 Koichi Fukase
Koichi Fukase Yoshiyuki Manabe1,2
Yoshiyuki Manabe1,2 Atsushi Shimoyama
Atsushi Shimoyama
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 05 December 2023
Sec. Organic Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1319883
This article is part of the Research Topic Chemical Reactivity and Solution Structure View all 6 articles
The presence of NHAc groups in the substrates (both glycosyl donors and acceptors) significantly reduced the reactivity of glycosylation. This decrease was attributed to the NHAc groups forming intermolecular hydrogen bonds by the NHAc groups, thereby reducing molecular mobility. Hence, a diacetyl strategy involving the temporary conversion of NHAc to diacetyl imide (NAc2) was developed for the synthesis of NHAc-containing glycans. This strategy has two significant advantages for oligosaccharide synthesis. The NAc2 protection of NHAc substantially enhances the rate of glycosylation reactions, resulting in improved yields. Moreover, NAc2 can be readily reverted to NHAc by the simple removal of one acetyl group under mild basic conditions, obviating the necessity for treating the polar amino group. We have achieved the efficient synthesis of oligosaccharides containing GlcNHAc and N-glycans containing sialic acid using the diacetyl strategy.
Natural glycans play diverse roles in biological events such as self and non-self-recognition, viral and bacterial infection, immunoregulation, cancer invasion, and cell development (Zhao et al., 2008; Macauley et al., 2014; Raman et al., 2016; Ballal and Inamdar, 2018; Shirakawa et al., 2021; Manabe and Fukase, 2023). Bacterial glycoconjugates, including lipopolysaccharide and peptidoglycan, have been known to activate innate immunity (Kusumoto et al., 2010; Shimoyama and Fukase, 2023). Owing to the high diversity and heterogeneity of natural glycans, the production of pure glycans through chemical synthesis has proven to be a potent method for understanding the biological functions of glycans.
Amino sugars are essential components of various glycans including N-glycans, O-glycans, glycolipids, glycosaminoglycans, and bacterial cell walls. Glucosamine, galactosamine, and sialic acids are common amino sugars that usually exist in N-acylated or N-sulfated forms. The choice of protecting groups for amines is crucial in synthesizing glycans containing amino sugars because it affects glycosylation reactivity. Various protecting groups, such as 2,2,2-trichloroethoxycarbonyl (Kusumoto et al., 1985; Imoto et al., 1987), allyloxycarbonyl (Boullanger et al., 1987; Boullanger et al., 1990), trichloroacetyl (Wolfrom and Bhat, 1967; Blatter et al., 1994), trifluoroacetyl (Meyer zu Reckendorf and Wassiliadou-Micheli, 1970), and phthaloyl (Lemieux et al., 1977) have been reported to protect amino sugars. The azide group is also a useful precursor of the amino group (Lemieux and Ratcliffe, 1979). Since most amino groups are acetylated in natural glycans, protected amino groups must be converted to acetamide (NHAc) through acetylation. These steps can be avoided when glycans with NHAc are used directly to construct glycan chains. However, fragments containing NHAc exhibit low reactivity in glycosylation although Tamura et al. showed that the formation of interglycosidic O-imidates with NHAc in GalNAc increases glycosylation yields (Tamura et al., 2008). Boons et al. demonstrated that NAc2 sialyl donors exhibit significantly higher reactivity than NHAc donors (Demchenko and Boons, 1998). Kononov et al. observed hydrogen bonding of NHAc in Neu5Ac donors, and reactivity improvement was achieved by protecting NHAc as NAc2 or adding external amides/imides to disrupt the hydrogen bond (Kononov et al., 2008; Kononov et al., 2009; Kononov et al., 2012). Crich et al. reported that intermolecular hydrogen bonding formed by NHAc in GlcNAc acceptors reduces reactivity (Crich and Dudkin, 2001). Auzanneau observed the aggregation of a tetrasaccharide through hydrogen bonding of NHAc in GlcNAc using NMR (Kuir et al., 2015).
We also observed that sialic acid and GlcNAc derivatives containing NHAc or fragments that include them displayed low reactivity (Nagasaki et al., 2016; Zhou et al., 2016; Tsutsui et al., 2020). This could be attributed to the formation of intermolecular hydrogen bonds between N-acetylneuraminic acid and GlcNAc derivatives, which restrict the molecular movement and hinder the access of acceptors to donors, as noted by Kononov and Crich. We therefore developed a “diacetyl strategy” by temporarily converting NHAc to diacetyl imide (NAc2). The diacetyl strategy offers two advantages for oligosaccharide synthesis. NAc2 protection of NHAc significantly enhanced the glycosylation reactions, leading to improved yields. Additionally, NAc2 can be easily converted back to NHAc by removing one acetyl group under mild basic conditions, eliminating the need to treat the polar amino group (Nagasaki et al., 2016; Zhou et al., 2016; Tsutsui et al., 2020; Shirakawa et al., 2021).
Much effort has been devoted to the development of efficient and stereoselective α-sialylation. This reaction was one of the most challenging in the field of oligosaccharide synthesis because α-sialylation poses steric and electronic difficulties. Recent advances in glycosylation chemistry such as the development of new leaving and protecting groups have enabled efficient construction of α-sialoside linkages.
As mentioned above, the reactivity of sialic acid derivatives possessing a C5-acetamide group is low. Thus, new sialyl donors with various C5 substituents, including protecting groups and functional groups such as carbamates, have been developed to improve the reactivity and stereoselectivity in α-sialylation. The kinetic solvent effect of nitriles has been used for α-sialylation, except in the case of cyclic carbamates and macrocyclic donors described below. Conducting reactions at lower temperatures generally enhances selectivity in kinetically controlled reactions; thus, the improvement in reactivity contributes to the enhancement of stereoselectivity. These substituents include N-Ac2 (Demchenko and Boons, 1998), N-TFA (Meo et al., 2001; Ando et al., 2005), N-Troc (Ando et al., 2005; 2003; Adachi et al., 2004), N-phthaloyl (Tanaka K. et al., 2005), azide (Yu et al., 2001), isothiocyanato (Mandhapati et al., 2015), and ureido groups (Tanase et al., 2016). 5,4-N,O-cyclic carbamates have been utilized for highly selective α-sialylation, especially in the α (2–8)- and α (2–9)-sialylation cases (Tanaka H. et al., 2006; 2008; 2009; De Meo et al., 2008; Hanashima et al., 2009; Chu et al., 2011). A highly selective sialylation method was developed using a macrocyclic α-sialyl donor bridged by alkyl chains at the C1 and C5 positions of sialic acids (Komura et al., 2019; Vibhute et al., 2021). In this method, nucleophilic attack is possible only from the α-face because of the blocking of the β-face by the tethered moiety.
Microflow synthesis is characterized by its ability to remove heat rapidly and efficiently, maintain precise temperature control, and facilitate effective mixing. Consequently, microflow synthesis has been employed to enhance the reaction rate and improve yields in a biphasic system involving water and organic solvents, primarily due to its efficient mixing capabilities (Liu et al., 2004; Tanaka K. et al., 2008; Otake et al., 2018; Aimi, 2021).
On the other hand, glycosylation reactions are conducted in a homogeneous reaction system in organic solvents under dry conditions. However, as pointed out by Kononov et al., protected sugar derivatives may not be uniformly dispersed in solution; instead, they often tend to form clusters because of interactions such as intermolecular hydrogen bonding (Kononov, 2015; Kononov et al., 2017; Podvalnyy et al., 2017; Orlova et al., 2017; Nagornaya et al., 2018; Myachin and Kononov, 2023a; Myachin and Kononov, 2023b). Microflow mixing of glycosyl donor and/or receptor clusters can afford a non-equilibrium mixing state similar to well-dispersed biphasic mixture. Therefore, the use of microreactors, as with biphasic reactions, can influence both the yield and selectivity.
We observed a similar phenomenon in the synthesis of partial structures of Helicobacter pylori-derived lipopolysaccharides. Lipopolysaccharides consist of a glycolipid portion called lipid A and a polysaccharide part, with the polysaccharide part being linked to lipid A through an acidic sugar termed Kdo (3-deoxy-D-manno-oct-2-ulosonic acid). In the synthesis of Kdo-lipid A, we developed Kdo glycosyl donor 1 protected with isopropylidene at the 4th and 5th hydroxyl groups and containing N-phenyltrifluoroacetimidate as a leaving group (Shimoyama et al., 2011a; Shimoyama et al., 2011b). Compound 1 adopts a boat conformation, with the isopropylidene group covering the β-face of Kdo. As a result, in glycosylation using 1, sugar acceptors attack from the α-direction, leading to a high selectivity for α-glycosides. However, a substantial amount of glycal 4 was generated as a byproduct through intramolecular elimination reactions, and a considerable excess of 1 (5 equivalents) was used to obtain the desired trisaccharide 3 in 70% yield.
For the total synthesis of Kdo-lipid A, microflow synthesis was employed to increase the efficiency of glycosylation. The solution of glycosyl donor 1 and glycosyl acceptor 2 in cyclopentyl methyl ether (CPME) and the solution of TBSOTf in CPME were mixed using a micro-mixer, allowing the reaction to proceed for 42 s in a tube reactor (Figure 1). This method enabled us to reduce the amount of glycosyl acceptor to 1.5 equivalents, yet we still obtained trisaccharide 3 with a 72% yield, which is almost equivalent to the batch process. This result suggests that efficient mixing disrupts the solute clusters and promotes intermolecular reactions.
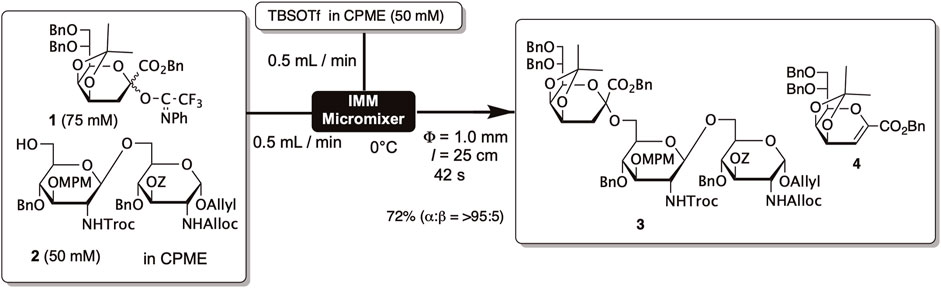
FIGURE 1. Glycosylation of Kdo donor by using the microreactor.
We have developed other microfluidic glycosylation reactions, including α-sialylation (Tanaka K. et al., 2005; Tanaka S et al., 2007; Uchinashi, Y. et al., 2014), β-mannosylation (Takaka, K. et al., 2009), and N-glycosylation of asparagine (Takaka, K. et al., 2009), many of which have successfully improved both selectivity and yield. The main factor thought to contribute to the improvement in selectivity and yield is efficient temperature control; however, enhanced mixing efficiency may also play a role in the improvement. We achieved quantitative sialylation using N-phenyltrifluoroacetimidate donors 5 and 6 with C5-phthalimide (Tanaka K. et al., 2005; Tanaka S et al., 2007) or azide (Uchinashi, Y. et al., 2014) with high α-selectivity under microfluidic conditions (Figure 2).
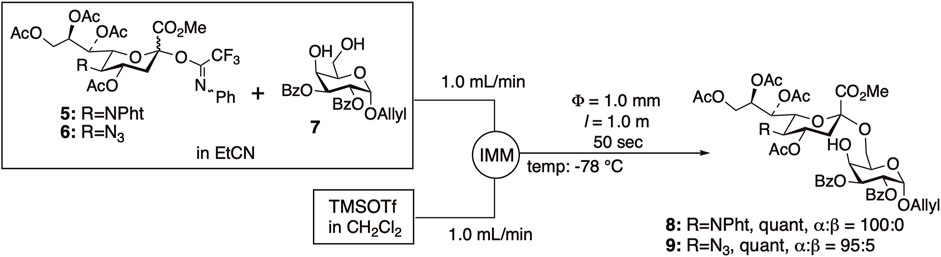
FIGURE 2. α-Sialylation under microfluidic conditions.
Despite previous studies describing low to modest efficiency, we re-examined the readily available C5-acetamide donor 10 for its application in α-sialylation under batch and microfluidic conditions (Figure 3). N-Phenyltrifluoroacetimidate donor 10 was efficiently mixed with an appropriate amount of TMSOTf to yield α (2–6) and α (2–3)-sialylation products of galactose and glucosamine acceptors with excellent yields and high α-selectivity (Uchinashi, Y. et al., 2011).
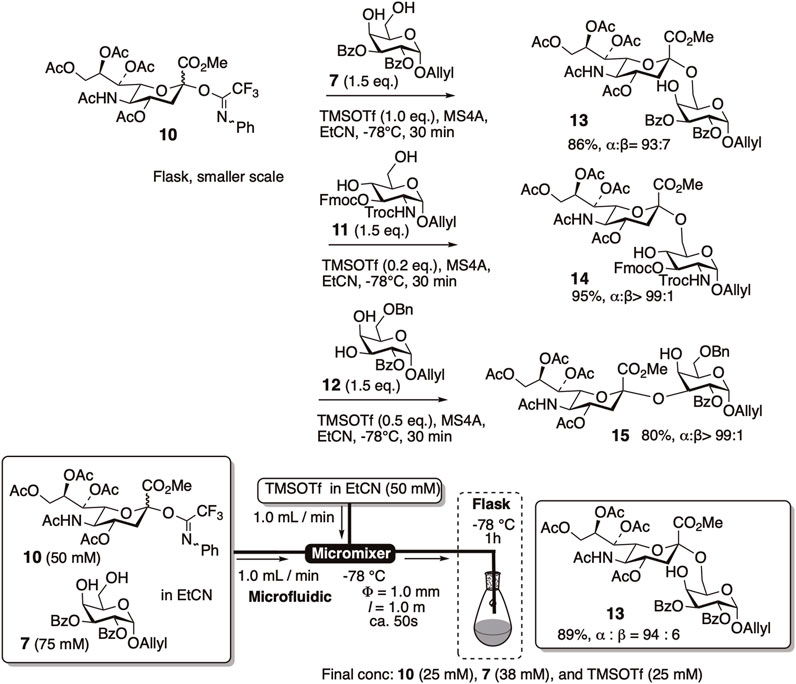
FIGURE 3. Re-investigation of α-sialylation using C5-acetamide donor 10 under batch and microfluidic conditions.
As described above, Crich et al. demonstrated that N-acetylated glucosamine acceptors exhibit lower reactivity than N-phthaolyl glucosamine and 2-azido-2deoxy glucose derivatives. They attributed the reduced reactivity to intermolecular hydrogen bonding, which was confirmed by the concentration-dependent chemical shift of NHAc in 1H-NMR. We also confirmed that the NHAc group of GlcNAc derivative 17 formed intermolecular hydrogen bonds based on the concentration- and temperature-dependent chemical shift of the amide proton in the 1H-NMR spectrum in CH2Cl2 (Figure 4) (Tsutsui et al., 2020). The supramolecular structure of GlcNAc 17 formed by intermolecular hydrogen bonds was observed using diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy (DOSY) measurements (Figure 5). The effective volume of GlcNAc 17 was 1.2 times larger than that of N,N-diacetylated glucosamine (GlcN(Ac)2) 18 according to the DOSY spectra, indicating that 17 formed a supramolecular structure through intermolecular hydrogen bonds, whereas 18 did not aggregate. This slight difference suggests that the hydrogen bonds formed by NHAc are likely dynamic but still have a significant impact on the reactivity of GlcNAc.
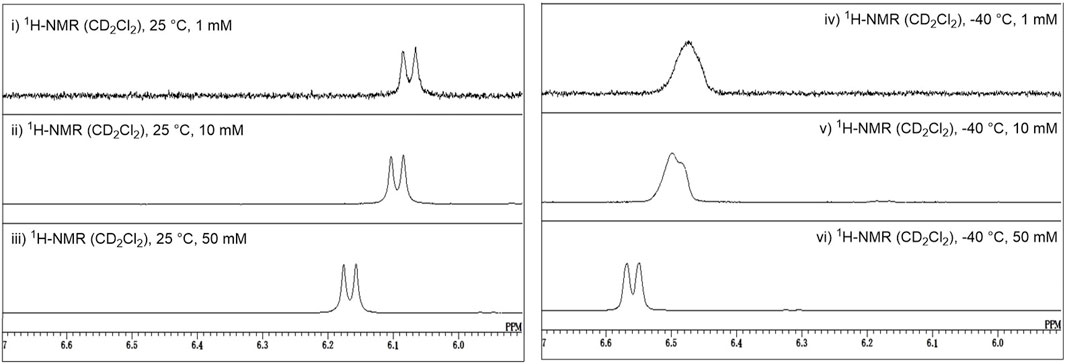
FIGURE 4. Concentration and temperature dependence of the 1H-NMR spectrum of acetamide acceptor 17, where downfield shifts of the amide proton are observed upon increasing the concentration and decreasing the temperature.
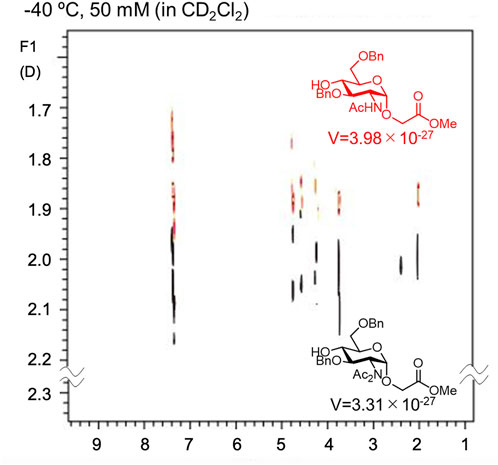
FIGURE 5. DOSY spectrum of N-acetylated (red) and N,N-diacetylated (black) glucosamine derivatives. The effective volume (V) was estimated by Stokes–Einstein equation.
Next, we demonstrated the effectiveness of the diacetyl strategy in the synthesis of α-gal trisaccharide and H antigen trisaccharide. As anticipated, diacetyl protection significantly enhanced the reactivity of all the tested glycosylations. After the preparation of thiodisaccharide 16, the glycosylation of glucosaminyl acceptors 17 or 18 with 16 was performed to evaluate the effectiveness of the diacetyl strategy (Figure 6). The reaction of N,N-diacetylated acceptor 18 with 16 proceeded more smoothly than that of 17 with 16, yielding trisaccharide 20 in excellent yield. These results indicated that the formation of hydrogen bonds by NHAc in 17 decreased its reactivity and demonstrated the effectiveness of the diacetyl strategy.
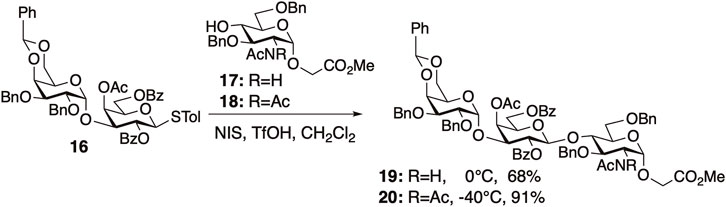
FIGURE 6. The [2 + 1] glycosylation using N-acetylated acceptor 17 or N,N-diacetylated acceptor 18 for the synthesis of protected α-gal 19 or 20.
Efficient α-gal synthesis was accomplished through one-pot and one-flow glycosylation employing the armed-disarmed strategy, benefiting from enhanced reactivity due to diacetylation. The one-flow method under microflow conditions enhanced the reproducibility of the consecutive glycosylations (Figure 7). This was the first report of one-flow glycosylation utilizing the armed-disarmed strategy, which offers distinct advantages in terms of reproducibility compared to one-pot glycosylation in a batch system.
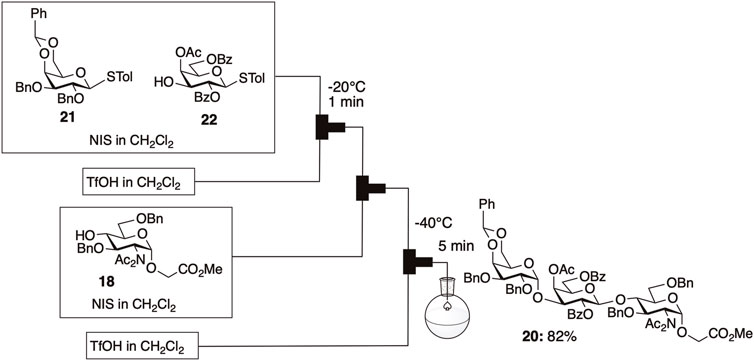
FIGURE 7. One-flow synthesis of protected α-gal 20 under microfluidic conditions.
In H antigen synthesis, enhancement of reactivity by NAc2 protection was observed in both glycosylations proximal and distal to GlcNAc (Figure 8). We investigated the glycosylation of GlcNAc 17 and GlcN(Ac)2 18 with a galactosyl donor 23. As expected, glycosylation with 17 was less reactive, requiring an increase in temperature to 0°C to give compound 24 in 24% yield. In contrast, glycosylation with 18 proceeded smoothly at −20°C, affording disaccharide 25 with perfect β-selectivity in 76% yield.
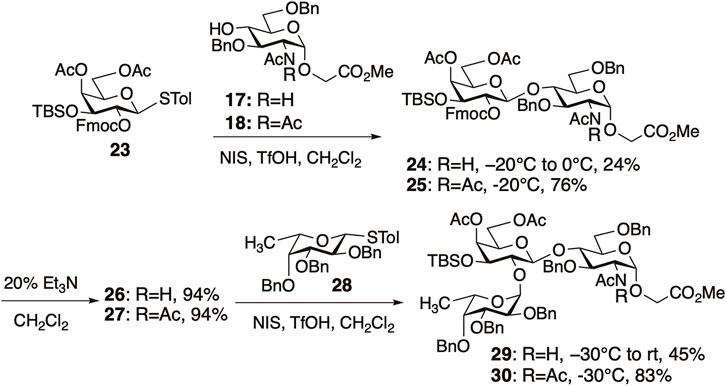
FIGURE 8. Synthesis of H-antigen trisaccharides.
Next, we compared the reactivity of the disaccharide acceptors 26 (N-acetylated) and 27 (N,N-diacetylated) after deprotection of the Fmoc groups in 24 and 25. The glycosylation of 26 with fucosyl donor 28 did not proceed at −30°C; however, when the reaction temperature was elevated to room temperature (rt), trisaccharide 29 was obtained in 45% yield. In contrast, the glycosylation of 27 proceeded smoothly at −30°C to give 30 in 83% yield. This illustrates that N,N-diacetyl protection enhances reactivity even when the reactive sites are distant from the NAc2 group, underscoring the versatility of the diacetyl strategy in synthesizing glycans containing GlcNAc.
We also reported a “diacetyl strategy” for the efficient synthesis of sialyl glycans. Disialylated tetrasaccharide (Neu5Ac (α2,3) Gal (β1,3) [Neu5Ac (α2,6)] GlcNAc) is a structural motif found in the N-glycans of human Factor X and fetuin, gangliosides in human colon adenocarcinoma, and human milk. For the synthesis of this motif, we investigated a convergent route based on the glycosylation of two sialylated disaccharides (Figure 9) (Zhou et al., 2016). The reactivity of the glycosylation reactions between two sialyl disaccharides 31 and 32 with NHAc at the C5 position of sialic acid residues was found to be extremely low, resulting in no desired product formation (0% yield). However, NAc2 protection of both sialyl fragments 34 and 35 significantly enhanced the reactivity, leading to the quantitative yield of the desired tetrasaccharide 36. Protection of the amide group of the sialic acid residues resulted in a substantial improvement in the glycosylation yield between the two sialylated disaccharides, suggesting that the presence of hydrogen bonding on the sialic acid residues decreases reactivity.
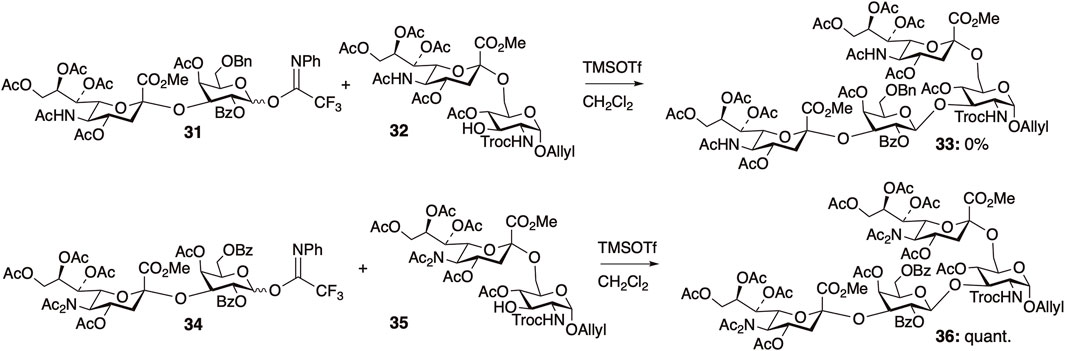
FIGURE 9. Synthesis of disialylated tetrasaccharide.
Asparagine-linked (N-linked) glycans in glycoproteins (N-glycans) are oligosaccharides present in eukaryotes and some prokaryotes that display a broad spectrum of structural diversity. These can be classified into three categories: high-mannose type, complex type, and hybrid type. Typically, N-glycans exhibit heterogeneity even at specific glycosylation sites. Complex N-glycans play crucial roles in various biological processes and diseases, including regulation of glycoprotein dynamics, cell development, immunity, and cancer invasion.
We aimed to synthesize sialylated N-glycans, including core fucose-containing disialylated biantennary N-glycans, disialylated biantennary N-glycans with asymmetrically deuterated sialic acid acetyl groups, and fully sialylated tetraantennary N-glycans, using a diacetyl strategy, and we successfully achieved their synthesis (Nagasaki et al., 2016; Shirakawa et al., 2021).
The synthesis of N-glycans has been extensively investigated by several research groups. Danishefsky (Wu et al., 2006; Wang et al., 2009; Walczak and Danishefsky, 2012; Walczak et al., 2013) and Unverzagt (Schuberth and Unverzagt, 2005; Eller et al., 2007; Mönnich et al., 2016; Luber et al., 2018) successfully chemically synthesized N-glycans, including those with core fucose and bisecting GlcNAc. Ito (Koizumi et al., 2013), Boons (Wang et al., 2013; Li et al., 2016; Gagarinov et al., 2017), Wang (Li et al., 2015), and Wong (Shivatare et al., 2013; 2016) constructed N-glycan libraries through a combination of chemical and enzymatic synthesis.
Our approach to N-glycan synthesis is characterized by two key aspects: 1) the practical synthesis of fragments using micro-flow reactions, as described above, and 2) the development of a convergent synthetic route for the efficient assembly of N-glycan skeletons. Another distinctive feature of our N-glycan synthesis is the early-stage incorporation of asparagine (Asn). Glycans were extended from the Asn-containing reducing-end fragment, which was obtained through N-glycosylation, as described above. This approach facilitates the straightforward preparation of glycoconjugates, including glycopeptides and glycoproteins, as it eliminates the need for Asn attachment following deprotection.
Mammalian core fucose is added to complex-type glycans by fucosyltransferase 8 (FUT8) via α (1–6) fucosyl linkages. Knockout mice lacking FUT8 exhibit severe developmental delays and a 70% mortality rate within the first 3 days of life. Core-fucosylated immunoglobulin G (IgG) is significantly less functional (100-fold weaker) in antibody-dependent cell-mediated cytotoxicity (ADCC) than non-core-fucosylated IgG. The importance of core-fucosylation is evident in various stages of carcinogenesis and tumor progression. Reduced core-fucosylation in human colon carcinomas leads to resistance against TRAIL-induced apoptosis and immune evasion. In hepatocellular carcinoma, elevated levels of core-fucosylated α-fetoprotein serve as a valuable marker. Additionally, upregulation of core-fucosylation on growth factor receptors, such as epidermal growth factor receptor (EGFR) and fibroblast growth factor receptor (FGFR), has been associated with receptor activation.
We first examined the synthesis of the non-reducing-end tetrasaccharide in the synthesis of the core-fucosylated N-glycan (Nagasaki et al., 2016) (Figure 10). When attempting glycosylation between the NHAc-containing sialyl disaccharide donor 37 and the disaccharide acceptor 39, only 52% of the desired tetrasaccharide 40 was obtained, even after raising the temperature to room temperature. On the other hand, using 38 where the NHAc at the 5-position of sialic acid was converted to NAc2, improved the reactivity. Glycosylation proceeded rapidly at 0°C, yielding the desired tetrasaccharide 41 in 96% yield. We observed a concentration-dependent chemical shift of the amide protons in NMR measurements at various concentrations of 37 (Figure 11), revealing that the NHAc in 37 forms intermolecular hydrogen bonds. This also highlights the significant impact of supramolecular structures formed through hydrogen bonding on reactivity.
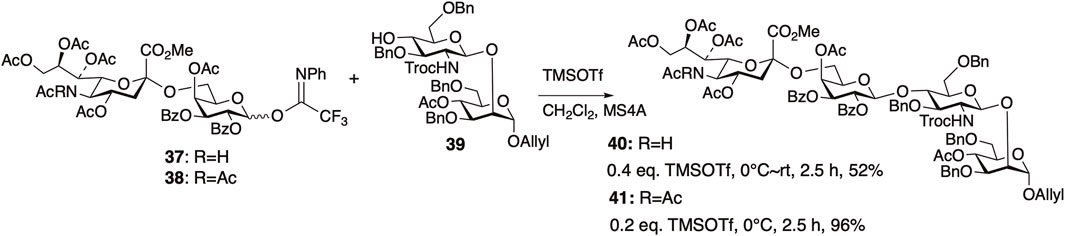
FIGURE 10. Comparison of reactivity of NHAc donor 37 and NAc2 donor 38.
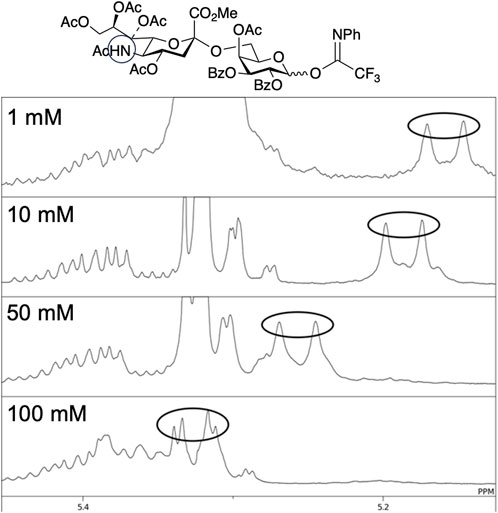
FIGURE 11. 1H NMR of the disaccharide donor 37 at various concentrations.
Solvent selection played a crucial role in the glycosylation of the reducing-end tetrasaccharide 42 with the non-reducing-end tetrasaccharide 43. When ether-based solvents were used, particularly cyclopentyl methyl ether (CPME), the desired octasaccharide was obtained in 91% yield. The ether solvents probably stabilize and prolong the lifetime of the intermediate oxocarbenium ion by coordination. However, the stereoselectivity of this glycosylation was moderate (α/β = 3/1). Following cleavage of the benzylidene group from the obtained octasaccharide, the α-isomer 44 was separated. Subsequently, glycosylation at the 6th position of the branched mannose in 44 was investigated. When CPME was used as the solvent, the desired product 45 was obtained in high yield. However, α/β selectivity remained low, and 45 was obtained as a 1/1 mixture.
After the cleavage of allyl ester of 45 with Pd(OAc)2, PPh3, sodium 2-ethylhexanoate in acetone, all acyl groups, N-Troc groups, and methyl esters were removed by aqueous LiOH and the resulting amino groups were acetylated. Both the α and β isomers were then separated using HPLC, and all benzyl-type protecting groups were removed under catalytic hydrogenation conditions to afford the core fucose-containing N-glycan (Figure 12). Protection of the acetamide in sialic acid as a diacetyl moiety may have facilitated the efficient glycosylation of each fragment coupling. The problem of low selectivity in the two glycosylation steps was resolved by remote participation using O-3 and O-6 acyl protection of the mannose residue in the synthesis of N-glycans containing bisecting GlcNAc, along with the synthesis of the previously mentioned deuterated disialylated biantennary N-glycans and the fully sialylated tetraantennary N-glycan (Manabe et al., 2018; Shirakawa et al., 2021).
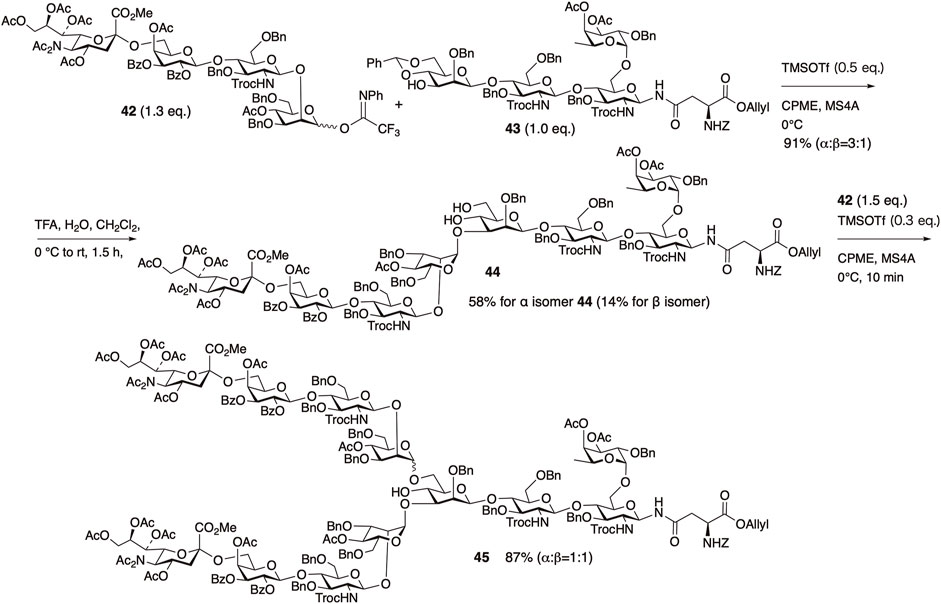
FIGURE 12. Synthesis of core fucosylated N-glycan.
The diacetyl strategy was applied to synthesize two asymmetrically deuterated sialyl N-glycans, 55 and 56. Using deuterium-labeled N-glycan 55, it was revealed that the neuraminidase derived from H1N1 preferentially cleaved sialic acids on α1,3-branched chains over α1,6-branched chains.
Deuterated N-glycans 55 and 56 were synthesized by glycosylation first at the 3-position of branching mannose in trisaccharide 48 with sialyl tetrasaccharides 46 or 47, followed by glycosylation at the 6-position of branching mannose (Figure 13) (Shirakawa et al., 2021). We employed the remote participation method previously described by Kim et al. for mannosylation, in which acyl protection of the mannosyl donors at the O-3 and O-6 positions enhances α-selectivity (Baek et al., 2009).
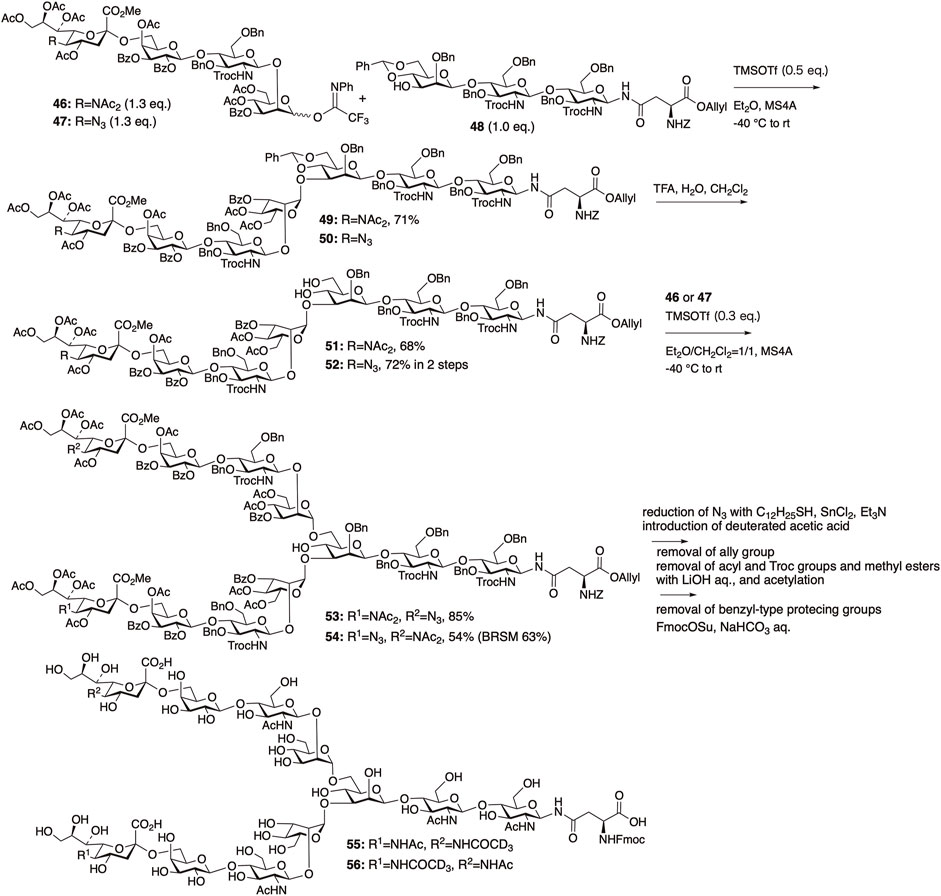
FIGURE 13. Synthesis of two biantennary N-glycans with asymmetrically deuterium-labeled sialic acid.
In fact, the glycosylation between 46 and 48 proceeded smoothly with a stoichiometric amount of TMSOTf in Et2O to afford the desired heptasaccharide 49 in 71% yield with perfect α-selectivity. Glycosylation of the azide-containing sialyl tetrasaccharide 47 with 48, followed by the deprotection of benzylidene in 50, afforded 52 in good yield.
Because of the low solubility of sialyl glycan 51 obtained by the deprotection of 49 in Et2O, the [7 + 4] glycosylation between 51 and 47 was carried out in a mixed solvent system of Et2O/CH2Cl2 = 1/1, yielding the desired undecasaccharide 53 in 85% yield with perfect α-selectivity. Glycosylation between 52 and 46 under similar conditions afforded 54 in 54% yield (BRSM: 63%). Thus, we successfully constructed two asymmetric disialyl undecasaccharides, 53 and 54, with one sialyl unit having an azide group available for deuterium labeling. The target deuterated N-glycans, 55 and 56, were obtained by introducing a deuterated acetyl group, followed by global deprotection. Deuterium-hydrogen exchange was observed during the alkaline treatment for the removal of acyl and Troc groups, resulting in a decrease in the deuterium ratio of 55 to 42% and that of 56 to 63%, respectively.
Among the naturally occurring N-glycans, tetrasialylated N-glycan 61 is essential for evaluating the impact of multivalency and steric hindrance associated with the multiantennary structure. Fully sialylated tetraantennary N-glycan 61 was synthesized in a manner similar to those of 55 and 56 (Figure 14) (Shirakawa et al., 2021). The glycosylation between trisaccharide 48 and heptasaccharide donor 57 in the mixed solvent of Et2O/CH2Cl2 = 1/1 proceeded with complete α-selectivity, followed by cleavage of the benzylidene group, yielding decasaccharide 58 in 33% yield (BRSM: 49%) in two steps. The selection of the Lewis acid, solvent, and temperature played a crucial role in the subsequent glycosidation between decasaccharide 58 and heptasaccharide donor 59. The glycosylation of 58 and 59 was achieved using TBDPSOTf at 0°C in a mixed solvent with a high ether ratio (Et2O/CH2Cl2 = 5/1), resulting in the formation of compound 60 with 36% yield. Following the deprotection of 60 and Fmoc introduction under conditions similar to the synthesis of 55 and 56, we obtained fully sialylated tetraantennary N-glycan 61.

FIGURE 14. Synthesis of tetraantennary sialyl N-glycan.
In conclusion, the diacetyl strategy was instrumental in overcoming the challenges in sialyl glycan synthesis, allowing for the successful chemical synthesis of the asymmetrically deuterium-labeled sialyl N-glycans and the tetraantennary sialyl N-glycan.
As mentioned above, the presence of NHAc groups in either glycosyl donors or acceptors significantly decreased the reaction rate of glycosylation. These phenomena are attributed to the decrease in the molecular motion of the substrates owing to intermolecular hydrogen bonding. Kononov et al. demonstrated that various protected saccharides form cluster structures termed supramers in solution, through intermolecular interactions mediated by hydrogen bonds and/or other non-covalent interactions between saccharides, and the formation of supramers significantly affects glycosylation reactivity. We observed that the effective volume of GlcNAc 17 was 1.2 times larger than that of N,N-diacetylated glucosamine (GlcN(Ac)2) 18 by DOSY experiments, indicating the formation of a supramer in the reaction solution through intermolecular hydrogen bonds. In the glycosylation of glycosyl donor 31 and glycosyl acceptor 32, both of which have an NHAc group, the yield of sialic acid-containing tetrasaccharide 33 was 0% (Figure 8). Glycosylation reactions with either the donor or acceptor possessing an NHAc group afforded glycosylation products, despite the low yield. The notable difference in yields indicated an exceedingly low probability of collision between glycosyl donor 31 and glycosyl acceptor 32. This outcome is suggested by the formation of supramers in both donor 31 and acceptor 32, in addition to the decreased substrate mobility caused by intermolecular hydrogen bonds.
Myachin and Kononov reviewed the influence of reactor shape and flow rate on the efficiency and selectivity of reactions based on the formation of supramers and their dynamic behavior (Myachin and Kononov, 2023a; Myachin and Kononov, 2023b). In the Kdo glycosylation mentioned above, despite the absence of amide groups in the substrates, the efficiency of the glycosylation reaction increased when using a microflow reactor (Figure 1). This could be interpreted as microflow mixing disrupting the supramer structures in the solution to promote glycosylation.
In contrast, imide protection is used in the diacetyl strategy to prevent the formation of cluster structures via intermolecular hydrogen bonding, thus avoiding a decrease in reactivity. In our N-glycan synthesis, the Asn residue was bound to the reducing-end sugar. The amide group of the Asn side chain did not significantly affect synthetic efficiency. In the synthesis of the tetraantennary sialyl N-glycan, solubility decreased as the molecular size of the sialyl fragments increased. Consequently, glycosylation yields with such fragments were low. To improve the solubility of such fragments, it is necessary to change synthetic strategies, such as the selection of protective groups. Research focusing on solution structures in glycan synthesis is still in its early stages, and further advancements are anticipated in the future.
KF: Funding acquisition, Investigation, Writing–original draft, Writing–review and editing, Project administration, Supervision. YM: Funding acquisition, Investigation, Writing–review and editing. AS: Funding acquisition, Investigation, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by JSPS KAKENHI grants 20241053 and 21106008 in Integrated Synthesis; 23241074, 26560447, and 15H05836 in Middle Molecular Strategy; 16H01885, 16H05924, 18H03934, 19KK0145, 20H00404, 20H05675, 20K05727, 20H04709, and 21H05074; JST CREST grants JPMJCR20R3; AMED grants 20ek0109444h0001 and 20fk0210079h0001; Japan Initiative for World-Leading Vaccine Research and Development Centers, a Naito research grant, and a Nikki Saneyoshi research grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adachi, M., Tanaka, H., and Takahashi, T. (2004). An effective sialylation method using N-Troc- and N-Fmoc-protected β-thiophenyl sialosides and application to the one-pot two-step synthesis of 2,6-sialyl-T antigen. Synlett, 609–614. doi:10.1055/s-2004-817759
Aimi, H. (2021). “Recent advances in the integrated microflow synthesis of organofluorine compounds,” in Middle molecular strategy. Flow synthesis to functional molecules (Springer Nature), 331–357.
Ando, H., Koike, Y., Ishida, H., and Kiso, M. (2003). Extending the possibility of an N-Troc-protected sialic acid donor toward variant sialo-glycoside synthesis. Tetrahedron Lett. 44, 6883–6886. doi:10.1016/S0040-4039(03)01707-6
Ando, H., Koike, Y., Koizumi, S., Ishida, H., and Kiso, M. (2005). 1,5-Lactamized sialyl acceptors for various disialoside syntheses: novel method for the synthesis of glycan portions of Hp-s6 and HLG-2 gangliosides. Angew. Chem. Int. Ed. 44, 6759–6763. doi:10.1002/anie.200501608
Baek, J. Y., Lee, B.-Y., Jo, M. G., and Kim, K. S. (2009). β-Directing effect of electron-withdrawing groups at O-3, O-4, and O-6 positions and α-directing effect by remote participation of 3-O-acyl and 6-O-acetyl groups of donors in mannopyranosylations. J. Am. Chem. Soc. 131, 17705–17713. doi:10.1021/ja907252u
Ballal, S., and Inamdar, S. R. (2018). An overview of lectin-glycan interactions: a key event in initiating fungal infection and pathogenesis. Arch. Microbiol. 200, 371–382. doi:10.1007/s00203-018-1487-1
Blatter, G., Beau, J. M., and Jacquinet, J. C. (1994). The use of 2-deoxy-2-trichloroacetamido-D-glucopyranose derivatives in syntheses of oligosaccharides. Carbohydr. Res. 260, 189–202. doi:10.1016/0008-6215(94)84038-5
Boullanger, P., Banoub, J., and Descotes, G. (1987). N-Allyloxycarbonyl derivatives of D-glucosamine as promotors of 1,2-trans-glucosylation in Koenigs–Knorr reactions and in Lewis acid catalyzed condensations. Can. J. Chem. 65, 1343–1348. doi:10.1139/v87-225
Boullanger, P., Jouineau, M., Bouammali, B., Lafont, D., and Descotes, G. (1990). The use of N-alkoxycarbonyl derivatives of 2-amino-2-deoxy-D-glucose as donors in glycosylation reactions. Carbohydr. Res. 202, 151–164. doi:10.1016/0008-6215(90)84077-8
Chu, K.-C., Ren, C.-T., Lu, C.-P., Hsu, C.-H., Sun, T.-H., Han, J.-L., et al. (2011). Efficient and stereoselective synthesis of α(2→9) oligosialic acids: from monomers to dodecamers. Angew. Chem. Int. Ed. 50, 9391–9395. doi:10.1002/anie.201101794
Crich, D., and Dudkin, V. (2001). Why are the hydroxy groups of partially protected N-acetylglucosamine derivatives such poor glycosyl acceptors, and what can be done about it? A comparative study of the reactivity of N-acetyl-N-phthalimido-and 2-azido-2-deoxy-glucosamine derivatives in glycosylation. 2-Picolinyl ethers as reactivity-enhancing replacements for benzyl ethers. J. Am. Chem. Soc. 123, 6819–6825. doi:10.1021/ja010086b
Demchenko, A. V., and Boons, G.-J. (1998). A novel and versatile glycosyl donor for the preparation of glycosides of N-acetylneuraminic acid. Tetrahedron Lett. 39, 3065–3068. doi:10.1016/S0040-4039(98)00359-1
De Meo, C., Farris, M., Ginder, N., Gulley, B., Priyadarshani, U., and Woods, M. (2008). Solvent effect in the synthesis of sialosyl α(2-6) galactosides: is acetonitrile the only choice? Eur. J. Org. Chem. 2008, 3673–3677. doi:10.1002/ejoc.200800278
Eller, S., Schuberth, R., Gundel, G., Seifert, J., and Unverzagt, C. (2007). Synthesis of pentaantennary N-glycans with bisecting GlcNAc and core fucose. Angew. Chem. Int. Ed. 46, 4173–4175. doi:10.1002/anie.200604788
Gagarinov, I. A., Li, T., Toraño, J. S., Caval, T., Srivastava, A. D., Kruijtzer, J. A. W., et al. (2017). Chemoenzymatic approach for the preparation of asymmetric Bi-Tri-and tetra-antennary N-glycans from a common precursor. J. Am. Chem. Soc. 139, 1011–1018. doi:10.1021/jacs.6b12080
Hanashima, S., Sato, K., Ito, Y., and Yamaguchi, Y. (2009). Silylene/oxazolidinone double-locked sialic acid building blocks for efficient sialylation reactions in dichloromethane. Eur. J. Org. Chem. 4215-4220, 4215–4220. doi:10.1002/ejoc.200900543
Imoto, M., Yoshimura, H., Shimamoto, T., Sakaguchi, N., Kusumoto, S., and Shiba, T. (1987). Total synthesis of Escherichia coli lipid A, the endotoxically active principle of cell-surface lipopolysaccharide. Bull. Chem. Soc. Jpn. 60, 2205–2214. doi:10.1246/bcsj.60.2205
Koizumi, A., Matsuo, I., Takatani, M., Seko, A., Hachisu, M., Takeda, Y., et al. (2013). Top-down chemoenzymatic approach to a high-mannose-type glycan library: synthesis of a common precursor and its enzymatic trimming. Angew. Chem. Int. Ed. 52, 7426–7431. doi:10.1002/anie.201301613
Komura, N., Kato, K., Udagawa, T., Asano, S., Tanaka, H. N., Imamura, A., et al. (2019). Constrained sialic acid donors enable selective synthesis of α-glycosides. Science 364, 677–680. doi:10.1126/science.aaw4866
Kononov, L. O. (2015). Chemical reactivity and solution structure: on the way to a paradigm shift? RSC Adv. 5, 46718–46734. doi:10.1039/C4RA17257D
Kononov, L. O., Fedina, K. G., Orlova, A. V., Kondakov, N. N., Abronina, P. I., Podvalnyy, N. M., et al. (2017). Bimodal concentration-dependent reactivity pattern of a glycosyl donor: is the solution structure involved? Carbohydr. Res. 437, 28–35. doi:10.1016/j.carres.2016.11.009
Kononov, L. O., Malysheva, N. N., Kononova, E. G., and Orlova, A. V. (2008). Intermolecular hydrogen-bonding pattern of a glycosyl donor: the key to understanding the outcome of sialylation. Eur. J. Org. Chem. 2008, 3251–3255. doi:10.1002/ejoc.200800324
Kononov, L. O., Malysheva, N. N., and Orlova, A. V. (2009). Stereoselectivity of glycosylation may change during the reaction course: highly α-stereoselective sialylation achieved by supramer approach. Eur. J. Org. Chem. 2009, 611–616. doi:10.1002/ejoc.200801017
Kononov, L. O., Malysheva, N. N., Orlova, A. V., Zinin, A. I., Laptinskaya, T. V., Kononova, E. G., et al. (2012). Concentration dependence of glycosylation outcome: a clue to reproducibility and understanding the reasons behind. Eur. J. Org. Chem. 2012, 1926–1934. doi:10.1002/ejoc.201101613
Kuir, D., Guillemineau, M., and Auzanneau, F. I. (2015). Aggregation of a tetrasaccharide acceptor observed by NMR: synthesis of pentasaccharide fragments of the le(a)Le(x) tumor-associated hexasaccharide antigen. J. Org. Chem. 80, 5004–5013. doi:10.1021/acs.joc.5b00405
Kusumoto, S., Fukase, K., and Shiba, T. (2010). Key structures of bacterial peptidoglycan and lipopolysaccharide triggering the innate immune system of higher animals: chemical synthesis and functional studies. Proc. Jpn. Acad. Ser. B 86, 322–337. doi:10.2183/pjab.86.322
Kusumoto, S., Yoshimura, H., Imoto, M., Shimamoto, T., and Shiba, T. (1985). Chemical synthesis of 1-dephospho derivative of lipid A. Tetrahedron Lett. 26, 909–912. doi:10.1016/S0040-4039(00)61962-7
Lemieux, R. U., and Ratcliffe, R. M. (1979). The azidonitration of tri-O-acetyl-D-galactal. Can. J. Chem. 57, 1244–1251. doi:10.1139/v79-203
Lemieux, R. U., Takeda, T., and Chung, B. Y. (1977). “Synthesis of 2-Amino-2-deoxy-β-D-glucopyranosides,” in Synthetic methods for carbohydrates (Washington, DC: ACS Symposium Series), 39, 90–115. 6. doi:10.1021/bk-1977-0039.ch006
Li, L., Liu, Y., Ma, C., Qu, J., Calderon, A. D., Wu, B., et al. (2015). Efficient chemoenzymatic synthesis of an N-glycan isomer library. Chem. Sci. 6, 5652–5661. doi:10.1039/C5SC02025E
Li, T., Huang, M., Liu, L., Wang, S., Moremen, K. W., and Boons, G.-J. (2016). Divergent chemoenzymatic synthesis of asymmetrical-core-fucosylated and core-unmodified N-glycans. Chem. Eur. J. 22, 18742–18746. doi:10.1002/chem.201604999
Liu, S., Fukuyama, T., Sato, M., and Ryu, I. (2004). Continuous microflow synthesis of butyl cinnamate by a mizoroki-heck reaction using a low-viscosity ionic liquid as the recycling reaction medium. Org. Process Res. Dev. 8, 477–481. doi:10.1021/op034200h
Luber, T., Niemietz, M., Karagiannis, T., Mönnich, M., Ott, D., Perkams, L., et al. (2018). A single route to mammalian N-glycans substituted with core fucose and bisecting GlcNAc. Angew. Chem. Int. Ed. 57, 14543–14549. doi:10.1002/anie.201807742
Macauley, M. S., Crocker, P. R., and Paulson, J. C. (2014). Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 14, 653–666. doi:10.1038/nri3737
Manabe, Y., and Fukase, K. (2023). Innovative vaccine strategy: self-adjuvanting conjugate vaccines. Methods Mol. Biol. 2613, 55–72. doi:10.1007/978-1-0716-2910-9_5
Manabe, Y., Shomura, H., Minamoto, N., Nagasaki, M., Takakura, Y., Tanaka, K., et al. (2018). Convergent synthesis of a bisecting N-acetylglucosamine (GlcNAc)-Containing N-glycan. Chem. Asian J. 13, 1544–1551. doi:10.1002/asia.201800367
Mandhapati, A. R., Rajender, S., Shaw, J., and Crich, D. (2015). The isothiocyanato moiety: an ideal protecting group for the stereoselective synthesis of sialic acid glycosides and subsequent diversification. Angew. Chem. Int. Ed. 54, 1275–1278. doi:10.1002/anie.201409797
Meo, C. D., Demchenko, A. V., and Boons, G. J. (2001). A stereoselective approach for the synthesis of alpha-sialosides. J. Org. Chem. 66, 5490–5497. doi:10.1021/jo010345f
Meyer zu Reckendorf, W., and Wassiliadou-Micheli, N. (1970). Glykoside von Aminozuckern, II. Zur Synthese von 2-Amino-2-desoxy-α-D-glucopyranosiden. Chem. Ber. 103, 1792–1796. doi:10.1002/cber.19701030614
Mönnich, M., Eller, S., Karagiannis, T., Perkams, L., Luber, T., Ott, D., et al. (2016). Highly efficient synthesis of multiantennary bisected N-glycans based on imidates. Angew. Chem. Int. Ed. 55, 10487–10492. doi:10.1002/anie.201604190
Myachin, I. V., and Kononov, L. O. (2023a). Phase-transfer catalyzed microfluidic glycosylation: a small change in concentration results in a dramatic increase in stereoselectivity. Catalysts 13, 313. doi:10.3390/catal13020313
Myachin, I. V., and Kononov, L. O. (2023b). Mixer design and flow rate as critical variables in flow chemistry affecting the outcome of a chemical reaction: a review. Inventions 8, 128. doi:10.3390/inventions8050128
Nagasaki, M., Manabe, Y., Minamoto, N., Tanaka, K., Silipo, A., Molinaro, A., et al. (2016). Chemical synthesis of a complex-type N-glycan containing a core fucose. J. Org. Chem. 81 (22), 10600–10616. doi:10.1021/acs.joc.6b02106
Nagornaya, M. O., Orlova, A. V., Stepanova, E. V., Zinin, A. I., Laptinskaya, T. V., and Kononov, L. O. (2018). The use of the novel glycosyl acceptor and supramer analysis in the synthesis of sialyl-α(2–3)-galactose building block. Carbohydr. Res. 470, 27–35. doi:10.1016/j.carres.2018.10.001
Orlova, A. V., Laptinskaya, T. V., Bovin, N. V., and Kononov, L. O. (2017). Differences in reactivity of N-acetyl- and N,N-diacetylsialyl chlorides caused by their different supramolecular organization in solutions. Russ. Chem. Bull. 66, 2173–2179. doi:10.1007/s11172-017-1999-x
Otake, Y., Nakamura, H., and Fuse, S. (2018). Rapid and mild synthesis of amino acid N-carboxy anhydrides: basic-to-acidic flash switching in a microflow reactor. Angew. Chem. Int. Ed. Engl. 57, 11389–11393. doi:10.1002/anie.201803549
Podvalnyy, N. M., Malysheva, N. N., Panova, M. V., Zinin, A. I., Chizhov, A. O., Orlova, A. V., et al. (2017). Stereoselective sialylation with O-trifluoroacetylated thiosialosides: hydrogen bonding involved? Carbohydr. Res. 451, 12–28. doi:10.1016/j.carres.2017.09.002
Raman, R., Tharakaraman, K., Sasisekharan, V., and Sasisekharan, R. (2016). Glycan-protein interactions in viral pathogenesis. Curr. Opin. Struct. Biol. 40, 153–162. doi:10.1016/j.sbi.2016.10.003
Schuberth, R., and Unverzagt, C. (2005). Synthesis of a N-glycan nonasaccharide of the bisecting type with additional core-fucose. Tetrahedron Lett. 46, 4201–4204. doi:10.1016/j.tetlet.2005.04.061
Shimoyama, A., Fujimoto, Y., and Fukase, K. (2011b). Stereoselective glycosylation of 3-Deoxy-D-manno-2-octulosonic acid with batch and microfluidic methods. Synlett 2011, 2359–2362. doi:10.1055/s-0030-1260313
Shimoyama, A., and Fukase, K. (2023). Chemical synthesis and immunomodulatory functions of bacterial lipid as. Methods Mol. Biol. 2613, 33–53. doi:10.1007/978-1-0716-2910-9_4
Shimoyama, A., Saeki, A., Tanimura, N., Tsutsui, H., Miyake, K., Suda, Y., et al. (2011a). Chemical synthesis of Helicobacter pylori lipopolysaccharide partial structures and their selective proinflammatory responses. Chem. Eur. J. 17, 14464–14474. doi:10.1002/chem.201003581
Shirakawa, A., Manabe, Y., and Fukase, K. (2021). Recent advances in the chemical biology of N-glycans. Molecules 26, 1040. doi:10.3390/molecules26041040
Shirakawa, A., Manabe, Y., Marchetti, R., Yano, K., Masui, S., Silipo, A., et al. (2021). Chemical synthesis of sialyl N-glycans and analysis of their recognition by neuraminidase. Angew. Chem. Int. Ed. Engl. 60, 24686–24693. doi:10.1002/anie.202111035
Shivatare, S. S., Chang, S.-H., Tsai, T.-I., Ren, C.-T., Chuang, H.-Y., Hsu, L., et al. (2013). Efficient convergent synthesis of Bi-Tri-and tetra-antennary complex type N-glycans and their HIV-1 antigenicity. J. Am. Chem. Soc. 135, 15382–15391. doi:10.1021/ja409097c
Shivatare, S. S., Chang, S.-H., Tsai, T.-I., Tseng, S. Y., Shivatare, V. S., Lin, Y.-S., et al. (2016). Modular synthesis of N-glycans and arrays for the hetero-ligand binding analysis of HIV antibodies. Nat. Chem. 8, 338–346. doi:10.1038/nchem.2463
Takaka, K., Mori, Y., and Fukase, K. (2009). Practical synthesis of a manβ(1-4)GlcNTroc fragment via microfluidic β-mannosylation. J. Carbohydr. Chem. 28, 1–11. doi:10.1080/07328300802571129
Tamura, J., Nakada, Y., Taniguchi, K., and Yamane, M. (2008). Synthesis of chondroitin sulfate E octasaccharide in a repeating region involving an acetamide auxiliary. Carbohydr. Res. 343, 39–47. doi:10.1016/j.carres.2007.09.009
Tanaka, H., Nishiura, Y., and Takahashi, T. (2006). Stereoselective synthesis of oligo-α-(2,8)-sialic acids. J. Am. Chem. Soc. 128, 7124–7125. doi:10.1021/ja0613613
Tanaka, H., Nishiura, Y., and Takahashi, T. (2008). An efficient convergent synthesis of GP1c ganglioside epitope. J. Am. Chem. Soc. 130, 17244–17245. doi:10.1021/ja807482t
Tanaka, H., Nishiura, Y., and Takahashi, T. (2009). Stereoselective synthesis of α(2,9) di-to tetrasialic acids, using a 5,4-N,O-carbonyl protected thiosialoside. J. Org. Chem. 74, 4383–4386. doi:10.1021/jo900176e
Tanaka, K., Goi, T., and Fukase, K. (2005). Highly efficient sialylation towards α(2-3)- and α(2-6)-neu5Ac-gal synthesis: significant ‘fixed dipole effect' of N-phthalyl group on α-selectivity. Synlett 2005, 2958–2962. doi:10.1055/s-2005-921889
Tanaka, K., Miyagawa, T., and Fukase, K. (2009). Chemical N-glycosylation by asparagine under integrated microfluidic/batch conditions. Synlett 2009, 1571–1574. doi:10.1055/s-0029-1217343
Tanaka, K., Motomatsu, S., Koyama, K., and Fukase, K. (2008). Efficient aldol condensation in aqueous biphasic system under microfluidic conditions. Tetrahedron Lett. 49, 2010–2012. doi:10.1016/j.tetlet.2008.01.057
Tanaka, S., Goi, T., Tanaka, K., and Fukase, K. (2007). Highly efficient α-sialylation by virtue of fixed dipole effects of N-phthalyl group: application to continuous flow synthesis of α(2-3)-and α(2-6)-neu5Ac-gal motifs by microreactor. J. Carbohydr. Chem. 26, 369–394. doi:10.1080/07328300701634796
Tanase, M., Imamura, A., Ando, H., Ishida, H., and Kiso, M. (2016). A 5-ureido-modified sialyl donor: a tool for the synthesis of α-sialosides. Org. Lett. 18, 1454–1457. doi:10.1021/acs.orglett.6b00403
Tsutsui, M., Sianturi, J., Masui, S., Tokunaga, K., Manabe, Y., and Fukase, K. (2020). Efficient synthesis of antigenic trisaccharides containing N-acetylglucosamine: protection of NHAc as NAc2. Eur. J. Org. Chem. 2020, 1802–1810. doi:10.1002/ejoc.201901809
Uchinashi, Y., Nagasaki, M., Zhou, J., Tanaka, K., and Fukase, K. (2011). Reinvestigation of the C5-acetamide sialic acid donor for α-selective sialylation: practical procedure under microfluidic conditions. Org. Biomol. Chem. 9, 7243–7248. doi:10.1039/C1OB06164J
Uchinashi, Y., Tanaka, K., Manabe, Y., Fujimoto, Y., and Fukase, K. (2014). Practical and efficient method for α-sialylation with an azide sialyl donor using a microreactor. J. Carbohydr. Chem. 33, 55–67. doi:10.1080/07328303.2014.880116
Vibhute, A. M., Komura, N., Tanaka, H. N., Imamura, A., and Ando, H. (2021). Advanced chemical methods for stereoselective sialylation and their applications in sialoglycan syntheses. Chem. Rec. 21, 3194–3223. doi:10.1002/tcr.202100080
Walczak, M. A., and Danishefsky, S. J. (2012). Solving the convergence problem in the synthesis of triantennary N-glycan relevant to prostate-specific membrane antigen (PSMA). J. Am. Chem. Soc. 134, 16430–16433. doi:10.1021/ja307628w
Walczak, M. A., Hayashida, J., and Danishefsky, S. J. (2013). Building biologics by chemical synthesis: practical preparation of di- and triantennary N-linked glycoconjugates. J. Am. Chem. Soc. 135, 4700–4703. doi:10.1021/ja401385v
Wang, P., Zhu, J., Yuan, Y., and Danishefsky, S. J. (2009). Total synthesis of the 2,6-sialylated immunoglobulin G glycopeptide fragment in homogeneous form. J. Am. Chem. Soc. 131, 16669–16671. doi:10.1021/ja907136d
Wang, Z., Chinoy, Z. S., Ambre, S. G., Peng, W., McBride, R., de Vries, R. P., et al. (2013). A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans. Science 341, 379–383. doi:10.1126/science.1236231
Wolfrom, M. L., and Bhat, H. B. (1967). Trichloroacetyl and trifluoroacetyl as N-blocking groups in nucleoside synthesis with 2-amino sugars. J. Org. Chem. 32, 1821–1823. doi:10.1021/jo01281a025
Wu, B., Hua, Z., Warren, J. D., Ranganathan, K., Wan, Q., Chen, G., et al. (2006). Synthesis of the fucosylated biantennary N-glycan of erythropoietin. Tetrahedron Lett. 47, 5577–5579. doi:10.1016/j.tetlet.2006.05.086
Yu, C.-S., Niikura, K., Lin, C.-C., and Wong, C. H. (2001). The thioglycoside and glycosyl phosphite of 5-azido sialic acid: excellent donors for the alpha-glycosylation of primary hydroxy groups. Angew. Chem. Int. Ed. 40, 2900–2903. doi:10.1002/1521-3773(20010803)40:15<2900::AID-ANIE2900>3.0.CO;2-4
Zhao, Y. Y., Takahashi, M., Gu, J. G., Miyoshi, E., Matsumoto, A., Kitazume, S., et al. (2008). Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 99, 1304–1310. doi:10.1111/j.1349-7006.2008.00839.x
Keywords: glycosylation, amide bond, amide, oligosaccharide, glycan, flow reaction
Citation: Fukase K, Manabe Y and Shimoyama A (2023) Diacetyl strategy for synthesis of NHAc containing glycans: enhancing glycosylation reactivity via diacetyl imide protection. Front. Chem. 11:1319883. doi: 10.3389/fchem.2023.1319883
Received: 11 October 2023; Accepted: 21 November 2023;
Published: 05 December 2023.
Edited by:
Behrooz Maleki, University of Mazandaran, IranReviewed by:
Wei Zhao, Nankai University, ChinaCopyright © 2023 Fukase, Manabe and Shimoyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Koichi Fukase, a29pY2hpQGNoZW0uc2NpLm9zYWthLXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.