 Tomáš Vašíček
Tomáš Vašíček Benjamin Arensmeyer
Benjamin Arensmeyer Alla Zamyatina
Alla Zamyatina- Department of Chemistry, Institute of Organic Chemistry, University of Natural Resources and Life Sciences, Vienna, Austria
Trehalose-containing glycans play an essential role in bacterial pathogenesis, host-pathogen interaction, and cell signaling. The investigation of trehalose uptake and metabolism in Mycobacteria using synthetic desymmetrized trehalose probes is an important approach for the development of diagnostic tools and potential therapeutics for tuberculosis. Trehalose-derived mycobacterial glycolipids activate the innate immune response through recognition by the C-type lectin Mincle, justifying efforts to develop novel trehalose-based Mincle-dependent adjuvants. The chemical synthesis of trehalose-based glycoconjugates, glycolipids, and small-molecule trehalose probes requires the challenging chemical desymmetrization of eight hydroxyl groups in a C2-symmetric disaccharide αGlc(1↔1)αGlc. Using a novel set of orthogonal protecting groups, we developed a flexible multiscale synthetic approach to a collection of differently and variably protected fully desymmetrized trehalose derivatives, ready for final chemical modification with relevant functional or reporter groups. Using a regioselective and site-specific protecting group strategy, we performed multiple symmetry-breaking operations, resulting in a library of trehalose-derived orthogonally protected building blocks as a versatile source for the synthesis of complex trehalose-containing glycans.
1 Introduction
The manipulation of protecting groups is a common and convenient way to achieve the synthesis of complex biomolecules, including carbohydrates. In carbohydrate chemistry, protecting groups are often applied to impart specific chemical properties to synthetic intermediates, including controlling stereoselectivity in glycosylation reactions and manipulating the reactivity of donor and acceptor molecules (Zhang et al., 1999; Jensen et al., 2004; Pedersen et al., 2007; Wu and Wong, 2011). One of the challenges in carbohydrate synthesis is the selection of multiple orthogonal protecting groups (Ágoston et al., 2016), that can ensure the regio- and chemo-selective deprotection of a single hydroxyl or amino group used as a branching point in the oligosaccharide synthesis (Boltje et al., 2010) or as a site for attachment of functional groups to assemble complex glycoconjugates. The orthogonally protected monosaccharide building blocks are commonly used as a starting point for the assembly of biomolecules, but naturally occurring disaccharides can also be subject to protective group manipulation to synthesize highly functionalized products. One of the most representative examples is a non-reducing non-mammalian disaccharide trehalose (αGlc(1↔1)αGlc).
Trehalose-containing glycans and glycolipids found in Mycobacteria (Nobre et al., 2014), fungi (Yang et al., 2012), and worms (Penkov et al., 2010) and more recently in Salmonella (Reinink et al., 2019) are important virulence factors (Thanna and Sucheck, 2016; Vanaporn and Titball, 2020) that play an essential role in host-pathogen interaction (Gilleron et al., 2004; Gilmore et al., 2012; Dai et al., 2020), cell signaling (Schoenen et al., 2014; Kodar et al., 2017), and bacterial pathogenesis (Kalscheuer et al., 2010) and are therefore important synthetic targets. The pattern-recognition receptor, monocyte-inducible C-type lectin (Mincle) expressed on macrophages and other immune cells, plays a key role in immunity to Mycobacterium tuberculosis which produces abundant trehalose-based glycolipids, such as trehalose dimycolate (TDM) (Ishikawa et al., 2009; Schoenen et al., 2010; Lee et al., 2012). Mincle has been identified as a specific receptor for TDM responsible for triggering the production of the pro-inflammatory mediators via the Syk/CARD9 signaling pathway (Yamasaki et al., 2008; Werninghaus et al., 2009; Ostrop et al., 2015). The structural basis for Mincle activation by trehalose glycolipids is a subject of intensive research (Feinberg et al., 2013; Furukawa et al., 2013; Feinberg et al., 2016) and the potential of Mincle to activate the innate immune response has opened up new opportunities for the development of novel trehalose-derived vaccine adjuvants (Desel et al., 2013; Decout et al., 2017; Tima et al., 2017; Desel et al., 2022). Synthetic mycobacterial sulfolipid antigens and analogs (sulfoglycolipids based on the unsymmetrically derivatised trehalose) may be important candidates for tuberculosis vaccines (Guiard et al., 2008; Seeliger et al., 2012). The study of the trehalose pathway and metabolism in Mycobacteria, an important approach to tuberculosis diagnosis and drug development, necessitates the synthetic preparation of various desymmetrized trehalose probes (Backus et al., 2011; Swarts et al., 2012; Dai et al., 2020; Banahene et al., 2023).
The chemical synthesis of trehalose derivatives as part of glycoconjugates and parasitic glycans or trehaloses equipped with reporter groups for biorthogonal chemistry require either a complicated chemical glycosylation to assemble the double anomeric α,α-1,1′-glycosidic linkage that is difficult to perform in a stereoselective manner or a challenging desymmetrization of some of the eight hydroxyl groups in a C2-symmetric non-reducing disaccharide (Lin et al., 2007; Geerdink and Minnaard, 2014). The latter approach was extensively exploited, allowing the synthesis of many important biomolecules such as mycobacterial sulfolipids (Geerdink and Minnaard, 2014; Sarpe and Kulkarni, 2014) and trehalose-containing lipids (Mishra et al., 2019; Jana and Kulkarni, 2020) as well as the preparation of trehaloses modified with biorthogonal or fluorescent moieties as probes for studying the mycobacterial trehalome (Swarts et al., 2012; Dai et al., 2020; Carlier et al., 2022). However, most of the synthetic routes were based on a partial desymmetrization by differentiation of specific hydroxyl groups or on a direct substitution of the particular positions with a specific functional group (phosphate, lipid chain, reporter group, etc.) to produce the desired biomolecule.
Given the continuous progress in the study of trehalose-dependent mycobacterial biology and the discovery of new trehalose-recognizing proteins (such as the LpqY-SugABC transporter) (Parker et al., 2020; Furze et al., 2021; Pohane et al., 2021), as well as the ongoing efforts to develop novel Mincle-dependent vaccine adjuvants, a practical, easily reproducible and scalable approach to the synthesis of multipurpose desymmetrized trehalose scaffolds is in great demand. With this in mind, we aimed to develop a straightforward and efficient multi-gram-scale synthetic approach towards fully desymmetrized orthogonally protected trehaloses, which can then be used as versatile building blocks in the assembly of complex trehalose-based glycans, or as scaffolds for the introduction of reporter groups for biorthogonal chemistry and the study of trehalose pathways in mycobacteria.
2 Results and discussion
To design the synthetic strategy, we considered both the chemical and structural properties of the non-reducing C2-symmetric disaccharide trehalose. Its tertiary structure, elucidated by X-ray- and molecular dynamics simulation studies, is governed by an exceptional rigidity of the α,α-(1↔1′) glycosidic linkage and very specific torsion angles around it (Brown et al., 1972; Dowd et al., 1992). A preferred gauche-gauche conformation with respect to the values of the torsion angles around the α,α-(1↔1′) glycosidic bond imposes a skewed relative orientation of two glucose rings and is largely dependent on the anomeric effect (Figure 1). (French et al., 2002; Nunes et al., 2010) This arrangement is valid for variably functionalized trehaloses, corresponds to a single conformational minimum, and is not influenced by the nature of the protecting or functional groups (Baddeley et al., 2003; Färnbäck et al., 2004). Therefore, the derivatization of multiple hydroxyl groups in trehalose with bulky substituents should not lead to significant deviations from a 4C1 conformation of the pyranose rings (as is characteristic of, e.g., triple TBDMS-substituted pyranoses (Pedersen et al., 2007; Bols and Pedersen, 2017)). Therefore, if the 4C1 conformation of both Glc rings is additionally supported by a conformational lock provided by 4,6-O-cyclic protecting groups, the steric clash that would occur between bulky protecting groups on C2-OH and C2′-OH should allow the regioselective mono-substitution of either of positions 2/2′ (Figure 1).

FIGURE 1. Desymmetrization design based on the conformational properties of α,α-trehalose.
Thus, our approach to trehalose desymmetrization was designed to rely on an initial C2-symmetry breaking operation by differentiating C-4/C-6 diols on both glucose units. To this end, we used two different cyclic protecting groups: benzylidene acetal to protect the C4´/C6′-OH on one glucose moiety and di-tert-butylsilylene (DTBS) protecting group to mask the C4/C6-OH groups on the other (Figure 1). The resulting partially desymmetrized intermediate 2 would then have two pairs of free hydroxyl groups, C2/C2′-OH and C3/C3′-OH, which would have to be differentiated by regioselective introduction of the orthogonal protecting groups. Considering the crystal structures of free- and differently substituted α,α-trehaloses (Brown et al., 1972; Baddeley et al., 2003; Färnbäck et al., 2004), we assumed that the application of bulky protecting groups would allow the differentiation of positions 2 and 2′ due to a likely steric clash between bulky substituents.
Along these lines, the C-4/C-6 diol in one of the glucose moieties was initially protected by reaction with benzaldehyde dimethylacetate in the presence of camphor sulfonic acid to give monobenzylidene acetal 1 in 75% yield (BRSM), while the unreacted trehalose was fully recovered. The synthesis of trehalose 4,6-O-benzylidene acetal 1 starting from trehalose has been reported previously (Cheng, 1976), albeit in low yield and without indication of the purity and spectroscopic data of the isolated material. Alternatively, 1 was obtained as a by-product in the synthesis of symmetric trehalose 4,6/4′6′- dibenzylidene acetal or by partial hydrolysis or reductive opening of one of the benzylidene groups in 2,3/2′3′-protected trehalose dibenzylidene acetals (Richardson and Tarelli, 1971; Hadfield et al., 1978; Lin et al., 2007; Sarpe and Kulkarni, 2013). Here, we report the first targeted and highly efficient synthesis of trehalose 4,6-O-monobenzylidene acetal 1 on a 10 g scale (Supplementary Scheme 1). The second α-D-Glc unit in 1 was modified at the C4/C6 hydroxyl groups by the introduction of a di-tert-butylsilylene (DTBS) protecting group to generate 2 (Scheme 1). To further desymmetrize the molecule and to conform with the principles of orthogonality, we selected several temporary protecting groups with the intention to regioselectively protect either of positions C2/C3 and C2´/C3´ in compound 2. Attempts to regioselectively introduce 2,2,2-trichloroethoxycarbonyl (Troc), 4-oxopentanoyl (Lev), triisopropylsilyl ether (TIPS), or 2-naphthylmethyl ether (Nap) protecting groups resulted in complex mixtures with a predominant formation of tetrasubstituted derivatives. It then became apparent that the use of a less reactive but more sterically hindered reagent could help to achieve the desired symmetry-breaking effect. Indeed, the reaction of 2 with tert-butyldimethysilyl chloride (TBDMSCl) in the presence of imidazole (Im) at 60°C led to the formation of the 3,3′-substituted disaccharide 3 in an excellent 89% yield (Scheme 1). As we aimed at the synthesis of trehalose derivatives lacking the C2-symmetry, the additional substitution of either C2-OH or C2′-OH groups was achieved by increasing the concentration of TBDMSCl in the reaction solution to 2.8 M, which afforded a mixture of tri-substituted TBDMS-protected regioisomers 4 and 5 in 78% yield in a ratio 2:1 (according to the 1H-NMR analysis). The remarkable regioselectivity of this transformation was confirmed by using a large excess of reagents (up to 20 equiv., i.e., 5 equiv. per OH group) and prolonged reaction times – even under these conditions, the tetrasubstituted product (2,2′,3,3′-O-TBDMS-protected) was not detected.
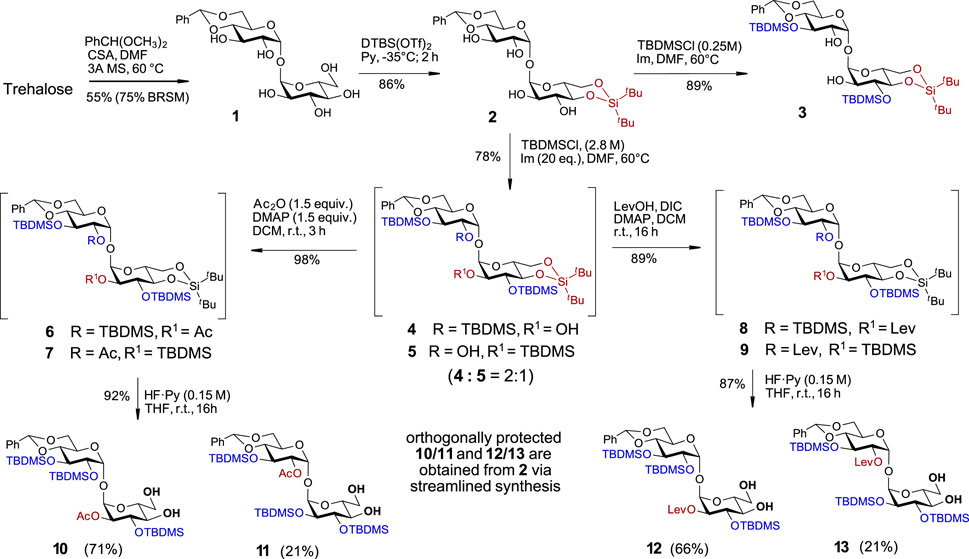
SCHEME 1. Initial symmetry-breaking transformations to differentiate C4,C6/C4′,C6′- and C2/C2′- hydroxyl groups.
To streamline the synthesis, the mixture of partially TBDMS-protected disaccharides 4 and 5 was further derivatized without intermediate separation of individual regioisomers. The unsubstituted C2/C2′-OH groups in 4 and 5 were protected with sterically undemanding substituents - acetate or levulinate ester groups. To this end, a mixture 4 + 5 was treated with acetic anhydride/DMAP to give a mixture of regioisomers 6 + 7 or was subjected to N,N′-diisopropylcarbodiimide (DIC)/DMAP–promoted acylation with levulinic acid to afford C-2/C-2′ Lev-protected regioisomers 8 and 9. In the next step, also performed with the mixtures of regioisomers 6 + 7 and 8 + 9, we removed the conformational lock in a form of the 4,6-O-DTBS protecting group with the intention of 'relaxing’ the 4C1 conformation on one side of the molecule to allow for further desymmetrization based on the different electronic and steric effects in a 4C1-locked and “unlocked” Glc moieties. To this end, we studied the conditions for the chemoselective cleavage of 4,6-O-DTBS protecting group in the presence of three secondary TBDMS groups. Screening different reagents (HF·Py, Et3N‧3HF, TBAF) and reaction solvents (THF, DMF) revealed that using HF·Py in THF guarantees the desired chemoselectivity, while the concentration of the fluoride reagent in the reaction solution, rather than the number of equivalents of HF·Py, is decisive for the chemoselective cleavage of the 4,6-O-DTBS group while leaving three TBDMS groups intact. Accordingly, treatment of the regioisomeric mixture 6 + 7 with a diluted solution of HF·Py (c 0.15 M in the reaction solution) in THF led to the chemoselective cleavage of the DTBS group to afford 4,6-diols 10 and 11, which were isolated in 71% and 21% yield, respectively (92% overall yield). Similarly, the mixture of levulinoyl derivatives 8 + 9 was treated with a diluted solution of HF·Py to generate the diols 12 and 13 in 66% and 21% isolated yields, respectively (87% overall yield) (Scheme 1). The excellent chemoselectivity of the DTBS group cleavage was confirmed by multigram upscaling of the transformation, where no TBDMS-deprotected or partially TBDMS-deprotected by-products were detected under the proposed reaction conditions.
With four partially desymmetrized 4,6-diols 10, 11, 12, and 13 in hand, we sought an efficient approach to regioselectively introduce an orthogonal protecting group onto the primary C6-OH. Due to a likely steric clash between the substituents at C2′ and C6 after the removal of the conformational lock provided by the 4,6-O-DTBS group (Figure 2), we had to rely on the use of sterically small substituents. However, the most reliable protocols for the differentiation of 4,6-diols include the use of bulky protecting groups which can be regioselectively attached to the primary C6-OH. To fulfil the criteria of orthogonality, we thought that a sterically small allyloxycarbonyl (Alloc) protecting group would be a good alternative and studied the possibilities for a regioselective protection of C6-OH using allyl chloroformate in the presence of several traditional hindered bases. Among bases tested, sym-collidine showed the best result, since the desired monosubstituted derivatives 14 and 15 were formed in 76% yields. Even when a large excess of AllocCl/collidine (5 eq. each) was applied, no detectable 4-O-Alloc-substituted by-products were formed confirming the true chemo- and regioselectivity of the transformation (Scheme 2). A similar result for the regioselective introduction of the allyloxycarbonyl group at the primary C6-OH was obtained from the reaction of 4,6-diols 11 and 13, which gave the 6-O-Alloc derivatives 20 (78%) and 21 (88%), respectively. The remaining unsubstituted C4′-OH and C4-OH groups in the 2/2′-levulinated disaccharides 15 and 21 were reacted with TrocCl in pyridine to give the orthogonally protected regioisomers 17 and 23 in excellent yields of 98% and 96%, respectively. The 2/2′-acetylated compounds 14 and 20 were treated with levulinic acid under standard acylation conditions (DIC/DMAP) to furnish the fully orthogonally protected 16 and 22. All transformations were performed in multigram scale to ensure high efficiency and reproducibility.

FIGURE 2. Toward orthogonally protected trehalose lacking the C2-symmetry, retrosynthetic scheme.
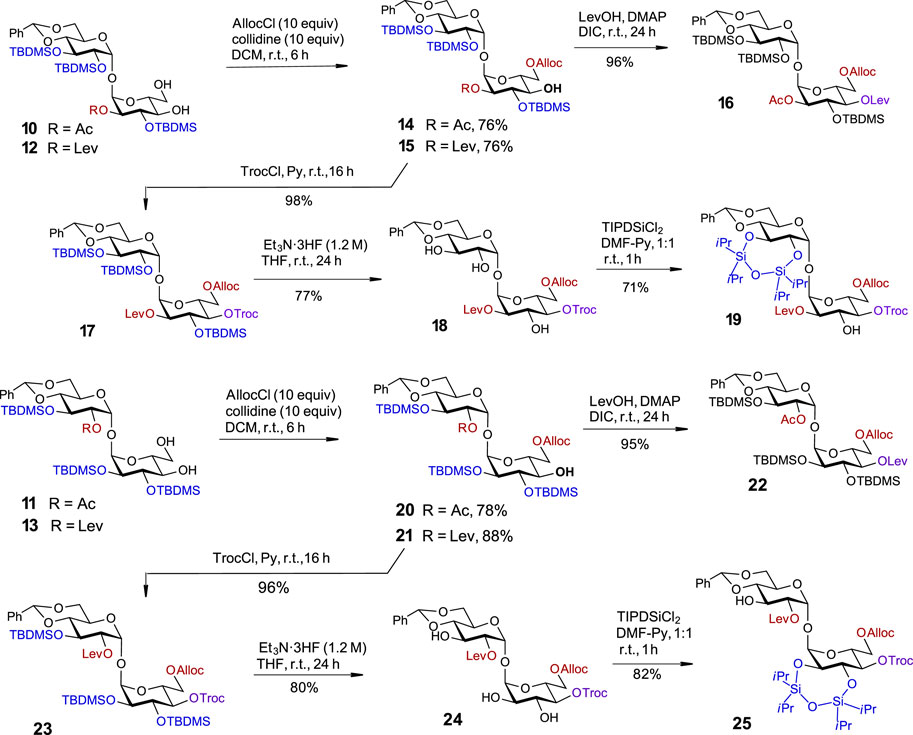
SCHEME 2. Third symmetry-breaking operation to fully differentiate two glucose moieties. Synthesis of partially desymmetrized orthogonally protected 17 and 23. Synthesis of fully desymmetrized orthogonally protected trehalose building blocks 19 and 25.
Given the propensity of the acetyl protecting group to migrate under a variety of chemical conditions (Lassfolk and Leino, 2023; Lassfolk and Leino, 2023), the orthogonally protected 2,2′-O-levulinoyl derivatives 17 and 23 were selected for the development of an advanced synthetic route. To pave the way for further desymmetrization, the TBDMS groups in 17 and 23 were removed, and the adjacent hydroxyl groups in 18 and 24 at C2´/C3´ and C2/C3, respectively, were protected as cyclic TIPDS ethers. To this end, the treatment of 2-O-Lev-protected 17 with a concentrated solution of Et3N‧3HF (c 1.2 M in the reaction solution) enabled the removal of all TBDMS groups and the formation of a triol 18 in 77% yield. A reaction of 18 with TIPSiCl2 in DMF-Py afforded the 2,3-O-TIPDS protected 19 in 71% yield. Similar transformations were performed with the 2′-O-Lev-protected regioisomer 23 which led to the formation of triol 24 in 80% yield. After the reaction of the latter with TIPSiCl2 in the presence of pyridine, the desymmetrized fully orthogonally protected trehalose 25 was obtained in 82% yield (Scheme 3).
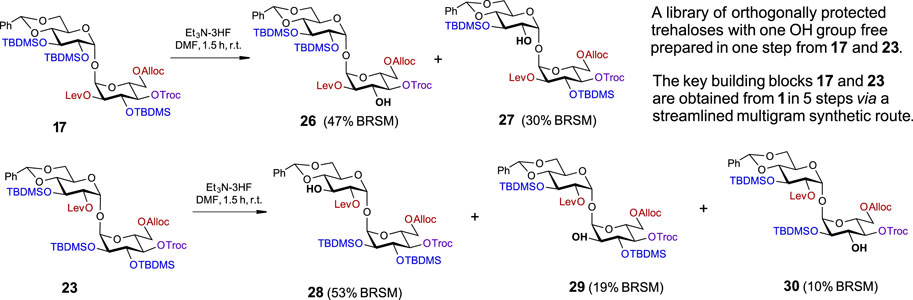
SCHEME 3. A one-step approach to a set of orthogonally protected trehaloses with one hydroxyl group free.
Using four major symmetry-breaking operations, we developed an uncomplicated, efficient, and easily reproducible synthetic route to a library of partially and fully desymmetrized orthogonally protected trehaloses (Figure 3). Both the fully orthogonally protected disaccharides 19 and 25 and the partially protected trehaloses 10/11, 12/13, 14/15, 16/17, 20/21, and 22/23 in Schemes 1, 2 represent valuable building blocks that can be used in the assembly of complex trehalose-containing glycans and glycoconjugates, depending on the needs of a particular synthetic route.

FIGURE 3. Synthetic steps to fully desymmetrized trehalose using four symmetry-breaking operations.
In order to gain access to a series of orthogonally protected trehalose derivatives with a single free OH group at a specific position in one step, we explored the possibility of regioselective deprotection of the TBDMS groups in the sterically tensed 17 and 23 (Scheme 3). As we have already established for the use of HF·Py for the chemoselective deprotection of the cyclic 4,6-O-DTBS group (Scheme 1), the total concentration of the fluoride reagent in the reaction solution was crucial for the chemo- and regioselectivity of transformations. Since the removal of all three TBDMS groups in 17 required the use of a 1.2 M solution of [3HF·Et3N] in THF, the reagent concentration was reduced to 0.8 M and then to 0.25 M, which required longer reaction times (Supplementary Table S1). Changing the solvent to DMF and using 0.2 M [3HF·Et3N] accelerated the reaction rate and gave an easily separable mixture of partially protected compounds 26 and 27 with a free OH group in position 3 or 2′ in 47% and 30% isolated yields (BRSM), respectively (Scheme 3). The sites of attachment of silyl groups were confirmed by 1H-29Si HMBC-NMR. Similarly, the treatment of a fully protected 23 with low concentration [3HF·Et3N] (0.20 M) in DMF allowed for the synthesis of C3′-OH compound 28 as the major product (53% BRSM) along with C2-OH derivative 29 (19% BRSM) and C3-OH product 30 (10% BRSM), while the unreacted starting material was completely recovered (Supplementary Table S2). In this way, five different orthogonally protected trehalose building blocks were straightforwardly prepared from intermediates 17 and 23.
In conclusion, using a novel set of orthogonal protecting groups, we developed a flexible multiscale synthetic route to a collection of differently and variably protected fully desymmetrized trehalose derivatives ready for final chemical derivatization with functional or reporter groups for biorthogonal chemistry. These trehalose building blocks serve as versatile precursors for a variety of biomolecules important for studying host-pathogen interactions, e.g., in Mycobacteria or other pathogens. The library of eleven desymmetrized variably orthogonally protected trehalose building blocks that can be routinely prepared via a streamlined synthetic route in multigram scale provides straightforward access to the synthesis of naturally occurring trehalose-based biomolecules and analogues thereof.
3 Experimental section
General synthetic methods. Reagents and solvents were purchased from commercial suppliers and used without further purification unless otherwise stated. Toluene was dried by distillation first over phosphorus pentoxide, then over calcium hydride, and was then stored over activated 4 Å molecular sieves (MS). Solvents were dried by storage over activated MS for at least 24 h prior to use (dichloromethane 4 Å, acetonitrile, and DMF 3 Å). Residual moisture was determined by coloumbometric titration on a Mitsubishi CA21 Karl Fischer apparatus and did not exceed 20 ppm. Reactions were monitored by TLC performed on silica gel 60 F254 HPTLC precoated glass plates with a 25 mm concentration zone (Merck). Spots were visualized by dipping into a sulfuric acid–p-anisaldehyde solution and subsequent charring at 250 °C. Solvents were removed under reduced pressure at ≤40 °C. Preparative HPLC was performed on a YMC Pack SIL-06 250 × 20 mm, S-5 μm, 6 nm column or on a YMC Pack SIL-06 250 × 10 mm, S-5 μm, 6 nm column. Preparative MPLC and column chromatography were performed using silica gel 60 (0.040–0.063 mm). NMR spectra were recorded on a Bruker Avance III 600 spectrometer (1H at 600.22 MHz; 13C at 150.93 MHz; 31P at 242.97 MHz) using standard Bruker NMR software. Chemical shifts are reported in ppm, where 1H NMR spectra recorded from samples in CDCl3 were referenced to internal TMS and 13C spectra were referenced to the corresponding solvent signal (77.16 ppm for CDCl3). NMR spectra recorded from samples in other solvents were referenced to residual solvent signals (for CD3OD 3.31 and 49.00 ppm; for CD2Cl2 5.32 and 53.84 ppm; for DMSO-d6 2.50 and 39.52 ppm; for 1H and 13C NMR, respectively). NMR spectra recorded in CDCl3-MeOD (4:1, v/v) were referenced to residual solvent signals of CDCl3 (7.26 ppm and 77.16 ppm; 1H and 13C NMR, respectively). NMR spectra recorded in CDCl3: MeOD (1:1 to 4:1, v/v) were referenced to residual solvent signals of MeOD (3.31 and 49.00 ppm, 1H and 13C NMR, respectively). 31P NMR spectra were referenced according to IUPAC recommendations from a referenced 1H-NMR spectrum. In all 1,1′-disaccharides, the NMR signals of the Glc ring on the left (benzylidene acetal-protected) is indicated by primes. Centrifugal partition chromatography (CPC) was performed on a SCPC-100 device with 100 mL column volume, a 3,000 rpm rotation speed, and 100 mg–1 g injection range (ARMEN, AlphaChrom). High-resolution mass spectrometry (HRMS) was carried out on acetonitrile solutions via LC-TOF MS (Agilent 1200SL HPLC and Agilent 6210 ESI-TOF, Agilent Technologies). Datasets were analyzed using Agilent Mass Hunter Software. MALDI-TOF MS was performed in negative-ion mode using a Bruker Autoflex Speed instrument with 6-aza-2-thiothymine (ATT) as matrix and ammonium sulfate as additive. Optical rotation was measured on an Anton Paar MCP 100 polarimeter featuring integrated Peltier temperature control. All [α]D20 values are reported in units of deg·dm−1 cm3 g−1; the corresponding concentrations are reported in g/100 mL.
4,6-O-Benzylidene-α-D-glucopyranosyl-(1↔1)-α-D-glucopyranoside (1). Trehalose dihydrate (10.0 g, 26.44 mmol) was dissolved in dry DMF (264 mL, c = 0.100 M) and stirred with crushed molecular sieves (3Å; 1.00 g) at r. t. for 12 h. Benzaldehyde dimethyl acetal (DMT) (8.04 mL, 52.87 mmol) and camphor-10-sulfonic acid (CSA) (1.840 g, 7.93 mmol) were added and the reaction mixture was stirred at 60°C for 2 h. The reaction mixture was brought to r. t. and Et3N (4.2 mL, 29.08 mmol) was added dropwise. The stirring was continued for 15 min, the solids were removed by filtration over a pad of Celite, and the solution was concentrated. The residue was repeatedly co-evaporated from toluene/MeOH (110 mL, 10/1) to completely remove the reaction solvent DMF (the residue was taken up in MeOH (10 mL) and diluted with toluene (100 mL), and the solution was concentrated to dryness). The residue was partitioned between H2O/EtOAc (400 mL, 3/1), the phases were separated, and the organic phase was washed with H2O (3 × 50 mL). The combined aqueous phases were reextracted with EtOAc (2 × 50 mL), and concentrated. The residue was dissolved in MeOH/H2O (60 mL, 8/2) and purified by ion-exchange chromatography using AG 1-X8 resin (OH)- - form (4 × 15 cm), eluent: MeOH/H2O (8/2) to remove the residual CSA. The fractions were collected and concentrated to dryness, and the residue was purified by i) column chromatography on silica gel or ii) centrifugal partition chromatography (CPC).
Purification by column chromatography on silica gel: the residue was taken up in (CH2Cl2-MeOH-NH4OH, 6/4/1) and loaded on a silica gel column (200 g, eluent: CH2Cl2-MeOH-NH4OH, 6/4/1 → 4/6/1) to afford 1 (5.9 g) and trehalose (2.9 g). The yield of 1 based on recovered starting material (BRSM) is 70%. Rf = 0.51 (CH2Cl2-MeOH-NH4OH, 6/4/0.8, v/v/v); Rf = 0.32 (iPrOH-H2O-NH4OH, 7/2/1, v/v/v);
Purification by centrifugal partition chromatography (CPC). The biphasic liquid system (800 mL) was prepared by equilibration in the separation funnel the three-component solvent mixture EtOAc/MeOH/H2O (20/7/20). The separation was performed in descending mode with the upper (organic) phase serving as the stationary phase and the lower (aqueous) phase serving as the mobile phase. The column was initially conditioned by pumping the upper phase in descending mode at a rotation speed of 500 rpm and a flow rate of 30 mL min−1. The 5 mL sample (9 g of crude material [1 + Tre] was dissolved in 45 mL of the solvent mixture (EtOAc-MeOH-H2O, 20/7/20, v/v/v; 23 mL of upper phase and 22 mL of lower phase; injection volume 5 mL/1 g) was injected into the CPC column. The separation was performed at a rotation speed of 2,000 rpm and a mobile phase (aqueous phase) flow rate of 6 mL min−1. Samples [1 + Tre] of 1 g (5 mL) each were injected repeatedly into the equilibrated CPC column to give, after separation, 1 (5.92 g) and trehalose (3,1 g). The yield of 1 based on recovered starting material (BRSM) is 75%.
4,6-O-Benzylidene-α-D-glucopyranosyl-(1↔1)-4,6-O-di-tert-butylsilylene-α-D-glucopyranoside (2). To a stirred solution of 1 (5.0 g; 11.62 mmol) in dry pyridine (116 mL) powdered activated molecular sieves (MS 3Å; 330 mg) were added and the suspension was stirred for 2 h at r. t. in the atmosphere of Ar. The mixture was cooled to −35°C and di-tert-butylsilyl bis(trifluoromethanesulfonate) (568 mL; 17.42 mmol) was added dropwise. The reaction mixture was stirred for 3.5 h at −35°C, then triethylamine (4,2 mL) and MeOH (8,4 mL) were successively added, and the mixture was stirred for 10 min. The reaction mixture was brought to r. t., the solids were removed by filtration over a pad of Celite, and the filtrate was concentrated. The residue was dissolved in EtOAc (350 mL), washed with sat. aq. NaHCO3 (3 × 100 mL), and brine (100 mL), dried over MgSO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene-EtOAc, 1/1 → 2/8) to afford 2 (5.7g, 86%) as a solid. Rf = 0.92 (CHCl3-MeOH-NH4OH, 8/2/0.25, v/v/v); Rf = 0.44 (EtOAc–toluene, 6/4, v/v);
4,6-O-Benzylidene-3-O-tert-butyldimethylsilyl-α-D-glucopyranosyl-(1↔1)-4,6-O-di-tert-butylsilylene-3-O-tert-butyldimethylsilyl-α-D-glucopyranoside (3). To a stirred solution of 2 (3.11 g, 5.45 mmol) in dry DMF (100 mL) powdered 3Å molecular sieves (300 mg), imidazole (2.24 g, 32.90 mmol) and tert-butyldimethylsilyl chloride (4.18 g, 27.7 mmol) were added under the atmosphere of Ar. The reaction mixture was stirred at 60°C for 90 min, then diluted with EtOAc (250 mL) and washed with aq. satd. NaHCO3 (2 × 50 mL) and brine (2 × 50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene - EtOAc, 95/5 → 90/10) to afford 3 (3.66 g, 84%) as a solid. Rf = 0.42 (toluene–EtOAc, 4/1, v/v); Rf = 0.70 (toluene–EtOAc, 3/2, v/v);
4,6-O-Benzylidene-2,3-di-O-tert-butyldimethylsilyl-α-D-glucopyranosyl-(1↔1)-3-O-tert-butyldimethylsilyl-4,6-O-di-tert-butylsilylene-α-D-glucopyranoside (4) and 4,6-O-Benzylidene-3-O-tert-butyldimethylsilyl-α-D-glucopyranosyl-(1↔1)-2,3-di-O-tert-butyldimethylsilyl-4,6-O-di-tert-butylsilylene-α-D-glucopyranoside (5). To a stirred solution of 2 (7.98 g, 13.98 mmol) in dry DMF (100 mL) imidazole (8.1 g, 279.6 mmol), tert-butyldimethylsilyl chloride (42.14 g, 279.6 mmol) and powdered molecular sieves (3Å, 800 mg, 10%) were added under atmosphere of Ar. The reaction mixture was stirred for 22 h at 60°C under atmosphere of Ar, diluted with EtOAc (500 mL), and washed with aq. satd. NaHCO3 (3 × 100 mL) and brine (2 × 100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene -EtOAc, 98/2 → 80/20) to afford a mixture of 4 and 5 (2:1 according to the 1H-NMR analysis), 9.91 g, 78%, as a solid. Rf = 0.43 and 0.38, respectively, (toluene–EtOAc, 95/5, v/v); Rf = 0.67 and 0.62 (hexane–EtOAc, 4/1, v/v); 1H NMR (chemical shifts for compound 4 are indicated by superscript m) (600 MHz, CDCl3): δ [ppm] = 7.47-7.33 (m, PhCH), 5.47, 5.42 (2xs, PhCH), 5.18 (d, 3J1,2 = 3.6 Hz, H-1), 5.10 (d, 1H, 3J1,2 = 3.5 Hz, H-1)m, 5.03 (d, 1H, 3J1′,2′ = 3.0 Hz, H-1′)m, 5.03 (d, 3J1′,2′ = 3.1 Hz, H-1′), 4.41-3.36 (4xH-3, 4xH-4, 4xH-5, 4xH-6), 3.67 (dd, 3J1′,2′ = 3.5 Hz, 3J2′,3′ = 8.7 Hz, H-2′)m, 3.65 (dd, 3J1′,2′ = 3.7 Hz, 3J2′,3′ = 8.7 Hz, H-2′), 3.59 (dd, 3J1′,2′ = 3.1 Hz, 3J2′,3′ = 8.9 Hz, H-2′), 3.58 (dd, 3J1,2 = 3.7 Hz, 3J2,3 = 9.4 Hz, H-2)m, 2.11 (d, 1H, C2-OH)m, 2.03 (d, 1H, C2′-OH), 1.06, 1.05, 1.02, 0.99, 0.97, 0.96, 0.94, 0.87, 0.80 (9xs, [CH3]3C, 3xTBDMS, 1xDTBS), 0.21, 0.17, 0.15, 0.13, 0.08, 0.00 (6xs, 12×CH3, 6xTBDMS); 13C NMR (151 MHz, CDCl3): δ [ppm] = 137.54, 137.42 (Cq, Ph), 128.95, 128.07, 128.00, 126.57, 126.42 (PhCH), 102.45, 102.14 (PhCH), 98.65, 96.62, 95.46, 94.16 (4xC-1), 82.50, 81.79, 78.58, 77.65, 74.70, 74.48, 73.78, 73.77, 73.28, 72.83, 72.24, 71.55, 68.08, 67.12, 63.86, 63.68 (4xC-2, 4xC-3, 4xC-4, 4xC-5), 69.10, 69.07, 66.96, 66.92 (4xC-6), 27.63, 27.61, 27.21, 27.12, 26.64, 26.58, 26.34, 26.13, 26.00, 25.87 (10x [CH3]3C, 6xTBDMS, 2xDTBS), 22.81, 22.78, 20.03, 19.86, 18.45, 18.29 ([CH3]3C, 6xTBDMS, 2xDTBS), −2.87, −3.25, −3.77, −3.90, −3.93, −4.04, −4.27, −4.42, −4.42, −4.54, −4.66, −4.95 (12xCH3, 6xTBDMS); HRMS (+ESI) m/z: calcd. for C45H85O11Si4 [M + H]+ 930.5429, found 930.5434. HRMS (+ESI) m/z: calcd. for C45H85O11Si4 [M + H]+: 930.5429, found 930.5434.
4,6-O-Benzylidene-2,3-di-O-tert-butyldimethylsilyl-α-D-glucopyranosyl-(1↔1)-3-O-tert-butyldimethylsilyl-4,6-O-di-tert-butylsilylene-2-O-(4-oxopentanoyl)-α-D-glucopyranoside (8) and 4,6-O-Benzylidene-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-2,3-di-O-tert-butyldimethylsilyl-4,6-O-di-tert-butylsilylene-α-D-glucopyranoside (9). To a stirred solution of [4 + 5] (9.91 g, 9.80 mmol) in dry CH2Cl2 (23 mL, c = 0.472 M), levulinic acid (3.33 mL, 29.40 mmol), N,N′-diisopropylcarbodiimide (6.72 mL, 39.19 mmol), and 4-(dimethylamino)-pyridine (5.30 g, 39.19 mmol) were added successively. The reaction mixture was stirred for 16 h, then diluted with EtOAc (500 mL) and washed with aq. satd. NaHCO3 (4 × 100 mL), aq. satd. CuSO4 (3 × 50 mL), and brine (3 × 100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene -EtOAc, 95/5 → 80/20) to afford [8 + 9] (9.73 g, 89%) as a mixture. Rf = 0.55 and 0.47 (hexane-EtOAc, 4/1, v/v, both regio-isomers); 1H NMR (the chemical shifts for compound 8 are indicated by superscript m) (600 MHz, CDCl3): δ [ppm] = 7.48-7.32 (m, PhCH), 5.49, 5.43m (2xs, PhCH), 5.19 (d, 1H, 3J1,2 = 3.6 Hz, H-1)m, 5.15 (d, 3J1′,2′ = 3.4 Hz, H-1′), 5.05 (d, 1H, 3J1′,2′ = 2.8 Hz, H-1′)m, 4.96 (d, 3J1,2 = 3.0Hz, H-1), 4.91 (dd, 3J2,3 = 9.6 Hz, 3J1,2 = 3.4 Hz, H-2), 4.79 (dd, 1H, 3J2,3 = 9.8 Hz, 3J1,2 = 3.6 Hz, H-2)m, 4.30-3.36 (4xH-3, 4xH-4, 4xH-5, 4xH-6), 3.66 (dd, 3J2′,3′ = 8.9 Hz, 3J1′,2′ = 2.7 Hz, H-2′)m, 3.58 (dd, 3J2,3 = 9.1 Hz, 3J1,2 = 3.1 Hz, H-2), 2.77- 2.62 (m, 2xCH2, Lev), 2.19, 2.02 (s, CH3, Lev), 1.07, 1.07, 1.02, 1.01, 0.97, 0.95, 0.94, 0.93 0.81, 0.81 (10×s, [CH3]3C, 6xTBDMS, 2xDTBS), 0.19, 0.18, 0.18, 0.17, 0.16, 0.16, 0.13, 0.04, 0.03, 0.03 (10×s, 12×CH3, 6xTBDMS); 13C NMR (151 MHz, CDCl3): δ [ppm] = 205.95m, 205.66 (CH3C = O, Lev), 172.38m, 172.33 (C=O, Lev), 137.35, 137.32m (Ph-Cq), 129.04m, 128.97, 128.08m, 128.04, 2 × 126.46 (CH, Ph), 102.60m, 102.33 (PhCH), 97.69, 97.69, 94.94, 93.42m (4xC-1), 82.69, 82.56, 78.51, 78.06, 74.32, 73.92, 73.80, 73.70, 73.48 71.42, 71.19, 69.01, 68.23, 67.57, 64.18, 63.47 (4xC-2, 4xC-3, 4xC-4, 4xC-5), 69.12, 69.07m, 66.88, 66.75m (4xC-6), 37.69, 37.53m (CH2, Lev), 29.82, 29.66m (CH3, Lev), 27.84m, 27.80 (CH2, Lev), 27.90, 27.67, 27.24, 27.21, 26.72, 26.65, 26.41, 26.14, 25.80, 25.66 (10x [CH3]3C], 6xTBDMS, 2xDTBS), 22.90, 22.84, 19.80, 19.80, 18.86, 18.82, 18.47, 18.39, 18.14, 18.11 (10x [CH3]3C, 6xTBDMS, 2xDTBS), −2.73, −3.16, −3.33, −3.87, −3.92, −3.98, 4.05, −4.42, −4.48, −4.55, −4.74, −4.95 (12xCH3, 6xTBDMS); HRMS (+ESI) m/z: calcd. for C50H91O13Si4 [M + H]+: 1011.5531, found 1011.5536; calcd. for C50H94NO13Si4 [M + NH4]+: 1028.5797, found 1028.5814; calcd. for C50H90KO13Si4 [M + K]+ 1049.5090, found 1049.5089.
4,6-O-Benzylidene-2,3-di-O-tert-butyldimethylsilyl-α-D-glucopyranosyl-(1↔1)-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-α-D-glucopyranoside (12) and 4,6-O-Benzylidene-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-2,3-di-O-tert-butyldimethylsilyl-α-D-glucopyranoside (13). To a stirred solution of [8 + 9] (2.94 g, 2.91 mmol) in dry THF (12 mL) in a PTFE reaction vessel, a solution of HF·pyridine (240 μL, 0.152 M) was added at 0°C. The reaction mixture was stirred for 16 h at r. t. and concentrated. The residue was dissolved in EtOAc (250 mL), washed with aq. satd. NaHCO3 (3 × 50 mL), aq. satd. CaCl2 (3 × 30 mL), and brine (2 × 50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene -EtOAc, 98/2 → 75/25) to afford 12 (1.670 g, 66%) as a solid and 13 (0.520 g, 21%) as a solid.
12: Rf = 0.56 (toluene–EtOAc, 3/2, v/v);
13: Rf = 0.33 (toluene–EtOAc, 3/2, v/v);
4,6-O-Benzylidene-2,3-di-O-tert-butyldimethylsilyl-α-D-glucopyrano-syl-(1↔1)-6-O-allyloxycarbonyl-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-α-D-glucopyranoside (15). To a stirred solution of 12 (4.930 g, 5.66 mmol c = 0.250M) in dry CH2Cl2 (22 mL), sym-collidine (7.50 mL, 56.6 mmol) and allyloxycarbonyl chloride (6.04 mL, 56.6 mmol) were added successively at 0 °C. The reaction mixture was stirred for 6 h at r. t., diluted with toluene (20 mL), and concentrated. The residue was redissolved in EtOAc (500 mL) and washed with aq. satd. NaHCO3 (3 × 150 mL), aq. satd. CuSO4 (3 × 50 mL), and brine (2 × 100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene -EtOAc, 90/10 → 80/20) to afford 15 (4.13 g, 76%) as a syrup. Rf = 0.22 (toluene–EtOAc, 9/1, v/v);
4,6-O-Benzylidene-2,3-di-O-tert-butyldimethylsilyl-α-D-glucopyrano-syl-(1↔1)-6-O-allyloxycarbonyl-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-4-O-(2,2,2-trichloroethoxycarbonylamino)-α-D-glucopyranoside (17). To a stirred solution of 15 (0.800 g, 0.837 mmol) in dry CH2Cl2 (8 mL, c = 0.110 M), pyridine (500 μL, 5.86 mmol) and 2,2,2-trichloroethyl chloroformate (600 μL, 4.19 mmol) were added successively at 0°C. The reaction mixture was stirred for 16 h at r. t., then diluted with toluene (20 mL) and concentrated. The residue was dissolved in EtOAc (200 mL) and washed with aq. satd. NaHCO3 (3 × 50 mL), aq. satd. CuSO4 (3 × 50 mL), and brine (2 × 50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene-EtOAc, 98/2 → 75/25) to afford 17 (0.927 g, 98%) as a solid. Rf = 0.58 (toluene–EtOAc, 90/10, v/v);
4,6-O-Benzylidene-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-2-O-(4-oxopentanoyl)-4-O-(2,2,2-trichloroethoxycarbonylamino)-α-D-glucopyranoside (18). To a stirred solution of triethylamine trihydrofluoride 3HF·Et3N (200 μL, 0.908 M) in dry DMF (200 µL), a solution of 22 (50.0 mg, 0.044 mmol) in dry DMF (100 µL) was added dropwise at 0°C (the final concentration of 3HF·Et3N in the reaction solution 1.2 M). The reaction mixture was stirred for 9 h at r. t. The reaction mixture was diluted with EtOAc (50 mL) and washed with aq. satd. NaHCO3 (2 × 10 mL), aq. satd. CaCl2 (2 × 10 mL), and brine (2 × 10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene-EtOAc, 80/20 → 40/60) to afford 18 (26.8 mg, 77%) as a syrup. Rf = 0.12 (toluene–EtOAc, 1:1, v/v);
4,6-O-Benzylidene-2,3-O-(tetraisopropyldisiloxane-1,3-diyl)-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-2-O-(4-oxopentanoyl)-4-O-(2,2,2-trichloroethoxycarbonyl)-α-D-glucopyranoside (19). To a stirred solution of 18 (130 mg, 0.165 mmol) in dry pyridine/DMF (500 μL, 1:1), 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (100 μL, 0.52M) was added dropwise. The reaction mixture was stirred for 60 min, diluted with hexane (1.0 mL), and purified by column chromatography on silica gel (hexane-acetone, 90/10 → 75/25) to afford 19 (121 mg, 71%) as a transparent sirup. Rf = 0.24 (hexane–acetone, 3:1, v/v); Rf = 0.42 (toluene–EtOAc, 4:1, v/v); Rf = 0.74 (toluene -MeOH, 90:10, v/v);
4,6-O-Benzylidene-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-2,3-di-O-tert-butyldimethylsilyl-α-D-glucopyranoside (21). To a stirred solution of 13 (2.164 g, 2.48 mmol) in dry CH2Cl2 (10 mL, c = 0.250M), sym-collidine (3.30 mL, 24.8 mmol) and allyloxycarbonyl chloride (2.70 mL, 5.23 mmol) were added successively at 0°C. The reaction mixture was stirred for 6 h at r. t., then diluted with toluene (20 mL) and concentrated. The residue was dissolved in EtOAc (400 mL) and washed with aq. satd. NaHCO3 (3 × 100 mL), aq. satd. CuSO4 (3 × 50 mL), and brine (2 × 100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene-EtOAc, 90/10 → 80/20) to afford 21 (2.09 g, 88%) as a transparent syrup. Rf = 0.75 (toluene–EtOAc, 3/2, v/v); Rf = 0.17 (toluene–EtOAc, 90/10, v/v);
4,6-O-Benzylidene-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-2,3-di-O-tert-butyldimethylsilyl-4-O-(2,2,2-trichloroethoxycarbonylamino)-α-D-glucopyranoside (23). To a stirred solution of 21 (2.09 g, 2.40 mmol) in dry CH2Cl2 (4 mL, c = 0.126 M), pyridine (1.25 mL, 15.29 mmol) and 2,2,2-trichloroethyl chloroformate (1.50 mL, 10.92 mmol) were added successively at 0°C. The reaction mixture was stirred for 16 h at r. t., then diluted with toluene (30 mL) and concentrated. The residue was dissolved in EtOAc (400 mL) and washed with aq. satd. NaHCO3 (3 × 100 mL), aq. satd. CuSO4 (2 × 50 mL), and brine (3 × 50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (stepwise gradient elution, toluene-EtOAc, 98/2 → 75/25) to afford 23 (2.32 g, 96%) as a solid. Rf = 0.28 (toluene–EtOAc, 90/10, v/v);
4,6-O-Benzylidene-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-4-O-(2,2,2-trichloroethoxycarbonyl)-α-D-gluco-pyranoside (24). To a stirred solution of triethylamine trihydrofluoride 3HF·Et3N (200 µL) in dry DMF (200 µL), a solution of 23 (50 mg, 0.044 mmol) in dry DMF (100 µL) was added dropwise at 0 °C. The reaction mixture was stirred for 9 h at r. t. under atmosphere of Ar, diluted with EtOAc (30 mL), and washed with aq. satd. NaHCO3 (2 × 10 mL), aq. satd. CaCl2 (2 × 10 mL), and brine (2 × 10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene-EtOAc, 80/20 → 40/60) to afford 24 (28.0 mg, 80%) as a transparent solid. Rf = 0.12 (toluene-EtOAc, 1:1, v/v); Rf = 0.02 (toluene-EtOAc, 8:2, v/v);
4,6-O-Benzylidene-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-2,3-O-(tetraisopropyldisiloxane-1,3-diyl)-4-O-(2,2,2-trichloroethoxycarbonyl)-α-D-glucopyranoside (25). To a stirred solution of 24 (20 mg, 0.025 mmol) in dry pyridine/DMF (200 μL, 1:1) 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (40 μL, 0.52M) was added dropwise. The reaction mixture was stirred for 60 min, diluted with hexane (500 µL), and purified by column chromatography on silica gel (hexane-acetone, 90/10 → 75/25) to give 25 (10.8 mg, BRSM 82%) as transparent oil. Unreacted 24 (10.2 mg) was recovered. Rf = 0.22 (hexane–acetone, 3:1, v/v); Rf = 0.58 (toluene–MeOH, 90:10, v/v);
4,6-O-Benzylidene-2,3-di-O-tert-butyldimethylsilyl-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-2-O-(4-oxopentanoyl)-4-O-(2,2,2-trichloroethoxycarbonyl)-α-D-glucopyranosid (26) and 4,6-O-Benzylidene-3-O-tert-butyldimethylsilyl-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-4-O-(2,2,2-trichloroethoxycarbonyl)-α-D-glucopyrano-side (27). To a stirred solution of triethylamine trihydrofluoride 3HF·Et3N (500 µL) in dry DMF (2500 µL) in a PTFE reaction vessel, a solution of 17 (500 mg, 0.442 mmol) in dry DMF (2000 µL) was added dropwise to reach the final concentration of HF·Et3N (0.2 M) in the reaction solution (pH = 3.5). The reaction mixture was stirred for 70 min at r. t. under atmosphere of Ar, then diluted with EtOAc (150 mL) and washed with aq. satd. CaCl2 (2 × 30 mL), aq. satd. NaHCO3 (3 × 30 mL), and brine (2 × 30 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene-EtOAc, 100/0 → 40/60) to give 26 (104 mg, 47%) and 27 (67 mg, 30%), overall yield 77%, the unreacted starting material 17 was fully recovered (256 mg).
26: Rf = 0.61 (toluene-EtOAc, 1/1, v/v); Rf = 0.32 (toluene-EtOAc, 4/1, v/v);
27: Rf = 0.73 (toluene-EtOAc, 1/1, v/v); Rf = 0.58 (toluene-EtOAc, 4/1, v/v);
4,6-O-Benzylidene-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-2,3-di-O-tert-butyldimethylsilyl-4-O-(2,2,2-trichloroethoxycarbonyl)-α-D-glucopyranoside (28) and 4,6-O-Benzylidene-3-O-tert-butyldimethylsilyl-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-3-O-tert-butyldimethylsilyl-4-O-(2,2,2-trichloroethoxycarbonyl)-α-D-glucopyranoside (29) and 4,6-O-Benzylidene-3-O-tert-butyldimethyl-silyl-2-O-(4-oxopentanoyl)-α-D-glucopyranosyl-(1↔1)-6-O-allyloxycarbonyl-3-O-tert-butyldimethylsilyl-4-O-(2,2,2-trichloroethoxycarbonyl)-α-D-glucopyranoside (30). To a stirred solution of triethylamine trihydrofluoride 3HF·Et3N (500 µL) in dry DMF (2500 µL) in a PTFE reaction vessel, a solution of 23 (500 mg, 0.442 mmol) in dry DMF (2000 µL) was added dropwise to reach the final concentration of HF·Et3N (0.2 M) in the reaction solution (pH = 3.5). The reaction mixture was stirred for 70 min at r. t. under atmosphere of Ar, then diluted with EtOAc (150 mL) and washed with aq. satd. CaCl2 (2 × 30 mL), aq. satd. NaHCO3 (3 × 30 mL), and brine (2 × 30 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (toluene-EtOAc, 100/0 → 40/60) to afford 28 (80 mg, 53%), 29 (29 mg, 19%), and 30 (16 mg, 10%), the overall yield being 82%. The unreacted starting material 23 was fully recovered (331 mg).
28: Rf = 0.67 (toluene–EtOAc, 1/1, v/v); Rf = 0.25 (toluene–EtOAc, 4/1, v/v);
29: Rf = 0.88 (toluene–EtOAc, 1/1, v/v); Rf = 0.49 (toluene–EtOAc, 4/1, v/v);
30: Rf = 0.7 (toluene–EtOAc, 3/1, v/v);
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
TV: Writing–original draft, Writing–review and editing, Investigation, Methodology. BA: Investigation, Writing–review and editing. AM: Investigation, Writing–review and editing. AZ: Conceptualization, Funding acquisition, Investigation, Methodology, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research was funded by the Austrian Science Foundation (FWF) (grant P-32397).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1332837/full#supplementary-material
References
Ágoston, K., Streicher, H., and Fügedi, P. (2016). Orthogonal protecting group strategies in carbohydrate chemistry. Tetrahedron Asymmetry 27, 707–728. doi:10.1016/j.tetasy.2016.06.010
Backus, K. M., Boshoff, H. I., Barry, C. S., Boutureira, O., Patel, M. K., D'Hooge, F. O., et al. (2011). Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat. Chem. Biol. 7, 228–235. doi:10.1038/nchembio.539
Baddeley, T., Clow, S. M., Cox, P. J., Davidson, I. G., Ryder, C., Ward, J. L., et al. (2003). Disordered 2,2',3,3',4,6,6'-hepta-O-pivaloyl-α,α-trehalose. Acta Crystallogr. Sect. E 59, 753–755. doi:10.1107/s1600536803009474
Banahene, N., Gepford, D. M., Biegas, K. J., Swanson, D. H., Hsu, Y.-P., Murphy, B. A., et al. (2023). A far-red molecular rotor fluorogenic trehalose probe for live mycobacteria detection and drug-susceptibility testing. Angew. Chem. Int. Ed. 62, e202213563. doi:10.1002/anie.202213563
Bols, M., and Pedersen, C. M. (2017). Silyl-protective groups influencing the reactivity and selectivity in glycosylations. Beilstein J. Org. Chem. 13, 93–105. doi:10.3762/bjoc.13.12
Boltje, T. J., Li, C., and Boons, G. J. (2010). Versatile set of orthogonal protecting groups for the preparation of highly branched oligosaccharides. Org. Lett. 12, 4636–4639. doi:10.1021/ol101951u
Brown, G. M., Rohrer, D. C., Berking, B., Beevers, C. A., Gould, R. O., and Simpson, R. (1972). The crystal structure of α,α-trehalose dihydrate from three independent X-ray determinations. Acta Cryst. Sect. B 28, 3145–3158. doi:10.1107/S0567740872007654
Carlier, M., Lesur, E., Baron, A., Lemétais, A., Guitot, K., Roupnel, L., et al. (2022). Synthesis of chemical tools to label the mycomembrane of corynebacteria using modified iron(iii) chloride-mediated protection of trehalose. Org. Biomol. Chem. 20, 1974–1981. doi:10.1039/D2OB00107A
Cheng, K. L. (1976). Synthesis of α-D-glucopyranosyl α-D-galoctopyranoside. Carbohydr. Res. 50, 152–157. doi:10.1016/S0008-6215(00)84093-8
Dai, T., Xie, J., Zhu, Q., Kamariza, M., Jiang, K., Bertozzi, C. R., et al. (2020). A fluorogenic trehalose probe for tracking phagocytosed Mycobacterium tuberculosis. J. Am. Chem. Soc. 142, 15259–15264. doi:10.1021/jacs.0c07700
Decout, A., Silva-Gomes, S., Drocourt, D., Barbe, S., André, I., Cueto, F. J., et al. (2017). Rational design of adjuvants targeting the C-type lectin Mincle. Proc. Nat. Acad. Sci. U. S. A. 114, 2675–2680. doi:10.1073/pnas.1612421114
Desel, C., Murray, P. J., Lehmann, C. H. K., Heger, L., Christensen, D., Andersen, P., et al. (2022). Monocytes elicit a neutrophil-independent Th1/Th17 response upon immunization with a mincle-dependent glycolipid adjuvant. Front. Immunol. 13, 880474. doi:10.3389/fimmu.2022.880474
Desel, C., Werninghaus, K., Ritter, M., Jozefowski, K., Wenzel, J., Russkamp, N., et al. (2013). The mincle-activating adjuvant TDB induces MyD88-dependent Th1 and Th17 responses through IL-1R signaling. PLoS ONE 8, e53531. doi:10.1371/journal.pone.0053531
Dowd, M. K., Reilly, P. J., and French, A. D. (1992). Conformational analysis of trehalose disaccharides and analogues using MM3. J. Comput. Chem. 13, 102–114. doi:10.1002/jcc.540130113
Färnbäck, M., Eriksson, L., and Widmalm, G. (2004). Octa-O-acetyl-α,α-trehalose ethanol disolvate. Acta Crystallogr. Sect. E 60, 1483–1485. doi:10.1107/s1600536804018707
Feinberg, H., Jégouzo, S. A. F., Rowntree, T. J. W., Guan, Y., Brash, M. A., Taylor, M. E., et al. (2013). Mechanism for recognition of an unusual mycobacterial glycolipid by the macrophage receptor Mincle. J. Biol. Chem. 288, 28457–28465. doi:10.1074/jbc.m113.497149
Feinberg, H., Rambaruth, N. D. S., Jégouzo, S. A. F., Jacobsen, K. M., Djurhuus, R., Poulsen, T. B., et al. (2016). Binding sites for acylated trehalose analogs of glycolipid ligands on an extended carbohydrate recognition domain of the macrophage receptor Mincle. J. Biol. Chem. 291, 21222–21233. doi:10.1074/jbc.M116.749515
French, A. D., Johnson, G. P., Kelterer, A. M., Dowd, M. K., and Cramer, C. J. (2002). Quantum mechanics studies of the intrinsic conformation of trehalose. J. Phys. Chem. A 106, 4988–4997. doi:10.1021/jp020126d
Furukawa, A., Kamishikiryo, J., Mori, D., Toyonaga, K., Okabe, Y., Toji, A., et al. (2013). Structural analysis for glycolipid recognition by the C-type lectins Mincle and MCL. Proc. Natl. Acad. Sci. U. S. A. 110, 17438–17443. doi:10.1073/pnas.1312649110
Furze, C. M., Delso, I., Casal, E., Guy, C. S., Seddon, C., Brown, C. M., et al. (2021). Structural basis of trehalose recognition by the mycobacterial LpqY-SugABC transporter. J. Biol. Chem. 296, 100307. doi:10.1016/j.jbc.2021.100307
Geerdink, D., and Minnaard, A. J. (2014). Total synthesis of sulfolipid-1. Chem. Commun. 50, 2286–2288. doi:10.1039/C3CC48087A
Gilleron, M., Stenger, S., Mazorra, Z., Wittke, F., Mariotti, S., Bohmer, G., et al. (2004). Diacylated sulfoglycolipids are novel mycobacterial antigens stimulating CD1-restricted T cells during infection with Mycobacterium tuberculosis. J. Exp. Med. 199, 649–659. doi:10.1084/jem.20031097
Gilmore, S. A., Schelle, M. W., Holsclaw, C. M., Leigh, C. D., Jain, M., Cox, J. S., et al. (2012). Sulfolipid-1 biosynthesis restricts Mycobacterium tuberculosis growth in human macrophages. ACS Chem. Biol. 7, 863–870. doi:10.1021/cb200311s
Guiard, J., Collmann, A., Gilleron, M., Mori, L., De Libero, G., Prandi, J., et al. (2008). Synthesis of diacylated trehalose sulfates: candidates for a tuberculosis vaccine. Angew. Chem. Int. Ed. 47, 9734–9738. doi:10.1002/anie.200803835
Hadfield, A. F., Hough, L., and Richardson, A. C. (1978). The synthesis of 6-deoxy-6-fluoro-α,α-trehalose and related analogues. Carbohydr. Res. 63, 51–60. doi:10.1016/S0008-6215(00)80928-3
Ishikawa, E., Ishikawa, T., Morita, Y. S., Toyonaga, K., Yamada, H., Takeuchi, O., et al. (2009). Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 206, 2879–2888. doi:10.1084/jem.20091750
Jana, S., and Kulkarni, S. S. (2020). Synthesis of trehalose glycolipids. Org. Biomol. Chem. 18, 2013–2037. doi:10.1039/D0OB00041H
Jensen, H. H., Nordstrom, L. U., and Bols, M. (2004). The disarming effect of the 4,6-acetal group on glycoside reactivity: torsional or electronic? J. Am. Chem. Soc. 126, 9205–9213. doi:10.1021/ja047578j
Kalscheuer, R., Weinrick, B., Veeraraghavan, U., Besra, G. S., and Jacobs, W. R. (2010). Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 107, 21761–21766. doi:10.1073/pnas.1014642108
Kodar, K., Harper, J. L., McConnell, M. J., Timmer, S. M., and Stocker, B. L. (2017). The Mincle ligand trehalose dibehenate differentially modulates M1-like and M2-like macrophage phenotype and function via Syk signaling. Immun. Inflamm. Dis. 5, 503–514. doi:10.1002/iid3.186
Lassfolk, R., and Leino, R. (2023). Mechanism of acyl group migration in carbohydrates. Chem. Eur. J. 29, e202301489. doi:10.1002/chem.202301489
Lee, W. B., Kang, J. S., Yan, J. J., Lee, M. S., Jeon, B. Y., Cho, S. N., et al. (2012). Neutrophils promote mycobacterial trehalose dimycolate-induced lung inflammation via the Mincle pathway. PLoS Pathog. 8, e1002614. doi:10.1371/journal.ppat.1002614
Lin, F. L., van Halbeek, H., and Bertozzi, C. R. (2007). Synthesis of mono- and dideoxygenated α,α-trehalose analogs. Carbohydr. Res. 342, 2014–2030. doi:10.1016/j.carres.2007.05.009
Mishra, V. K., Buter, J., Blevins, M. S., Witte, M. D., Van Rhijn, I., Moody, D. B., et al. (2019). Total synthesis of an immunogenic trehalose phospholipid from Salmonella typhi and elucidation of its sn-regiochemistry by mass spectrometry. Org. Lett. 21, 5126–5131. doi:10.1021/acs.orglett.9b01725
Nobre, A., Alarico, S., Maranha, A., Mendes, V., and Empadinhas, N. (2014). The molecular biology of mycobacterial trehalose in the quest for advanced tuberculosis therapies. Microbiology 160, 1547–1570. doi:10.1099/mic.0.075895-0
Nunes, S. C. C., Jesus, A. J. L., Moreno, M. J., and Eusebio, M. E. (2010). Conformational preferences of α,α-trehalose in gas phase and aqueous solution. Carbohydr. Res. 345, 2048–2059. doi:10.1016/j.carres.2010.07.001
Ostrop, J., Jozefowski, K., Zimmermann, S., Hofmann, K., Strasser, E., Lepenies, B., et al. (2015). Contribution of MINCLE-SYK signaling to activation of primary human APCs by mycobacterial cord factor and the novel adjuvant TDB. J. Immunol. 195, 2417–2428. doi:10.4049/jimmunol.1500102
Parker, H. L., Tomás, R. M. F., Furze, C. M., Guy, C. S., and Fullam, E. (2020). Asymmetric trehalose analogues to probe disaccharide processing pathways in mycobacteria. Org. Biomol. Chem. 18, 3607–3612. doi:10.1039/D0OB00253D
Pedersen, C. M., Nordstrom, L. U., and Bols, M. (2007). "Super armed" glycosyl donors: conformational arming of thioglycosides by silylation. J. Am. Chem. Soc. 129, 9222–9235. doi:10.1021/ja071955l
Penkov, S., Mende, F., Zagoriy, V., Erkut, C., Martin, R., Pässler, U., et al. (2010). Maradolipids: diacyltrehalose glycolipids specific to dauer larva in Caenorhabditis elegans. Angew. Chem. Int. Ed. 49, 9430–9435. doi:10.1002/anie.201004466
Pohane, A. A., Carr, C. R., Garhyan, J., Swarts, B. M., and Siegrist, M. S. (2021). Trehalose recycling promotes energy-efficient biosynthesis of the mycobacterial cell envelope. mBio 12, e02801-20. doi:10.1128/mbio.02801-20
Reinink, P., Buter, J., Mishra, V. K., Ishikawa, E., Cheng, T.-Y., Willemsen, P. T. J., et al. (2019). Discovery of Salmonella trehalose phospholipids reveals functional convergence with mycobacteria. J. Exp. Med. 216, 757–771. doi:10.1084/jem.20181812
Richardson, A. C., and Tarelli, E. (1971). Chemical modification of trehalose. Part IX. The monobenzylidene acetal. J. Chem. Sci. C. Org. 10, 3733–3735. doi:10.1039/J39710003733
Sarpe, V. A., and Kulkarni, S. S. (2013). Desymmetrization of trehalose via regioselective DIBAL reductive ring opening of benzylidene and substituted benzylidene acetals. Org. Biomol. Chem. 11, 6460–6465. doi:10.1039/C3OB41389F
Sarpe, V. A., and Kulkarni, S. S. (2014). Expeditious synthesis of Mycobacterium tuberculosis sulfolipids SL-1 and Ac2SGL analogues. Org. Lett. 16, 5732–5735. doi:10.1021/ol5027987
Schoenen, H., Bodendorfer, B., Hitchens, K., Manzanero, S., Werninghaus, K., Nimmerjahn, F., et al. (2010). Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol. 184, 2756–2760. doi:10.4049/jimmunol.0904013
Schoenen, H., Huber, A., Sonda, N., Zimmermann, S., Jantsch, J., Lepenies, B., et al. (2014). Differential control of mincle-dependent cord factor recognition and macrophage responses by the transcription factors C/EBPβ and HIF1α. J. Immunol. 193, 3664–3675. doi:10.4049/jimmunol.1301593
Seeliger, J. C., Holsclaw, C. M., Schelle, M. W., Botyanszki, Z., Gilmore, S. A., Tully, S. E., et al. (2012). Elucidation and chemical modulation of sulfolipid-1 biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 287, 7990–8000. doi:10.1074/jbc.m111.315473
Swarts, B. M., Holsclaw, C. M., Jewett, J. C., Alber, M., Fox, D. M., Siegrist, M. S., et al. (2012). Probing the mycobacterial trehalome with bioorthogonal chemistry. J. Am. Chem. Soc. 134, 16123–16126. doi:10.1021/ja3062419
Thanna, S., and Sucheck, S. J. (2016). Targeting the trehalose utilization pathways of Mycobacterium tuberculosis. MedChemComm 7, 69–85. doi:10.1039/C5MD00376H
Tima, H. G., Al Dulayymi, J. R., Denis, O., Lehebel, P., Baols, K. S., Mohammed, M. O., et al. (2017). Inflammatory properties and adjuvant potential of synthetic glycolipids homologous to mycolate esters of the cell wall of Mycobacterium tuberculosis. J. Innate Immun. 9, 162–180. doi:10.1159/000450955
Vanaporn, M., and Titball, R. W. (2020). Trehalose and bacterial virulence. Virulence 11, 1192–1202. doi:10.1080/21505594.2020.1809326
Werninghaus, K., Babiak, A., Groß, O., Hölscher, C., Dietrich, H., Agger, E. M., et al. (2009). Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRγ–Syk–Card9–dependent innate immune activation. J. Exp. Med. 206, 89–97. doi:10.1084/jem.20081445
Wu, C.-Y., and Wong, C.-H. (2011) Programmable one-pot glycosylation, In Reactivity tuning in oligosaccharide assembly. Berlin, Heidelberg: Springer Berlin Heidelberg. doi:10.1007/128_2010_109
Yamasaki, S., Ishikawa, E., Sakuma, M., Hara, H., Ogata, K., and Saito, T. (2008). Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 9, 1179–1188. doi:10.1038/ni.1651
Yang, S.-X., Wang, H.-P., Gao, J.-M., Zhang, Q., Laatsch, H., and Kuang, Y. (2012). Fusaroside, a unique glycolipid from Fusarium sp., an endophytic fungus isolated from Melia azedarach. Org. Biomol. Chem. 10, 819–824. doi:10.1039/C1OB06426F
Keywords: carbohydrates, glycochemistry, synthesis, protecting group manipulations, regioselectivity
Citation: Vašíček T, Arensmeyer B, Monti A and Zamyatina A (2024) Versatile approach towards fully desymmetrized trehalose with a novel set of orthogonal protecting groups. Front. Chem. 11:1332837. doi: 10.3389/fchem.2023.1332837
Received: 03 November 2023; Accepted: 18 December 2023;
Published: 11 January 2024.
Edited by:
Anthony J. Burke, University of Coimbra, PortugalReviewed by:
Carlos Afonso, University of Lisbon, PortugalSusana M. M. Lopes, University of Coimbra, Portugal
Copyright © 2024 Vašíček, Arensmeyer, Monti and Zamyatina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alla Zamyatina, YWxsYS56YW15YXRpbmFAYm9rdS5hYy5hdA==