
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 24 May 2024
Sec. Medicinal and Pharmaceutical Chemistry
Volume 12 - 2024 | https://doi.org/10.3389/fchem.2024.1403127
 Rubina Munir1*
Rubina Munir1* Sumera Zaib2*
Sumera Zaib2* Muhammad Zia-ur-Rehman3
Muhammad Zia-ur-Rehman3 Hira Javed2Ayesha Roohi1Muhammad Zaheer3Nabiha Fatima1
Hira Javed2Ayesha Roohi1Muhammad Zaheer3Nabiha Fatima1 Mashooq Ahmad Bhat4
Mashooq Ahmad Bhat4 Imtiaz Khan5*
Imtiaz Khan5*An important component of the pathogenicity of potentially pathogenic bacteria in humans is the urease enzyme. In order to avoid the detrimental impact of ureolytic bacterial infections, the inhibition of urease enzyme appears to be an appealing approach. Therefore, in the current study, morpholine-thiophene hybrid thiosemicarbazone derivatives (5a-i) were designed, synthesized and characterized through FTIR, 1H NMR, 13C NMR spectroscopy and mass spectrometry. A range of substituents including electron-rich, electron-deficient and inductively electron-withdrawing groups on the thiophene ring was successfully tolerated. The synthesized derivatives were evaluated in vitro for their potential to inhibit urease enzyme using the indophenol method. The majority of compounds were noticeably more potent than the conventional inhibitor, thiourea. The lead inhibitor, 2-(1-(5-chlorothiophen-2-yl)ethylidene)-N-(2-morpholinoethyl)hydrazinecarbothioamide (5g) inhibited the urease in an uncompetitive manner with an IC50 value of 3.80 ± 1.9 µM. The findings of the docking studies demonstrated that compound 5g has a strong affinity for the urease active site. Significant docking scores and efficient binding free energies were displayed by the lead inhibitor. Finally, the ADME properties of lead inhibitor (5g) suggested the druglikeness behavior with zero violation.
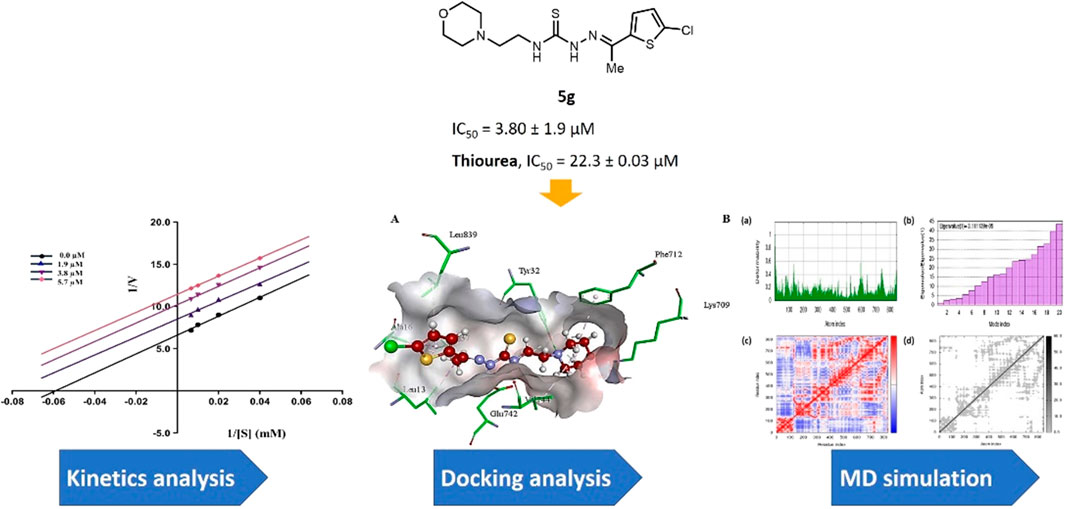
GRAPHICAL ABSTRACT | A concise library of morpholine-thiophene hybrid thiosemicarbazones was synthesized for the identification of potent inhibitors of urease enzyme to treat ureolytic bacterial infections.
Urease (amidohydrolase and phosphotriesterase; EC 3.5.1.5) is a metallopeptidase enzyme extensively found in prokaryotes as well as some eukaryotes and catalyzes the conversion of urea to ammonia and carbamate (Xiao et al., 2013; Mobley and Hausinger, 1989; Konieczna et al., 2012; D’Agostino and Carradori, 2024). Urease plays an important role in enzymology and speeds up the catalytic activity by 1014 times. The active site of urease contains three water molecules and two Ni atoms connected through a hydroxide bond (Mobley and Hausinger, 1989). The urease from H. pylori (Helicobacter pylori) is the most commonly investigated bacterial urease due to its infectious association with various diseases like gastritis, stomach cancer, and peptic ulcer (Kafarski and Talma, 2018; Hameed et al., 2019). The potential of the bacteria to produce ammonia which nullifies the tough acidic environment around them allows these bacteria to thrive in the acidic environment of the stomach (Zambelli et al., 2011). Urease is a key target for the design of drugs against H. pylori, therefore, urease inhibition treatments are considered to be a potentially effective way to treat disorders caused by urease-producing bacteria (Kafarski and Talma, 2018). The scientific community has paid a close attention towards the discovery of urease inhibitors because of their potential application against H. pylori urease. Several urease inhibitors have been identified until now, but only a limited number have progressed to the latter phases of drug development (Ibrar et al., 2013; Svane et al., 2020). These inhibitors include oxadiazoles (Mentese et al., 2017), coumarins (Ayaz et al., 2006), hydrazides (Amtul et al., 2002), pyrimidines (Rauf et al., 2012), triazoles (Mentese et al., 2017), amides (Rauf et al., 2016), triazolothiadiazoles (Hanif et al., 2012), thioureas (Khan et al., 2006), thiosemicarbazides (Rego et al., 2018a), phosphoramidates (Kot et al., 2001), hydroxamic acids (Muri et al., 2003), flavonoid glycosides (Babu et al., 2017), Schiff bases, and many others (Abid et al., 2010; Kazmi et al., 2019; Yang et al., 2023; Al-Fakhrany and Elekhnawy, 2023; Sepehri and Khedmati, 2023; Al-Rooqi et al., 2023; Khan et al., 2024; Akash et al., 2024; Viana et al., 2024; Wang et al., 2024; Valenzuela-Hormazabal, et al., 2024; Saeed K. et al., 2024; Ullah et al., 2024].
In parallel, thiosemicarbazone is a sulfur-nitrogen donor ligand which serves as a robust precursor to heterocyclic compounds (Castro et al., 2023; Palakkeezhillam et al., 2023). Thiosemicarbazones are synthesized by the condensation reaction of thiosemicarbazide with suitable aldehydes or ketones (Devi et al., 2022). The presence of amide, imine and thione groups make them potential polydentate ligands (Yousef and El-Reash, 2020). In 1960s, the first biological application of thiosemicarbazones was found against tuberculosis and leprosy (Volynets et al., 2019). Heterocyclic thiosemicarbazones have the ability to diffuse through semipermeable membrane into the cell lining (Nkungli et al., 2023). These are known for chelating property towards metals like nickel, zinc, cadmium, cobalt, copper and show a wide range of applications in analytical chemistry, pharmacological chemistry and nuclear medicine (Dilworth and Hueting, 2012; Raicopol et al., 2020). Various studies have demonstrated that the biological and chemical activities of thiosemicarbazones and their complexes may be tuned by altering the metal center attached to sulfur and/or hydrazine as well as the substituent attached to amide nitrogen (Summers, 2019). In recent years, thiosemicarbazone derivatives have been documented for their profound biological activities including antidiabetic (Basri et al., 2023), anticancer (Sibuh et al., 2021; Bai et al., 2022), antibacterial (Hassan et al., 2020), antifungal (Bajaj et al., 2021), anti-inflammatory (Saeed A. et al., 2024), anti-Alzheimer’s (Khan et al., 2023; Jalil et al., 2024), anti-melanoma (Kurt et al., 2024), antitubercular (Gobis et al., 2022) and antioxidant (Yang et al., 2020) activities. The diverse variety of biological potential shown by this class of compounds is possibly due to the multifunctional nature of thiosemicarbazone motif which carries C=S and NH electron donor groups as well as the hydrophobic aryl substituent (Pati et al., 2018). The structural similarity of thiosemicarbazide scaffold with thiourea makes them an ideal candidate for urease inhibitory investigation (Islam et al., 2019; Islam et al., 2023).
In parallel, morpholine heterocycle has been ranked as an important pharmacophore encountered in numerous drugs used for the treatment of bacterial infections and other diseases (Bektaş et al., 2013; Bektaş et al., 2017; Kumari and Singh, 2020; Sıcak et al., 2023). The incorporation of morpholine ring into various drug molecules leads to enhanced potency through the formation of molecular interactions with the target protein or by modulating the pharmacokinetic profile. The World Drug Index has declared more than 100 drugs featuring morpholine ring (Kumari and Singh, 2020). Thiophene containing compounds, on the other hand, have also been reported to exhibit potent inhibitory efficacy against urease (Khan et al., 2010; Noreen et al., 2017; Akyüz et al., 2022; Güven et al., 2023). Both morpholine and thiophene rings have been precedented in literature showing significant urease inhibitory efficacy (Khan et al., 2010; Bektaş et al., 2013; Bektaş et al., 2017; Rego et al., 2018b; Akyüz et al., 2022). Similarly, thiosemicarbazones bearing diverse structural features have also been documented as potent anti-urease agents (Islam et al., 2019; Islam et al., 2023). In view of the anti-urease potential of all three distinct pharmacophores, we aimed to explore their combined urease inhibitory effect as a single entity to successfully deliver the lead drug candidates (Figure 1). Therefore, the structural optimization led us to design and synthesize a concise series of morpholine- and thiophene-containing thiosemicarbazone conjugates which were evaluated against urease enzyme. The obtained in vitro inhibitory data was reinforced with the molecular docking analysis elaborating the ligand-enzyme interaction within the active site of urease enzyme. Molecular dynamics simulations as well as ADMET properties were also computed.
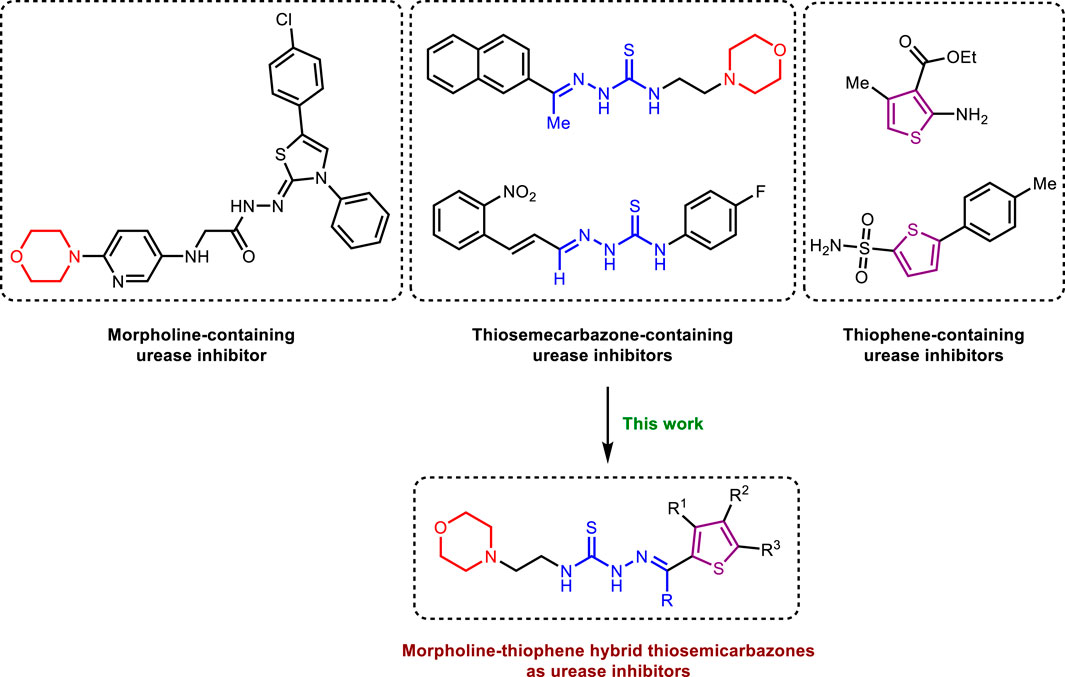
Figure 1. Literature reported urease inhibitors and rationale for strategic development of morpholine-thiophene hybrid thiosemicarbazones.
The synthesis of title thiosemicarbazones was achieved in a facile manner by following the route illustrated in Scheme 1. Base-catalyzed reaction of 4-(2-aminoethyl)morpholine (1) with carbon disulfide followed by desulfurization using copper(II) sulfate provided access to 4-(2-isothiocyanatoethyl)morpholine (2) (Mandapati et al., 2017). Subsequently, hydrazination of isothiocyanate (2) with hydrazine monohydrate at room temperature furnished N-(2-morpholinoethyl)hydrazinecarbothioamide (3) which was condensed in the final step with (un)substituted formyl/acetylthiophenes (4a-i) to afford thiosemicarbazones (5a-i) in good to excellent isolated yields.
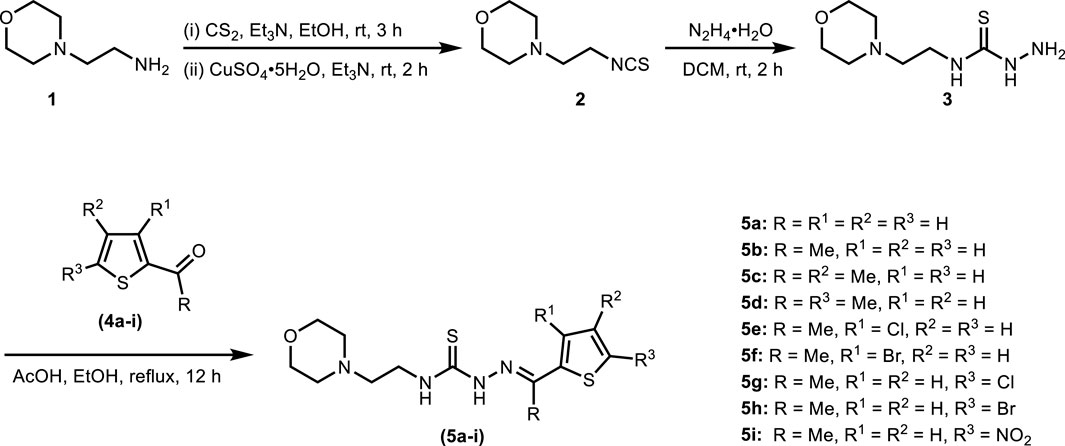
Scheme 1. Synthetic layout for morpholine-thiophene hybrid thiosemicarbazones.
The FTIR spectra of the synthesized thiosemicarbazones displayed N-H stretching bands at 3,345–3,312 cm−1, C=N around 1,530–1,506 cm−1 and C=S stretching vibrations at 1,142–1,102 cm−1. 1H NMR spectra exhibited two peaks concerning the secondary thioamide protons referring to = N-NH and C-NH. The = N-NH signal appeared as a singlet peak downfield between 10.45 and 11.57 ppm. The C-NH proton splits into a triplet due to its spin-spin coupling with the neighbouring methylene protons and was observed around 8.15 ppm. Hydrogens of morpholine ring gave two sets of triplets near 2.44 ppm and 3.60 ppm, the later signal though merged with the peak of the methylene in most of the compounds. The thiophene ring protons resonated between 6.78 and 8.09 ppm depending upon the substituent on the ring.
13C NMR spectral data endorsed the structures of the synthesized compounds where C=S functional group appeared at 178 ppm. Signals of morpholine ring carbon atoms resonated at 53.5 and 66.7 ppm while resonances for linear methylene carbon atoms were observed at 40.4 and 56.5 ppm. Thiophene ring carbon atoms resonated between 108.9 and 178.2 ppm. Likewise, the elemental analyses and mass spectral data of all the derivatives (5a-i) were in agreement with the anticipated structures.
The synthesized morpholine-thiophene hybrid thiosemicarbazones (5a-i) were tested to determine their urease inhibitory effect. In comparison to the standard inhibitor, thiourea, which showed inhibitory activity with an IC50 value of 22.31 ± 0.03 µM, these compounds were discovered to be effective inhibitors of the urease demonstrating outstanding inhibition with IC50 in the range of 3.80–5.77 µM. The inhibitory results of the tested compounds are shown in Table 1.
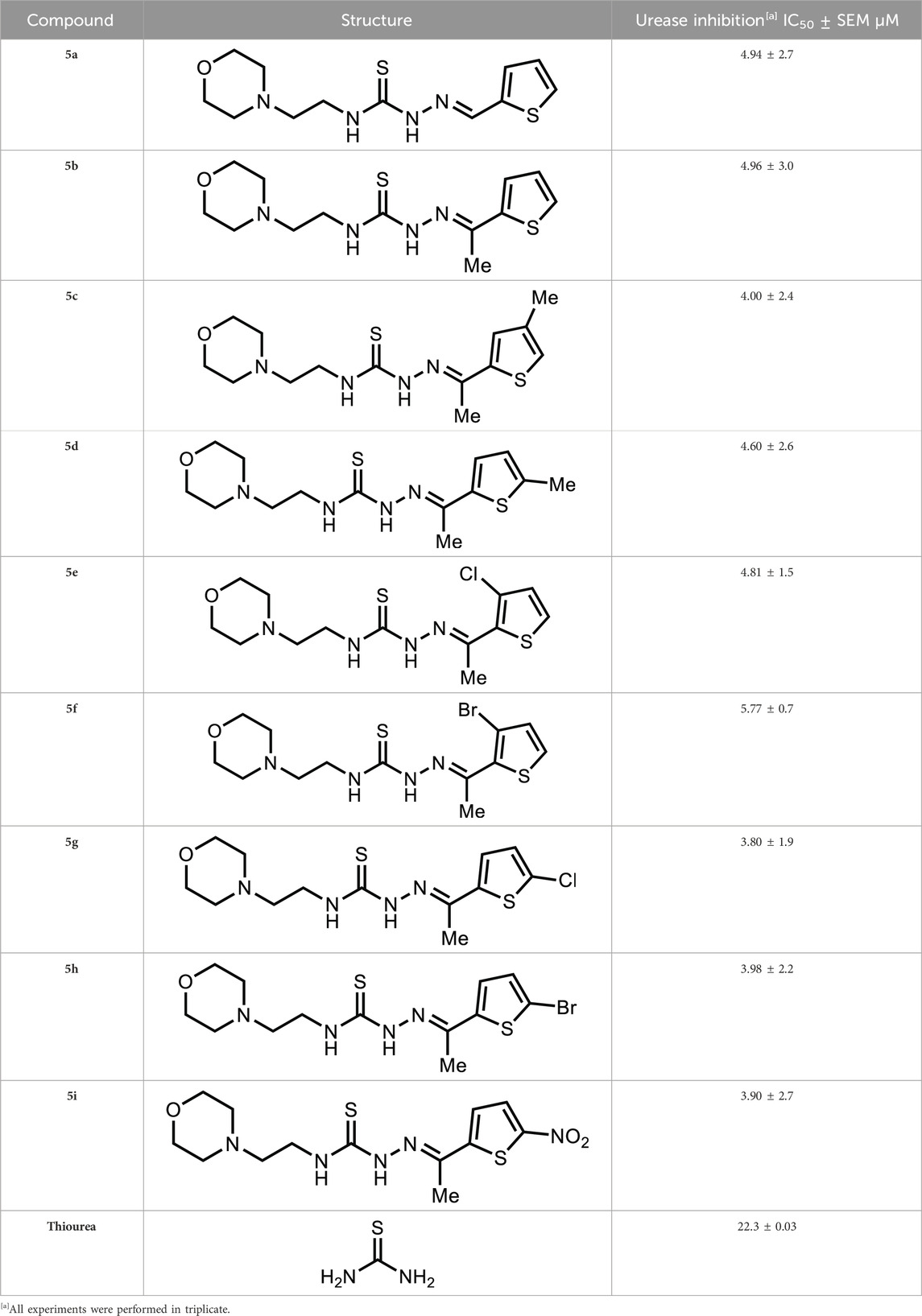
Table 1. In vitro urease inhibitory activity of target compounds (5a-i).
As depicted in Table 1, a varied degree of inhibitory potential was observed for tested compounds (5a-i). All the compounds showed remarkable inhibitory potential identifying compound 5g as the lead inhibitor exhibiting strong inhibitory potential with an IC50 value of 3.80 ± 1.9 µM, more than the standard inhibitor.
In vitro analysis of compounds against urease showed that compound 5a ((E)-N-(2-morpholinoethyl)-2-(thiophen-2-ylmethylene)hydrazinecarbothioamide) exhibited remarkable inhibitory potential with an IC50 value of 4.94 ± 2.7 µM, 4.5-fold strong inhibitory potential than thiourea. Similar inhibition potential was observed for compound 5b where an additional methyl group was present at the imine moiety. Compound 5b demonstrated an IC50 value of 4.96 ± 3.0 µM. However, the addition of methyl substituent at the thiophene ring on position 4 (5c) showed inhibitory potential with an IC50 value of 4.00 ± 2.4 µM. Switching the position of methyl substituent at thiophene ring from 4 to 5 (5d) exhibited slightly lower inhibitory potential than 5c but the strength was still 4.8-fold better than thiourea. Furthermore, the replacement of methyl substituent with a chloro group at thiophene ring (position 3) demonstrated similar inhibition results with an IC50 value of 4.81 ± 1.5 µM as shown by compound 5e. In compound 5f, the replacement of chloro with bromo demonstrated the least potential among all compounds (5a-i) with an IC50 value of 5.77 ± 0.7 µM. However, the inhibitory strength was still stronger than thiourea. Compound 5g exhibited inhibitory potential with the lowest IC50 value. The presence of the chloro group at the fifth position of thiophene ring demonstrated the highest inhibitory strength with an IC50 value of 3.80 ± 1.9 µM. The presence of other electron-withdrawing groups (Br, NO2) at the same position also showed remarkable inhibition of urease with IC50 values of 3.98 ± 2.2 and 3.90 ± 2.7 µM, respectively, as depicted by compounds 5h and 5i. In general, all the tested compounds exhibited several folds higher inhibitory power compared to thiourea, standard inhibitor. Although a varied degree of inhibition was noticed among the tested series of thiosemicarbazones, the extent of biological efficacy was found least dependent on the type of substituent on thiophene ring. Therefore, considering the incorporation of further ring variations as well as changing the ethylmorpholine unit on the left hand side of the synthesized molecules could exert beneficial role in designing new families of thiosemicarbazones for potent urease inhibitory properties.
The kinetics experiments of the most potent inhibitor 5g having the least IC50 value were performed and the results obtained were utilized to construct Lineweaver-Burk plot using GraphPad Prism version 10.2.1. These experiments helped in the assessment of Km and Vmax values to predict the mechanism of urease inhibition by plotting reciprocal of reaction rate 1/V (y-axis) against reciprocal of substrate concentrations 1/[S] (x-axis). As illustrated in Figure 2, compound 5g inhibits urease in an uncompetitive manner as all the slopes (Km/Vmax) are parallel to each other at different inhibitor concentrations. These results indicate that both the Vmax and Km decrease as the concentration of inhibitor increases.
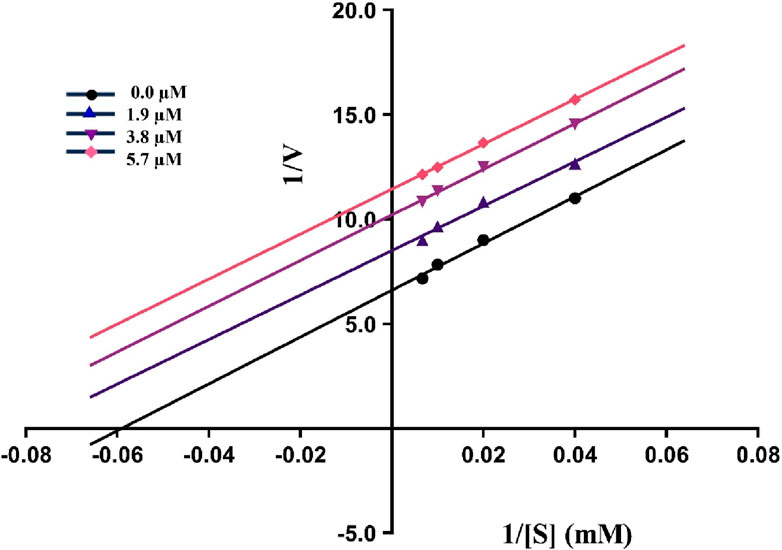
Figure 2. The Lineweaver-Burk plot of 5g at various substrate (urea) and inhibitor concentrations showing uncompetitive inhibition as indicated by parallel slopes.
The lead compound exhibiting the best inhibitory potential with low IC50 value was docked within the jack bean urease active site. Similar to literature examples (Elbastawesy et al., 2021; Hina et al., 2023), PDB ID: 3LA4 was selected as a computational urease model to perform the docking studies for compounds investigated in this research. Compound 5g demonstrated binding affinity in micromolar range along with various interactions with the active site amino acid residues, as shown in Table 2.
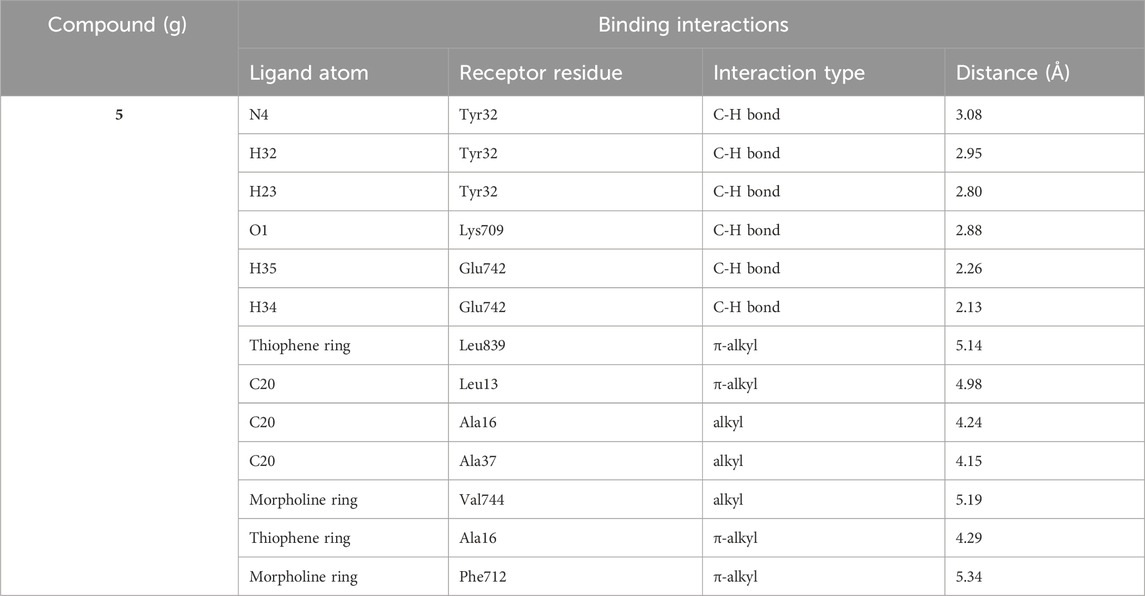
Table 2. The binding interactions between receptor residues and potent inhibitor 5g.
The analysis of intermolecular interactions of potent inhibitor 5g within the binding pocket of urease revealed that several amino acids including Leu13, Ala16, Tyr32, Ala37, Lys709, Phe712, Glu742, Val744 and Leu839 are significantly involved in the formation of key contacts. Compound 5g showed conventional hydrogen bonding with Tyr32, Glu742 and Lys709 present at a distance of 3.08, 2.26 and 2.88 Å, respectively. However, carbon hydrogen bond interactions were formed by the morpholine ring of 5g with Tyr32 (2.79, 2.80, 2.95 Å). Additionally, the morpholine ring of 5g also developed alkyl and π-alkyl interactions with Val744 (5.19 Å) and Phe717 (5.34 Å), respectively. Amino acid residues such as Ala16 and Leu839 interact with the thiophene ring via π-alkyl interactions having bond lengths of 4.29 and 5.14 Å, respectively. On the other hand, methyl group of 5g showed alkyl and π-alkyl interactions with Leu13 (4.98 Å), Ala16 (4.24 Å) and Ala37 (4.15 Å), as shown in Figure 3.
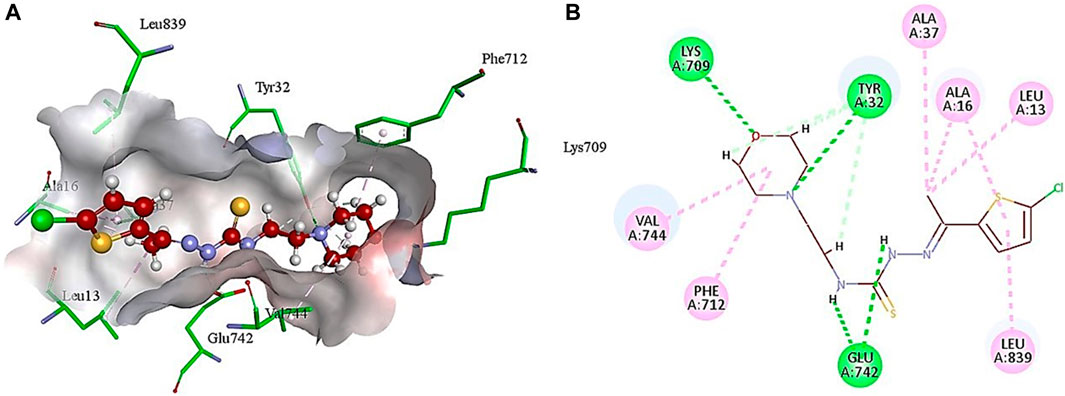
Figure 3. Illustration of 3D (A) and 2D (B) interactions of compound 5g with urease.
The other compounds were also docked in the same active site, and the sequence of the decrease in estimated affinity (5g > 5i > 5h > 5c > 5d> 5e > 5a>5b > 5f > positive control) verifies the IC50 values obtained in an in vitro assay against urease (Supplementary Figure S1).
The conventional inhibitor (thiourea) showed only three interactions (hydrogen bonds) with two amino acid residues (Ser421 and Thr715) when docked in the same binding pocket of urease. Therefore, it depicts more IC50 value as compared to morpholine-thiophene hybrid thiosemicarbazone derivatives. Moreover, all the synthesized morpholine-thiophene hybrid thiosemicarbazones (5a-i) showed maximum intermolecular interactions with the active site residues of urease. That is why, their IC50 values have no major differences. The thiophene ring as the main attachment with various substituents interacts with urease active site residues via alkyl, π-alkyl, π-anion, and π-cation interactions as shown in Supplementary Figure S2.
The presence of methyl substituent on the thiophene ring (5c and 5d) facilitates the formation of alkyl interaction. On the other hand, the bromo substituent on thiophene ring in 5f and 5h participates in the formation of alkyl interaction with amino acid residues of urease. In addition, nitro substitution on thiophene ring enables π-cation, π-anion and attractive charges interactions. The presence of chloro group as a substituent on the thiophene ring in 5e and 5g has not shown any interaction with the amino acid residues of the binding site. Apart from these, hydrogen bond interactions were observed as the common contacts within all compounds docked with the binding site residues of urease, but the number of these interactions varied as 5g showed 7 H-bond interactions. The presence of more hydrogen bond interactions makes 5g the most potent and lead inhibitor showing the lowest IC50 value of 3.80 ± 1.9 µM.
These findings also correlate with the literature precedent (Hassan and Švajdlenka, 2017) where hydrogen bonding involving amino acid residues Tyr32 and Lys709 are mainly responsible for the anti-urease activity. Another similar report (Kataria and Khatkar, 2019) also revealed that compounds having the best antibacterial activity develop key interactions with the same amino acids (Val744, Lys709, Try32, Ala16, Ala37, Glu742, Leu839, and Leu13) as mentioned in Figure 3. However, the computational analysis of standard inhibitor (thiourea) does not match with the literature example (Saeed et al., 2013). Regardless of this study, several other reports showed the inhibition of urease by various compounds thus making crucial interactions with the same amino acids (Hassan and Švajdlenka, 2017; Kataria and Khatkar, 2019; Hina et al., 2023), as mentioned in Figure 3. These results showed that compound 5g has the potential to exhibit antibacterial activity by the inhibition of urease.
LeadIT software was used to evaluate the Hydrogen bond and DEhydration energy (HYDE) of potent inhibitor 5g. HYDE studies showed that the lead inhibitor (5g) has the highest affinity towards urease. Figure 4 demonstrates the HYDE of atoms of compound 5g indicating their involvement in the binding affinity. The lower the HYDE of an atom, the higher is its involvement in the estimated affinity.
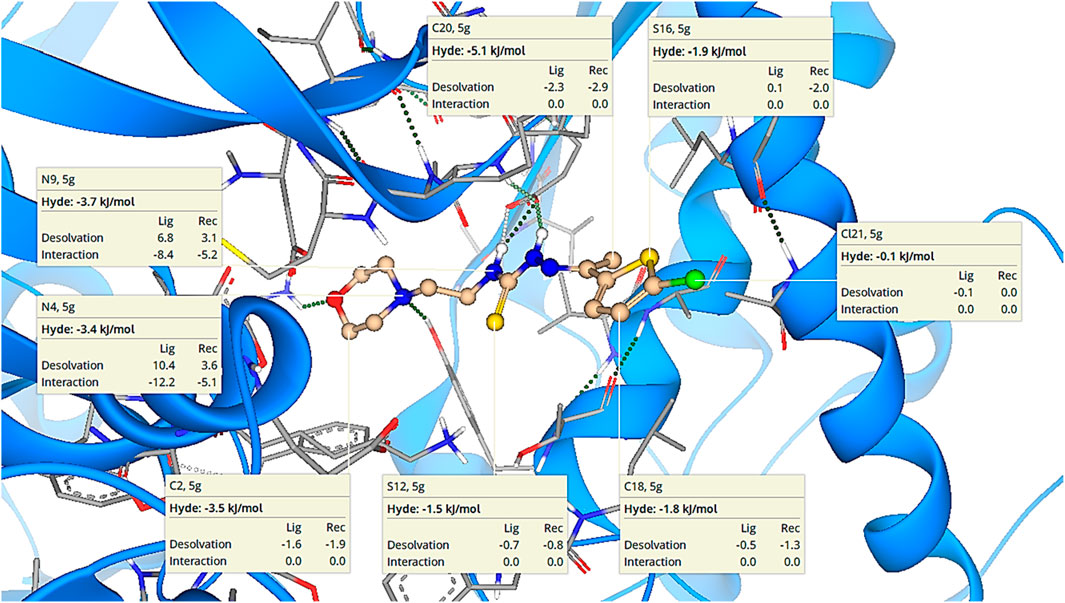
Figure 4. Visual representation of HYDE of compound 5g showing the favorable atoms involved in affinity inside the active site of urease (PDB: 3LA4).
The ADMET properties of compound 5g were determined via calculating various parameters, including physicochemical properties, lipophilicity, water solubility, pharmacokinetics, druglikeness, and medicinal chemistry. These properties determine the likelihood of a drug. In our case, these properties indicated that compound 5g is the lead inhibitor with 0 violation, as shown in Table 3. The pharmacokinetic properties and the lowest IC50 value support compound 5g to serve as a lead inhibitor. Furthermore, the comparative analysis of pharmacokinetic parameters of 5g from four different software (SwissADME, pkCSM, preADMET and vNN-ADMET) is shown in Table 4 (Dulsat et al., 2023). These results interpreted that compound 5g can be absorbed from gastrointestinal tract but unable to cross the barrier between blood and brain tissues. In addition, compound does not inhibit the P-glycoprotein (P-gp) but may act as a substrate of P-gp. The in depth toxicological analysis of 5g using admetSAR was also obtained in the form of probability score (Table 5). The prediction having probability of 0.7 or higher are more significant and included in the analysis of 5g. According to this, 5g exhibits non-carcinogenic and non-nephrotoxic character, whereas, it can induce respiratory and reproductive toxicity. Moreover, the compound does not cause any irritation to eyes or integumentary lining.
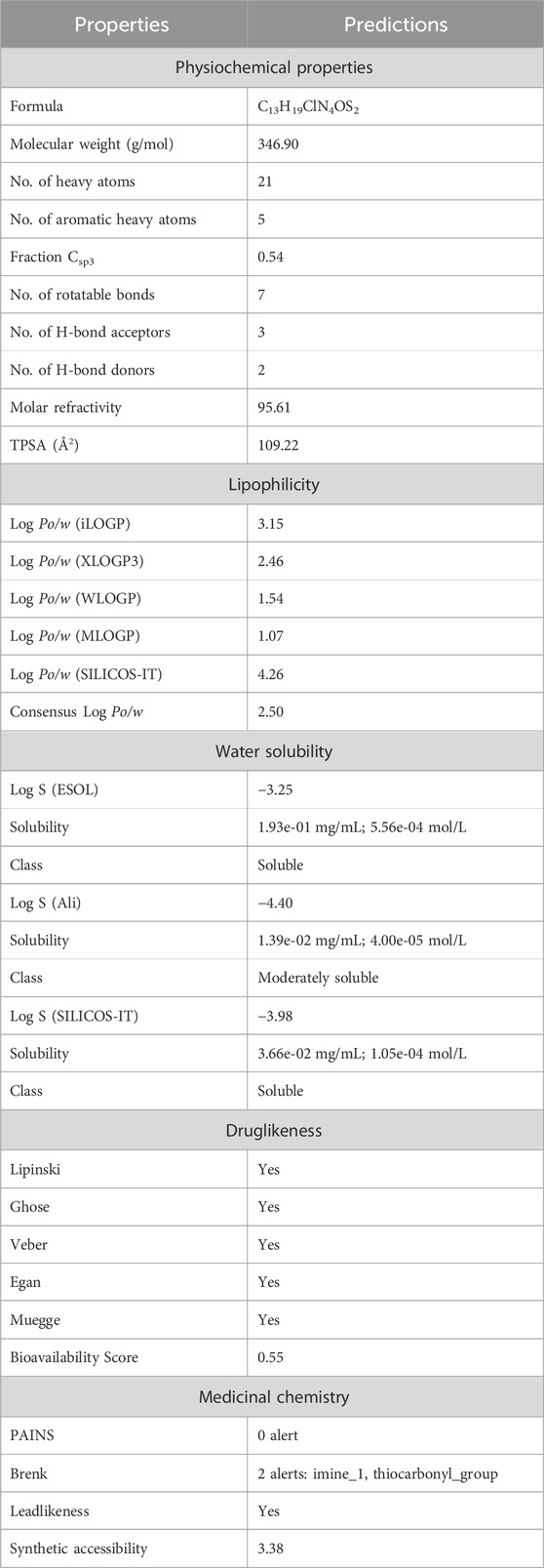
Table 3. ADME properties of compound 5g from SwissADME.
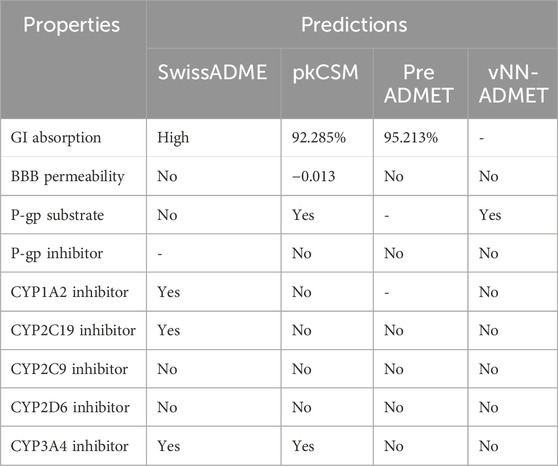
Table 4. Pharmacokinetics properties of 5g using different software.
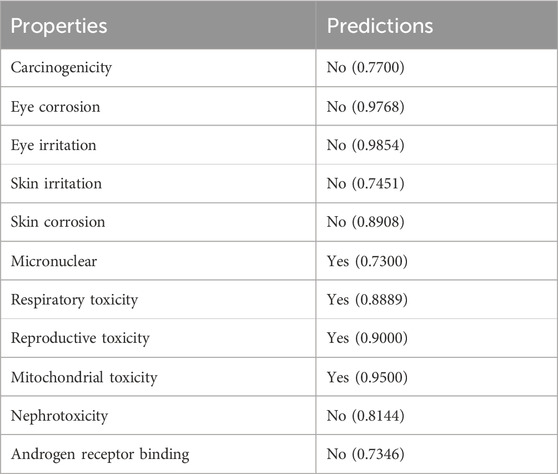
Table 5. Toxicological evaluation of 5g using admetSAR.
Biotransformation 3.0 showed the reactive potential of 5g with bacterial enzyme UDP-glucuronosyltransferase (Table 6). This enzyme can biotransform compound 5g into another compound via N-glucuronidation of tertiary aliphatic amine. The same software was used to access the abiotic transformation of 5g and it was disclosed that compound could undergo reduction, ozonation and chlorination under abiotic conditions. Xenosite analysis interpreted that 5g can undergo epoxidation, N-dealkylation, and have reactive site to bind with the DNA (Table 6). The predictions of Xenosite are scored on the basis of color such as red color scored for 1, yellow between 1 and 0.5, green color for 0.5, blue between 0.5 and zero and white for zero. The higher the score, the greater will be the chance of that site to be reactive (Dang et al., 2020).
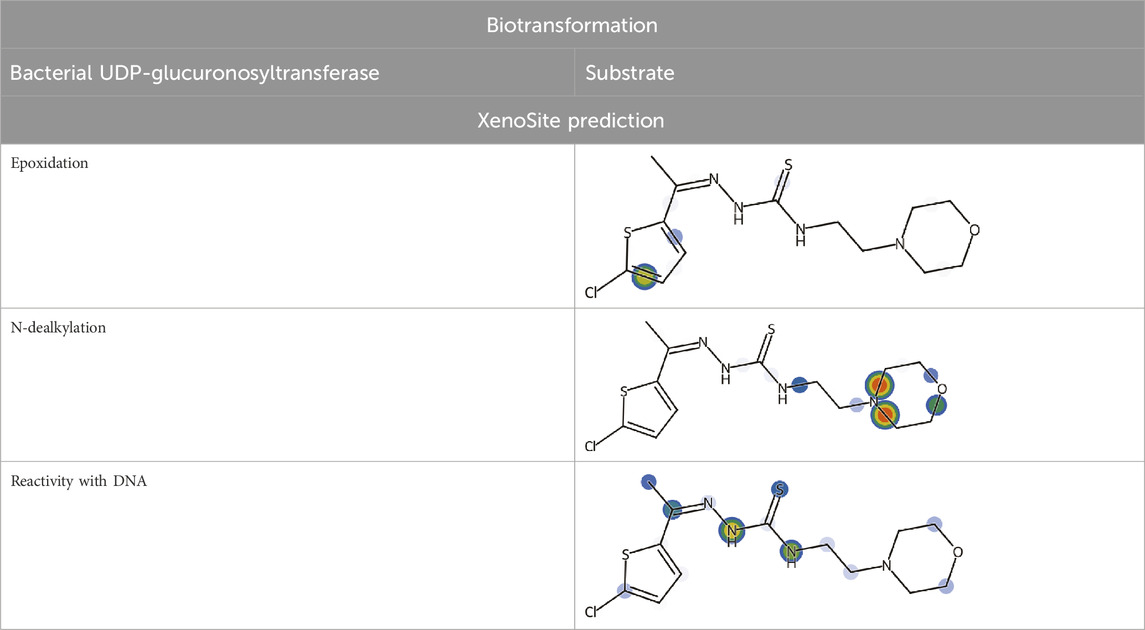
Table 6. Biotransformation and XenoSite prediction of 5g.
The molecular dynamics simulation was performed by using an iMOD server. This online server is used for the estimation of the stability of the ligand-protein complex. The results of molecular dynamics simulation of 5g are shown in Figure 5. The lower peaks are associated with lower deformability, while higher peaks represent higher deformability, as shown in Figure 5A. However, the eigenvalue represents the energy required for the deformation of the structure with an inverse relation, as shown in Figure 5B. Moreover, three different colors represent the covariance, such as correlated as red, non-correlated as blue, and anti-correlated as white (Figure 5C). Figure 5D represents the elastic model network that is associated with spring formation. The gray color indicates the extent of spring formation, and most of the atoms are forming spring.
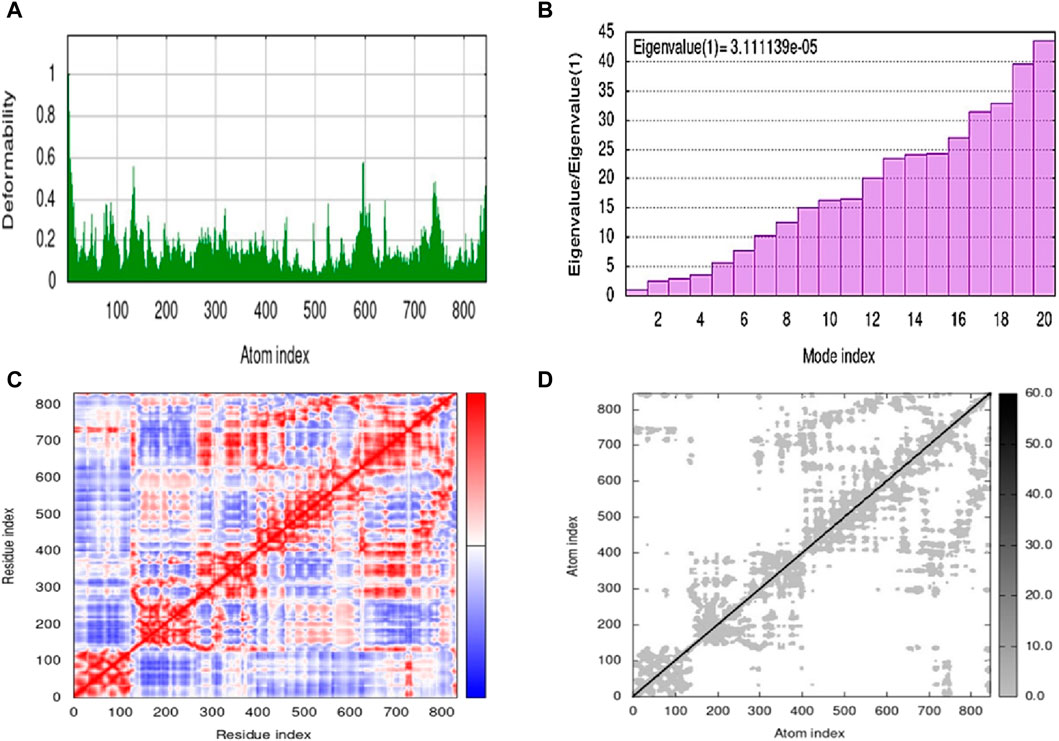
Figure 5. Molecular dynamics simulation of 5g by iMOD. (A) deformability plot; (B) eigenvalue; (C) covariance map; and (D) elastic network model.
The chemicals and solvents used to perform synthetic chemistry were of analytical grade and obtained from local dealers of Merck, Fisher and Fluka. Thin layer chromatography was carried out on aluminum plates coated with silica gel 60 F254 (Merck) in an appropriate eluting system and UV lamp was employed for visualization of spots. Melting points were recorded in open capillaries on Gallenkamp melting point apparatus and are uncorrected. 1H NMR spectra were recorded in DMSO-d6 on Bruker Avance NMR spectrometer at 300 MHz while 13C NMR at 75 MHz. Chemical shifts are reported as δ values in parts per million (ppm) compared to TMS as internal standard or the residual deuterated solvent used. Coupling constant (J) is given in Hertz. FTIR spectra (neat) were recorded on Bruker FTIR spectrophotometer. Mass spectra were obtained using LCMS 6495c Agilent whereas elemental analysis was attained on LECO 630–200-200 TRUSPEC CHNS micro analyzer and the experimental values are within ±0.4% of the calculated values.
Synthesis of compounds 2 and 3 was achieved by following the reported synthetic procedures (Mandapati et al., 2017; He et al., 2019).
To a stirred solution of N-(2-morpholinoethyl)hydrazinecarbothioamide 3 (1 mmol) in absolute ethanol (20 mL) was added thiophene-2-carbaldehyde (or un/substituted acetylthiophene) 4 (1 mmol) and two to three drops of glacial acetic acid. The reaction mixture was refluxed for 10–12 h and then allowed to stand for 48 h in a refrigerator. The precipitated crude product was filtered and recrystallized from methanol to give thiosemicarbazones (5a-i).
Yield 79%. Light brown crystalline solid. Mp 136°C–138 °C. FT-IR (ῡ, cm−1): 3,345 (N-H), 3,146 (Ar-H), 2,991 (C-H), 1,525 (C=N), 1,142 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.44 (t, J = 4.2 Hz), 2.53 (t, J = 6.3 Hz), 3.62–3.65 (m, 6H), 7.12 (t, J = 4.2 Hz, 1H), 7.44 (d, J = 3.3 Hz, 1H), 7.68 (d, J = 5.1 Hz, 1H), 8.14 (t, J = 4.8 Hz, 1H), 8.26 (s, 1H), 11.57 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 40.4, 53.5, 56.5, 66.8, 128.5, 129.2, 131.3, 137.7, 139.2, 176.8; Anal. Calcd. for C12H18N4OS2: C, 48.30; H, 6.08; N, 18.77; S, 21.49%; Found: C, 48.48; H, 6.12; N, 18.85; S, 21.63%.
Yield 93%. Yellow crystalline solid. Mp 142°C–144 °C. FT-IR (ῡ, cm−1): 3,326 (N-H), 3,160 (Ar-H), 2,968 (C-H), 1,521 (C=N), 1,102 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.34 (s, 3H), 2.44 (t, J = 3.9 Hz, 4H), 2.53 (t, J = 6.3 Hz, 2H), 3.63–3.67 (m, 6H), 7.09 (d, J = 4.5 Hz, 1H), 7.50 (t, J = 3.9 Hz, 1H), 7.63 (dd, J = 5.1, 0.9 Hz, 1H), 8.16 (t, J = 4.8 Hz, 1H), 10.47 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 15.1, 40.4, 53.5, 56.5, 66.8, 128.3, 128.6, 129.0, 143.4, 145.1, 177.7; LCMS m/z [M + H]+: 313.50; Anal. Calcd. for C13H20N4OS2: C, 49.97; H, 6.45; N, 17.93; S, 20.52%; Found: C, 50.09; H, 6.56; N, 18.01; S, 20.60%.
Yield 85%. Yellow crystalline solid. Mp 112°C–114 °C. FT-IR (ῡ, cm−1): 3,319 (N-H), 3,179 (Ar-H), 2,961 (C-H), 1,506 (C=N), 1,139 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.20 (s, 3H), 2.30 (s, 3H), 2.44 (t, J = 3.9 Hz, 4H), 2.53 (t, J = 6.3 Hz, 2H), 3.62–3.65 (m, 6H), 7.20 (t, J = 0.9 Hz, 1H), 7.33 (d, J = 1.2 Hz, 1H), 8.15 (t, J = 4.8 Hz, 1H), 10.45 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 15.0, 15.9, 40.4, 53.5, 56.5, 66.8, 124.2, 130.6, 138.1, 143.0, 144.9, 177.6; LCMS m/z [M + H]+: 327.50; Anal. Calcd. for C14H22N4OS2: C, 51.50; H, 6.79; N, 17.16; S, 19.64%; Found: C, 51.54; H, 6.87; N, 17.30; S, 19.70%.
Yield 77%. Light brown crystalline solid. Mp 156°C–158 °C. FT-IR (ῡ, cm−1): 3,312 (N-H), 3,145 (Ar-H), 2,988 (C-H), 1,527 (C=N), 1,109 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.27 (s, 3H), 2.42–2.45 (m, 7H), 2.53 (t, J = 6.3 Hz, 2H), 3.60–3.67 (m, 6H), 6.78 (dd, J = 3.6, 0.9 Hz, 1H), 7.28 (d, J = 3.6 Hz, 1H), 8.14 (t, J = 4.8 Hz, 1H), 10.46 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 14.6, 15.7, 40.4, 53.5, 56.5, 66.7, 126.6, 128.7, 141.1, 142.7, 145.0, 177.6; LCMS m/z [M + H]+: 327.50; Anal. Calcd. for C14H22N4OS2: C, 51.50; H, 6.79; N, 17.16; S, 19.64%; Found: C, 51.62; H, 6.91; N, 17.32; S, 19.76%.
Yield 65%. Light brown crystalline solid. Mp 116°C–118 °C. FT-IR (ῡ, cm−1): 3,321 (N-H), 3,198 (Ar-H), 2,991 (C-H), 1,525 (C=N), 1,105 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.40–2.42 (m, 7H), 2.53 (t, J = 6.3 Hz, 2H), 3.60 (t, J = 4.5 Hz, 4H), 3.65 (q, J = 5.6 Hz, 2H), 7.13 (d, J = 5.4 Hz, 1H), 7.74 (d, J = 5.4 Hz, 1H), 8.15 (t, J = 4.8 Hz, 1H), 10.64 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 16.9, 40.6, 53.5, 56.7, 66.7, 123.0, 127.7, 130.5, 135.2, 142.8, 178.0; LCMS m/z [M + H]+: 347.40; Anal. Calcd. for C13H19ClN4OS2: C, 45.01; H, 5.52; N, 16.15; S, 18.49%; Found: C, 45.09; H, 5.58; N, 16.21; S, 18.61%.
Yield 80%. Light brown solid. Mp 124°C–126 °C. FT-IR (ῡ, cm−1): 3,315 (N-H), 3,097 (Ar-H), 2,983 (C-H), 1,530 (C=N), 1,107 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.40–2.42 (m, 7H), 2.53 (t, J = 6.3 Hz, 2H), 3.59 (t, J = 4.5 Hz, 4H), 3.67 (q, J = 4.8 Hz, 2H), 7.18 (d, J = 5.4 Hz, 1H), 7.73 (d, J = 5.1 Hz, 1H), 8.20 (t, J = 4.8 Hz, 1H), 10.65 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 17.3, 40.4, 53.5, 56.8, 66.6, 108.9, 128.4, 133.1, 136.8, 142.8, 178.2; LCMS m/z [M + H]+: 391.30; Anal. Calcd. for C13H19BrN4OS2: C, 39.90; H, 4.89; N, 14.32; S, 16.39%; Found: C, 40.04; H, 4.97; N, 14.40; S, 16.53%.
Yield 90%. Yellow crystalline solid. Mp 148°C–150 °C. FT-IR (ῡ, cm−1): 3,321 (N-H), 3,198 (Ar-H), 2,959 (C-H), 1,525 (C=N), 1,105 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.28 (s, 3H), 2.44 (t, J = 4.2 Hz, 4H), 2.53 (t, J = 6.3 Hz, 2H), 3.60–3.67 (m, 6H), 7.11 (d, J = 3.9 Hz, 1H), 7.35 (d, J = 4.2 Hz, 1H), 8.15 (t, J = 4.8 Hz, 1H), 10.58 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 14.1, 40.4, 53.5, 56.5, 66.7, 128.1, 130.9, 142.6, 144.0, 177.7; LCMS m/z [M + H]+: 347.40; Anal. Calcd. for C13H19ClN4OS2: C, 45.01; H, 5.52; N, 16.15; S, 18.49%; Found: C, 45.13; H, 5.76; N, 16.29; S, 18.63%.
Yield 61%. Light brown solid. Mp 148°C–150 °C. FT-IR (ῡ, cm−1): 3,315 (N-H), 3,146 (Ar-H), 2,965 (C-H), 1,527 (C=N), 1,110 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.29 (s, 3H), 2.45 (t, J = 3.6 Hz, 4H), 2.53 (t, J = 6.3 Hz, 2H), 3.60–3.68 (m, 6H), 7.22 (d, J = 3.9 Hz, 1H), 7.31 (d, J = 3.9 Hz, 1H), 8.16 (t, J = 4.5 Hz, 1H), 10.57 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 14.2, 40.4, 53.5, 56.5, 66.7, 114.7, 128.9, 131.6, 143.9, 145.3, 177.7; LCMS m/z [M + H]+: 391.30; Anal. Calcd. for C13H19BrN4OS2: C, 39.90; H, 4.89; N, 14.32; S, 16.39%; Found: C, 39.77; H, 4.81; N, 14.30; S, 16.25%.
Yield 92%. Orange crystalline solid. Mp 192°C–194 °C. FT-IR (ῡ, cm−1): 3,317 (N-H), 3,110 (Ar-H), 2,959 (C-H), 1,525 (C=N), 1,107 (C=S); 1H NMR (DMSO-d6, 300 MHz) δ 2.35 (s, 3H), 2.45 (t, J = 4.5 Hz, 4H), 2.55 (t, J = 6.0 Hz, 2H), 3.65–3.68 (m, 6H), 7.55 (d, J = 4.5 Hz, 1H), 8.09 (d, J = 4.5 Hz, 1H), 8.32 (t, J = 4.8 Hz, 1H), 10.86 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 14.2, 40.7, 53.6, 56.5, 66.7, 127.4, 130.9, 142.6, 151.0, 151.1, 177.8; Anal. Calcd. for C13H19N5O3S2: C, 43.68; H, 5.36; N, 19.59; S, 17.94%; Found: C, 43.82; H, 5.44; N, 19.63; S, 18.00%.
The indophenol method with a little modification was used to determine the urease inhibitory activity of synthetic compounds (5a-i) (Weatherburn, 1967; Yaseen et al., 2016). Thiourea was used as a standard inhibitor. The assay mixture was comprised of 40 μL of buffer (100 mmol/L urea, 0.01 mol/L K2HPO4, 1 mmol/L EDTA and 0.01 mol/L LiCl2, pH 8.2), 10 μL of jack bean urease (5 U/mL) and 10 μL of test compound (1 mM). The reaction mixture was incubated for 30 min with 10 μL urea (1 mM) at 37 °C in 96-well plate. The phenol reagent (40 μL, 1% w/v phenol, 0.005% w/v sodium nitroprusside) and alkali reagent (40 μL, 0.5% w/v NaOH, 0.1% active chloride NaOCl) were added to each well and after 10 min of incubation at 37 °C, the absorbance was measured at 630 nm using a microplate reader (Bio-Tek ELx 800™, Instruments, Inc. United States). The experiments to determine the inhibitory activity of compounds were performed in triplicates. The percentage inhibition was calculated by using the formula given below.
Compounds showing >50% inhibition against urease were further evaluated for the determination of IC50 values at various concentrations (0.1, 0.3, 1, 3, 10, 30, 100, 300, 1,000 μM) against jack bean urease. The experiments were performed in triplicate. IC50 values were calculated by using the non-linear curve fitting program PRISM 5.0 (GraphPad, San Diego, California, United States) to analyze the results.
The most potent inhibitor 5g was examined for its mechanism of inhibition against urease by Michaelis-Menten kinetics. The rate of enzyme inhibition was accessed across numerous concentrations of substrate (25, 50, 100, 150 mM) and compound 5g (0, 1.9, 3.8, 5.7 μM) against urease. Afterwards, the data was plotted to obtain the Lineweaver-Burk plot by using GraphPad Prism version 10.2.1.
For the molecular docking studies, the crystallographic structure of jack bean urease (PDB ID: 3LA4) was retrieved from the RSCB PDB database (Balasubramanian and Ponnuraj, 2010). The structures of compound and enzyme were prepared by protonation with the Protonate3D (Labute, 2007) algorithm implemented in MOE molecular modeling tool (http://www.chemcomp.com/MOEMolecular_Operating_Environment.htm). However, the calculations for the docking investigations were performed using LeadIT from BioSolveIT, GmbH, Germany (www.biosolveit.de/LeadIT). The FlexX application of LeadIT was used for the docking of compounds. Based on the binding free energies, the conformation of the ligand-receptor complex was determined. No modifications were made to the default docking parameters (www.biosolveit.de/LeadIT). The highest affinity for interfacing with the receptor and the most stable poses were determined to possess the lowest free binding energies. Complexes were visualized using Discovery Studio Visualizer v4.
An online platform (SwissADME; http://www.swissadme.ch/index.php) was used to determine the pharmacokinetic properties of potent inhibitor 5g. Several properties such as physical and chemical characteristics, bioavailability, solubility, druglikeness, lipophilicity, pharmacokinetics, and medicinal chemistry of compound 5g were determined to represent the identified inhibitor as a safer therapeutic agent (Daina et al., 2017). The detailed toxicity analysis was conducted via another user-friendly interface admetSAR (http://lmmd.ecust.edu.cn/admetsar2/). It predicts the hepatotoxicity, carcinogenicity, nephrotoxicity, acute oral toxicity, non-target receptor binding, skin sensitization and ocular sensitivity (Yang et al., 2019). The preADMET (https://preadmet.webservice.bmdrc.org) analysis of 5g based on pharmacokinetics was also included to validate the predictions. Apart from these software, vNN ADMET (https://vnnadmet.bhsai.org/vnnadmet/login.xhtml) was used which utilizes the vNN approach (a set of 15 predictive models) for ADMET analysis. These models serve as rapid evaluators of crucial characteristics in drug candidates such as drug-related liver injury (Schyman et al., 2017). On the other hand, BioTransformer 3.0 (https://biotransformer.ca) represents a freely accessible online platform designed to integrate machine learning methodologies alongside rule-based systems. The primary objective of this tool is to forecast the metabolism of compounds within various tissues of humans, human gastrointestinal tract and soil and water microbiota from external environment (Wishart et al., 2022). XenoSite (https://xenosite.org) is also an online tool that identifies actual atoms of a drug having probability of transformation during the metabolism in human body (Dang et al., 2020).
The best pose of the docked complex of 5g was studied using molecular dynamics (MD) analysis based on the docking results. The MD simulations were performed by the iMOD server (http://imods.chaconlab.org/) at 300 K constant temperature and 1 atm constant pressure (López-Blanco et al., 2014). The improved normal mode analysis (NMA) approach in inner coordinates has a user-friendly interface provided by the iMod server. All of the major browsers as well as contemporary mobile devices, are quite receptive and instantaneous with the online interface (Santra and Maiti, 2022).
In summary, the present work reports the design, synthesis and characterization of morpholine-thiophene hybrid thiosemicarbazones. The structures of these compounds were elucidated by FTIR, 1H NMR, 13C NMR spectroscopy and mass spectrometry. The synthetic compounds were tested against urease enzyme to combat ureolytic bacterial infections. The evaluation of these compounds against urease revealed a multi-fold superior inhibitory potential compared to the conventional inhibitor, thiourea. Among the tested compounds, 5g was recognized as the lead inhibitor, exhibiting remarkable inhibitory potential with an IC50 value of 3.80 ± 1.9 µM. Enzyme’s kinetics experiments revealed that the potent analogue exhibits uncompetitive inhibition, suggesting a unique mode of action against the urease enzyme. In docking analysis, 5g displayed a diverse variety of interactions with the active site amino acid residues of urease. In silico ADMET profile of 5g showed the druglikeness and leadlikeness properties with zero violations. Notably, the structural stability of the 5g complex was demonstrated through molecular dynamics simulations, further supporting its candidacy for preclinical studies. Altogether, the results achieved in the current research work contribute significantly towards the plethora of synthetic inhibitors and could potentially be targeted as a new template for the development of therapeutic candidates for ureolytic bacterial infections.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
RM: Conceptualization, Data curation, Methodology, Project administration, Supervision, Writing–original draft. SZ: Methodology, Software, Validation, Visualization, Writing–original draft. MZ-u-R: Formal Analysis, Resources, Writing–original draft. HJ: Investigation, Writing–original draft. AR: Formal Analysis, Writing–original draft. MZ: Formal Analysis, Writing–original draft. NF: Investigation, Writing–original draft. MB: Formal Analysis, Funding acquisition, Writing–review and editing. IK: Formal Analysis, Project administration, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors are grateful to King Saud University, Riyadh, Saudi Arabia for funding the work through the Researchers Supporting Project (Project No. RSPD 2024R740).
We are thankful to the research supporting project number (RSPD 2024R740), King Saud University, Riyadh, Saudi Arabia. RM would like to extend gratitude to Kinnaird College for Women for the provision of facilities for synthetic work. Authors appreciate BioSolveIT for providing necessary access to software to carry out the research work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1403127/full#supplementary-material
Abid, O. U. R., Babar, T. M., Ali, F. I., Ahmed, S., Wadood, A., Rama, N. H., et al. (2010). Identification of novel urease inhibitors by high-throughput virtual and in vitro screening. ACS Med. Chem. Lett. 1 (4), 145–149. doi:10.1021/ml100068u
Akash, M., Zaib, S., Ahmad, M., Sultan, S., and Al-Hussain, S. A. (2024). Synthesis and biological evaluation of pyridylpiperazine hybrid derivatives as urease inhibitors. Front. Chem. 12, 1371377. doi:10.3389/fchem.2024.1371377
Akyüz, G., Emirik, M., Sökmen, B. B., and Menteşe, E. (2022). Synthesis and docking studies of novel benzimidazole derivatives containing thiophene and triazole rings as potential urease inhibitors. Russ. J. Bioorg. Chem. 48, S87–S95. Suppl 1. doi:10.1134/S106816202301003X
Al-Fakhrany, O. M., and Elekhnawy, E. (2023). Helicobacter pylori in the post-antibiotics era: from virulence factors to new drug targets and therapeutic agents. Archives Microbiol. 205, 301. doi:10.1007/s00203-023-03639-0
Al-Rooqi, M. M., Mughal, E. U., Raja, Q. A., Hussein, E. M., Naeem, N., Sadiq, A., et al. (2023). Flavonoids and related privileged scaffolds as potential urease inhibitors: a review. RSC Adv. 13, 3210–3233. doi:10.1039/d2ra08284e
Amtul, Z., Siddiqui, R. A., and Choudhary, M. I. (2002). Chemistry and mechanism of urease inhibition. Curr. Med. Chem. 9 (14), 1323–1348. doi:10.2174/0929867023369853
Ayaz, M., Lodhi, M. A., Riaz-ul-Haq, M., Malik, A. A., Choudhary, M. I., and Iqbal Choudhary, M. (2006). Novel urease inhibitors from Daphne oleoids. J. Enzyme Inhibition Med. Chem. 21 (5), 527–529. doi:10.1080/14756360600774470
Babu, T. M. C., Rajesh, S. S., Bhaskar, B. V., Devi, S., Rammohan, A., Sivaraman, T., et al. (2017). Molecular docking, molecular dynamics simulation, biological evaluation and 2D QSAR analysis of flavonoids from Syzygium alternifolium as potent anti-Helicobacter pylori agents. RSC Adv. 7 (30), 18277–18292. doi:10.1039/C6RA27872H
Bai, X. G., Zheng, Y., and Qi, J. (2022). Advances in thiosemicarbazone metal complexes as anti-lung cancer agents. Front. Pharmacol. 13, 1018951. doi:10.3389/fphar.2022.1018951
Bajaj, K., Buchanan, R. M., and Grapperhaus, C. A. (2021). Antifungal activity of thiosemicarbazones, bis (thiosemicarbazones), and their metal complexes. J. Inorg. Biochem. 225, 111620. doi:10.1016/j.jinorgbio.2021.111620
Balasubramanian, A., and Ponnuraj, K. (2010). Crystal structure of the first plant urease from jack bean: 83 years of journey from its first crystal to molecular structure. J. Mol. Biol. 400 (3), 274–283. doi:10.1016/j.jmb.2010.05.009
Basri, R., Ullah, S., Khan, A., Mali, S. N., Abchir, O., Chtita, S., et al. (2023). Synthesis, biological evaluation and molecular modelling of 3-Formyl-6-isopropylchromone derived thiosemicarbazones as α-glucosidase inhibitors. Bioorg. Chem. 139, 106739. doi:10.1016/j.bioorg.2023.106739
Bektaş, H., Albay, C., Menteşe, E., and Demirbaş, N. (2017). New pyridine derivatives as antiurease inhibitors: synthesis and their evaluation for antimicrobial activities. Roum. J. Chem. 62 (3), 199–205. doi:10.1016/j.ejmech.2017.12.089
Bektaş, H., Ceylan, Ş., Demirbaş, N., Alpay-Karaoğlu, Ş., and Sökmen, B. B. (2013). Antimicrobial and antiurease activities of newly synthesized morpholine derivatives containing an azole nucleus. Med. Chem. Res. 22, 3629–3639. doi:10.1007/s00044-012-0318-1
Castro, A., Andrade, I. M. G., Coelho, M. C., da Costa, D. P., Moreira, D. D. N., Maia, R. A., et al. (2023). Multicomponent synthesis of spiro 1, 3, 4-thiadiazolines with anticancer activity by using deep eutectic solvent under microwave irradiation. J. Heterocycl. Chem. 60 (3), 392–405. doi:10.1002/jhet.4591
D’Agostino, I., and Carradori, S. (2024) Chapter 5.1 - urease. Academic Press, 393–410. doi:10.1016/B978-0-12-823974-2.00035-8
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7 (1), 42717. doi:10.1038/srep42717
Dang, N. L., Matlock, M. K., Hughes, T. B., and Swamidass, S. J. (2020). The metabolic rainbow: deep learning phase I metabolism in five colors. J. Chem. Inf. Model. 60 (3), 1146–1164. doi:10.1021/acs.jcim.9b00836
Devi, J., Kumar, B., and Taxak, B. (2022). Recent advancements of organotin (IV) complexes derived from hydrazone and thiosemicarbazone ligands as potential anticancer agents. Inorg. Chem. Commun. 139, 109208. doi:10.1016/j.inoche.2022.109208
Dilworth, J. R., and Hueting, R. (2012). Metal complexes of thiosemicarbazones for imaging and therapy. Inorganica Chim. Acta 389, 3–15. doi:10.1016/j.ica.2012.02.019
Dulsat, J., López-Nieto, B., Estrada-Tejedor, R., and Borrell, J. I. (2023). Evaluation of free online ADMET tools for academic or small biotech environments. Molecules 28, 776. doi:10.3390/molecules28020776
Elbastawesy, M. A., El-Shaier, Y. A., Ramadan, M., Brown, A. B., Aly, A. A., and Abuo-Rahma, G. E. D. A. (2021). Identification and molecular modeling of new quinolin-2-one thiosemicarbazide scaffold with antimicrobial urease inhibitory activity. Mol. Divers. 25 (1), 13–27. doi:10.1007/s11030-019-10021-0
Gobis, K., Szczesio, M., Olczak, A., Mazerant-Politowicz, I., Ziembicka, D., Pacholczyk-Sienicka, B., et al. (2022). N′-Substituted 4-phenylpicolinohydrazonamides with thiosemicarbazone moiety as new potential antitubercular agents: synthesis, structure and evaluation of antimicrobial activity. Materials 15 (16), 5513. doi:10.3390/ma15165513
Güven, O., Menteşe, E., Emirik, M., Sökmen, B. B., and Akyüz, G. (2023). Benzimidazolone-piperazine/triazole/thiadiazole/furan/thiophene conjugates: synthesis, in vitro urease inhibition, and in silico molecular docking studies. Arch. Pharm. 356 (11), 2300336. doi:10.1002/ardp.202300336
Hameed, A., Al-Rashida, M., Uroos, M., Qazi, S. U., Naz, S., Ishtiaq, M., et al. (2019). A patent update on therapeutic applications of urease inhibitors (2012–2018). Expert Opin. Ther. Pat. 29 (3), 181–189. doi:10.1080/13543776.2019.1584612
Hanif, M., Saleem, M., Hussain, M. T., Rama, N. H., Zaib, S., Aslam, M. A. M., et al. (2012). Synthesis, urease inhibition, antioxidant and antibacterial studies of some 4-amino-5-aryl-3H-1, 2, 4-triazole-3-thiones and their 3, 6-disubstituted 1,2,4-triazolo[3,4-b]1,3,4-thiadiazole derivatives. J. Braz. Chem. Soc. 23, 854–860. doi:10.1590/S0103-50532012000500010
Hassan, M., Ghaffari, R., Sardari, S., Farahani, Y. F., and Mohebbi, S. (2020). Discovery of novel isatin-based thiosemicarbazones: synthesis, antibacterial, antifungal, and antimycobacterial screening. Res. Pharm. Sci. 15 (3), 281. doi:10.4103/1735-5362.288435
Hassan, S. T., and Švajdlenka, E. (2017). Biological evaluation and molecular docking of protocatechuic acid from Hibiscus sabdariffa L. as a potent urease inhibitor by an ESI-MS based method. Molecules 22 (10), 1696. doi:10.3390/molecules22101696
He, Z., Qiao, H., Yang, F., Zhou, W., Gong, Y., Zhang, X., et al. (2019). Novel thiosemicarbazone derivatives containing indole fragment as potent and selective anticancer agent. Eur. J. Med. Chem. 184, 111764. doi:10.1016/j.ejmech.2019.111764
Hina, S., Zaib, S., Uroos, M., Zia-ur-Rehman, M., Munir, R., Riaz, H., et al. (2023). N-Arylacetamide derivatives of methyl 1, 2-benzothiazine-3-carboxylate as potential drug candidates for urease inhibition. R. Soc. Open Sci. 10 (4), 230104. doi:10.1098/rsos.230104
Ibrar, A., Khan, I., and Abbas, N. (2013). Structurally diversified heterocycles and related privileged scaffolds as potential urease inhibitors: a brief overview. Arch. Pharm. 346 (6), 423–446. doi:10.1002/ardp.201300041
Islam, M., Khan, A., Khan, M., Halim, S. A., Ullah, S., Hussain, J., et al. (2023). Synthesis and biological evaluation of 2-nitrocinnamaldehyde derived thiosemicarbazones as urease inhibitors. J. Mol. Struct. 1284, 135387. doi:10.1016/j.molstruc.2023.135387
Islam, M., Khan, A., Shehzad, M. T., Hameed, A., Ahmed, N., Halim, S. A., et al. (2019). Synthesis and characterization of new thiosemicarbazones, as potent urease inhibitors: in vitro and in silico studies. Bioorg. Chem. 87, 155–162. doi:10.1016/j.bioorg.2019.03.008
Jalil, S., Basri, R., Aziz, M., Shafiq, Z., Ejaz, S. A., Hameed, A., et al. (2024). Pristine 2-Chloroquinoline-based-Thiosemicarbazones as multitarget agents against alzheimer’s disease: in vitro and in silico studies of monoamine oxidase (MAO) and cholinesterase (ChE) inhibitors. J. Mol. Struct. 1306, 137841. doi:10.1016/j.molstruc.2024.137841
Kafarski, P., and Talma, M. (2018). Recent advances in design of new urease inhibitors: a review. J. Adv. Res. 13, 101–112. doi:10.1016/j.jare.2018.01.007
Kataria, R., and Khatkar, A. (2019). Molecular docking, synthesis, kinetics study, structure–activity relationship and ADMET analysis of morin analogous as Helicobacter pylori urease inhibitors. BMC Chem. 13 (1), 45–17. doi:10.1186/s13065-019-0562-2
Kazmi, M., Khan, I., Khan, A., Halim, S. A., Saeed, A., Mehsud, S., et al. (2019). Developing new hybrid scaffold for urease inhibition based on carbazole-chalcone conjugates: synthesis, assessment of therapeutic potential and computational docking analysis. Bioorg. Med. Chem. 27 (22), 115123. doi:10.1016/j.bmc.2019.115123
Khan, K. M., Ullah, Z., Lodhi, M. A., Ali, M., Choudhary, M. I., Rahman, A. u., et al. (2006). Successful computer guided planned synthesis of (4R)-thiazolidine carboxylic acid and its 2-substituted analogues as urease inhibitors. Mol. Divers. 10 (2), 223–231. doi:10.1007/s11030-005-9000-6
Khan, K. M., Wadood, A., Ali, M., Ul-Haq, Z., Lodhi, M. A., Khan, M., et al. (2010). Identification of potent urease inhibitors via ligand-and structure-based virtual screening and in vitro assays. J. Mol. Graph. Model. 28 (8), 792–798. doi:10.1016/j.jmgm.2010.02.004
Khan, S., Hussain, R., Khan, Y., Iqbal, T., Anwar, S., Aziz, T., et al. (2024). In vitro enzymatic, in silico ADME and molecular docking based analysis for the identification of novel bis-indole containing triazine-thiazole hybrids derivatives as promising urease inhibitors. Z. für Naturforsch. C 0. doi:10.1515/znc-2024-0061
Khan, S., Ullah, H., Hussain, R., Khan, Y., Khan, M. U., Khan, M., et al. (2023). Synthesis, in vitro bio-evaluation, and molecular docking study of thiosemicarbazone-based isatin/bis-Schiff base hybrid analogues as effective cholinesterase inhibitors. J. Mol. Struct. 1284, 135351. doi:10.1016/j.molstruc.2023.135351
Konieczna, I., Zarnowiec, P., Kwinkowski, M., Kolesinska, B., Fraczyk, J., Kaminski, Z., et al. (2012). Bacterial urease and its role in long-lasting human diseases. Curr. Protein Peptide Sci. 13 (8), 789–806. doi:10.2174/138920312804871094
Kot, M., Zaborska, W., and Orlinska, K. (2001). Inhibition of jack bean urease by N-(n-butyl) thiophosphorictriamide and N-(n-butyl) phosphorictriamide: determination of the inhibition mechanism. J. Enzyme Inhibition 16 (6), 507–516. doi:10.1080/14756360127569
Kumari, A., and Singh, R. K. (2020). Morpholine as ubiquitous pharmacophore in medicinal chemistry: deep insight into the structure-activity relationship (SAR). Bioorg. Chem. 96, 103578. doi:10.1016/j.bioorg.2020.103578
Kurt, B. Z., Altundağ, Ö., Gökçe, M., Cakmak, U., Tuncay, F. O., Kolcuoğlu, Y., et al. (2024). Synthesis of naproxen thiadiazole urea hybrids and determination of their anti-melanoma, anti-migration, tyrosinase inhibitory activity, and molecular docking studies. J. Mol. Struct. 1295, 136618. doi:10.1016/j.molstruc.2023.136618
Labute, P. (2007) Protonate 3D, chemical computing group. Available at: https://www.chemcomp.com/journal/proton.htm.
LeadIT (2017) Version 2.3.2; BioSolveIT GmbH: sankt augustin, Germany. Available at: www.biosolveit.de/LeadIT.
López-Blanco, J. R., Aliaga, J. I., Quintana-Ortí, E. S., and Chacón, P. (2014). iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 42 (W1), W271–W276. doi:10.1093/nar/gku339
Mandapati, U., Pinapati, S., and Rudraraju, R. (2017). Copper promoted desulfurization towards the synthesis of isothiocyanates. Tetrahedron Lett. 58 (2), 125–128. doi:10.1016/j.tetlet.2016.11.086
Menteşe, E., Bektaş, H., Sokmen, B. B., Emirik, M., Çakır, D., and Kahveci, B. (2017). Synthesis and molecular docking study of some 5, 6-dichloro-2-cyclopropyl-1H-benzimidazole derivatives bearing triazole, oxadiazole, and imine functionalities as potent inhibitors of urease. Bioorg. Med. Chem. Lett. 27 (13), 3014–3018. doi:10.1016/j.bmcl.2017.05.019
Mobley, H. L., and Hausinger, R. P. (1989). Microbial ureases: significance, regulation, and molecular characterization. Microbiol. Rev. 53 (1), 85–108. doi:10.1128/mr.53.1.85-108.1989
MOE (2019) Chemical computing group’s molecular operating environment (MOE) MOE 20190201. Available at: http://www.chemcomp.com/MOEMolecular_Operating_Environment.htm.
Muri, E. M. F., Mishra, H., Avery, M. A., and Williamson, J. S. (2003). Design and synthesis of heterocyclic hydroxamic acid derivatives as inhibitors of Helicobacter pylori urease. Synth. Commun. 33 (12), 1977–1995. doi:10.1081/SCC-120021024
Nkungli, N. K., Fouegue, A. D. T., Tasheh, S. N., Bine, F. K., Hassan, A. U., and Ghogomu, J. N. (2023). In silico investigation of falcipain-2 inhibition by hybrid benzimidazole-thiosemicarbazone antiplasmodial agents: a molecular docking, molecular dynamics simulation, and kinetics study. Mol. Divers. 28, 475–496. doi:10.1007/s11030-022-10594-3
Noreen, M., Rasool, N., Gull, Y., Nasim, F. U. H., Zahoor, A. F., Yaqoob, A., et al. (2017). A facile synthesis of new 5-aryl-thiophenes bearing sulfonamide moiety via Pd(0)-catalyzed Suzuki-Miyaura cross coupling reactions and 5-bromothiophene-2-acetamide: as potent urease inhibitor, antibacterial agent and hemolytically active compounds. J. Saudi Chem. Soc. 21, S403–S414. doi:10.1016/j.jscs.2014.04.007
Palakkeezhillam, V. N. V., Haribabu, J., Manakkadan, V., Rasin, P., Varughese, R. E., Gayathri, D., et al. (2023). Synthesis, spectroscopic characterizations, single crystal X-ray analysis, DFT calculations, in vitro biological evaluation and in silico evaluation studies of thiosemicarbazones based 1,3,4-thiadiazoles. J. Mol. Struct. 1273, 134309. doi:10.1016/j.molstruc.2022.134309
Pati, M. L., Niso, M., Spitzer, D., Berardi, F., Contino, M., Riganti, C., et al. (2018). Multifunctional thiosemicarbazones and deconstructed analogues as a strategy to study the involvement of metal chelation, Sigma-2 (σ2) receptor and P-gp protein in the cytotoxic action: in vitro and in vivo activity in pancreatic tumors. Eur. J. Med. Chem. 144, 359–371. doi:10.1016/j.ejmech.2017.12.024
Raicopol, M. D., Chira, N. A., Pandele, A. M., Hanganu, A., Ivanov, A. A., Tecuceanu, V., et al. (2020). Electrodes modified with clickable thiosemicarbazone ligands for sensitive voltammetric detection of Hg (II) ions. Sensors Actuators B Chem. 313, 128030. doi:10.1016/j.snb.2020.128030
Rauf, A., Liaqat, S., Qureshi, A. M., Yaqub, M., Rehman, A. U., Hassan, M. U., et al. (2012). Synthesis, characterization, and urease inhibition of 5-substituted-8-methyl-2H-pyrido[1,2-a] pyrimidine-2,4(3H)-diones. Med. Chem. Res. 21, 60–74. doi:10.1007/s00044-010-9491-2
Rauf, M. K., Zaib, S., Talib, A., Ebihara, M., Badshah, A., Bolte, M., et al. (2016). Solution-phase microwave assisted parallel synthesis, biological evaluation and in silico docking studies of N,N′-disubstituted thioureas derived from 3-chlorobenzoic acid. Bioorg. Med. Chem. 24 (18), 4452–4463. doi:10.1016/j.bmc.2016.07.042
Rego, Y. F., Queiroz, M. P., Brito, T. O., Carvalho, P. G., de Queiroz, V. T., de Fátima, Â., et al. (2018b). A review on the development of urease inhibitors as antimicrobial agents against pathogenic bacteria. J. Adv. Res. 13, 69–100. doi:10.1016/j.jare.2018.05.003
Rego, Y. F., Queiroz, M. P., Brito, T. O., Carvalho, P. G., de Queiroz, V. T., de Fátima, Â., et al. (2018a). A review on the development of urease inhibitors as antimicrobial agents against pathogenic bacteria. J. Adv. Res. 13, 69–100. doi:10.1016/j.jare.2018.05.003
Saeed, A., Ahmed, A., Haider, M. B., Ismail, H., Hayat, K., Shabir, G., et al. (2024b). Novel pyrazoline linked acyl thiourea pharmacophores as antimicrobial, urease, amylase and α-glucosidase inhibitors: design, synthesis, SAR and molecular docking studies. RSC Adv. 14, 1018–1033. doi:10.1039/d3ra06812a
Saeed, A., Zaib, S., Pervez, A., Mumtaz, A., Shahid, M., and Iqbal, J. (2013). Synthesis, molecular docking studies, and in vitro screening of sulfanilamide-thiourea hybrids as antimicrobial and urease inhibitors. Med. Chem. Res. 22, 3653–3662. doi:10.1007/s00044-012-0376-4
Saeed, K., Rafiq, M., Khalid, M., Hussain, A., Siddique, F., Hanif, M., et al. (2024a). Synthesis, characterization, computational assay and anti-inflammatory activity of thiosemicarbazone derivatives: highly potent and efficacious for COX inhibitors. Int. Immunopharmacol. 126, 111259. doi:10.1016/j.intimp.2023.111259
Santra, D., and Maiti, S. (2022). Molecular dynamic simulation suggests stronger interaction of Omicron-spike with ACE2 than wild but weaker than Delta SARS-CoV-2 can be blocked by engineered S1-RBD fraction. Struct. Chem. 33 (5), 1755–1769. doi:10.1007/s11224-022-02022-x
Schyman, P., Liu, R., Desai, V., and Wallqvist, A. (2017). vNN web server for ADMET predictions. Front. Pharmacol. 8, 889. doi:10.3389/fphar.2017.00889
Sepehri, S., and Khedmati, M. (2023). An overview of the privileged synthetic heterocycles as urease enzyme inhibitors: structure–activity relationship. Arch Pharm Chem. Life Sci. 356 (9), 2300252. doi:10.1002/ardp.202300252
Sibuh, B. Z., Gupta, P. K., Taneja, P., Khanna, S., Sarkar, P., Pachisia, S., et al. (2021). Synthesis, in silico study, and anti-cancer activity of thiosemicarbazone derivatives. Biomedicines 9 (10), 1375. doi:10.3390/biomedicines9101375
Sıcak, Y., Aktar, B. S. K., Yılmaz, G. T., Ozturk, F. A., Ozturk, M., Tok, T. T., et al. (2023). Design, synthesis, pharmacological activities, structure–activity relationship, and in silico studies of novel 5-Substituted-2-(morpholinoimino)-thiazolidin-4-ones. ACS omega 8 (41), 38641–38657. doi:10.1021/acsomega.3c05928
Summers, K. L. (2019). A structural chemistry perspective on the antimalarial properties of thiosemicarbazone metal complexes. Mini Rev. Med. Chem. 19 (7), 569–590. doi:10.2174/1389557518666181015152657
Svane, S., Sigurdarson, J. J., Finkenwirth, F., Eitinger, T., and Karring, H. (2020). Inhibition of urease activity by different compounds provides insight into the modulation and association of bacterial nickel import and ureolysis. Sci. Rep. 10 (1), 8503. doi:10.1038/s41598-020-65107-9
Ullah, S., Halim, S. A., Ibrar, A., Khan, I., Ataya, F. S., Fouad, D., et al. (2024). Urease inhibitory potential of pyridine-containing triazolothiadiazole and triazolothiadiazine scaffolds for the treatment of ulceration and kidney stone: in vitro screening, kinetics mechanism, and in silico computational analysis. J. Biomol. Struct. Dyn., 1–10. doi:10.1080/07391102.2023.2291542
Valenzuela-Hormazabal, P., Sepúlveda, R. V., Alegría-Arcos, M., Valdés-Muñoz, E., Rojas-Pérez, V., González-Bonet, I., et al. (2024). Unveiling novel urease inhibitors for Helicobacter pylori: a multi-methodological approach from virtual screening and ADME to molecular dynamics simulations. Int. J. Mol. Sci. 25, 1968. doi:10.3390/ijms25041968
Viana, L. P. S., Naves, G. M., Medeiros, I. G., Guimarães, A. S., Sousa, E. S., Santos, J. C. C., et al. (2024). Synergizing structure and function: cinnamoyl hydroxamic acids as potent urease inhibitors. Bioorg. Chem. 146, 107247. doi:10.1016/j.bioorg.2024.107247
Volynets, G. P., Tukalo, M. A., Bdzhola, V. G., Derkach, N. M., Gumeniuk, M. I., Tarnavskiy, S. S., et al. (2019). Benzaldehyde thiosemicarbazone derivatives against replicating and nonreplicating Mycobacterium tuberculosis. J. Antibiotics 72 (4), 218–224. doi:10.1038/s41429-019-0140-9
Wang, Y.-N., Li, S.-Y., Yuan, L.-C., Bu, S.-F., Zeng, Y., Xiao, Z.-P., et al. (2024). Synthesis and biological evaluation of triazolones/oxadiazolones as novel urease inhibitors. Bioorg. Med. Chem. 102, 117656. doi:10.1016/j.bmc.2024.117656
Weatherburn, M. W. (1967). Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 39 (8), 971–974. doi:10.1021/ac60252a045
Wishart, D. S., Tian, S., Allen, D., Oler, E., Peters, H., Lui, V. W., et al. (2022). BioTransformer 3.0—a web server for accurately predicting metabolic transformation products. Nucleic Acids Res. 50 (W1), W115–W123. doi:10.1093/nar/gkac313
Xiao, Z. P., Peng, Z. Y., Dong, J. J., Deng, R. C., Wang, X. D., Ouyang, H., et al. (2013). Synthesis, molecular docking and kinetic properties of β-hydroxy-β-phenylpropionyl-hydroxamic acids as Helicobacter pylori urease inhibitors. Eur. J. Med. Chem. 68, 212–221. doi:10.1016/j.ejmech.2013.07.047
Yang, H., Lou, C., Sun, L., Li, J., Cai, Y., Wang, Z., et al. (2019). admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 35 (6), 1067–1069. doi:10.1093/bioinformatics/bty707
Yang, L., Liu, H., Xia, D., and Wang, S. (2020). Antioxidant properties of camphene-based thiosemicarbazones: experimental and theoretical evaluation. Molecules 25 (5), 1192. doi:10.3390/molecules25051192
Yang, W., Peng, Z., and Wang, G. (2023). An overview: metal-based inhibitors of urease. J. Enzyme Inhibition Med. Chem. 38 (1), 361–375. doi:10.1080/14756366.2022.2150182
Yaseen, S., Rauf, M. K., Zaib, S., Badshah, A., Tahir, M. N., Ali, M. I., et al. (2016). Synthesis, characterization and urease inhibition, in vitro anticancer and antileishmanial studies of Co (III) complexes with N, N, N′-trisubstituted acylthioureas. Inorganica Chim. Acta 443, 69–77. doi:10.1016/j.ica.2015.12.027
Yousef, T. A., and El-Reash, G. A. (2020). Synthesis, and biological evaluation of complexes based on thiosemicarbazone ligand. J. Mol. Struct. 1201, 127180. doi:10.1016/j.molstruc.2019.127180
Keywords: thiosemicarbazone, thiophene, morpholine, urease, binding interactions, pharmacokinetics
Citation: Munir R, Zaib S, Zia-ur-Rehman M, Javed H, Roohi A, Zaheer M, Fatima N, Bhat MA and Khan I (2024) Exploration of morpholine-thiophene hybrid thiosemicarbazones for the treatment of ureolytic bacterial infections via targeting urease enzyme: Synthesis, biochemical screening and computational analysis. Front. Chem. 12:1403127. doi: 10.3389/fchem.2024.1403127
Received: 18 March 2024; Accepted: 06 May 2024;
Published: 24 May 2024.
Edited by:
Jean-Yves Winum, Université de Montpellier, FranceReviewed by:
Sergio Hernan Szajnman, University of Buenos Aires, ArgentinaCopyright © 2024 Munir, Zaib, Zia-ur-Rehman, Javed, Roohi, Zaheer, Fatima, Bhat and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rubina Munir, b3JnYW5pc3Q5NEBnbWFpbC5jb20=; Sumera Zaib, c3VtZXJhLnphaWJAdWNwLmVkdS5waw==; Imtiaz Khan, a2ltdGlhekBob3RtYWlsLmNvLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.