 Muhammad Akash1
Muhammad Akash1 Nehal Rana2Sana Aslam3*
Nehal Rana2Sana Aslam3* Matloob Ahmad1*Muhammad Jawwad Saif4Aneeza Asghar2
Matloob Ahmad1*Muhammad Jawwad Saif4Aneeza Asghar2 Sadia Sultan5,6
Sadia Sultan5,6 Sami A. Al-Hussain7Afifa Liaqat2
Sami A. Al-Hussain7Afifa Liaqat2 Sumera Zaib2*
Sumera Zaib2* Magdi E. A. Zaki7*
Magdi E. A. Zaki7*- 1Department of Chemistry, Government College University Faisalabad, Faisalabad, Pakistan
- 2Department of Basic and Applied Chemistry, Faculty of Science and Technology, University of Central Punjab, Lahore, Pakistan
- 3Department of Chemistry, Government College Women University Faisalabad, Faisalabad, Pakistan
- 4Department of Applied Chemistry, Government College University Faisalabad, Faisalabad, Pakistan
- 5Faculty of Pharmacy, Universiti Teknologi MARA, Puncak Alam, Malaysia
- 6Atta-ur-Rahman Institute for Natural Products Discovery (AuRIns), Universiti Teknologi MARA, Puncak Alam, Malaysia
- 7Department of Chemistry, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh, Saudi Arabia
The urease enzyme is recognized as a valuable therapeutic agent for treating the virulent Helicobacter pylori bacterium because of its pivotal role in aiding the colonization and growth of the bacterium within the gastric mucosa. In order to control the harmful consequences of bacterial infections, urease inhibition presents itself as a promising and effective approach. The current research aimed to synthesize pyridylpiperazine-based carbodithioate derivatives 5a–5n and 7a–7n that could serve as potential drug candidates for preventing bacterial infections through urease inhibition. The synthesized carbodithioate derivatives 5a–5n and 7a–7n were explored to assess their ability to inhibit the urease enzyme after their structural explication by gas chromatography–mass spectrometry (GC-MS). In the in vitro evaluation with thiourea as a standard drug, it was observed that all the synthesized compounds exhibited significant inhibitory activity compared to the reference drug. Among the compounds tested, 5j (bearing an o-tolyl moiety) emerged as the most effective inhibitor, displaying strong urease inhibition with an IC50 value of 5.16 ± 2.68 μM. This IC50 value is notably lower than that of thiourea (23 ± 0.03 μM), indicating the significantly most potent potential of inhibition. In molecular docking of 5j within the active site of urease, numerous noteworthy interactions were identified.
1 Introduction
Urease is an enzyme with two Ni2+ ions at its active site, which facilitates the breakdown of urea into carbonic acid and ammonia by the formation of carbamic acid. Within living organisms, this is the ultimate stage of nitrogen metabolism. It can be found in a diverse range of organisms, including algae, plants, and fungi (Svane et al., 2020). The principal physiological function of urease is to furnish organisms with nitrogen in the form of ammonia to support their growth. Despite that, excessive urease activity can result in the liberation of unusually high levels of ammonia into the atmosphere, potentially causing environmental and economic challenges (Hanif et al., 2012). Urease plays a significant role in the pathogenesis of diseases caused by Helicobacter pylori (Mahernia et al., 2015). H. pylori is a Gram-negative, microaerophilic bacterium that has a significant global presence, affecting more than half of the world population. It typically establishes itself during childhood and, if not addressed, can potentially endure throughout an individual’s lifetime (Tempera et al., 2022).
Urease is a key enzyme that benefits the H. pylori bacterium by enabling its survival in the acidic conditions of the stomach. Consequently, this bacterial presence can lead to gastrointestinal (GIT) diseases, peptic ulcer, gastritis, and even gastric cancer (Mahernia et al., 2015). Urease activity is a critical factor in H. pylori infection, as bacteria lacking functional urease lose their capacity to establish an infection and colonize the host (Gull et al., 2016). Over the past few decades, various treatment regimens have been proposed for curing H. pylori infection. Among these, the triple-therapy regimens comprising proton pump, amoxicillin, and clarithromycin inhibitors have been the most commonly prescribed method for H. pylori eradication. Despite achieving a success rate of 80%, it is imperative to acknowledge certain drawbacks, including unwanted side effects and the emergence of antibiotic resistance, all of which somewhat limit its clinical utility (Zhou et al., 2017). However, in the early stages of the infection, targeting the activity of urease can eliminate the bacterium. The research interest in designing novel urease inhibitors has surged due to the role of urease in bacterial infections (Imran et al., 2020). Therefore, the primary approach for managing infections caused by microorganisms that produce urease involves the use of urease inhibitors (Alqahtani et al., 2022).
In recent decades, researchers have successfully crystallized urease enzymes derived from various bacterial and plant sources. These crystals were obtained both in their isolated form and in conjunction with inhibitors. As a result, we now possess a comprehensive understanding of both the functions and structural characteristics of urease enzymes at the molecular level (Hameed et al., 2019). The primary approach has extensively focused on identifying urease inhibitors that either directly bind to the di-nickeled ions located in the active site enzyme or disrupt its catalytic cycle. Despite the existence of a considerable number of known urease inhibitors, this approach has yielded limited success to date. Only a small subset of these compounds has been evaluated in therapeutic studies, and they have raised concerns related to their effectiveness and safety when used in vivo (Tarsia et al., 2019).
Additionally, some of these compounds have proved ineffective in therapy due to either their low stability, limited bioavailability, or the need for exceptionally high doses. Furthermore, these inhibitors often struggle to efficiently penetrate the plasma membrane of Gram-negative bacteria to access their target within the cytoplasm (Zambelli et al., 2014). To date, there exists only a single clinically approved effective inhibitor, acetohydroxamic acid, although it causes associated adverse side effects. Enzymes have emerged as crucial targets for drug development, and enzyme inhibitors have shown significant success as drugs (Yang et al., 2018). A prevalent approach in designing drugs that target enzymes involves the identification or development of structural analogs resembling the enzyme substrates, effectively mimicking their reactivity. Nevertheless, this approach can face limitations when enzyme active sites are not readily accessible to solvents or when enzyme substrates exhibit a high degree of specificity (Rutherford, 2014). In our recent studies, the pyridylpiperazine hybrid was found to be a highly active scaffold against the urease enzyme (Figure 1) (Akash et al., 2024). In another report, S-benzyl-substituted carbodithioate emerged as a highly potent urease inhibitor (Figure 1) (Khan et al., 2024). The coupling of two or more pharmacologically important scaffolds is a diverse approach that can produce highly potent hybrid molecules. In continuation of our previous research dealing with the development of urease enzyme inhibitors (Akash et al., 2024), we herein report a novel series of pyridylpiperazine-based carbodithioates as potent urease inhibitors.
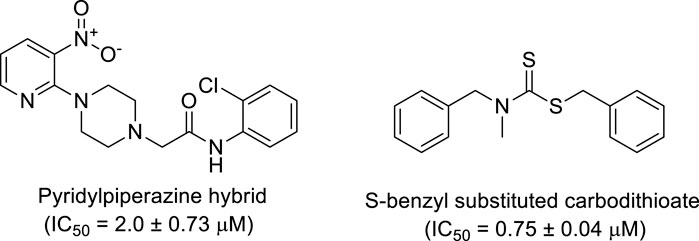
Figure 1. Chemical structures of the pyridylpiperazine hybrid and S-benzyl-substituted carbodithioate—highly potent urease inhibitors.
2 Results and discussions
2.1 Chemistry
A number of 2-oxo-2-(arylamino)ethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioates 5a–5n and 4-((aryl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioates 7a–7n were synthesized, as shown in Scheme 1. The treatment of 2-chloro-3-nitropyridine 1 carried out with excess of piperazine 2 in acetonitrile on reflux for 12 h furnished 1-(3-nitropyridin-2-yl)piperazine 3 in 65% yield. The reaction of pyridinylpiperazine 3 with 2-chloro-N-arylacetamides 4a–4n in the presence of CS2 and NaOAc in methanol under reflux for 8–16 h produced 2-oxo-2-(arylamino)ethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate 5a–5n (Figure 2) in average in good yield (62%–88%). In a similar manner, the reaction between pyridinylpiperazine 3 and 4-(chloromethyl)-N-arylbenzamide 6a–6n in the presence of CS2 and NaOAc in methanol under reflux for 12–24 h produced 4-((aryl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate 7a–7n (Figure 3) in moderate yield (49%–71%). All the synthesized compounds 5a–5n and 7a–7n were extracted from the reaction mixture via precipitation and purified by column chromatography. The structures were elucidated via spectroscopic techniques. The HRMS proved the predicted chemical formula the molecular ion peak of each compound. The 1H NMR spectrum of 5a–5n and 7a–7n justifies the presence of methylene protons at a shift value (δ) of 4.25–4.35 ppm and 4.66–4.69 ppm, respectively. Meanwhile, the 13C NMR spectra of 5a–5n and 7a–7n revealed the presence of S-C=S in all the compounds at a shift value (δ) of 194.4–195.3 ppm.
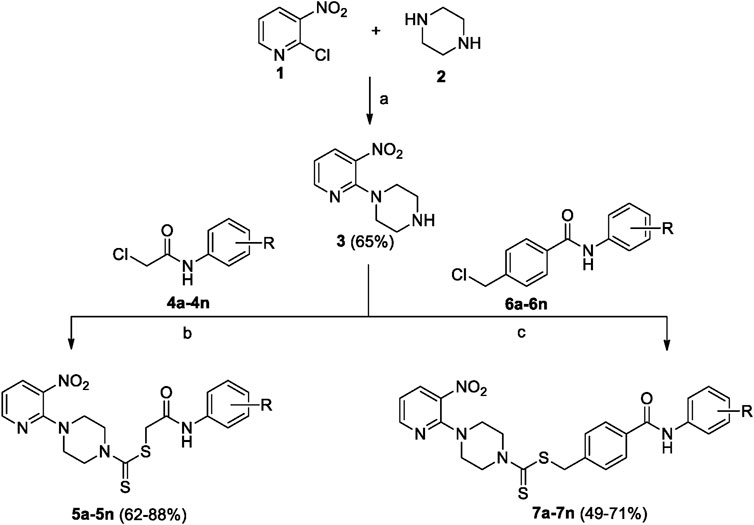
Scheme 1. 2-Oxo-2-(arylamino)ethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioates 5a–5n and 4-((aryl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioates 7a–7n. Reagents and conditions: (a) acetonitrile, reflux, 12 h; (b) CS2, NaOAc acetonitrile, reflux, 8–16 h; and (c) CS2, NaOAc, acetonitrile, reflux, 12–24 h.
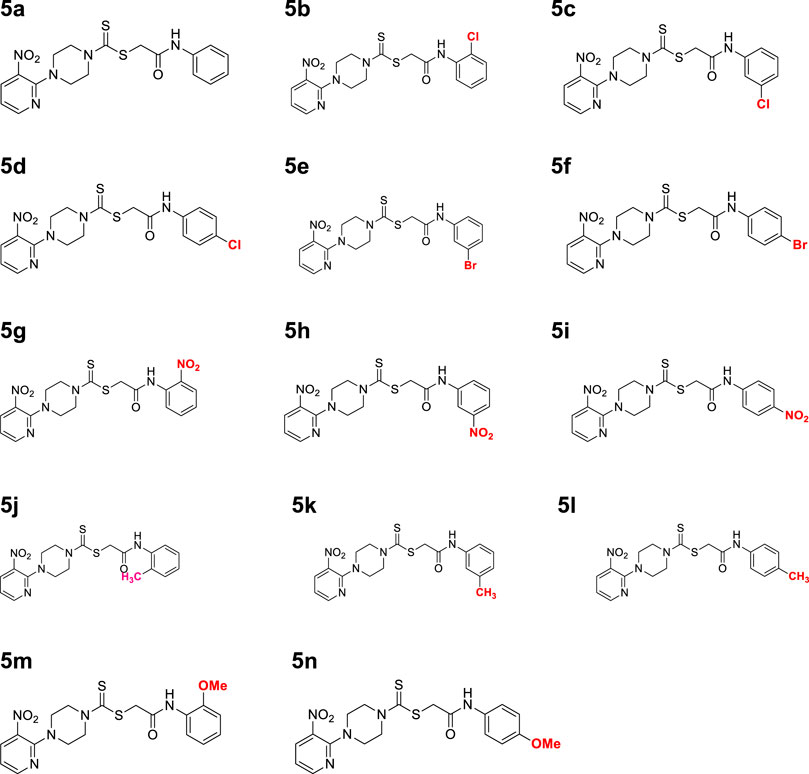
Figure 2. Chemical structures of compounds 5a–5n.
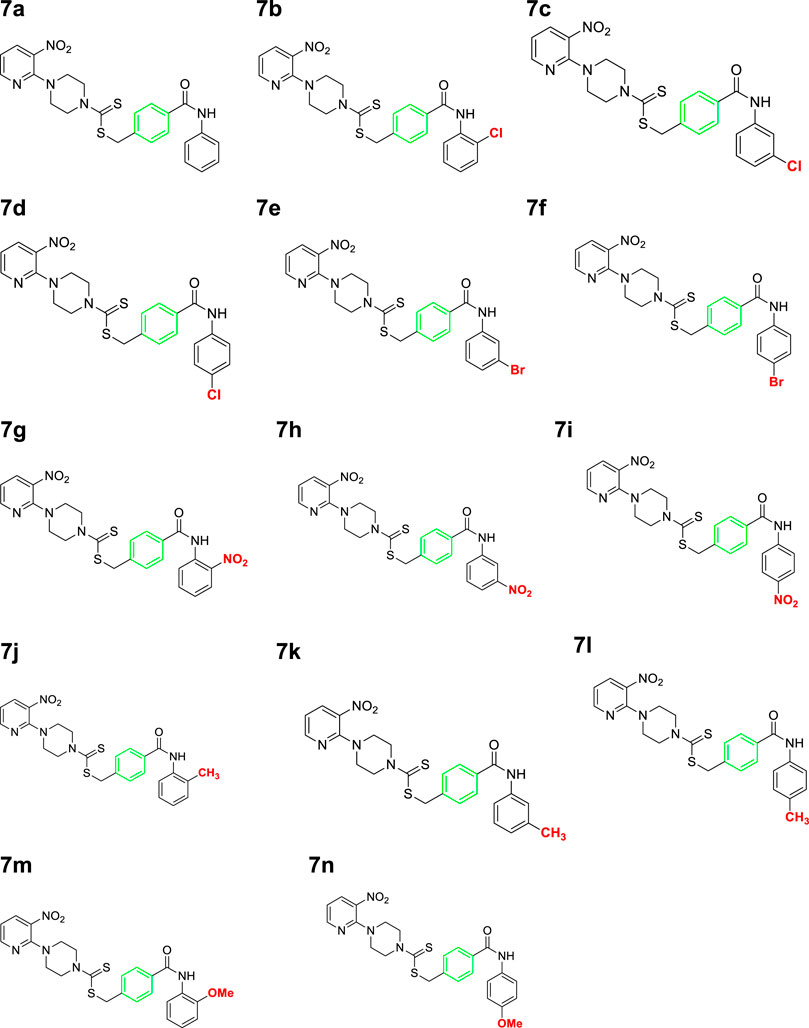
Figure 3. Chemical structures of compounds 7a–7n.
2.2 In vitro inhibition and structure–activity relationship analysis
Using thiourea as a standard (IC50 = 23.00 ± 0.03 μM), the compounds that we synthesized were estimated against urease for their inhibitory potential. All the experimental data are given in Table 1. Generally, the compounds (5a, 5f, 5g, 7e, 7f, 5j–5n, 7a, 7c, 7e, 7f, 7i–7k, and 7m) showed potent activity against the urease enzyme in a range of 5.16–21.34 μM. The in vitro analysis showed that compound 5j was the most effective inhibitor of urease in the series, possessing an IC50 value of 5.16 ± 2.68 μM, which is 22-fold more potent than the standard (thiourea; IC50 = 23.00 ± 0.03 μM). The compounds 5j, 5k, and 5l possess a methyl group at the ortho, meta, and para positions, respectively; however, 5j, with an IC50 value of 5.16 ± 2.68 μM, revealed stronger inhibitory activity against urease than 5k (IC50 = 18.30 ± 0.17 μM) and 5l (IC50 = 10.61 ± 0.36 μM). A notable decrease was observed in the activity of compounds 5b (IC50 = 41.03 ± 0.23 μM), 5c (IC50 = 37.33 ± 0.19 μM), and 5d (IC50 = 55.69 ± 0.20 μM) when the chloro (-Cl) group was added instead of the methyl (-CH3) group in piperazine-1-carbodithioate. The presence of the nitro (-NO2) group in compound 5g with IC50 = 16.84 ± 0.12 μM as a substituent at the ortho position exhibited effective inhibitory potential, while the nitro group at the meta and para positions in compounds 5h with IC50 = 26.98 ± 2.15 μM and 5i with IC50 = 45.41 ± 0.23 μM showed poor inhibitory potential compared to the positive control (thiourea). Furthermore, the compounds 5n (17.77 ± 0.29 μM) with the methoxy group at position 4 and 5a (IC50 = 18.67 ± 0.52 μM) with no substituent revealed almost the same inhibitory activity, while compound 5m with the methoxy group at position 2 showed effective inhibitory activity with an IC50 value of 08.17 ± 0.37 μM. Effective inhibition was shown by 7j (IC50 = 12.26 ± 0.27 μM) and 7k (IC50 = 10.65 ± 0.26 μM) when the tolylcarbamoyl group was attached at the ortho and meta positions, respectively; however, a decrease in activity was observed when the same substituent was attached at the para position in compound 7l with IC50 = 37.54 ± 0.26 μM. When methoxyphenyl was inserted at position 2 on 7m (IC50 = 18.67 ± 0.27 μM), it showed good inhibition; however, when the same group was introduced at position 4 in 7n (IC50 = 24.02 ± 0.27 μM), the inhibition activity was not effective. The addition of the nitrophenyl group at position 2 and position 3 in 7g (IC50 = 56.89 ± 0.31 μM) and 7h (IC50 = 47.94 ± 0.25 μM) showed poor inhibition, while when the nitrophenyl group was introduced at position 4 instead of position 2 or 3 in compound 7i with IC50 of 10.51 ± 0.34 μM, it showed effective inhibitory activity against urease. Compound 7a without any substitution exhibited IC50 of 11.69 ± 0.26 μM and revealed effective inhibitory activity. However, when the chlorophenyl group was attached at position 3 in 7c (IC50 = 10.80 ± 0.52 μM), the same inhibitory potential was observed, while when the same group was added at positions 2 and 4 in compounds 7b (IC50 = 32.53 ± 0.31 μM) and 7d (IC50 = 43.11 ± 0.31 μM), respectively, the inhibitory activity decreased. The same inhibitory potential against urease was shown by 7e (IC50 = 16.50 ± 0.28 μM) and 7f (IC50 = 16.83 ± 0.29 μM) when the bromophenyl group was inserted at position 3 in 7e and at position 4 in 7f.
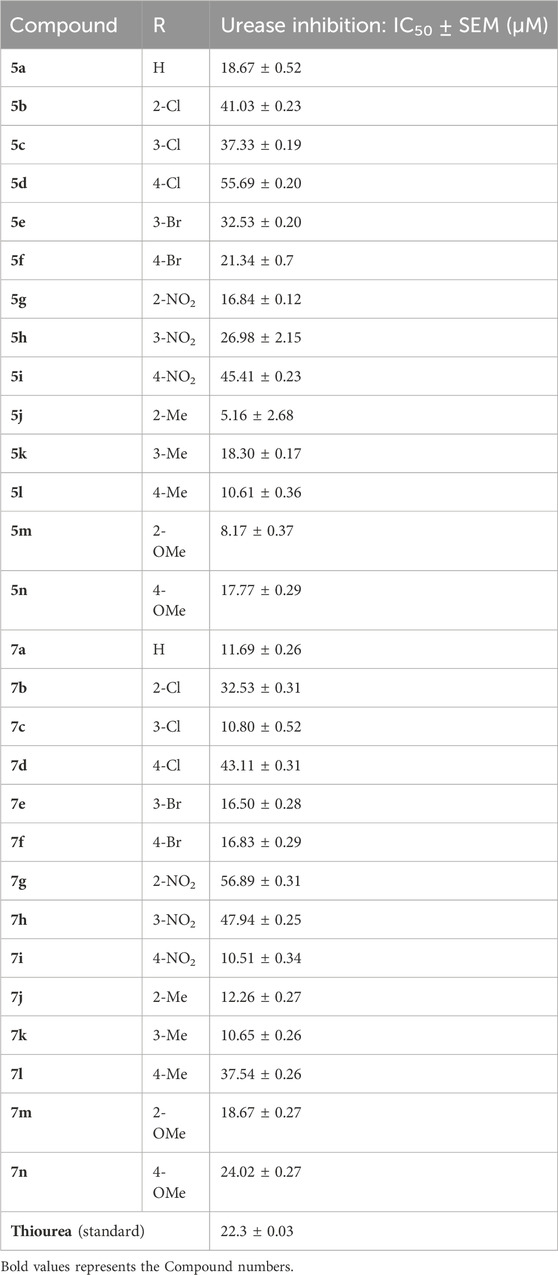
Table 1. Inhibitory concentration of the synthesized compounds and thiourea against urease was determined by calculating the IC50 values.
Comparing both the series and the impact of substituents on the activity of the overall nucleus showed that halogen substitutions are favorable for the 7 series compared to the 5 series. Among halogen substituents, chlorine substitution at the meta position of 7c was more impactful than that in 5c. Similarly, the presence of bromine at either the meta or para position of 7e and 7f showed similar and good inhibitory results compared to that in 5e and 5f.
Nitro-group substitutions at the ortho and meta positions were found to be favorable for increasing the urease inhibitory activities of 5g and 5h, while at the para position, the nitro group enhances the inhibitory potential of 7i. Methyl substitution at the ortho position was favorable for 5j, while when placed at the meta position, it increased the inhibitory potential of 7k but not of 5k. Contrarily, the methyl group at the para position increases the urease inhibition for 5l. Lastly, methoxy substitution showed better outcomes in 5m and 5n than in 7m (Table 1).
2.3 Kinetic studies
By using Lineweaver–Burk graphs, we determined the mechanism of action of the leading compound 5j against urease. We evaluated the effect of the inhibitor on Km and Vmax using reciprocal 1/S and 1/V of the product concentration and calculated the inhibition type. The slope Km/Vmax of each line was schemed against different concentrations of the substrate and inhibitor. In kinetic studies of the potent compound, different concentrations of the compound and substrate were used. Different concentrations of 1 mM 5j were 0, 2.55, 5.11, and 7.65 μM, and different concentrations of the substrate were 0, 25, 50, 100, and 150 mM. Compound 5j is shown in Figure 4, which illustrates a mixed type of inhibition against urease.
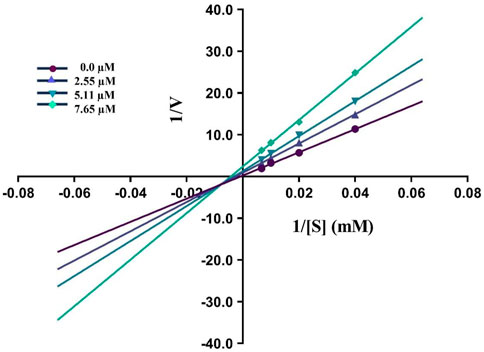
Figure 4. Demonstrated the Inhibition type of compound 5j against urease.
2.4 Molecular docking and intermolecular interactions
Based on the results of dilutions, the five compounds 5j, 5l, 5m, 7i, and 7k were selected to perform molecular docking against urease (PDB ID: 3LA4). Compounds 5j, 5l, 5m, 7i, and 7k exhibited binding affinities of −7.1, −6.6, −6.6, −7.4, and −6.7 kcal/mol, respectively, with the first pose. On the other hand, the binding energy of thiourea was found to be −3.2 kcal/mol when docked against the same binding site of urease. However, for further visualization through Discovery Studio, we selected the first pose of all the compounds and thiourea with the lowest binding affinity. The 2D and 3D interactions for compounds 5j, 5l, 5m, 7i, and 7k against urease are shown in Figure 5. Compounds 5j, 5l, and 5m revealed conventional hydrogen, alkyl, C-H, and π–alkyl bonds with different residues of amino acids, as shown in Table 2.
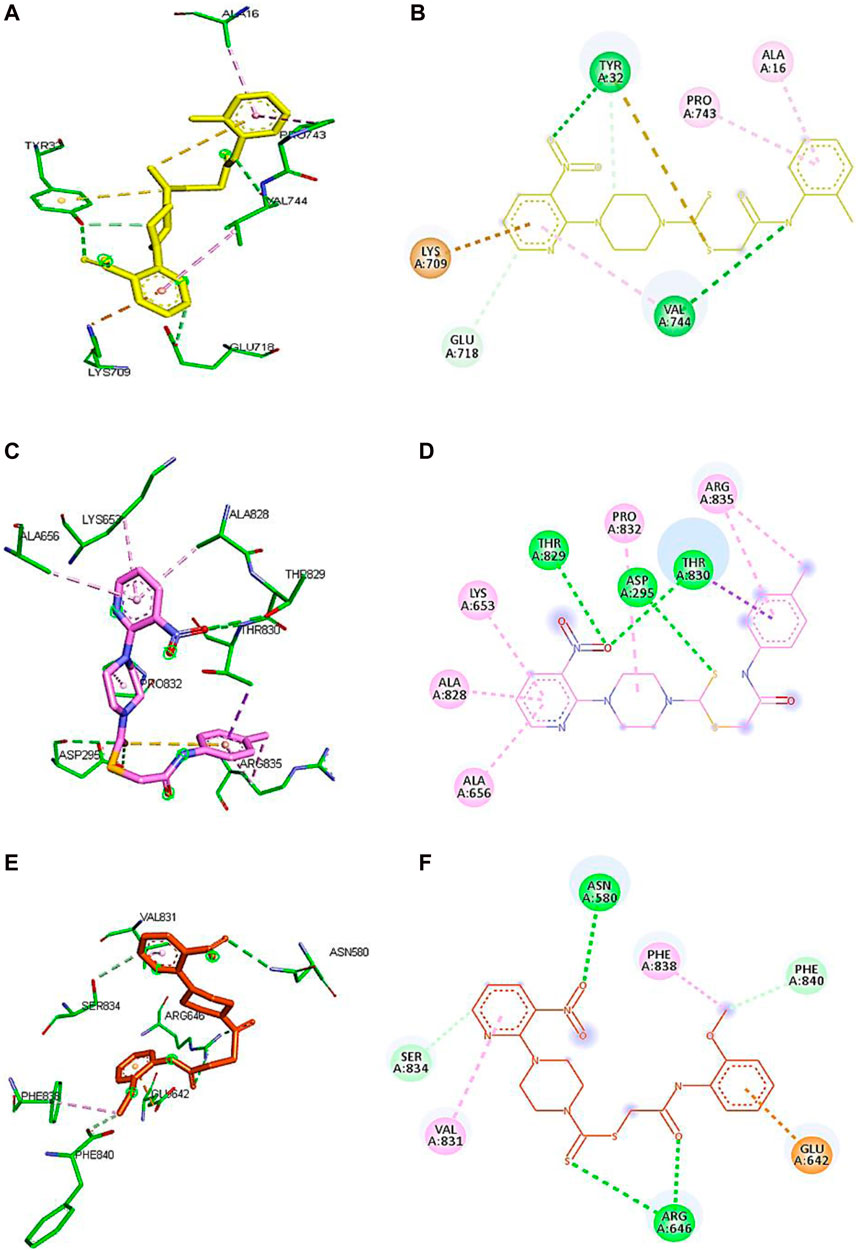
Figure 5. Demonstration of 3D and 2D interactions of compounds 5j, 5m, and 5l with urease. Green represents the amino acid residues, while yellow, purple, and red show the ligand in 3D structure (A,C,E). It explains the correlation between the ligand and residues of the amino acid by different types of interactions; (B,D,F) 2D image of the ligand and amino acid residues.
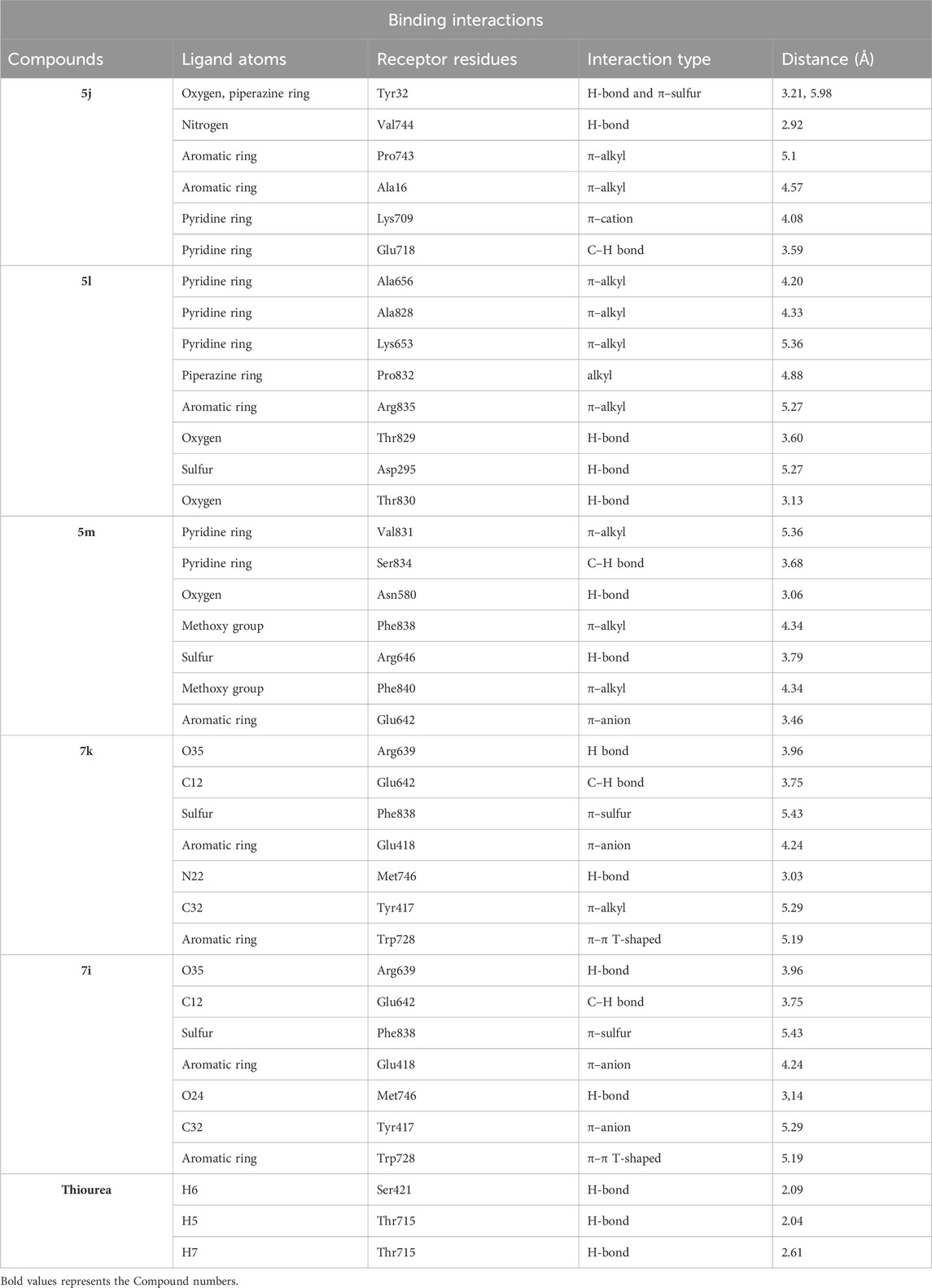
Table 2. Binding interactions and type of compounds against urease. Different interactions between amino acid residues and ligand are reported below.
Compound 5j interacts with the urease binding site by hydrophilic, hydrophobic, and electrostatic interactions. The pyridine ring of 5j forms π–cation and carbon–hydrogen bonds with Lys709 (4.08 Å) and Glu718 (3.59 Å) of the urease active site, respectively. The aromatic ring of 5j exhibits π–alkyl interactions with Pro743 and Ala16 of the active pocket. Lastly, hydrogen bond interactions are also present between Tyr32 (3.21 Å) and oxygen and nitrogen and Val744 (2.92 Å). Similarly, the pyridine ring of 5l forms π–alkyl interactions with Ala656 (4.20 Å), Ala828 (4.33 Å), and Lys653 (5.36 Å). Apart from the pyridine ring, the aromatic ring of 5l also develops π–alkyl interactions with Arg835 (5.27 Å) of urease. Moreover, Thr829 (3.60 Å), Asp295 (5.27 Å), and Thr830 (3.13 Å) of urease are found to be involved in hydrogen bond interactions with the oxygen and sulfur atoms of 5l, as shown in Figure 6. In the case of 5m, the pyridine ring is involved in the π–alkyl and hydrogen bond interaction formation with Val831 (5.36 Å) and Ser834 (3.68 Å), respectively. Other hydrogen bond interactions are formed by Asn580 (3.06 Å) and Arg646 (3.79 Å) of urease with the oxygen and sulfur of 5m, respectively. Furthermore, the substituent of 5m, the methoxy group, also forms π–alkyl interactions with Phe838 (4.34 Å) and Phe840 (4.34 Å) of the active site of urease.
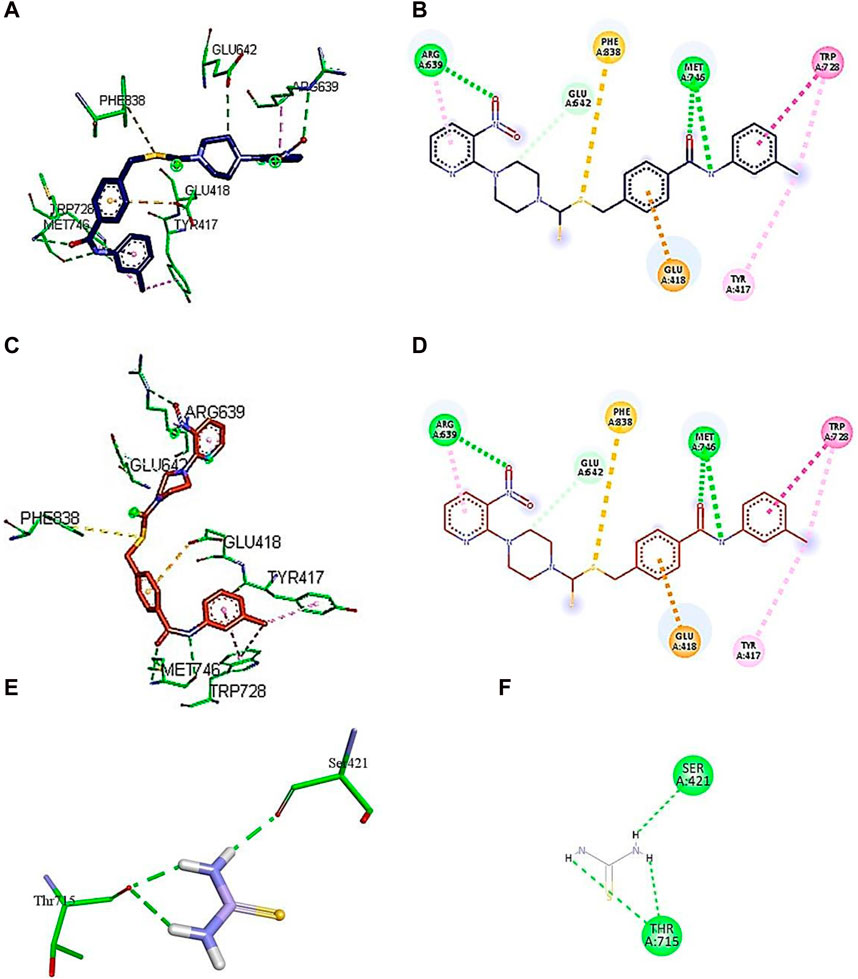
Figure 6. Demonstration of 3D and 2D interactions of compounds 7k and 7i and thiourea with urease. Green represents the amino acid residues, while blue, red, and purple show the ligand in 3D structure (A, C,E). It explains the correlation between the ligand and residues of the amino acid by different types of interactions; (B,D,F) 2D image of the ligand and amino acid residues.
A conventional hydrogen bond was revealed between MET746 oxygen and nitrogen of compound 7k with a distance of 3.14 and 3.03 Å, respectively, and the same type of interaction was found between ARG639 and the oxygen atom, with a distance of 3.97 Å. The carbon–hydrogen bond was observed between GLU642 and the benzene ring of the compound (3.75 Å). The π–anion interactions were observed between the sulfur atom and PHE838 (5.43 Å). The π–sulfur interactions were shown between the benzene ring of the compound and GLU418 with a distance of 4.24 Å. The π–π T-shaped interactions were revealed between the benzene ring and TRP728 (5.09 Å). Moreover, the π–alkyl interactions were observed between TRP728 (5.19 Å) and the carbon atom, TYR417 (5.29 Å) and the carbon atom, and ARG639 (3.96 Å) and the benzene ring of the compound. A conventional hydrogen bond was observed between MET746 oxygen and nitrogen of compound 7i with a distance of 3.14 and 3.03 Å, respectively, and the same type of interaction was found between ARG639 and the oxygen atom with a distance of 3.97 Å. The carbon–hydrogen bond was observed between GLU642 and the benzene ring of the compound (3.75 Å). The π–anion bond was observed between the sulfur atom and PHE838 (5.43 Å). The π–sulfur interactions were observed between the benzene ring of the compound and GLU418 with a distance of 4.24 Å. The π–π T-shaped interactions were revealed the between benzene ring and TRP728 (5.09 Å). Moreover, the π–alkyl interactions were observed between TRP728 (5.19 Å) and the carbon atom, TYR417 (5.29 Å) and the carbon atom, and ARG639 (3.96 Å) and the benzene ring of the compound, as shown in Figure 6. The interactions of thiourea were also analyzed, which showed that it only develops conventional hydrogen bond interactions with Ser421 (2.09 Å) and Thr715 (2.04 and 2.61 Å).
In light of the above discussion and literature review, it has been observed that interactions with Ala16, Tyr32, Asp235, Val744, Thr830, Val831, Pro832, Ser834, Arg835, and Phe840 residues in the urease binding site are crucial. It is because these are involved in the inhibition of urease by interacting with numerous active compounds (Khan et al., 2019; Hina et al., 2023; Akash et al., 2024).
2.5 Molecular dynamics simulation
iMODs was used to estimate the protein–ligand docking to determine the deformability in the main chain and the deformed nature of the residues. Some higher peaks for compound 5j and eigenvalues state the stiffness of the model shown in Figure 7A. Its eigenvalue was low, explaining that low energy is required to deform the structure. The graphs shown in Figure 7B represent the eigenvalues of 5j, i.e., 3.085545e-05. The elastic network model is shown in Figure 7C, where each dot represents one spring and pair of atoms. It further explains the stiffness and spring stiffness of compound 5j to the protein. In addition, Figure 7D represents the covariance matrix, in which the motion of atoms can be seen as correlated and anti-correlated because the red and blue regions are more obvious than the white regions.
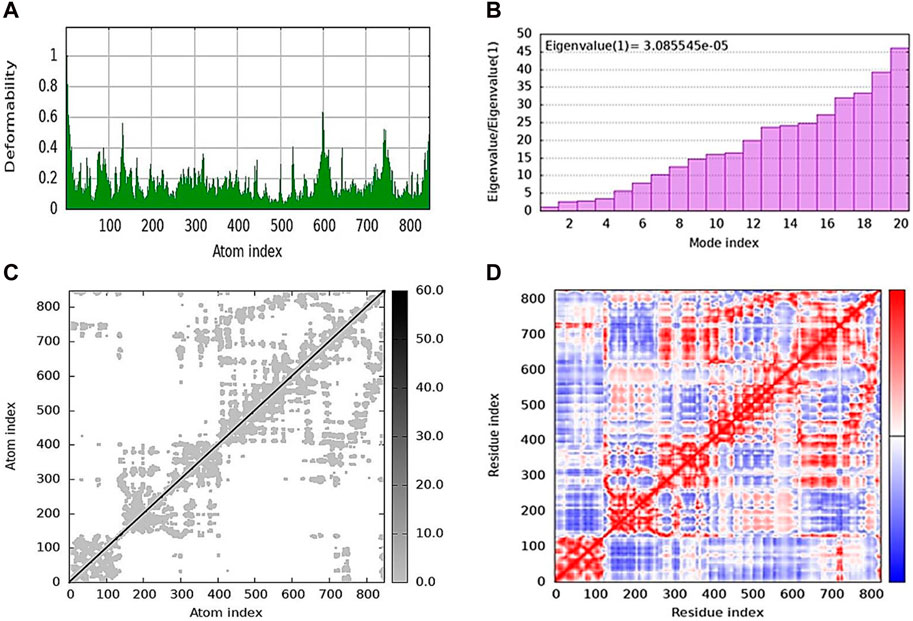
Figure 7. Molecular dynamics simulation of 5j obtained from the iMOD server. (A) Deformability plot indicating that most residues are stable. (B) Eigenvalue is low, predicting the requirement of low energy to deform the protein. (C) Elastic network model showing the stiffness of the protein chain with darker gray. (D) Covariance matrix analysis showing that most of the atoms in the protein structure have correlated (red color) and anti-correlated (blue) motions.
2.6 Computational ADMET analysis
SwissADME was applied to assess the drug-likeness characteristics of compounds 5j, 5l, 5m, 7k, and 7i with respect to urease. The pharmacokinetic properties of 5j, 5l, 5m, 7k, and 7i were examined by interpreting the results based on the compound structure. In this case, the evaluation indicated that all these compounds could serve as a lead since they met all the specified parameters. Compounds 5j, 5l, and 5m showed molecular weights of 431.53, 292.78, and 447.53 g/mol, respectively, their topological polar surface areas (TPSAs) were 151.68, 69.70, and 160.91, respectively, and the consensus log P o/w was lower than 5. They further showed synthetic accessibility of 3.43, 2.86, and 3.44, as shown in Table 3. The ADMET analysis of compounds 7k and 7i showed that they have a molecular weight of 507.63 and 538.60 g/mol, respectively. It further showed that that the TPSAs were 151.68 and 197.50 Å2, respectively, and the consensus log P o/w was lower than 5, as shown in Table 3.
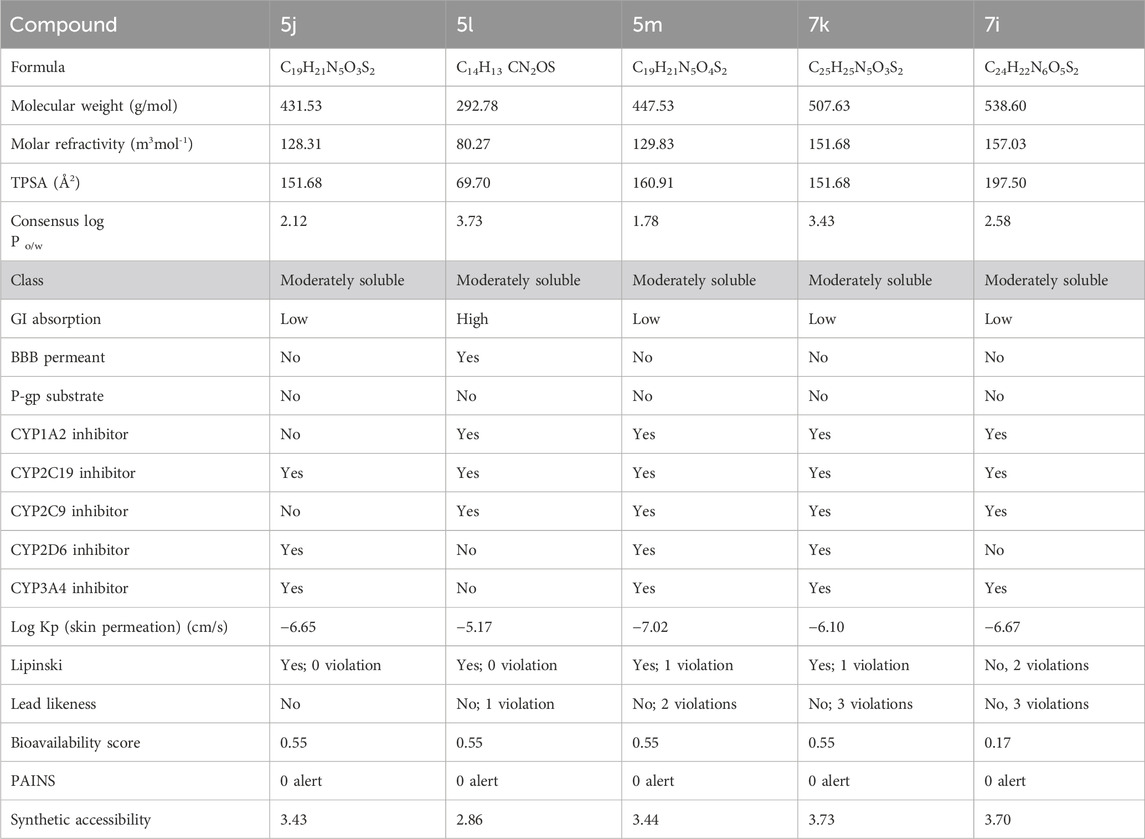
Table 3. ADMET analysis for determining the drug likeliness and lead likeliness properties of compounds 5j, 5l, 5m, 7k, and 7i against urease.
3 Materials and methods
3.1 General
All the chemicals, reagents, and solvents were purchased from Alfa Aesar (Kandel, Germany) and utilized without any further purification. The 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were recorded in dimethyl sulfoxide (DMSO) using a Bruker DPX spectrophotometer (Bruker; Zürich, Switzerland). The chemical shifts were recorded in ppm reference to tetramethylsilane. Thin-layer chromatography (CHCl3/MeOH) was used in combination with a Spectroline E-Series UV lamp to monitor the progress of chemical reactions (Alfa Aesar, Kandel, Germany). Melting points were recorded on a Gallenkamp instrument (Fisons; Uckfield, United Kingdom). Compounds 2, 5a–5o, and 7a–k were produced according to Scheme 1.
3.2 Procedure for 1-(3-nitropyridin-2-yl)piperazine (3)
A homogenous solution of piperazine (2) (40.5 g, 472 mmol) was prepared in acetonitrile (100 ml) in a round-bottom flask (solution A). On the other hand, 15 g (94.30 mmol) of 2-chloro-3-nitropyridine (1) was dissolved in 50 ml of acetonitrile in a beaker (solution B). Solution B was added dropwise to solution A under constant stirring. The reaction mixture was refluxed for 12 h, and the reaction progress was monitored using TLC. On completion of the reaction, the reaction mixture was cooled to room temperature, and 100 ml of ice-cold distilled water was added to it. From the mixture, 1-(3-nitropyridin-2-yl)piperazine (3) was extracted with chloroform and further purified by column chromatography with CHCl3/MeOH. On evaporating the solvent, yellow crystals of (3) were produced with 65% yield.
3.2.1 1-(3-Nitropyridin-2-yl)piperazine (3)
Yield, 65%; brownish–yellow solid; mp: 77°C–79°C. 1H NMR (δ): 8.29–8.30 (m, 1H, Ar-H), 8.15 (dd, 1H, J = 7.95 Hz, 1.8 Hz, Ar-H), 6.82–6.84 (m, 1H, Ar-H), 4.2 (t, 4H, J = 5 Hz, piperazinyl), 3.23–3.25 (m, 4H, piperazinyl), and 2.50–2.51 (m, 1H, NH). 13C NMR (δ): 152.6679 (NCH), 152.5184 (N=C-N), 136.4210 (C(CH)2), 132.7990 (CH(C)C), 114.3262 (CHCH), 48.7025 (NCH2CH2), and 45.0161(NHCH2CH2). Elemental analysis of C9H12N4O2: calculated: C, 51.92; H, 5.81; and N, 26.9; found: C, 51.97; H, 5.77; and N, 26.88%.
3.3 Procedure for the synthesis of 2-oxo-2-(arylamino)ethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioates 5a–5n
A mixture of 1-(3-nitropyridin-2-yl) piperazine (3) (0.15 mmol) and NaOAc (0.30 mmol) was prepared in 15 ml of acetonitrile. The mixture was stirred for 15 min, and CS2 (0.30 mmol) was added dropwise under constant stirring for 30 min at room temperature. On the other hand, a solution of 2-chloro-N-arylacetamides 4a–4o (0.15 mmol) was prepared in 5 ml of acetonitrile and added to the above mixture. The resulting mixture was refluxed for 8–16 h under stirring and monitored using TLC. Finally, the addition of ice-cold water resulted in yellow–orange precipitates of 2-oxo-2-(arylamino)ethyl 4-(3-nitropyridin-2-yl) piperazine-1-carbodithioate derivatives 5a–5o. The ppts were collected and purified by column chromatography.
3.3.1 2-Oxo-2-(phenylamino)ethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5a)
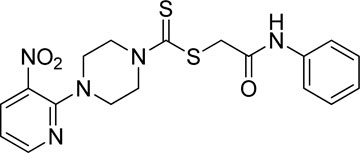
Yield, 75%; yellow solid; mp: 119°C–120°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.27 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.65 Hz, 1.42 Hz, Ar-H), 7.57 (d, 2H, J = 7.25 Hz, Ar-H), 7.31 (t, 2H, J = 6.45 Hz, Ar-H), 7.05 (t, 1H, J = 6.85 Hz, Ar-H), 6.96 (q, 1H, J = 6.70 Hz, Ar-H), 4.32 (Br s, 2H, piperazinyl), 4.26 (s, 2H, methylene), 4.14 (Br s, 2H, piperazinyl), and 3.6 (Br s, 4H, piperazinyl). Elemental analysis of C18H19N5O3S2: calculated: C, 51.78; H, 4.59; and N, 16.77; found: C, 51.75; H, 4.63; and N, 16.75%.
3.3.2 2-((2-Chlorophenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5b)
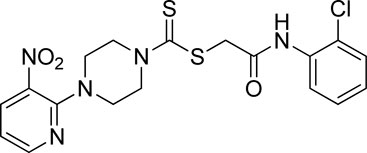
Yield, 68%; yellow solid; mp: 114°C–116°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 9.74 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.35 Hz, Ar-H), 7.76 (d, 1H, J = 6.75 Hz, Ar-H), 7.49 (dd, 1H, J = 6.7 Hz, 1.20 Hz, Ar-H), 7.33 (t, 1H, J = 6.45 Hz, Ar-H), 7.18 (t, 1H, J = 7.75, Ar-H), 6.96 (q, 1H, J = 7.70 Hz, Ar-H), 4.35 (s, 2H, methylene), 4.33 (Br s, 2H, piperazinyl), 4.14 (Br s, 2H, piperazinyl), and 3.6 (Br s, 4H, piperazinyl). 13C NMR (DMSO-d6 125 MHz) δ (ppm): 194.9 (N(C=S)S), 166.4 (C=O), 152.6 (Ar-C), 152.0 (Ar-C), 136.3 (Ar-C), 135.1 (Ar-C), 132.6 (Ar-C), 129.9 (Ar-C), 127.9 (Ar-C), 126.7 (Ar-C), 126.3 (Ar-C), 125.8 (Ar-C), 114.4 (Ar-C), 50.8 (piperazine, NCH2), 49.0 (piperazine, NCH2), 46.4 (piperazine, 2 x NCH2), and 40.8 (SCH2(CO)). Elemental analysis of C18H18ClN5O3S2: calculated: C, 47.84; H, 4.01; and N, 15.50; found: C, 47.88; H, 4.04; and N, 15.47%.
3.3.3 2-((3-Chlorophenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5c)
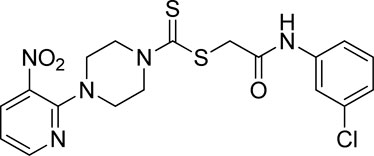
Yield, 65%; yellow solid; mp: 121°C–122°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.48 (s, 1H, NH), 8.44–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.45 Hz, Ar-H), 7.78 (s, 1H, Ar-H), 7.44 (d, 1H, J = 6.90 Hz, Ar-H), 7.34 (t, 1H, J = 6.70 Hz, Ar-H), 7.11 (d, 1H, J = 6.70 Hz, Ar-H), 6.96 (q, 1H, J = 6.7 Hz, Ar-H), 4.31 (Br s, 2H, piperazinyl), 4.29 (Br s, 2H, methylene), 4.13 (Br s, 2H, piperazinyl), and 3.6 (Br s, 4H, piperazinyl). Elemental analysis of C18H18ClN5O3S2: calculated: C, 47.84; H, 4.01; and N, 15.50; found: C, 47.80; H, 4.05; and N, 15.55%.
3.3.4 2-((4-Chlorophenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5d)
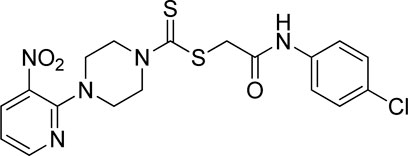
Yield, 72%; yellow solid; mp: 143°C–144°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.41 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.40 Hz, Ar-H), 7.61 (d, 2H, J = 7.4 Hz, Ar-H), 7.36 (d, 2H, J = 7.4, Ar-H), 6.96 (q, 1H, J = 6.70 Hz, Ar-H), 4.31 (Br s, 2H, piperazinyl), 4.28 (s, 2H, methylene), 4.13 (Br s, 2H, piperazinyl), and 3.6 (Br s, 4H, piperazinyl). Elemental analysis of C18H18ClN5O3S2: calculated: C, 47.84; H, 4.01; and N, 15.50; found: C, 47.89; H, 3.98; and N, 15.54%.
3.3.5 2-((3-Bromophenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5e)
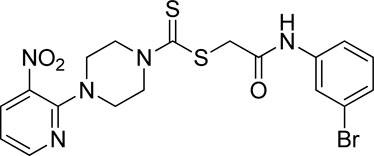
Yield, 72%; yellow solid; mp: 110°C–112°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.46 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.40 Hz, Ar-H), 7.93 (s, 1H, Ar-H), 7.48 (d, 1H, J = 6.65 Hz, Ar-H), 7.23–7.29 (m, 2H, Ar-H), 6.96 (q, 1H, J = 6.7 Hz, Ar-H), 4.31 (Br s, 2H, piperazinyl), 4.26 (s, 2H, methylene), 4.13 (Br s, 2H, piperazinyl), and 3.6 (Br s, 4H, piperazinyl). Elemental analysis of C18H18BrN5O3S2: calculated: C, 43.55; H, 3.65; and N, 14.11; found: C, 43.51; H, 3.70; and N, 14.13%.
3.3.6 2-((4-Bromophenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5f)
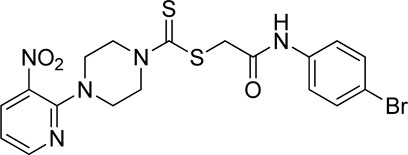
Yield, 76%; yellow solid; mp: 136°C–138°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.48 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.33 (dd, 1H, J = 6.65 Hz, 1.40 Hz, Ar-H), 7.95 (s, 1H, Ar-H), 7.48 (d, 1H, J = 6.70 Hz, Ar-H), 7.28–7.33 (m, 2H, Ar-H), 6.98 (q, 1H, J = 6.65 Hz, Ar-H), 4.33 (Br s, 2H, piperazinyl), 4.29 (s, 2H, methylene), 4.15 (Br s, 2H, piperazinyl), and 3.62 (Br s, 4H, piperazinyl). Elemental analysis of C18H18BrN5O3S2: calculated: C, 43.55; H, 3.65; and N, 14.11; found: C, 43.51; H, 3.68; and N, 14.13%.
3.3.7 2-((2-Nitrophenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5g)
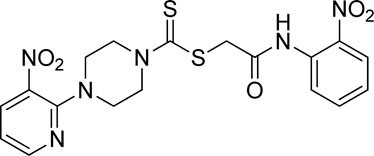
Yield, 82%; yellow solid; mp: 149°C–151°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.65 (s, 1H, NH), 8.44–8.46 (m, 1H, Ar-H), 8.32 (dd, 1H, J = 6.75 Hz, 1.35 Hz, Ar-H), 8.03 (d, 1H, J = 6.70 Hz, Ar-H), 7.85 (d, 1H, J = 6.95 Hz, Ar-H), 7.78 (t, 1H, J = 6.85 Hz, Ar-H), 7.35 (t, 1H, J = 6.75, Ar-H), 6.95 (d, 1H, J = 6.65 Hz, Ar-H), 4.34 (Br s, 2H, methylene), 4.26 (Br s, 2H, piperazinyl), 4.17 (Br s, 2H, piperazinyl), and 3.59 (Br s, 4H, piperazinyl). 13C NMR (DMSO-d6 125 MHz) δ (ppm): 194.4 (N(C=S)S), 166.5 (C=O), 152.6 (Ar-C), 152.0 (Ar-C), 141.4 (Ar-C), 136.3 (Ar-C), 134.9 (Ar-C), 132.7 (Ar-C), 131.9 (Ar-C), 125.6 (2 × Ar-C), 124.9 (Ar-C), 114.4 (Ar-C), 50.9 (piperazine, NCH2), 49.1 (piperazine, NCH2), 46.4 (piperazine, 2 × NCH2), and 40.8 (SCH2(CO)). Elemental analysis of C18H18N6O5S2: calculated: C, 46.74; H, 3.92; and N, 18.17; found; C, 46.77; H, 3.89; and N, 18.14%.
3.3.8 2-((3-Nitrophenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5h)
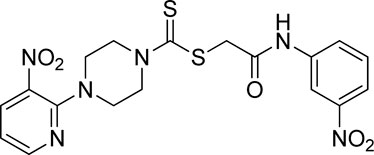
Yield, 85%; yellow solid; mp: 174°C–175°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.63 (s, 1H, NH), 8.45–8.47 (m, 1H, Ar-H), 8.31 (dd, 1H, J = 6.7 Hz, 1.40 Hz, Ar-H), 8.01 (dd, 1H, J = 6.85 Hz, 1.25 Hz, Ar-H), 7.88 (d, 1H, J = 6.90 Hz, Ar-H), 7.73 (t, 1H, J = 6.75 Hz, Ar-H), 7.37 (t, 1H, J = 6.90, Ar-H), 6.96 (d, 1H, J = 6.7 Hz, Ar-H), 4.32 (Br s, 2H, methylene), 4.29 (Br s, 2H, piperazinyl), 4.15 (Br s, 2H, piperazinyl), and 3.61 (Br s, 4H, piperazinyl). Elemental analysis of C18H18N6O5S2: calculated: C, 46.74; H, 3.92; and N, 18.17; found: C, 46.79; H, 3.97; and N, 18.21%.
3.3.9 2-((4-Nitrophenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5i)
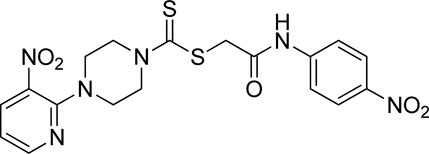
Yield, 88%; yellow solid; mp: 168°C–169°C. 1H NMR (500 MHz) δ (ppm): 10.92 (s, 1H, NH), 8.46–8.47 (m, 1H, Ar-H), 8.31 (dd, 1H, J = 6.7 Hz, 1.40 Hz, Ar-H), 8.23 (d, 2H, J = 7.7 Hz, Ar-H), 7.83 (d, 2H, J = 7.7 Hz, Ar-H), 6.96 (q, 1H, J = 6.7 Hz, Ar-H), 4.36 (s, 2H, methylene), 4.31 (Br s, 2H, piperazinyl), 4.14 (Br s, 2H, piperazinyl), and 3.6 (Br s, 4H, piperazinyl). Elemental analysis of C18H18N6O5S2: calculated: C, 46.74; H, 3.92; and N, 18.17; found: C, 46.78; H, 3.94; and N, 18.12%.
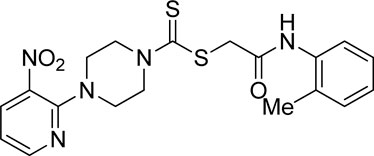
3.3.10 2-Oxo-2-(o-tolylamino)ethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5j)
Yield, 71%; yellow solid; mp: 141°C–143°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 9.56 (s, 1H, NH), 8.46–8.47 (m, 1H, Ar-H), 8.31 (dd, 1H, J = 6.7 Hz, 1.45 Hz, Ar-H), 7.37 (d, 1H, J = 6.6 Hz, Ar-H), 7.20 (d, 1H, J = 6.2 Hz, Ar-H), 7.16 (t, 1H, J = 6.85 Ar-H), 7.09 (t, 1H, J = 6.2 Hz, Ar-H), 6.96 (q, 1H, J = 6.7 Ar-H), 4.33 (Br s, 2H, piperazinyl), 4.30 (s, 2H, methylene), 4.14 (Br s, 2H, piperazinyl), 3.6 (Br s, 4H, piperazinyl), and 2.22 (Br s, 3H, methyl). Elemental analysis of C19H21N5O3S2: calculated: C, 52.88; H, 4.91; and N, 16.23; found: C, 52.84; H, 4.93; and N, 16.26%.
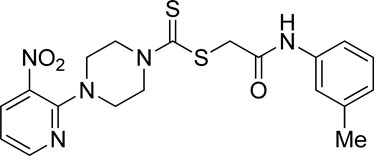
3.3.11 2-Oxo-2-(m-tolylamino)ethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5k)
Yield, 75%; yellow solid; mp: 101°C–103°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.19 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.45 Hz, Ar-H), 7.42 (s, 1H, Ar-H), 7.37 (d, 1H, J = 6.90 Ar-H), 7.18 (t, 1H, J = 6.5 Hz, Ar-H), 6.96 (q, 1H, J = 6.7 Hz, Ar-H), 6.87 (d, 1H, J = 6.95 Hz, Ar-H), 4.32 (Br s, 2H, piperazinyl), 4.28 (s, 2H, methylene), 4.14 (Br s, 2H, piperazinyl), 3.6 (Br s, 4H, piperazinyl), and 2.27 (Br s, 3H, methyl). 13C NMR (DMSO-d6 125 MHz) δ (ppm): 195.3 (N(C=S)S), 165.6 (C=O), 152.5 (Ar-C), 152.0 (Ar-C), 139.3 (Ar-C), 138.4 (Ar-C), 136.3 (Ar-C), 132.7 (Ar-C), 129.0 (Ar-C), 124.5 (Ar-C), 120.1 (Ar-C), 116.8 (Ar-C), 114.4 (Ar-C), 50.6 (piperazine, NCH2), 48.8 (piperazine, NCH2), 46.8 (piperazine, 2 × NCH2), 41.6 (SCH2(CO)), and 21.6 (CH3). Elemental analysis of C19H21N5O3S2: calculated: C, 52.88; H, 4.91; N, 16.23; found: C, 52.93; H, 4.95; and N, 16.19%.
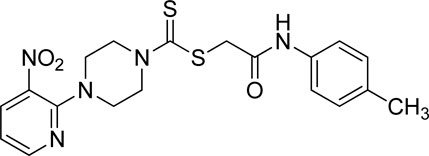
3.3.12 2-Oxo-2-(p-tolylamino)ethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5l)
Yield, 69%; yellow solid; mp: 145°C–146°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.17 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.1 Hz, Ar-H), 7.46 (d, 2H, J = 7.00 Hz Ar-H), 7.11 (d, 2H, J = 7.35 Hz, Ar-H), 6.96 (q, 1H, J = 6.70 Hz, Ar-H), 4.31 (Br s, m, 2H, piperazinyl), 4.26 (s, 2H, methylene), 4.12 (Br s, m, 2H, piperazinyl), 3.6 (Br s, 4H, piperazinyl), and 2.24 (Br s, 3H, methyl). Elemental analysis of C19H21N5O3S2: calculated: C, 52.88; H, 4.91; and N, 16.23; found: C, 52.92; H, 4.86; and N, 16.26%.
3.3.13 2-((2-Methoxyphenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5m)
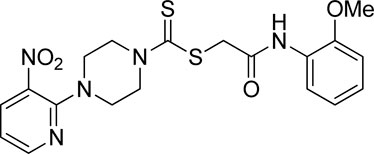
Yield, 62%; yellow solid; mp: 137°C–138°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 9.40 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.40 Hz, Ar-H), 7.98 (d, 1H, J = 6.6 Hz, Ar-H), 7.03–7.08 (m, 2H, Ar-H), 6.96 (q, 1H, J = 6.7 Hz, Ar-H), 6.90 (t, 1H, J = 6.70 Ar-H), 4.33 (s, 2H, methylene), 4.29 (Br s, 2H, piperazinyl, 4.14 (Br s, 2H, piperazinyl), 3.38 (s, 3H, methyl), and 3.6 (Br s, 4H, piperazinyl). 13C NMR (DMSO-d6 125 MHz) δ (ppm): 195.0 (N(C=S)S), 165.9 (C=O), 152.6 (Ar-C), 152.0 (Ar-C), 149.5 (Ar-C), 136.38 (Ar-C), 132.7 (Ar-C), 127.4 (Ar-C), 124.8 (Ar-C), 121.3 (Ar-C), 120.8 (Ar-C), 114.4 (Ar-C), 111.6 (Ar-C), 56.2 (OCH3), 50.9 (piperazine, NCH2), 49.0 (piperazine, NCH2), 46.4 (piperazine, 2 × NCH2), and 41.1 (SCH2(CO)). Elemental analysis of C19H21N5O4S2: calculated: C, 50.99; H, 4.73; and N, 15.65; found: C, 51.02; H, 4.69; and N, 15.68%.
3.3.14 2-((4-Methoxyphenyl)amino)-2-oxoethyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (5n)
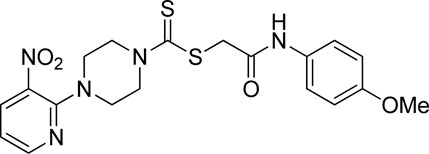
Yield, 66%; yellow solid; mp: 151°C–153°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.12 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.40 Hz, Ar-H), 7.48 (d, 2H, J = 7.55 Hz, Ar-H), 6.96 (q, 1H, J = 6.70 Hz Ar-H), 6.88 (d, 2H, J = 7.5 Hz, Ar-H), 4.32 (Br s, 2H, piperazinyl), 4.25 (s, 2H, methylene), 4.13 (Br s, 2H, piperazinyl), 3.71 (s, 3H, methyl), and 3.60 (Br s, 4H, piperazinyl). Elemental analysis of C19H21N5O4S2: calculated: C, 50.99; H, 4.73; and N, 15.65; found: C, 51.03; H, 4.69; and N, 15.67%.
3.4 Procedure for the synthesis of 4-((aryl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioates 7a–7n
A mixture of 1-(3-nitropyridin-2-yl)piperazine (3) (0.15 mmol), NaOAc (0.30 mmol), and CS2 (0.30 mmol) was prepared in 15 ml of acetonitrile in a round-bottom flask (solution A). On the other hand, a solution of 4-(chloromethyl)-N-arylbenzamide (6a–6n) (0.15 mmol) was prepared in 5 ml of acetonitrile (solution B). Solution B was added to solution A, and the resulting mixture was refluxed for 12–24 h with constant stirring. The reaction was monitored with TLC. Finally, the addition of water resulted in the precipitates of 4-((aryl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate derivatives 7a–7n. The ppts were collected and purified by column chromatography.
3.4.1 4-(Phenylcarbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7a)
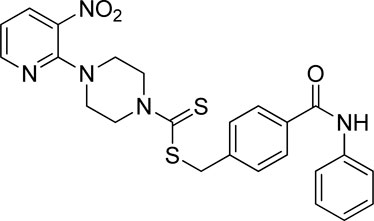
Yield, 63%; yellow solid; mp: 142°C–143°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.08 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.65 Hz, 1.4 Hz, Ar-H), 7.89 (d, 2H, J = 6.75 Hz, Ar-H), 7.76 (d, 2H, J = 7.35 Hz, Ar-H), 7.55 (d, 2H, J = 6.67 Hz, Ar-H), 7.36 (t, 2H, J = 6.5 Hz, Ar-H), 7.10 (t, 1H, J = 7.05, Ar-H), 6.95 (q, 1H, J = 6.70 Hz, Ar-H), 4.66 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.07 (Br s, 2H, piperazinyl), and 3.57 (Br s, 4H, piperazinyl). Elemental analysis of C24H23N5O3S2: calculated: C, 57.93; H, 5.47; and N, 14.07; found: C, 57.89; H, 5.45; and N, 14.11%.
3.4.2 4-((2-Chlorophenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7b)
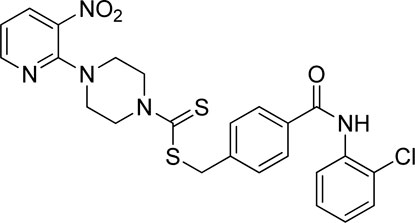
Yield, 61%; yellow solid; mp: 109°C–110°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.02 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.29–8.31 (dd, 1H, J = 6.7 Hz, 1.35 Hz, Ar-H), 7.94 (d, 2H, J = 7.00 Hz, Ar-H), 7.60 (dd, 1H, J = 6.60 Hz, 1.30 Hz, Ar-H), 7.55 (t, 3H, J = 6.90 Hz, Ar-H), 7.38 (t, 1H, J = 6.60 Hz, Ar-H), 7.29 (t, 1H, J = 6.50 Hz, Ar-H), 6.95 (d, 1H, J = 6.70 Hz, Ar-H), 4.68 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.09 (Br s, 2H, piperazinyl), and 3.58 (Br s, 4H, piperazinyl). 13C NMR (DMSO-d6 125 MHz) δ (ppm): 195.2 (N(C=S)S), 165.6 (C=O), 152.5 (Ar-C), 152.0 (Ar-C), 141.19 (Ar-C), 136.3 (Ar-C), 133.3 (Ar-C), 132.7 (Ar-C), 130.0 (Ar-C), 129.8 (2 × Ar-C), 129.7 (Ar-C), 128.8 (2 × Ar-C), 128.2 (Ar-C), 127.9 (2 × Ar-C), 114.4 (Ar-C), 50.8 (piperazine, NCH2), 49.0 (piperazine, NCH2), 46.4 (piperazine, 2 × NCH2), and 40.8 (SCH2). Elemental analysis of C24H22ClN5O3S2: calculated: C, 54.59; H, 4.20; and N, 13.26; found: C, 54.55; H, 4.24; and N, 13.30%.
3.4.3 4-((3-Chlorophenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7c)
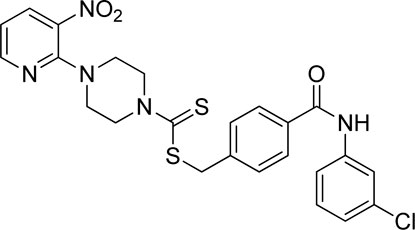
Yield, 56%; yellow solid; mp: 140°C–141°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.38 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.29 (dd, 1H, J = 6.5 Hz, 1.4 Hz, Ar-H), 7.97 (s, 1H, Ar-H), 7.90 (d, 2H, J = 6.85 Hz, Ar-H), 7.70–72 (m, 1H, Ar-H), 7.56–7.57 (m, 2H, Ar-H), 7.38 (t, 1H, J = 6.5 Hz, Ar-H), 7.15 (dd, 1H, J = 6.7 Hz, 1.05 Hz, Ar-H), 6.95 (q, 1H, J = 6.65 Hz, Ar-H), 4.68 (s, 2H, methylene), 4.35 (Br s, 2H, piperazinyl), 4.09 (Br s, 2H, piperazinyl), and 3.59 (Br s, 4H, piperazinyl). Elemental analysis of C24H22ClN5O3S2: calculated: C, 54.59; H, 4.20; and N, 13.26; found: C, 54.64; H, 4.23; and N, 13.27%.
3.4.4 4-((4-Chlorophenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7d)
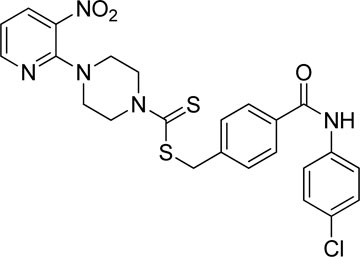
Yield, 53%; yellow solid; mp: 147°C–148°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.34 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.29 (dd, 1H, J = 6.75 Hz, 1.4 Hz, Ar-H), 7.89 (d, 2H, J = 6.85 Hz, Ar-H), 7.80 (d, 2H, J = 7.35 Hz, Ar-H), 7.56 (d, 2H, J = 6.8 Hz, Ar-H), 7.40 (d, 2H, J = 7.35 Hz, Ar-H), 6.95 (q, 1H, J = 6.7 Hz, Ar-H), 4.67 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.08 (Br s, 2H, piperazinyl), and 3.58 (Br s, 4H, piperazinyl). 13C NMR (DMSO-d6 125 MHz) δ (ppm): 195.1 (N(C=S)S), 165.8 (C=O), 152.5 (Ar-C), 152.0 (Ar-C), 141.0 (Ar-C), 138.5 (Ar-C), 136.3 (Ar-C), 134.0 (Ar-C), 132.6 (Ar-C), 129.6 (2 × Ar-C), 128.9 (2 × Ar-C), 128.2 (2 × Ar-C), 127.7 (Ar-C), 122.3 (2 × Ar-C), 114.4 (Ar-C), 50.7 (piperazine, NCH2), 48.7 (piperazine, NCH2), 46.4 (piperazine, 2 × NCH2), and 40.3 (SCH2). Elemental analysis of C24H22ClN5O3S2: calculated: C, 54.59; H, 4.20; and N, 13.26; found: C, 54.63; H, 4.17; and N, 13.31%.
3.4.5 4-((3-Bromophenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7e)
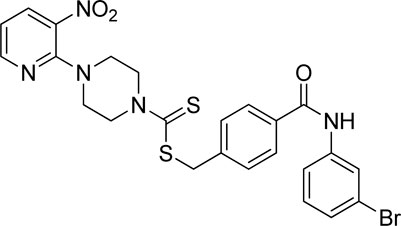
Yield, 55%; yellow solid; mp: 141°C–142°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.36 (s, 1H, NH), 8.43–8.44 (m, 1H, Ar-H), 8.28 (d, 1H, J = 6.65 Hz, Ar-H), 8.08 (s, 1H, Ar-H), 7.88 (d, 2H, J = 6.60 Hz, Ar-H), 7.71 (d, 1H, J = 6.65 Hz, Ar-H), 7.55 (d, 2H, J = 6.60 Hz, Ar-H), 7.27–32 (m, 2H, Ar-H), 6.95 (q, 1H, J = 6.75 Hz, Ar-H), 4.66 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.07 (Br s, 2H, piperazinyl), and 3.57 (Br s, 4H, piperazinyl). Elemental analysis of C24H22BrN5O3S2: calculated: C, 50.35; H, 3.87; and N, 12.23; found: C, 50.39; H, 3.86; and N, 12.28%.
3.4.6 4-((4-Bromophenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7f)
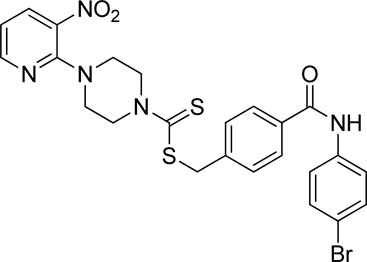
Yield, 59%; yellow solid; mp: 162°C–164°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.36 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.4 Hz, Ar-H), 7.89 (d, 2H, J = 6.90 Hz, Ar-H), 7.76 (d, 2H, J = 7.4 Hz, Ar-H), 7.52–7.56 (m, 4H, Ar-H), 6.95 (q, 1H, J = 6.8 Hz, Ar-H), 4.67 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.08 (Br s, 2H, piperazinyl), and 3.58 (Br s, 4H, piperazinyl). Elemental analysis of C24H22BrN5O3S2: calculated: C, 50.35; H, 3.87; and N, 12.23; found: C, 50.38; H, 3.91; and N, 12.25%.
3.4.7 4-((2-Nitrophenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7g)
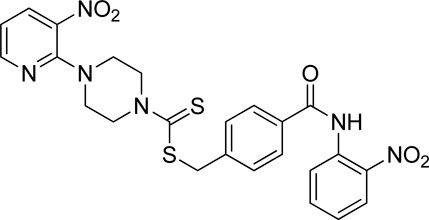
Yield 71%; yellow solid; mp: 133°C–134°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.78 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.31 (dd, 1H, J = 6.65 Hz, 1.35 Hz, Ar-H), 8.28 (d, 2H, J = 7.85 Ar-H), 8.07 (d, 2H, J = 6.70 Hz, Ar-H), 7.93 (d, 2H, J = 6.95 Hz, Ar-H), 7.59 (d, 2H, J = 6.8 Hz, Ar-H), 6.98 (q, 1H, J = 6.65 Hz, Ar-H), 4.67 (s, 2H, methylene), 4.35 (Br s, 2H, piperazinyl), 4.09 (Br s, 2H, piperazinyl), and 3.59 (Br s, 4H, piperazinyl). Elemental analysis of C24H22N6O5S2: calculated: C, 53.52; H, 4.12; and N, 15.60; found: C, 53.55; H, 4.15; and N, 15.55%.
3.4.8 4-((3-Nitrophenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7h)
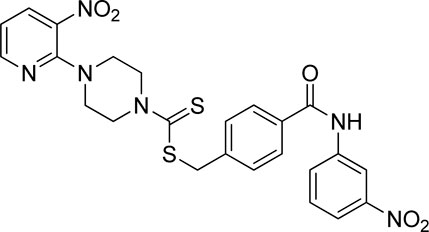
Yield, 49%; yellow solid; mp: 165°C–166°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.66 (s, 1H, NH), 8.80 (s, 1H, Ar-H), 8.44–8.45 (m, 1H, Ar-H), 8.29–8.31 (dd, 1H, J = 6.7 Hz, 1.40 Hz, Ar-H), 8.18 (d, 1H, J = 6.90 Hz, Ar-H), 7.94–7.97 (m, 3H, Ar-H), 7.65 (t, 1H, J = 6.80 Hz, Ar-H), 7.59 (d, 2H, J = 6.75 Hz, Ar-H), 6.95 (q, 1H, J = 6.70 Hz, Ar-H), 4.68 (s, 2H, methylene), 4.35 (Br s, 2H, piperazinyl), 4.09 (Br s, 2H, piperazinyl), and 3.59 (Br s, 4H, piperazinyl). Elemental analysis of C24H22N6O5S2: calculated: C, 53.52; H, 4.12; and N, 15.60; found: C, 53.57; H, 4.16; and N, 15.56%.
3.4.9 4-((4-Nitrophenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7i)
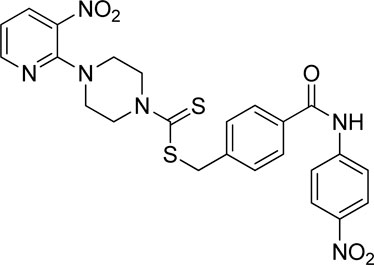
Yield, 63%; yellow solid; mp: 191°C–192°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.77 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.29 (dd, 1H, J = 6.70 Hz, 1.40 Hz, Ar-H), 8.25 (d, 2H, J = 7.70 Ar-H), 8.04 (d, 2H, J = 6.85 Hz, Ar-H), 7.92 (d, 2H, J = 7 Hz, Ar-H), 7.58 (d, 2H, J = 6.70 Hz, Ar-H), 6.95 (q, 1H, J = 6.70 Hz, Ar-H), 4.68 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.08 (Br s, 2H, piperazinyl), and 3.58 (Br s, 4H, piperazinyl). Elemental analysis of C24H22N6O5S2: calculated: C, 53.52; H, 4.12; and N, 15.60; found: C, 53.55; H, 4.15; and N, 15.55%.
3.4.10 4-(o-Tolylcarbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7j)
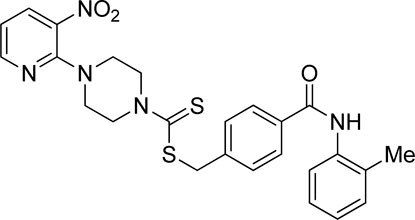
Yield, 55%; yellow solid; mp: 113°C–115°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.09 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.75 Hz, 1.40 Hz, Ar-H), 7.92 (d, 2H, J = 6.85 Hz, Ar-H), 7.55 (d, 2H, J = 6.90 Hz, Ar-H), 7.33 (d, 1H, J = 6.5 Hz, Ar-H), 7.26 (d, 1H, J = 6.35 Hz, Ar-H), 7.21 (t, 1H, J = 6.2 Hz, Ar-H), 7.16 (t, 1H, J = 6.2 Hz, Ar-H), 6.95 (q, 1H, J = 6.70 Hz, Ar-H), 4.67 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.08 (Br s, 2H, piperazinyl), 3.58 (Br s, 4H, piperazinyl), and 2.29 (s, 3H, methyl). 13C NMR (DMSO-d6 125 MHz) δ (ppm): 195.1 (N(C=S)S), 165.6 (C=O), 152.6 (Ar-C), 152.0 (Ar-C), 140.7 (Ar-C), 136.8 (Ar-C), 136.3 (Ar-C), 134.1 (Ar-C), 133.9 (Ar-C), 132.6 (Ar-C), 130.7 (Ar-C), 129.6 (2 × Ar-C), 128.2 (2 × Ar-C), 127.4 (Ar-C), 126.4 (2 × Ar-C), 114.4 (Ar-C), 50.7, 49 (piperazine, 2 × NCH2), 46.4 (piperazine, 2 × NCH2), 40.4 (SCH2), and 18.3 (CH3). Elemental analysis of C25H25N5O3S2: calculated: C, 59.15; H, 4.96; and N, 13.80; found: C, 59.19; H, 4.98; and N, 13.77%.
3.4.11 4-(m-Tolylcarbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7k)
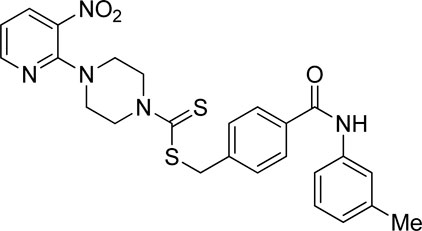
Yield, 57%; yellow solid; mp: 103°C–104°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.13 (s, 1H, NH), 8.45–8.46 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.65 Hz, 1.40 Hz, Ar-H), 7.88 (d, 2H, J = 6.85 Hz, Ar-H), 7.60 (s, 1H, Ar-H), 7.55 (t, 3H, J = 6.90 Hz, Ar-H), 7.22 (t, 1H, J = 6.5 Hz, Ar-H), 6.96 (q, 1H, J = 6.40 Hz, Ar-H), 6.93 (d, 1H, J = 6.30 Hz, Ar-H), 4.67 (Br s, 2H, methylene), 4.35 (Br s, 2H, piperazinyl), 4.08 (Br s, 2H, piperazinyl), 3.58 (Br s, 4H, piperazinyl), and 2.3 (s, 3H, methyl). Elemental analysis of C25H25N5O3S2: calculated: C, 59.15; H, 4.96; and N, 13.80; found: C, 59.13; H, 4.99; and N, 13.85%.
3.4.12 4-(P-tolylcarbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7l)
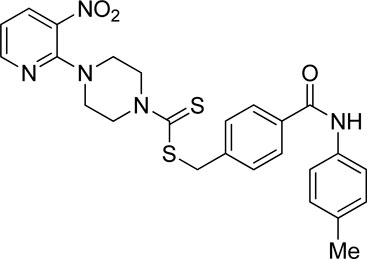
Yield, 51%; yellow solid; mp: 158°C–160°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.13 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.75 Hz, 1.40 Hz, Ar-H), 7.88 (d, 2H, J = 7.00 Ar-H), 7.63 (d, 2H, J = 7.00 Hz, Ar-H), 7.54 (d, 2H, J = 6.95 Hz, Ar-H), 7.15 (d, 2H, J = 7.05 Hz, Ar-H), 6.95 (q, 1H, J = 6.70, Ar-H), 4.69 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.08 (Br s, 2H, piperazinyl), 3.58 (Br s, 4H, piperazinyl), and 2.28 (s, 3H, methyl). Elemental analysis of C25H25N5O3S2: calculated: C, 59.15; H, 4.96; and N, 13.80; found: C, 59.20; H, 4.98; and N, 13.75%.
3.4.13 4-((2-Methoxyphenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7m)
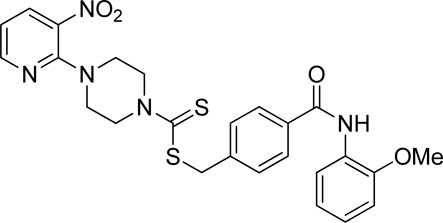
Yield, 67%; yellow solid; mp: 166°C–167°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 9.37 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.29–8.30 (dd, 1H, J = 6.40 Hz, 1.45 Hz, Ar-H), 7.90 (d, 2H, J = 6.85 Hz, Ar-H), 7.79 (dd, 1H, J = 6.60 Hz, 1.40 Hz, Ar-H), 7.54 (d, 2H, J = 6.9 Hz, Ar-H), 7.16–7.19 (m, 1H, Ar-H), 7.09 (d, 1H, J = 7.00 Hz, Ar-H), 6.94–6.98 (m, 2H, Ar-H), 4.67 (s, 2H, methylene), 4.35 (Br s, 2H, piperazinyl), 4.08 (Br s, 2H, piperazinyl), 3.83 (s, 3H, methyl), and 3.58 (Br s, 4H, piperazinyl). Elemental analysis of C25H25N5O4S2: calculated: C, 57.34; H, 4.81; and N, 13.30; found: C, 57.3; H, 4.84; and N, 13.32%.
3.4.14 4-((4-Methoxyphenyl)carbamoyl)benzyl 4-(3-nitropyridin-2-yl)piperazine-1-carbodithioate (7n)
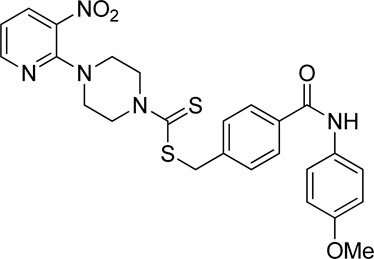
Yield, 51%; yellow solid; mp: 187°C–189°C. 1H NMR (DMSO-d6 500 MHz) δ (ppm): 10.08 (s, 1H, NH), 8.44–8.45 (m, 1H, Ar-H), 8.30 (dd, 1H, J = 6.7 Hz, 1.35 Hz, Ar-H), 7.88 (d, 2H, J = 6.8 Hz, Ar-H), 7.65 (d, 2H, J = 7.5 Hz, Ar-H), 7.54 (d, 2H, J = 7.00 Hz, Ar-H), 6.95 (q, 1H, J = 6.75 Hz, Ar-H), 6.91 (d, 2H, J = 7.5 Hz, Ar-H), 4.66 (s, 2H, methylene), 4.34 (Br s, 2H, piperazinyl), 4.08 (Br s, 2H, piperazinyl), 3.74 (s, 3H, methyl), and 3.58 (Br s, 4H, piperazinyl). 13C NMR (DMSO-d6 125 MHz) δ (ppm): 195.1 (N(C=S)S), 165.4 (C=O), 156.0 (Ar-C), 152.6 (Ar-C), 152.0 (Ar-C), 140.5 (Ar-C), 136.3 (Ar-C), 134.4 (Ar-C), 132.2 (2 × Ar-C), 129.5 (2 × Ar-C), 128.1 (2 × Ar-C), 122.4 (2 × Ar-C), 114.4 (Ar-C), 114.2 (Ar-C), 55.6 (OCH3), 50.6 (piperazine, NCH2), 49.8 (piperazine, NCH2), 46.4 (piperazine, 2 × NCH2), and 40.4 (SCH2). Elemental analysis of C25H25N5O4S2: calculated: C, 57.34; H, 4.81; and N, 13.30; found: C, 57.38; H, 4.76; and N, 13.34%.
3.5 Urease inhibition activity assay
The inhibitory potential of the compounds was assessed using a slightly modified indophenol method (Hina et al., 2023). In 96-well plates, a mixture consisting of 50 µL urease, 30 µL buffer solution (phosphate buffer, sodium salicylate, sodium nitroprusside, and EDTA, pH 8.0), 10 µL urea substrate (100 mM), and 10 µL of the test compound (1 mM) was subjected to pre-incubation for 10 min at room temperature. Subsequently, an alkali reagent (70 µL) was promptly introduced into each well. Then, 30 min later, the absorbance at 630 nm for all samples was measured using a BioTek ELx800 instrument from BioTek Instruments, Inc., United States. The data were collected for all experiments performed in triplicate, and the percentage of inhibitory activities was assessed utilizing the following formula:
3.5.1 Kinetic study
Experiments using Michaelis–Menten kinetics were conducted to identify the type of enzyme inhibition exhibited by urease. Detailed kinetic analyses were undertaken using the most potent compound (5j) to investigate its potential mechanism of action in inhibiting the respective enzyme. The type of inhibitory action was determined by using four different concentrations of the substrate (0, 2.5, 5, 10, and 15 mM) in the absence and presence of different concentrations of inhibitor 5j (0, 2.55, 5.11, and 7.65 μM).
3.6 Protocol for in silico studies
3.6.1 Structure selection and preparation
The structure of the ligands was sketched using ChemDraw Ultra 12.0 from the ChemOffice suite, ensuring that they were given accurate 2D orientations. Subsequently, the structure of each compound was scrutinized for any bond-order connection errors. To explore the notable interactions between the inhibitors and the enzyme urease, we utilized docking procedures. We obtained the crystallographic structure of Jack bean urease (3LA4) from the RCSB Protein Data Bank library. Moreover, these structures were prepared for further analysis in docking studies (Cunha et al., 2021). Before conducting docking studies, the compounds and the urease enzyme were prepared in the following manner: to prevent the binding pockets from collapsing during the energy minimization calculations, a slight force was applied to reinforce the backbone atoms. As a result, the ligands and water molecules were eliminated, and polar hydrogens were added to the crystallographic structure (Labute, 2007).
3.6.2 Molecular docking analysis and simulations
Docking studies of inhibitory compound 5j were performed against the urease enzyme. In order to optimize the effectiveness of the docking results, the urease structure was prepared by eliminating any unwanted ligands and water molecules. The docking analysis for receptor–ligand interactions utilized a grid box with dimensions of 80 × 72 × 66 Å along the x-, y-, and z-axes, respectively. This grid had a grid point spacing of 0.375 Å and was centered at coordinates 7.836 Å in the x-axis, 10.509 Å in the y-axis, and 22.951 Å in the z-axis. The finest binding conformational pose of the protein–ligand docked complexes was obtained by utilizing a default exhaustiveness value of 8. AutoDock Vina software was used to conduct the docking analysis and generate binding affinities. To visualize the residues of amino acids interacting at the active site of the protein, molecular visualization was performed using Edu-PyMOL.
Molecular dynamics simulation (MDS) analysis was used to investigate the stability of the interaction between the protein and the ligand. Furthermore, MD simulation studies were used to scrutinize the structural transitions within the macromolecules, elucidating the functional significance of the resulting complex. In this simulation, atomic movements over time were recorded in accordance with Newton’s fundamental motion equation, providing insights into how the ligand binds within the biological environment (Hassan et al., 2022).
3.6.3 ADMET analysis
The absorption–distribution–metabolism–excretion–toxicity (ADMET) analysis of the potent inhibitor 5j was performed via admetSAR (http://lmmd.ecust.edu.cn/admetsar2), ProTox-II (https://tox-new.charite.de/protox_II/), and eMolTox (https://xundrug.cn/moltox). All these tools are freely accessible and not only determine the physicochemical properties of a compound but also interpret whether a compound has the ability to be a drug or not. In addition, ProTox-II also predicts the LD50 value and toxicity class of the query SMILE, and eMolTox evaluates the toxic substructures existing in the input compound (Cheng et al., 2012; Banerjee et al., 2018; Ji et al., 2018).
4 Conclusion
The inhibitory activity against urease was evaluated for the compounds (5a–5n and 7a–7n). All compounds exhibited notably stronger inhibitory potential than the positive control (thiourea). Notably, 5j emerged as the primary inhibitor, demonstrating remarkable efficacy with an IC50 value of 5.16 ± 2.68 µM. The docking analysis revealed diverse interactions between 5j and the active site amino acids. The in silico ADMET profile exhibited diverse drug-like characteristics of 5j. The lead inhibitor revealed notable docking scores and effective binding free energies, showing a strong binding interaction.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author contributions
MAk: formal analysis, investigation, methodology, writing–original draft, and writing–review and editing. NR: formal analysis, writing–original draft, and writing–review and editing. SAs: conceptualization, investigation, methodology, supervision, writing–original draft, and writing–review and editing. MAh: conceptualization, investigation, writing–original draft, and writing–review and editing. MS: data curation, investigation, software, writing–original draft, and writing–review and editing. AA: formal analysis, writing–original draft, and writing–review and editing. SS: data curation, formal analysis, writing–original draft, and writing–review and editing. SAi: software, validation, writing–original draft, and writing–review and editing. AL: software, writing–original draft, and writing–review and editing. SZ: formal analysis, investigation, methodology, software, writing–original draft, and writing–review and editing. MZ: conceptualization, formal analysis, funding acquisition, methodology, writing–original draft, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors thank the Higher Education Commission of Pakistan for support through Research Project No. 6-13/Pak-US/HEC/2015/13 and Government College University Faisalabad.
Acknowledgments
The authors thank the Higher Education Commission of Pakistan for support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1423385/full#supplementary-material
References
Akash, M., Zaib, S., Ahmad, M., Sultan, S., and Al-Hussain, S. A. (2024). Synthesis and biological evaluation of pyridylpiperazine hybrid derivatives as urease inhibitors. Front. Chem. 12, 1371377. doi:10.3389/fchem.2024.1371377
Alqahtani, Y. S., Alyami, B. A., Alqarni, A. O., Mahnashi, M. H., Ali, A., Javed, Q., et al. (2022). Cephalosporin as potent urease and tyrosinase inhibitor: exploration through enzyme inhibition, kinetic mechanism, and molecular docking studies. Biomed. Res. Int. 2022, 1–11. doi:10.1155/2022/1092761
Banerjee, P., Eckert, A. O., Schrey, A. K., and Preissner, R. (2018). ProTox-II: a webserver for the prediction of toxicity of chemicals. Nuc. Acid. Res. 46 (W1), W257–W263. doi:10.1093/nar/gky318
Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., et al. (2012). admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 52 (11), 3099–3105. doi:10.1021/ci300367a
Cunha, E. S., Chen, X., Sanz-Gaitero, M., Mills, D. J., and Luecke, H. (2021). Cryo-EM structure of Helicobacter pylori urease with an inhibitor in the active site at 2.0 Å resolution. Nat. Comm. 12 (1), 230. doi:10.1038/s41467-020-20485-6
Gull, N. R., Noreen, M., Altaf, A., Musharraf, S., Zubair, M., Nasim, F.-U.-H., et al. (2016). Synthesis of N-6-Arylbenzo [d] thiazole-2-acetamide derivatives and their biological activities: an experimental and computational approach. Molecules 21 (3), 266. doi:10.3390/molecules21030266
Hameed, A., Al-Rashida, M., Uroos, M., Qazi, S. U., Naz, S., Ishtiaq, M., et al. (2019). A patent update on therapeutic applications of urease inhibitors (2012–2018). Exp. Opin. Ther. Pat. 29 (3), 181–189. doi:10.1080/13543776.2019.1584612
Hanif, M., Shoaib, K., Saleem, M., Hasan Rama, N., Zaib, S., and Iqbal, J. (2012). Synthesis, urease inhibition, antioxidant, antibacterial, and molecular docking studies of 1, 3, 4-oxadiazole derivatives. Int. Sch. Res. Not. 2012, 1–9. doi:10.5402/2012/928901
Hassan, S. S. U., Abbas, S. Q., Ali, F., Ishaq, M., Bano, I., Hassan, M., et al. (2022). A Comprehensive in silico exploration of pharmacological properties, bioactivities, molecular docking, and anticancer potential of vieloplain F from Xylopia vielana targeting B-Raf Kinase. Molecules 27 (3), 917. doi:10.3390/molecules27030917
Hina, S., Zaib, S., Uroos, M., Zia-ur-Rehman, M., Munir, R., Riaz, H., et al. (2023). N-Arylacetamide derivatives of methyl 1, 2-benzothiazine-3-carboxylate as potential drug candidates for urease inhibition. Roy. Soc. Op. Sci. 10 (4), 230104. doi:10.1098/rsos.230104
Imran, M., Waqar, S., Ogata, K., Ahmed, M., Noreen, Z., Javed, S., et al. (2020). Identification of novel bacterial urease inhibitors through molecular shape and structure based virtual screening approaches. RSC Adv. 10 (27), 16061–16070. doi:10.1039/D0RA02363A
Ji, C., Svensson, F., Zoufir, A., and Bender, A., 2018. eMolTox: prediction of molecular toxicity with confidence. Bioinform 34(14), 2508–2509. doi:10.1093/bioinformatics/bty135
Khan, B. A., Hamdani, S. S., Jabeen, A., Ashfaq, M., Ahmed, M. N., Bogdanov, G., et al. (2024). Exploring S-benzyl carbodithioates as urease inhibitors: synthesis, single crystal XRD, Hirshfeld surface analysis, enzyme inhibition and molecular modeling. J. Molec. Str. 1311, 138444. doi:10.1016/j.molstruc.2024.138444
Khan, M., Ahad, G., Manaf, A., Naz, R., Hussain, S. R., Deeba, F., et al. (2019). Synthesis, in vitro urease inhibitory activity, and molecular docking studies of (perfluorophenyl) hydrazone derivatives. Med. Chem. Res. 28, 873–883. doi:10.1007/s00044-019-02341-5
Labute, P. (2007). Protonate 3D: assignment of macromolecular protonation state and geometry. Montreal: Chemical Computing Group Inc.
Mahernia, S., Bagherzadeh, K., Mojab, F., and Amanlou, M. (2015). Urease inhibitory activities of some commonly consumed herbal medicines. Iran. J. Pharm. Res. 14 (3), 943–947.
Rutherford, J. C. (2014). The emerging role of urease as a general microbial virulence factor. PLoS Pathog. 10 (5), e1004062. doi:10.1371/journal.ppat.1004062
Svane, S., Sigurdarson, J. J., Finkenwirth, F., Eitinger, T., and Karring, H., 2020. Inhibition of urease activity by different compounds provides insight into the modulation and association of bacterial nickel import and ureolysis. Sci. Rep. 10(1), 8503. doi:10.1038/s41598-020-65107-9
Tarsia, C., Danielli, A., Florini, F., Cinelli, P., Ciurli, S., and Zambelli, B. (2018). Targeting Helicobacter pylori urease activity and maturation: in-cell high-throughput approach for drug discovery. Biochim. Biophys. Act. Gen. Sub. 1862 (10), 2245–2253. doi:10.1016/j.bbagen.2018.07.020
Tempera, P. J., Michael, M., Tageldin, O., and Hasak, S. (2022). Gastric cancer due to chronic H. pylori infection: what we know and where we are going. Diseases 10 (3), 57. doi:10.3390/diseases10030057
Yang, X., Koohi-Moghadam, M., Wang, R., Chang, Y. Y., Woo, P. C., Wang, J., et al. (2018). Metallochaperone UreG serves as a new target for design of urease inhibitor: a novel strategy for development of antimicrobials. PLoS Bio. 16 (1), e2003887. doi:10.1371/journal.pbio.2003887
Zambelli, B., Berardi, A., Martin-Diaconescu, V., Mazzei, L., Musiani, F., Maroney, M. J., et al. (2014). Nickel binding properties of Helicobacter pylori UreF, an accessory protein in the nickel-based activation of urease. J. Bio. Inorg. Chem. 19, 319–334. doi:10.1007/s00775-013-1068-3
Keywords: pyridine and piperazine derivatives, heterocyclic carbodithioates, urease inhibitors, molecular docking, synthesis and biological evaluation
Citation: Akash M, Rana N, Aslam S, Ahmad M, Saif MJ, Asghar A, Sultan S, Al-Hussain SA, Liaqat A, Zaib S and Zaki MEA (2024) Pyridylpiperazine-based carbodithioates as urease inhibitors: synthesis and biological evaluation. Front. Chem. 12:1423385. doi: 10.3389/fchem.2024.1423385
Received: 25 April 2024; Accepted: 08 July 2024;
Published: 06 August 2024.
Edited by:
Mehlika Dilek Altıntop, Anadolu University, TürkiyeReviewed by:
Derya Osmaniye, Anadolu University, TürkiyeKeriman Ozadali Sari, Hacettepe University, Türkiye
Pawel Kafarski, Wrocław University of Technology, Poland
Copyright © 2024 Akash, Rana, Aslam, Ahmad, Saif, Asghar, Sultan, Al-Hussain, Liaqat, Zaib and Zaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sana Aslam, ZHIuc2FuYUBnY3d1Zi5lZHUucGs=; Matloob Ahmad, bWF0bG9vYi5haG1hZEBnY3VmLmVkdS5waw==; Sumera Zaib, c3VtZXJhLnphaWJAdWNwLmVkdS5waw==; Magdi E. A. Zaki, bWV6YWtpQGltYW11LmVkdS5zYQ==