 Chanmi Lee1†
Chanmi Lee1† Dohun Lee
Dohun Lee Sung You Hong
Sung You Hong Byunghyuck Jung
Byunghyuck Jung Sangwon Seo
Sangwon Seo- 1Department of Physics and Chemistry, DGIST, Daegu, Republic of Korea
- 2School of Undergraduate Studies, DGIST, Daegu, Republic of Korea
- 3Department of Chemistry, UNIST, Ulsan, Republic of Korea
Over the past few years, earth-abundant transition metal-catalyzed hydrosilylation has emerged as an ideal strategy for the synthesis of organosilanes. The success in this area of research has expanded to the advancements of alkyne dihydrosilylation reactions, offering broadened synthetic applications through the selective installation of two silyl groups. In particular, catalysts based on Fe, Co, and Ni have engendered enabling platforms for mild transformations with a range of distinct regioselectivity. This mini-review summarizes recent advances in this research field, highlighting the unique features of each system from both synthetic and mechanistic perspectives.
Introduction
Owing to the importance of organosilanes in broad research fields, the development of general methods for their preparation has garnered significant attention in the past few decades (Roesky, 2017). Among those that engendered considerable efforts, the hydrosilylation of unsaturated C–C bonds has been established as a powerful strategy accommodating 100% atom economy (Marciniec, 2009), in which the addition of a silyl group and hydrogen atom across the π-system gives direct access to organosilanes from readily available alkenes and alkynes. These alluring features called for extensive investigations toward the relevant reaction developments and, consequently, a diverse range of catalytic systems have appeared for hydrosilylation reactions. In particular, the advent of earth-abundant metal-catalyzed methods (Greenhalgh et al., 2015; Sun and Deng, 2016; Chen et al., 2018; Obligacion and Chirik, 2018) has contributed substantially to allaying the cost and environmental issues associated with the conventional strategies that were dominated by precious metal catalysts. The synthetic merits of such advancements can be reflected by their wide applications in the industrial production of organosilicon compounds, with the contemporary catalysts offering the opportunity for chemo-, regio-, and stereocontrol of the reactions.
Hydrosilylation reactions are generally classified into Markovnikov and anti-Markovnikov hydrosilylation in terms of regiochemistry, the former of which installs a silyl group on the more substituted carbon while the latter inserts it on the less substituted carbon (Scheme 1A). The modification of ligands is often crucial to attain control over the regioselectivity, as well as for the realization of enantioselective reactions where branched silane products are produced to introduce a stereocenter. A large body of studies has been carried out by relying on this common tactic, and the related discoveries of alkene and alkyne hydrosilylation have been discussed intensively in several reviews (Greenhalgh et al., 2015; Nakajima and Shimada, 2015; Sun and Deng, 2016; Zaranek et al., 2016; Chen et al., 2018; Obligacion and Chirik, 2018; de Almeida et al., 2021; He et al., 2022). On the other hand, the selectivity control becomes more complicated and challenging when it comes to the development of alkyne dihydrosilylation, which represents the net addition of two silyl groups and two hydrogen atoms across a C–C triple bond (Scheme 1B). The possible regiochemistry includes 1,1-, 1,2-, 2,1-, and 2,2-additions of silanes, and the control of stereochemistry should also be considered in all cases especially if two different silane sources were to be inserted. These challenges in selectivity control impeded the advances of such transformations in some ways and, as a result, only a few limited examples were reported (Shimada et al., 2002) until the recent emergence of earth-abundant metal-catalyzed strategies. This mini-review covers the recent advances in this field, specifically highlighting how Fe, Co, and Ni catalysts have been utilized to address the associated challenges for the development of alkyne dihydrosilylation reactions.
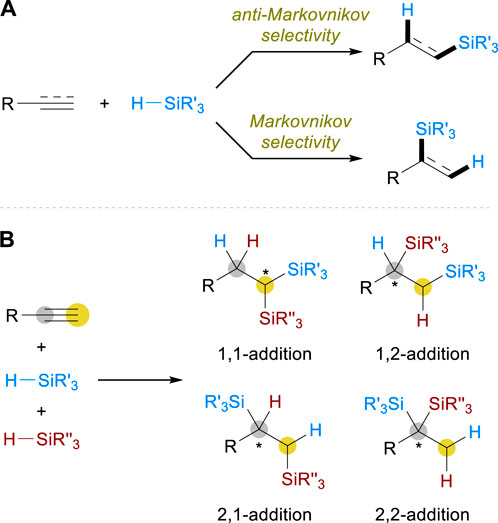
Scheme 1. Regio- and stereoselectivity in (A) Alkene/alkyne monohydrosilylation and (B) Alkyne dihydrosilylation.
Fe-catalyzed dihydrosilylation of alkynes
Iron offers prominence as an ideal catalyst for sustainable chemical reactions, benefiting from its abundance, low costs, and low toxicity (Bauer and Knölker, 2015; Fürstner, 2016). Dihydrosilylation of alkynes, if combined with Fe catalysis, would thus represent a powerful method for the access to disilylalkanes with an excellent atom economy. While Fe-catalyzed protocols have been broadly investigated for monohydrosilylation of alkynes (Greenhalgh et al., 2014; Challinor et al., 2016; Docherty et al., 2017; Hu et al., 2020), the dihydrosilylation congener is less common and the only example was reported by Zhu in 2019 (Hu et al., 2019). Such a development was achieved by employing an iron catalyst bearing 2,9-diaryl-1,10-phenanthroline ligand (Fe1), in combination with EtMgBr as an activator, PhSiH3 as a hydrosilylation agent, and THF solvent to allow for the conversion of a wide range of aliphatic alkynes 1 (Scheme 2A). In this system, the reaction was proven to be highly efficient and regioselective, furnishing 1,1-disilylalkanes 2 through anti-Markovnikov hydrosilylation followed by the second addition at the carbon α to the silyl group of vinylsilane intermediates. A number of functional groups were tolerated on the terminal alkyne substrates, including aryl chloride (2a), sulfide (2b), ether (2c), acetal (2d), and an alkyl silane (n-C12H25SiH3) could also be employed as an alternative hydrosilylation source (2e). The reaction with a secondary silane (Ph2SiH2) only led to the corresponding monohydrosilylation products (3a-b).
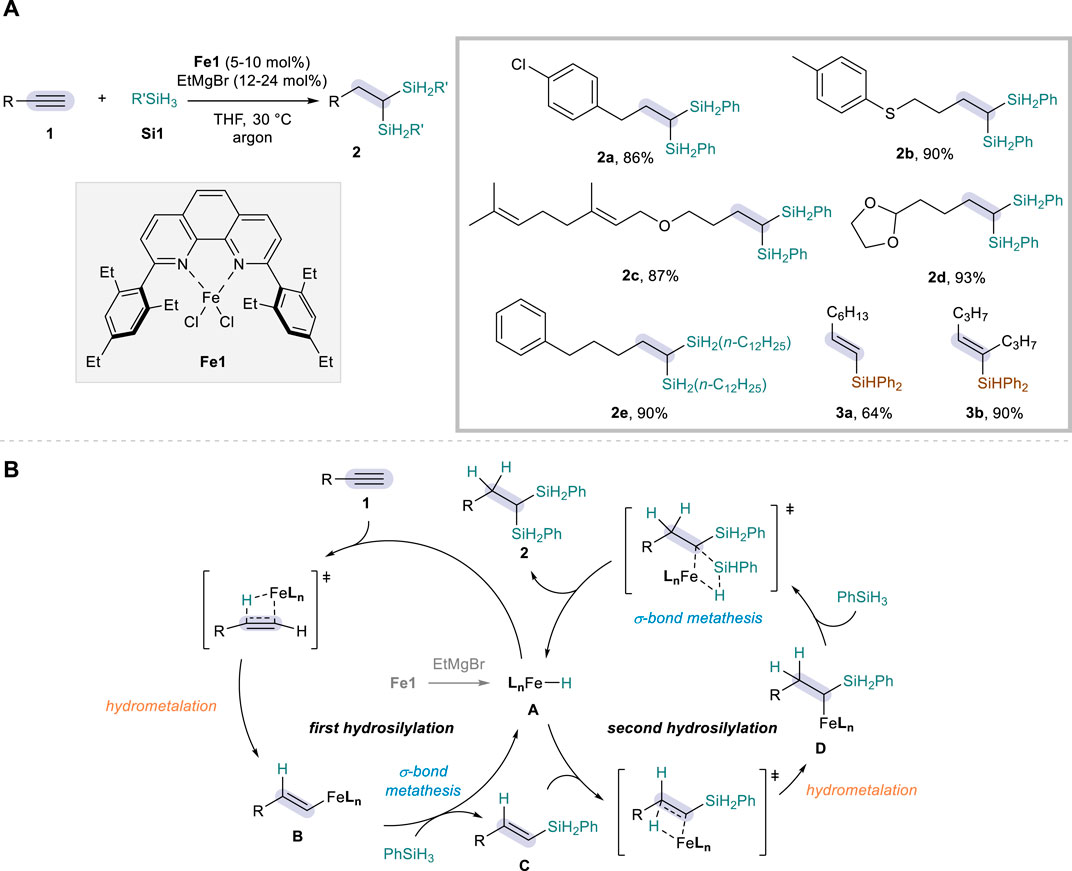
Scheme 2. Fe-catalyzed dihydrosilylation of alkynes: (A) Selected examples of reaction scope. (B) Proposed mechanism.
The reaction was proposed to proceed via activation of Fe(II) to a low-valent Fe(I) species A with EtMgBr playing a role as the reductant, after which the catalytic cycle kicks off to promote the hydrosilylation reactions (Scheme 2B). Based on the precedents of Fe-catalyzed alkene hydrosilylation (Hu et al., 2018), the authors proposed that the reaction takes place through sequential hydrosilylations involving hydrometalation (1 to B & C to D) and σ-bond metathesis (B to C & D to 2). The excellent regioselectivity in the second hydrosilylation was attributed to the α-silicon effect of vinylsilane intermediate C, which stabilizes the insertion of electropositive metal at the α-position that holds a higher charge density. This mechanism proposal was supported by control experiments showing the rapid formation of (E)-vinylsilanes C within 10 min under the standard conditions, as well as by the successful conversion of such an intermediate to the dihydrosilylation products 2. Deuterium labeling experiments further showed that hydrogen is specifically delivered from the silane source.
Co-catalyzed dihydrosilylation of alkynes
It is not exaggerated to state that the development of alkyne dihydrosilylation has been mostly dominated by cobalt catalysis, since the recent advances have seen the advent of a much wider spectrum of dihydrosilylation transformations based on the use of such an earth-abundant metal. In particular, Lu and co-workers led this research field at the forefront, elegantly showing numerous protocols with diverse selectivity. Building upon their previous strategy for the Co-catalyzed alkene hydrosilylation (Cheng et al., 2017), the Lu group reported the first example of Co-catalyzed alkyne dihydrosilylation reactions in 2019 (Guo et al., 2019). The reaction utilizes a dual catalytic system hinging on CoBr2·Xantphos (Co1) and CoBr2·OIP (Co2; OIP = oxazoline-iminopyridine), affording 1,1-disilylalkanes (4) not only in high efficiency and regioselectivity but also in excellent enantioselectivity (Scheme 3A). Similar to the above-mentioned Fe strategy, additive amounts of a reductant (NaBHEt3) were required to activate the Co catalysts, and a variety of primary (Si1) and secondary silanes (Si2) could be employed together as the silane sources for the sequential additions (4a–4c). The scope was broad with respect to the aliphatic alkynes (1), tolerating a range of functional groups such as heterocyclic motif (4d), chloro (4e), silyl ether (4f), and ether (4g) to achieve the products in excellent enantioselectivity. The synthetic virtues of this protocol were proven by the compatibility for gram-scale reactions and further by the site-selective post-modification of the secondary silane moiety of product 4 (Scheme 3B). For instance, the reaction with (4-methoxyphenyl)magnesium bromide gave product 5 via arylation at the secondary silane motif, while the use of Co1 catalyst in further hydrosilylation with propyne furnished product 6 in a good diastereoselectivity.
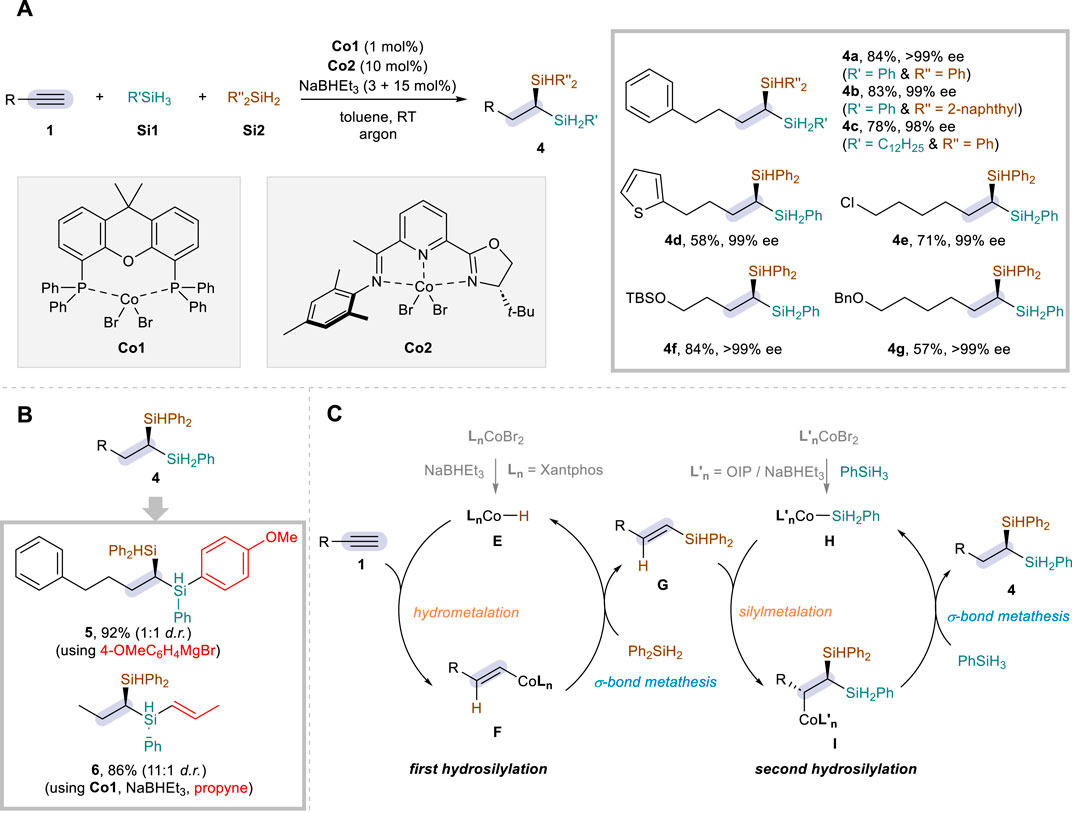
Scheme 3. Co-catalyzed enantioselective dihydrosilylation of aliphatic alkynes via dual catalysis. (A) Selected examples of reaction scope. (B) Synthetic application. (C) Proposed mechanism.
The reaction was proposed to operate through two distinctive catalytic cycles, and control experiments corroborated that secondary silane Si2 is inserted first and CoBr2·Xantphos (Co1) is essential for this process. Notably, NaBHEt3 was suggested to react with Co1 to form a CoH species E, which then joins the first catalytic cycle to enable the hydrosilylation of aliphatic alkyne 1 with Si2 via hydrometalation and σ-bond metathesis of F to form the C–Si bond (Scheme 3C). On the other hand, the second catalytic cycle was proposed to proceed with OIP-based Co–Si species H (Wang et al., 2017), allowing for the asymmetric hydrosilylation of vinylsilane G with primary silane Si1 through silylmetalation followed by C–H bond formation. The proposed mechanism was in good agreement with DFT calculations, and the regioselectivity of the second hydrosilylation was elucidated to be directed predominantly by the electronic effects of the silyl group of G.
Based on the same mechanistic platform, the Lu group further improved the dihydrosilylation reaction to a single catalytic system (Cheng et al., 2019) that relies on CoCl2·IIP complex (Co3; IIP = imidazoline iminopyridine), once again facilitating the dihydrosilylation of aliphatic alkynes 1 but rather with primary silanes Si1 as a sole hydrosilylation reagent (Scheme 4). An enhanced functional group tolerance was observed when compared to the dual catalytic strategy, leading to the corresponding 1,1-disilylalkanes 2 in excellent selectivity and improved efficiency with reduced catalyst loadings (2f–2h, Scheme 4A). Remarkably, the reaction was also effective for the incorporation of ethyne as the alkyne coupling partner, successfully transforming the gaseous reactant to the corresponding 1,1-disilylalkane product 2i. Furthermore, the asymmetric sequential dihydrosilylation was also possible by employing a one-pot procedure (Scheme 4B). For instance, both enantiomers of 1,1-disilylalkane 7a could be obtained by simply changing the addition sequence of the two silane sources under identical reaction conditions. Although this protocol was mechanistically drawn upon the previous dual catalytic system, the authors suggested that the second hydrosilylation step could also proceed via CoH-based hydrometalation/σ-bond metathesis.
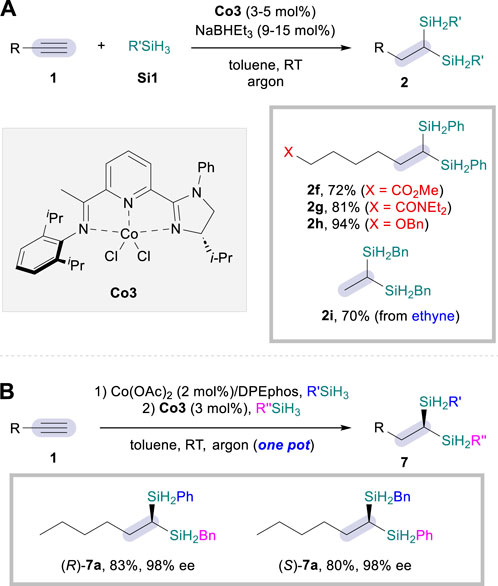
Scheme 4. Co-catalyzed dihydrosilylation of aliphatic alkynes. (A) Single catalysis system. (B) Enantioselective one-pot sequential procedure.
While proven to be highly efficient for the access to 1,1-disilylalkanes, the aforementioned protocols were limited to the dihydrosilylation of aliphatic terminal alkynes. The use of rare-earth La (Chen et al., 2020) or borane catalysts (Wang et al., 2021) provided alternative strategies that render such a limitation mitigated to certain extents, furnishing geminal disilylalkanes from aryl alkynes via initial Markovnikov or anti-Markovnikov hydrosilylation, respectively. In 2022, Banach et al. (2022) demonstrated that Co catalysts are also effective for the dihydrosilylation of alkynes bearing an aryl substituent (8), albeit requiring elevated reaction temperatures. The initial screening involved the use of Fe catalysts and Fe/Co dual conditions, but Co4 with an N,N,N-tridentate hydrazone ligand was shown to be most efficient for the reaction employing Si1, NaHBEt3, and THF solvent (Scheme 5). A range of aryl alkynes could be reacted through sequential Markovnikov-selective hydrosilylations, furnishing geminal aryl (disilyl)alkanes 9 in excellent regioselectivity but with moderate efficiency. Common substituents such as methyl (9b), methoxy (9c), and fluoro (9d) could be tolerated, and so were thiophenyl alkyne (9e) and aliphatic silane (9f). While the mechanistic picture was not drawn in detail, the authors utilized 1H and 11B NMR experiments to suggest that NaHBEt3 participates not only as a reductant for the formation of CoH species but also as a base to modify the ligand and influence the catalytic activity of the metal center.
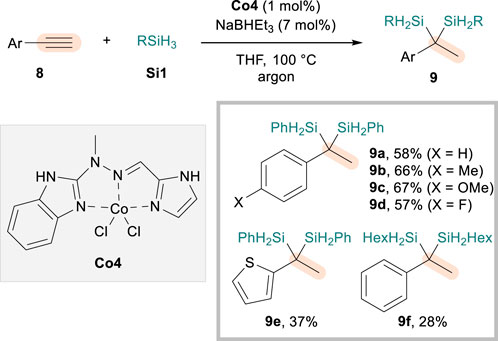
Scheme 5. Co-catalyzed dihydrosilylation of aryl alkynes.
In 2023, Lu and co-workers reported a novel method to selectively achieve not only α,α- but also α,β-dihydrosilylation of aryl alkynes (Cheng et al., 2023). These regiodivergent reactions commonly employ NaBHEt3 as an activator, CoBr2·OIP (Co5) as a precatalyst, secondary silanes Si2, and THF solvent for the first hydrosilylation step in reaction with aryl alkynes 8, but differed reaction conditions are needed to diversify the regioselectivity of the second hydrosilylation of α-substituted vinylsilane intermediates 10 (Scheme 6). The α,β-dihydrosilylation was achieved by utilizing CoBr2·Xantphos (Co1) under neat conditions in the second addition step, which led to the vicinal disilylalkanes 11 via anti-Markovnikov hydrosilylation of vinylsilane intermediates 10 with Ph2SiH2 (Scheme 6A). Under these reaction conditions, a variety of aryl alkynes were reacted for the α,β-dihydrosilylation, showing a good functional group tolerance (11a–11c) and compatibility with other secondary silane sources (11d–11e). On the other hand, the α,α-dihydrosilylation was realized by further addition of NaBHEt3, in combination with toluene solvent and primary silanes Si1 as an alternative second hydrosilylation reagent since the sterically more encumbered secondary silanes hinder the incorporation of silyl groups at the same carbon (Scheme 6B). A range of aryl alkynes 8 could be transformed to the corresponding geminal disilylalkanes 12, with a similar level of functional group tolerance in comparison to the α,β-dihydrosilylation system.
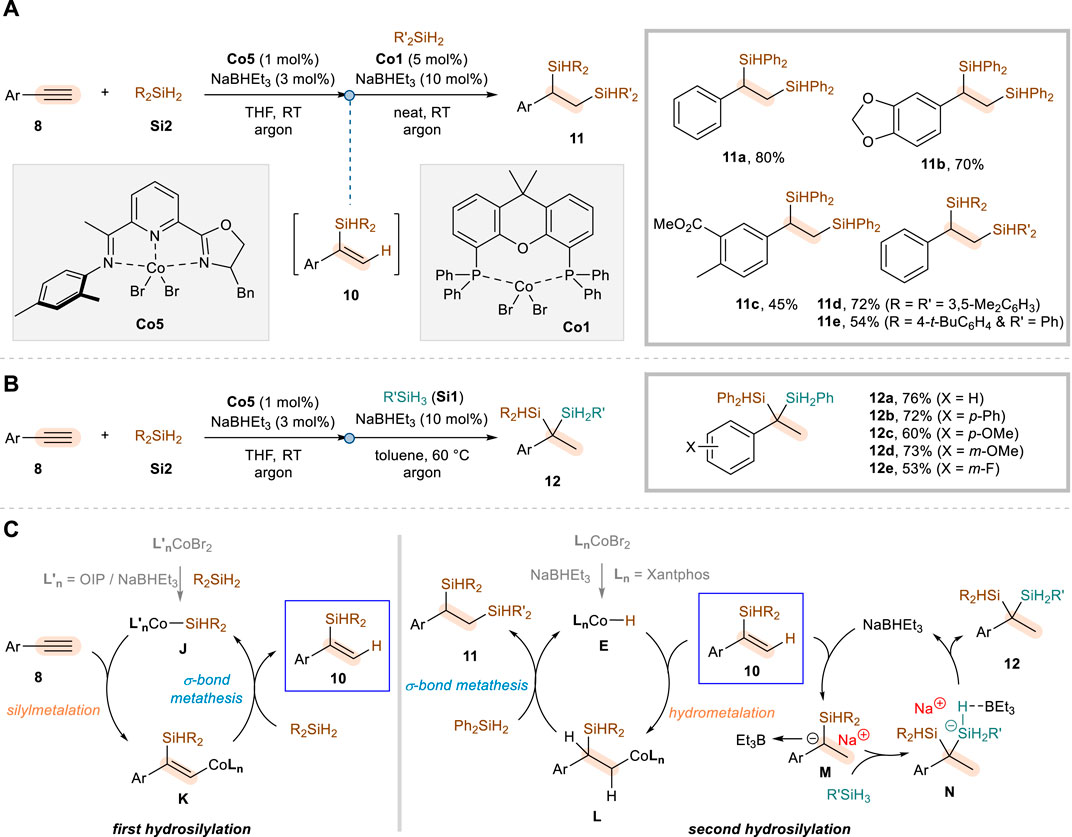
Scheme 6. Co-catalyzed regiodivergent dihydrosilylation of aryl alkynes. (A) α,β-selective dihydrosilylation. (B) α,α-selective dihydrosilylation. (C) Proposed mechanism.
For the initial α-selective hydrosilylation of terminal aryl alkynes, the catalysis involving Co–Si species was proposed in which the migratory insertion of the alkyne into the Co–Si bond (J to K) is followed by σ-bond metathesis (K to 10) with a secondary silane (Scheme 6C). The succeeding anti-Markovnikov hydrosilylation leading to α,β-dihydrosilylation products 11 was then proposed to involve a CoH species E, which is initially generated from the reaction between CoBr2·Xantphos (Co1) and NaBHEt3. The hydrometalation of the α-substituted vinylsilane intermediate 10 proceeds with a selective insertion of the cobalt at the β-position (L), thus eventually furnishing the vicinal bis(silane) products 11 through C–Si bond-forming σ-bond metathesis. In contrast, the authors proposed that the second hydrosilylation leading to α,α-dihydrosilylation products 12 is rather catalyzed by NaBHEt3, generating α-silyl-α-phenylcarbanion M through a hydride transfer. It was presumed that coordination by triethylboron and the subsequent reaction with a primary silane (N) would then follow to give the geminal bis(silane) products 12 and regenerate NaBHEt3.
Ni-catalyzed dihydrosilylation of alkynes
One of the common requirements observed in most of the earth-abundant metal-catalyzed alkyne dihydrosilylation strategies is the need of an air- and moisture-sensitive reductant to activate the precatalyst into a low-valent catalytic species. Such a prerequisite, as a corollary, lowers the synthetic maneuverability owing to the consequent need of reaction set-up under an inert atmosphere. In this regard, Ni is a good candidate as a potential catalyst that could preclude the use of strong reductants, since the formation of low-valent Ni species is well precedented even with silanes as a mild reductant (Eberhardt and Guan, 2016). Nonetheless, Ni-catalyzed hydrosilylation of alkynes is relatively rare, and the few reported examples of monohydrosilylation reactions are mainly based on the use of a porous organic polymer (POP) as a heterogeneous ligand (Zhou et al., 2018). Very recently, Jung and co-workers reported a Ni-catalyzed protocol for dihydrosilylation of alkynes, a highly practical method that circumvents the need of strong reductants while being operative even under aqueous and aerobic conditions (Lee et al., 2024). The reaction was found to be highly effective using Ni(acac)2 as a precatalyst, 2,9-dimethyl-1,10-phenanthroline ligand L1, Ph2SiH2 as the sole silane source, and H2O solvent at 30°C, resulting in the formation of 1,1-disilylalkanes 13 with excellent regioselectivity (Scheme 7A). Although the reaction was also compatible with organic solvents or under neat conditions, the reaction rate was found to be escalated when H2O solvent was employed.
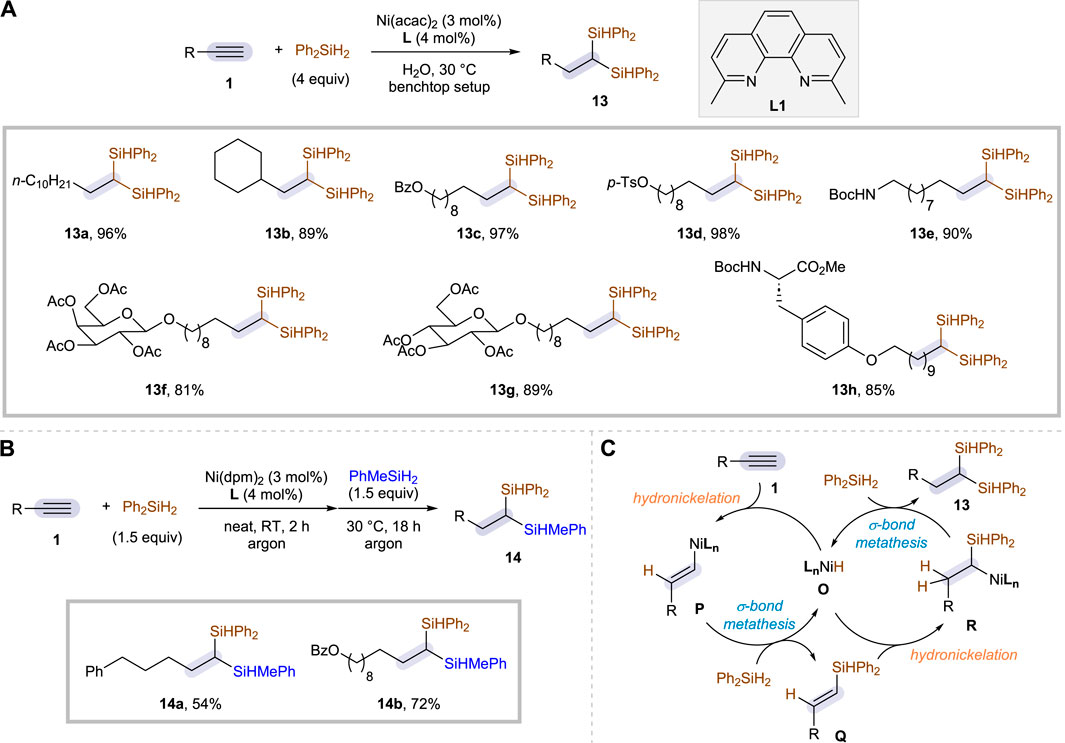
Scheme 7. Ni-catalyzed dihydrosilylation of aliphatic alkynes in aqueous and aerobic conditions. (A) Selected examples of reaction scope. (B) Sequential dihydrosilylation. (C) Proposed mechanism.
The substrate scope is broad and includes not only simple aliphatic alkynes (13a–13e) but also those substituted with a biologically relevant moiety (13f–13h): galactose, glucose, and tyrosine derivatives could all be incorporated successfully. Moreover, the installations of two distinctive silyl groups were possible through sequential additions of Ph2SiH2 and Ph(Me)SiH2, affording the corresponding 1,1-disilylalkane products 14 in good yields (Scheme 7B). A range of functional groups could be tolerated once again, but the attempts for asymmetric induction with chiral ligands were unsuccessful. It is worthwhile to mention that 1,1-addition of silanes is generally challenging if both silane sources being inserted are secondary silanes. Indeed, a primary silane was required as the second silane reagent for the previous reactions leading to 1,1-disilylalkanes, enabling the transformation by easing the steric effects. In contrast, the Ni system was shown to be surprisingly efficient for the geminal insertions of two secondary silanes. The authors conducted a series of control experiments and DFT calculations, and subsequently proposed a mechanism consisting of two identical catalytic cycles, one catalyzing the hydrosilylation of alkyne 1 and the other one promoting the conversion of (E)-vinylsilane intermediate Q (Scheme 7C). The precatalyst was suggested to be converted to NiH species O as an active catalyst when treated with Ph2SiH2, which then undergoes a series of hydronickelation (1 to P and Q to R) followed by C–Si bond-forming σ-bond metathesis (P to Q and R to 13). This operation mode was computed to be consistent with the observed regioselectivity.
Conclusion and outlook
In this mini-review, we have highlighted recent developments of earth-abundant transition metal-catalyzed strategies for dihydrosilylation of alkynes. Given the significant electronic and steric differences between alkynes and vinylsilanes, monohydrosilylation is more common with alkynes, as the second addition would necessitate differed catalytic conditions. Recent advancements have addressed this challenge by either utilizing dual catalyst systems or enhancing the performance of a single catalyst to enable both additions. Catalytic systems based on Fe, Co, and Ni have been reported, each exhibiting distinctive features in terms of substrate compatibility, regioselectivity, practicality, and reaction mechanism. The Fe system has been proven selective for the dihydrosilylation of aliphatic terminal alkynes leading to 1,1-disilylalkanes, utilizing EtMgBr as an activator and PhSiH3 as a silane source. The Co systems are more diverse and have offered enantioselective methods for the synthesis of 1,1-disilylalkanes, as well as regiodivergent strategies for the dihydrosilylation of aryl alkynes, be it based on single or dual catalysts. In the Co systems, NaBHEt3 has been commonly employed as an activator and a wider spectrum of silane reagents have been applied. The Ni system has been developed more recently and offers a practical method that operates under atmospheric conditions while circumventing the need of strong reductants. The efficiency is not deteriorated by the presence of air or water in this system, thus rendering the synthetic maneuver extremely simple for access to 1,1-disilylalkanes.
Despite these advancements, this research area is still in its infancy and the reaction development for unseen selectivity is highly desired. For instance, the dihydrosilylation of aliphatic alkynes has been mainly developed for the 1,1-addition and, as such, the investigation toward 1,2- or 2,1- or 2,2-additions could be an interesting research topic, especially for the development of enantioselective reactions. Lu and co-workers showed that the dihydrosilylation of aryl alkynes could be achieved in a regiodivergent manner, and the study toward its asymmetric version would also be of great significance. The reaction efficiency should also be improved for sterically encumbered substrates, including internal alkynes, which have been shown operational only for the monohydrosilylation. Furthermore, there is a demand for detailed investigations of the reaction mechanisms. Two commonly proposed operation modes are: 1) migratory insertion of alkyne or vinylsilane into M–H bond followed by C–Si bond-forming σ-bond metathesis, and 2) migratory insertion of alkyne or vinylsilane into M–Si bond followed by C–H bond-forming σ-bond metathesis. These mechanisms have been proposed mainly based on DFT calculations, kinetic studies, and control experiments, thus lacking evidence that provides more direct insights. The isolation of key intermediates would be an appealing strategy for the sophisticated elucidation of the involved reaction mechanisms.
Author contributions
CL: Writing–original draft. DL: Writing–original draft. SYH: Conceptualization, Writing–review and editing, Project administration. BJ: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–original draft, Writing–review and editing. SS: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the DGIST Start-up Fund Program of the Ministry of Science and ICT (2023040012), and also by Alchemist Project (no. 20025719) funded by the Korean Ministry of Trade, Industry, and Energy (MOTIE).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Banach, Ł., Brykczyńska, D., Gorczyński, A., Wyrzykiewicz, B., Skrodzki, M., and Pawluć, P. (2022). Markovnikov-selective double hydrosilylation of challenging terminal aryl alkynes under cobalt and iron catalysis. Chem. Commun. 58, 13763–13766. doi:10.1039/D2CC04015H
Bauer, I., and Knölker, H.-J. (2015). Iron catalysis in organic synthesis. Chem. Rev. 115, 3170–3387. doi:10.1021/cr500425u
Challinor, A. J., Calin, M., Nichol, G. S., Carter, N. B., and Thomas, S. P. (2016). Amine-activated iron catalysis: air- and moisture-stable alkene and alkyne hydrofunctionalization. Adv. Synth. Catal. 358, 2404–2409. doi:10.1002/adsc.201600570
Chen, J., Guo, J., and Lu, Z. (2018). Recent advances in hydrometallation of alkenes and alkynes via the first row transition metal catalysis. Chin. J. Chem. 36, 1075–1109. doi:10.1002/cjoc.201800314
Chen, W., Song, H., Li, J., and Cui, C. (2020). Catalytic selective dihydrosilylation of internal alkynes enabled by rare-earth ate complex. Angew. Chem. Int. Ed. 59, 2365–2369. doi:10.1002/anie.201913773
Cheng, B., Lu, P., Zhang, H., Cheng, X., and Lu, Z. (2017). Highly enantioselective cobalt-catalyzed hydrosilylation of alkenes. J. Am. Chem. Soc. 139, 9439–9442. doi:10.1021/jacs.7b04137
Cheng, Z., Li, M., Zhang, X.-Y., Sun, Y., Yu, Q.-L., Zhang, X.-H., et al. (2023). Cobalt-catalyzed regiodivergent double hydrosilylation of arylacetylenes. Angew. Chem. Int. Ed. 62, e202215029. doi:10.1002/anie.202215029
Cheng, Z., Xing, S., Guo, J., Cheng, B., Hu, L.-F., Zhang, X.-H., et al. (2019). Highly regioselective sequential 1,1-dihydrosilylation of terminal aliphatic alkynes with primary silanes. Chin. J. Chem. 37, 457–461. doi:10.1002/cjoc.201900079
de Almeida, L. D., Wang, H., Junge, K., Cui, X., and Beller, M. (2021). Recent advances in catalytic hydrosilylations: developments beyond traditional platinum catalysts. Angew. Chem. Int. Ed. 60, 550–565. doi:10.1002/anie.202008729
Docherty, J. H., Peng, J., Dominey, A. P., and Thomas, S. P. (2017). Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem. 9, 595–600. doi:10.1038/nchem.2697
Eberhardt, N. A., and Guan, H. (2016). Nickel hydride complexes. Chem. Rev. 116, 8373–8426. doi:10.1021/acs.chemrev.6b00259
Fürstner, A. (2016). Iron catalysis in organic synthesis: a critical assessment of what it takes to make this base metal a multitasking champion. ACS Cent. Sci. 2, 778–789. doi:10.1021/acscentsci.6b00272
Greenhalgh, M. D., Frank, D. J., and Thomas, S. P. (2014). Iron-Catalysed chemo-regio-and stereoselective hydrosilylation of alkenes and alkynes using a bench-stable iron(II) pre-catalyst. Adv. Synth. Catal. 356, 584–590. doi:10.1002/adsc.201300827
Greenhalgh, M. D., Jones, A. S., and Thomas, S. P. (2015). Iron-Catalysed hydrofunctionalisation of alkenes and alkynes. ChemCatChem 7, 190–222. doi:10.1002/cctc.201402693
Guo, J., Wang, H., Xing, S., Hong, X., and Lu, Z. (2019). Cobalt-catalyzed asymmetric synthesis of gem-Bis(silyl)alkanes by double hydrosilylation of aliphatic terminal alkynes. Chem 5, 881–895. doi:10.1016/j.chempr.2019.02.001
He, P., Hu, M.-Y., Zhang, X.-Y., and Zhu, S.-F. (2022). Transition-metal-catalyzed stereo- and regioselective hydrosilylation of unsymmetrical alkynes. Synthesis 54, 49–66. doi:10.1055/a-1605-9572
Hu, M.-Y., He, P., Qiao, T.-Z., Sun, W., Li, W.-T., Lian, J., et al. (2020). Iron-catalyzed regiodivergent alkyne hydrosilylation. J. Am. Chem. Soc. 142, 16894–16902. doi:10.1021/jacs.0c09083
Hu, M.-Y., He, Q., Fan, S.-J., Wang, Z.-C., Liu, L.-Y., Mu, Y.-J., et al. (2018). Ligands with 1,10-phenanthroline scaffold for highly regioselective iron-catalyzed alkene hydrosilylation. Nat. Commun. 9, 221. doi:10.1038/s41467-017-02472-6
Hu, M.-Y., Lian, J., Sun, W., Qiao, T.-Z., and Zhu, S.-F. (2019). Iron-catalyzed dihydrosilylation of alkynes: efficient access to geminal bis(silanes). J. Am. Chem. Soc. 141, 4579–4583. doi:10.1021/jacs.9b02127
Lee, C., Jeon, J. H., Lee, S., Choe, W., Kwak, J., Seo, S., et al. (2024). Nickel-catalyzed mono- and dihydrosilylation of aliphatic alkynes in aqueous and aerobic conditions. ACS Catal. 14, 5077–5087. doi:10.1021/acscatal.4c00109
Marciniec, B. (2009) Hydrosilylation: a comprehensive review on recent advances. Dordrecht: Springer.
Nakajima, Y., and Shimada, S. (2015). Hydrosilylation reaction of olefins: recent advances and perspectives. RSC Adv. 5, 20603–20616. doi:10.1039/C4RA17281G
Obligacion, J. V., and Chirik, P. J. (2018). Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Rev. Chem. 2, 15–34. doi:10.1038/s41570-018-0001-2
Shimada, T., Mukaide, K., Shinohara, A., Han, J. W., and Hayashi, T. (2002). Asymmetric synthesis of 1-Aryl-1,2-ethanediols from arylacetylenes by palladium-catalyzed asymmetric hydrosilylation as a key step. J. Am. Chem. Soc. 124, 1584–1585. doi:10.1021/ja017617g
Sun, J., and Deng, L. (2016). Cobalt complex-catalyzed hydrosilylation of alkenes and alkynes. ACS Catal. 6, 290–300. doi:10.1021/acscatal.5b02308
Wang, C., Teo, W. J., and Ge, S. (2017). Cobalt-catalyzed regiodivergent hydrosilylation of vinylarenes and aliphatic alkenes: ligand- and silane-dependent regioselectivities. ACS Catal. 7, 855–863. doi:10.1021/acscatal.6b02518
Wang, G., Su, X., Gao, L., Liu, X., Li, G., and Li, S. (2021). Borane-catalyzed selective dihydrosilylation of terminal alkynes: reaction development and mechanistic insight. Chem. Sci. 12, 10883–10892. doi:10.1039/D1SC02769G
Zaranek, M., Marciniec, B., and Pawluć, P. (2016). Ruthenium-catalysed hydrosilylation of carbon–carbon multiple bonds. Org. Chem. Front. 3, 1337–1344. doi:10.1039/C6QO00261G
Keywords: alkyne functionalization, dihydrosilylation, earth-abundant metal catalysis, organosilanes, sustainable synthesis
Citation: Lee C, Lee D, Hong SY, Jung B and Seo S (2024) Recent advances in earth-abundant transition metal-catalyzed dihydrosilylation of terminal alkynes. Front. Chem. 12:1411140. doi: 10.3389/fchem.2024.1411140
Received: 02 April 2024; Accepted: 23 April 2024;
Published: 27 May 2024.
Edited by:
Sehoon Park, Guangdong Technion-Israel Institute of Technology (GTIIT), ChinaReviewed by:
Chinmoy Kumar Hazra, Indian Institute of Technology Delhi, IndiaJianbo Zhang, Northwestern Polytechnical University, China
Copyright © 2024 Lee, Lee, Hong, Jung and Seo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byunghyuck Jung, Ynl1bmdoeXVjay5qdW5nQGRnaXN0LmFjLmty; Sangwon Seo, c2FuZ3dvbi5zZW9AZGdpc3QuYWMua3I=
†These authors have contributed equally to this work