 Anton Tamtögl
Anton Tamtögl Helen Chadwick
Helen Chadwick Barbara A. J. Lechner
Barbara A. J. Lechner Marco Sacchi
Marco Sacchi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
EDITORIAL article
Front. Chem., 18 April 2024
Sec. Chemical Physics and Physical Chemistry
Volume 12 - 2024 | https://doi.org/10.3389/fchem.2024.1411748
This article is part of the Research TopicDynamics at Surfaces: Understanding Energy Dissipation and Physicochemical Processes at the Atomic and Molecular LevelView all 13 articles
Editorial on the Research Topic
Dynamics at surfaces: understanding energy dissipation and physicochemical processes at the atomic and molecular level
The processes of energy dissipation at solid interfaces (see Figure 1) are integral to numerous physical phenomena ranging from catalytic reactions and astrochemistry to lubrication and materials science including the development of nanostructures (Ertl, 2009; Yang and Wodtke, 2016; Park et al., 2019; Ollier et al., 2023). Despite its ubiquity and importance in both technological applications and natural systems these surface dynamical processes remain poorly understood (Yang and Wodtke, 2016; Park et al., 2019; Sacchi and Tamtögl, 2023; Yu et al., 2023). For advancements in fields like catalysis, electrochemistry, and photoactivated processes, a comprehensive understanding, including energy transfer from gas or liquid phase molecules to surfaces and how energy is further dissipated through various means, such as phonons and via electron-phonon coupling, is essential (Chadwick and Beck, 2017; Tamtögl et al., 2020). Over recent decades, both experimental and theoretical advancements have significantly enriched the field, enabling more detailed investigations of surface structures and surface dynamical processes (Meyer and Reuter, 2014; Nattino et al., 2016; Alducin et al., 2017; Maurer et al., 2019; Dou and Subotnik, 2020; Holst et al., 2021).
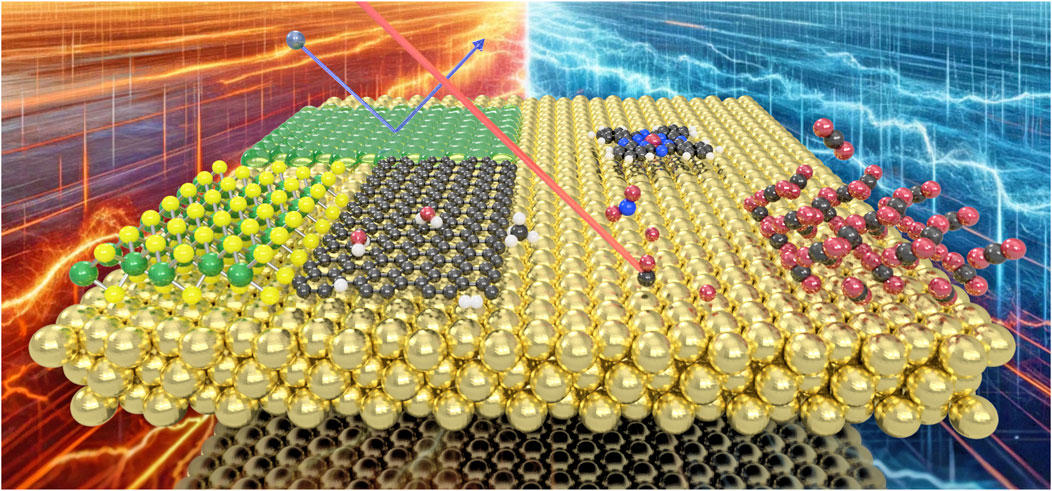
Figure 1. Energy dissipation processes are ubiquitous on surfaces and interfaces, from molecular motion to the formation of thin-films, determining adsorption, desorption and dissociation processes of molecules as well as the energetics upon molecular scattering from surfaces.
Within the current Research Topic a diverse editorial team with different approaches to the topic, including molecular and state-to-state scattering (H. Chadwick), scanning probe techniques (B. A. J. Lechner) theoretical methods (M. Sacchi) and atom/beam scattering (A. Tamtögl) collected a total of 10 original articles and one mini review including the recent progress in understanding energy dissipation in surface dynamical processes (see Figure 1 for a few illustrated examples).
The study by Sabik et al. investigates the surface dynamical motion of cobalt phthalocyanine molecules on silver using helium spin-echo (HeSE) spectroscopy, revealing that the activation energy for lateral diffusion decreases with temperature, leading to a transition from single jumps to predominantly long jumps at higher temperatures. It highlights the importance of considering a wide temperature range to capture the complete dynamics of molecular motion on surfaces.
Using the same method, Kyrkjebø et al. illustrate a stark contrast in water mobility across graphene-covered and bare Ir(111) surfaces. On graphene-covered Ir(111), water molecules exhibit significantly hindered diffusion, attributed to the trapping at specific sites within the surface’s corrugated structure. Their findings not only advance our understanding of water-surface interactions but also implicate potential impacts on the development of anti-icing and anti-corrosion materials.
Via atom-surface scattering, Maier et al. study the surface properties of 1T-TaS2 and TlBiTe2, contrasting electron-phonon coupling between these materials, and potential implications for phase transitions driven by phonons. The study of thermal expansion and interaction potentials offers valuable insights into the complex behaviour of these compounds, contributing to a broader knowledge of charge-density wave systems and topological insulators.
The scattering of keV protons through graphene is studied by Bühler et al. who challenge prior assumptions of the process. By incorporating the lattice thermal motion in simulations, they uncover that previously observed phenomena, such as the outer rainbow scattering, are artefacts of statistical averaging. At the same time, they illustrate new avenues for detailed studies of proton-graphene interactions and the orientations of graphene membranes.
Dorst et al. study the recombinative desorption of O atoms from Ag(111) by combining ion imaging techniques with temperature-programmed desorption. The hyperthermal velocity distribution of the resulting O2 is consistent with activated recombinative desorption but lower than state-of-the-art calculations currently predict. These results, therefore, provide a valuable benchmark for refining theoretical models of metal oxidation processes.
The influence of vibrational excitation on the sublimation of CO2 is investigated by Jansen and Juurlink, where they use a laser to excite the antisymmetric stretch vibration (ν3) of the CO2 impinging on the CO2 ice. They report that exciting ν3 has a negligible effect on either the sticking of CO2 to the ice, or the resulting structure of the CO2 ice despite the additional vibrational energy being greater than the CO2 desorption energy.
Floß et al. studied the surface-induced vibrational energy redistribution of methane scattering from Ni(111) and Au(111). Quantum state and angle-resolved measurements reveal a stark contrast in the vibrational energy conversion from ν3 to ν1 modes of methane, underlining the catalytic superiority of Ni(111) over the more inert Au(111). It thus shows a direct correlation between surface-induced vibrational energy redistribution efficiency and catalytic activity.
Tetenoire et al. elucidated the complex interplay between electrons and phonons in driving the photoinduced desorption and oxidation of CO on ruthenium surfaces. They demonstrate that phononic excitations play a pivotal role in CO desorption, while both electronic and phononic excitations significantly contribute to CO oxidation. Their research opens new avenues for optimising photochemical reactions on metal surfaces.
Xavier Jr and co-workers investigate graphene nanoribbons (GNRs) as potential catalysts for catalytic methane decomposition using density functional theory. They find that armchair edges offer lower energy barriers for hydrogen desorption, compared to zizag edges on GNRs, indicating a better regeneration potential. Highlighting GNRs as comparable to metallic catalysts for methane decomposition, their research may pave the way for sustainable hydrogen production and emphasises the significance of nanomaterials in catalytic processes for green technology.
The study by Prabhu and Groot demonstrates direct synthesis of metallic 1T Co-promoted MoS2 without intercalating agents via growth in a highly reducing environment. High-pressure in-situ reactor scanning tunnelling microscopy measurements, reveal the transformation from a disordered CoMoSx phase at low temperatures to crystalline 1T slabs at around 600 K. It highlights the importance of reducing conditions in materials growth thus avoiding the need of additional chemicals.
In their mini review, Ueta et al. summarise recent studies on ortho-para conversion of hydrogen in molecular chemisorption and isolated matrix systems. These have found that nuclear-spin conversion can occur on a timescale of seconds, even for non-magnetic surfaces, and that the surface can provide a pathway for dissipating the accompanying change in rotational energy.
The collection of 11 articles within this Research Topic, though only a fraction of the extensive work in the field, highlights that understanding energy dissipation and transfer at interfaces is an extremely active area of research being studied with state-of-the-art methods both experimentally and theoretically. The importance of understanding these surface dynamical processes at the molecular level, focusing on phenomena such as photoinduced reactions, vibrational energy redistribution, and molecular diffusion on surfaces cannot be overstated. Advancements in both experimental setups and theoretical models have opened up new avenues. For example, experiments include the dynamics of larger and more complex molecules and studies of more complex surfaces compared to flat metal substrates, including two-dimensional materials and heterostructures. Similarly, enhanced computing power and the utilisation of computational clusters have enabled more sophisticated ab initio calculations, incorporating phenomena like non-adiabatic effects and quantum friction (Alducin et al., 2017; Chadwick and Beck, 2017; Yu et al., 2023). Furthermore, the integration of machine learning approaches promises to refine theoretical analysis further (Jiang et al., 2016; Kapil et al., 2022). Thus, the studies do not only shed light on the underlying atomic-level interactions but also pave the way for optimising materials for specific technological applications, from optoelectronics to hydrogen production.
However, challenges remain, e.g., in extending ab initio methods to larger systems and longer timescales and in conducting experiments under conditions that more closely mimic “real-life” parameters in catalysis to name a few (Yang and Wodtke, 2016). Moreover, while theory does well in studying specific nanosystems, there is still a need for experimental development to measure dynamical processes at tailored nanostructures or in confinement (Sacchi and Tamtögl, 2023; Yu et al., 2023). By overcoming these challenges and unravelling the mechanisms governing energy dissipation at interfaces, our community can unlock a new era of material fabrication and device control. For example, imagine designing catalysts with unparalleled efficiency, tailoring self-assembly processes for nanomaterial fabrication, or even manipulating environmental interactions on a molecular level. Future research will thus be pivotal for advancing various applications, including catalysis, energy production, and materials science by providing insights into the interaction mechanisms between molecules and surfaces, the influence of surface properties on these interactions, and the development of novel materials with enhanced functionalities.
AT: Writing–original draft, Writing–review and editing. HC: Writing–original draft, Writing–review and editing. BL: Writing–review and editing. MS: Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded in whole, or in part, by the Austrian Science Fund (FWF): https://doi.org/10.55776/P34704.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alducin, M., Díez Muiño, R., and Juaristi, J. I. (2017). Non-adiabatic effects in elementary reaction processes at metal surfaces. Prog. Surf. Sci. 92, 317–340. doi:10.1016/j.progsurf.2017.09.002
Chadwick, H., and Beck, R. D. (2017). Quantum state–resolved studies of chemisorption reactions. Annu. Rev. Phys. Chem. 68, 39–61. doi:10.1146/annurev-physchem-052516-044910
Dou, W., and Subotnik, J. E. (2020). Nonadiabatic molecular dynamics at metal surfaces. J. Phys. Chem. A 124, 757–771. doi:10.1021/acs.jpca.9b10698
Holst, B., Alexandrowicz, G., Avidor, N., Benedek, G., Bracco, G., Ernst, W. E., et al. (2021). Material properties particularly suited to be measured with helium scattering: selected examples from 2D materials, van der Waals heterostructures, glassy materials, catalytic substrates, topological insulators and superconducting radio frequency materials. Phys. Chem. Chem. Phys. 23, 7653–7672. doi:10.1039/D0CP05833E
Jiang, B., Li, J., and Guo, H. (2016). Potential energy surfaces from high fidelity fitting of ab initio points: the permutation invariant polynomial - neural network approach. Int. Rev. Phys. Chem. 35, 479–506. doi:10.1080/0144235X.2016.1200347
Kapil, V., Schran, C., Zen, A., Chen, J., Pickard, C. J., and Michaelides, A. (2022). The first-principles phase diagram of monolayer nanoconfined water. Nature 609, 512–516. doi:10.1038/s41586-022-05036-x
Maurer, R. J., Freysoldt, C., Reilly, A. M., Brandenburg, J. G., Hofmann, O. T., Björkman, T., et al. (2019). Advances in density-functional calculations for materials modeling. Annu. Rev. Mater. Res. 49, 1–30. doi:10.1146/annurev-matsci-070218-010143
Meyer, J., and Reuter, K. (2014). Modeling heat dissipation at the nanoscale: an embedding approach for chemical reaction dynamics on metal surfaces. Angew. Chem. Int. Ed. 53, 4721–4724. doi:10.1002/anie.201400066
Nattino, F., Migliorini, D., Kroes, G.-J., Dombrowski, E., High, E. A., Killelea, D. R., et al. (2016). Chemically accurate simulation of a polyatomic molecule-metal surface reaction. J. Phys. Chem. Lett. 7, 2402–2406. doi:10.1021/acs.jpclett.6b01022
Ollier, A., Kisiel, M., Lu, X., Gysin, U., Poggio, M., Efetov, D. K., et al. (2023). Energy dissipation on magic angle twisted bilayer graphene. Commun. Phys. 6, 344–349. doi:10.1038/s42005-023-01441-4
Park, G. B., Krüger, B. C., Borodin, D., Kitsopoulos, T. N., and Wodtke, A. M. (2019). Fundamental mechanisms for molecular energy conversion and chemical reactions at surfaces. Rep. Prog. Phys. 82, 096401. doi:10.1088/1361-6633/ab320e
Sacchi, M., and Tamtögl, A. (2023). Water adsorption and dynamics on graphene and other 2D materials: computational and experimental advances. Adv. Phys. X 8, 2134051. doi:10.1080/23746149.2022.2134051
Tamtögl, A., Sacchi, M., Avidor, N., Calvo-Almazán, I., Townsend, P. S. M., Bremholm, M., et al. (2020). Nanoscopic diffusion of water on a topological insulator. Nat. Commun. 11, 278. doi:10.1038/s41467-019-14064-7
Yang, X., and Wodtke, A. M. (2016). Surface reaction dynamics. Chem. Soc. Rev. 45, 3573–3575. doi:10.1039/C6CS90049F
Keywords: surface chemistry, catalysis, Ab initio (calculations), energy transfer, scattering spectroscopy, nanotechnology/nanomaterials, thin film growth and stability, surface diffusion
Citation: Tamtögl A, Chadwick H, Lechner BAJ and Sacchi M (2024) Editorial: Dynamics at surfaces: understanding energy dissipation and physicochemical processes at the atomic and molecular level. Front. Chem. 12:1411748. doi: 10.3389/fchem.2024.1411748
Received: 03 April 2024; Accepted: 10 April 2024;
Published: 18 April 2024.
Edited and reviewed by:
Malgorzata Biczysko, Shanghai University, ChinaCopyright © 2024 Tamtögl, Chadwick, Lechner and Sacchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anton Tamtögl, dGFtdG9lZ2xAdHVncmF6LmF0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.