 Lamya H. Al-Wahaibi1
Lamya H. Al-Wahaibi1 Hesham A. Abou-Zied2Mostafa H. Abdelrahman3Martha M. Morcoss4Laurent Trembleau5*
Hesham A. Abou-Zied2Mostafa H. Abdelrahman3Martha M. Morcoss4Laurent Trembleau5* Bahaa G. M. Youssif6*
Bahaa G. M. Youssif6* Stefan Bräse7*
Stefan Bräse7*- 1Department of Chemistry, College of Sciences, Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia
- 2Medicinal Chemistry Department, Faculty of Pharmacy, Deraya University, Minia, Egypt
- 3Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Al-Azhar University, Assiut, Egypt
- 4Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Nahda University, Beni-Suef, Egypt
- 5School of Natural and Computing Sciences, University of Aberdeen, Aberdeen, United Kingdom
- 6Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut, Egypt
- 7Institute of Biological and Chemical Systems, IBCS-FMS, Karlsruhe Institute of Technology, Karlsruhe, Germany
The present study details the design, synthesis, and bio-evaluation of indoles 3–16 as dual inhibitors of aromatase and inducible nitric oxide synthase (iNOS)with antiproliferative activity. The study evaluates the antiproliferative efficacy of 3–16 against various cancer cell lines, highlighting hybrids 12 and 16 for their exceptional activity with GI50 values of 25 nM and 28 nM, respectively. The inhibitory effects of the most active hybrids 5, 7, 12, and 16, on both aromatase and iNOS were evaluated. Compounds 12 and 16 were investigated for their apoptotic potential activity, and the results showed that the studied compounds enhance apoptosis by activating caspase-3, 8, and Bax and down-regulating the anti-apoptotic Bcl-2. Molecular docking studies are intricately discussed to confirm most active hybrids’ 12- and 16-binding interactions with the aromatase active site. Additionally, our novel study discussed the ADME characteristics of derivatives 8–16, highlighting their potential as therapeutic agents with reduced toxicity.
1 Introduction
Aromatase, a member of the cytochrome P450 family (CYP19), is required for estrogen biosynthesis. CYP19 catalyzes a series of three hydroxylation reactions to transform C19 androgens (androstenedione and testosterone) into aromatic C18 estrogens (estradiol and estrone) (Graham Lorence et al., 1995). It is well established that elevated estrogen levels, both before and after menopause, promote hormone-dependent breast cancer (HDBC) and the metastasis of cancer cells to other parts of the body in women (Zhao et al., 2016). Aromatase inhibition reduces estrogen levels, which proves essential for controlling hormone-sensitive breast cancer. Aromatase is an important enzyme in the production of estrogen, especially after menopause. Aromatase inhibitors have been shown to be effective chemopreventive medicines in HDBC (Riemsma et al., 2010; Maghraby et al., 2023).
Aromatase inhibitors (AIs) are classified structurally into steroidal and nonsteroidal derivatives, which appear to differ in their ability to interact with the enzyme (Mokbel, 2002; Dixon et al., 2003). The most studied nonsteroidal inhibitor was aminoglutethimide [AG, 3-(4-aminophenyl)-3-ethylpiperidine-2,6-dione, compound I], Figure 1 (Brodie et al., 1993), 4-substituted anilines (Hartmann et al., 1992; Chen et al., 2021), imidazole antifungals as well as analogs (Karjalainen et al., 2000; Saxena et al., 2015). AG, The first commercially available nonsteroidal inhibitor, was released in 1981 (Santen and Henderson, 1982). However, due to its lack of specificity against other cytochrome P450 enzymes and inherent toxicity, the use of AG has been limited (Foti, 2023). Many research teams’ investigations have discovered several heterocyclic azoles (Recanatini et al., 2002; Rani et al., 2021; Kharb et al., 2020; Avvaru et al., 2018), such as Fadrozole (II), Anastrozole (III), Letrozole (IV), and Vorozole (V) (Figure 1). These nonsteroidal aromatase inhibitors exhibited improved selectivity and a significant increase in potency.
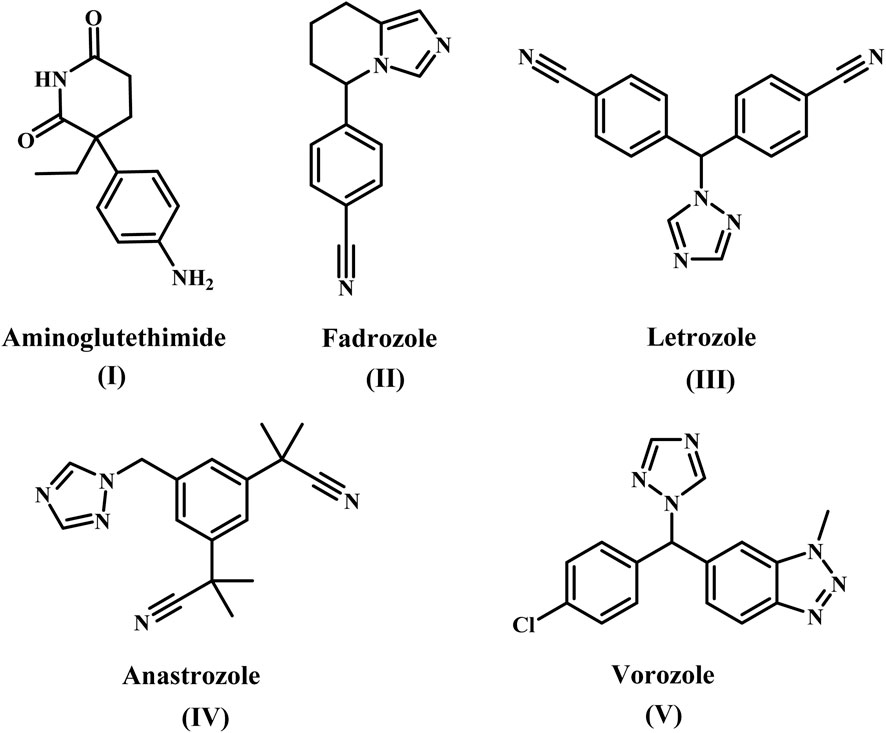
Figure 1. Structure of some nonsteroidal aromatase inhibitors (I-V).
Indole is a valuable scaffold in medicinal chemistry because it is the building block for many important natural substances, such as serotonin, tryptophan, and tryptamine (Kaushik et al., 2013). Even though indole has a variety of therapeutic applications, the indole nucleus received increased attention in the AI class after the approval of Zindoxifene (compound VI, Figure 2), a strong anti-estrogen (Fürstner et al., 1994). Given the importance of indole heterocycles in medicinal chemistry, Prior et al. synthesized and tested a series of novel 2-aryl indoles for aromatase inhibition (Prior et al., 2017). With an IC50 value of 1.61 μM, compound VII (Figure 2) containing a nitrile (CN) group at indole’s C-3 position was the most potent. The CN group lost its potency twice when it was moved to the indole’s C-5 position (IC50 = 3.34 μM). According to SAR, analogs with EWG substituted at position C-3 rather than C-5 exhibited excellent aromatase inhibition activity.
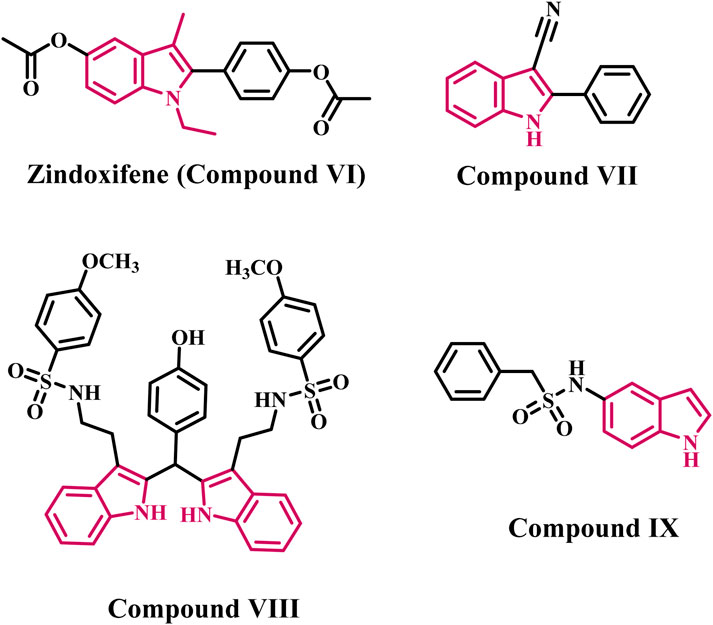
Figure 2. Structures of indole-based aromatase inhibitors (VI-IX).
Pingaew et al. synthesized a series of bis- and tris-indoles with sulfonamides substituted with EWG (electron withdrawing group) and EDG (electron donating group) to develop effective AI (Pingaew et al., 2018). The bis-indole derivative (compound VIII, Figure 2) was discovered to be the most potent derivative, with an IC50 value of 0.7 µM. The biological results showed that activity decreased when EWG was used instead of the methoxy group. Furthermore, it was discovered that bis-indole derivatives were more effective than tris-indoles.
Amoroso and colleagues developed indole aryl sulfonamides as effective AIs (Fantacuzzi et al., 2020). The most potent AI was derivative IX (Figure 2), which had an indole ring at the C-5 position and an IC50 value of 0.16 µM. The potency is lost when aryl sulfonamide is directly substituted with the 3-/6-position of indole. According to biological findings, the attachment position of the indole ring plays an important role in the aromatase inhibition activity of indole aryl sulfonamides.
On the other hand, nitric oxide (NO) is a small biomolecule that has various effects on tumor biology (Pervin et al., 2007). In general, depending on the tumor microenvironment and concentration, NO can influence cell proliferation, migration, and apoptosis (Burke et al., 2013; Shami et al., 1998). Endothelial (eNOS) and neuronal (nNOS) are two constitutive NOS isoforms; eNOS is crucial for maintaining vascular homeostasis, and its uncoupling is linked to the emergence of cardiovascular disorders (Pourbagher-Shahri et al., 2021). Conversely, the nNOS is primarily linked to learning and memory, smooth muscle relaxation, and synaptic plasticity (Wang et al., 1999). Aside from the constitutive NOS isoforms, an inducible NOS isoform (iNOS) is expressed in response to pro-inflammatory stimuli and is thus vital in the immune system. However, iNOS can be overexpressed in a variety of pathological conditions that result in uncontrolled NO production, including inflammatory bowel disease, rheumatoid arthritis, and cancer; thus, iNOS inhibition could be a therapeutic strategy for these diseases (Cinelli et al., 2020; Maccallini et al., 2020). In particular, iNOS overexpression is thought to be a predictor of poor outcomes in TNBC (Triple Negative Breast Cancer) patients, as it is associated with decreased relapse-free survival (Glynn et al., 2010), and iNOS can modulate many potential therapeutic targets for TNBC (Walsh et al., 2016). TNBC proliferation and migration were suppressed in vitro and in vivo by iNOS inhibition using L-NAME (compound X) and 1400 W (compound XI) (phase 1/2 clinical trials, Figure 3) (Granados-Principal et al., 2015). Furthermore, iNOS regulates tumor progression in an inflammatory environment by modulating the EGFR/MAPK pathway (Garrido et al., 2017). As a result, safer iNOS inhibitors with improved pharmacokinetic properties are desired to obtain new valuable therapeutic tools for treating breast cancer.
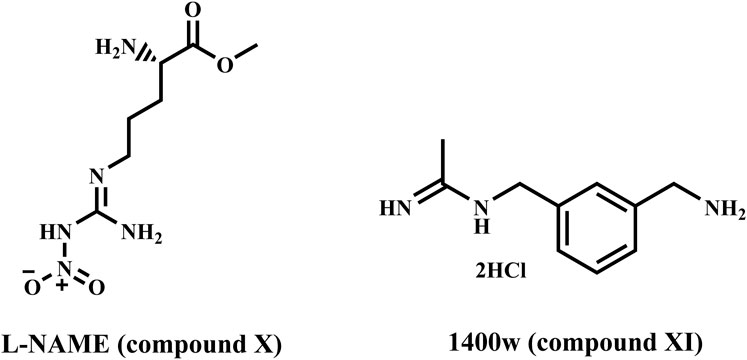
Figure 3. Structures of compounds X and XI.
Additionally, apoptosis (programmed cell death) is a crucial control mechanism that triggers cell death when DNA damage surpasses the capability of the repair processes (Denmeade and Isaacs, 1996). Apoptosis, which is a normal factor of development, helps to control cell number and proliferation. Defects in apoptotic signalling contribute to several human disorders, including cancer. These flaws allow tumor cells to live longer than expected, reducing their reliance on exogenous survival factors and shielding them from oxidative stress and hypoxia, resulting in tumor growth and expansion (Cotter, 2009). These flaws allow for the accumulation of genetic changes that promote angiogenesis, disrupt cell proliferation, interfere with differentiation, and increase invasiveness during tumor formation. Therefore, restoring normal apoptotic equilibrium is a reliable practice for cancer treatment (Plati et al., 2008).
In our ongoing search for anticancer and chemopreventive agents (Al-Wahaibi et al., 2020; Al-Wahaibi et al., 2023a; Al-Wahaibi et al., 2023b; Al-Wahaibi et al., 2023c; Gomaa et al., 2022), we synthesized and tested a series of indole-based scaffold derivatives (3–16) against aromatase and iNOS. The newly synthesized compounds fall into two categories (Figure 4): scaffold A compounds that are indole-2-carboxamide derivatives (3, 4, 8, and 13) and scaffold B compounds that are pyrazino [1,2-a]indol-1(2H)-ones (5–7, 9, 10–12, and 14–16). The newly synthesized compounds will be tested in vitro against a panel of four cancer cell lines as antiproliferative agents. All the newly synthesized compounds were then tested for aromatase inhibitory activity. The most potent aromatase inhibitors will be tested for their ability to inhibit iNOS. Moreover, the apoptotic potential of the most active derivatives was evaluated. Finally, docking analysis and ADME experiments were conducted for the most potent derivatives.
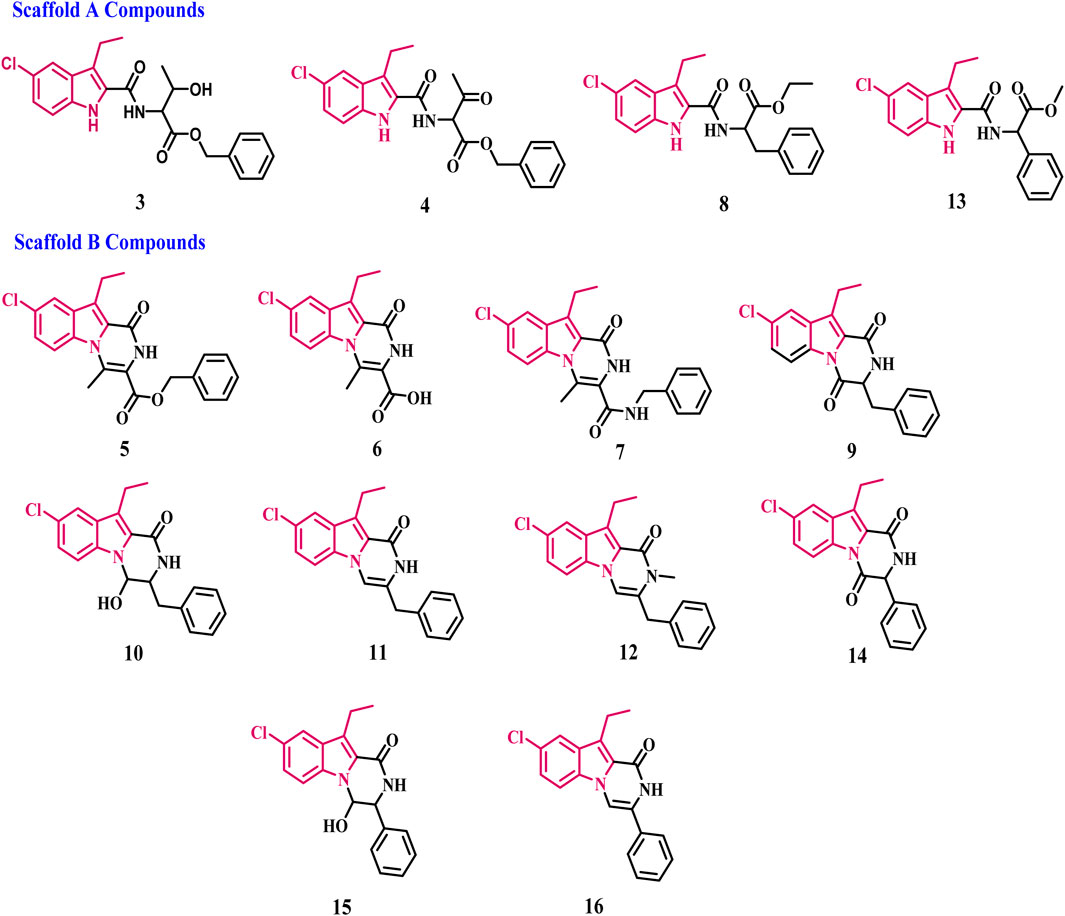
Figure 4. Structures of new compounds 3–16.
2 Results and discussion
2.1 Chemistry
Scheme 1 depicts the synthesis of the key intermediates and target compounds 3–7. Compound 1 was reacted with L-threonine benzyl ester 2 in the presence of BOP and DIPEA as coupling reagents, yielding compound 3 as a white solid after chromatography on silica gel with EtOAc/hexane (2:3) as an eluent. The oxidation of compound 3 with Dess-Martin periodinane reagent produced a crude product 4 that was used in the following step without further purification. Refluxing 4 in toluene with PTSA yields crude product purified by flash chromatography using EtOAc and n-hexane to afford 5 in 82% yield as a white solid. The structure of 5 was validated using 1H NMR, 13C NMR, and HRMS spectroscopy. The 1H NMR spectrum of 5 revealed the pyrazine NH characteristic signal in the form of a singlet of one proton at δ 8.41 ppm corresponding to NH and a singlet signal of benzyl methylene protons at δ 5.36 ppm (CH2Ph). The spectrum also revealed a singlet signal of 3H at δ 3.19 ppm, corresponding to the CH3 group, as well as protons from the ethyl group in the form of a quartet signal (2H) at δ 3.30 ppm and a triplet signal (3H) at δ 1.30 ppm. The 13C NMR spectrum of 5 revealed two signals at δ 162 and δ 156 ppm for (ester C=O) and (NH-C=O), respectively, and aromatic carbons. The 5 structure was also validated by HRESI-MS with the [M + H]+ ion at m/z 393.1013. Compound 5 was hydrolysed with aqueous NaOH to yield carboxylic acid 6. Carboxamide 7 was synthesized by coupling carboxylic acid 6 with benzylamine in the presence of DIPEA in DCM using BOP as the coupling reagent.
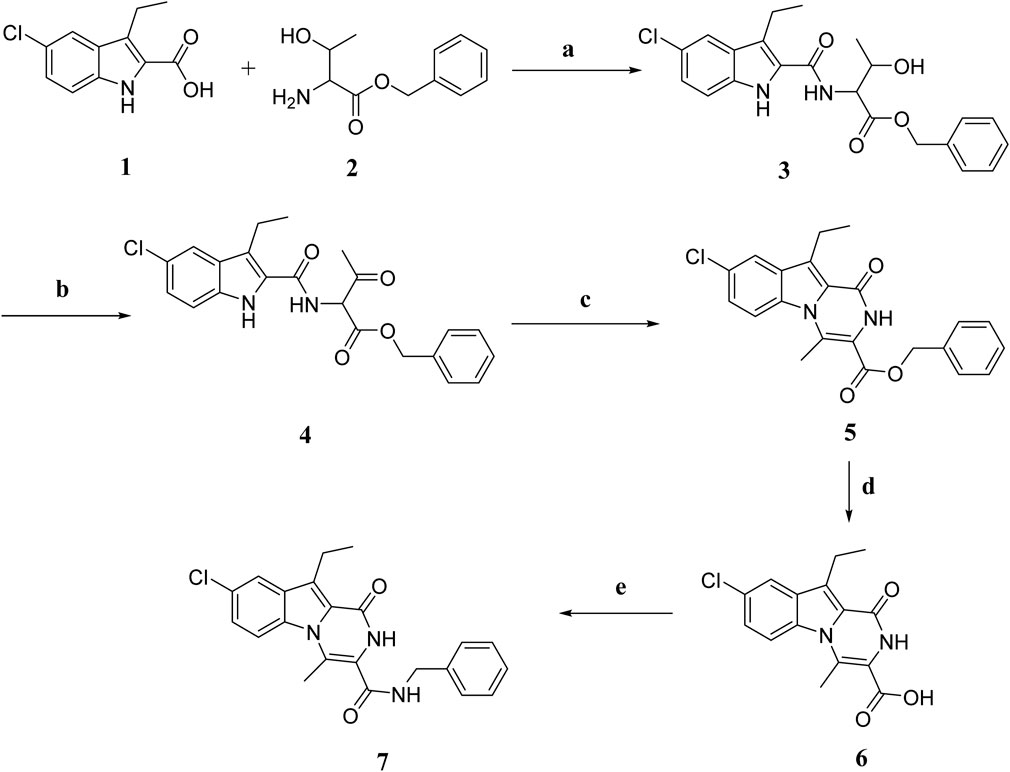
Scheme 1. Synthesis of compounds 3–7. Regents and reaction conditions: (A) POB, DIPEA, r. t., overnight, 91%; (B) Dess-Martin periodinane, DCM, r. t. 5h; (C) PTAS, Toluene, reflux overnight, 82%; (D) 5% NaOH, 45°C, overnight, 96%; (E) benzylamine, POB, DIPEA, r. t. overnight, 81%.
Compounds 6 and 7’s structures were confirmed using 1H NMR, 13C NMR, and HRESI-MS spectroscopy. Compound 7’s 1H NMR spectrum revealed the presence of two singlet signals, one at 10.74 ppm of pyrazine NH and the other at δ 9.05 ppm of NHCH2Ph, the characteristic signals of ethyl group in the form of a quartet at δ 3.25 ppm (2H) and triplet at δ 1.25 ppm (3H), and a singlet signal of CH3 group at δ 2.73 ppm (3H). Additionally, the spectrum revealed the presence of aromatic proton-specific signals. HRESI-MS m/z of 7 calcd for [M-H]- C22H19ClN3O2: 392.1171, found: 392.1168.
The synthesis of compounds 8–12 is outlined in Scheme 2. Compound 8 was synthesized using compound 1 and ethyl phenylalanine (compound 1a) as described in the general procedure for the synthesis of compound 3 (Scheme 1), yielding 8 as an oily product after purification by flash chromatography on silica gel short column using EtOA and n-hexane. Compound 9 is obtained by cyclizing 8 in toluene with PTSA. It is then purified using flash chromatography with EtOAc and n-hexane to yield 9 as a white solid in 75% yield. Various spectroscopic analysis methods were used to confirm the structure of compound 9. The 1H NMR spectrum 9 revealed a pyrazine NH characteristic signal at δ 8.31 ppm and a methine proton multiplet signal at δ 4.81–4.79 ppm (NHCH). The spectrum also revealed two doublet signals of one proton each at δ 3.29 and 3.10 ppm, corresponding to the CH2Ph group and ethyl and aromatic protons. The 13C NMR spectrum of 9 revealed two signals for (CHC = O) and (NH-C = O), respectively, at δ 165.56 and δ 157.82 ppm and aromatic carbons. Compound 10 was obtained in quantitative yield by reducing 9 with NaBH4 in aqueous ethanol. The 1H NMR spectrum of 10 revealed the presence of a doublet signal at δ 7.69 (1H) corresponding to CHOH and a singlet signal at δ 4.89 of the hydroxyl group (CHOH). Moreover, the 13C NMR revealed the presence of only one carbonyl carbon signal at δ 161.50 of (NH-C=O). HRESI-MS m/z of 10 calcd for [M + H]+ C20H20ClN2O2: 355.1208, found: 355.1210 validated compound 10. Compound 12 was synthesized from 10 in two steps: dehydration with PTSA in toluene to yield 11, then alkylation with MeI in DMF in the presence of NaI to afford 12 in 88% yield as a white solid. The compound 12 1H NMR spectrum revealed a singlet signal at δ 3.86 (2H, PhCH2), while the CH3 group signal overlapped with that of the ethyl group signal as a multiplet signal at δ 3.26–3.19 (5H, CH3, CH2CH3) and a triplet signal at δ 1.25 (3H, CH2CH3). HRESI-MS with the [M + H]+ ion at m/z 351.1257 also validated the 12 structure.
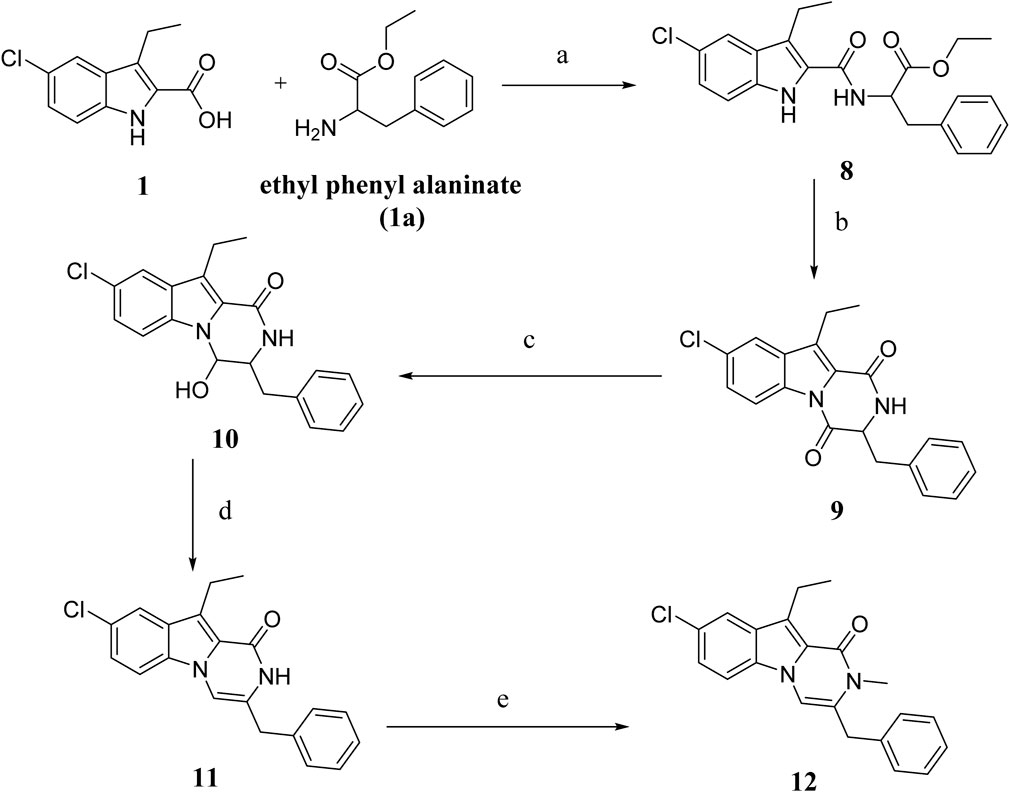
Scheme 2. Synthesis of compounds 8–12 Regents and reaction conditions: (A) POB, DIPEA, r. t., overnight, 83%; (B) PTAS, Toluene, reflux overnight, 75%; (C) NaBH4, aq. EtOH, r. t. 1h, 93%; (D) PTAS, Toluene, reflux 4 h, 60%; (E) CH3I, NaI, NaH, DMF, r. t. overnight, 88%.
Scheme 3 depicts the synthesis of compounds 13–16. Compound 13 was synthesized from compound 1 and methyl-2-amino-2-phenylacetate (compound 1b) using the typical process for compound 8 synthesis (Scheme 2), producing 13 as a solid product. By cyclizing 13 in toluene with PTSA, compound 14 is obtained as a white solid. Compound 15 was obtained in 89% yield by reducing 14 in aqueous ethanol with NaBH4, followed by dehydration with PTSA in toluene to get 16. Various spectroscopic methods of analyses were employed to confirm the structures of compounds 13–16 (Supplementary Material).
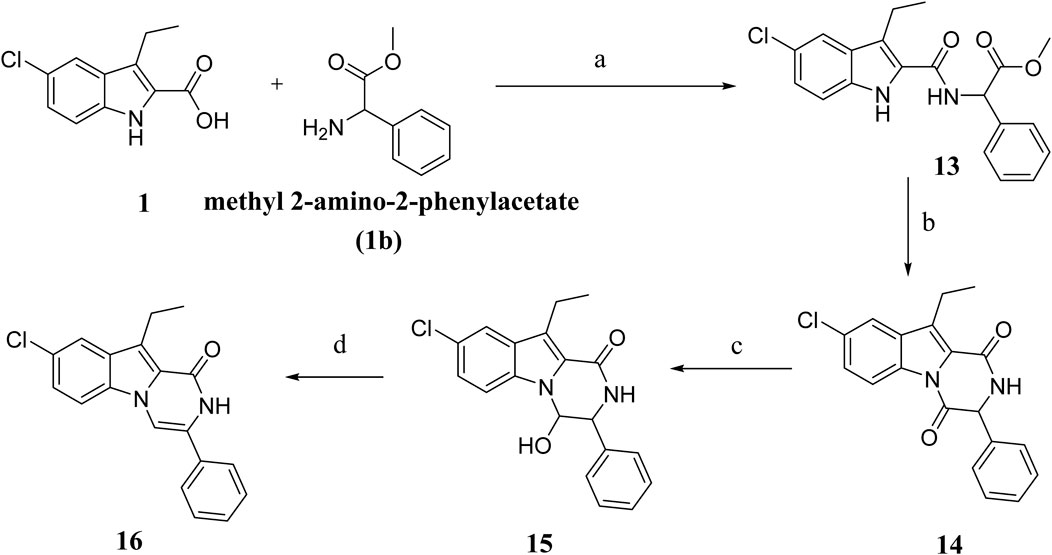
Scheme 3. Synthesis of compounds 13–16 Regents and reaction conditions: (A) POB, DIPEA, r. t., overnight, 94%; (B) PTAS, Toluene, reflux overnight, 85%; (C) NaBH4, aq. EtOH, r. t. 1h, 89%; (D) PTAS, Toluene, reflux 4 h, 88%.
2.2 Biology
2.2.1 Cell viability assay
This test investigates the effects of the new compounds 3–16 on normal cell lines to determine their safety. The normal human mammary gland epithelial (MCF-10 A) cell line was used to test the viability of the tested compounds. After 4 days of incubation on MCF-10 A cells within 50 µM of each investigated compound, cell viability was determined using the MTT assay (Gomaa et al., 2020; Marzouk et al., 2020). Table 1 outcomes demonstrate that none of the compounds examined were cytotoxic, and all compounds showed more than 89% cell viability at 50 µM.
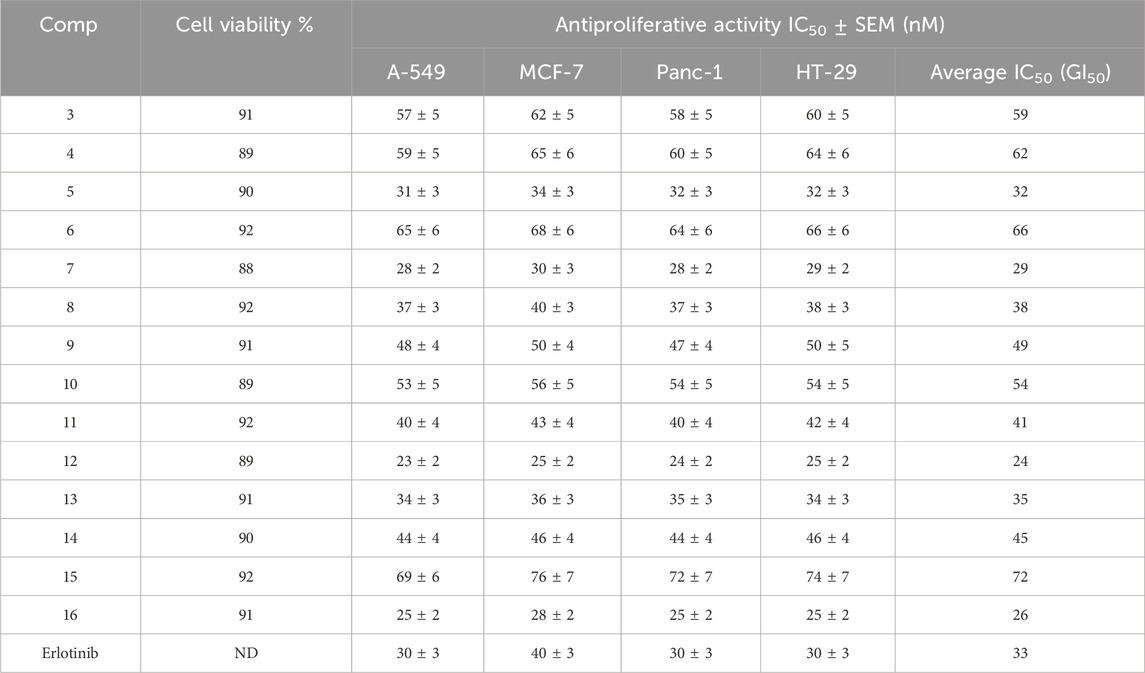
Table 1. Cell viability assay and antiproliferative action of compounds 3–16.
2.2.2 Antiproliferative assay
The MTT assay was used to investigate the antiproliferative activity of new compounds 3–16 versus four human cancer cell lines using erlotinib as a control: colon (HT-29), pancreatic (Panc-1), lung (A-549), and breast (MCF-7) cancer cell lines (Youssif et al., 2022; Mahmoud et al., 2022). Table 1 displays the median inhibitory concentration (IC50) and GI50 (average IC50) against the four cancer cell lines.
Generally, the investigated compounds 3–16 had significant antiproliferative activity with GI50 values ranging from 24 nM to 72 nM versus the four cancer cell lines evaluated, compared to the standard Erlotinib, which had a GI50 value of 33 nM. Compounds 5, 7, 8, 12, 13, and 16 were the most potent six derivatives, with GI50 values ranging from 24 nM to 38 nM, making compounds 5, 7, 12, and 16 (GI50 = 32, 29, 24, and 26, respectively) more potent than Erlotinib (GI50 = 33 nM), and even more potent than Erlotinib against breast (MCF-7) cancer cell lines. Compound 12 (R = 3-benzyl) was the most potent of the newly synthesized derivatives 3–16, with a GI50 value of 24 nM, 1.4-fold higher than the reference Erlotinib (GI50 = 33 nM). Based on the results, the type of substitution detected on the nitrogen atom at position two of the pyrazino [1,2-a]indol-1(2H)-one moiety appears to be crucial for activity. Compound 11 (R = 3-benzyl), which has the same backbone as compound 12 but with a free NH group at position 2, displayed a GI50 of 41 nM (1.7-fold less potent than 12), demonstrating that the N-methyl group at position two is important for the antiproliferative activity.
Moreover, the 3-position substitution of the pyrazino [1,2-a]indol-1(2H)-one moiety is critical for activity. Compounds 7 (R = 3-benzylcarboxamide) and 16 (R = 3-phenyl) had GI50 values of 29 nM and 26 nM, respectively, indicating they were at least 1.4-fold more effective than compound 11. These data indicate that the third position substitution significantly affects the antiproliferative activity of these compounds, with the highest activity shown in compounds with a benzyl substitution, followed by carboxamide and phenyl substitutions.
Another area of interest is the difference in efficacy between open-ring compounds and their closed-ring counterparts. Open-ring compounds 8 and 13 showed promising antiproliferative effects with GI50 values of 38 nM and 35 nM, respectively, being equipotent to the reference Erlotinib (GI50 = 33 nM), whereas compounds 3 and 4 showed weak activity with GI50 values of 59 nM and 62 nM, respectively. Based on these findings, it is possible to conclude that the kind of functional groups and the type of substituents dictate the nature of these compounds’ activity. Finally, Compounds 6, 10, and 15 exhibited low potency, as seen by their GI50 values of 66, 54, and 72 nM, respectively. This suggests that having a high level of hydrophilicity does not confer any advantage for antiproliferative activity.
2.2.3 Assay for aromatase inhibitory action
Using ketoconazole and letrozole as reference medications, compounds 5, 7, 8, 12, 13, and 16 were examined for their ability to inhibit aromatase, a possible target for their antiproliferative effect (Maghraby et al., 2023). Table 2 displays the results as IC50 values. The examined compounds 5, 7, 8, 12, 13, and 16 displayed significant aromatase inhibitory effect with IC50 values ranging from 10 nM to 33 nM, in comparison to the reference drugs ketoconazole (IC50 > 100 nM) and letrozole (IC50 = 2 nM). The assay’s results align with the findings of the antiproliferative assay, which demonstrated that compounds 7, 12, and 16, the most potent antiproliferative derivatives, were also the most potent aromatase inhibitors, with IC50 values of 15 nM, 10 nM, and 12 nM, respectively.
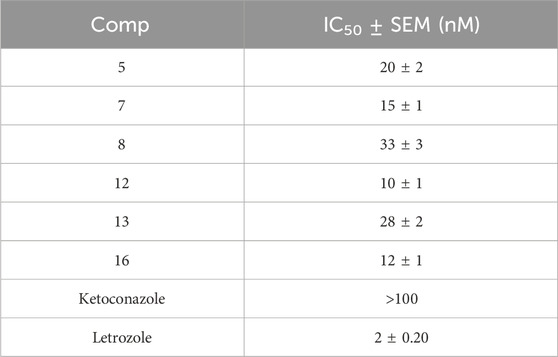
Table 2. Anti-aromatase activity of compounds 5, 7, 8, 12, 13, and 16.
Once again, compound 12 was the most potent aromatase inhibitor with an IC50 value of 10 ± 1 nM, at least 10-fold more potent than the standard Ketoconazole but 5-fold less potent than the Letrozole. Compound 16 was the second most active aromatase inhibitor, with an IC50 value of 12 ± 1 nM, like compound 12. Finally, compounds 5, 8, and 13 demonstrated good inhibitory action with IC50 values of 20 ± 2, 33 ± 3, and 28 ± 2 nM, respectively. These findings indicate that antiproliferative and anti-aromatase activity are linked and that any parameter that affects one has the same effect on the other.
2.2.4 Nitric oxide synthase inhibition
The L-citrulline assay method with fluorometric detection was used to test compounds 5, 7, 12, and 16 (most potent anti-aromatase derivatives) for their ability to inhibit iNOS (Carrión et al., 2023). The investigated compounds were tested at 1 µM against the iNOS and 10 µM against the eNOS to determine their isoform selectivity. Because of its critical role in cardiovasculature, inhibiting the constitutive enzyme eNOS should be avoided. Table 3 shows the results, which were expressed as enzyme percent inhibition compared to 1400 W as the positive control (100% inhibition at 10 µM).
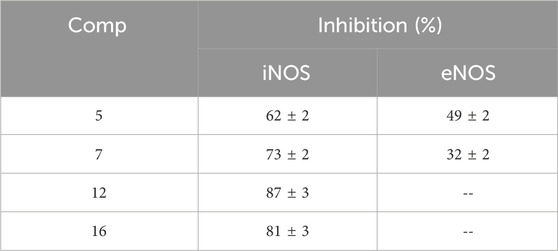
Table 3. Enzyme percent inhibition of compounds 5, 7, 12, and 16 against iNOS and eNOS.
Results revealed that compounds 7, 12, and 16 were effective iNOS inhibitors, with enzyme percent inhibition values of 73, 87, and 81, respectively. Compound 7 had moderate inhibitory action against eNOS isoform, whereas compounds 12 and 16 were completely inactive against eNOS. Moreover, compound 5 was active against eNOS with a percent % inhibition value of 49%, even though it maintained a good inhibition of iNOS (62%), Table 3.
As a result, compounds 12 and 16 were chosen for IC50 evaluation (Table 4). Interestingly, derivative 12 had an IC50 value against iNOS of 0.075 µM, compared to reference 1,400 W (0.082 µM), as well as excellent isoform selectivity against eNOS (>670). Additionally, despite being less potent than reference 1400W, compound 16 demonstrated its efficacy against iNOS (IC50 = 0.098 µM) while being inactive against eNOS. Based on their inhibition potency, derivatives 12 and 16 were thought to be possible aromatase inhibitors with iNOS inhibitory action.
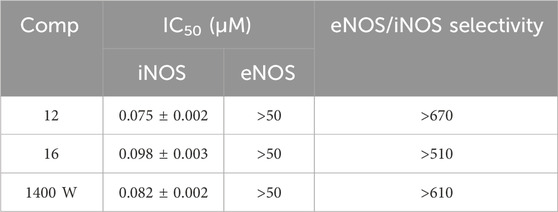
Table 4. IC50 values of compounds 12 and 16 against iNOS and eNOS.
2.2.5 Apoptosis assays
Compounds 12 and 16, the most potent derivatives in all in vitro investigations, were examined for their ability to initiate the apoptosis cascade and demonstrate proapoptotic activity.
2.2.5.1 Caspase-3 and caspase-8 expression levels
When cells get specific signal instructions, they undergo apoptosis, which results in several significant changes. Caspases, considered the main workers in apoptosis, are activated early in the process, cleaving critical cellular components such as nuclear proteins like DNA repair enzymes or structural proteins found in the cytoskeleton that are required for optimal cellular function. Caspases can activate DNases, which break down nuclear DNA (Chauhan et al., 2001; Kroemer and Reed, 2000). Compounds 12 and 16 were tested as caspase-3/8 activators against the human epithelial (A-594) cancer cell line (Youssif et al., 2019), with the results presented in Table 5.
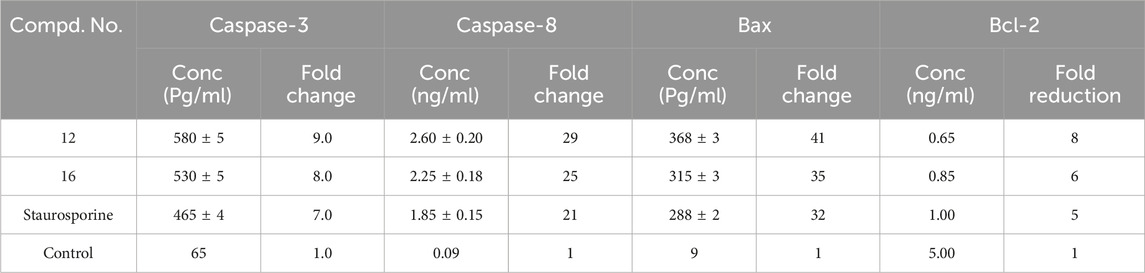
Table 5. Apoptotic markers assays of compounds 12 and 16.
In comparison to the untreated control cells, compound 12 treatment at its IC50 concentration increased the expression levels of active caspase-3 and caspase-8 in A-594 cells by 9 and 29 folds, respectively (Table 5; Figure 5). Compound 16 treatment causes induction of caspase-3 and caspase-8 levels to be 8 and 25 times greater than those of untreated control cells. In all cases, compounds 12 and 16 were more effective caspase-3 and 8 activators than the control Staurosporine.
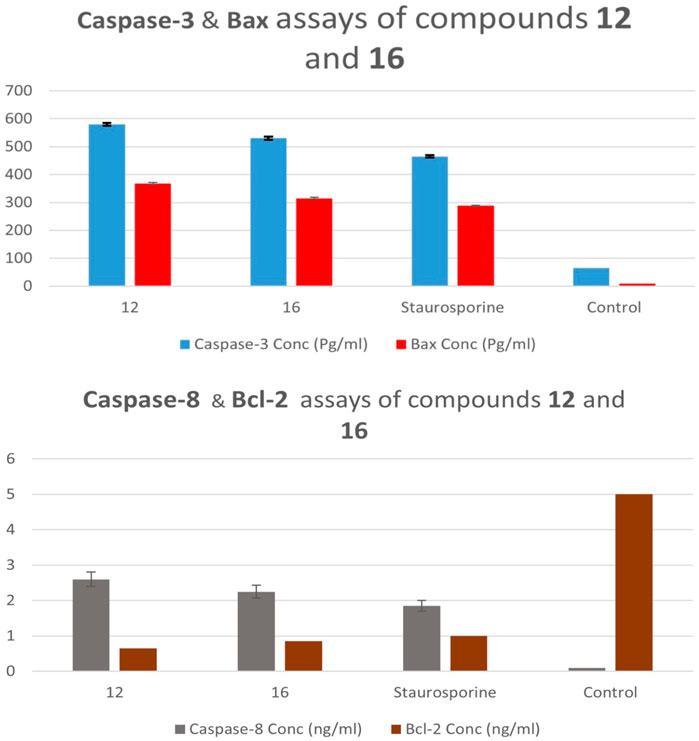
Figure 5. Caspase 3, 8, Bax, and Bcl-2 expression levels of compounds 12 and 16.
2.2.5.2 Proapoptotic BAX and anti-apoptotic Bcl-2 expression levels
Tumor cells can develop resistance to apoptosis through mutation or downregulation of proapoptotic proteins (e.g., Bax and Bak) or over-expression of anti-apoptotic proteins (e.g., Bcl-2 and Bcl-xL), which promote cell survival and are considered a hallmark in more than half of all cancers. This makes cancer cells more resistant to apoptotic stimuli and conventional cytotoxic anticancer drugs (Miyashita et al., 1994).
In the current study, treating lung (A-549) cancer cell lines with compounds 12 and 16 at IC50 values increased pro-apoptotic Bax expression levels by 41 and 35 folds, respectively, while decreasing anti-apoptotic Bcl-2 expression levels by approximately 8 and 6 folds, Table 5 and Figure 5. As a result, Compounds 12 and 16 significantly increased the Bax/Bcl-2 ratio compared to the untreated control cells.
2.3 Docking study into aromatase
This research conducted a computational docking analysis to explore the binding interactions of the most active derivatives 7, 12, and 16 with the human placental aromatase cytochrome P450 (CYP19A1). Aromatase, a crucial enzyme in estrogen biosynthesis, belongs to the cytochrome P450 family and features a heme group at its catalytic centre, essential for its enzymatic activity. The active site is located within a deep hydrophobic pocket, surrounded by key residues such as Met374, Arg115, Ala306, Val370, and Val373, which are critical for substrate binding and catalysis. These residues not only stabilize the binding of substrates and inhibitors but also play a significant role in the enzyme’s selectivity and function. The careful arrangement of these residues around the heme group ensures precise catalytic activity, making aromatase a pivotal target in breast cancer therapy. The Discovery Studio software was used for this study, combined with the crystallographic structure of human placental aromatase cytochrome P450, which was co-crystallized with testosterone (PDB: 5JKW) (Ibrahim et al., 2020; Shaykoon et al., 2020; Bhat et al., 2022).
The molecular systems underwent energy minimization using the OPLS-AA force field, a method essential for ensuring conformational stability (Bernacki et al., 2005). Before starting the docking experiment, careful preparation of the protein structure was carried out to enhance accuracy. This included protonation steps, which significantly improved the reliability of the following docking analysis. The effectiveness of the docking process was validated by re-docking the co-crystallized testosterone into the active site of the aromatase enzyme, which yielded an S score of −8.43 kcal/mol. The S score is a measure used to assess the binding strength between compounds and their target protein, with lower scores indicating stronger binding affinities. Notably, this result was marked by a crucial hydrogen bond interaction between the hydroxyl group of testosterone and the amino acids Met374 and Arg115. Additionally, there were multiple interactions with Ala306, Val370, and Val373, highlighting their importance in ligand stabilization (Figure 6).
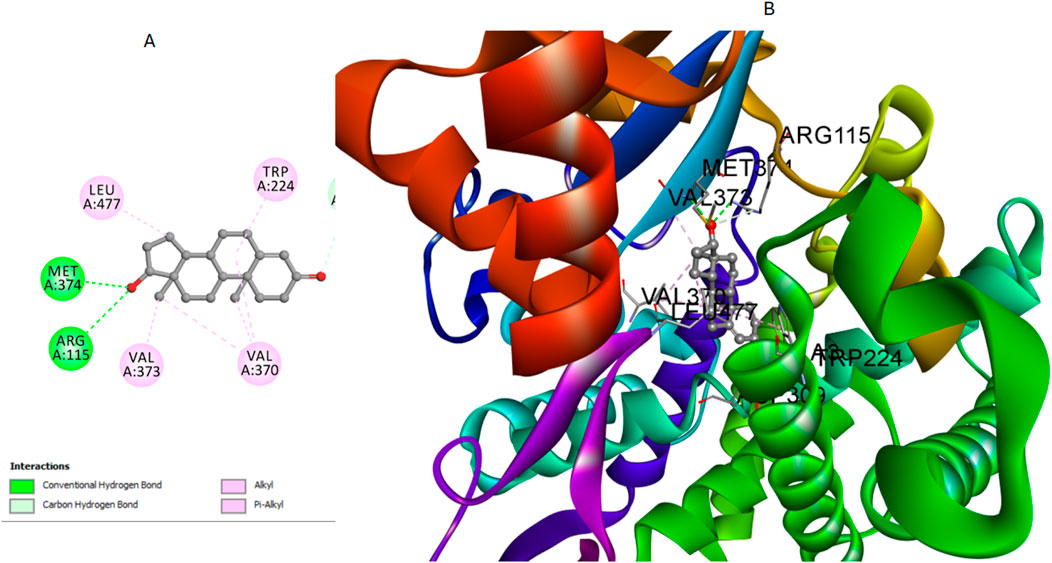
Figure 6. Cartoon and surface representations of the aromatase binding pocket docked with testosterone (PDB: 5JKW). The model shows the hydrogen bond interaction between the hydroxyl group of testosterones and the amino acids Met374 and Arg115, with additional interactions with Ala306, Val370, and Val373.
To further illustrate the binding interactions, cartoon and surface representations of the aromatase binding pocket are provided, highlighting the spatial arrangement of these critical residues and their interaction with the docked ligands. These visual models help in understanding how the structural features of the protein contribute to its interaction with different ligands (Figure 6).
The analysis of docking scores revealed that hybrid 12, which had the highest in vitro aromatase activity, showed a score of −7.86 kcal/mol. In contrast, hybrid 7, with the lowest in vitro aromatase activity, registered a score of −6.95 kcal/mol. Additionally, the docking score for compound 16, when tested against aromatase, was determined to be −7.55 kcal/mol.
Regarding the interactions between the compounds and the aromatase enzyme, compound 12 demonstrated significant interactions. Hydrogen bonds are fundamental in stabilizing the ligand within the active site of the enzyme, and their strength is largely dependent on both the bond distance and the bond angle. The carbonyl oxygen in its pyrazine ring acted as a hydrogen bond acceptor, forming a bond with crucial Met374 residue. The hydrogen bond distance between the carbonyl oxygen of compound 12 and the hydrogen of the Met374 side chain was measured at 2.50 Å. The bond angle formed was calculated to be approximately 167°, indicating a nearly linear and therefore strong hydrogen bond, which is optimal for interaction stability. Additionally, the aromatic rings of compound 12 were involved in several hydrophobic interactions with key residues in the binding pocket. Notably, these interactions included pi-pi T-shaped interactions with Phe134 and alkyl/pi-alkyl interactions with Val370, Leu372, and Leu477. These non-covalent interactions collectively enhance the stability and binding affinity of compound 12 within the aromatase active site, Figure 7. The combination of these different interactions played a key role in the strong binding affinity of compound 12 within the active site of the aromatase enzyme.
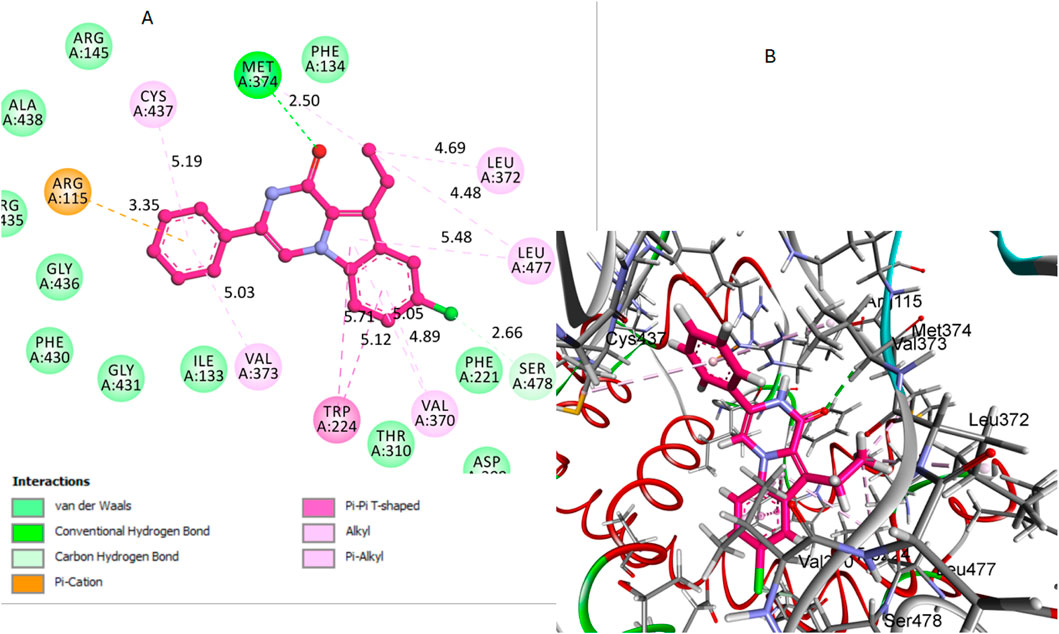
Figure 7. Docking representation models of compound 12 within the binding site of aromatase (PDB: 5JKW) (A) 2D-docked model of compound 12 (B) 3D-docked model of compound 12. The carbonyl oxygen of pyrazine forms a hydrogen bond with Met374, and aromatic rings engage in hydrophobic interactions with Val370, Leu372, and Leu477.
Compound 16, like compound 12, showed significant interactions with the aromatase enzyme. In compound 16, the carbonyl oxygen of the pyrazine moiety acted as a hydrogen bond acceptor with the Arg115 residue. This hydrogen bond plays a crucial role in anchoring compound 16 within the active site. The hydrogen bond distance between the carbonyl oxygen of compound 16 and the hydrogen of the Arg115 side chain was measured at 2.66 Å. The bond angle formed was approximately 170°, which indicates a strong and nearly optimal hydrogen bond for stabilizing the compound within the active site. Additionally, its aliphatic methyl group was involved in a pi-alkyl bond interaction with the Phe134 residue, as illustrated in Figure 8. These interactions will likely play a crucial role in stabilizing the compound within the binding pocket of the aromatase enzyme.
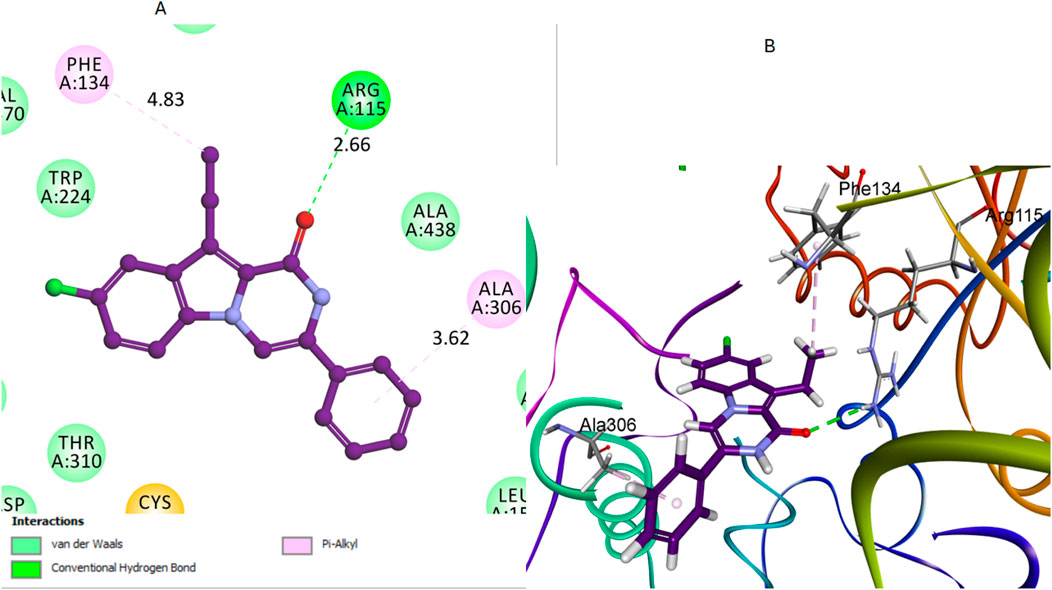
Figure 8. Docking representation models of compound 16 within the binding site of aromatase (PDB: 5JKW) (A) 2D-docked model of compound 16 (B) 3D-docked model of compound 16. The carbonyl oxygen of pyrazine forms a hydrogen bond with Arg115, and the aliphatic methyl group interacts with Phe134.
Similarly, pyrazoline oxygen of compound 7 establishes a hydrogen bond with the crucial residue Arg115 when it interacts with the aromatase enzyme, Figure 9. This hydrogen bond is significant as it represents the primary interaction stabilizing compound 7 within the active site. The hydrogen bond distance between the carbonyl oxygen of compound 7 and the hydrogen of the Arg115 side chain was measured at 2.76 Å. The bond angle was approximately 160°, which supports a moderately strong hydrogen bond for stabilizing the compound within the active site. However, this is the only significant interaction observed for compound 7. The length of its amide bridge may not be optimal for a snug fit in the aromatase active site. Compound 7 also lacks the extensive hydrophobic interactions in compounds 12 and 16.
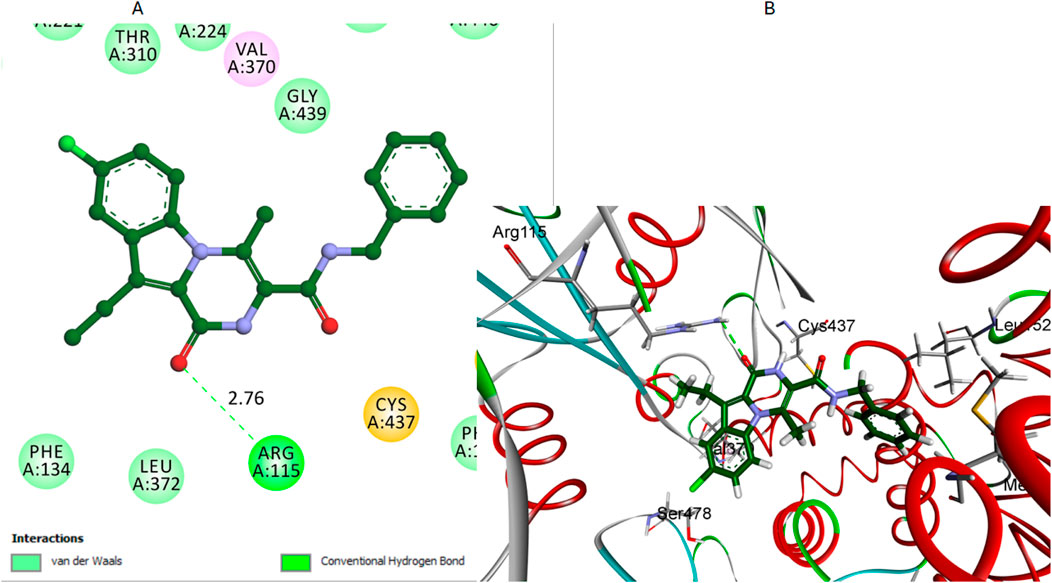
Figure 9. Docking representation models of compound 7 within the binding site of aromatase (PDB: 5JKW) (A) 2D-docked model of compound 7 (B) 3D-docked model of compound 7. The carbonyl oxygen of pyrazine establishes a hydrogen bond with Arg115.
In conclusion, these results offer a basic insight into the potential inhibitory effects of the examined compounds and establish a foundation for further experimental validation and optimization in drug discovery. The detailed elucidation of these molecular interactions is crucial in directing the rational design of new therapeutic agents, particularly those targeting cancer-related pathways.
2.4 ADMET studies
This supplementary section presents a comprehensive overview of the ADME (Absorption, Distribution, Metabolism, Excretion) studies for the newly synthesized compounds, specifically compounds 8–16. These studies are essential to evaluate the compounds’ pharmacokinetics and potential therapeutic efficacy (Al-Wahaibi et al., 2023d). The absorption studies show that all compounds have good absorption levels but vary in their Polar Surface Area (PSA), influencing their solubility and permeability. In terms of distribution, all compounds exhibit high plasma protein binding (>90%), which is crucial for their distribution in the bloodstream and free concentration. Additionally, the Blood-Brain Barrier (BBB) permeability ranges from very high to medium, indicating potential central nervous system activity for some compounds. The metabolism analysis reveals that several compounds (10–16) are inhibitors of the CYP2D6 enzyme, which could lead to significant drug-drug interactions and affect drug metabolism. The excretion studies indicate that most compounds have low solubility levels, potentially impacting their excretion rates. The Lipophilicity (AlogP98) values, ranging from moderate to high, suggest implications for membrane permeability and possible bioaccumulation Table 6. Overall, these ADME profiles offer valuable insights into the pharmacokinetic behaviors of the compounds. The high plasma protein binding and variation in BBB permeability necessitate carefully evaluating their therapeutic window and CNS effects (Figure 10).
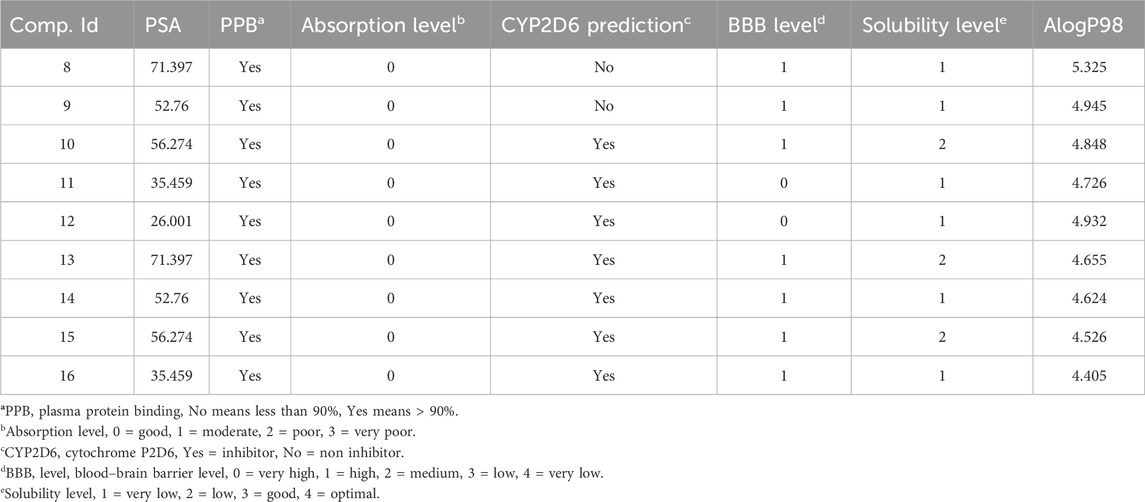
Table 6. Comprehensive prediction of synthesized hybrids’ absorption, distribution, metabolism, excretion, and toxicity (ADME) profiles 8–16.
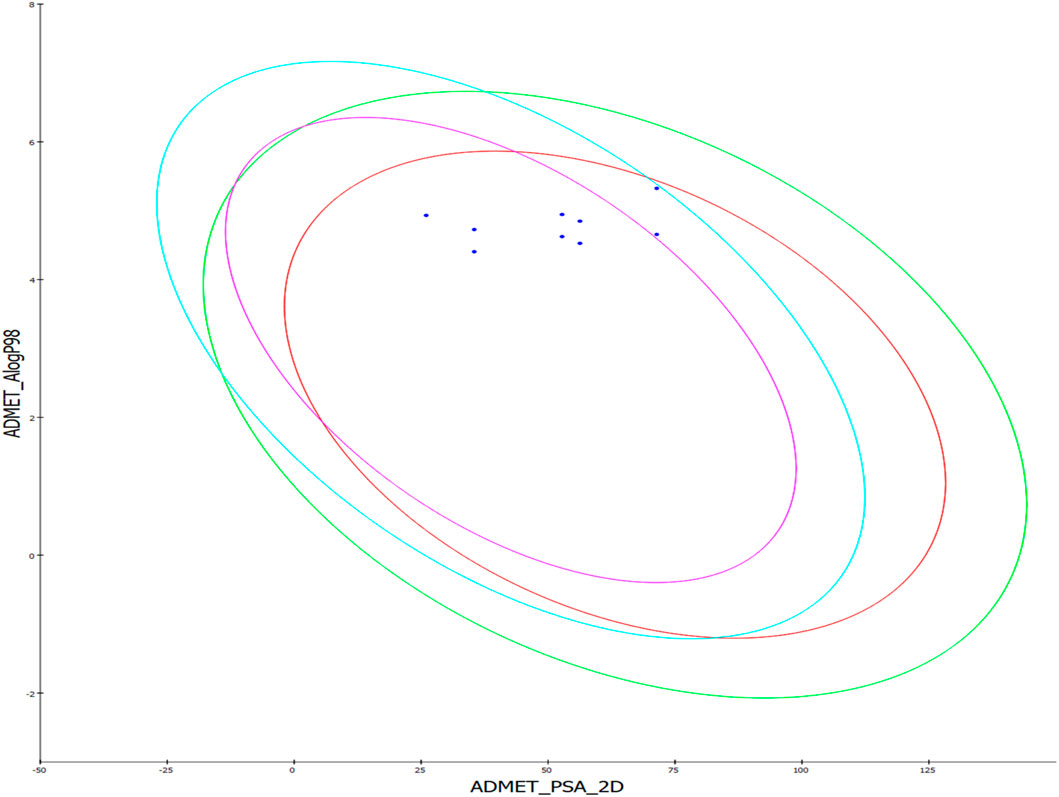
Figure 10. The predicted ADME study of hybrids 8–16. Comprehensive profiles of absorption, distribution, metabolism, and excretion properties are depicted, including plasma protein binding, blood-brain barrier permeability, CYP2D6 inhibition, solubility, and lipophilicity (AlogP98).
The inhibition of CYP2D6 by several compounds highlights the need to assess potential drug interactions further. The solubility and lipophilicity profiles underscore the importance of optimization in future drug formulation strategies. This data is instrumental in guiding these compounds’ clinical development and safety assessments, paving the way for their potential therapeutic application.
2.5 Structure activity relationship (SAR)
1. Open-ring (Scaffold A) compounds, namely, compounds 8 and 13, exhibited significant antiproliferative effects, comparable to the reference Erlotinib. However, compounds 3 and 4 displayed limited activity, indicating that the nature of functional groups and substituents determine the landscape of these compounds’ therapeutic effects.
2. For Scaffold B (closed-ring) compounds, the antiproliferative effects is determined by the following factors:
a) The carbonyl oxygen in pyrazine ring (position-1) is essential for activity. It acted as a hydrogen bond acceptor, forming a bond with crucial amino acid Arg115.
b) The type of substitution detected on the nitrogen atom at position two of the pyrazino [1,2-a]indol-1(2H)-one moiety appears to be crucial for activity demonstrating that the N-methyl group (R1 = Me) at position two is important for the antiproliferative activity.
c) The third position substitution significantly affects the antiproliferative activity of these compounds, with the highest activity shown in compounds with a benzyl substitution (R2 = CH2Ph), followed by carboxamide (R2 = CONHCH2PH) and phenyl (R2 = Ph) substitutions
d) A high level of hydrophilicity (R2 = COOH and/or R3 = OH) is not favoured for antiproliferative action.
3 Conclusion
In the present work, we report the design and synthesis of new indole-based derivatives able to inhibit the aromatase enzyme and/or iNOS isozyme. Compounds 12 and 16 were found to be the most intriguing since they are strong aromatase inhibitors with IC50 values of 10 and 12 nM, respectively. Moreover, they are potent and selective iNOS inhibitors (IC50 = 0.075 and 0.098 µM, respectively), and they were potential antiproliferative agents against the MCF-7 breast cancer cell line (IC50 = 25 and 28 nM, respectively). Notably, at 50 µM concentrations, 12 and 16 demonstrated no effect on non-tumor cells MCF-10A, indicating the promise tumor-cell selectivity of these derivatives. The molecular docking studies have successfully demonstrated the specific binding interactions of compounds 12 and 16, with the aromatase active site. This detailed analysis is pivotal in understanding their mechanism of action in breast cancer therapy. Furthermore, the comprehensive assessment of the absorption, distribution, metabolism, and excretion (ADME) properties of these hybrids underscores their potential as therapeutic agents. These insights, particularly their reduced toxicity profiles, are instrumental in their candidacy for effective breast cancer treatments. Based on the findings, 12 and 16 are promising prospects for the development of new agents for cancer therapy. Moreover, additional investigation into the mechanism of action, in vivo carcinogenic animal models, and lead optimization remains ongoing in our lab.
4 Experimental
4.1 Chemistry
General Details: See Supplementary Appendix SA1.
Synthesis of Benzyl 2-(5-chloro-3-ethyl-1H-indol-2-carboxamido)-3-hydroxybutanoate (3).
To a stirred solution of compound 1 (0.8 g, 3.59 mmol, 1eq) in DMF (30 mL), BOP (2.07 g, 4.67 mmol, 1.3 eq), L-threonine benzyl ester 2 (1.06 g, 4.31 mmol, 1.2 eq) were added and reaction mixture was cooled to 0°C before dropwise addition of DIPEA (1.87 mL, 10.77 mmol, 3 eq). After stirring overnight at rt, the reaction mixture was diluted with EtOAc and successively washed with H2O. The organic layer was washed with 5% aqueous HCl, saturated NaHCO3, and finally with brine. The organic phase was dried over MgSO4, and the solvent was removed under reduced pressure to yield 1.35 g (91%) of 3 as a white solid after chromatographed on silica gel using EtOAc/hexane (2:3) as an eluent. HRESI-MS m/z calcd for [M + H]+ C22H24ClN2O4: 415.1419, found: 415.1418.
4.1.1 Synthesis of benzyl 2-(5-chloro-3-ethyl-1h-indol-2-carboxamido)-3-oxobutanoate (4)
To a solution of compound 3 (0.34 g, 0.82 mmol) in DCM (30 mL), Dess-Martin periodinane (0.45 g, 1.00 mmol) was added with stirring at 0°C for 30 min, then the temperature was warmed to rt for 5 h. A saturated solution of Na2S2O3 (10 mL) and NaHCO3 (10 mL) was added to the reaction mixture by stirring at rt for 15 min. The organic layer was separated, dried over MgSO4, and evaporated under reduced pressure to yield a crude product 4, which was used for the next step without further purification.
4.1.2 Synthesis of benzyl 8-chloro-10-ethyl-4-methyl-1-oxo-1,2-dihydropyrazino [1,2-a]indole-3-carboxylate (5)
A mixture of 4 (0.85 mmol, 1equiv) and PTSA (0.33 g, 1.7 mmol, 2 eq) in toluene (40 mL) was refluxed with stirring overnight. After concentration of the solvent in vacuo, the residue was extracted with EtOAc, washed with a saturated solution of NaHCO3 brine, dried over MgSO4, and concentrated under reduced pressure to yield a crude product which purified by flash chromatography using EtOAc, hexanes (1:4) to yield 5 (0.3 g, 82%) as white solid. Mp 180–182°C. 1H NMR (400 MHz, Chloroform-d) δ 8.41 (s, 1H, pyrazine NH), 7.98 (d, J = 9.2 Hz, 1H, Ar-H), 7.77 (d, J = 2.1 Hz, 1H, Ar-H), 7.46–7.30 (m, 6H, Ar-H), 5.36 (s, 2H, PhCH2), 3.30 (q, J = 7.5 Hz, 2H, CH2CH3), 3.19 (s, 3H, CH3), 1.30 (t, J = 7.5 Hz, 3H, CH2CH3). 13C NMR (101 MHz, Chloroform-d) δ 161.87 (ester C=O), 155.99 (C=O), 134.77, 132.35, 130.85, 129.18, 128.86, 128.82, 128.69, 128.66, 125.52, 125.27, 124.73, 120.18, 116.72, 110.65, 67.88, 17.54, 16.63, 15.52. HRESI-MS m/z calcd for [M-H]- C22H18ClN2O3: 393.1011, found: 393.1013.
4.1.3 Synthesis of 8-chloro-10-ethyl-4-methyl-1-oxo-1,2-dihydropyrazino [1,2-a]indole-3-carboxylic acid (6)
To a solution of 5 (0.25 g, 0.64 mmol) in ethanol (15 mL), 5% NaOH (4 mL) was added. The reaction mixture was kept at 40°C while stirring overnight. The residue after removal of ethanol was taken into water and acidified with 5% HCl. The formed precipitate was filtered, washed with water, and dried under vacuum to afford 6 (0.18, 96%) as a white solid. Mp 270–272°C. 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H, pyrazine NH), 8.22 (d, J = 9.3 Hz, 1H, Ar-H), 7.86 (s, 1H, Ar-H), 7.30 (d, J = 9.2 Hz, 1H, Ar-H), 3.25 (q, J = 7.5 Hz, 2H, CH2CH3), 2.07 (s, 3H, CH3), 1.18 (t, J = 7.5, 3H, CH2CH3). 13C NMR (101 MHz, DMSO-d6) δ 164.05 (COOH), 155.67, 131.76, 129.81, 126.57, 125.99, 124.13, 121.45, 120.81, 119.45, 117.99, 117.87, 17.27, 16.47, 15.78.
4.1.4 Synthesis of N-benzyl-8-chloro-10-ethyl-4-methyl-1-oxo-1,2-dihydropyrazino [1,2-a]indole-3-carboxamide (7)
To a stirred solution of 6 (0.12 g, 0.39 mmol, 1eq) in DMF (20 mL), BOP (0.22 g, 0.51 mmol, 1.3 eq), benzylamine (0.05 mL, 0.47 mmol, 1.2 eq) was added and the reaction mixture was cooled to 0°C before dropwise addition of DIPEA (0.1 mL, 0.78 mmol, 2 eq). After stirring overnight at rt, the reaction mixture was diluted with EtOAc and successively washed with H2O. The organic layer was washed with 5% aqueous HCl, saturated NaHCO3, and finally with brine. The organic phase was dried over MgSO4, and the solvent was removed under reduced pressure to yield the crude product, which was purified by short column chromatography on silica gel using EtOAc as an eluent to provide 7 as g a white solid (0.13 g, 81%). Mp 290–292°C. 1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H, pyrazine NH), 9.05 (t, J = 5.9 Hz, 1H, NHCH2), 8.16 (d, J = 9.2 Hz, 1H, Ar-H), 7.91 (d, J = 2.2 Hz, 1H, Ar-H), 7.43–7.23 (m, 6H, Ar-H), 4.44 (d, J = 5.8 Hz, 2H, NHCH2), 3.25 (q, J = 7.4 Hz, 2H, CH2CH3), 2.73 (s, 3H, CH3), 1.20 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (101 MHz, DMSO-d6) δ 163.01 (amide C=O), 157.03 (C=O), 139.14, 131.55, 129.44, 128.73, 128.05, 127.40, 126.83, 125.03, 124.70, 121.82, 119.68, 118.51, 117.48, 117.40, 43.33, 17.30, 16.67, 16.44. HRESI-MS m/z calcd for [M-H]- C22H19ClN3O2: 392.1171, found: 392.1168.
4.1.5 Synthesis of ethyl 2-(5-chloro-3-ethyl-1h-indole-2-carboxamido)-3-phenylpropanoate (8)
This compound was prepared as described in the general procedure for synthesizing compound 3 using compound 1 and ethyl phenylalanine. Yield % 83. Oil, b. p 178–180°C. 1H NMR (400 MHz, Chloroform-d) δ 9.93 (s, 1H, indole NH), 7.55 (d, J = 2.1 Hz, 1H, Ar-H), 7.36–7.25 (m, 4H, Ar-H), 7.23–7.14 (m, 3H, Ar-H), 6.70 (d, J = 7.2 Hz, 1H, amide NH), 5.18–5.09 (m, 1H, NHCH), 4.25 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.41–3.25 (m, 2H, PhCH2), 2.86–2.71 (m, 2H, CH2CH3), 1.29 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.13 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR (101 MHz, Chloroform-d) δ 171.52 (ester C=O), 161.82 (C=O), 135.59, 133.94, 129.36, 128.73, 128.68, 127.33, 127.11, 125.42, 125.07, 119.42, 119.33, 113.12, 61.90, 53.74, 37.76, 18.11, 15.21, 14.14. HRESI-MS m/z calcd for [M + H]+ C22H24ClN2O3: 399.1470, found: 399.1472.
4.1.6 Synthesis of 3-benzyl-8-chloro-10-ethyl-2,3-dihydropyrazino [1,2-a] indole-1,4-dione (9)
This compound was prepared as described in the general procedure for synthesizing compound 5 using compound 4. Yield % 75, mp 196–198°C. 1H NMR (400 MHz, DMSO-d6) δ 8.61 (d, J = 2.5 Hz, 1H, Ar-H), 8.31 (d, J = 8.7 Hz, 1H, NH), 7.77 (d, J = 2.2 Hz, 1H, Ar-H), 7.51 (dd, J = 8.8, 2.1 Hz, 1H, Ar-H), 7.02 (s, 5H, Ar-H), 4.81–4.79 (m, 1H, NHCH), 3.29 (dd, J = 13.6, 3.6 Hz, 1H, PhCHa), 3.10 (dd, J = 13.6, 5.0 Hz, 1H, PhCHb), 3.04–2.81 (m, 2H, CH2CH3), 0.98 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (101 MHz, DMSO-d6) δ 165.56 (CHC = O), 157.82 (C=O), 135.19, 131.68, 130.94, 130.00, 129.73, 129.55, 128.37, 127.98, 127.26, 124.00, 120.42, 117.66, 57.98, 16.68, 14.92. HRESI-MS m/z calcd for [M + H]+ C20H18ClN2O2: 353.1051, found: 353.1052.
4.1.7 Synthesis of 3-benzyl-8-chloro-10-ethyl-4-hydroxy-3,4-dihydropyrazino [1,2-a] indol-1(2H)-one (10)
To a stirred solution of 10 (1 g, 2.80 mmol, 1 equiv) in ethanol (50 mL), a solution of NaBH4 (0.21 g, 2 equiv) in water (15 mL) was added at 0°C. The cooling bath was removed, and the reaction mixture was stirred for 1 h at rt. After the ethanol concentration in vacuo, the residue was extracted with EtOAc, washed with brine, dried over MgSO4, and concentrated under reduced pressure to afford 11 as a white solid. Yield % 93, mp 160–162°C. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H, NH), 7.69 (d, J = 8.3 Hz, 1H, CHOH), 7.61 (d, J = 2.1 Hz, 1H, Ar-H), 7.38 (d, J = 8.7 Hz, 1H, Ar-H), 7.30–7.20 (m, 4H, Ar-H), 7.15 (dd, J = 8.4, 2.2 Hz, 2H, Ar-H), 4.89 (s, 1H, OH), 4.24–4.14 (m, 1H, NHCH), 2.99–2.85 (m, 3H, CH2CH3, PhCH2a), 2.79 (dd, J = 13.8, 8.4 Hz, 1H, PhCH2b), 1.05 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (101 MHz, DMSO-d6) δ 161.50 (C=O), 139.54, 134.03, 129.51, 128.86, 128.61, 128.56, 126.42, 124.10, 124.01, 120.89, 119.22, 113.98, 62.88, 53.06, 37.07, 17.54, 16.02. HRESI-MS m/z calcd for [M + H]+ C20H20ClN2O2: 355.1208, found: 355.1210.
4.1.8 3-Benzyl-8-chloro-10-ethylpyrazino[1,2-a]indol-1(2H)-one (11)
A mixture of 11 (0.05 g, 0.14 mmol, 1equiv) and PTSA (0.05 g, 2 equiv) in toluene (10 mL) was refluxed for 4 h. After removing the solvent in vacuo, the residue was extracted with EtOAc, washed with saturated solution of NaHCO3, brine, dried over MgSO4, and concentrated under reduced pressure to yield a crude product which was purified by flash chromatography using EtOAc/hexanes (1:4) as eluent to provide 12 as a brown solid. Yield % 60, mp 261–263°C. 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H, pyrazine NH), 7.94 (d, J = 8.9 Hz, 1H, Ar-H), 7.85 (d, J = 2.0 Hz, 1H, Ar-H), 7.71 (d, J = 1.7 Hz, 1H, Ar-H), 7.41–7.27 (m, 5H, Ar-H), 7.26–7.19 (m, 1H, Ar-H), 3.69 (s, 2H, PhCH2), 3.18 (q, J = 7.4 Hz, 2H, CH2CH3), 1.18 (t, J = 7.4 Hz, 4H, CH2CH3). 13C NMR (101 MHz, DMSO-d6) δ 158.57 (C=O), 138.74, 129.50, 129.03, 128.83, 127.62, 127.01, 126.46, 124.84, 124.32, 123.31, 119.77, 119.66, 113.66, 103.81, 35.82, 17.36, 16.67. HRESI-MS m/z calcd for [M + H]+ C20H18ClN2O: 337.1102, found: 337.1103.
4.1.9 3-Benzyl-8-chloro-10-ethyl-2-methylpyrazino[1,2-a]indol-1(2H)-one (13)
A solution of 12 (1 equiv) in DMF (0.2 M) was added dropwise at 0°C to a suspension of NaH (1.5 equiv, 60% dispersion in mineral oil) in DMF (0.2 M). After stirring for 0.5 h, the resulting mixture was treated dropwise with a MeI (1.2 equiv). The cooling bath was removed, and the mixture was stirred overnight at rt. The reaction mixture was diluted with EtOAc and successively washed twice with water. The EtOAc layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure to yield 13 as a white solid after column chromatography using EtOAc/hexane (2:3) as an eluent. Yield % 88, mp 161–163°C. 1H NMR (400 MHz, Chloroform-d) δ 7.70 (s, 1H, Ar-H), 7.38 (d, J = 9.0 Hz, 1H, Ar-H), 7.33–7.15 (m, 6H, Ar-H), 6.90 (s, 1H, Ar-H), 3.86 (s, 2H, PhCH2), 3.26–3.19 (m, 5H, CH3, CH2CH3), 1.25 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR (101 MHz, Chloroform-d) δ 158.81 (C=O), 136.66, 129.00, 128.81, 128.22, 128.16, 127.25, 124.51, 124.29, 122.62, 120.82, 119.93, 111.38, 105.40, 37.11, 29.26, 17.68, 16.06. HRESI-MS m/z calcd for [M + H]+ C21H20ClN2O: 351.1259, found: 351.1257.
4.1.10 Methyl 2-(5-chloro-3-ethyl-1h-indole-2-carboxamido)-2-phenylacetate (13)
This compound was prepared as described in the general procedure for synthesizing compound 3 using compound 1 and methyl-2-amino-2-phenylacetate. Yield % 94, mp 210–212°C. 1H NMR (400 MHz, Chloroform-d) δ 9.77 (s, 1H, indole NH), 7.59 (d, J = 2.0 Hz, 1H, Ar-H), 7.49–7.44 (m, 2H, Ar-H), 7.44–7.34 (m, 4H, Ar-H), 7.13 (dd, J = 8.7, 2.0 Hz, 1H, Ar-H), 7.01 (d, J = 8.7 Hz, 1H, Ar-H, amide NH), 5.79 (d, J = 6.4 Hz,1H, PhCH), 3.78 (s, 3H, OCH3), 3.06 (q, J = 7.6 Hz, 2H, CH2CH3), 1.40 (t, J = 7.6 Hz, 3H, CH2CH3). 13C NMR (101 MHz, Chloroform-d) δ 171.37 (ester C=O), 161.42 (C=O), 136.46, 133.89, 129.09, 128.69, 127.13, 126.96, 125.48, 125.16, 119.55, 119.28, 113.13, 56.86, 53.16, 18.50, 15.43. RESI-MS m/z calcd for [M + H]+ C20H20ClN2O3: 371.1157, found: 371.1156.
4.1.11 8-Chloro-10-ethyl-3-phenyl-2,3-dihydropyrazino [1,2-a] indole-1,4-dione (14)
This compound was prepared as described in the general procedure for synthesizing compound 5 using compound 13. Yield % 85, mp 202–204°C. 1H NMR (400 MHz, Chloroform-d) δ 8.33 (d, J = 8.8 Hz, 1H, Ar-H), 7.68 (d, J = 2.0 Hz, 1H, Ar-H), 7.49–7.35 (m, 6H, Ar-H), 6.35 (s, 1H, NH), 5.46 (d, J = 2.2 Hz, 1H, PhCH), 3.24 (q, J = 7.4 Hz, 2H, CH2CH3), 1.33 (t, J = 7.5 Hz, 3H, CH2CH3). 13C NMR (101 MHz, Chloroform-d) δ 162.97, 158.14, 136.68, 133.61, 132.64, 131.07, 130.90, 129.33, 129.26, 128.65, 127.07, 122.30, 120.27, 117.85, 61.72, 17.51, 14.37. HRESI-MS m/z calcd for [M-H]- C19H14ClN2O2: 337.0749, found: 337.0753.
4.1.12 8-Chloro-10-ethyl-4-hydroxy-3-phenyl-3,4-dihydropyrazino [1,2-a] indol-1(2H)-one (15)
This compound was prepared as described in the general procedure for synthesizing compound 10 using compound 14. Yield % 89, mp 213–215°C. 1H NMR (400 MHz, DMSO-d6) δ 8.54 (d, J = 4.7 Hz, 1H, NH), 7.70 (d, J = 2.1 Hz, 1H, Ar-H), 7.51 (d, J = 8.9 Hz, 1H, Ar-H), 7.29–7.12 (m, 6H, Ar-H), 6.98 (d, J = 5.7 Hz, 1H, CHOH), 6.02 (d, J = 5.8 Hz, 1H, OH), 4.86–4.79 (m, 1H, PhCH), 3.11 (q, J = 7.5 Hz, 2H, CH2CH3), 1.19 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (101 MHz, DMSO-d6) δ 160.55 (C=O), 140.14, 133.89, 129.00, 128.39, 128.04, 126.53, 125.22, 124.83, 124.43, 123.68, 119.70, 112.95, 76.41, 61.00, 17.35, 15.94. HRESI-MS m/z calcd for [M-H]- C19H16ClN2O2: 339.0906, found: 339.0904.
4.1.13 8-Chloro-10-ethyl-3-phenylpyrazino [1,2-a]indol-1(2H)-one (16)
This compound was prepared as described in the general procedure for synthesizing compound 11 using compound 15. Yield % 88, mp 263–265°C. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H, pyrazine NH), 8.20 (d, J = 8.9 Hz, 1H, Ar-H), 8.15 (d, J = 1.6 Hz, 1H, Ar-H), 7.89 (d, J = 2.1 Hz, 1H, Ar-H), 7.80–7.72 (m, 2H, Ar-H), 7.50–7.33 (m, 4H, Ar-H), 3.23 (q, J = 7.4 Hz, 2H, CH2CH3), 1.23 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (101 MHz, DMSO-d6) δ 158.65 (C=O), 132.50, 130.11, 129.10, 128.75, 128.01, 126.91, 126.23, 124.96, 124.51, 123.17, 120.30, 119.80, 114.29, 104.24, 17.42, 16.62. HRESI-MS m/z calcd for [M + H]+ C19H16ClN2O: 323.0946, found: 323.0947.
4.2 Biology
4.2.1 Assay of cell viability effect
The normal human mammary gland epithelial (MCF-10A) cell line was used to test the viability of the tested compounds. After 4 days of incubation on MCF-10A cells within 50 µM of each investigated compound, cell viability was determined using the MTT assay (Gomaa et al., 2020; Marzouk et al., 2020). Refer to Supplementary Appendix SA1 (supp. File) for more details.
4.2.2 Antiproliferative assay
The MTT assay was used to investigate the antiproliferative activity of 3–16 versus four human cancer cell lines using erlotinib as a control (Youssif et al., 2022; Mahmoud et al., 2022). See Supplementary Appendix SA1 for more details.
4.2.3 Aromatase inhibitory assay
Compounds 5, 7, 8, 12, 13, and 16 were tested for their capacity to inhibit aromatase, a potential target for their antiproliferative activity, using ketoconazole and letrozole as reference drugs (Maghraby et al., 2023). See Supplementary Appendix SA1 for more information.
4.2.4 Assay of nitric oxide synthase inhibition
The L-citrulline assay method with fluorometric detection was used to test compounds 5, 7, 12, and 16 (most potent anti-aromatase derivatives) for their ability to inhibit iNOS (Carrión et al., 2023). Refer to Supplementary Appendix SA1 for more details.
4.3 In silico studies
4.3.1 Docking study
In our molecular docking study, we employed BIOVIA Discovery Studio 2021 software (version 21.1.0.20.298) (Pawar and Rohane, 2021). We utilized the Protein Preparation Wizard to prepare the selected proteins for docking analysis. Following protein preparation, we meticulously mapped the ligands onto a three-dimensional model and conducted energy minimization using LigPrep. We generated a receptor grid tailored to the selected binding site to optimize potential binding interactions using the Receptor Grid Generation Tool. Subsequently, the Glide tool was employed to comprehensively assess both docking scores and the diverse binding modes exhibited by the ligands. This rigorous approach facilitated a thorough exploration of the binding affinities and interaction patterns between the ligands and the selected proteins, enhancing our understanding of their potential as inhibitors in breast cancer therapy.
4.3.2 In silico ADMET analysis
In our study, ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) studies were conducted using BIOVIA I Discovery Studio 2021 (Mohammed Ali et al., 2023). The chemical structures of all compounds were imported, and ADMET descriptors were predicted using integrated models. These models encompassed assessments based on Lipinski’s Rule of Five and evaluations of absorption, distribution, metabolism, excretion, and toxicity. The resulting data were meticulously analysed to determine the drug-likeness and safety profiles of the compounds under investigation.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
LA-W: Conceptualization, Funding acquisition, Project administration, Resources, Writing–original draft. HA-Z: Software, Writing–original draft. MA: Investigation, Methodology, Validation, Visualization, Writing–original draft. MM: Data curation, Formal Analysis, Software, Writing–review and editing. LT: Methodology, Supervision, Validation, Writing–review and editing. BY: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Validation, Visualization, Writing–original draft, Writing–review and editing. SB: Visualization, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by Princess Nourah Bint Abdulrahman University Researchers Supporting Project Number (PNURSP2024R3), Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Acknowledgments
The authors acknowledge the support by Princess Nourah Bint Abdulrahman University Researchers Supporting Project Number (PNURSP2024R3), Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1432920/full#supplementary-material
References
Al-Wahaibi, L. H., Abou-Zied, H. A., Hisham, M., Beshr, E. A., Youssif, B. G., Bräse, S., et al. (2023d). Design, synthesis, and biological evaluation of novel 3-cyanopyridone/pyrazoline hybrids as potential apoptotic antiproliferative agents targeting EGFR/BRAFV600E inhibitory pathways. Molecules 28 (18), 6586. doi:10.3390/molecules28186586
Al-Wahaibi, L. H., Gouda, A. M., Abou-Ghadir, O. F., Salem, O. I., Ali, A. T., Farghaly, H. S., et al. (2020). Design and synthesis of novel 2, 3-dihydropyrazino [1, 2-a] indole-1, 4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 104, 104260. doi:10.1016/j.bioorg.2020.104260
Al-Wahaibi, L. H., Mohammed, A. F., Abdel Rahman, F.E.-Z. S., Abdelrahman, M. H., Gu, X., Trembleau, L., et al. (2023a). Design, synthesis, apoptotic, and antiproliferative effects of 5-chloro-3-(2-methoxyvinyl)-indole-2-carboxamides and pyrido [3, 4-b] indol-1-ones as potent EGFRWT/EGFRT790M inhibitors. J. enzyme inhibition Med. Chem. 38 (1), 2218602. doi:10.1080/14756366.2023.2218602
Al-Wahaibi, L. H., Mohammed, A. F., Abdelrahman, M. H., Trembleau, L., and Youssif, B. G. (2023b). Design, synthesis, and antiproliferative activity of new 5-Chloro-indole-2-carboxylate and pyrrolo [3, 4-b] indol-3-one derivatives as potent inhibitors of egfrt790m/brafv600e pathways. Molecules 28 (3), 1269. doi:10.3390/molecules28031269
Al-Wahaibi, L. H., Mohammed, A. F., Abdelrahman, M. H., Trembleau, L., and Youssif, B. G. (2023c). Design, synthesis, and biological evaluation of indole-2-carboxamides as potential multi-target antiproliferative agents. Pharmaceuticals 16 (7), 1039. doi:10.3390/ph16071039
Avvaru, S. P., Noolvi, M. N., Aminbhavi, T. M., Chkraborty, S., Dash, A., and Shukla, S. S. (2018). Aromatase inhibitors evolution as potential class of drugs in the treatment of postmenopausal breast cancer women. Mini Rev. Med. Chem. 18 (7), 609–621. doi:10.2174/1389557517666171101100902
Bernacki, K., Kalyanaraman, C., and Jacobson, M. P. (2005). Virtual ligand screening against Escherichia coli dihydrofolate reductase: improving docking enrichment using physics-based methods. SLAS Discov. 10 (7), 675–681. doi:10.1177/1087057105281220
Bhat, M. A., Tüzün, B., Alsaif, N. A., Khan, A. A., and Naglah, A. M. (2022). Synthesis, characterization, molecular modeling against EGFR target and ADME/T analysis of novel purine derivatives of sulfonamides. J. Mol. Struct. 1257, 132600. doi:10.1016/j.molstruc.2022.132600
Brodie, A., Brodie, H., Callard, G., Robinson, C., Roselli, C., and Santen, R. (1993). Preface. J. Steroid Biochem. Mol. Biol. 44, 321–696. doi:10.1016/0960-0760(93)90234-n
Burke, A. J., Sullivan, F. J., Giles, F. J., and Glynn, S. A. (2013). The yin and yang of nitric oxide in cancer progression. Carcinogenesis 34 (3), 503–512. doi:10.1093/carcin/bgt034
Carrión, M. D., Rubio-Ruiz, B., Franco-Montalban, F., Amoia, P., Zuccarini, M. C., De Simone, C., et al. (2023). New amidine-benzenesulfonamides as iNOS inhibitors for the therapy of the triple negative breast cancer. Eur. J. Med. Chem. 248, 115112. doi:10.1016/j.ejmech.2023.115112
Chauhan, D., Hideshima, T., Rosen, S., Reed, J. C., Kharbanda, S., and Anderson, K. C. (2001). Apaf-1/cytochrome c-independent and Smac-dependent induction of apoptosis in multiple myeloma (MM) cells. J. Biol. Chem. 276 (27), 24453–24456. doi:10.1074/jbc.c100074200
Chen, P.-Y., Hsu, C.-W., Ho, T.-I., and Ho, J.-H. (2021). The selective synthesis of N-arylbenzene-1, 2-diamines or 1-arylbenzimidazoles by irradiating 4-methoxy-4′-substituted-azobenzenes in different solvents. RSC Adv. 11 (12), 6662–6666. doi:10.1039/d0ra10068d
Cinelli, M. A., Do, H. T., Miley, G. P., and Silverman, R. B. (2020). Inducible nitric oxide synthase: regulation, structure, and inhibition. Med. Res. Rev. 40 (1), 158–189. doi:10.1002/med.21599
Cotter, T. G. (2009). Apoptosis and cancer: the genesis of a research field. Nat. Rev. Cancer 9 (7), 501–507. doi:10.1038/nrc2663
Denmeade, S. R., and Isaacs, J. T. (1996). Programmed cell death (apoptosis) and cancer chemotherapy. Cancer control. 3 (4), 303–309. doi:10.1177/107327489600300401
Dixon, J., Jackson, J., Renshaw, L., and Miller, W. (2003). Neoadjuvant tamoxifen and aromatase inhibitors: comparisons and clinical outcomes. J. Steroid Biochem. Mol. Biol. 86 (3-5), 295–299. doi:10.1016/s0960-0760(03)00370-4
Fantacuzzi, M., De Filippis, B., Gallorini, M., Ammazzalorso, A., Giampietro, L., Maccallini, C., et al. (2020). Synthesis, biological evaluation, and docking study of indole aryl sulfonamides as aromatase inhibitors. Eur. J. Med. Chem. 185, 111815. doi:10.1016/j.ejmech.2019.111815
Foti, R. S. (2023). Cytochrome P450 and other drug-metabolizing enzymes as therapeutic targets. Drug Metabolism Dispos. 51 (8), 936–949. doi:10.1124/dmd.122.001011
Fürstner, A., Jumbam, D. N., and Seidel, G. (1994). Syntheses of Zindoxifene and analogues by titanium-induced oxo-amide coupling. Chem. Berichte 127 (6), 1125–1130. doi:10.1002/cber.19941270624
Garrido, P., Shalaby, A., Walsh, E. M., Keane, N., Webber, M., Keane, M. M., et al. (2017). Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 8 (46), 80568–80588. doi:10.18632/oncotarget.19631
Glynn, S. A., Boersma, B. J., Dorsey, T. H., Yi, M., Yfantis, H. G., Ridnour, L. A., et al. (2010). Increased NOS2 predicts poor survival in estrogen receptor–negative breast cancer patients. J. Clin. investigation 120 (11), 3843–3854. doi:10.1172/jci42059
Gomaa, H. A., El-Sherief, H. A., Hussein, S., Gouda, A. M., Salem, O. I., Alharbi, K. S., et al. (2020). Novel 1, 2, 4-triazole derivatives as apoptotic inducers targeting p53: synthesis and antiproliferative activity. Bioorg. Chem. 105, 104369. doi:10.1016/j.bioorg.2020.104369
Gomaa, H. A., Shaker, M. E., Alzarea, S. I., Hendawy, O., Mohamed, F. A., Gouda, A. M., et al. (2022). Optimization and SAR investigation of novel 2, 3-dihydropyrazino [1, 2-a] indole-1, 4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 120, 105616. doi:10.1016/j.bioorg.2022.105616
Graham Lorence, S., Peterson, J. A., Amarneh, B., Simpson, E. R., and White, R. E. (1995). A three-dimensional model of aromatase cytochrome P450. Protein Sci. 4 (6), 1065–1080. doi:10.1002/pro.5560040605
Granados-Principal, S., Liu, Y., Guevara, M. L., Blanco, E., Choi, D. S., Qian, W., et al. (2015). Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 17 (1), 25. doi:10.1186/s13058-015-0527-x
Hartmann, R. W., Batzl, C., Pongratz, T. M., and Mannschreck, A. (1992). Synthesis and aromatase inhibition of 3-cycloalkyl-substituted 3-(4-aminophenyl) piperidine-2, 6-diones. J. Med. Chem. 35 (12), 2210–2214. doi:10.1021/jm00090a010
Ibrahim, T. S., Bokhtia, R. M., Al-Mahmoudy, A. M., Taher, E. S., AlAwadh, M. A., Elagawany, M., et al. (2020). Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2, 4-dione as HIV-1 fusion inhibitors. Bioorg. Chem. 99, 103782. doi:10.1016/j.bioorg.2020.103782
Karjalainen, A., Kalapudas, A., Södervall, M., Pelkonen, O., and Lammintausta, R. (2000). Synthesis of new potent and selective aromatase inhibitors based on long-chained diarylalkylimidazole and diarylalkyltriazole molecule skeletons. Eur. J. Pharm. Sci. 11 (2), 109–131. doi:10.1016/s0928-0987(00)00074-9
Kaushik, N. K., Kaushik, N., Attri, P., Kumar, N., Kim, C. H., Verma, A. K., et al. (2013). Biomedical importance of indoles. Molecules 18 (6), 6620–6662. doi:10.3390/molecules18066620
Kharb, R., Haider, K., Neha, K., and Yar, M. S. (2020). Aromatase inhibitors: role in postmenopausal breast cancer. Arch. Der Pharm. 353 (8), 2000081. doi:10.1002/ardp.202000081
Kroemer, G., and Reed, J. C. (2000). Mitochondrial control of cell death. Nat. Med. 6 (5), 513–519. doi:10.1038/74994
Maccallini, C., Gallorini, M., Cataldi, A., and Amoroso, R. (2020). Targeting iNOS as a valuable strategy for the therapy of glioma. ChemMedChem 15 (4), 339–344. doi:10.1002/cmdc.201900580
Maghraby, M. T., Mazyad Almutairi, T., Bräse, S., Salem, O. I., Youssif, B. G., and Sheha, M. M. (2023). New 1, 2, 3-triazole/1, 2, 4-triazole hybrids as aromatase inhibitors: design, synthesis, and apoptotic antiproliferative activity. Molecules 28 (20), 7092. doi:10.3390/molecules28207092
Mahmoud, M. A., Mohammed, A. F., Salem, O. I., Gomaa, H. A., and Youssif, B. G. (2022). New 1, 3, 4-oxadiazoles linked with the 1, 2, 3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch. Pharm. 355 (6), 2200009. doi:10.1002/ardp.202200009
Marzouk, A. A., Abdel-Aziz, S. A., Abdelrahman, K. S., Wanas, A. S., Gouda, A. M., Youssif, B. G., et al. (2020). Design and synthesis of new 1, 6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: molecular docking and antiproliferative evaluation. Bioorg. Chem. 102, 104090. doi:10.1016/j.bioorg.2020.104090
Miyashita, T., Krajewski, S., Krajewska, M., Wang, H. G., Lin, H., Liebermann, D. A., et al. (1994). Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 9 (6), 1799–1805.
Mohammed Ali, H. S., Altayb, H. N., Bayoumi, A. A. M., El Omri, A., Firoz, A., and Chaieb, K. (2023). In silico screening of the effectiveness of natural compounds from algae as SARS-CoV-2 inhibitors: molecular docking, ADMT profile and molecular dynamic studies. J. Biomol. Struct. Dyn. 41 (7), 3129–3144. doi:10.1080/07391102.2022.2046640
Mokbel, K. (2002). The evolving role of aromatase inhibitors in breast cancer. Int. J. Clin. Oncol. 7, 0279–0283. doi:10.1007/s101470200040
Pawar, S. S., and Rohane, S. H. ((2021). Review on discovery studio: an important tool for molecular docking, Sch. J., 14, 1, 3. doi:10.5958/0974-4150.2021.00014.6
Pervin, S., Singh, R., Hernandez, E., Wu, G., and Chaudhuri, G. (2007). Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: involvement of mammalian target of rapamycin/eIF4E pathway. Cancer Res. 67 (1), 289–299. doi:10.1158/0008-5472.can-05-4623
Pingaew, R., Mandi, P., Prachayasittikul, V., Prachayasittikul, S., Ruchirawat, S., and Prachayasittikul, V. (2018). Synthesis, molecular docking, and QSAR study of sulfonamide-based indoles as aromatase inhibitors. Eur. J. Med. Chem. 143, 1604–1615. doi:10.1016/j.ejmech.2017.10.057
Plati, J., Bucur, O., and Khosravi-Far, R. (2008). Dysregulation of apoptotic signaling in cancer: molecular mechanisms and therapeutic opportunities. J. Cell. Biochem. 104 (4), 1124–1149. doi:10.1002/jcb.21707
Pourbagher-Shahri, A. M., Farkhondeh, T., Talebi, M., Kopustinskiene, D. M., Samarghandian, S., and Bernatoniene, J. (2021). An overview of NO signaling pathways in aging. Molecules 26 (15), 4533. doi:10.3390/molecules26154533
Prior, A. M., Yu, X., Park, E.-J., Kondratyuk, T. P., Lin, Y., Pezzuto, J. M., et al. (2017). Structure-activity relationships and docking studies of synthetic 2-arylindole derivatives determined with aromatase and quinone reductase 1. Bioorg. and Med. Chem. Lett. 27 (24), 5393–5399. doi:10.1016/j.bmcl.2017.11.010
Rani, S., Raheja, K., Luxami, V., and Paul, K. (2021). A review on diverse heterocyclic compounds as the privileged scaffolds in non-steroidal aromatase inhibitors. Bioorg. Chem. 113, 105017. doi:10.1016/j.bioorg.2021.105017
Recanatini, M., Cavalli, A., and Valenti, P. (2002). Nonsteroidal aromatase inhibitors: recent advances. Med. Res. Rev. 22 (3), 282–304. doi:10.1002/med.10010
Riemsma, R., Forbes, C. A., Kessels, A., Lykopoulos, K., Amonkar, M. M., Rea, D. W., et al. (2010). Systematic review of aromatase inhibitors in the first-line treatment for hormone sensitive advanced or metastatic breast cancer. Breast cancer Res. Treat. 123, 9–24. doi:10.1007/s10549-010-0974-0
Santen, R. J., and Henderson, I. C. (1982). A comprehensive guide to the therapeutic use of aminoglutethimide. Switzerland: S. Karger AG.
Saxena, A., Devillers, J., Bhunia, S., and Bro, E. (2015). Modelling inhibition of avian aromatase by azole pesticides. SAR QSAR Environ. Res. 26 (7-9), 757–782. doi:10.1080/1062936x.2015.1090749
Shami, P., Sauls, D., and Weinberg, J. (1998). Schedule and concentration-dependent induction of apoptosis in leukemia cells by nitric oxide. Leukemia 12 (9), 1461–1466. doi:10.1038/sj.leu.2401131
Shaykoon, M. S., Marzouk, A. A., Soltan, O. M., Wanas, A. S., Radwan, M. M., Gouda, A. M., et al. (2020). Design, synthesis and antitrypanosomal activity of heteroaryl-based 1, 2, 4-triazole and 1, 3, 4-oxadiazole derivatives. Bioorg. Chem. 100, 103933. doi:10.1016/j.bioorg.2020.103933
Walsh, E. M., Keane, M. M., Wink, D. A., Callagy, G., and Glynn, S. A. (2016). Review of triple negative breast cancer and the impact of inducible nitric oxide synthase on tumor biology and patient outcomes. Crit. Reviews™ Oncog. 21 (5-6), 333–351. doi:10.1615/critrevoncog.2017021307
Wang, Y., Newton, D. C., and Marsden, P. A. (1999). Neuronal NOS: gene structure, mRNA diversity, and functional relevance. Crit. Reviews™ Neurobiol. 13 (1), 21–43. doi:10.1615/critrevneurobiol.v13.i1.20
Youssif, B. G., Gouda, A. M., Moustafa, A. H., Abdelhamid, A. A., Gomaa, H. A., Kamal, I., et al. (2022). Design and synthesis of new triarylimidazole derivatives as dual inhibitors of BRAFV600E/p38α with potential antiproliferative activity. J. Mol. Struct. 1253, 132218. doi:10.1016/j.molstruc.2021.132218
Youssif, B. G., Mohamed, A. M., Osman, E. E. A., Abou-Ghadir, O. F., Elnaggar, D. H., Abdelrahman, M. H., et al. (2019). 5-Chlorobenzofuran-2-carboxamides: from allosteric CB1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 177, 1–11. doi:10.1016/j.ejmech.2019.05.040
Keywords: indole, pyrazine, aromatase, nitric oxide, synthase, inhibitors
Citation: Al-Wahaibi LH, Abou-Zied HA, Abdelrahman MH, Morcoss MM, Trembleau L, Youssif BGM and Bräse S (2024) Design and synthesis new indole-based aromatase/iNOS inhibitors with apoptotic antiproliferative activity. Front. Chem. 12:1432920. doi: 10.3389/fchem.2024.1432920
Received: 14 May 2024; Accepted: 26 August 2024;
Published: 06 September 2024.
Edited by:
Manuel Aureliano, University of Algarve, PortugalReviewed by:
Custódia Fonseca, University of Algarve, PortugalKhaled Mohamed Darwish, Suez Canal University, Egypt
Copyright © 2024 Al-Wahaibi, Abou-Zied, Abdelrahman, Morcoss, Trembleau, Youssif and Bräse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bahaa G. M. Youssif, Ymd5b3Vzc2lmMkBnbWFpbC5jb20=; Laurent Trembleau, bC50cmVtYmxlYXVAYWJkbi5hYy51aw==; Stefan Bräse, YnJhZXNlQGtpdC5lZHU=