
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 10 May 2024
Sec. Medicinal and Pharmaceutical Chemistry
Volume 12 - 2024 | https://doi.org/10.3389/fchem.2024.1387923
 Lamya H. Al-Wahaibi1
Lamya H. Al-Wahaibi1 Ali M. Elshamsy2
Ali M. Elshamsy2 Taha F. S. Ali3
Taha F. S. Ali3 Bahaa G. M. Youssif4*
Bahaa G. M. Youssif4* S. Bräse5*Mohamed Abdel-Aziz3*
S. Bräse5*Mohamed Abdel-Aziz3* Nawal A. El-Koussi2,6*
Nawal A. El-Koussi2,6*A novel series of dihydropyrimidine/sulphonamide hybrids 3a–j with anti-inflammatory properties have been developed and tested as dual mPGES-1/5-LOX inhibitors. In vitro assay, results showed that compounds 3c, 3e, 3h, and 3j were the most effective dual inhibitors of mPGES-1 and 5-LOX activities. Compound 3j was the most potent dual inhibitor with IC50 values of 0.92 µM and 1.98 µM, respectively. In vivo, anti-inflammatory studies demonstrated that compounds 3c, 3e, 3h, and 3e had considerable anti-inflammatory activity, with EI% ranging from 29% to 71%. Compounds 3e and 3j were equivalent to celecoxib after the first hour but exhibited stronger anti-inflammatory effects than celecoxib after the third and fifth hours. Moreover, compounds 3e and 3j significantly reduced the levels of pro-inflammatory cytokines (PGE2, TNF-α, and IL-6) with gastrointestinal safety profiles. Molecular docking simulations explored the most potent derivatives’ binding affinities and interaction patterns within mPGES-1 and 5-LOX active sites. This study disclosed that compound 3j is a promising anti-inflammatory lead with dual mPGES-1/5-LOX inhibition that deserves further preclinical investigation.
Inflammation is a complex cascade of events that acts as the body’s natural response to injury. It is a crucial aspect of the healing process, helping to fight off infection-causing bacteria, viruses, and other microorganisms. If the acute inflammation fails to fight off the stimulus in time, it may become associated with chronic diseases, such as arthritis, cardiovascular disorders, respiratory diseases, neurodegenerative disorders, and cancer (Megha et al., 2021; Placha and Jampilek, 2021; Harvanová et al., 2023).
Prostaglandin E2 (PGE2) is a key pro-inflammatory prostanoid involved in many physiological processes, such as pain, inflammation, and fever. That’s why PGE2 is overproduced in several inflammatory diseases, such as chronic infections, rheumatoid arthritis, bronchial asthma, and various cancers (Ihsan, 2023). It is produced from arachidonic acid through enzymatic reactions, with microsomal prostaglandin E synthase-1 (mPGES-1) playing a crucial role in its biosynthesis (Zhang et al., 2022). mPGES-1 is highly upregulated in inflammation, making it a potential target for developing selective anti-inflammatory therapies that specifically inhibit PGE2 production without affecting other prostaglandins, potentially reducing the risk of gastrointestinal and cardiovascular side effects accompanied by traditional COX inhibitors (Bergqvist et al., 2020). Efforts to progress selective mPGES-1 inhibitors have led to two candidates, LY3023703 (whose trials were halted due to hepatotoxicity) (Jin et al., 2018) and GRC27864 (currently in Phase 2 trials) (Sant et al., 2018).
Another vital enzyme in the inflammatory process is 5-lipoxygenase (5-LOX), which converts arachidonic acid into bioactive leukotrienes. Leukotrienes play roles in various inflammatory conditions, including psoriasis, allergic asthma, and rheumatoid arthritis (Sinha et al., 2019; Meshram et al., 2020). Inhibiting the 5-LOX pathway is seen as a promising tactic for emerging potent anti-inflammatory drugs, although currently, only one 5-LOX inhibitor (Zileuton) is available for treating allergic asthma (Wenzel and Kamada, 1996).
Dihydropyrimidines are an important scaffold in medicinal chemistry because of their diverse variety of biological activities, which include anticancer (Janković et al., 2019; Dowarah et al., 2021), anti-inflammatory (Alfayomy et al., 2021), antioxidant (Vyas et al., 2023), antiviral (Spunde et al., 2022), antibacterial (Zhuang and Ma, 2020), antidiabetic (Jin et al., 2019), and antihypertensive activities (Mahgoub et al., 2021). Over the last few years, compounds possessing dihydropyrimidine moiety have been reported to show potent inhibitory activity against the mPGES-1 enzyme (Figure 1). Compounds I and II were discovered by Lauro et al. as potential mPGES-1 inhibitors by virtual screening with IC50 values of 4.16 ± 0.47 μM and 7.56 ± 0.94 μM, respectively (Lauro et al., 2014). Terracciano et al. synthesized compound III to optimize further these structures, which demonstrated 10-fold higher activity than compound I with IC50 value in the sub-micromolar range (IC50 = 0.41 ± 0.02 μM) (Terracciano et al., 2015). Some dihydropyrimidines were also reported to inhibit the 5-LOX enzyme (Figure 1), such as compound IV, designed and synthesized by Lokwani et al. It exhibited 51.84% inhibition of the enzyme at a concentration of 100 μg/mL with an IC50 equal to 19.12 μM, which was in line with the computational study in which the carbonyl moiety acted as a metal binding group and established interactions with the ferrous ion in the active site (Lokwani et al., 2015). Another compound, V, was developed by Venugopala et al., and it showed promising results demonstrating 81.19% ± 0.94% inhibition at 2.46 µM concentration (Venugopala et al., 2015).
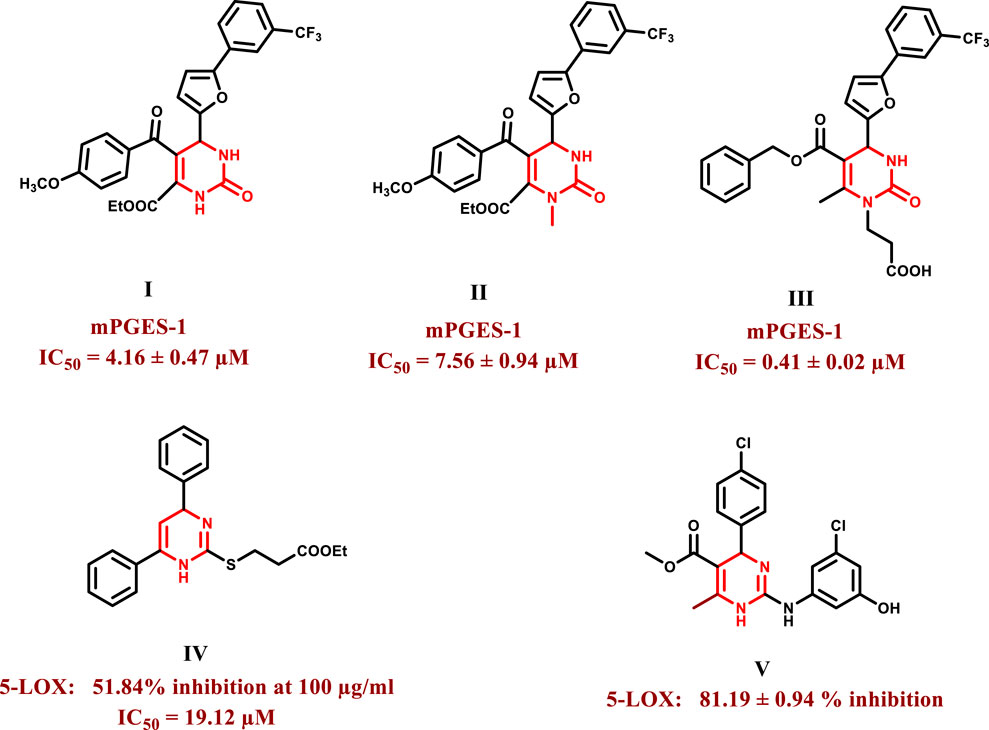
Figure 1. Structures of some dihydropyrimidines as mPGES-1 and 5-LOX inhibitors.
Sulfonamides have drawn much interest due to their widespread application as a privileged scaffold in drug design, with many clinically approved drugs containing this moiety, such as antibacterial (sulfamethoxazole), antidiabetic (gliclazide), anti-inflammatory (celecoxib), diuretic (Bumetanide), antiviral (Dasabuvir), and anticonvulsant drugs (Sultiame) (Apaydın and Török, 2019; Yousif et al., 2022). An example of an anti-inflammatory sulphonamide acting as anti-mPGES-1 is compound VI, which Kim et al. synthesized with the ability to inhibit PGE2 production in A549 cells at an IC50 of 0.24 μM which was about 9-fold more active than the standard inhibitor MK-886 (Figure 2) (Kim et al., 2021). Elkady et al. reported that replacement of the carboxylic group of NSAIDs with a substituted benzene sulphonamide group yielded compounds with dual mPGES-1/5-LOX inhibition and decreased COX inhibition compared to the parent drugs, such as indomethacin derivative VII, which showed IC50 values of 6.4 µM and 2.9 µM for mPGES-1 and 5-LOX respectively (more than six fold more potent mPGES-1 inhibitor than indomethacin) and lonazolac derivative VIII which showed IC50 values of 2.3 µM and 2.9 µM for mPGES-1 and 5-LOX respectively (19 and 20 folds more potent than lonazolac calcium against mPGES-1 and 5-LOX respectively) (Figure 2) (Elkady et al., 2012).
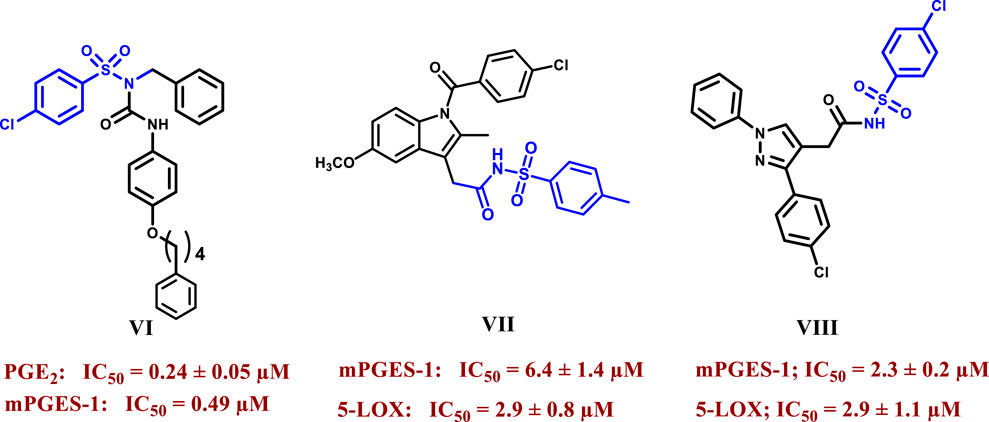
Figure 2. Structures of selected sulphonamides showing mPGES-1 and/or 5-LOX inhibitory activities.
As part of our ongoing search for a highly safe and effective anti-inflammatory drug (Elbastawesy et al., 2015; Abdelazeem et al., 2017; Abdelrahman et al., 2017; Youssif et al., 2019; Abdel-Aziz et al., 2021; Hendawy et al., 2021; Mohassab et al., 2021; Abdel et al., 2022; Shawky et al., 2023), we aimed to fill the research gap on the limited investigation of dihydropyrimidines’ potential as dual mPGES-1 and 5-LOX inhibitors in the current study. Our main objective was to explore the anti-inflammatory properties of novel dihydropyrimidine/sulfonamide hybrids (3a–j), taking advantage of the known anti-inflammatory potencies of both components. By combining these two important scaffolds into a single molecule, we aimed to investigate the potential synergistic effects and enhanced anti-inflammatory activity. Although previous research has examined the inhibitory potential of each scaffold individually, investigating these hybrid compounds is relatively new and holds promising prospects for developing more effective anti-inflammatory agents. Moreover, the synthesized dihydropyrimidine/sulfonamide derivatives were designed with various substitutions of electron-donating and electron-withdrawing groups to investigate their structure-activity relationship (SAR). The most effective derivatives were further subjected to molecular docking and dynamic simulations to explore their interactions within the active sites of mPGES-1 and 5-LOX (Figure 3).
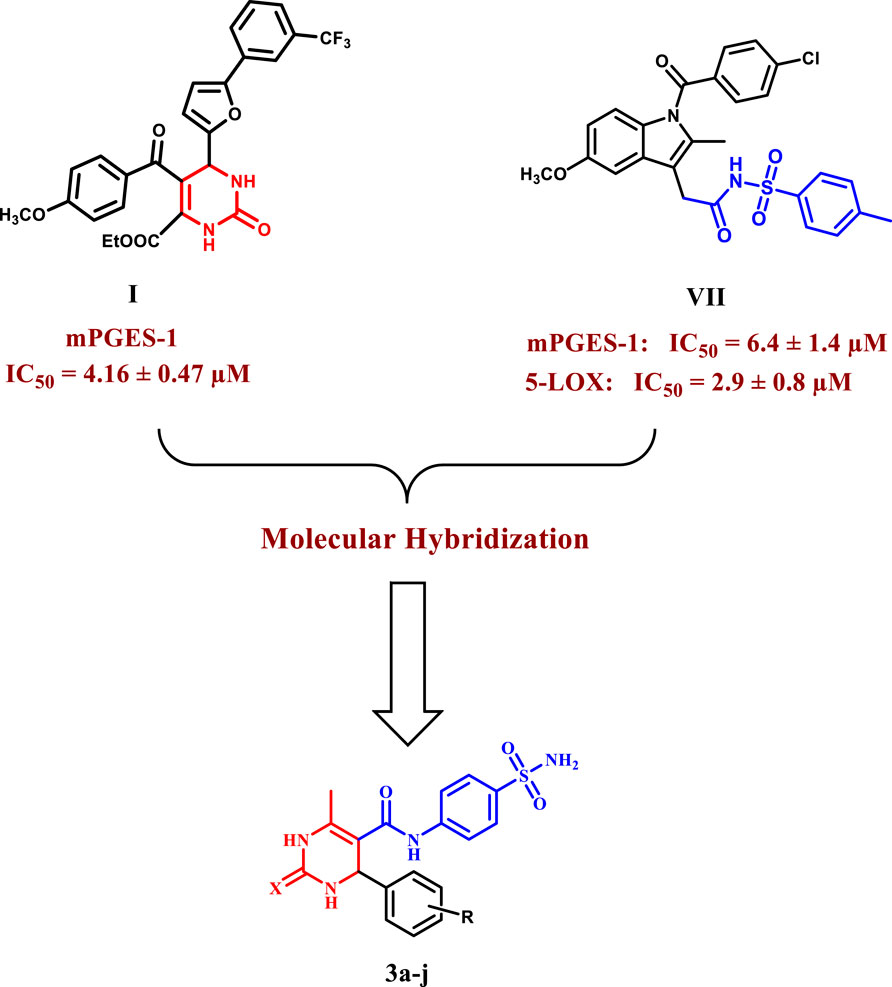
Figure 3. Design of the target compounds 3a–j as dual mPGES-1/5-LOX inhibitors.
Scheme 1 shows the chemical synthesis of target compounds 3a–j. The first step entails a 16-h reaction of sulfanilamide with a slight excess of 2, 2, 6-trimethyl-4H-1, 3-dioxin-4-one (Dioxinone) in a small amount of refluxing THF in the presence of anhydrous sodium acetate. Compound 2’s structure was confirmed by its reported melting point (Fares et al., 2020). Pyrimidine-5-carboxamides 3a–j were synthesized via acid-catalysed Biginelli cyclo-condensation of the intermediate 2 with various substituted benzaldehydes in the presence of urea or thiourea. The superlative yields were obtained by heating the reaction mixture in acetonitrile under reflux with a catalytic amount of trifluoroacetic acid for 18 h.
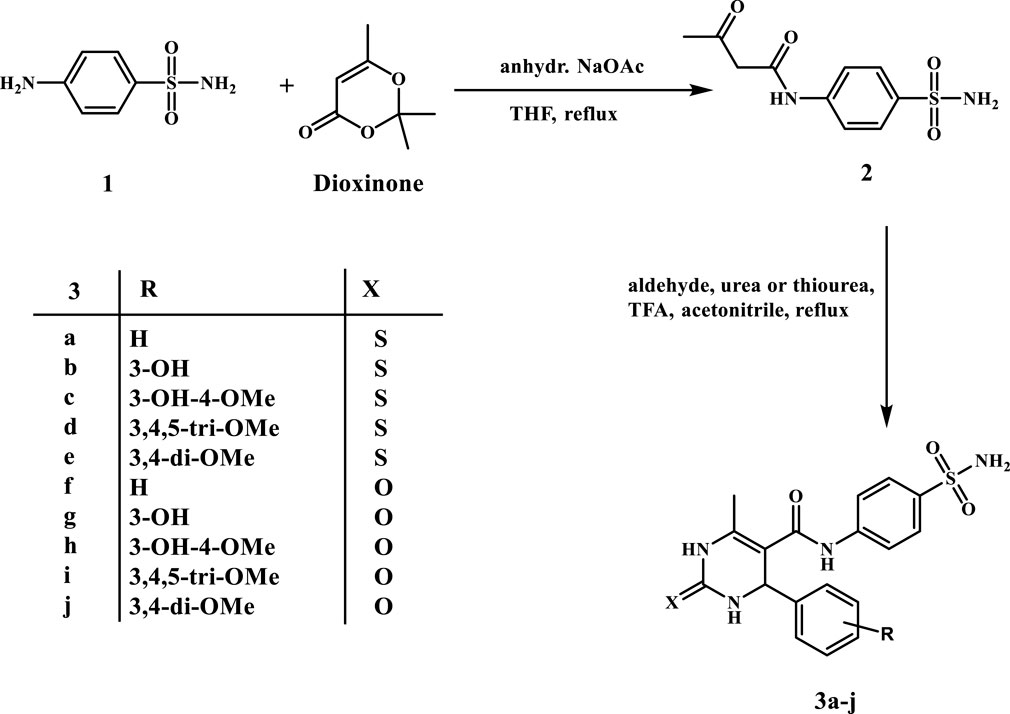
Scheme 1. Synthesis of pyrimidine-5-carboxamides 3a–j.
1H NMR, 13C NMR, mass spectra and elemental microanalysis confirmed the chemical structures of the target compounds 3a–j. All compounds showed a doublet at δ 5.31–5.44 ppm (CH) and a singlet at δ 2.05–2.09 ppm (CH3), confirming the formation of the dihydropyrimidine derivative. In compounds 3b–e and 3g–j, the sulfamoyl NH2 group showed as a singlet at δ 7.21–7.23 ppm or as a multiplet with the aromatic protons of the unsubstituted phenyl ring in compounds 3a and 3f. 13C NMR DEPTQ-135 spectra of the title compounds showed characteristic (CH3) peak at δ 16.54–16.62 ppm and (CH) peak at δ 54.60–55.00 ppm, while the aromatic carbons of the two phenyl rings appeared at δ 110.37–153.00 ppm. Furthermore, compounds 3a–e containing dihydropyrimidine-thione scaffold showed a highly downfield shifted peak at 173.85–174.38 ppm, which corresponds to the C2 thione moiety, while compounds 3f-j containing a dihydropyrimidinone nucleus showed characteristic C2 carbonyl peak at δ 152 ppm. Their ESI+ and ESI- mass spectra further confirmed the compounds, which showed characteristic [M + Na]+ and [M-H]− peaks for the synthesized compounds.
A cell-free assay was conducted to evaluate the capacity of compounds 3a–g to act as inhibitors of mPGES-1. In this assay, microsomal fractions from IL-1β-stimulated A549 cells served as the enzyme source (Gürses et al., 2021). During the initial screening phase, compounds 3a–j were examined for their effects on mPGES-1 at a concentration of 10 µM. The residual activity percentage (RA %) was determined for each target compound, as depicted in Table 1. Remarkably, compounds 3c, 3e, 3h, and 3j extremely inhibited mPGES-1 activity with RA% ranging from 24.7 to 33.6, but none of the other compounds were significantly active at 10 µM. A more thorough analysis of the IC50 values for 3c, 3e, 3h, and 3j revealed values between 0.92 and 1.5 µM (Table 1), significantly outperforming the reference MK886 (IC50 = 2.2 µM). Compound 3j (R = 3, 4-di-OMe, X = O) was the most active analog, with an IC50 value of 0.92 µM being 2.4-fold more potent than the reference MK886. Compound 3e (R = 3,4-di-OMe, X = S), which substitutes sulfur for oxygen at position C2 of 3j, had an IC50 of 0.97 µM, demonstrating that both oxygen and sulfur atoms at position C2 were tolerated for inhibitory activity against mPGES-1. Compounds 3h (R = 3-OH-4-OMe, X = O) and 3c (R = 3-OH-4-OMe, X = S) demonstrated comparable IC50 values of 1.32 µM and 1.53 µM, respectively. These compounds were 1.5-fold less potent than 3j, implying that the 3,4-di-OMe group may significantly influence the mPGES-1 inhibitory activity in this chemotype. Another intriguing finding was that changing the 3, 4-diOMe group in 3j to the 3,4,5-trimethoxy group caused the analog 3i (R = 3,4,5-tri-OMe, X = O) to be three times less potent than 3j, indicating the importance of the methoxy group numbers for activity.
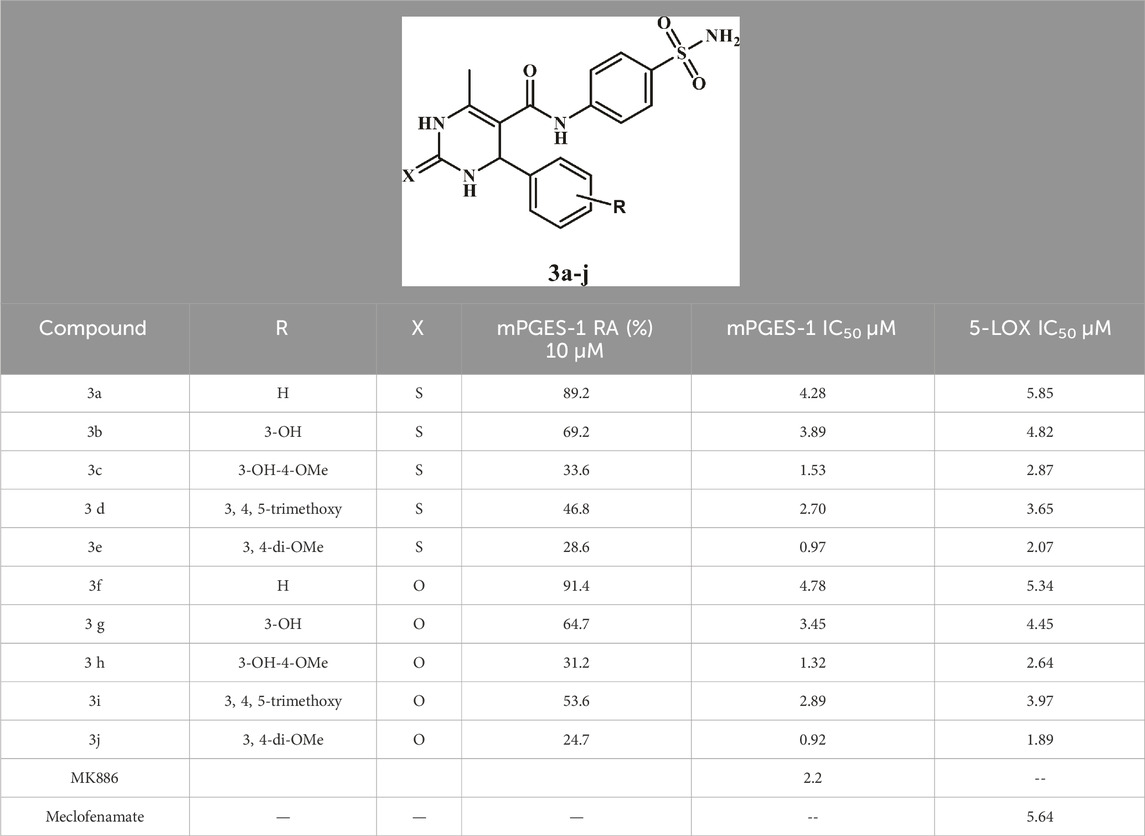
Table 1. Inhibition of mPGES-1/5-LOX assay of compounds 3a–j.
The unsubstituted derivatives 3a (R = H, X = S) and 3f (R = H, X = O), with IC50 values of 4.28 µM and 4.78 µM, respectively, were the least potent, indicating that the substitution at C4 Phenyl group is essential for activity and that the activity was increased in the following order: 3, 4-diOMe > 3-OH-4-OMe >3, 4, 5-trimethoxy > 3-OH > H.
The capacity of compounds 3a–j to inhibit the enzyme 5-lipoxygenase (5-LOX) has been investigated (Youssif et al., 2019). The IC50 of each compound is listed in Table 1.
The results of this assay matched the results of the m-PGES-1 inhibitory assay, in which compound 3j (R = 3,4-di-OMe, X = O), the most potent m-PGES-1 inhibitor, was found to be the most active as a 5-LOX inhibitor, with an IC50 value of 1.89 µM compared to the reference IC50 value of 5.60 µM. Once again, compound 3e (R = 3, 4-di-OMe, X = S) was ranked second in activity as a 5-LOX inhibitor with an IC50 value of 2.07 µM. According to the data on biological activity, 3j is the most effective dual inhibitor of mPGES-1 and 5-LOX activities. Compounds 3c, 3e, and 3h are potent inhibitors of mPGES-1 and 5-LOX, while the remaining compounds have moderate to weak inhibitory activity against both targets.
Compounds 3c, 3e, 3h, and 3j, the most effective dual m-PGES-1/5-LOX inhibitors, were chosen to be investigated for in vivo anti-inflammatory activity using the carrageen-induced paw edema bioassay method devised by Winter et al. (Winter et al., 1962). The compounds’ efficacy was measured as edema inhibition percentage (EI %) after 1, 3, and 5 h of carrageenan injection vs. the conventional medicine Celecoxib. Results are cited in Table 2. The findings revealed that the studied compounds have significant anti-inflammatory properties, with EI% ranging from 29% to 71%.
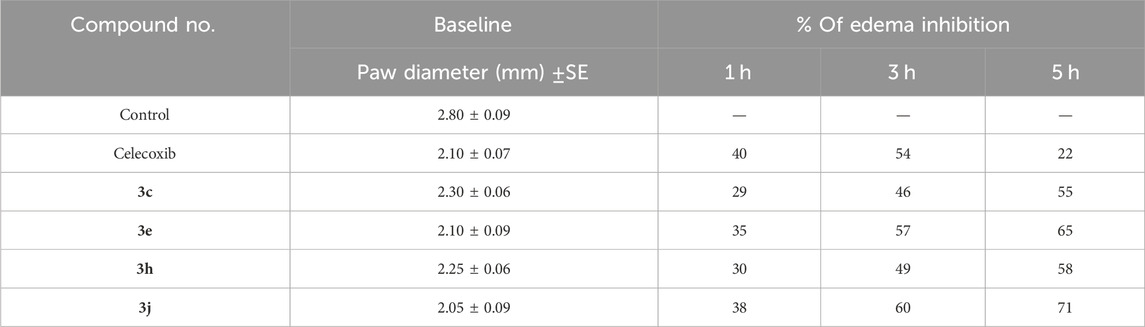
Table 2. Anti-inflammatory impact of 3c, 3e, 3h, and 3j.
After 5 h of treatment, all evaluated compounds showed greater anti-inflammatory effects than Celecoxib. They showed a rapid onset of action and a long-lasting effect until the fifth hour after the compounds were delivered. Compounds 3e and 3j were comparable to celecoxib after the first hour but had greater anti-inflammatory effects than celecoxib after the third and fifth hours (Table 2). According to our findings, the novel scaffold is a plausible lead for building highly effective m-PGES-1/5-LOX dual inhibitors as prospective anti-inflammatory medicines.
Inhibiting PGE2 is a crucial strategy in anti-inflammatory therapy, playing a pivotal role in managing inflammation and its associated conditions. PGE2, a potent inflammatory mediator, is highly detected in inflammatory diseases (Fattahi and Mirshafiey, 2012; Hassan et al., 2019). Moreover, recent research has demonstrated the importance of PGE2 reduction in anti-inflammatory actions (Cardoso et al., 2020). To assess the potential of compounds 3e, 3h, and 3j to inhibit PGE2, the levels of PGE2 in serum samples taken 4 hours after administering subcutaneous carrageenan injections were measured. The percentage of PGE2 inhibition was determined, and the values are presented in Table 3.
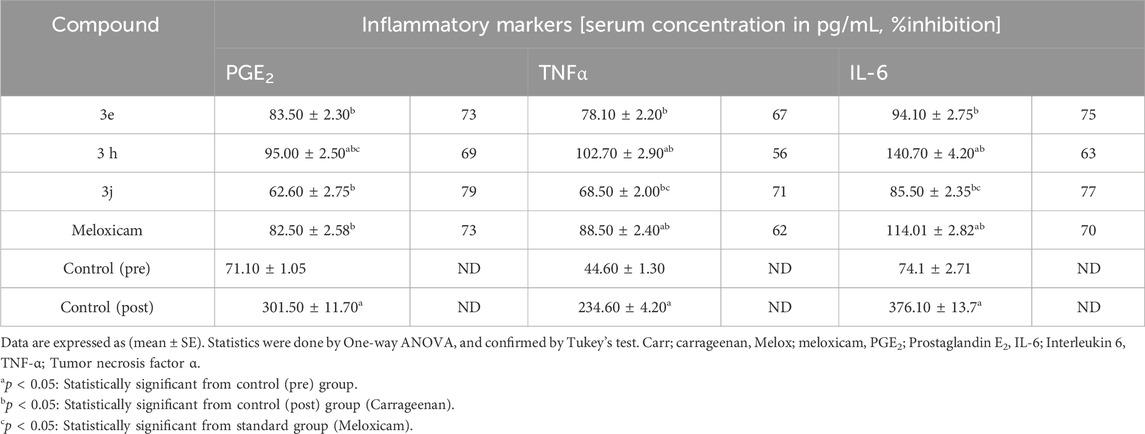
Table 3. Rat serum concentrations of PGE2, TNF-α and IL-6 for compounds 3e, 3h, 3j and Meloxicam.
The results of this testing were in line with the in vitro findings. Compared to the reference drug meloxicam, which displayed a 72.60% inhibition of PGE2, all three compounds examined exhibited marked reductions in serum PGE2 levels, ranging from 68.50% to 79.20%. Notably, compounds 3e and 3j demonstrated the highest activity, inhibiting PGE2 by 72.70% and 79.20%, respectively. It is worth mentioning that these same compounds were also the most potent dual mPGES-1/5-LOX inhibitors.
TNF-α and IL-6, the pro-inflammatory cytokines, are pivotal in promoting inflammation and are often associated with developing chronic illnesses (Hunter and Jones, 2015). Decreased plasma levels of these mediators play a significant role in achieving an overall anti-inflammatory effect, which, in turn, helps mitigate the progression and severity of various chronic conditions (Desai and Furst, 2006). In the current study, we assessed the serum concentrations of TNF-α and IL-6 in the blood samples collected from rats following administration of compounds 3e, 3h, and 3j, as presented in Table 3. All tested compounds significantly reduced the concentrations of TNF-α (% inhibition = 56–71) and IL-6 (% inhibition = 63–77) in rat serum. Notably, compound 3j demonstrated the highest efficacy, with a TNF-α % inhibition of 71%, surpassing that of the reference drug meloxicam (%TNF-α inhibition = 62) and exhibiting a higher drop in serum IL-6 levels (% inhibition = 77), in comparison to meloxicam (% IL-6 inhibition = 70).
The two most common side effects of long-term NSAID use are gastrointestinal erosion and ulcers (Hendawy et al., 2021). As a result, we were curious about the ulcerogenic potential of the most efficacious drugs, 3e and 3j, when given orally. The ulcerogenic effects of 3e and 3j were assessed by macroscopic inspection of rat intestinal mucosa after oral administration of 10 mg/kg of 3e, 3j, indomethacin, and celecoxib (Manivannan and Chaturvedi, 2011).
Compound 3j did not generate ulceration in the isolated rat stomach, whereas compound 3e produced mild hyperemia but no widespread ulceration (Table 4). Compounds 3e and 3j were found to have a potent m-PGES-1/5-LOX inhibitory profile with no (or weak) gastrointestinal side effects.
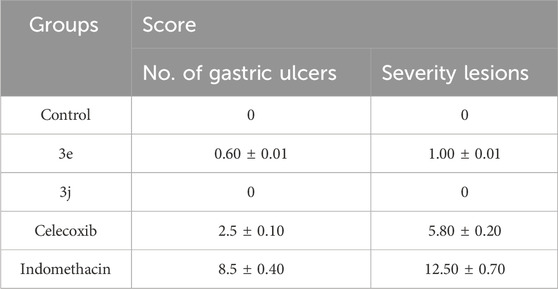
Table 4. Ulcerogenic effects of compounds 3e and 3j.
To explore the potential interactions of compound 3j with the target proteins mPGES-1 and 5-LO, we created their structural models and performed molecular docking simulations using the crystalline structures of these proteins (PDB ID, 4 bpm and 6 n2w, respectively) as reported by Li et al, (2014) and Gilbert et al, (2020). For mPGES-1 (PDB ID, 4 bpm), our strategy involved docking compound 3j at the site occupied by a co-crystallized inhibitor, rather than the glutathione (GSH) binding site, due to the latter’s strong affinity and resistance to displacement by other inhibitors, as discussed by Li et al, (2014) and Koeberle and Werz, (2018). The docking protocol was validated by re-docking the co-crystalized ligands into the active sites of both enzymes (i.e., mPGES-1 and 5-LO, respectively). The resulting top-scoring poses of both ligands were in good alignment with the co-crystalized ones with slight deviations (RMSDs = 1.27 and 1.04, respectively). Superposition of the co-crystallized ligands of both enzymes is illustrated in Figure 4. The docking results, illustrated in Figure 5, reveal that the preferred orientation of 3j was comparable to the binding behavior of the co-crystallized inhibitor, engaging in a hydrogen bond with SER-127 and hydrophobic contacts with LEU-132 and PRO-124, alongside an additional hydrogen bond with PRO-124’s backbone.
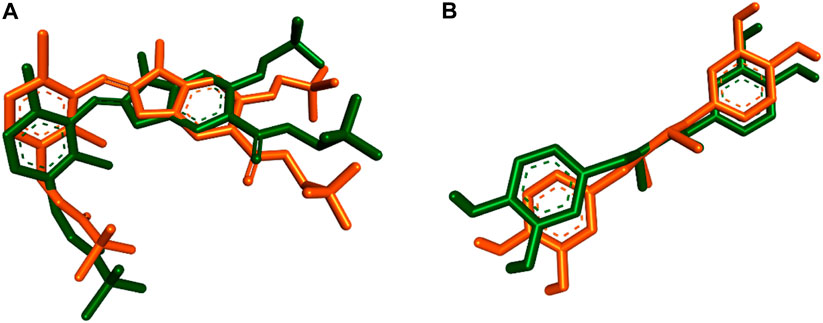
Figure 4. (A) and (B) Superposition of both the redocked poses and co-crystallized inhibitors inside mPGES-1 (PDB ID, 4 bpm) and 5-LOX (PDB ID, 6 n2w) respectively.
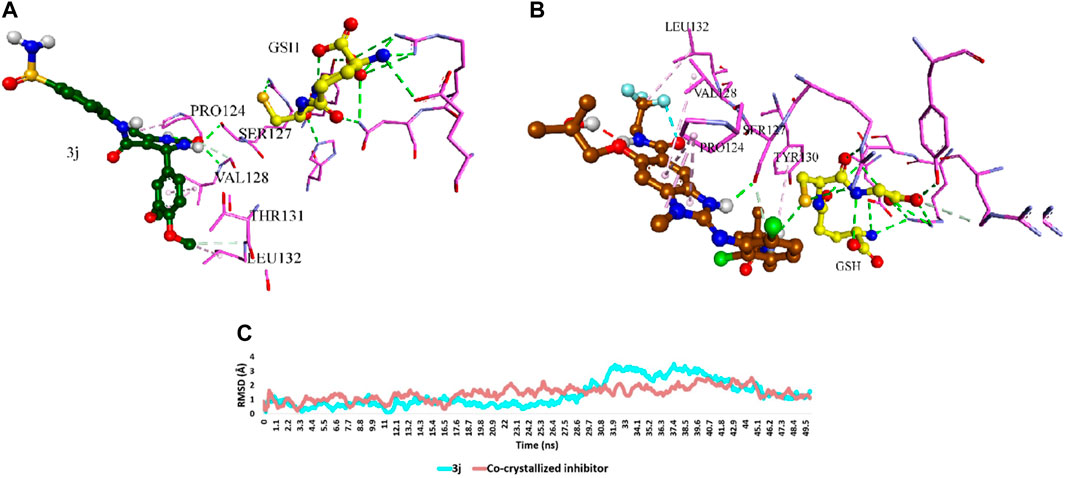
Figure 5. (A) and (B) Binding mode of 3j inside the co-crystallized inhibitor-binding site of mPGES-1 (PDB ID: 4bpm) in comparison with that of the co-crystallized inhibitor, respectively. (C) RMSDs of 3j inside the co-crystallized inhibitor-binding site of mPGES-1 in comparison with that of the co-crystallized inhibitor over 50 ns-long MD simulations.
Regarding 5-LO (PDB ID: 6n2w), 3j was docked into the enzyme’s redox site, achieving a binding posture partially akin to that of the native inhibitor, as depicted in Figure 6. Here, 3j predominantly formed hydrophobic interactions with residues LEU-368, PHE-359, LEU-414, and TRP-599, while also establishing hydrogen bonds with GLY-430 and HIS-432.
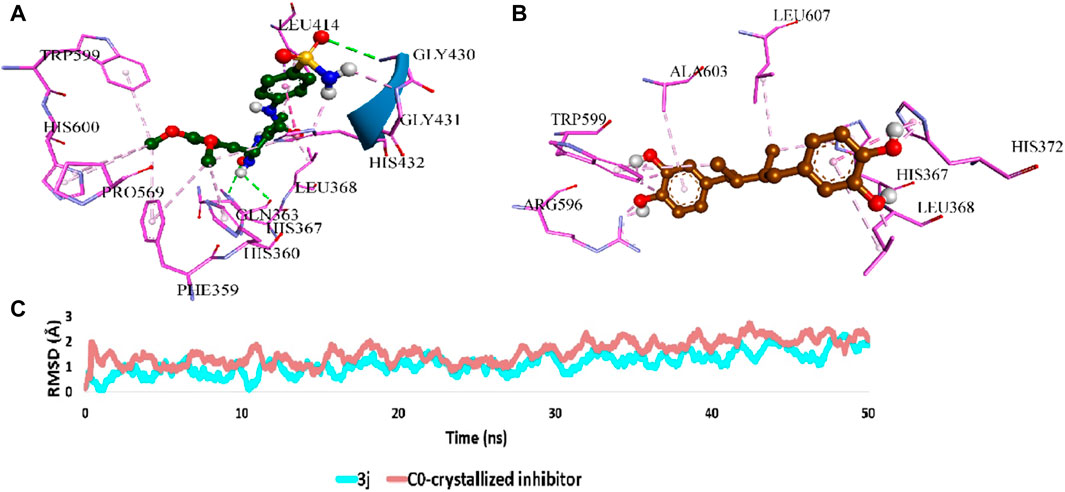
Figure 6. (A) and (B) Binding mode of 3j inside the redox binding site of 5-LO (PDB ID, 6n2w) in comparison with that of the co-crystallized inhibitor, respectively. (C) RMSDs of 3j inside the redox binding site of 5-LO in comparison with that of the co-crystallized inhibitor over 50 ns-long MD simulations.
To validate the docking poses of the most potent compound 3j inside the active sites of both mPGES-1 (PDB ID: 4bpm) and 5-LO (PDB ID: 6n2w), respectively, they were subjected to 50 ns-long molecular dynamic simulations (MDS). As shown in Figures 5C, 6C, 3j exhibited acceptable stability inside each binding site throughout the simulation with an average RMSD of 2.2 Å and 1.4 Å, respectively relative to the initial docking poses.
Accordingly, the calculated electrostatic and van der Waals interaction energies of 3j within the active site of each enzyme showed an average total interaction energies of around −61.19 and −26.52 kcal/mol, respectively (Figure 7). Moreover, their co binding free energies (ΔGBinding) using MM-PBSA were found to be −16.8422 and −7.998 kcal/mol, respectively indicating strong affinities towards the corresponding active sites, particularly with 5-LOX (Table 5).
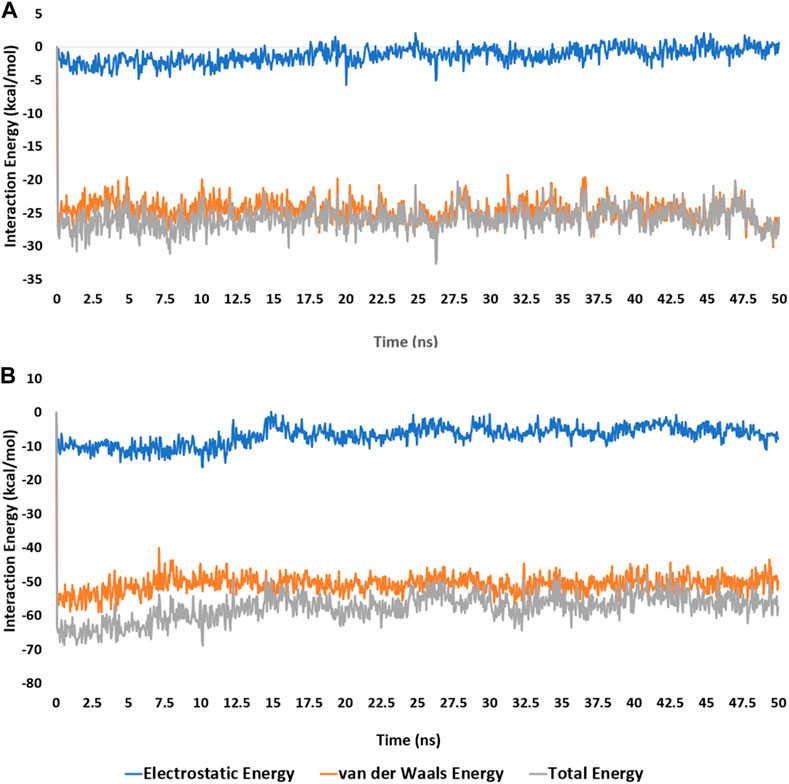
Figure 7. Electrostatic and van der Waals Interaction energies of compound 3j inside the active sites of mPGES-1 and 5-LO over 50 ns-long MD simulations [(A) and (B), respectively].
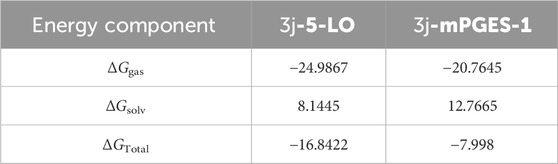
Table 5. Calculated binding free energies (ΔGBinding; MM-PBSA) of compound 3j in complex with 5-LO and mPGES-1. The values were calculated in kcal/mol.
Compound 3j established stable multiple hydrophilic and hydrophobic interactions, particularly H-bonds that were found to be around 2 H-bonds inside 5-LOX, and around one H-bond inside mPGES-1 throughout the simulation course (Figure 8).
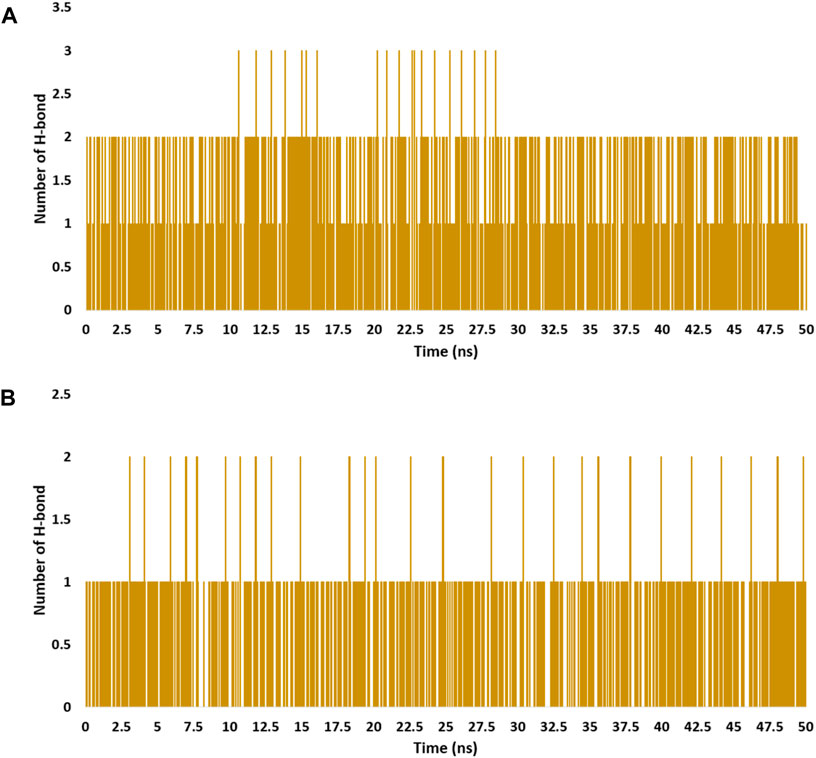
Figure 8. Number of H-bonds detected for 3j inside the active sites of mPGES-1 and 5-LO over 50 ns-long MD simulations [(A) and (B), respectively]. Cut-off distance for H-bonds was set to 3.0 Å.
In conclusion, compound 3j exhibited acceptable levels of binding stability inside the active sites of both 5-LO and mPGES-1 throughout a 50-ns long MDS indicating a possible inhibitory activity against both enzymes.
SAR studies could be summarized as follows.
• Substitution on the C4 phenyl ring on the DHPM scaffold proved advantageous for both mPGES-1 and 5-LOX inhibitory activities, with the unsubstituted compounds 3a and 3f being the least active of the series.
• The number of methoxy groups greatly affected the activity, with the highest potency exhibited by the 3, 4 dimethoxy derivatives 3e and 3j.
• On the other hand, the trimethoxy derivatives 3d and 3i were less potent against both mPGES-1 and 5-LOX (possibly due to the increased steric hindrance).
• The introduction of sulfonamide group was beneficial as it provided auxiliary interactions with GLY-430 and HIS-432 in the 5-LOX redox binding site through hydrogen bonding. which further stabilized its binding.
• The enzymes tolerated both urea and thiourea moieties well, with urea derivatives having slightly better activity against both enzymes.
• The DHPM anchored compound 3j to the mPGES-1 active site through formation of important hydrogen bonding with SER-127 and PRO-124
SAR of compounds 3a–j as dual mPGES-1/5-LOX inhibitors is outlined in Figure 9.
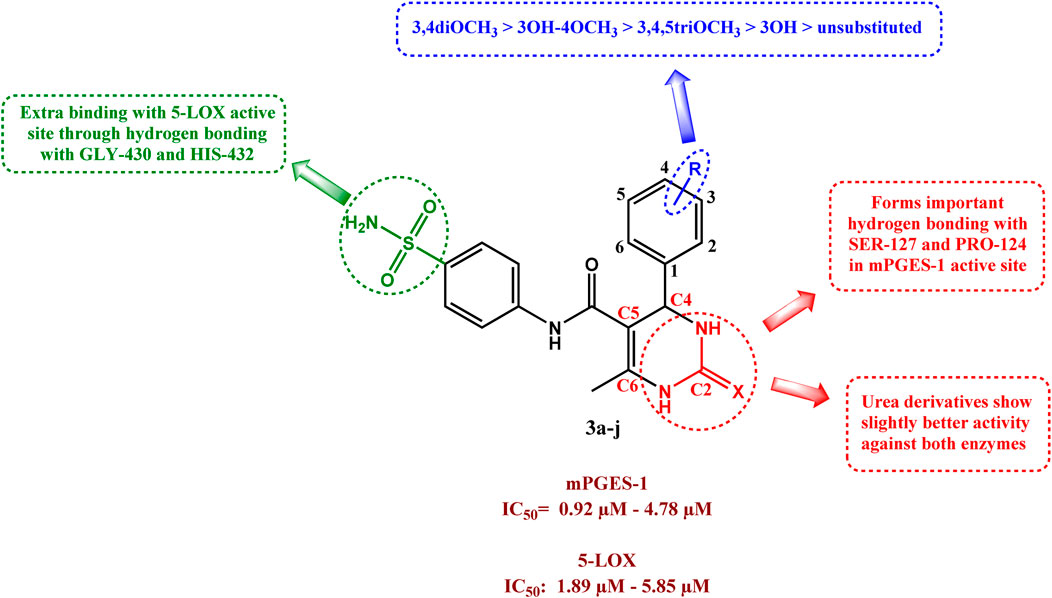
Figure 9. SAR of compounds 3a–j as dual mPGES-1/5-LOX inhibitors.
As potential anti-inflammatory agents, a novel class of dual mPGES-1/5-LOX inhibitors 3a–j has been developed and tested in vitro. Compounds 3c, 3e, and 3j were discovered to be effective mPGES-1 and 5-LOX inhibitors. The most potent dual inhibitor of mPGES-1 and 5-LOX activity was 3j. Compounds 3c, 3e, and 3j showed promising anti-inflammatory action with rapid onset of action and long-lasting effects up to 5 h with no or weak gastrointestinal unwanted side effects. The levels of pro-inflammatory cytokines (PGE2, TNF-α, IL-6) also decreased significantly. Furthermore, molecular docking studies predicted the binding affinities and interaction patterns of these compounds with both mPGES-1 and 5-LOX, which revealed that these compounds established key interactions with both targets with better affinities than the cocrystallized ligands. The most potent derivatives will be subjected to more detailed biological assays to evaluate their anti-inflammatory activity to obtain a lead compound for future optimization.
General details: (See supplementary data)
Sulfanilamide, 2, 2, 6-trimethyl-4H-1, 3-dioxin-4-one (Dioxinone), and all solvents were purchased from Sigma Aldrich, Combi-Blocks, Fisher Scientific and they were used without purification unless mentioned.
A mixture of the appropriate aldehyde (2 mmol), urea or thiourea (3 mmol, 0.228 g), and compound 2 (2 mmol, 0.512 g) were heated in acetonitrile containing a catalytic amount of trifluoroacetic acid (0.4 mmol, 30 µL) for 8 h. The excess solvent was evaporated, and the reaction mixture was left overnight. The solid precipitate that formed was filtered off and then washed with cold acetonitrile and distilled water before being recrystallized from the appropriate solvent.
White powder (acetonitrile) (0.442 g, 55% yield), m. p: 266°C–269°C; 1H NMR (400 MHz, DMSO-d6) δ 10.06 (d, J = 10.2 Hz, 2H), 9.51 (s, 1H), 7.71 (s, 4H), 7.36 (t, J = 7.4 Hz, 2H), 7.29–7.21 (m, 5H), 5.43 (d, J = 3.0 Hz, 1H), 2.09 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 174.23 (s), 165.36 (s), 143.05 (s), 141.94 (s), 138.39 (s), 136.66 (s), 128.69 (s), 127.75 (s), 126.53 (s), 126.29 (s), 119.11 (s), 106.74 (s), 55.00 (s), 16.59 (s); MS (ESI+) m/z 424.6 [M + Na]+, 826.5 [2M + Na]+; MS (ESI−) m/z 400.7 [M-H]-. Anal. Calcd. For C18H18N4O3S2 (402.49): C, 53.72; H, 4.51; N, 13.92. Found: C, 53.61; H, 4.82; N, 14.04.
White powder (acetonitrile) (0.334 g, 40% yield), m. p: 288°C–291°C; 1H NMR (400 MHz, DMSO-d6) δ 10.03 (s, 2H), 9.46 (s, 2H), 7.72 (s, 4H), 7.22 (s, 2H), 7.12 (t, J = 7.7 Hz, 1H), 6.65 (d, J = 7.9 Hz, 3H), 5.35 (d, J = 2.7 Hz, 1H), 2.07 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ174.19 (s), 165.39(s), 157.60 (s), 144.59 (s), 142.00 (s), 138.35 (s), 136.39 (s), 129.60 (s), 126.53 (s), 119.13 (s), 116.74 (s), 114.70 (s), 113.15 (s), 106.88 (s), 54.97 (s), 16.56 (s); MS (ESI+) m/z 858.4 [2M + Na]+; MS (ESI−) m/z 416.6 [M-H]-, 834.5 [2M-H]-. Anal. Calcd. For C18H18N4O4S2 (418.49): C, 51.66; H, 4.34; N, 13.39. Found: C, 51.92; H, 4.50; N, 13.67.
White powder (acetic acid) (0.224 g, 25% yield), m. p: 296°C; 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 2H), 9.42 (s, 1H), 9.02 (s, 1H), 7.72 (s, 4H), 7.22 (s, 2H), 6.86 (d, J = 8.4 Hz, 1H), 6.71 (d, J = 2.1 Hz, 1H), 6.62 (d, J = 8.3 Hz, 1H), 5.31 (d, J = 2.9 Hz, 1H), 3.72 (s, 3H), 2.08 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 173.85 (s), 165.40 (s), 147.33 (s), 146.59 (s), 142.03 (s), 138.31 (s), 136.28 (s), 135.86 (s), 126.51 (s), 119.09 (s), 117.01 (s), 113.79 (s), 112.11 (s), 107.00 (s), 55.67 (s), 54.65 (s), 16.54 (s); MS (ESI+) m/z 470.6 [M + Na]+, 918.3 [2M + Na]+; MS (ESI−) m/z 446.7 [M-H]-, 894.4 [2M-H]-. Anal. Calcd. For C19H20N4O5S2 (448.51): C, 50.88; H, 4.49; N, 12.49. Found: C, 51.18; H, 4.64; N, 12.73.
White powder (ethanol) (0.384 g, 39% yield), m. p: 270°C–272°C; 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 10.07 (s, 1H), 9.46 (s, 1H), 7.73 (s, 4H), 7.23 (s, 2H), 6.56 (s, 2H), 5.40 (d, J = 2.7 Hz, 1H), 3.69 (s, 6H), 3.62 (s, 3H), 2.08 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 174.38 (s), 165.59 (s), 153.00 (s), 141.93 (s), 138.64 (s), 138.51 (s), 137.07 (s), 136.55 (s), 126.61 (s), 119.23 (s), 106.65 (s), 103.54 (s), 60.02 (s), 55.88 (s), 54.93 (s), 16.62 (s); MS (ESI+) m/z 514.6 [M + Na]+, 1006.3 [2M + Na]+; MS (ESI−) m/z 490.6 [M-H]-, 982.4 [2M-H]-. Anal. Calcd. For C21H24N4O6S2 (492.57): C, 51.21; H, 4.91; N, 11.37. Found: C, 51.37; H, 5.11; N, 11.68.
White powder (ethanol) (0.323g, 35% yield), m. p: 272°C–274°C; 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 2H), 9.45 (s, 1H), 7.72 (s, 4H), 7.22 (s, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.84 (s, 1H), 6.78 (d, J = 10.3 Hz, 1H), 5.38 (d, J = 2.8 Hz, 1H), 3.71 (s, 3H), 3.67 (s, 3H), 2.09 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 174.05 (s), 165.48 (s), 148.68 (s), 148.43 (s), 141.97 (s), 138.39 (s), 136.54 (s), 135.40 (s), 126.54 (s), 119.12 (s), 118.31 (s), 111.88 (s), 110.37 (s), 106.74 (s), 55.57 (s), 55.43 (s), 54.60 (s), 16.58 (s); MS (ESI+) m/z 484.6 [M + Na]+, 946.2 [2M + Na]+; MS (ESI−) m/z 460.6 [M-H]-, 922.4 [2M-H]-. Anal. Calcd. For C20H22N4O5S2 (462.54): C, 51.94; H, 4.79; N, 12.11. Found: C, 52.24; H, 5.05; N, 12.26.
White powder (methanol) (0.463 g, 60% yield), m. p: 258°C–260°C; 1H NMR (400 MHz, DMSO-d6) δ 9.88 (s, 1H), 8.82 (s, 1H), 7.71 (s, 4H), 7.66 (s, 1H), 7.37–7.19 (m, 7H), 5.44 (d, J = 2.4 Hz, 1H), 2.07 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 165.66 (s), 152.51 (s), 144.28 (s), 142.22 (s), 139.76 (s), 138.09 (s), 128.52 (s), 127.38 (s), 126.48 (s), 126.17 (s), 118.96 (s), 104.95 (s), 54.94 (s), 17.15 (s). Anal. Calcd. For C18H18N4O4S (386.43): C, 55.95; H, 4.7; N, 14.5. Found: C, 56.08; H, 4.99; N, 14.63.
White powder (methanol) (0.539 g, 67% yield), m. p: 270°C–272°C; 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 9.39 (s, 1H), 8.78 (s, 1H), 7.76–7.68 (m, 4H), 7.60 (s, 1H), 7.21 (s, 2H), 7.09 (t, J = 7.8 Hz, 1H), 6.72–6.65 (m, 2H), 6.62 (d, J = 8.1 Hz, 1H), 5.36 (d, J = 2.1 Hz, 1H), 2.05 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 165.69 (s), 157.52 (s), 152.61 (s), 145.88 (s), 142.30 (s), 139.54 (s), 138.06 (s), 129.44 (s), 126.49 (s), 118.99 (s), 116.64 (s), 114.31 (s), 113.02 (s), 105.12 (s), 54.85 (s), 17.14 (s). Anal. Calcd. For C18H18N4O5S (402.43): C, 53.72; H, 4.51; N, 13.92. Found: C, 53.46; H, 4.60; N, 14.20.
White powder (methanol) (0.467 g, 54% yield), m. p: 275°C–279°C; 1H NMR (400 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.95 (s, 1H), 8.75 (s, 1H), 7.75–7.68 (m, 4H), 7.54 (s, 1H), 7.21 (s, 2H), 6.82 (d, J = 8.3 Hz, 1H), 6.73 (s, 1H), 6.62 (d, J = 8.3 Hz, 1H), 5.32 (d, J = 2.3 Hz, 1H), 3.71 (s, 3H), 2.05 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 166.17 (s), 152.95 (s), 147.49 (s), 146.98 (s), 142.79 (s), 139.83 (s), 138.48 (s), 137.62 (s), 126.93 (s), 119.42 (s), 117.21 (s), 114.12 (s), 112.52 (s), 105.74 (s), 56.13 (s), 55.00 (s), 17.58 (s). Anal. Calcd. For C19H20N4O6S (432.45), C, 52.77; H, 4.66; N, 12.96. Found, C, 52.99; H, 4.75; N, 13.18.
White powder (methanol) (0.476 g, 50% yield), m. p: 260°C–263°C; 1H NMR (400 MHz, DMSO-d6) δ 9.90 (s, 1H), 8.78 (s, 1H), 7.72 (s, 4H), 7.60 (s, 1H), 7.21 (s, 2H), 6.56 (s, 2H), 5.39 (d, J = 2.3 Hz, 1H), 3.68 (s, 6H), 3.60 (s, 3H), 2.05 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 165.86 (s), 152.87 (s), 152.48 (s), 142.19 (s), 139.78 (s), 139.54 (s), 138.19 (s), 136.80 (s), 126.52 (s), 119.02 (s), 104.73 (s), 103.40 (s), 59.97 (s), 55.83 (s), 54.95 (s), 17.14 (s); MS (ESI+) m/z 498.6 [M + Na]+, 974.3 [2M + Na]+; MS (ESI−) m/z 474.6 [M-H]-. Anal. Calcd. For C21H24N4O7S (476.5): C, 52.93; H, 5.08; N, 11.76. Found: C, 53.03; H, 5.33; N, 11.87.
White powder (ethanol) (0.420 g, 47% yield), m. p: 266°C–267°C; 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 8.77 (s, 1H), 7.71 (s, 4H), 7.58 (s, 1H), 7.21 (s, 2H), 6.90 (d, J = 8.3 Hz, 1H), 6.86 (s, 1H), 6.80 (d, J = 8.3 Hz, 1H), 5.39 (d, J = 2.4 Hz, 1H), 3.71 (s, 3H), 3.66 (s, 3H), 2.06 (s, 3H); 13C NMR DEPTQ-135 (100 MHz, DMSO-d6) δ 165.78 (s), 152.48 (s), 148.63 (s), 148.17 (s), 142.25 (s), 139.60 (s), 138.10 (s), 136.67 (s), 126.49 (s), 118.96 (s), 118.09 (s), 111.78 (s), 110.31 (s), 104.96 (s), 55.55 (s), 55.41 (s), 54.56 (s), 17.14 (s); MS (ESI+) m/z 468.7 [M + Na]+, 914.5 [2M + Na]+; MS (ESI−) m/z 444.8 [M-H]-. Anal. Calcd. For C20H22N4O6S (446.48), C, 53.8; H, 4.97; N, 12.55. Found, C, 53.63; H, 5.13; N, 12.68.
Microsomal measures of A549 cells expressing mPGES-1 were made in accordance with prior research findings (Koeberle et al., 2008). These cells were cultivated in Dulbecco’s Modified Eagle Medium and resuspended in a homogenization buffer. Subsequently, the microsomes were subjected to pre-incubation with either assessed compounds or a carrier solution containing 0.1 percent DMSO. The enzymatic process was halted by introducing FeCl3, citric acid, and 11-PGE2 as an internal standard. The quantities of Prostaglandin E2 (PGE2) were measured via RP-HPLC methodologies.
The study used an enzyme immune assay (EIA) kit (catalogue no. 760700, Cayman Chemical, Ann Arbor, Michigan, USA) to evaluate the inhibitory activity of target analogues against soya bean 5-LOX, ensuring compliance with manufacturer’s instructions and protocols, and calculating IC50 values (Roschek et al., 2009).
Compounds 3c, 3e, 3h, and 3j were chosen for in vivo anti-inflammatory testing using the carrageen-induced paw edema bioassay method described by Winter et al, (1962). The compounds’ efficacy was measured as edema inhibition percentage (EI%) after 1, 3, and 5 h of carrageenan injection vs. the conventional medicine Celecoxib.
In this study, specializing ELISA kits were used to determine the concentration of inflammatory cytokines PGE2, IL-6, and TNF-α. The study’s findings were examined in accordance with the instructions provided by the manufacturer, and measurements were taken based on the optical density at 450 nm.
The ulcerogenic effects of compounds 3e and 3j were evaluated by macroscopic examination of rat intestinal mucosa after oral administration of 10 mg/kg of these compounds and indomethacin and celecoxib (Manivannan and Chaturvedi, 2011). See Appendix A for details.
OpenBabel v.3.1.1 (O’Boyle et al., 2011) was used to convert the structures’ SMILES codes to three-dimensional configurations that were subsequently subjected to a minimization of energy using the steepest descent technique with the same software. The minimization was performed by the force field MMFF94. Using AutoDockTools v.4.2, all torsions of the selected structures were assigned and their Gasteiger charges were provided for all studied atoms in structures (Morris et al., 2009).
For docking screening, the mPGES-1 and 5-LO crystal structures (PDB codes: 4bpm and 6n2w, respectively) (Li et al., 2014; Koeberle and Werz, 2018) were used. PDBfixer (Eastman et al., 2013) was used to edit the downloaded structure, adding missing residues and atoms, and removing co-crystalized H2O and heteroatoms. Through AutoDock Tools v.4.2, polar hydrogen and Gasteiger charges were subsequently made available for both proteins.
The docking process was carried out using the PyRx platform’s built-in AutoDock Vina software (Eastman et al., 2013; Dallakyan et al., 2015). According to the co-crystalized ligands of both enzymes, the docking search grid boxes were determined to perfectly enclose them with a 20 Å3 total size.
The grid box’s coordinates were set to be x = −9.682; y = 4.274; z = −23.145 and x = 45.424; y = 92.375; z = 34.811, respectively. The level of exhaustion was held at 24. Ten poses were generated for each docking experiment. Docking poses were analyzed and visualized using Pymol software (Seeliger and de Groot, 2010).
The NAMD 3.0.0 program, which makes use of the Charmm-36 force field, was used to do molecular dynamics simulations (Phillips et al., 2005; Ribeiro et al., 2018). The QwikMD toolbox in VMD software was used to build protein systems (Humphrey et al., 1996). The procedure encompassed the examination of the protein structure to identify any hydrogens that were absent, the modification of the protonation states of the amino acids to achieve a pH of 7.4, and the elimination of co-crystallized water molecules. Following this, the entire configuration was enclosed within an orthorhombic container including TIP3P water molecules, along with the addition of sodium (Na+) and chloride (Cl-) ions at a concentration of 0.15 M, creating a solvent buffer with a size of 20 Å. Subsequently, the constructed systems underwent energy minimization and equilibration for a duration of 5 nanoseconds. In the context of protein-ligand complexes, the initial configurations with the highest scores were utilized as a basis for subsequent simulation. The VMD plugin Force Field Toolkit (ffTK) was utilized to calculate the properties and topologies of the compounds. Subsequently, the resulting parameters and topology files were introduced into VMD to facilitate the accurate reading of the protein-ligand complexes and subsequent execution of the simulation procedures.
The Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) technique, which was introduced into the AMBER18 MMPBSA. py module, was used to estimate the binding free energy for the docked complex. The results of this calculation may be found in the following sentence (Miller et al., 2012). The trajectories were processed into one hundred frames, and the net energy of the system was predictable using the below formula:
In order to accurately compute each of the previously mentioned variables, it is necessary to consider a wide variety of energy components. Some of these components include electrostatic energy, van der Waals energy, the polar contribution to solvation energy, as well as the internal energy derived from molecular mechanics.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because simple experiment was applied to animals.
LA-W: Funding acquisition, Project administration, Writing–original draft. AE: Formal Analysis, Methodology, Software, Writing–original draft, Writing–review and editing. TA: Supervision, Validation, Writing–original draft, Writing–review and editing. BY: Formal Analysis, Investigation, Methodology, Software, Validation, Writing–original draft, Writing–review and editing. SB: Writing–original draft, Writing–review and editing. MA-A: Investigation, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. NE-K: Investigation, Supervision, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP 2024R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP 2024R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelazeem, A. H., El-Saadi, M. T., Said, E. G., Youssif, B. G., Omar, H. A., and El-Moghazy, S. M. (2017). Novel diphenylthiazole derivatives with multi-target mechanism: synthesis, docking study, anticancer and anti-inflammatory activities. Bioorg Chem. 75, 127–138. doi:10.1016/j.bioorg.2017.09.009
Abdel-Aziz, S. A., Taher, E. S., Lan, P., Asaad, G. F., Gomaa, H. A., El-Koussi, N. A., et al. (2021). Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives bearing 1, 3-thiazole moiety as novel anti-inflammatory EGFR inhibitors with cardiac safety profile. Bioorg Chem. 111, 104890. doi:10.1016/j.bioorg.2021.104890
Abdel, S. A., Taher, E. S., Lan, P., El-Koussi, N. A., Salem, O. I., Gomaa, H. A., et al. (2022). New pyrimidine/thiazole hybrids endowed with analgesic, anti-inflammatory, and lower cardiotoxic activities: design, synthesis, and COX-2/sEH dual inhibition. Arch. Pharm. Weinh. 355 (7), 2200024. doi:10.1002/ardp.202200024
Abdelrahman, M. H., Youssif, B. G., Abdelazeem, A. H., Ibrahim, H. M., Abd El Ghany, A. M., Treamblu, L., et al. (2017). Synthesis, biological evaluation, docking study and ulcerogenicity profiling of some novel quinoline-2-carboxamides as dual COXs/LOX inhibitors endowed with anti-inflammatory activity. Eur. J. Med. Chem. 127, 972–985. doi:10.1016/j.ejmech.2016.11.006
Alfayomy, A. M., Abdel-Aziz, S. A., Marzouk, A. A., Shaykoon, M. S. A., Narumi, A., Konno, H., et al. (2021). Design and synthesis of pyrimidine-5-carbonitrile hybrids as COX-2 inhibitors: anti-inflammatory activity, ulcerogenic liability, histopathological and docking studies. Bioorg Chem. 108, 104555. doi:10.1016/j.bioorg.2020.104555
Apaydın, S., and Török, M. (2019). Sulfonamide derivatives as multi-target agents for complex diseases. Bioorg Med. Chem. Lett. 29 (16), 2042–2050. doi:10.1016/j.bmcl.2019.06.041
Bergqvist, F., Morgenstern, R., and Jakobsson, P.-J. (2020). A review on mPGES-1 inhibitors: from preclinical studies to clinical applications. Prostagl. other lipid Mediat. 147, 106383. doi:10.1016/j.prostaglandins.2019.106383
Cardoso, C. S., Silva, D. P. B., Silva, D. M., Florentino, I. F., Fajemiroye, J. O., Moreira, L. K. S., et al. (2020). Mechanisms involved in the antinociceptive and anti-inflammatory effects of a new triazole derivative: 5-[1-(4-fluorophenyl)-1H-1,2,3-triazol-4-yl]-1H-tetrazole (LQFM-096). Inflammopharmacology 28 (4), 877–892. doi:10.1007/s10787-020-00685-8
Dallakyan, S., and Olson, A. J. (2015). “Small-molecule library screening by docking with PyRx,” in Chemical Biology: methods and protocols. Editors J. E. Hempel, C. H. Williams, and C. C. Hong (New York, NY: Springer New York), 243–250.
Desai, S. B., and Furst, D. E. (2006). Problems encountered during anti-tumour necrosis factor therapy. Best. Pract. Res. Clin. Rheumatol. 20 (4), 757–790. doi:10.1016/j.berh.2006.06.002
Dowarah, J., Patel, D., Marak, B. N., Yadav, U. C. S., Shah, P. K., Shukla, P. K., et al. (2021). Green synthesis, structural analysis and anticancer activity of dihydropyrimidinone derivatives. RSC Adv. 11 (57), 35737–35753. doi:10.1039/d1ra03969e
Eastman, P., Friedrichs, M. S., Chodera, J. D., Radmer, R. J., Bruns, C. M., Ku, J. P., et al. (2013). OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput. 9 (1), 461–469. doi:10.1021/ct300857j
Elbastawesy, M., Youssif, B., Abdelrahman, M. H., and Hayallah, A. (2015). Synthesis and biological evaluation of some new coumarin derivatives as potential antimicrobial, analgesic and anti-inflammatory agents. Der Pharma Chem. 7, 337–349.
Elkady, M., Nieß, R., Schaible, A. M., Bauer, J., Luderer, S., Ambrosi, G., et al. (2012). Modified acidic nonsteroidal anti-inflammatory drugs as dual inhibitors of mPGES-1 and 5-LOX. J. Med. Chem. 55 (20), 8958–8962. doi:10.1021/jm3010543
Fares, M., Eldehna, W. M., Bua, S., Lanzi, C., Lucarini, L., Masini, E., et al. (2020). Discovery of potent dual-tailed benzenesulfonamide inhibitors of human carbonic anhydrases implicated in glaucoma and in vivo profiling of their intraocular pressure-lowering action. J. Med. Chem. 63 (6), 3317–3326. doi:10.1021/acs.jmedchem.9b02090
Fattahi, M. J., and Mirshafiey, A. (2012). Prostaglandins and rheumatoid arthritis. Arthritis. 2012, 1–7. doi:10.1155/2012/239310
Gilbert, N. C., Gerstmeier, J., Schexnaydre, E. E., Börner, F., Garscha, U., Neau, D. B., et al. (2020). Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nat. Chem. Biol. 16 (7), 783–790. doi:10.1038/s41589-020-0544-7
Gürses, T., Olğaç, A., Garscha, U., Gür Maz, T., Bal, N. B., Uludağ, O., et al. (2021). Simple heteroaryl modifications in the 4,5-diarylisoxazol-3-carboxylic acid scaffold favorably modulates the activity as dual mPGES-1/5-LO inhibitors with in vivo efficacy. Bioorg Chem. 112, 104861. doi:10.1016/j.bioorg.2021.104861
Harvanová, G., Duranková, S., and Bernasovská, J. (2023). The role of cytokines and chemokines in the inflammatory response. Alergol. Pol. 10 (3), 210–219. doi:10.5114/pja.2023.131708
Hassan, G. S., Abdel Rahman, D. E., Abdelmajeed, E. A., Refaey, R. H., Alaraby Salem, M., and Nissan, Y. M. (2019). New pyrazole derivatives: synthesis, anti-inflammatory activity, cycloxygenase inhibition assay and evaluation of mPGES. Eur. J. Med. Chem. 171, 332–342. doi:10.1016/j.ejmech.2019.03.052
Hendawy, O., Gomaa, H. A., Alzarea, S. I., Alshammari, M. S., Mohamed, F. A., Mostafa, Y. A., et al. (2021). Novel 1, 5-diaryl pyrazole-3-carboxamides as selective COX-2/sEH inhibitors with analgesic, anti-inflammatory, and lower cardiotoxicity effects. Bioorg Chem. 116, 105302. doi:10.1016/j.bioorg.2021.105302
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph 14 (1), 33–38. doi:10.1016/0263-7855(96)00018-5
Hunter, C. A., and Jones, S. A. (2015). IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16 (5), 448–457. doi:10.1038/ni.3153
Ihsan, E. (2023). Eicosanoids in human physiology: polyunsaturated fatty acid sources, biosynthesis, functions, and therapeutic implications. Int. Anatol. Acad. 9 (2), 117–133.
Janković, N., Trifunović Ristovski, J., Vraneš, M., Tot, A., Petronijević, J., Joksimović, N., et al. (2019). Discovery of the Biginelli hybrids as novel caspase-9 activators in apoptotic machines: lipophilicity, molecular docking study, influence on angiogenesis gene and miR-21 expression levels. Bioorg Chem. 86, 569–582. doi:10.1016/j.bioorg.2019.02.026
Jin, J., Ye, X., Boateng, D., Dai, K., Ye, F., Du, P., et al. (2019). Identification and characterization of potent and selective inhibitors targeting protein tyrosine phosphatase 1B (PTP1B). Bioorg Med. Chem. Lett. 29 (16), 2358–2363. doi:10.1016/j.bmcl.2019.06.011
Jin, Y., Regev, A., Kam, J., Phipps, K., Smith, C., Henck, J., et al. (2018). Dose-dependent acute liver injury with hypersensitivity features in humans due to a novel microsomal prostaglandin E synthase 1 inhibitor. Br. J. Clin. Pharmacol. 84 (1), 179–188. doi:10.1111/bcp.13423
Kim, M., Kim, G., Kang, M., Ko, D., Nam, Y., Moon, C. S., et al. (2021). Discovery of N-amido-phenylsulfonamide derivatives as novel microsomal prostaglandin E(2) synthase-1 (mPGES-1) inhibitors. Bioorg Med. Chem. Lett. 41, 127992. doi:10.1016/j.bmcl.2021.127992
Koeberle, A., Siemoneit, U., Bühring, U., Northoff, H., Laufer, S., Albrecht, W., et al. (2008). Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J. Pharmacol. Exp. Ther. 326 (3), 975–982. doi:10.1124/jpet.108.139444
Koeberle, A., and Werz, O. (2018). Natural products as inhibitors of prostaglandin E(2) and pro-inflammatory 5-lipoxygenase-derived lipid mediator biosynthesis. Biotechnol. Adv. 36 (6), 1709–1723. doi:10.1016/j.biotechadv.2018.02.010
Lauro, G., Strocchia, M., Terracciano, S., Bruno, I., Fischer, K., Pergola, C., et al. (2014). Exploration of the dihydropyrimidine scaffold for the development of new potential anti-inflammatory agents blocking prostaglandin E2 synthase-1 enzyme (mPGES-1). Eur. J. Med. Chem. 80, 407–415. doi:10.1016/j.ejmech.2014.04.061
Li, D., Howe, N., Dukkipati, A., Shah, S. T. A., Bax, B. D., Edge, C., et al. (2014). Crystallizing membrane proteins in the lipidic mesophase. Experience with human prostaglandin E2 synthase 1 and an evolving strategy. Cryst. Growth Des. 14 (4), 2034–2047. doi:10.1021/cg500157x
Lokwani, D., Azad, R., Sarkate, A., Reddanna, P., and Shinde, D. (2015). Structure Based Library Design (SBLD) for new 1,4-dihydropyrimidine scaffold as simultaneous COX-1/COX-2 and 5-LOX inhibitors. Bioorg Med. Chem. 23 (15), 4533–4543. doi:10.1016/j.bmc.2015.06.008
Mahgoub, S., Kotb El-Sayed, M. I., El-Shehry, M. F., Mohamed, A. S., Mansour, Y. E., and Fatahala, S. S. (2021). Synthesis of novel calcium channel blockers with ACE2 inhibition and dual antihypertensive/anti-inflammatory effects: a possible therapeutic tool for COVID-19. Bioorg Chem. 116, 105272. doi:10.1016/j.bioorg.2021.105272
Manivannan, E., and Chaturvedi, S. C. (2011). Analogue-based design, synthesis and molecular docking analysis of 2,3-diaryl quinazolinones as non-ulcerogenic anti-inflammatory agents. Bioorg Med. Chem. 19 (15), 4520–4528. doi:10.1016/j.bmc.2011.06.019
Megha, K., Joseph, X., Akhil, V., and Mohanan, P. (2021). Cascade of immune mechanism and consequences of inflammatory disorders. Phytomedicine 91, 153712. doi:10.1016/j.phymed.2021.153712
Meshram, D., Bhardwaj, K., Rathod, C., Mahady, G. B., and Soni, K. K. (2020). The role of leukotrienes inhibitors in the management of chronic inflammatory diseases. Recent Pat. Inflamm. Allergy Drug Discov. 14 (1), 15–31. doi:10.2174/1872213x14666200130095040
Miller, B. R., McGee, T. D., Swails, J. M., Homeyer, N., Gohlke, H., and Roitberg, A. E. (2012). MMPBSA.py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8 (9), 3314–3321. doi:10.1021/ct300418h
Mohassab, A. M., Hassan, H. A., Abdelhamid, D., Gouda, A. M., Gomaa, H. A., Youssif, B. G., et al. (2021). New quinoline/1, 2, 4-triazole hybrids as dual inhibitors of COX-2/5-LOX and inflammatory cytokines: design, synthesis, and docking study. J. Mol. Struct. 1244, 130948. doi:10.1016/j.molstruc.2021.130948
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30 (16), 2785–2791. doi:10.1002/jcc.21256
O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011). Open Babel: an open chemical toolbox. J. Cheminform 3 (1), 33. doi:10.1186/1758-2946-3-33
Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., et al. (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem. 26 (16), 1781–1802. doi:10.1002/jcc.20289
Placha, D., and Jampilek, J. (2021). Chronic inflammatory diseases, anti-inflammatory agents and their delivery nanosystems. Pharmaceutics 13 (1), 64. doi:10.3390/pharmaceutics13010064
Ribeiro, J. V., Bernardi, R. C., Rudack, T., Schulten, K., and Tajkhorshid, E. (2018). QwikMD - gateway for easy simulation with VMD and NAMD. Biophys. J. 114 (3), 673a–4a. doi:10.1016/j.bpj.2017.11.3632
Roschek, B., Fink, R. C., Li, D., McMichael, M., Tower, C. M., Smith, R. D., et al. (2009). Pro-inflammatory enzymes, cyclooxygenase 1, cyclooxygenase 2, and 5-lipooxygenase, inhibited by stabilized rice bran extracts. J. Med. Food 12 (3), 615–623. doi:10.1089/jmf.2008.0133
Sant, S., Tandon, M., Menon, V., Gudi, G., Kattige, V., Khairatkar, J. N., et al. (2018). GRC 27864, novel, microsomal prostaglandin E synthase-1 enzyme inhibitor: phase 1 study to evaluate safety, PK and biomarkers in healthy, adult subjects. Osteoarthr. Cartil. 26, S351–S352. doi:10.1016/j.joca.2018.02.698
Seeliger, D., and de Groot, B. L. (2010). Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 24 (5), 417–422. doi:10.1007/s10822-010-9352-6
Shawky, A. M., Almalki, F. A., Abdalla, A. N., Youssif, B. G., Abdel-Fattah, M. M., Hersi, F., et al. (2023). Discovery and optimization of 2, 3-diaryl-1, 3-thiazolidin-4-one-based derivatives as potent and selective cytotoxic agents with anti-inflammatory activity. Eur. J. Med. Chem. 259, 115712. doi:10.1016/j.ejmech.2023.115712
Sinha, S., Doble, M., and Manju, S. L. (2019). 5-Lipoxygenase as a drug target: a review on trends in inhibitors structural design, SAR and mechanism based approach. Bioorg Med. Chem. 27 (17), 3745–3759. doi:10.1016/j.bmc.2019.06.040
Spunde, K., Vigante, B., Dubova, U. N., Sipola, A., Timofejeva, I., Zajakina, A., et al. (2022). Design and synthesis of hepatitis B virus (HBV) capsid assembly modulators and evaluation of their activity in mammalian cell model. Pharm. (Basel) 15 (7), 773. doi:10.3390/ph15070773
Terracciano, S., Lauro, G., Strocchia, M., Fischer, K., Werz, O., Riccio, R., et al. (2015). Structural insights for the optimization of dihydropyrimidin-2(1H)-one based mPGES-1 inhibitors. ACS Med. Chem. Lett. 6 (2), 187–191. doi:10.1021/ml500433j
Venugopala, K. N., Govender, R., Khedr, M. A., Venugopala, R., Aldhubiab, B. E., Harsha, S., et al. (2015). Design, synthesis, and computational studies on dihydropyrimidine scaffolds as potential lipoxygenase inhibitors and cancer chemopreventive agents. Drug Des. Devel Ther. 9, 911–921. doi:10.2147/dddt.s73890
Vyas, A., Sahu, B., Pathania, S., Nandi, N. K., Chauhan, G., Asati, V., et al. (2023). An insight on medicinal attributes of pyrimidine scaffold: an updated review. J. Heterocycl. Chem. 60 (7), 1081–1121. doi:10.1002/jhet.4593
Wenzel, S. E., and Kamada, A. K. (1996). Zileuton: the first 5-lipoxygenase inhibitor for the treatment of asthma. Ann. Pharmacother. 30 (7-8), 858–864. doi:10.1177/106002809603000725
Winter, C. A., Risley, E. A., and Nuss, G. W. (1962). Carrageenin-induced edema in hind paw of the rat as an assay for antiinflammatory drugs. Proc. Soc. Exp. Biol. Med. 111, 544–547. doi:10.3181/00379727-111-27849
Yousif, N. M. M., El-Gazzar, B. A. A.-R., Hafez, N. H., Fayed, A. A., ElRashedy, A., and Yousif, M. N. (2022). Recent advances in the chemistry and biological activity of sulfonamide derivatives. Mini-Rev Org. Chem. 19 (6), 695–707. doi:10.2174/1570193x19666220105145504
Youssif, B. G., Mohamed, M. F., Al-Sanea, M. M., Moustafa, A. H., Abdelhamid, A. A., and Gomaa, H. A. (2019). Novel aryl carboximidamide and 3-aryl-1, 2, 4-oxadiazole analogues of naproxen as dual selective COX-2/15-LOX inhibitors: design, synthesis and docking studies. Bioorg Chem. 85, 577–584. doi:10.1016/j.bioorg.2019.02.043
Zhang, Y.-Y., Yao, Y.-D., Luo, J.-F., Liu, Z.-Q., Huang, Y.-M., Wu, F.-C., et al. (2022). Microsomal prostaglandin E2 synthase-1 and its inhibitors: molecular mechanisms and therapeutic significance. Pharmacol. Res. 175, 105977. doi:10.1016/j.phrs.2021.105977
Keywords: pyrimidine, sulphonamide, inflammation, prostaglandin, lipoxygenase
Citation: Al-Wahaibi LH, Elshamsy AM, Ali TFS, Youssif BGM, Bräse S, Abdel-Aziz M and El-Koussi NA (2024) Design and synthesis of new dihydropyrimidine/sulphonamide hybrids as promising anti-inflammatory agents via dual mPGES-1/5-LOX inhibition. Front. Chem. 12:1387923. doi: 10.3389/fchem.2024.1387923
Received: 18 February 2024; Accepted: 25 April 2024;
Published: 10 May 2024.
Edited by:
Kai Lv, Chinese Academy of Medical Sciences, ChinaReviewed by:
Gianluigi Lauro, University of Salerno, ItalyCopyright © 2024 Al-Wahaibi, Elshamsy, Ali, Youssif, Bräse, Abdel-Aziz and El-Koussi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bahaa G. M. Youssif, Ymd5b3Vzc2lmMkBnbWFpbC5jb20=; S. Bräse, YnJhZXNlQGtpdC5lZHU=; Mohamed Abdel-Aziz, YWJ1bG5pbEBob3RtYWlsLmNvbQ==; Nawal A. El-Koussi, bmF3YWwuYWJkZWxoYWxpbUBkZXJheWEuZWR1LmVn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.