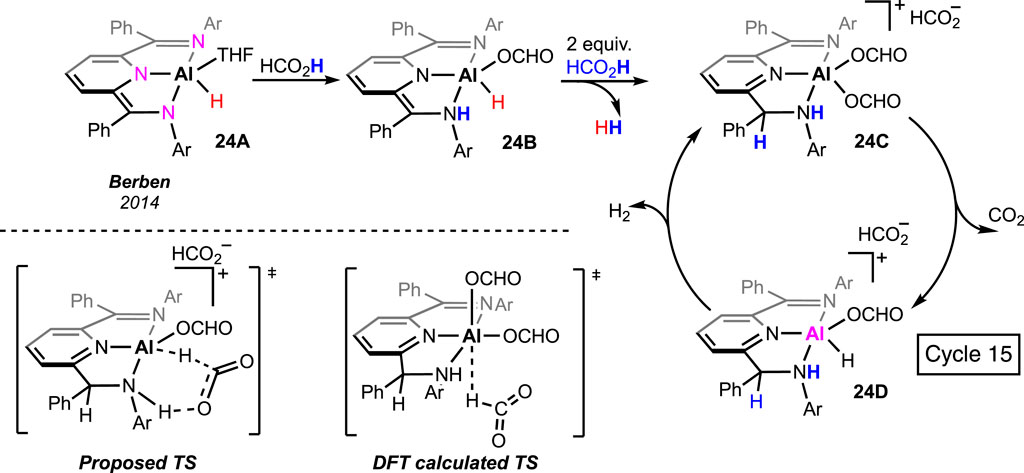
Scheme 15. Formic acid dehydrogenation by aluminum pincer complexes.
7 Challenges and future direction
Over the past decade, the discovery of pincer complexes played a pivotal role in advancing the catalytic FA dehydrogenation processes. More importantly, experimental observations along with theoretical calculation successfully provided a number of distinct catalytic pathways. Responses and outcomes from the variation in electronics and sterics around the active site offered a detailed understanding and the technicalities associated with the process. For instance, functional groups on the ligand backbone or the donor atoms have shown surprising effects on productivity. These insights tremendously helped to identify the existing challenges and their origins. Needless to say, through pincer systems, various strategies were successfully implied, including (i) incorporation of the internal base through ligand modification in numerous ways; (ii) use of Lewis acids, ionic liquids, and amino acids as effective additives; (iii) applying bimetallic frustrated Lewis pair complexes; and (iv) catalyst immobilization on solid surfaces. Although significant progress was made and important details were uncovered, there is still vast scope for improvement.
(1) Except for a few catalytic cycles, the release of CO2 from the metal–formate complex was found to be the rate-limiting step. Therefore, to achieve more efficient dehydrogenation, lowering the activation barrier through the stabilization of the corresponding transition states will be a rational choice. As an example, Schneider and Hazari et al. demonstrated the importance of Lewis acids in facilitating decarboxylation. To the best of our knowledge, fewer attempts have been made to examine CO2 elimination, and it needs further attention. In this regard, the isolation of various M-OCHO complexes and investigating their reactivity toward decarboxylation will be a strategic approach.
(2) The use of organic and costly solvents is another disadvantage of the existing methods. Most catalysts displayed greater efficiency in a highly polar medium. Nevertheless, protic solvents are proven to be ineffective, likely due to the solubility and stability issues of the catalysts. Keeping the objective of cost-effective and environment-friendly methodologies in mind, reactions in water, alcohol, or under solvent-less conditions will be desired. Among existing pincer catalysts, Ru-pincer 6A is capable of dehydrogenating neat FA. Furthermore, in the case of immobilized Ir-pincer 12A, cesium formate was found to be an effective alternative to conduct solvent-less FAD. Clearly, this shows the scope for improvement in managing catalyst-related solubility and stability issues. A straightforward viewpoint could be the introduction of ionic or polar functionality on the ligand backbones.
(3) Although plenty of catalysts have shown their capability in producing CO-free FA decomposition, to obtain pure H2, the separation of CO2 from the product gas mixture requires an additional step. In this respect, a noteworthy discovery was made by Junge and Beller. Following their method, a supply of carbon dioxide-free H2 directly from the reaction mixture was accomplished (pure up to >99%). Applying lysine and its salt to trap the carbon dioxide was the key. Undoubtedly, this strategy unfolded the importance of CO2-capturing materials and their exploration in formic acid dehydrogenation (Wei et al., 2022).
Along with homogeneous catalysts, a variety of heterogeneous systems have been successful in FAD (Pan et al., 2016; Agapova et al., 2019). In this domain, palladium is the most applied transition metal in addition to gold and silver. However, to the best of our knowledge, the use of palladium, gold, and silver complexes under homogeneous conditions is scarce in producing H2 from FA. The use of main-group metals is also rare. Therefore, exploring other metals and different ligand platforms needs to be continued, which eventually leads to novel and improved catalysts.
Author contributions
NK: writing–original draft and writing–review and editing. AA: writing–original draft and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors acknowledge the Research Grant for Engineering Mathematics and Science (RGEMS-2021015) from VIT-AP University.
Acknowledgments
AA thanks Professor Hairong Guan (University of Cincinnati, United States) and Professor Chinmoy Kumar Hazra (Indian Institute of Technology, Delhi, India) for their help and support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agapova, A., Alberico, E., Kammer, A., Junge, H., and Beller, M. (2019). Catalytic dehydrogenation of formic acid with ruthenium-PNP-pincer complexes: comparing N-methylated and NH-ligands. ChemCatChem 11, 1910–1914. doi:10.1002/cctc.201801897
Alig, L., Fritz, M., and Schneider, S. (2018). First-row transition metal (De)Hydrogenation catalysis based on functional pincer ligands. Chem. Rev. 119, 2681–2751. doi:10.1021/acs.chemrev.8b00555
Alrais, L., Gholap, S. S., Dutta, I., Abou-Hamad, E., Chen, B. W. J., Zhang, J., et al. (2024). Highly efficient immobilized PN3P-pincer iridium catalyst for dehydrogenation of neat formic acid. Appl. Catal. B. 342, 123439. doi:10.1016/j.apcatb.2023.123439
Álvarez, A., Bansode, A., Urakawa, A., Bavykina, A. V., Wezendonk, T. A., Makkee, M., et al. (2017). Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 117, 9804–9838. doi:10.1021/acs.chemrev.6b00816
Andérez-Fernández, M., Vogt, L. K., Fischer, S., Zhou, W., Jiao, H., Garbe, M., et al. (2016). A stable manganese pincer catalyst for the selective dehydrogenation of methanol. Angew. Chem. Int. Ed. 129, 574–577. doi:10.1002/ange.201610182
Anderson, N. H., Boncella, J., and Tondreau, A. M. (2019). Manganese-mediated formic acid dehydrogenation. Chem. Eur. J. 25, 10557–10560. doi:10.1002/chem.201901177
Anderson, N. H., Boncella, J. M., and Tondreau, A. M. (2017). Reactivity of silanes with (tBuPONOP)Ruthenium dichloride: facile synthesis of chloro-silyl ruthenium compounds and formic acid decomposition. Chem. Eur. J. 23, 13617–13622. doi:10.1002/chem.201703722
Bernskoetter, W. H., and Hazari, N. (2017). Reversible hydrogenation of carbon dioxide to formic acid and methanol: Lewis acid enhancement of base metal catalysts. Acc. Chem. Res. 50, 1049–1058. doi:10.1021/acs.accounts.7b00039
Bielinski, E. A., Lagaditis, P. O., Zhang, Y., Mercado, B. Q., Würtele, C., Bernskoetter, W. H., et al. (2014). Lewis acid-assisted formic acid dehydrogenation using a pincer-supported iron catalyst. J. Am. Chem. Soc. 136, 10234–10237. doi:10.1021/ja505241x
Curley, J. B., Smith, N. E., Bernskoetter, W. H., Ertem, M. Z., Hazari, N., Mercado, B. Q., et al. (2021). Understanding the reactivity and decomposition of a highly active iron pincer catalyst for hydrogenation and dehydrogenation reactions. ACS Catal. 11, 10631–10646. doi:10.1021/acscatal.1c03347
Curley, J. B., Smith, N. E., Bernskoetter, W. H., Hazari, N., and Mercado, B. Q. (2018). Catalytic formic acid dehydrogenation and CO2 hydrogenation using iron PNRP pincer complexes with isonitrile ligands. Organometallics 37, 3846–3853. doi:10.1021/acs.organomet.8b00534
Dutta, I., Alobaid, N. A., Menicucci, F. L., Chakraborty, P., Guan, C., Han, D., et al. (2023). Dehydrogenation of formic acid mediated by a Phosphorus–Nitrogen PN3P-manganese pincer complex: catalytic performance and mechanistic insights. Int. J. Hydrog. Energy 48, 26559–26567. doi:10.1016/j.ijhydene.2022.04.220
Enthaler, S., Brück, A., Kammer, A., Junge, H., Irran, E., and Gülak, S. (2014). Exploring the reactivity of nickel pincer complexes in the decomposition of formic acid to CO2/H2 and the hydrogenation of NaHCO3 to HCOONa. ChemCatChem 7, 65–69. doi:10.1002/cctc.201402716
Felderhoff, M., Weidenthaler, C., von Helmolt, R., and Eberle, U. (2007). Hydrogen storage: the remaining scientific and technological challenges. Phy. Chem. Chem. Phys. 9, 2643. doi:10.1039/b701563c
Filonenko, G. A., van Putten, R., Schulpen, E. N., Hensen, E. J. M., and Pidko, E. A. (2014). Highly efficient reversible hydrogenation of carbon dioxide to formates using a ruthenium PNP-pincer catalyst. ChemCatChem 6, 1526–1530. doi:10.1002/cctc.201402119
Grasemann, M., and Laurenczy, G. (2012). Formic acid as a hydrogen source – recent developments and future trends. Energy Environ. Sci. 5, 8171. doi:10.1039/c2ee21928j
He, T., Pachfule, P., Wu, H., Xu, Q., and Chen, P. (2016). Hydrogen carriers. Nat. Rev. Mater. 1, 16059. doi:10.1038/natrevmats.2016.59
Jongbloed, L. S., de Bruin, B., Reek, J. N. H., Lutz, M., and van der Vlugt, J. I. (2016). Reversible cyclometalation at RhI as a motif for metal–ligand bifunctional bond activation and base-free formic acid dehydrogenation. Catal. Sci. Technol. 6, 1320–1327. doi:10.1039/c5cy01505g
Kar, S., Rauch, M., Leitus, G., Ben-David, Y., and Milstein, D. (2021). Highly efficient additive-free dehydrogenation of neat formic acid. Nat. Catal. 4, 193–201. doi:10.1038/s41929-021-00575-4
Katerina, S., Tang, C., Vogt, L. K., Junge, H., Dyson, P. J., Beller, M., et al. (2018). Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem. Rev. 118, 372–433. doi:10.1021/acs.chemrev.7b00182
Kothandaraman, J., Czaun, M., Goeppert, A., Haiges, R., Jones, J.-P., May, R. B., et al. (2015). Amine-free reversible hydrogen storage in formate salts catalyzed by ruthenium pincer complex without pH control or solvent change. ChemSusChem 8, 1442–1451. doi:10.1002/cssc.201403458
Lentz, N., Aloisi, A., Thuéry, P., Nicolas, E., and Cantat, T. (2021). Additive-free formic acid dehydrogenation catalyzed by a cobalt complex. Organometallics 40, 565–569. doi:10.1021/acs.organomet.0c00777
Li, Z., and Xu, Q. (2017). Metal-nanoparticle-catalyzed hydrogen generation from formic acid. Acc. Chem. Res. 50, 1449–1458. doi:10.1021/acs.accounts.7b00132
Lu, Q.-Q., Yu, H.-Z., and Fu, Y. (2016). Computational study of formic acid dehydrogenation catalyzed by AlIII -Bis(imino)pyridine. Chem. Eur. J. 22, 4584–4591. doi:10.1002/chem.201504573
Marino, T., and Prejanò, M. (2021). Dehydrogenation of formic acid to CO2 and H2 by manganese(I)–Complex: theoretical insights for green and sustainable route. Catalysts 11, 141. doi:10.3390/catal11010141
Mellmann, D., Sponholz, P., Junge, H., and Beller, M. (2016). Formic acid as a hydrogen storage material – development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 45, 3954–3988. doi:10.1039/c5cs00618j
Mellone, I., Gorgas, N., Bertini, F., Maurizio, P., Kirchner, K., and Luca, G. (2016). Selective formic acid dehydrogenation catalyzed by Fe-PNP pincer complexes based on the 2,6-diaminopyridine scaffold. Organometallics 35, 3344–3349. doi:10.1021/acs.organomet.6b00551
Myers, T. W., and Berben, L. A. (2014). Aluminium–ligand cooperation promotes selective dehydrogenation of formic acid to H2 and CO2. Chem. Sci. 5, 2771–2777. doi:10.1039/c4sc01035c
Onishi, N., Kanega, R., Kawanami, H., and Himeda, Y. (2022). Recent progress in homogeneous catalytic dehydrogenation of formic acid. Molecules 27, 455. doi:10.3390/molecules27020455
Osipova, E. S., Sedlova, D. V., Gutsul, E. I., Nelyubina, Y. V., Dorovatovskii, P. V., Epstein, L. M., et al. (2023). Reactivity of heterobimetallic ion pairs in formic acid dehydrogenation. Organometallics 42, 2651–2660. doi:10.1021/acs.organomet.3c00125
Pan, Y., Pan, C.-L., Liu, S.-L., Li, H., Min, S., Guo, X., et al. (2016). Selective hydrogen generation from formic acid with well-defined complexes of ruthenium and phosphorus–nitrogen PN3 pincer ligand. Chem. Asian J. 11, 1357–1360. doi:10.1002/asia.201600169
Pandey, B., Krause, J. A., and Guan, H. (2022). Iron dihydride complex stabilized by an all-phosphorus-based pincer ligand and carbon monoxide. Inorg. Chem. 61, 11143–11155. doi:10.1021/acs.inorgchem.2c01027
Pandey, B., Krause, J. A., and Guan, H. (2023a). Methyl effects on the stereochemistry and reactivity of PPP-ligated iron hydride complexes. Inorg. Chem. 62, 967–978. doi:10.1021/acs.inorgchem.2c03803
Pandey, B., Krause, J. A., and Guan, H. (2023b). On the demise of PPP-ligated iron catalysts in the formic acid dehydrogenation reaction. Inorg. Chem. 62, 18714–18723. doi:10.1021/acs.inorgchem.3c03125
Piccirilli, L., Lobo Justo Pinheiro, D., and Nielsen, M. (2020). Recent progress with pincer transition metal catalysts for sustainability. Catalysts 10, 773. doi:10.3390/catal10070773
Piccirilli, L., Rabell, B., Padilla, R., Riisager, A., Das, S., and Nielsen, M. (2023). Versatile CO2 hydrogenation–dehydrogenation catalysis with a Ru–PNP/Ionic liquid system. J. Am. Chem. Soc. 145, 5655–5663. doi:10.1021/jacs.2c10399
Schlapbach, L., and Züttel, A. (2001). Hydrogen-storage materials for mobile applications. Nature 414, 353–358. doi:10.1038/35104634
Schüth, F. (2009). Challenges in hydrogen storage. Eur. Phy. J. Spec. Top. 176, 155–166. doi:10.1140/epjst/e2009-01155-x
Singh, A. K., Singh, S., and Kumar, A. (2016). Hydrogen energy future with formic acid: a renewable chemical hydrogen storage system. Catal. Sci. Technol. 6, 12–40. doi:10.1039/c5cy01276g
Szabó, K. J., and Wendt, O. F. (2014). Pincer and pincer-type complexes: applications in organic synthesis and catalysis. John Wiley & Sons.
Tanaka, R., Yamashita, M., Chung, L. W., Morokuma, K., and Nozaki, K. (2011). Mechanistic studies on the reversible hydrogenation of carbon dioxide catalyzed by an Ir-PNP complex. Organometallics 30, 6742–6750. doi:10.1021/om2010172
Tondreau, A. M., and Boncella, J. M. (2016). 1,2-Addition of formic or oxalic acid to –N{CH2CH2(PiPr2)}2-supported Mn(I) dicarbonyl complexes and the manganese-mediated decomposition of formic acid. Organometallics 35, 2049–2052. doi:10.1021/acs.organomet.6b00274
Turner, J. A. (2004). Sustainable hydrogen production. Science 305, 972–974. doi:10.1126/science.1103197
Vogt, M., Nerush, A., Diskin-Posner, Y., Yehoshoa, B.-D., and Milstein, D. (2014). Reversible CO2 binding triggered by metal–ligand cooperation in a rhenium(i) PNP pincer-type complex and the reaction with dihydrogen. Chem. Sci. 5, 2043–2051. doi:10.1039/c4sc00130c
Wang, W.-H., Himeda, Y., Muckerman, J. T., Manbeck, G. F., and Fujita, E. (2015). CO2 hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chem. Rev. 115, 12936–12973. doi:10.1021/acs.chemrev.5b00197
Wei, D., Sang, R., Moazezbarabadi, A., Junge, H., and Beller, M. (2022). Homogeneous carbon capture and catalytic hydrogenation: toward a chemical hydrogen battery system. JACS Au 2, 1020–1031. doi:10.1021/jacsau.1c00489
Wei, D., Sang, R., Sponholz, P., Junge, H., and Beller, M. (2022a). Reversible hydrogenation of carbon dioxide to formic acid using a Mn-pincer complex in the presence of lysine. Nat. Energy 7, 438–447. doi:10.1038/s41560-022-01019-4
Wei, D., Shi, X., Sponholz, P., Junge, H., and Beller, M. (2022b). Manganese promoted (Bi)carbonate hydrogenation and formate dehydrogenation: toward a circular carbon and hydrogen economy. ACS Cent. Sci. 8, 1457–1463. doi:10.1021/acscentsci.2c00723
Yin, Z., Palmore, G. T. R., and Sun, S. (2019). Electrochemical reduction of CO2 catalyzed by metal nanocatalysts. Trends Chem. 1, 739–750. doi:10.1016/j.trechm.2019.05.004
Zell, T., Butschke, B., Ben-David, Y., and Milstein, D. (2013). Efficient hydrogen liberation from formic acid catalyzed by a well-defined iron pincer complex under mild conditions. Chem. Eur. J. 19, 8068–8072. doi:10.1002/chem.201301383
Zhang, Y., MacIntosh, A. D., Wong, J. L., Bielinski, E. A., Williard, P. G., Mercado, B. Q., et al. (2015). Iron catalyzed CO2 hydrogenation to formate enhanced by Lewis acid co-catalysts. Chem. Sci. 6, 4291–4299. doi:10.1039/c5sc01467k
Zhou, W., Wei, Z., Anke, S., Jiao, H., Junge, K., Junge, H., et al. (2019). Cobalt-catalyzed aqueous dehydrogenation of formic acid. Chem. Eur. J. 25, 8459–8464. doi:10.1002/chem.201805612
Keywords: formic acid dehydrogenation, pincer complexes, ligand cooperativity, decarboxylation, transition metal hydride, transition metal formate
Citation: Kumar NS and Adhikary A (2024) Transition metal pincer catalysts for formic acid dehydrogenation: a mechanistic perspective. Front. Chem. 12:1452408. doi: 10.3389/fchem.2024.1452408
Received: 20 June 2024; Accepted: 12 July 2024;
Published: 26 August 2024.
Edited by:
Graham De Ruiter, Technion Israel Institute of Technology, IsraelReviewed by:
Martin Nielsen, Technical University of Denmark, DenmarkSubhash Garhwal, Massachusetts Institute of Technology, United States
Copyright © 2024 Kumar and Adhikary. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anubendu Adhikary, YW51YmVuZHUuYUB2aXRhcC5hYy5pbg==