 Muhammad Akash
Muhammad Akash Sumera Zaib
Sumera Zaib Matloob Ahmad
Matloob Ahmad Sadia Sultan3,4
Sadia Sultan3,4- 1Department of Chemistry, Government College University Faisalabad, Faisalabad, Pakistan
- 2Department of Basic and Applied Chemistry, Faculty of Science and Technology, University of Central Punjab, Lahore, Pakistan
- 3Faculty of Pharmacy, Universiti Teknologi MARA, Puncak Alam, Selangor Darul Ehsan, Malaysia
- 4Atta-ur-Rahman Institute for Natural Products Discovery (AuRIns), Universiti Teknologi MARA, Puncak Alam, Selangor Darul Ehsan, Malaysia
- 5Department of Chemistry, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh, Saudi Arabia
Urease, a nickel-dependent enzyme found in various life forms, catalyzes urea breakdown, concluding nitrogen metabolism by generating ammonia and carbamate. This process causes a rise in pH, supports the survival of pathogens, and can lead to infections such as gastric disorders like ulcers and cancer in humans. Helicobacter pylori employs urease for survival in the acidic environment of the stomach and in protein synthesis. To treat such infections and inhibit the growth of pathogens, it is mandatory to obstruct urease activity; therefore, derivatives of 1-(3-nitropyridin-2-yl)piperazine were synthesized (5a-o; 7a-k). All these newly synthesized compounds were investigated for urease inhibition by in vitro inhibition assays. The results showed that 5b and 7e are the most active inhibitors, having IC50 values of 2.0 ± 0.73 and 2.24 ± 1.63 µM, respectively. These IC50 values are lower than the IC50 value of the standard thiourea, which was 23.2 ± 11.0 µM. The hemolysis potential of 5b, 5c, 5i, 7e, and 7h was also determined; 7e and 7h exhibited good biocompatibility in human blood cells. Through in silico analysis, it was shown that both these potent inhibitors develop favorable interactions with the active site of urease, having binding energies of −8.0 (5b) and −8.1 (7e) kcal/mol. The binding energy of thiourea was −2.8 kcal/mol. Moreover, 5b and 7e have high gastrointestinal permeability as predicted via computational analysis. On the other hand, the IC50 value and binding energy of precursor compound 3 was 3.90 ± 1.91 µM and −6.1 kcal/mol, respectively. Consequently, 5b and 7e can serve as important inhibitors of urease.
1 Introduction
Urease (urea amidohydrolase E.C. 3.5.1.5) is a nickel-dependent enzyme prevalent across diverse life forms, encompassing animals, plants, fungi, and bacteria (Mazzei et al., 2020). It facilitates the breakdown of urea into ammonia and carbamate, which is a biochemical process marking the end stage of nitrogen metabolism (Naseer et al., 2022; Lin et al., 2012). It results in the alkylation effect (rise in pH); hence, it supports the survival of numerous pathogenic microorganisms in the host body (Konieczna et al., 2012; Maier and Benoit, 2019; Ahmad et al., 2023). In humans, gastrointestinal infections are mostly caused by the alkaline pH, which leads to significant complications such as gastric ulcers and gastric cancer (Sohrabi et al., 2022). Pathogen-mediated gastric disorders are mainly attributed to Helicobacter pylori, which is a Gram-negative bacterium (Sukri et al., 2023). This bacterium utilizes urease activity not only to survive in the acidic environment of the stomach but also to synthesize proteins (Sohrabi et al., 2022). Owing to this significance, urease represents nearly 10% of the total content of proteins (Mazzei et al., 2020). In addition to H. pylori, urease activity is also crucial for the survival of Staphylococcus aureus (Zhou et al., 2019), Mycobacterium tuberculosis, Yersinia enterocolitica (Righetto et al., 2020), and Cryptococcus neoformans, which cause kidney diseases (rodents), tuberculosis (humans), yersiniosis (humans) (Bhagat and Virdi, 2009), and cryptococcosis (humans) (Cox et al., 2000), respectively. Therefore, urease is a significant target to limit the survival of various pathogenic organisms. However, in this research paper, our core focus is to discover potential urease inhibitors that will help limit the survival of these pathogens.
The structure of urease is made up of four domains: the N-terminal αβ domain; αβ domain; β domain; and (αβ)8 TIM barrel domain, which consists of an active site and flap region. The active site encompasses two nickel atoms that are spaced apart by a distance of approximately 4 Å (Balasubramanian and Ponnuraj, 2010). One nickel interacts with a terminal water molecule and two histidine residues; the second nickel atom interacts with aspartate, two histidine atoms, and a solvent molecule. In addition, the active site binds with the substrate and initiates catalysis due to the presence of two histidine residues in it (Carter et al., 2009). Several inhibitors have been discovered such as sulphamethazine, sulphamethoxazole (Hamad et al., 2020), hydroxamic acids (Liu et al., 2018), benzimidazole (Zaman et al., 2019), bis-indole, and quinazoline-4(3H)-one (Uddin et al., 2020). Nevertheless, heterocycles with nitrogen atoms like pyridine and piperazine are very interesting due to their increased pharmacological activities (Moghadam et al., 2022; Naseer et al., 2022). Apart from pharmaceutical roles, inhibition of urease in agriculture is also essential to limit ammonia volatilization in the environment. Urease inhibitors play a mandatory role in prolonging the urea hydrolysis process, diminishing nitrogen (N) losses, and enhancing bioavailability (Matczuk and Siczek, 2021).
Literature studies revealed that pyrido-N-substituted piperazines are key scaffolds for various enzymatic inhibitions (Swanson et al., 2005; Kumar et al., 2014). Pyridine is a backbone in the skeleton of marketed drugs (Jayasinghe et al., 2003; Chaves et al., 2021) (Figure 1). Piperazine is also a key ingredient for various biologically active scaffolds. They are core fragments of a variety of drugs (Campoli-Richards et al., 1988; Croxtall, 2012) (Figure 2). In recent literature, pyridine carbothioamide (Naseer et al., 2022) and piperazine-based semicarbazone (Moghadam et al., 2022) were found to be effective urease inhibitors (Figure 3). The coupling of two or more pharmacologically important scaffolds is a diverse approach that can produce hybrid molecules with enhanced biological activity. In continuation of our previous research work dealing with the development of various enzyme inhibitors, for example, α-amylase and α-glucosidase inhibitors (Khan et al., 2021; Khan et al., 2022; Saddique et al., 2022), we herein report a novel series of pyridine-based piperazines as potent urease inhibitors.
FIGURE 1. Structures of pyridine-based drugs.

FIGURE 2. Structures of piperazine-based drugs.
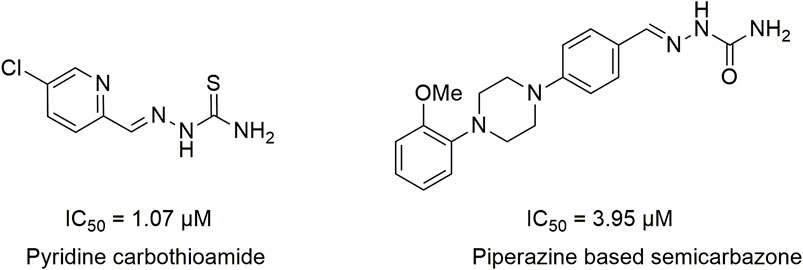
FIGURE 3. Structures of pyridine carbothioamides and piperazine-based semicarbazones as potent urease inhibitors.
2 Results and discussion
2.1 Chemistry
Several 2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)-N-arylacetamide derivatives 5a-5o and N-aryl-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide derivatives 7a-7k were synthesized, as shown in Scheme 1. The reaction of 2-chloro-3-nitropyridine (1) with excess piperazine (2) under reflux for 12 h in acetonitrile produced pyridinylpiperazine (3) in 65% yield. The presence of a nitro group at the 3-position withdraws the electron at the 2-position, making it a strong electrophilic center that facilitates the attack of the nucleophilic nitrogen atom of piperazine under nucleophilic aromatic substitution reaction. The reaction of piperazine (3) and 2-chloro-N-arylacetamides 4a-4o in the presence of potassium carbonate in acetonitrile under reflux for 18–36 h afforded compounds 5a-5o in 50%–70% yield. Similarly, piperazine (3) and 2-chloro-N-arylpropanamides 6a-6k reacted in the presence of potassium carbonate in acetonitrile under reflux for 24–48 h furnished compounds 7a-7k in 30%–55% yield. The compounds 4a-4o and 6a-6k were accessible from already reported methodologies (Cormier et al., 2012; Lavorato et al., 2017; Faiz et al., 2019; Abdel-Latif et al., 2020). All the synthesized compounds, 3, 5a-5o, and 7a-7k, were purified by column chromatography and structures were established using spectroscopic methodologies. The HRMS justifies the chemical formula of each compound in terms of [M]+, [M+H]+, and [M+2H]+2 peaks. The nitrogenous heterocyclic compounds having basic nitrogen (nitrogen with available lone-pair) may protonate, causing peaks greater than [M+H]+ like [M+2H]+ (Joyce and Richards, 2011). The 1HNMR spectra of 3, 5a-5o, and 7a-7k revealed the presence of methylene as well as all aromatic protons. Similarly, 13CNMR spectra of 3, 5a-5o, and 7a-7k revealed the presence of C=O in all compounds. The FT-IR spectra of 3, 5a-5o, and 7a-7k include the peaks of functionalities like NH (3,343–3,185 cm−1), C=O (1,698–1,664), NO2 (1,597–1,586 cm−1), and CH2 (1,513–1,481 cm−1), which is comparable with literature (Krishnakumar and Muthunatesan, 2006; George et al., 2018; Crocker et al., 2022).
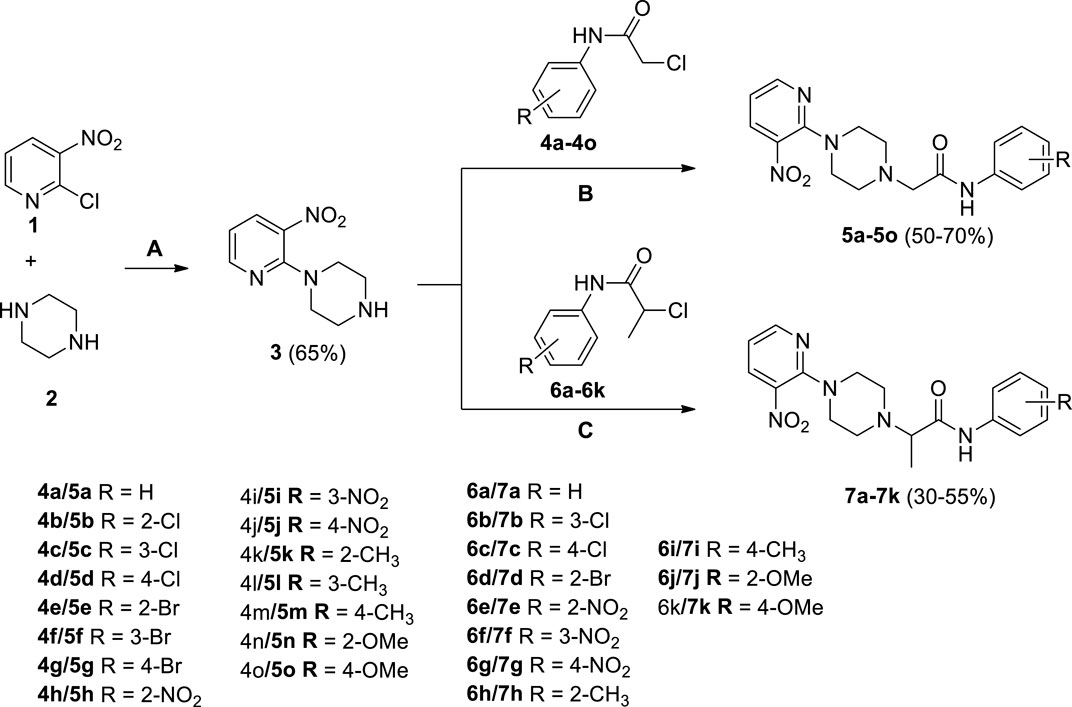
SCHEME 1. Synthesis of 2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)-N-arylacetamide derivatives 5a-5o and N-aryl-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide derivatives 7a-7k. Reagents and conditions: (A) acetonitrile, reflux, 12 h (B) K2CO3, acetonitrile, reflux, 18–36 h (C) K2CO3, acetonitrile, reflux, 24–48 h.
2.2 Evaluation of structure-activity relationship
The inhibitory potential of piperazine (3), 1-(3-nitropyridin-2-yl)piperazine derivatives (5a-o and 7a-k) and standard (thiourea) against jack bean urease was determined by the indophenol method, as shown in Table 1. The thiourea was selected as a positive standard because it has been well-characterized and well-documented as a standard inhibitor of urease in literature. According to the results elucidated in Table 1, all the compounds showed good inhibitory activities against urease, with their IC50 values ranging between 2.0 ± 0.73 μM and 14.12 ± 0.67 μM. Moreover, piperazine (3) and thiourea have IC50 values of 3.90 ± 1.91 and 23.2 ± 11.0 μM, respectively.
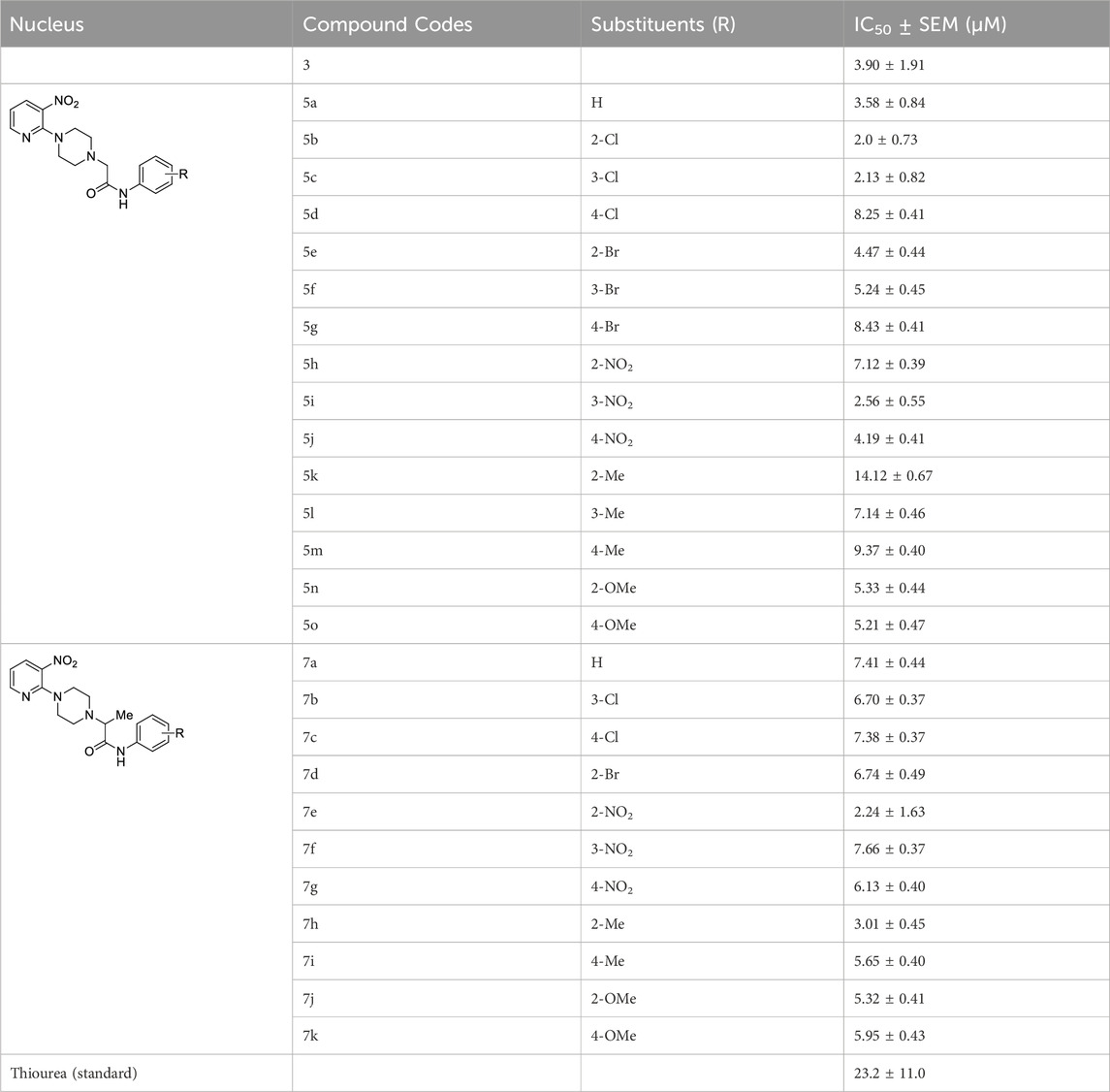
TABLE 1. The IC50 values of all the compounds have been elucidated in tabular form.
1-(3-Nitropyridin-2-yl)piperazine is the static motif in all the test compounds, which were further diversified by the attachment of N-phenylpropionamide and N-phenylisobutyramide. This diversification resulted in two sets of compounds: the first set of compounds (5a-o) contain 2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)-N-arylacetamide nucleus (nucleus A) while the second set of compounds (7a-k) contain N-aryl-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide nucleus (nucleus B). 5b and 5c in the first set are potent inhibitors because they have the lowest and comparable IC50 values, which are 2.13 ± 0.82 µM and 2.0 ± 0.73 µM, respectively. This ideal activity of 5b is attributed to the presence of an electron-withdrawing group (EWG) chlorine (Cl) at ortho-position (ortho-position) of the aryl group of N-phenylpropionamide. The urease inhibitory activity has a tendency to decrease (IC50 = 4.47 ± 0.44 µM) when Cl is substituted with another halogen bromine (Br) (5e). This decrease in activity is because of the decrease in electronegativity and the increase in size of Br. In addition, attachment of another electrophilic group, NO2 (5h), causes a decrease in the urease inhibitory activity (IC50 = 7.12 ± 0.39 µM). Similarly, the presence of methyl (5k) and methoxy (5n) also resulted in the decreased inhibitory potential of nucleus A against urease due to their electron-donating nature.
The aryl group of nucleus A was also substituted at meta-position with electron-donating groups (EDGs) and EWGs. In the absence of any substitution at the aryl group (5a), the inhibitory activity of nucleus A against urease was observed to be 3.58 ± 0.84 µM. The urease inhibitory potential of 5c and 5i were comparable due to the presence of electrophilic groups, namely, Cl (IC50 = 2.13 ± 0.82 µM) and NO2 (IC50 = 2.56 ± 0.55 µM) at meta-positions, accordingly. Contrarily, a decrease in inhibitory potential, with an IC50 value of 5.24 ± 0.45 µM, was observed when Br was substituted at the meta-position of the aryl group (5f). The substitution of EDG such as methyl at the meta-position of aryl group (5l) resulted in the inactivation of nucleus A (IC50 = 7.14 ± 0.46 µM).
The substitution of EDGs and EWGs at para-position of the aryl group of nucleus A did not result in potent inhibitors of the urease enzyme. 5d with Cl substitution had an IC50 value of 8.25 ± 0.41 µM, which increased by replacing Cl with another EWG, namely, NO2 (IC50 = 4.19 ± 0.41 µM) (5j). Contrarily, substituting Cl with Br (5g) did not significantly alter the inhibitory potential of nucleus A (IC50 = 8.43 ± 0.41 µM). The presence of methyl group (5m) inactivated nucleus A (IC50 = 9.37 ± 0.40 µM); on the other hand, the substitution of methoxy at para-position (5o) increased the urease inhibitory potential of nucleus A (IC50 = 5.21 ± 0.47 µM).
In the case of nucleus B, the substitution of the aryl group at ortho-position increased the urease inhibitory potential of 7a, 7d, 7e, 7h, and 7j. An amplification tendency in the inhibitory activity of nucleus B (IC50 = 6.74 ± 0.49 µM) was observed upon substitution with an EWG, namely, Br at ortho-position (7d) as compared to 7a (IC50 = 7.41 ± 0.44 µM). The addition of ortho-NO2 (7e) or ortho-methyl (7h) resulted in a increased inhibitory activity of nucleus B against urease [IC50 = 4.19 ± 0.41 µM (7e); IC50 = 3.01 ± 0.45 µM (7h)]. Similarly, the presence of methoxy substituent at ortho-position (7j), also enhanced the inhibition of nucleus B (IC50 = 5.32 ± 0.41 µM).
Compounds 7c, 7g, 7i, and 7k are para-substituted variants of nucleus B, and their IC50 ranged between 5.65 and 7.38 µM. The urease inhibitory potential of EWGs Cl (7c) and NO2 (7g) were 7.38 ± 0.37 µM and 6.13 ± 0.40 µM, respectively. The inhibitory activity of nuclease B was further increased when methyl (7i) and methoxy (7k) substituents were introduced at the ortho-position of the aryl group. The IC50 value of 7i was 5.65 ± 0.40 µM, while the IC50 value of 7k was 5.95 ± 0.43 µM. Only two meta-substituted variants at the aryl group of nucleus B were obtained; it is quite interesting to know that both these substituents are EWGs: Cl-substituted 7b (IC50 = 6.70 ± 0.37 µM) and NO2-substituted 7f (IC50 = 7.66 ± 0.37 µM) (Figure 4).
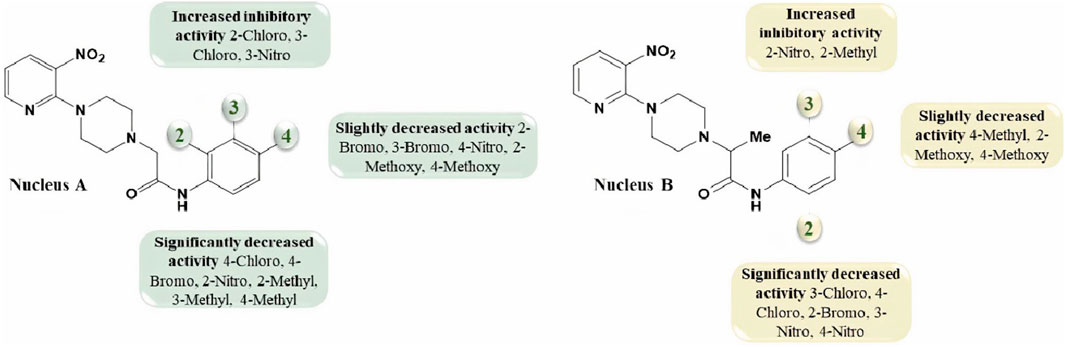
FIGURE 4. The variations in the urease inhibitory activity of nucleus A and B due to different substituents.
2.3 Hemolysis potential
1-(3-nitropyridin-2-yl)piperazine 3 and its derivatives (5b, 5c, 5i; 7e, 7h) were analyzed for hemolysis activity due to their remarkable urease inhibition potential. Results revealed that, out of all screened compounds, 7e and 7h exhibited good biocompatibility in human blood cells depending upon hemolysis percentage at 0.5 mg/mL and 1 mg/mL nearer to that of PBS. These results showed that compounds 7e and 7h are favorable for injection at low concentrations due to good biocompatibility and the results were found statistically significant after the application of one-way ANOVA as the value of p was found to be less than 0.05. Experimental data on hemolysis percentage have been depicted in Figure 5.
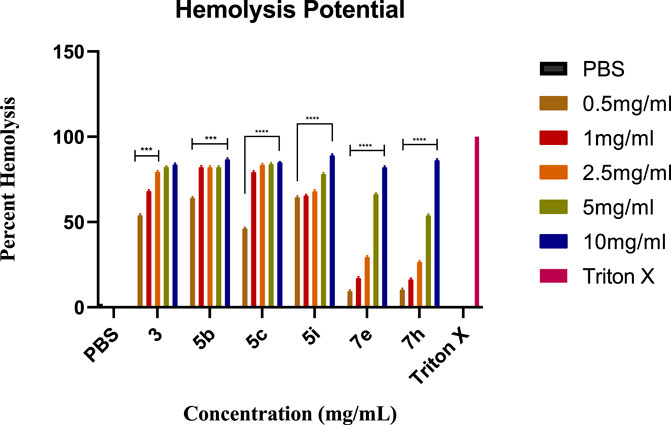
FIGURE 5. The graph represents the comparison of hemolysis activity of Triton X-100, PBS, 3, 5b, 5c, 5i, 7e, and 7h at different concentrations.
2.4 ADMET investigation
admetSAR, ProTox-II, and eMolTox were used for the in silico pre-clinical evaluation of the precursor compound 3 and the most potent compounds, which were selected based on the lowest IC50. 5b was chosen from the first set of compounds while 7e was scrutinized from the second set of compounds to investigate their druggable properties. The ADMET evaluation of compound 3 predicted that it follows Lipinski’s rule of five, having a molecular weight of 208.096 g/mol, five hydrogen bond acceptors, one hydrogen bond donor, two rotatable bonds, and a topological polar surface area (TPSA) of 71.3 Å2. Moreover, it has a probability of 0.9693 for crossing the blood-brain barrier(BBB). Compound 3 is also a non-inhibitor and non-substrate of cytochrome P450 (CYP) enzymes in the hepatic tissue, except for CYP450 1A2, as shown in Table 2. The toxic substructure prediction interpreted that compound 3 may produce toxicophores (Supplementary Table S1). Moreover, acute rat toxicity can be reduced in vivo trials by administering the dose via oral route because this route has the highest LD50 value of 332.2 mg/kg and belongs to predicted toxicity class 4 (Supplementary Table S2).
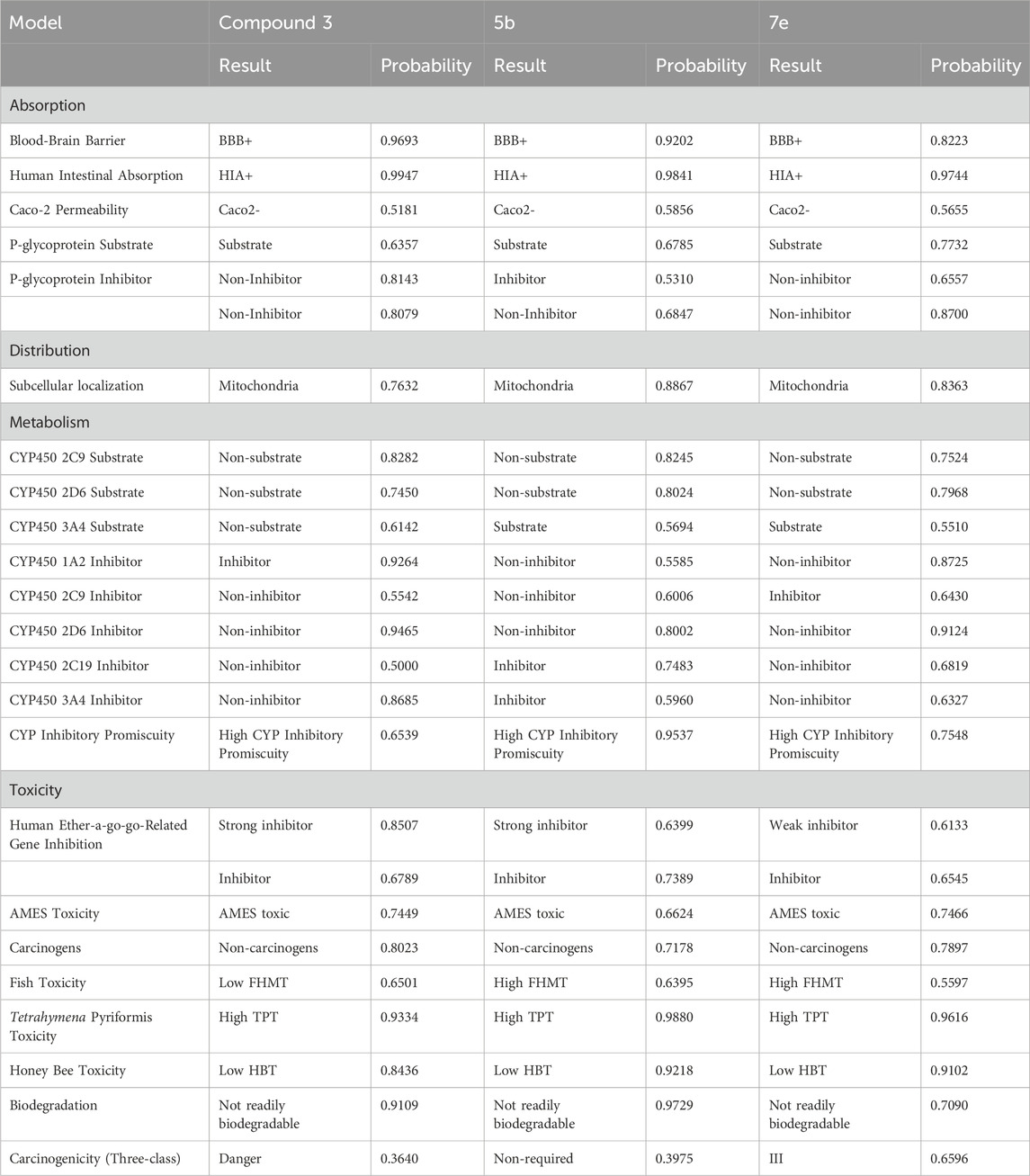
TABLE 2. The pre-clinical investigation of 3, 5b, and 7e.
The results obtained via eMolTox revealed that 5b has a molecular weight of 375.11 g/mol, five rotatable bonds, six hydrogen bond acceptors, and one hydrogen bond donor. Moreover, the probability of 5b penetrating the brain is 0.9202, and easily crosses the gastrointestinal lining with a probability of 0.9841, as shown in Table 2. Its topological polar surface area is 91.61 Å2 and LogP is 2.404. In addition, it may be toxic to the nervous system, being a modulator of P2X purinoceptor 7, and can also produce toxicophores (substructures) (Supplementary Table S1). Furthermore, GUSAR analysis predicted that 5b can be administered to rats via the subcutaneous route because this route has the highest LD50 (1,360 mg/kg) with a predicted toxicity class 5 (Supplementary Table S2).
Pre-clinical analysis of the most potent inhibitor of the second set of compounds predicted that 7e has six rotatable bonds, 29 heavy atoms, 12 aromatic heavy atoms, 134.75 Å2 TPSA, and 2.047 LogP. It belongs to the soluble class of compounds and has high gastrointestinal absorption with a probability of 0.9744. In addition, the blood-brain barrier penetration probability is 0.8223 and it is a non-carcinogenic and non-inhibitor of renal organic cation transporter and P-glycoprotein (P-gp), as represented in Table 2. Apart from the ADMET analysis, the toxicophores were also predicted via eMolTox, which includes substructures having the ability to covalently bind with protein and DNA. Moreover, toxic substructures may also be produced (Supplementary Table S1). Acute rat toxicity was predicted via GUSAR, which showed that oral administration of 7e to rats will be non-toxic because of the highest LD50 value (911.1 mg/kg) (Supplementary Table S2).
Although compounds 5b and 7e are the lead inhibitors of urease in this research, their gastrointestinal absorption determined by in silico analysis needs to be limited. This can be reduced by selecting the best drug delivery system, such as nanoparticles (Gupta et al., 2023).
2.5 Molecular docking studies
The molecular docking of the most potent inhibitors of both sets of compounds was performed by AutoDockTools 1.5.7 and SeeSAR version 12.1.0. The most druggable binding site of the urease (PDB id: 4H9M) was identified via the binding site mode of SeeSAR and has a DoGSiteScore of 0.57. After docking by AutoDockTools 1.5.7, nine poses for each of the potent inhibitors were generated and the best poses with the lowest binding energy were selected for the visualization of intermolecular interactions. The binding energy of the best pose of precursor compound 3, 5b, 7e, and thiourea was −6.1, −8.0, −8.1, and −2.8 kcal/mol, respectively, as shown in Table 3.
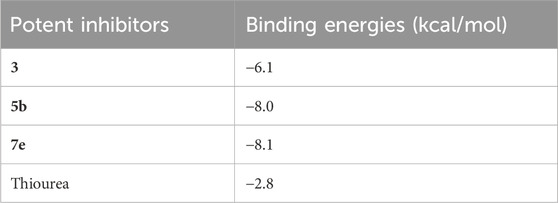
TABLE 3. The binding energies of the best-docked poses of 5b and 7e with urease.
The intermolecular interactions of 3, 5b, and 7e were visualized by BIOVIA Discovery Studio molecular visualizer 2021. According to the results, compound 3 develops hydrogen and hydrophobic interactions with the binding site residues of urease. Lys716 forms two conventional hydrogen interactions: one with O8 (2.99 Å) and the other with O9 (2.95 Å) of compound 3. Similarly, Thr33 also forms conventional hydrogen interactions with the O8 (3.05 Å) of the ligand. Compound 3 also interacts with the Glu742 and Val744 by forming carbon hydrogen interactions. The pyridine ring of compound 3 forms a π-donor hydrogen and two π-alkyl interactions with Tyr32 (4.13 Å), Val36 (4.76 Å) and Val744 (4.81 Å) of urease. Additionally, Val744 also develops alkyl interaction with the piperazine ring (5.03 Å) of compound 3, as represented in Figure 6.
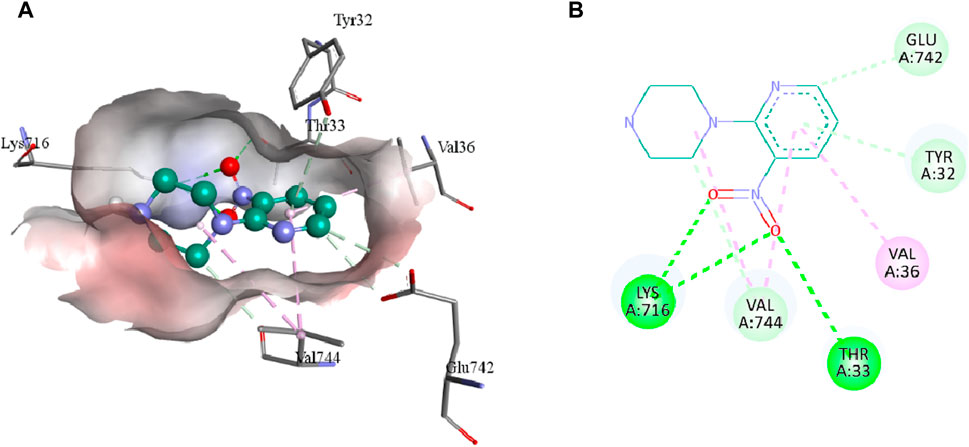
FIGURE 6. The 3D (A) and 2D (B) representation of molecular interactions of compound 3 with the binding site residues of urease. Compound 3 is represented by a green color while the dotted lines are showing the molecular interactions. Green: conventional hydrogen interaction; Light blue: carbon hydrogen interaction; Pink: hydrophobic interaction.
5b interacts with the active site residues of urease by developing alkyl, π-alkyl, π-donor hydrogen interactions, carbon hydrogen interactions, and conventional hydrogen interactions (Table 4). Lys653, Ala656, and Ala828 interact by π-alkyl interactions (5.47 Å; 4.04 Å; 4.72 Å) with the aromatic ring of 5b. Another π-alkyl interaction exists between Arg835 of the active pocket and the pyridine ring. The alkyl interaction is formed by Pro832 with the piperazine ring (5.01 Å) which also develops carbon hydrogen bonds with Asp295 (3.69 Å), Thr830 (3.69 Å), and Val831 (3.43 Å). In addition, the formation of conventional hydrogen interactions with the 5b atoms is attributed to the presence of Arg132 (3.27 Å), Ser834 (3.21 Å), Arg835 (2.91 Å) and Asn836 (3.06 Å). Asn836 (3.50 Å) also develops a carbon hydrogen interaction with the pyridine ring of the ligand, as indicated in Figure 7.
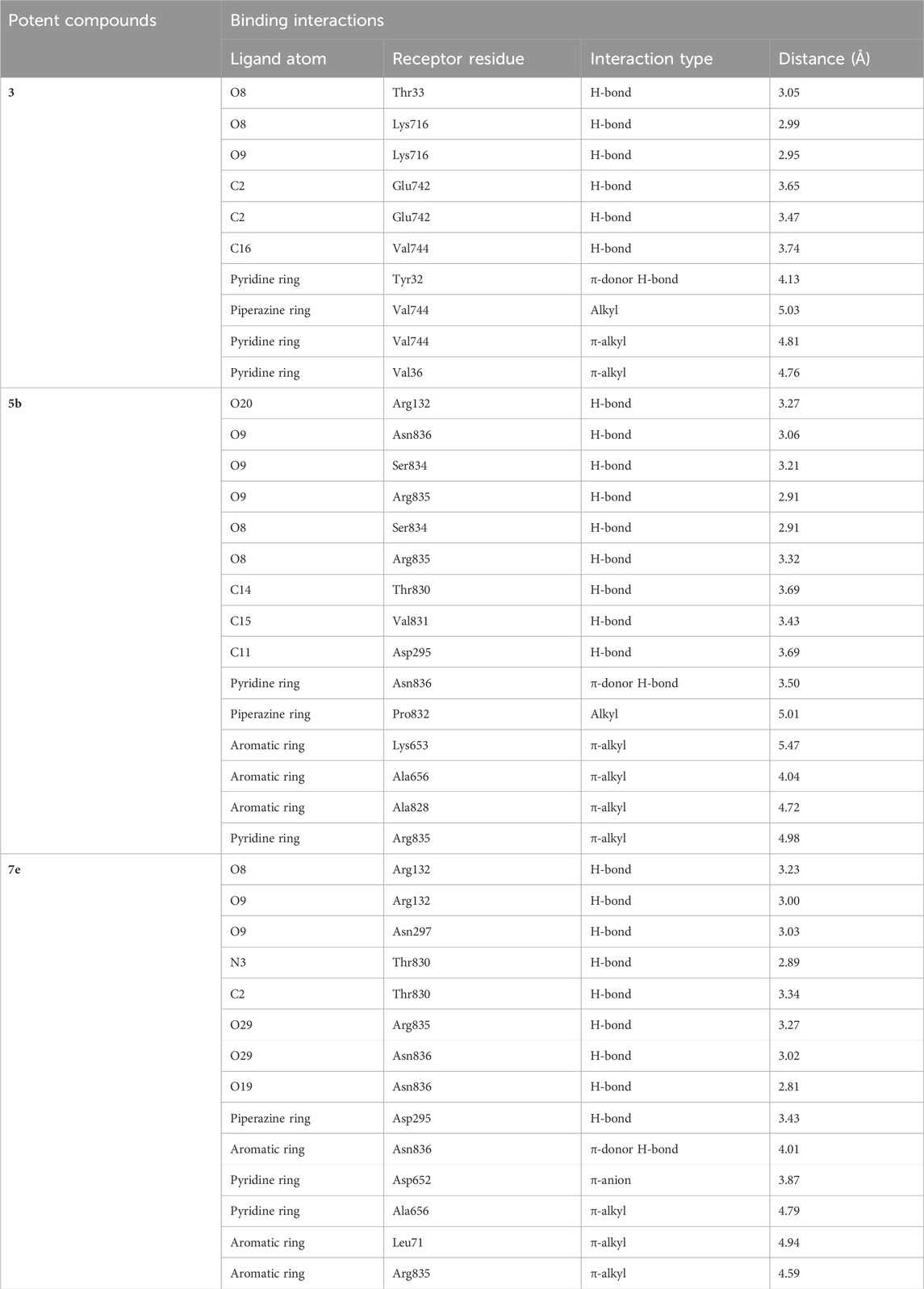
TABLE 4. The detailed description of intermolecular interactions between urease residues and compound 3 and potent ligands 5b and 7e along with their bond lengths.
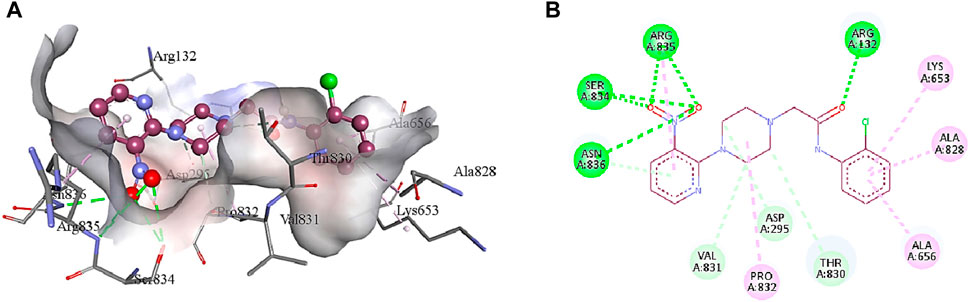
FIGURE 7. The interactions between the binding site of urease and 5b can be visualized in 3D (A) and 2D (B) format. Dotted lines of various colors indicated these intermolecular interactions. Green represents conventional hydrogen bonds; light blue indicates carbon hydrogen bonds; pink highlights the alkyl and π-alkyl bonds.
Compound 7e forms the conventional hydrogen, π-alkyl, π-anion, π-donor hydrogen, and carbon hydrogen interactions with the residues of the active pocket of urease (Table 4). Both the NO2 groups develop conventional hydrogen interactions with the Arg132 (3.23 Å), Asn297 (3.03 Å), Arg835 (3.27 Å), and Asn836 (3.02 Å); Asp652 forms π-anion interaction (3.87 Å) with the pyridine ring of 7e. Another conventional hydrogen interaction is formed by the Thr830 with the nitrogen of the pyridine ring (2.89 Å), with which Thr830 also interacts by carbon hydrogen interaction (3.34 Å). The same pyridine ring of 7e also develops π-alkyl interaction with the Ala656 (4.79 Å) of the urease active site. Additionally, Leu71 and Asn836 interact with the aromatic ring of 7e via π-alkyl interactions. Lastly, Asn295 forms a carbon hydrogen interaction with the piperazine ring of the ligand (3.43 Å), as indicated in Figure 8.
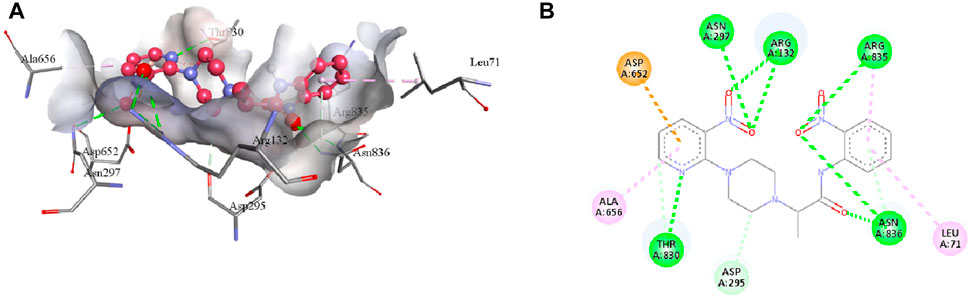
FIGURE 8. The 3D (A) and 2D (B) visualization of molecular interactions between 7e and the active site residues of urease. These intermolecular interactions are indicated by dotted lines of varying colors. Green represents conventional hydrogen bonds; light blue indicates carbon hydrogen and π-donor hydrogen bonds; pink highlights the π-alkyl bonds; orange is an indicator of π-anion interaction.
3 Materials and methods
3.1 General
All the chemicals, reagents, and solvents were purchased from Alfa Aesar (Kandal, Germany) and were utilized without any further purification. The 1HNMR (400 MHz) and 13CNMR (100 MHz) were recorded in dimethyl sulfoxide (DMSO) Bruker DPX spectrophotometer (Bruker; Zürich, Switzerland). The chemical shifts were recorded in ppm reference to tetramethylsilane. The HRMS of all the synthesized compounds were recorded with LCMS/MS Q-TOF (Agilent Technologies 6520, Senta Clara, Ca, United States). Thin-layer chromatography (CHCl3/MeOH) was used in combination with a Spectroline E-Series UV lamp to monitor the progress of chemical reactions (Alfa Aesar, Kandal, Germany). Compounds 3, 5a-5o, and 7a-k were produced according to Scheme 1.
3.2 Procedure for the synthesis of 1-(3-nitropyridin-2-yl)piperazine (3)
40.5 g (472 mmol) of piperazine (2) was dissolved in 100 mL of acetonitrile on stirring. A solution of 15 g (94.30 mmol) of 2-chloro-3-nitropyridine (1) in 50 mL of acetonitrile was added to it. The mixture was refluxed for 12 h on constant stirring. The reaction was monitored every hour with the help of TLC. The yellow solid (3) produced was extracted with chloroform and purified by column chromatography using silica (0.040–0.063 mm) in chloroform/methanol (90:10) with 65% (12.76 g) yield.
3.2.1 1-(3-Nitropyridin-2-yl)piperazine (3)
Yield 65%; Brownish-yellow solid; m.p. 77°C–79°C. IR: ν (cm−1): 3,174 (NH), 1,590 (NO2), 1,556 (C=C), 1,435 (C=N), 1,230 (C-N). 1H NMR (400 MHz) δ (ppm): 8.29–8.30 (m, 1H, Ar-H), 8.15 (dd, 1H, J = 7.95 Hz, 1.8 Hz, Ar-H), 6.82-6.84 (m, 1H, Ar-H), 3.23–3.25 (m, 4H, piperazinyl), 2.71–2.73 (m, 4H, piperazinyl). 13C NMR (100 MHz) δ (ppm): 152.7, 152.5, 136.4, 132.8, 114.3, 48.7 (2C), 45.0(2C). HRMS: m/z [M+2H]+2 calculated: 210.1106; found: 210.2011.
3.3 Procedure for the synthesis of 2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)-N-arylacetamide derivatives 5a-5o
A solution of 2-chloro-N-arylacetamides 4a-4o (0.15 mmol) was prepared in 10 mL of acetonitrile and added to the stirred mixture of 1-(3-nitropyridin-2-yl)piperazine (3) (0.15 mmol) and potassium carbonate (0.3 mmol) in 15 mL of acetonitrile. The reaction mixture was refluxed for 18–36 h on stirring by monitoring with TLC. On the addition of water, colored (yellow to red) precipitates of 2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)-N-arylacetamide derivatives 5a-5o were formed. The produced solid was collected and purified by column chromatography. The calculated yield was found to be 50%–70%.
3.3.1 2-(4-(3-Nitropyridin-2-yl)piperazin-1-yl)-N-phenylacetamide (5a)
Yield 64%; Orange solid; m.p. 79°C–80°C. IR: ν (cm−1): 3,301 (NH), 1,677 (C=O), 1,589 (NO2), 1,552 (C=C), 1,500 (CH2), 1,436 (C=N), 1,237 (C-N). 1H NMR (400 MHz) δ (ppm): 9.86 (s, 1H, NH), 8.31–8.33 (m, 1H, Ar-H), 8.18 (dd, 1H, J= 8.1 Hz, 1.65 Hz, Ar-H), 7.52 (d, 2H, J= 7.9 Hz, Ar-H), 7.28 (d, 2H, J= 7.6 Hz, Ar-H), 7.06 (t, 1H, J= 7.35 Hz, Ar-H), 6.86–6.88 (m, 1H, Ar-H), 3.38 (Br s, 4H, piperazinyl), 3.19 (s, 2H, methylene), 2.60 (Br s, 4H, piperazinyl). HRMS: m/z [M+H]+ calculated: 342.1561; found: 342.1817.
3.3.2 N-(2-Chlorophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5b)
Yield 69%; Yellow solid; m.p. 108°C–109°C. IR: ν (cm−1): 3,248 (NH), 1,690 (C=O), 1,593 (NO2), 1,556 (C=C), 1,507 (CH2), 1,433 (C=N), 1,232 (C-N). 1H NMR (400 MHz) δ (ppm): 9.94 (s, 1H, NH), 8.41–8.42 (m, 1H, Ar-H), 8.25–8.27 (m, 1H, Ar-H), 8.21 (dd, 1H, J= 8.25 Hz, 1.6 Hz, Ar-H), 7.52 (dd, 1H, J= 8.05, 1.45 Hz), 7.34–7.37 (m, 1H, Ar-H), 7.13–7.17 (m, 1H, Ar-H), 6.92–6.94 (m, 1H, Ar-H), 3.45 (Br s, 4H, piperazinyl), 3.25 (s, 2H, methylene), 2.68 (Br s, 4H, piperazinyl). 13C NMR (δ): 168.4 (C=O), 152.0 (Ar-C), 151.9 (Ar-C), 135.8 (Ar-C), 134.1 (Ar-C), 132.7 (Ar-C), 129.2 (Ar-C), 127.8 (Ar-C), 125.2 (Ar-C), 123.3 (Ar-C), 121.8 (Ar-C), 114.2 (Ar-C), 60.9 [N(CH2)CO], 52.2 (piperazine, 2 x NCH2), 47.7 (piperazine, 2 x NCH2). 1HRMS: m/z [M+2H]+ calculated: 377.1244; found: 377.3016.
3.3.3 N-(3-Chlorophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5c)
Yield 55%; Yellow solid; m.p. 71°C–73°C. IR: ν (cm−1): 3,276 (NH), 1,664 (C=O), 1,593 (NO2), 1,552 (C=C), 1,491 (CH2), 1,429 (C=N), 1,236 (C-N). 1H NMR (400 MHz) δ (ppm): 9.97 (s, 1H, NH), 8.39–8.40 (m, 1H, Ar-H), 8.23 (dd, 1H, J = 8.15 Hz, 1.7 Hz, Ar-H), 7.85–7.86 (m, 1H, Ar-H), 7.55–7.57 (m, 1H, Ar-H), 7.33 (t, 1H, J = 8.15 Hz, Ar-H), 7.10–7.13 (m, 1H, Ar-H), 6.88–6.91 (m, 1H, Ar-H), 3.42 (Br s, 4H, piperazinyl), 3.20 (s, 2H, methylene), 2.61 (Br s, 4H, piperazinyl). 13C NMR (δ): 169.1 (C=O), 152.5 (Ar-C), 152.4 (Ar-C), 140.4 (Ar-C), 136.3 (Ar-C), 133.4 (Ar-C), 133.0 [ClC(CH)2], 130.8 (Ar-C), 123.6 (Ar-C), 119.6 (Ar-C), 118.5 (Ar-C), 114.3 (Ar-C), 61.8 [N(CH2)CO], 52.8 (piperazine, 2 × NCH2), 47.8 (piperazine, 2 × NCH2). HRMS: m/z [M+2H]+ calculated: 377.1244; found: 377.2965.
3.3.4 N-(4-Chlorophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5d)
Yield 61%; Yellow solid; m.p. 99°C–100°C. IR: ν (cm−1): 3,334 (NH), 1,685 (C=O), 1,595 (NO2), 1,554 (C=C), 1,487 (CH2), 1,433 (C=N), 1,239 (C-N). 1H NMR (400 MHz) δ (ppm): 9.95 (s, 1H, NH), 8.32–8.34 (m, 1H, Ar-H), 8.18 (dd, 1H, J= 8.1 Hz, 1.75 Hz), 7.56–7.59 (m, 2H, Ar-H), 7.30–7.32 (m, 2H, Ar-H), 6.85–6.88 (m, 1H, Ar-H), 3.38 (Br s, 4H, piperazinyl), 3.15 (s, 2H, methylene), 2.56 (Br s, 4H, piperazinyl). 13C NMR (δ): 169.3 (C=O), 152.5 (Ar-C), 152.4 (Ar-C), 137.4 (Ar-C), 136.3 (Ar-C), 133.0 (Ar-C), 129.0 (3 × Ar-C), 128.0 (Ar-C), 122.0 (Ar-C), 114.5 (Ar-C), 61.5 [N(CH2)CO], 52.6 (piperazine, 2 × NCH2), 47.8 (piperazine, 2 × NCH2). HRMS: m/z [M+2H]+ calculated: 377.1244; found: 377.3023.
3.3.5 N-(2-Bromophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5e)
Yield 51%; Yellow solid; m.p. 109°C–110°C. IR: ν (cm−1): 3,260 (NH), 1,694 (C=O), 1,587 (NO2), 1,551 (C=C), 1,507 (CH2), 1,433 (C=N), 1,232 (C-N). 1H NMR (400 MHz) δ (ppm): 9.99 (s, 1H, NH), 8.30–8.32 (m, 1H, Ar-H), 8.17 (dd, 1H, J= 8.1 Hz, 1.7 Hz, Ar-H), 8.87–8.88 (m, 1H, Ar-H), 7.43–7.45 (m, 1H, Ar-H), 7.23–7.25 (m, 2H, Ar-H), 6.85–6.87 (m, 1H, Ar-H), 3.37 (Br s, 4H, piperazinyl), 3.16 (s, 2H, methylene), 2.56 (Br s, 4H, piperazinyl). 1H NMR (400 MHz) δ (ppm): 9.97 (s, 1H, NH), 8.29-8.31 (m, 1H, Ar-H), 8.15 (dd, 1H, J= 8.1 Hz, 1.7 Hz, Ar-H), 8.85–8.88 (m, 1H, Ar-H), 7.44–7.46 (m, 1H, Ar-H), 7.22–7.25 (m, 2H, Ar-H), 6.84–6.86 (m, 1H, Ar-H), 3.35 (Br s, 4H, piperazinyl), 3.15 (s, 2H, methylene), 2.56 (Br s, 4H, piperazinyl l). HRMS: m/z [M+H]+ calculated: 420.0666; found: 420.0865.
3.3.6 N-(3-Bromophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5f)
Yield 50%; Orange-yellow solid; m.p. 101°C–102°C. IR: ν (cm−1): 3,254 (NH), 1,670 (C=O), 1,593 (NO2), 1,552 (C=C), 1,489 (CH2), 1,429 (C=N), 1,236 (C-N). 1H NMR (400 MHz) δ (ppm): 9.97 (s, 1H, NH), 8.29-8.31 (m, 1H, Ar-H), 8.15 (dd, 1H, J= 8.1 Hz, 1.7 Hz, Ar-H), 8.85–8.88 (m, 1H, Ar-H), 7.44–7.46 (m, 1H, Ar-H), 7.22–7.25 (m, 2H, Ar-H), 6.84–6.86 (m, 1H, Ar-H), 3.35 (Br s, 4H, piperazinyl), 3.15 (s, 2H, methylene), 2.56 (Br s, 4H, piperazinyl l)13C NMR (δ): 169.2 (C=O), 151.9 (2 × Ar-C), 139.0 (Ar-C), 136.0 (Ar-C), 132.6 (Ar-C), 130.7 (Ar-C), 126.8 (Ar-C), 122.4 (Ar-C), 121.4 (Ar-C), 118.8 (Ar-C), 114.2 (Ar-C), 60.6 [N(CH2)CO], 51.8 (piperazine, 2 × NCH2), 47.2 (piperazine, 2 × NCH2).
3.3.7 N-(4-Bromophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5g)
Yield 53%; Yellow solid; m.p. 115°C–116°C. IR: ν (cm−1): 3,334 (NH), 1,686 (C=O), 1,593 (NO2), 1,552 (C=C), 1,485 (CH2), 1,431 (C=N), 1,239 (C-N). 1H NMR (400 MHz) δ (ppm): 9.96 (s, 1H, NH), 8.31 (m, 1H, Ar-H), 8.18–8.21 (m, 1H, Ar-H), 7.46 (t, 4H, J= 9.15 Hz, Ar-H), 6.86–6.88 (m, 1H, Ar-H), 3.37 (Br s, 4H, piperazinyl), 3.31 (s, 2H, methylene), 2.57 (Br s, 4H, piperazinyl). HRMS: m/z [M+H]+ calculated: 420.0666; found: 420.1088.
3.3.8 N-(2-Nitrophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5h)
Yield 65%; Pale-yellow solid; m.p. 113°C–115°C. IR: ν (cm−1): 3,185 (NH), 1,687 (C=O), 1,597 (NO2), 1,556 (C=C), 1,492 (CH2), 1,435 (C=N), 1,226 (C-N). 1H NMR (400 MHz) δ (ppm): 11.55 (s, 1H, NH), 8.58 (dd, 1H, J= 8.5 Hz, 1.33 Hz, Ar-H), 8.42–8.43 (m, 1H, Ar-H), 8.26 (dd, 1H, J= 8.05 Hz, 1.7 Hz, Ar-H), 8.19 (dd, 1H, J= 8.4 Hz, 1.55 Hz, Ar-H), 7.76–7.79 (m, 1H, Ar-H), 7.30–7.33 (m, 1H, Ar), 6.92–6.96 (m, 1H, Ar-H), 3.48 (Br s, 4H, piperazinyl), 3.26 (s, 2H, methylene), 2.68 (Br s, 4H, piperazinyl). 13C NMR (δ): 170.1 (C=O), 152.6 (Ar-C), 152.5 (Ar-C), 137.8 (Ar-C), 136.3 (Ar-C), 136.1 (Ar-C), 133.9 (Ar-C), 133.2 (Ar-C), 126.2 (Ar-C), 124.3 (Ar-C), 122.3 (Ar-C), 114.6 (Ar-C), 61.8 [N(CH2)CO], 52.9 (piperazine, 2 × NCH2), 48.1 (piperazine, 2 × NCH2). HRMS: m/z [M+H]+ calculated: 387.1411; found: 387.3554.
3.3.9 N-(3-Nitrophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5i)
Yield 58%; Orange-yellow solid; m.p. 139°C–141°C. IR: ν (cm−1): 3,276 (NH), 1,690 (C=O), 1,597 (NO2), 1,552 (C=C), 1,481 (CH2), 1,431 (C=N), 1,232 (C-N). 1H NMR (400 MHz) δ (ppm): 10.28 (s, 1H, NH), 8.69–8.70 (m, 1H, Ar-H), 8.40–8.41 (m, 1H, Ar-H), 8.24 (dd, 1H, J= 8.05 Hz, 1.7 Hz, Ar-H), 8.02–8.04 (m, 1H, Ar-H), 7.91–7.94 (m, 1H, Ar-H), 7.61 (t, 1H, J= 8.2 Hz, Ar-H), 6.89-6.91 (m, 1H, Ar-H), 3.46 (Br s, 4H, piperazinyl), 3.25 (s, 2H, methylene), 2.63 (Br s, 4H, piperazinyl). 13C NMR (δ): 169.6 (C=O), 152.5 (Ar-C), 152.5 (Ar-C), 148.4 (Ar-C), 140.2 (Ar-C), 136.3 (Ar-C), 133.0 (Ar-C), 130.5 (Ar-C), 126.2 (Ar-C), 118.5 (Ar-C), 114.3 (Ar-C), 61.8 [N(CH2)CO], 52.8 (piperazine, 2 × NCH2), 47.8 (piperazine, 2 × NCH2). HRMS: m/z [M+H]+ calculated: 387.1411; found: 387.1437.
3.3.10 N-(4-Nitrophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5j)
Yield 60%; Pale-yellow solid; m. p. 145°C–146°C. IR: ν (cm−1): 3,211 (NH), 1,694 (C=O), 1,589 (NO2), 1,552 (C=C), 1,500 (CH2), 1,448 (C=N), 1,236 (C-N). 1H NMR (400 MHz) δ (ppm): 10.28 (s, 1H, NH), 8.69–8.70 (m, 1H, Ar-H), 8.40–8.41 (m, 1H, Ar-H), 8.24 (dd, 1H, J= 8.05 Hz, 1.7 Hz, Ar-H), 8.02–8.04 (m, 1H, Ar-H), 7.91–7.94 (m, 1H, Ar-H), 7.61 (t, 1H, J= 8.2 Hz, Ar-H), 6.89–6.91 (m, 1H, Ar-H), 3.49 (Br s, 4H, piperazinyl), 3.27 (s, 2H, methylene), 2.63 (Br s, 4H, piperazinyl). HRMS: m/z [M+H]+ calculated: 387.1339; found: 387.1550.
3.3.11 2-(4-(3-Nitropyridin-2-yl)piperazin-1-yl)-N-(ortho-tolyl)acetamide (5k)
Yield 58%; Yellow solid; m.p. 105°C–106°C. IR: ν (cm−1): 3,304 (NH), 1,685 (C=O), 1,586 (NO2), 1,554 (C=C), 1,513 (CH2), 1,452 (C=N), 1,238 (C-N). 1H NMR (400 MHz) δ (ppm): 9.43 (s, 1H, NH), 8.41–8.43 (m, 1H, Ar-H), 8.26 (dd, 1H, J= 8.1 Hz, 1.7 Hz, Ar-H), 7.72 (dd, 1H, J= 8.05 Hz, 1.35 Hz, Ar-H), 7.16–7.24 (m, 2H, Ar-H), 7.04–7.08 (m, 1H, Ar-H), 6.91–6.93 (m, 1H, Ar-H), 3.51 (Br s, 4H, piperazinyl), 3.19 (s, 2H, methylene), 2.66 (Br s, 4H, piperazinyl), 2.24 (s, 3H, methyl). HRMS: m/z [M+2H]+2 calculated: 357.1790; found: 357.1865.
3.3.12 2-(4-(3-Nitropyridin-2-yl)piperazin-1-yl)-N-(meta-tolyl)acetamide (5L)
Yield 64%; Orange-yellow solid; m.p. 95°C–96°C. IR: ν (cm−1): 3,301 (NH), 1,686 (C=O), 1,591 (NO2), 1,551 (C=C), 1,507 (CH2), 1,433 (C=N), 1,231 (C-N). 1H NMR (400 MHz) δ (ppm): 9.71 (s, 1H, NH), 8.39–8.40 (m, 1H, Ar-H), 8.24 (dd, 1H, J= 8.05 Hz, 1.7 Hz, Ar-H), 7.43–7.47 (m, 2H, Ar-H), 7.18 (t, 1H, J= 7.75 Hz, Ar-H), 6.87–6.89 (m, 2H, Ar-H), 3.44 (Br s, 4H, piperazinyl), 3.17 (s, 2H, methylene), 2.61 (Br s, 4H, piperazinyl), 2.27 (s, 3H, methyl). HRMS: m/z [M+H]+ calculated: 356.1717; found: 356.2223.
3.3.13 2-(4-(3-Nitropyridin-2-yl)piperazin-1-yl)-N-(para-tolyl)acetamide (5m)
Yield 54%; Yellow solid; m.p. 112°C–113°C. IR: ν (cm−1): 3,295 (NH), 1,683 (C=O), 1,588 (NO2), 1,551 (C=C), 1,505 (CH2), 1,448 (C=N), 1,236 (C-N). 1H NMR (400 MHz) δ (ppm): 9.74 (s, 1H, NH), 8.29–8.30 (m, 1H, Ar-H), 8.16 (dd, 1H, J= 8.05 Hz, 1.65 Hz, Ar-H), 7.36–7.38 (m, 2H, Ar-H), 7.07 (d, 2H, J= 8.25 Hz, Ar-H), 6.84–6.86 (m, 1H, Ar-H), 3.46 (Br s, 4H, piperazinyl), 3.13 (s, 2H, methylene), 2.55 (Br s, 4H, piperazinyl), 2.18 (s, 3H, methyl). 13C NMR (δ): 169.1 (C=O), 152.5 (Ar-C), 152.4 (Ar-C), 136.4 (Ar-C), 135.5 (Ar-C), 134.0 (2 × Ar-C), 133.0 (Ar-C), 129.6 (2 × Ar-C), 120.6 (Ar-C), 114.6 (Ar-C), 61.4 [N(CH2)CO], 55.9 (CH3), 52.5 (piperazine, 2 × NCH2), 47.8 (piperazine, 2 × NCH2). HRMS: m/z [M+H]+ calculated: 356.1644; found: 356.2183.
3.3.14 N-(2-Methoxyphenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5n)
Yield 53%; Orange-yellow solid; m.p. 120°C–121°C. IR: ν (cm−1): 3,301 (NH), 1,683 (C=O), 1,593 (NO2), 1,556 (C=C), 1,504 (CH2), 1,437 (C=N), 1,221 (C-N). 1H NMR (400 MHz) δ (ppm): 9.73 (s, 1H, NH), 8.42–8.43 (m, 1H, Ar-H), 8.27 (dd, 1H, J= 8.1 Hz, 1.65 Hz, Ar-H), 8.18 (d, 1H, J= 7.9 Hz, Ar-H), 7.06–7.07 (m, 2H, Ar-H), 6.92–6.95 (m, 2H, Ar-H), 3.88 (s, 3H, methoxy), 3.44 (Br s, 4H, piperazinyl), 3.19 (s, 2H, methylene), 2.64 (Br s, 4H, piperazinyl). 13C NMR (δ): 167.8 (C=O), 152.0 (Ar-C), 151.9 (Ar-C), 148.2 (Ar-C), 135.8 (Ar-C), 132.6 (Ar-C), 126.0 (Ar-C), 123.9 (Ar-C), 120.5 (Ar-C), 119.0 (Ar-C), 114.1 (Ar-C), 110.9 (Ar-C), 61.0 [N(CH2)CO], 55.9 (CH3), 52.8 (piperazine, 2 × NCH2), 47.8 (piperazine, 2 × NCH2). HRMS: m/z [M+2H]+2 calculated: 373.1739; found: 373.1768.
3.3.15 N-(4-Methoxyphenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)acetamide (5o)
Yield 64%; Orange-yellow solid; m.p. 107°C–108°C. IR: ν (cm−1): 3,310 (NH), 1,677 (C=O), 1,590 (NO2), 1,552 (C=C), 1,508 (CH2), 1,437 (C=N), 1,236 (C-N). 1H NMR (400 MHz) δ (ppm): 9.65 (s, 1H, NH), 8.39−8.40 (m, 1H, Ar-H), 8.23−8.25 (m, 1H, Ar-H), 7.52−7.55 (m, 2H, Ar-H), 6.86−6.91 (m, 3H, Ar-H), 3.48 (s, 3H, methoxy), 3.44 (Br s, 4H, piperazinyl), 3.15 (s, 2H, methyl), 2.60 (Br s, 4H, piperazinyl). 13C NMR (δ) 168.1 (C=O), 155.9 (Ar-C), 152.5 (Ar-C), 152.4 (Ar-C), 136.3 (Ar-C), 133.0 (Ar-C), 132.1 (Ar-C), 121.7 (2 × Ar-C), 114.3 (Ar-C), 114.2 (2 × Ar-C), 61.8 [N(CH2)CO], 55.6(OCH3), 52.8 (piperazine, 2 × NCH2), 47.9 (piperazine, 2 × NCH2). HRMS: m/z [M+H]+ calculated: 372.1666; found: 372.1987.
3.4 Procedure for the synthesis of N-aryl-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide derivatives 7a-7k
A solution of 2-chloro-N-arylpropanamides 6a-6k (0.15 mmol) was prepared in 10 mL of acetonitrile and added to the stirred mixture of 1-(3-nitropyridin-2-yl)piperazine (3) (0.15 mmol) and potassium carbonate (0.3 mmol) in 15 mL of acetonitrile. The reaction mixture was refluxed for 24–48 h on stirring by monitoring with TLC. On the addition of water colored (yellow to red) precipitates of N-aryl-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide derivatives 7a-7k was formed. The produced solid was collected, and purified by column chromatography. The calculated yield was found to be 30%–55%.
3.4.1 2-(4-(3-Nitropyridin-2-yl)piperazin-1-yl)-N-phenylpropanamide (7a)
Yield 43%; Yellow solid; m.p. 92°C–94°C. IR: ν (cm−1): 3,323 (NH), 1,690 (C=O), 1,593 (NO2), 1,556 (C=C), 1,487 (CH2), 1,433 (C=N), 1,238 (C-N). 1H NMR (400 MHz) δ (ppm): 9.93 (s, 1H, NH), 8.29–8.31 (m, 1H, Ar-H), 8.15–8.17 (m, 1H, Ar-H), 7.51 (d, 2H, J= 8 Hz, Ar-H), 7.28 (t, 2H, J= 7.75 Hz, Ar-H), 7.04–7.07 (m, 1H, Ar-H), 6.84–6.86 (m, 1H, Ar-H), 3.35 (Br s, 4H, piperazinyl), 3.25 (q, 1H, J = 6.70 Hz, CH), 2.54–2.63 (m, 4H, piperazinyl), 1.18 (d, 3H, J= 6.9 Hz, methyl). HRMS: m/z [M+H]+ calculated: 356.1717; found: 356.2021.
3.4.2 N-(3-Chlorophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7b)
Yield 38%; Yellow solid; m.p. 107°C–109°C. IR: ν (cm−1): 3,291 (NH), 1,694 (C=O), 1,590 (NO2), 1,556 (C=C), 1,504 (CH2), 1,429 (C=N), 1,232 (C-N). 1H NMR (400 MHz) δ (ppm): 10.06 (s, 1H, NH), 8.32–8.33 (m, 1H, Ar-H), 8.19 (dd, 1H, J= 8.15 Hz, 1.7 Hz, Ar-H), 7.98 (dd, 1H, J= 8.1 Hz, 1.6 Hz, Ar-H), 7.47 (dd, 1H, J= 8.1 Hz, 1.45 Hz, Ar-H), 7.29–7.33 (m, 1H, Ar-H), 7.12–7.15 (m, 1H, Ar-H), 6.87–6.90 (m, 1H, Ar), 3.40 (Br s, 4H, piperazinyl), 3.27 (q, 1H, J = 6.70 Hz, CH), 2.57–2.65 (m, 4H, piperazinyl), 1.18 (d, 3H, J= 7 Hz, methyl).
3.4.3 N-(4-Chlorophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7c)
Yield 42%; Red-orange solid; m.p. 57°C–59°C. IR: ν (cm−1): 3,278 (NH), 1,673 (C=O), 1,595 (NO2), 1,556 (C=C), 1,487 (CH2), 1,429 (C=N), 1,239 (C-N). 1H NMR (400 MHz) δ (ppm): 10.04 (s, 1H, NH), 8.29–8.30 (m, 1H, Ar-H), 8.15 (dd, 1H, J= 8.1 Hz, 1.7 Hz, Ar-H), 7.53–7.57 (m, 2H, Ar-H), 7.53–7.56 (m, 2H, Ar-H), 6.83–6.85 (m, 1H, Ar-H), 3.34 (Br s, 4H, piperazinyl), 3.25 (q, 1H, J = 6.90 Hz, CH), 2.52–2.61 (m, 4H, piperazinyl), 1.16 (d, 3H, J= 6.85, methyl). 13C NMR (δ): 172.3 (C=O), 152.5 (Ar-C), 152.4 (Ar-C), 137.3 (Ar-C), 136.3 (Ar-C), 133.0 (Ar-C), 129.0 (2 × Ar-C), 128.1 (Ar-C), 122.0 (2 × Ar-C), 114.5 (Ar-C), 63.6 [N(CH)CO], 49.2 (piperazine, 2 × NCH2), 48.1 (piperazine, 2 × NCH2), 13.4 (CH3). HRMS: m/z [M+H]+ calculated: 390.1327; found: 390.2126.
3.4.4 N-(2-Bromophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7d)
Yield 45%; Yellow solid; m.p. 91°C–94°C. IR: ν (cm−1): 3,289 (NH), 1,692 (C=O), 1,587 (NO2), 1,556 (C=C), 1,503 (CH2), 1,429 (C=N), 1,234 (C-N). 1H NMR (400 MHz) δ (ppm): 10.04 (s, 1H, NH), 8.32–8.34 (m, 1H, Ar-H), 8.19 (dd, 1H, J= 8.05 Hz, 2.05 Hz, Ar-H), 8.00 (dd, 1H, J= 8.15 Hz, 1.55 Hz, Ar-H), 7.62 (dd, 1H, J= 8 Hz, 1.3 Hz, Ar-H), 7.33–7.36 (m, 1H, Ar-H), 7.05–7.08 (m, 1H, Ar-H), 6.87–6.90 (m, 1H, Ar-H), 3.43 (Br s, 4H, piperazinyl), 3.36 (q, 1H, J = 6.75 Hz, CH), 2.58–2.65 (m, 4H, piperazinyl), 1.18 (d, 3H, J= 6.95 Hz, methyl). HRMS: m/z [M+H]+ calculated: 434.0822; found: 434.2365.
3.4.5 N-(2-Nitrophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7e)
Yield 55%; Yellow solid; m.p. 124°C–125°C. IR: ν (cm−1): 3,293 (NH), 1,698 (C=O), 1,592 (NO2), 1,560 (C=C), 1,489 (CH2), 1,420 (C=N), 1,221 (C-N). 1H NMR (400 MHz) δ (ppm): 11.6 (s, 1H, NH), 8.55 (dd, 1H, J= 8.45 Hz, 1.35 Hz, Ar-H), 8.40–8.41 (m, 1H, Ar-H), 8.24–8.28 (m, 1H, Ar-H), 8.17 (dd, 1H, J= 8.4 Hz, 1.55 Hz, Ar-H), 7.74–7.78 (m, 1H, Ar-H), 7.29–7.32 (m, 1H, Ar-H), 6.90–6.93 (m, 1H, Ar-H), 3.53–3.60 (m, 4H, piperazinyl), 3.45 (q, 1H, J = 6.70 Hz, CH), 2.59–2.68 (m, 4H, piperazinyl), 1.19 (d, 3H, J= 7 Hz, methyl). 13C NMR (δ): 172.7 (C=O), 152.0 (Ar-C), 151.9 (Ar-C), 137.5 (Ar-C), 135.8 (Ar-C), 135.5 (Ar-C), 133.3 (Ar-C), 132.5 (Ar-C), 125.5 (Ar-C), 123.7 (Ar-C), 121.9 (Ar-C), 114.0 (Ar-C), 63.5 [N(CH2)CO], 48.7 (piperazine, 2 × NCH2), 47.6 (piperazine, 2 × NCH2), 10.2 (CH3). HRMS: m/z [M+H]+ calculated: 401.1568; found: 401.1834.
3.4.6 N-(3-Nitrophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7f)
Yield 47%; Orange solid; m.p. 120°C–121°C. IR: ν (cm−1): 3,221 (NH), 1,692 (C=O), 1,593 (NO2), 1,556 (C=C), 1,502 (CH2), 1,429 (C=N), 1,232 (C-N). 1H NMR (400 MHz) δ (ppm): 10.36 (s, 1H, NH), 8.56–8.57 (m, 1H, Ar-H), 8.25-8.28 (m, 1H, Ar-H), 8.12–8.14 (m, 1H, Ar-H), 7.85–7.87 (m, 1H, Ar-H), 7.74–7.82 (m, 1H, Ar-H), 7.51–7.55 (m, 1H, Ar-H), 6.82–6.84 (m, 1H, Ar-H), 3.34 (Br s, 4H, piperazinyl), 3.29 (q, 1H, J = 6.65 Hz, CH), 2.57-2.61 (m, 4H, piperazinyl), 1.18 (d, 3H, J= 6.9 Hz, methyl). HRMS: m/z [M+H]+ calculated: 401.1568; found: 401.1877.
3.4.7 N-(4-Nitrophenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7g)
Yield 51%; Yellow solid; m.p. 159°C–161°C. IR: ν (cm−1): 3,343 (NH), 1,698 (C=O), 1,590 (NO2), 1,556 (C=C), 1,498 (CH2), 1,440 (C=N), 1,232 (C-N). 1H NMR (400 MHz) δ (ppm): 10.47 (s, 1H, NH), 8.38–8.39 (m, 3H, Ar-H), 8.21–8.24 (m, 2H, Ar-H), 6.88–6.90 (m, 1H, Ar-H), 3.48–3.57 (m, 4H, piperazinyl), 3.40 (q, 1H, J = 6.75 Hz, CH), 2.57–2.67 (m, 4H, piperazinyl), 1.21 (d, 3H, J= 6.85 Hz, methyl). 13C NMR (δ):172.7 (C=O), 152.5 (Ar-C), 152.4 (Ar-C), 145.3 (Ar-C), 142.8 (Ar-C), 136.3 (Ar-C), 133.0 (Ar-C), 125.3 (2 × Ar-C), 119.7 (2 × Ar-C), 114.3 (Ar-C), 63.8 (Ar-C), 49.2 (piperazine, 2 × NCH2), 48.2 (piperazine, 2 × NCH2), 12.7 (CH3). HRMS: m/z [M+2H]+2 calculated: 402.1495; found: 402.1550.
3.4.8 N-(ortho-Tolyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7h)
Yield 33%; Yellow solid; m.p. 90°C–92°C. IR: ν (cm−1): 3,301 (NH), 1,687 (C=O), 1,590 (NO2), 1,556 (C=C), 1,506 (CH2), 1,433 (C=N), 1,220 (C-N). 1H NMR (400 MHz) δ (ppm): 9.56 (s, 1H, NH), 8.31–8.32 (m, 1H, Ar-H), 8.15–8.18 (m, 1H, Ar-H), 7.44–7.45 (m, 1H, Ar-H), 7.12–7.19 (m, 2H, Ar-H), 7.04–7.07 (m, 1H, Ar-H), 6.85–6.88 (m, 1H, Ar-H), 3.37 (Br s, 4H, piperazinyl), 3.26 (q, 1H, J = 6.65 Hz, CH), 2.56–2.66 (m, 4H, piperazinyl), 2.15 (s, 3H, methyl), 1.19 (d, 3H, J= 7 Hz, methyl). HRMS: m/z [M+2H]+2 calculated: 371.1946; found: 371.2119.
3.4.9 N-(para-Tolyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7i)
Yield 41%; Orange-yellow solid; m.p. 88°C–89°C. IR: ν (cm-1): 3,278 (NH), 1,670 (C=O), 1,593 (NO2), 1,554 (C=C), 1,506 (CH2), 1,429 (C=N), 1,236 (C-N). 1H NMR (400 MHz) δ (ppm): 9.7 (s, 1H, NH), 8.38–8.40 (m, 1H, Ar-H), 8.22 (dd, 1H, J= 8 Hz, 1.8 Hz, Ar-H), 7.50–7.56 (m, 2H, Ar-H), 7.09–7.11 (m, 2H, Ar-H), 6.87–6.90 (m, 1H, Ar-H), 3.35 (Br s, 4H, piperazinyl), 3.30 (m, 1H, J = 6.90 Hz, CH), 2.55–2.67 (m, 4H, piperazinyl), 2.24 (s, 3H, methyl), 1.9 (d, 3H, J= 6.85 Hz, methyl). HRMS: m/z [M+H]+ calculated: 370.1874; found: 370.2142.
3.4.10 N-(2-Methoxyphenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7j)
Yield 30%; Yellow solid; m.p. 108°C–110°C. IR: ν (cm−1): 3,304 (NH), 1,683 (C=O), 1,593 (NO2), 1,556 (C=C), 1,515 (CH2), 1,433 (C=N), 1,236 (C-N). 1H NMR (400 MHz) δ (ppm): 9.92 (s, 1H, NH), 8.41–8.43 (m, 1H, Ar-H), 8.2–8.29 (m, 1H, Ar-H), 8.15–8.17 (m, 1H, Ar-H), 7.03–7.07 (m, 2H, Ar-H), 6.91–6.95 (m, 2H, Ar-H), 3.88 (s, 3H, methoxy), 3.50–3.56 (m, 4H, piperazinyl), 3.37–3.42 (m, 1H, CH), 2.58–2.67 (m, 4H, piperazinyl), 1.8 (d, 3H, J= 7 Hz, methyl).
3.4.11 N-(4-Methoxyphenyl)-2-(4-(3-nitropyridin-2-yl)piperazin-1-yl)propanamide (7k)
Yield 32%; Red solid; m.p. 62°C-64°C. IR: ν (cm−1): 3,301 (NH), 1,675 (C=O), 1,593 (NO2), 1,556 (C=C), 1,507 (CH2), 1,435 (C=N), 1,232 (C-N). 1H NMR (400 MHz) δ (ppm): 9.72 (s, 1H, NH), 8.37–8.39 (m, 1H, Ar-H), 8.21–8.23 (m, 1H, Ar-H), 7.52–7.55 (m, 2H, Ar-H), 6.86–6.89 (m, 3H, Ar-H), 3.71 (s, 3H, methoxy), 3.43 (t, 4H, J= 4.95 Hz, piperazinyl), 3.27–3.31 (m, 1H, CH), 2.56 (t, 4H, J= 5 Hz, piperazinyl), 1.9 (d, 3H, J= 6.9 Hz, methyl) 13C NMR (δ): 171.0 (C=O), 155.8 (Ar-C), 152.5 (Ar-C), 152.4 (Ar-C), 136.3 (Ar-C), 132.9 (Ar-C), 132.3 (Ar-C), 121.6 (2 × Ar-C), 114.8 (Ar-C), 114.2 (2× Ar-C), 63.6 [N(CH)CO], 55.6 (OCH3), 52.9 (piperazine, 2 × NCH2), 49.4 (piperazine, 2 × NCH2), 13.3 (CH3). HRMS: m/z [M+H]+ calculated: 386.1750; found: 386.3397.
3.5 In vitro inhibition assay of urease
The inhibitory activity of 1-(3-nitropyridin-2-yl)piperazine derivatives (5a-o; 7a-k) was assessed in vitro against urease with minor modifications using the indophenol method (Al Azzam et al., 2022; Da Costa et al., 2022). At first, 1 mM solution of a test compound was prepared in 10% DMSO followed by the preparation of the reaction mixture. The reaction mixture was prepared by adding 30 μL of reaction buffer (100 mmol/L of urea, 1 mmol/L of EDTA, 10 mmol/L of LiCl2, 10 mmol/L of KH2PO4; pH: 8.2), 50 μL of R1 (120 mmol/L phosphate buffer, 60 mmol/L sodium salicylate, 5 mmol/L sodium nitroprusside, 1 mmol/L EDTA and 5 KU/L urease), 10 μL of substrate (1 mM urea), and 10 μL of test compound in triplicate in a 96-well plate. After 5 min of incubation at ambient temperature, 70 μL of R2 (120 mmol/L phosphate buffer, 400 mmol/L sodium hydroxide, and 10 mmol/L sodium hypochlorite) was added to the reaction mixture (Rauf et al., 2013). After another incubation period of 10 min at room temperature, the absorbance was measured using a microplate reader (Bio-Tek, United States) at a particular wavelength of 630 nm. In this assay, thiourea was used as a positive control (standard). The following equation was used to determine the percentage of inhibition for each test compound.
The IC50 values of test compounds (5a-o; 7a-k) at various concentrations were determined by analyzing the data, obtained from in vitro urease inhibitory assay, by GraphPad Prism version 8.0.2 (GraphPad, California, United States).
3.6 In vitro hemocompatibility screening
Hemolysis is the rupturing of the cell membrane of red blood cells that causes the expulsion of hemoglobin into the blood plasma. To analyze the effects of 1-(3-nitropyridin-2-yl)piperazine 3 and its derivatives (5b, 5c, 5i; 7e, 7h) on human blood, a hemolysis analysis was carried out (Supplementary Figure S69). About 5 mL fresh sample of human blood was collected from a 24-year-old volunteer and transferred to a vacutainer tube containing EDTA to prevent coagulation. The ultracentrifugation of the blood sample was carried out at 3,000 rpm for 20 min and the separated plasma was discarded. The blood cells were washed about three times by using a double volume of phosphate buffer solution (PBS) (pH = 7.4). Stock solutions of 1-(3-nitropyridin-2-yl)piperazine 3 and its derivatives (5b, 5c, 5i; 7e, 7h) were prepared (5 mg/mL) and test samples were diluted as follows: 0.5 mg/mL, 1 mg/mL, 2.5 mg/mL, 5 mg/mL, and 10 mg/mL. For negative control reference, 1 mL PBS was added to the blood test sample while 100 µL (4%), Triton X-100 was added as a positive control reference. After that, 400 µL of blood test cells and 1 mL of PBS were taken in each Eppendorf tube and a 100 µL of 1-(3-nitropyridin-2-yl)piperazine 3 and its derivatives (5b, 5c, 5i; 7e, 7h) were added separately to make the final volume. All test samples were incubated at 37°C for 1 h and then kept in an ice bath for 1 min. Finally, test samples were centrifuged at 3,000 rpm for 5 min. To quantify hemoglobin in test samples, the absorbance of the supernatant was measured at 540 nm (Munawwar et al., 2023). From the absorbance value, the hemolysis percentage was determined by the following formula:
3.7 In silico investigation
3.7.1 Pre-clinical investigation
The toxicological and pharmacological properties of the precursor compound and potent inhibitors were determined by freely accessible software, namely, admetSAR (http://lmmd.ecust.edu.cn/admetsar1/home/, accessed on 10 September 2023) and eMolTox (http://xundrug.cn/moltox, accessed on 8 July 2023). Furthermore, GUSAR (http://www.way2drug.com/gusar/acutoxpredict.html, accessed on 11 September 2023) was used to evaluate the optimal route of administration of potent inhibitors to rats for in vivo studies in the future based on lethal dose 50 (LD50) values (Al Azzam et al., 2022). Similarly, eMolTox functions as an online platform designed to forecast the possible toxicological effects linked to the molecular configuration of a compound using an aggregate predictor method (Da Costa et al., 2022).
3.7.2 Docking studies
AutoDockVina was employed to perform the molecular docking of the potent inhibitor with the most druggable active site of urease (Kumar et al., 2022). The most druggable active site was identified based on DoGSiteScore obtained from the analysis of target protein in the binding site mode of SeeSAR version 12.1.0, as shown in Figure 9 (Volkamer et al., 2012; BioSolveIT GmbH, 2022). Initially, the pdb structure of jack bean urease (PDB id: 4H9M), obtained from the RCSB repository, was prepared by eliminating water molecules and ligands in Discovery Studio molecular visualizer 2021. Similarly, Discovery Studio was used to convert the format of compound 3 and potent inhibitors from sdf to pdb format after minimizing their energy via Chem3D Pro 12.0.2.1076. Chem3D Pro 12.0.2.1076 utilizes Merck Molecular Force Field 2 (MM2) algorithm that iteratively adjusts the coordinates of atoms in the molecule to minimize its energy (Al Azzam et al., 2022; Moulishankar and Thirugnanasambandam, 2023). Afterward, AutoDockTools 1.5.7 was utilized to construct pdbqt files of the receptor (4H9M) and potent inhibitors followed by the formation of a grid box encompassing a druggable binding site (Fatima et al., 2022). After giving specific commands, nine different poses as output files were generated, and the best pose was selected based on the lowest binding energy. The grid dimensions were adjusted as indicated in Table 5. Later, the intermolecular interactions between the best-docked pose and the active site of the receptor were determined by BIOVIA Discovery Studio 2021. This software determines the favorable interactions of hydrogen bonds, salt bridges, and hydrophobic interactions (Baskaran et al., 2020).
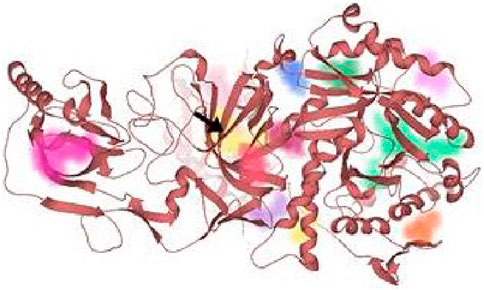
FIGURE 9. All the probable binding sites in urease. Among these binding pockets, the yellow one is the most druggable allosteric site.

TABLE 5. Dimensions of grid adjusted for molecular docking.
4 Conclusion
Urease plays a pivotal role in urea degradation and generates ammonia and carbamate. This enzymatic process significantly raises the pH, thereby facilitating the survival of pathogenic microorganisms. Consequently, targeting urease activity is as a crucial strategy in the treatment of such pathogen-mediated diseases. So, precursor compound 3 and derivatives of 1-(3-nitropyridin-2-yl)piperazine were evaluated by in silico and in vitro analysis. 5b and 7e exhibit the most inhibitory activity against, with impressive IC50 values of 2.0 ± 0.73 and 2.24 ± 1.63 µM, respectively. Their IC50 values were lower than the precursor compound 3, whose IC50 value was 3.90 ± 1.91 µM. In silico analyses illustrated that both of these potent inhibitors establish favorable interactions within the active site of urease; residues Arg132, Asp295, Thr830, Arg835, and Asn836 emerge as pivotal constituents of the active site. However, none of the binding site residues interacting with precursor compound 3 resemble the interacting residues of 5b and 7e. The binding energies of 5b and 7e were −8.0 kcal/mol (5b) and −8.1 kcal/mol (7e), accordingly. Pre-clinical investigation predicted that both 5b and 7e may exhibit gastrointestinal permeability and cross the blood-brain barrier. In addition, both inhibitors may produce toxic substructures and have LD50 values of 1,360 (5b) and 911.1 mg/kg (7e). The hemolysis evaluation predicted that 7e and 7h are more biocompatible in the human blood stream. Therefore, 5b and 7e can be pivotal therapeutic interventions for the management of infections caused by microbes whose survival is dependent on urease activity. Further, in vivo studies are crucial to validate their clinical efficacy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethics Review Committee Government College University Faisalabad. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
MuA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Writing–original draft, Writing–review and editing. SZ: Formal Analysis, Investigation, Validation, Writing–original draft, Writing–review and editing. MaA: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing–original draft, Writing–review and editing. SS: Data curation, Formal Analysis, Writing–original draft, Writing–review and editing. SA-H: Funding acquisition, Resources, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors are thankful to the Government College University Faisalabad for financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1371377/full#supplementary-material
References
Abdel-Latif, E., Fahad, M. M., and Ismail, M. A. (2020). Synthesis of N-aryl 2-chloroacetamides and their chemical reactivity towards various types of nucleophiles. Synth. Commun. 50, 289–314. doi:10.1080/00397911.2019.1692225
Ahmad, S., Abdul Qadir, M., Ahmed, M., Imran, M., Yousaf, N., Wani, T. A., et al. (2023). New acetamide-sulfonamide-containing scaffolds: antiurease activity screening, structure-activity relationship, kinetics mechanism, molecular docking, and md simulation studies. Molecules 28, 5389. doi:10.3390/molecules28145389
Al Azzam, K. M., Negim, E. S., and Aboul-Enein, H. Y. (2022). ADME studies of TUG-770 (a GPR-40 inhibitor agonist) for the treatment of type 2 diabetes using SwissADME predictor: in silico study. J. App. Pharm. Sci. 12 (4), 159–169. doi:10.7324/JAPS.2022.120418
Balasubramanian, A., and Ponnuraj, K. (2010). Crystal structure of the first plant urease from jack bean: 83 years of journey from its first crystal to molecular structure. J. Mol. Biol. 400, 274–283. doi:10.1016/j.jmb.2010.05.009
Baskaran, K. P., Arumugam, A., Kandasamy, R., and Alagarsamy, S. (2020). In silico method for prediction of maximum binding affinity and ligand-protein interaction studies on Alzheimer’s disease. Int. J. Res. Granthaalayah. 8, 362–370. doi:10.29121/granthaalayah.v8.i11.2020.2472
Bhagat, N., and Virdi, J. S. (2009). Molecular and biochemical characterization of urease and survival of Yersinia enterocolitica biovar 1A in acidic pH in vitro. BMC Microbiol. 9, 262–314. doi:10.1186/1471-2180-9-262
Campoli-Richards, D. M., Monk, J. P., Price, A., Benfield, P., Todd, P. A., and Ward, A. (1988). Ciprofloxacin: a review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs 35, 373–447. doi:10.2165/00003495-198835040-00003
Carter, E. L., Flugga, N., Boer, J. L., Mulrooney, S. B., and Hausinger, R. P. (2009). Interplay of metal ions and urease. Metallomics 1, 207–221. doi:10.1039/b903311d
Chaves, O. A., Sacramento, C. Q., Ferreira, A. C., Mattos, M., Fintelman-Rodrigues, N., Temerozo, J. R., et al. (2021). Atazanavir is a competitive inhibitor of SARS-CoV-2 Mpro, impairing variants replication in vitro and in vivo. Pharmaceuticals 15, 21. doi:10.3390/ph15010021
Cormier, R., Burda, W. N., Harrington, L., Edlinger, J., Kodigepalli, K. M., Thomas, J., et al. (2012). Studies on the antimicrobial properties of N-acylated ciprofloxacins. Bioorg. Med. Chem. Lett. 22, 6513–6520. doi:10.1016/j.bmcl.2012.05.026
Cox, G. M., Mukherjee, J., Cole, G. T., Casadevall, A., and Perfect, J. R. (2000). Urease as a virulence factor in experimental cryptococcosis. Infect. Immun. 68, 443–448. doi:10.1128/iai.68.2.443-448.2000
Crocker, M. S., Deng, Z., and Johnston, J. N. (2022). Preparation of N-aryl amides by epimerization-free umpolung amide synthesis. J. Am. Chem. Soc. 144, 16708–16714. doi:10.1021/jacs.2c05986
Croxtall, J. D. (2012). Aripiprazole: a review of its use in the management of schizophrenia in adults. CNS Drugs 26, 155–183. doi:10.2165/11208400-000000000-00000
Da Costa, G. V., Neto, M. F., Da Silva, A. K., De Sá, E. M., Cancela, L. C., Vega, J. S., et al. (2022). Identification of potential insect growth inhibitor against Aedes aegypti: a bioinformatics approach. Intern. J. molec. Sci. 23, 8218. doi:10.3390/ijms23158218
Faiz, S., Zahoor, A. F., Ajmal, M., Kamal, S., Ahmad, S., Abdelgawad, A. M., et al. (2019). Design, synthesis, antimicrobial evaluation, and laccase catalysis effect of novel benzofuran–oxadiazole and benzofuran–triazole hybrids. J. Het. Chem. 56, 2839–2852. doi:10.1002/jhet.3674
Fatima, K., Khalid, S., Qadeer, K., Yasin, H., Arsalan, A., Abrar, H., et al. (2022). Urease inhibition and DPPH radical scavenging potential of phytoconstituent from Alstonia scholaris and molecular docking interactions of bioactive luteolin with target proteins. Pak. J. Pharm. Sci. 35, 219–225. doi:10.36721/PJPS.2022.35.1.SUP.219-225.1
George, J., Prasana, J. C., Muthu, S., Kuruvilla, T. K., Sevanthi, S., and Saji, R. S. (2018). Spectroscopic (FT-IR, FT Raman) and quantum mechanical study on N-(2, 6-dimethylphenyl)-2-{4-[2-hydroxy-3-(2-methoxyphenoxy) propyl] piperazin-1-yl} acetamide. J. Molec. Struct. 1171, 268–278. doi:10.1016/j.molstruc.2018.05.106
Gupta, A., Shetty, S., Mutalik, S., Chandrashekar H, R., K, N., Mathew, E. M., et al. (2023). Treatment of H. pylori infection and gastric ulcer: need for novel Pharmaceutical formulation. Heliyon 9 (10), e20406. doi:10.1016/j.heliyon.2023.e20406
Hamad, A., Khan, M. A., Ahmad, I., Imran, A., Khalil, R., Al-Adhami, T., et al. (2020). Probing sulphamethazine and sulphamethoxazole based Schiff bases as urease inhibitors; synthesis, characterization, molecular docking and ADME evaluation. Bioorg. Chem. 105, 104336. doi:10.1016/j.bioorg.2020.104336
Jayasinghe, L., Jayasooriya, C. P., Hara, N., and Fujimoto, Y. (2003). A pyridine ring-containing ecdysteroid from Diploclisia glaucescens. Tetrahed. Lett. 44, 8769–8771. doi:10.1016/j.tetlet.2003.09.177
Joyce, J. R., and Richards, D. S. (2011). Kinetic control of protonation in electrospray ionization. J. Am. Soc. Mass Spectrom. 22 (2), 360–368. doi:10.1007/s13361-010-0037-0
Khan, I. A., Ahmad, M., Ashfaq, U. A., Sultan, S., and Zaki, M. E. (2021). Discovery of amide-functionalized benzimidazolium salts as potent α-glucosidase inhibitors. Molecules 26, 4760. doi:10.3390/molecules26164760
Khan, I. A., Saddique, F. A., Aslam, S., Ashfaq, U. A., Ahmad, M., Al-Hussain, S. A., et al. (2022). Synthesis of novel N-Methylmorpholine-Substituted benzimidazolium salts as potential α-glucosidase inhibitors. Molecules 27, 6012. doi:10.3390/molecules27186012
Konieczna, I., Zarnowiec, P., Kwinkowski, M., Kolesinska, B., Fraczyk, J., Kaminski, Z., et al. (2012). Bacterial urease and its role in long-lasting human diseases. Curr. Protein. Pept. Sci. 13, 789–806. doi:10.2174/138920312804871094
Krishnakumar, V., and Muthunatesan, S. (2006). FT-IR, FT-Raman spectra and scaled quantum mechanical study of 2, 3-dihydroxy pyridine and 2, 4-dihyroxy-3-nitropyridine. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 65 (3-4), 818–825. doi:10.1016/j.saa.2005.12.039
Kumar, G., Chauhan, B., Singh, S., and Negi, M. (2022). In-silico prediction and docking studies of novel synthesized benzoxazole derivatives as anti-tubercular activity. J. Pharm. Neg. Res., 9216–9226. doi:10.47750/pnr.2022.13.S09.1078
Kumar, K. S. S., Hanumappa, A., Hegde, M., Narasimhamurthy, K. H., Raghavan, S. C., and Rangappa, K. S. (2014). Synthesis and antiproliferative effect of novel 4-thiazolidinone-pyridine-and piperazine-based conjugates on human leukemic cells. Eur. J. Med. Chem. 81, 341–349. doi:10.1016/j.ejmech.2014.05.009
Lavorato, S. N., Duarte, M. C., Andrade, P. H. R. D., Coelho, E. A. F., and Alves, R. J. (2017). Synthesis, antileishmanial activity and QSAR studies of 2-chloro-N-arylacetamides. Braz. J. Pharm. Sci. 53. doi:10.1590/s2175-97902017000116067
Lin, W., Mathys, V., Ang, E. L. Y., Koh, V. H. Q., Martínez Gómez, J. M., Ang, M. L. T., et al. (2012). Urease activity represents an alternative pathway for Mycobacterium tuberculosis nitrogen metabolism. Infect. Immun. 80, 2771–2779. doi:10.1128/iai.06195-11
Liu, Q., Shi, W. K., Ren, S. Z., Ni, W. W., Li, W. Y., Chen, H. M., et al. (2018). Arylamino containing hydroxamic acids as potent urease inhibitors for the treatment of Helicobacter pylori infection. Eur. J. Med. Chem. 156, 126–136. doi:10.1016/j.ejmech.2018.06.065
Maier, R. J., and Benoit, S. L. (2019). Role of nickel in microbial pathogenesis. Inorg 7, 80. doi:10.3390/inorganics7070080
Matczuk, D., and Siczek, A. (2021). Effectiveness of the use of urease inhibitors in agriculture: a review. Intern. Agrophys. 35, 197–208. doi:10.31545/intagr/139714
Mazzei, L., Musiani, F., and Ciurli, S. (2020). The structure-based reaction mechanism of urease, a nickel dependent enzyme: tale of a long debate. J. Biol. Inorg. Chem. 25, 829–845. doi:10.1007/s00775-020-01808-w
Moghadam, E. S., Al-Sadi, A. M., Talebi, M., Amanlou, M., Shongwe, M., Amini, M., et al. (2022). Piperazine-based semicarbazone derivatives as potent urease inhibitors: design, synthesis, and bioactivity screening. Lett. Drug. Des. Disc. 19, 1111–1120. doi:10.2174/1570180819666220405234009
Moulishankar, A., and Thirugnanasambandam, S. (2023). Quantitative structure activity relationship (QSAR) modeling study of some novel thiazolidine 4-one derivatives as potent anti-tubercular agents. J. Recept. Signal Transduct. Res. 43 (3), 83–92. doi:10.1080/10799893.2023.2281671
Munawwar, A., Sajjad, A., Rasul, A., Sattar, M., and Jabeen, F. (2023). Dissecting the role of SMYD2 and its inhibitor (LLY-507) in the treatment of chemically induced non-small cell lung cancer (NSCLC) by using Fe3O4 nanoparticles drug delivery system. Pharm 16, 986. doi:10.3390/ph16070986
Naseer, A., Osra, F. A., Awan, A. N., Imran, A., Hameed, A., Ali Shah, S. A., et al. (2022). Exploring novel pyridine carboxamide derivatives as urease inhibitors: synthesis, molecular docking, kinetic studies and ADME profile. Pharmaceuticals 15, 1288. doi:10.3390/ph15101288
Rauf, M. K., Talib, A., Badshah, A., Zaib, S., Shoaib, K., Shahid, M., et al. (2013). Solution-phase microwave assisted parallel synthesis of N, N′-disubstituted thioureas derived from benzoic acid: biological evaluation and molecular docking studies. Eur. J. Med. Chem. 70, 487–496. doi:10.1016/j.ejmech.2013.10.012
Righetto, R. D., Anton, L., Adaixo, R., Jakob, R. P., Zivanov, J., Mahi, M. A., et al. (2020). High-resolution cryo-EM structure of urease from the pathogen Yersinia enterocolitica. Nat. Commun. 11, 5101. doi:10.1038/s41467-020-18870-2
Saddique, F. A., Ahmad, M., Ashfaq, U. A., Muddassar, M., Sultan, S., and Zaki, M. E. (2022). Identification of cyclic sulfonamides with an N-arylacetamide group as α-glucosidase and α-amylase inhibitors: biological evaluation and molecular modeling. Pharm 15, 106. doi:10.3390/ph15010106
Sohrabi, M., Nazari Montazer, M., Farid, S. M., Tanideh, N., Dianatpour, M., Moazzam, A., et al. (2022). Design and synthesis of novel nitrothiazolacetamide conjugated to different thioquinazolinone derivatives as anti-urease agents. Sci. Rep. 12, 2003. doi:10.1038/s41598-022-05736-4
Sukri, A., Hanafiah, A., Patil, S., and Lopes, B. S. (2023). The potential of alternative therapies and vaccine candidates against Helicobacter pylori. Pharmaceuticals 16, 552. doi:10.3390/ph16040552
Swanson, D. M., Dubin, A. E., Shah, C., Nasser, N., Chang, L., Dax, S. L., et al. (2005). Identification and biological evaluation of 4-(3-trifluoromethylpyridin-2-yl) piperazine-1-carboxylic acid (5-trifluoromethylpyridin-2-yl) amide, a high affinity TRPV1 (VR1) vanilloid receptor antagonist. J. Med. Chem. 48, 1857–1872. doi:10.1021/jm0495071
Uddin, I., Ullah, H., Bibi, A., Taha, M., Khan, F., Rahim, F., et al. (2020). Synthesis, in vitro alpha glucosidase, urease activities and molecular docking study of bis-indole bearing Schiff base analogs. Chem. Dat. Collect. 28, 100396. doi:10.1016/j.cdc.2020.100396
Volkamer, A., Kuhn, D., Rippmann, F., and Rarey, M. (2012). DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment. Bioinformat 28, 2074–2075. doi:10.1093/bioinformatics/bts310
Zaman, K., Rahim, F., Taha, M., Ullah, H., Wadood, A., Nawaz, M., et al. (2019). Synthesis, in vitro urease inhibitory potential and molecular docking study of Benzimidazole analogues. Bioorg. Chem. 89, 103024. doi:10.1016/j.bioorg.2019.103024
Zhou, C., Bhinderwala, F., Lehman, M. K., Thomas, V. C., Chaudhari, S. S., Yamada, K. J., et al. (2019). Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 15, e1007538. doi:10.1371/journal.ppat.1007538
Keywords: pyridylpiperazine hybrid derivatives, heterocyclic compounds, urease inhibition, molecular docking, ADMET studies
Citation: Akash M, Zaib S, Ahmad M, Sultan S and Al-Hussain SA (2024) Synthesis and biological evaluation of pyridylpiperazine hybrid derivatives as urease inhibitors. Front. Chem. 12:1371377. doi: 10.3389/fchem.2024.1371377
Received: 16 January 2024; Accepted: 23 February 2024;
Published: 13 March 2024.
Edited by:
Manuel Aureliano, University of Algarve, PortugalReviewed by:
Marcela Cristina De Moraes, Fluminense Federal University, BrazilLuiz Antonio Escorteganha Pollo, Federal University of Santa Catarina, Brazil
Copyright © 2024 Akash, Zaib, Ahmad, Sultan and Al-Hussain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matloob Ahmad, bWF0bG9vYi5haG1hZEBnY3VmLmVkdS5waw==; Sami A. Al-Hussain, c2FodXNzYWluQGltYW11LmVkdS5zYQ==