
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 10 November 2023
Sec. Organic Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1292541
This article is part of the Research Topic Women in Chemistry 2023 View all 11 articles
 Alejandro Cortés-Villena1
Alejandro Cortés-Villena1 Iván Soriano-Díaz1
Iván Soriano-Díaz1 Moisés Domínguez2
Moisés Domínguez2 Matías Vidal2
Matías Vidal2 Pablo Rojas2
Pablo Rojas2 Carolina Aliaga2,3*
Carolina Aliaga2,3* Angelo Giussani1
Angelo Giussani1 Antonio Doménech-Carbó4Enrique Ortí1*
Antonio Doménech-Carbó4Enrique Ortí1* Raquel E. Galian1*
Raquel E. Galian1* Julia Pérez-Prieto1*
Julia Pérez-Prieto1*Donor–acceptor-substituted biphenyl derivatives are particularly interesting model compounds, which exhibit intramolecular charge transfer because of the extent of charge transfer between both substituents. The connection of a 4-[1,1′-biphenyl]-4-yl-2-pyrimidinyl) moiety to differently disubstituted amino groups at the biphenyl terminal can offer push–pull compounds with distinctive photophysical properties. Herein, we report a comprehensive study of the influence of the torsion angle of the disubstituted amino group on the emissive properties of two pull–push systems: 4-[4-(4-N,N-dimethylaminophenyl)phenyl]-2,6-diphenylpyrimidine (D1) and 4-[4-(4-N,N-diphenylaminophenyl)phenyl]-2,6-diphenylpyrimidine (D2). The torsion angle of the disubstituted amino group, either N,N-dimethyl-amine or N,N-diphenyl-amine, at the biphenyl end governs their emissive properties. A drastic fluorescence quenching occurs in D1 as the solvent polarity increases, whereas D2 maintains its emission independently of the solvent polarity. Theoretical calculations on D1 support the presence of a twisted geometry for the lowest energy, charge-transfer excited state (S1,90), which corresponds to the minimum energy structure in polar solvents and presents a small energy barrier to move from the excited to the ground state, thereby favoring the non-radiative pathway and reducing the fluorescence efficiency. In contrast, this twisted structure is absent in D2 due to the steric hindrance of the phenyl groups attached to the amine group, making the non-radiative decay less favorable. Our findings provide insights into the crucial role of the substituent in the donor moiety of donor–acceptor systems on both the singlet excited state and the intramolecular charge-transfer process.
Push–pull structures are conjugated organic molecules integrated by electron-donating and electron-withdrawing moieties separated by a π-system, which broadens the charge distribution across the molecule, endowing it with exotic optical and electronic properties (Bureš, 2014). This class of materials has been extensively used as sensitizers in dye-sensitized solar cells (DSSCs) (Verbitskiy et al., 2014; Verbitskiy et al., 2015; Tan et al., 2016; Kozlov et al., 2017; Sun et al., 2018; Verbitskiy et al., 2021b) and hole-transporting materials in perovskite-based solar cells (Maddala et al., 2021; Bouihi et al., 2022; Manda et al., 2022) owing to their high molar absorption coefficient and efficient hole mobility, respectively. Moreover, they can be integrated into organic light-emitting diodes (OLEDs) (Wong et al., 2002; Wu et al., 2002; Nakao et al., 2017; Ryutaro et al., 2018; Verbitskiy et al., 2021a) because of their high fluorescence quantum yields (ΦF).
Pyrimidine derivatives have been used as the electron-withdrawing group in push–pull systems due to the significant π-deficient character of diazine rings (Bureš, 2014; Achelle et al., 2023). This character of the pyrimidine ring can be further increased by protonation, complexation, or alkylation of the nitrogen electron lone pair. Pyrimidine derivatives substituted with electron-donating fragments through π-conjugated linkers are highly fluorescent and sensitive to external stimuli (Achelle et al., 2023).
In general, push–pull organic chromophores play a strategic role in the development of new and sophisticated applications in photonics (Verbitskiy et al., 2021b). The optimization of the organic structure with the appropriate design of a π-electron structure has made it possible to bring these systems closer to the market. Thus, it is possible to exploit the unique properties of push–pull benzenoid derivatives and heterocyclic rings to obtain novel systems that can efficiently convert the emission of a cheap, easily available IR laser into that of a more technological valuable visible laser. Exploiting substituent effects and properly adjusting the π-electron structure can not only modulate the emission frequency, so that a whole range of laser wavelengths is accessible, but also improve upconversion efficiencies to meet market requirements (Fecková et al., 2020).
Particularly, push–pull organic systems exhibit strong fluorosolvatochromism as a consequence of the large dipole moment in the excited state (Hadad et al., 2011; Verbitskiy et al., 2014; Liu et al., 2022), thus making them interesting candidates for their application as chemical and biochemical environmental probes (Qin et al., 2021). The large Stokes shift observed upon increasing solvent polarity stems from an intramolecular relaxation process in an electronic excited state, which sometimes leads to a new energetic minimum far below the former structure in the excited state. This relaxation process typically accompanies not only changes in bond lengths and bond angles but also structural changes due to rotation around a single bond (Haberhauer, 2017).
The importance of electron-donor and acceptor groups, conjugation in the excited state, and the nature of π-bridges (particularly thiophene) has been investigated in the literature for indolo [2,3-b]quinoxaline-based dyes, aryl-substituted indolo [2,3-a]carbazole derivatives synthesized and indeno [1,2-b]indole donor derivatives (Venkateswararao et al., 2014; Yang et al., 2015; Qian et al., 2017). The solvent polarity effect has also been reported for a large number of organic fluorophores, such as tetrazole-substituted pyrene and carbazole-substituted quinoline dyes (Slodek et al., 2019; Zych et al., 2019). Complex systems based on the D–D–π–A architecture such as new indolo [3,2,1-jk]carbazole derivatives have also been reported by Schab-Balcerzak, E. et al. for dye-sensitized solar cells. A phenothiazine unit and an acetylene linkage containing either an aldehyde or cyanoacrylic acid as electron-withdrawing groups were used, and a significant solvent effect was only observed for the cyanoacrylic acid, demonstrating the high sensitivity of the ICT state to the electronic properties of the linkage group (Gnida et al., 2022). Another D–π–D–π–A architecture reported by Schab-Balcerzak, E. et al. using a phenothiazine-based cyanoacrylic acid containing an imidazole ring substituted with the alkyl group with different chain lengths was employed. Time-resolved fluorescence studies were performed using different solvent polarities, indicating the presence of an ICT state more stabilized in polar solvents such as DMF (Zimosz et al., 2022).
The structure–fluorosolvatochromism relationship of pyrimidine-based chromophores has already been reported from an experimental point of view and using Taguchi methodology (Denneval et al., 2014; Achelle and Robin-le Guen, 2017). As a benchmark molecule that undergoes such a process, 4-(dimethylamino)benzonitrile (DMABN) has been widely used to disentangle empirical findings from a theoretical perspective. Yet, several models have been proposed for the explanation of the lower energy band exhibited by this relatively simple molecule; however, they are still under debate (Grabowski et al., 1979; Zachariasse et al., 1996; Gómez et al., 2021). First, the twisted intramolecular charge-transfer (TICT) model was proposed by Grabowski et al. to elucidate the dual fluorescence (from locally excited and intramolecular charge-transfer states) observed for DMABN in polar solvents (Grabowski et al., 1979; Grabowski et al., 2003; Sasaki et al., 2016). This model states that after the electronic excitation of DMABN in polar solvents, a charge transfer concomitantly with a ca. 90° twist of the single bond occurs, thereby electronically disconnecting the donor and acceptor moieties in the excited state. The resulting charge-transfer state is more stable than the preceding localized state (Galván et al., 2010; Segarra-Martí and Coto, 2014). The driving force for this sort of stabilization emerges from the minimization of the Coulomb interaction of the two unpaired electrons. Since the rotation leads to π-orbital decoupling, the fluorescence associated with a TICT process is typically weakened and redshifted (Haberhauer, 2017). On the other hand, Zachariasse et al. proposed the planarized intramolecular charge-transfer (PLICT) model to explain the opposite effect in which a rotation of ca. 90° leads to planarization of the donor and acceptor moieties in the excited state instead (Zachariasse et al., 1996; Zachariasse et al., 1997; Il’ichev et al., 1998). A key difference compared to the TICT process is that the fluorescence from a PLICT state to the ground state is now allowed, and thus high quantum yields are expected (Haberhauer et al., 2016).
Consequently, given the vast amount of the literature on DMABN as a model molecule that is still under discussion, we were motivated to shed light on the mechanism underlying fluorosolvatochromism that occurs in biphenylpyrimidine derivatives. The impact of the donor substituent on the optical and excited state properties of two π-extended biphenylpyrimidines was studied. Particularly, push–pull systems which comprise a dialkylamino or diarylamino and a pyrimidine as electron-donor and -acceptor moieties, respectively, were studied by steady-state and time-resolved fluorescence techniques combined with theoretical calculations that significantly contribute to support the experimental findings.
This research contributes to expanding knowledge about the photochemical behavior and properties of simple push–pull systems and understanding of the relationship between the molecular structure of the amino donor, the solvent environment, and the fluorescence properties. This analysis can help select the right conditions for various applications in chemistry and materials science, such as sensing, imaging, and optoelectronic devices.
The synthesis and characterization of the respective 2,4,6-triarylpyrimidine derivatives, 4-[4-(4-N,N-dimethylaminophenyl)phenyl]-2,6-diphenylpyrimidine (D1) and 4-[4-(4-N,N-diphenylaminophenyl)phenyl]-2,6-diphenylpyrimidine (D2), by Rodríguez Aguilar and coworkers were reported elsewhere (Rodríguez-Aguilar et al., 2018). In brief, a microwave vial (10 mL) was charged with bromophenyl-4-pyrimidines (0.7 mmol), Pd(PPh3)4 (41 mg, 0.035 mmol, and 5 mol%), K2CO3 (97 mg and 0.7 mmol), N,N-dimethylformamide (5 mL), and 4-(N,N-dimethyl)phenylboronic acid (139 mg and 0.84 mmol) for D1 or the 4-(N,N-diphenyl)phenylboronic acid (243 mg and 0.84 mmol) for D2. The resulting reaction mixture was heated for 1 h at 100°C. Upon the end of the reaction (as observed on TLC, n-hexanes/EtOAc, 5:1), the crude was diluted with water (25 mL) and extracted with EtOAc (3 × 15 mL). The combined organic extracts were dried over Na2SO4, and all the volatile components were removed by rotary evaporation. The respective products were purified by column chromatography (n-hexane: EtOAc, 20:1 → 5:1).
All commercially available solvents used for the spectroscopy investigation were purchased from Alfa Aesar and used as received without further purification. The short names of the solvents are included in Table 1.
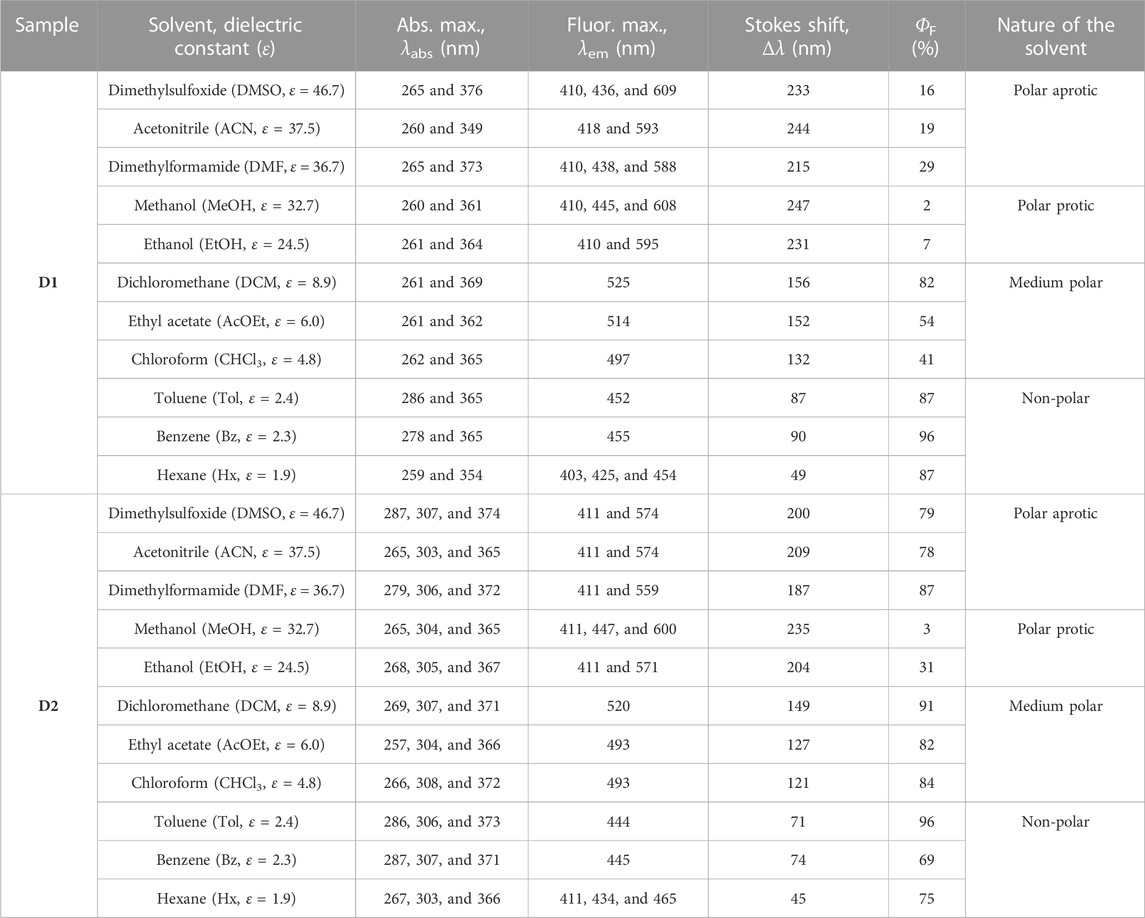
TABLE 1. Optical data (absorption and fluorescence maxima, Stokes shift, and absolute fluorescence quantum yield (ΦF)) of D1 and D2 systems in solvents with a decreasing dielectric constant (ε) under anaerobic conditions. Fluorescence and ΦF measurements were recorded upon excitation at 365 nm.
A measure of 3 mL contained of optically matched D1 or D2 solutions at 0.1 OD at an excitation wavelength (365 nm) was purged with nitrogen directly from the cylinder (to degas air from the sample) for 5 min by placing a syringe needle through the septum into the bottom and another into the air space above the sample as a vent with a flow rate of dry nitrogen so that bubbles are observable in the sample, unless specified. In case, the solvent was evaporated upon bubbling, the cuvettes were refilled with extra purged solvent. Immediately after this procedure, samples were placed in the instrument for measurement. Cuvettes of 1 cm optical path length were used for spectroscopy studies.
Steady-state UV-vis absorption spectra were recorded on a UV/Vis/NIR PerkinElmer LAMBDA 1050 spectrophotometer equipped with deuterium and tungsten halogen light sources and Peltier-controlled InGaAs and PbS detectors covering from 175 to 3,300 nm. The absorption spectra were collected in the range of 250–700 nm.
Stationary fluorescence spectra were recorded on a FLS1000 photoluminescence spectrometer (Edinburgh Instruments) equipped with a 450 W ozone-free continuous xenon arc lamp and a photomultiplier (PMT-980) detector in a cooled housing with an extended spectral range of 185–980 nm. A 365 nm excitation wavelength was used in all measurements. The emission range was registered between 380 and 800 nm.
Absolute fluorescence quantum yields (ΦF) were recorded on a FLS1000 photoluminescence spectrometer equipped with an integrating sphere system with a reflectance higher than 99% in the range of 400–1,500 nm. Sample solutions with optical density (OD) of 0.1 at a 365 nm excitation wavelength were used to minimize re-absorption effects. For reference, the neat solvent with the same volume was used.
Time-resolved fluorescence measurements were recorded on a FLS1000 photoluminescence spectrometer through the time-correlated single-photon counting (TCSPC) technique coupled with a 375 nm ps pulsed diode laser (EPL-375, pulse width: 75 ps, peak power: 140 mW, and repetition rate: 10 MHz, Edinburgh Instruments) and a microchannel plate (MCP-900) detector in a cooled housing with a spectral range of 200–850 nm in the nanosecond domain. A Ludox solution (0.1 OD at an excitation wavelength) was used as an instrument response function (IRF). The IRF was approximately 110 ps in our setup. All spectra were recorded using a 1-cm-path-length quartz cuvette at room temperature.
The characterization of redox properties was performed on an Autolab potentiostat (Autolab 128N potentiostat/galvanostat) using a three-electrode system. Cyclic voltammetry (CV) experiments were carried out in 0.1 M tetrabutylammonium tetrafluoroborate (TBABF4) solution in dried acetonitrile (ACN) using Pt as a working electrode, Pt wire as an auxiliary electrode, and Ag/AgCl as a reference electrode separated from the tested solution by means of a Luggin capillary. The measurements were performed at room temperature (298 ± 1 K) partially deaerating the electrolyte solution by bubbling nitrogen for 2 min. Partially oxygenated solutions were optionally used to facilitate the use of the O2/O2•- couple as the internal standard for electrode potential measurements. The concentration of the compounds was approximately 0.2 mM in dried acetonitrile. Experiments were performed under air conditions, with a scan rate of 100 mV/s. To evaluate the electrochemical bandgaps, potentials were referred to the Fc/Fc+ couple using 0.2 mM ferrocene solutions in 0.1 M TBABF4/ACN.
The thermal properties of D1 and D2 were examined under a nitrogen atmosphere, with a heating rate of 10°C/min up to 950°C.
The singlet ground state (S0) was optimized using density functional theory (DFT) calculations. The first excited singlet state (S1) was also optimized at the time-dependent DFT (TD-DFT) level of theory. All the calculations were performed based on the exchange-correlation Becke’s three-parameter (B3LYP) functional (Lee et al., 1988; Becke, 1993) and the split-valence triple-zeta 6-311G** basis set (Francl et al., 1982), without imposing any symmetry restriction. Calculations were carried out using Gaussian 16 software (Rev. A.03) (Frisch et al., 2016). Solvent effects (hexane, toluene, ACN, and DMSO) were implicitly considered by employing the polarizable continuum model (PCM) method (Tomasi and Persico, 1994). For this reason, all the excited state geometries were obtained at the LR-PCM TD-DFT level of theory. Nevertheless, solvent effects on the photophysical processes (absorption and emission) can be inadequately reproduced using LR-PCM (Chibani et al., 2014). To solve this problem, single vertical point calculations were employed, at the optimized LR-TD-DFT geometries, using the corrected linear response (CLR-PCM) method (Improta et al., 2006; Improta et al., 2007). This approach allows to relax the interaction between the solvent and the solute molecule in the excited-state minima, thus improving the description of the excited states from an energetical point of view. All the energies discussed in the main text refer to PCM-CLR TD-DFT energies. The optimizations must be carried out at the LR-PCM TD-DFT level of theory because CLR-PCM TD-DFT is only implemented for single-point calculations. The ORCA 5.0.1 code was additionally used (Neese, 2022), employing the same computational approach (B3LYP/6-311G**) and simulating the solvent effects with CPCM methodology (Cammi et al., 2000) to characterize the minimum energy paths (MEPs) connecting two singlet excited-state minima belonging to the same potential energy surface (PES). This was accomplished by employing the climbing image nudged elastic band (CI-NEB) method (Henkelman et al., 2000). The energy of the so-obtained MEPs was then re-computed using Gaussian 16 as described previously. The computation of natural transition orbitals (NTOs) and the Mulliken population analysis were performed as implemented in Gaussian 16 (Martin, 2003; Soriano-Díaz et al., 2023).
4-[4-(4-N,N-dimethylaminophenyl)phenyl]-2,6-diphenylpyrimidine (D1) and 4-[4-(4-N,N-diphenylaminophenyl)phenyl]-2,6-diphenylpyrimidine (D2) (Scheme 1) were synthesized using previously reported procedures (Rodríguez-Aguilar et al., 2018), and their procedure can be found in the Materials and Methods section. Their optical and photophysical properties were investigated in a battery of solvents with different polarities.
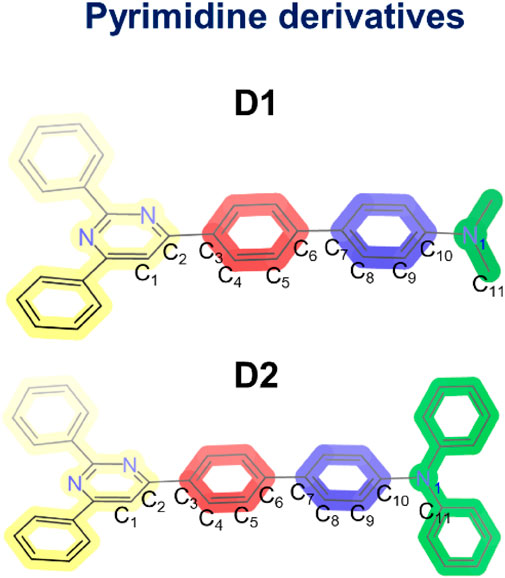
SCHEME 1. Molecular structures of the two pyrimidine derivatives (D1 and D2) and atomic numbering used to define the dihedral angles in the optimized geometries.
The optical properties (steady-state absorption and photoluminescence) of compounds D1 and D2 were measured in diluted solutions of 0.1 OD at the excitation wavelength in solvents with an increasing dielectric constant at room temperature and under anaerobic conditions. The recorded absorption and emission spectra are shown in Figure 1, and the relevant optical data are summarized in Table 1. It is evident that these systems exhibit slight differences in the ground-state absorption features, which involve two major absorption bands, namely, 260–285 nm and 355–380 nm for high- and low-energy bands, respectively, and a small shoulder at approximately 303–307 nm for D2. The shape and energy of the absorption bands were proved to be weakly dependent on the solvent polarity (Figure 1B).
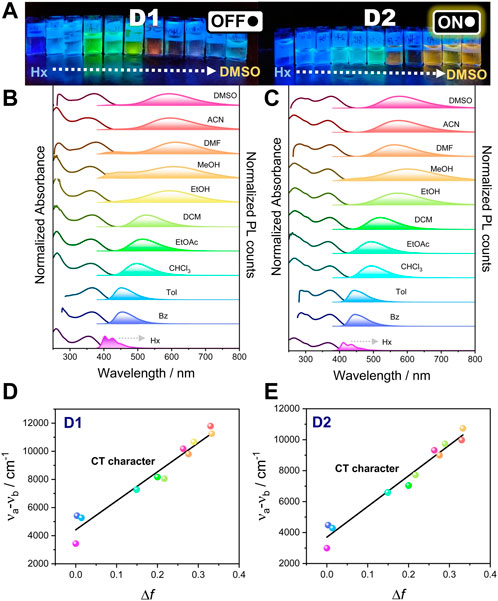
FIGURE 1. (A) Photographs of D1 and D2 pyrimidine derivative solutions in solvents with an increasing dielectric constant from hexane (left) to DMSO (right) under UV irradiation (365 nm). Normalized steady-state absorption and fluorescence spectra of D1 (B) and D2 (C) in the solvents shown previously under anaerobic conditions. From top to bottom, solvents are ordered by decreasing dielectric constant (DMSO, ACN, DMF, MeOH, EtOH, DCM, EtOAc, CHCl3, Tol, Bz, and Hx). For fluorescence measurements, an excitation wavelength of 365 nm was used. Lippert–Mataga plot for D1 (D) and D2 (E).
The shape and energy of the high-energy absorption band in both systems are nearly independent of the solvent polarity, whereas the low-energy band is shown to be weakly dependent on the solvent polarity, being redshifted in DMSO compared to Hx (Δλ is 22 and 18 nm for D1 and D2, respectively), which suggests a low molecular dipole moment in the ground state. However, the observation of a blueshift in polar protic solvents, particularly more intensified in MeOH, is attributed to the hydrogen-bond formation ability (α) of alcohol with the molecule in the ground state, which stabilizes this state and leads to an increase in the energy gap as noted previously (Al-Ahmed et al., 2021). Interestingly, in the case of ACN, which is a highly polar solvent, there is also a significant blueshift in the low-energy absorption band compared to DMF, for which the dielectric constant is slightly lower. Then, the observed blueshift is justified by the better hydrogen-bond formation ability of ACN (α = 19) than DMF (α = 0).
In contrast to the absorption spectra, the fluorescence spectra under a 365 nm excitation wavelength show a strong dependence on the solvent polarity and a remarkable positive solvatochromism upon increasing the solvent dielectric constant. This fluorosolvatochromism suggests a potential intramolecular charge transfer between the donor and acceptor units in the emitting excited state, indicating that the excited state has a larger dipole moment than the ground state. The digital photographs of dye-containing solutions exhibited a wide range of colors, from deep blue to orange. In hexane, the fluorescence spectra of both compounds show that the fingerprint shape were associated with a structured emission as a result of the small solute–solvent interaction (Subuddhi et al., 2006). As the polarity of the solvent increases, the fluorescence spectrum loses its vibrational fine structure, and a broadening and bathochromic shift is observed when moving from hexane to DMSO (Table 1), thus supporting a charge-transfer character of the emitting excited state that explains the large Stokes shift observed (50–250 nm). It is worth mentioning that in polar solvents (from ethanol to DMSO), we observed the appearance of two emission bands, the higher-energy band being more pronounced in D1 than in D2 (Figure 1C). The coexistence of two emission bands (small contribution of the higher-energy band) can be rationally understood by the presence of two emitting species as previously observed on the vast number of push–pull systems found in the literature (Fecková et al., 2021). The higher-energy band may be ascribed to the locally excited (LE) state because of the negligible redshift as compared to the fluorescence in hexane. In contrast, the redshifted, higher-intensity emission band is attributed to the charge-transfer emitting state, which proves the large Stokes shift.
To further evaluate this, analysis using the Lippert–Mataga equation was employed in which the Stokes shift (Δν) is plotted as a function of the orientation polarizability (Δf) of the solvents (Equations 1, 2) (Lippert, 1955; Mataga et al., 1955):
where νa and νb are the wavenumbers (cm−1) of absorption and fluorescence peaks, respectively, h is the Planck’s constant, c is the speed of light in vacuum, μE and μG are dipole moments in the excited and ground states, respectively, a is the radius of the Onsager cavity, and Δf is the orientation polarizability of the solvent given in the following equation:
where ε and η are the dielectric constant and refractive index of the solvent, respectively. The clear linear trend in both compounds (except for hexane) indicates the increase in dipole moment in the excited state compared to the ground state and supports the ICT nature of the excited state (Figures 1D,E). The deviation of the linearity in hexane in both compounds supports the existence of partial contribution of the LE state. A higher slope for D1 than for D2 suggests that it exhibits a more pronounced charge-transfer process.
Regarding the emission efficiency, both compounds exhibited high emission ΦF in non-polar solvents such as hexane (Hx), toluene (Tol), and benzene (Bz). However, ΦF of D1 was significantly reduced in polar solvents such as dimethylformamide (DMF), acetonitrile (ACN), and dimethylsulfoxide (DMSO), whereas D2 approximately maintains the high ΦF observed in non-polar solvents (see photographs in Figure 1A and values in Table 1).
The solvent-dependent emissive properties shown in Table 1 can be classified according to the dielectric constants as follows: 1) non-polar; 2) medium polar; 3) protic polar; and 4) aprotic polar solvents (Table 1). In non-polar solvents, the fluorescence quantum yield is high because of the negligible non-radiative constants obtained for both D1 and D2. In the case of solvents with intermediate polarity, the solvent polarizability (π*) and dielectric constant (ε), could help understand the solvent-dependent photophysical behavior. For example, chloroform and ethyl acetate have π* values of 0.58 and 0.55, respectively, i.e., a π* value similar to toluene (0.54); however, they present an intermediate dielectric constant that deactivates D1 in a non-radiative pathway, reducing the fluorescence quantum yield. However, DCM with higher ε in the block showed a high fluorescence quantum yield for both compounds, which can be attributed to the higher polarizability (0.82) of the solvent (Marcus, 1993).
It has been shown that the α values of chloroform and DCM play a key role in the aggregation of molecules in crystal structures and the ability to form H-bonds (Mansoor and Shafi, 2015). Chloroform and DCM present α values of 0.44 and 0.30, respectively, confirming the higher H-bond formation ability of chloroform, which consequently reduces the ICT character (Marcus, 1993). For chlorinated solvents with intermediate polarity values, the PL quantum yield could also be affected by π*. The higher π* for DCM (0.82) compared to chloroform (0.58) facilitates a larger ICT character, increasing the fluorescence quantum yield up to 82%. A similar behavior was observed for the D–π–D–π–A architecture where the greater the stabilized ICT state, the higher the fluorescence quantum yield (Zimosz et al., 2022).
In the polar protic solvent, the H-bond ability of alcohols and thus the α value play a key role in the ICT character and, consequently, the emissive properties (Marcus, 1993). Both molecules displayed low fluorescence ΦF in polar protic solvents such as ethanol (EtOH) and methanol (MeOH), as previously observed for related aminopyrimidines (Herbich et al., 1992; Herbich and Waluk, 1994). Hence, ΦF is strongly influenced by the polarity and hydrogen-bonding ability of solvents used (Pannipara et al., 2014; Kalyagin et al., 2022). Furthermore, MeOH and EtOH present the values of 0.98 and 0.86, respectively, indicating a higher hydrogen-bond formation ability for MeOH, which reduces the donor character of the substituted-amine group and, consequently, decreases the fluorescence quantum yield (2%–3%) for both systems (Anderson et al., 2019). However, a quantum yield of 31% is observed in EtOH for D2, suggesting a less-effective interaction of EtOH with the electron pair of the amine nitrogen due to the steric hindrance introduced by the phenyl substituents compared with the methyl groups in D1 (7%) (Kinoshita et al., 2000).
The significant redshift (35 nm) of the emission maximum in D1 compared to D2 in polar aprotic solvents such as DMSO could be rationalized due to a larger molecule planarization and better stabilization of the polar excited state by the solvent molecules of D1. Similar behavior has been observed in other systems such as N,N′-disubstituted dihydrodibenzo[a,c]phenazines (Chen et al., 2017).
Time-resolved fluorescence measurements were performed to analyze the dependence of the emissive properties of D1 and D2 on the dielectric constant of the solvent. The fluorescence decay profiles were recorded in all the aforementioned solvents (Figure 2 and Supplementary Table S1). Fluorescence decay traces showed a correlation of the lifetime parameter with solvent polarity only for D2, with lifetimes spanning from 1.51 ns to 4.64 ns from hexane to ACN. Nearly all the decays displayed a clear mono-exponential fitting, except for D1 in DMF, ACN, and DMSO, which showed a biexponential behavior. It is worth noting that the fluorescence lifetime is drastically reduced in polar protic solvents for both compounds, and as a consequence, ΦF decreases below 10% for D1 (EtOH and MeOH) and to 31% and 3% for D2 in EtOH and MeOH, respectively (Liu et al., 2022). Kinetic parameters (radiative and non-radiative rate constants) of both D1 and D2 systems were extracted from ΦF and time-resolved measurements. As shown in Supplementary Table S2, the radiative rate constant (kr) decreased as the solvent polarity increased for D1, whereas the non-radiative rate constant (knr) increased considerably, thus leading to a lower ΦF. In contrast, both the radiative and non-radiative pathways decreased upon increasing the solvent polarity for D2 (Supplementary Table S2 and Supplementary Figure S1), thus resulting in a high ΦF even in polar solvents. These data agree with the high dependence of the emissive properties on the nature of the N-donor substituent of the biphenylpyrimidine push–pull systems.
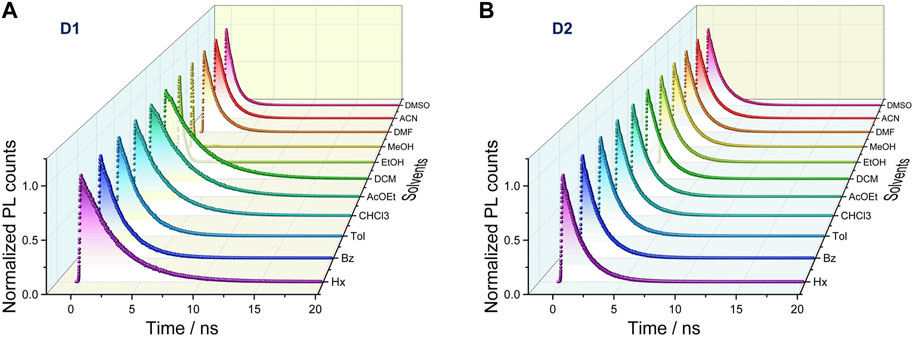
FIGURE 2. Time-resolved fluorescence spectra recorded in their maxima in different solvents under anaerobic conditions as a function of the dielectric constant for D1 (A) and D2 (B). An excitation wavelength of 375 nm was employed using a pulsed laser (10 MHz).
The electrochemical properties of D1 and D2 were then characterized through cyclic voltammetry (CV) measurements, and the energies of the highest occupied (HOMO) and lowest unoccupied molecular orbital (LUMO) were estimated using the energy level of ferrocene (Fc, 4.8 eV) as an external standard and calibrated by comparing with the E1/2(Fc/Fc+) half-wave electrode potential. The reduction potential of triarylpyrimidines has been reported to be dependent on the nature of the aryl groups. The introduction of electron-donor groups would increase the reduction potential to some extent (Itami et al., 2004). Supplementary Figure S2 shows the comparison of the cyclic voltametric responses of D1 and D2 in partially deaerated ACN solutions. The voltammograms showed an essentially reversible couple near −0.65 V vs. Ag/AgCl, corresponding to the well-known one-electron reduction of dissolved oxygen, accompanied by an apparently irreversible cathodic signal at approximately −2.1 and –2.1 V and an apparently irreversible anodic wave at approximately 1 and 0.9 V for D1 and D2, respectively. The value recorded for the reduction potential is close to that previously reported and is attributed to the introduction of one electron into the pyrimidine ring, whereas the oxidation potential arises from the strong electron-donating ability of the dimethyl/phenylamino group (Itami et al., 2004; Qiao et al., 2020).
To estimate the electrochemical bandgaps of D1 and D2, we employed the following widely used equations:
where EHOMO and ELUMO represent the energies in the vacuum scale of HOMO and LUMO, respectively; Eox and Ered are the half-peak electrode potentials corresponding to the oxidation and reduction of the tested compounds, respectively, and the E1/2(Fc/Fc+) half-wave electrode potential of the Fc/Fc+ couple is ca. 0.5 V under these conditions; all potentials relative to the reference electrode were used for voltametric measurements. Then, the electrochemical bandgap (∆ECV) was calculated using Eq. (5):
Since the oxidation and reduction of D1 and D2 are not electrochemically reversible processes, the half-wave potentials at 100 mV/s were used as approximate estimates of Eox and Ered. The corresponding values led to the bandgaps of 3.1 ± 0.1 and 3.0 ± 0.1 eV for D1 and D2, respectively. These values are consistent with the respective optical bandgaps (∆EOpt) determined for D1 and D2 (2.9 and 2.8 eV) derived from the absorption onset. The energy levels obtained by CV for D1 and D2 are very close to each other, with the HOMO level of D1 slightly more stabilized than that of D2. Table 2 summarizes the electrochemical data.
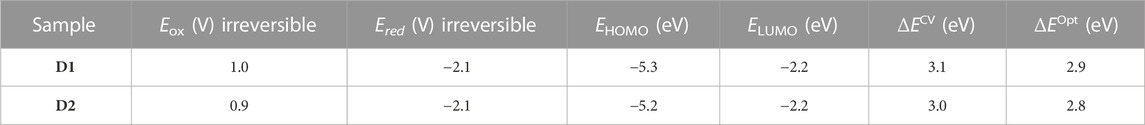
TABLE 2. Electrochemical data, HOMO and LUMO energies, and energy gaps obtained by cyclic voltammetry and optical measurements.
According to thermal properties (Supplementary Figure S3), both D1 and D2 compounds present good thermal stability at the usual annealing temperature (>100°C) required for optical device preparation, as demonstrated by thermogravimetric (TGA) and differential scanning calorimetry (DSC) analyses. The higher decomposition temperature observed for D2 agrees with the presence of the phenyl groups in the amine-donor moiety.
In order to explain the different optical behavior observed for D1 and D2, the excited-state properties of these two molecules were theoretically investigated using DFT calculations for the ground state and TD-DFT calculations for the excited states. The geometry of the two systems was optimized, both in the ground electronic state (S0) and in the lowest singlet excited state (S1) at the DFT B3LYP/6-311G** and TD-DFT B3LYP/6-311G** levels of theory, respectively. Four solvents were chosen (hexane and toluene to describe the non-polar environments and ACN and DMSO to describe the polar solvents) to interpret the photophysical properties of D1 and D2.
Supplementary Figure S4 shows the optimized geometries calculated for the electronic ground state (S0) of the two molecules in the four different solvents. The C1C2C3C4, C5C6C7C8, and C9C10N1C11 dihedral angles, which account for the internal twisting of the pyrimidine ring, the central biphenyl unit, and the amino group, respectively, and define the deviations from molecular planarity, are used for characterizing the obtained minima (Scheme 1 shows the atomic numbering). Independent of the solvent, the C1C2C3C4 and C5C6C7C8 angles present similar values for both D1 and D2 (approximately 20° and 35°, respectively), whereas the C9C10N1C11 angle shows significantly different values of approximately 8° and 35° for D1 and D2, respectively. This difference is most probably the result of the steric hindrance introduced by the phenyl groups borne by the terminal amine group in D2. The conjugated core of molecules D1 and D2 therefore presents a maximum deviation of 35° from planarity.
To disentangle the absorption spectra, the electronic S0→Sn transitions were calculated at the S0 minimum-energy geometry using TD-DFT calculations. Independent of the solvent, the low- and high-energy bands experimentally observed at approximately 370 and 270 nm were assigned to transitions to the S1 and S4 singlet excited states, respectively. The nature of the S1 state for the four solvents considered in our calculations (hexane, toluene, ACN, and DMSO) is mainly of a charge-transfer character for both D1 and D2. The state is mainly described by a one-electron excitation from HOMO to LUMO, which are respectively localized over the amine, with a large contribution of the biphenyl linker, and the pyrimidine part of the molecule (Figure 3 and Supplementary Figures S5, S6). The charge transfer upon excitation is supported by the localization of HOMO and LUMO orbitals over the four fragments defined in Scheme 1 that has been evaluated performing a Mulliken population analysis (Supplementary Table S3).
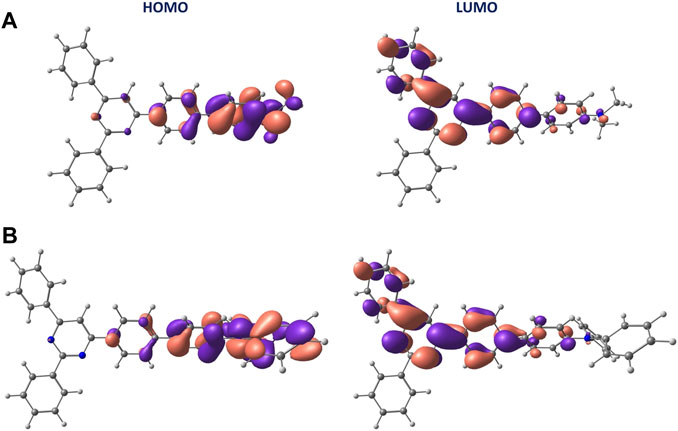
FIGURE 3. Isosurface contour plots (±0.03 a. u.) calculated at the B3LYP/6-311G**(PCM) level for the HOMO and LUMO orbitals in ACN of D1 (A) and D2 (B).
The charge-transfer nature of the S1 state is reflected in the value calculated for the dipole moment, equal to 36 and 32 D for D1 and D2, respectively, which is considerably larger than that computed for the S0 state (6 and 3 D, respectively). The nature of the S4 state of D1 for the four solvents considered is of a π–π* state localized on the pyrimidine part of the molecule, being described by a one-electron promotion from HOMO-1 to LUMO orbitals (Supplementary Figure S5 and Supplementary Table S4). In contrast, the S4 state of D2 shows a charge-transfer character (similar to S1) and mainly results from the HOMO→LUMO+2 excitation (Supplementary Figure S6 and Supplementary Table S5). The nature of the S1 and S4 states of D1 and D2 is confirmed by computing the corresponding natural transition orbitals (NTOs), both at the ground-state minimum-energy geometry and at the S1 minima (Supplementary Figures S7, S8). The topology of NTOs closely resembles the topology characterizing the MOs involved in the respective electronic transition.
The absorption maxima of D1, computed as the vertical energy to the S1/S4 states at the optimized S0 minimum, are calculated at 447/315, 462/316, 483/316, and 492/316 nm (2.77/3.94, 2.68/3.92, 2.57/3.92, and 2.52/3.92 eV) in hexane, toluene, ACN, and DMSO, respectively. For D2, these values are 467/333, 480/336, 489/340, and 498/341 nm (2.66/3.72, 2.58/3.69, 2.54/3.65, and 2.49/3.64 eV), respectively. Then, theoretical calculations accurately predict that both absorptions weakly depend on the solvent polarity, being slightly redshifted passing from D1 to D2. As previously suggested, this agrees with the small dipole moment calculated in S0 for D2 (3 D) and the slightly larger value obtained for D1 (6 D). Compared with the experimental results (Table 1), the theoretical data were redshifted for all the solvents considered. The reported redshifted trend in the theoretical energies is not surprising, considering the charge-transfer character of the excited states and the well-documented underestimation that B3LYP gives in such cases (Dreuw and Head-Gordon, 2004).
To gain insights into the changes in the fluorescence properties that D1 and D2 undergo with the solvent, the geometry of both molecules in the emitting S1 state was optimized at the TD-DFT 6-311G** level in the four solvents selected. The geometry optimization of S1 evolves differently for D1 and D2. For D1, two minima were found for S1 (Supplementary Figure S9). In the first minimum, hereafter S1,0, the amine group is placed in the same plane of the adjacent phenyl ring, displaying a C9C10N1C11 dihedral angle at approximately 0°. In the second minimum, hereafter S1,90, the amine group is instead perpendicular to the plane of the adjacent phenyl ring, displaying a C9C10N1C11 dihedral angle close to 90°. Regarding the C5C6C7C8 twisting angle of the central biphenyl unit, its value decreases with respect to the S0 minimum by approximately 8° and 16° in the S1,0 and S1,90 minima, respectively, independent of the polarity of the solvent.
On the other hand, the C1C2C3C4 angle defining the twisting of the pyrimidine environment decreases to almost zero in all cases, except for the S1,0 minima in non-polar solvents, where it only decreases by a few degrees. The charge transfer between the amine and pyrimidine environments associated with the S0→S1 transition therefore determines that the conjugated skeleton of D1 is, in general, more planar in S1 than in S0. For D2, only one minimum was obtained, hereafter S1,40, in which the amine group is significantly rotated (C9C10N1C11 dihedral angle equal to approximately 50° in hexane and toluene and at approximately 40° in ACN and DMSO), but it is not perpendicular to the phenyl ring. The non-planarity of such a structure is compatible with the steric hindrance offered by the phenyl rings in D2. All attempts to obtain a S1,90 structure for D2 were unsuccessful. Regarding the C1C2C3C4 and C5C6C7C8 angles, the former decreases to almost zero, and the latter is reduced by approximately 15° under both polar and non-polar conditions. The variation in the three aforementioned dihedral angles passing from S0 to S1 is schematically summarized in Figure 4. The specific values of these dihedral angles shed light on the crucial role of the molecular geometry in their optical and electronic properties, including intramolecular charge transfer. According to the values obtained, the biphenyl spacer has a minimal impact on the connection with the acceptor triphenylpyrimidine moiety, thus facilitating ICT, but has a relevant impact on the conformation adopted by the amine-donor group passing from S0 to S1, depending on the substituent donor nature. For D1, a fully twisted 90° conformation is achieved upon excitation to the charge-transfer S1 state (S1,90), whereas only an intermediate twisting is possible for D2 (S1,40).
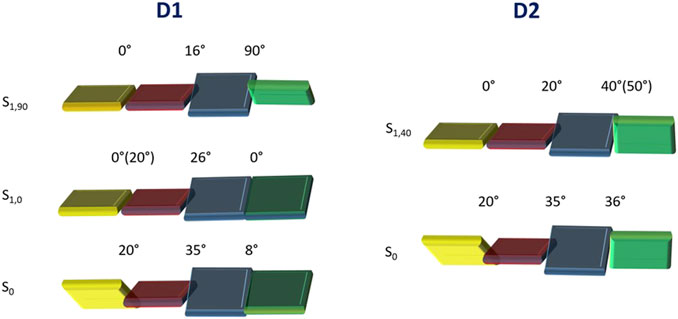
FIGURE 4. Schematic representation showing the twisting of the different molecular fragments constituting compounds D1 and D2. Averaged values of the C1C2C3C4, C5C6C7C8, and C9C10N1C11 dihedral angles are given for the optimized S0 and S1 geometries of D1 and D2. When the dihedral angle in non-polar solvents differs significantly, the value is reported in parenthesis. In order to better identify the dihedral angels, the same color code presented in Scheme 1 was adopted.
From the characterized S1 minima, the vertical energy differences with respect to the ground state were computed and compared with the recorded fluorescence emission. For D1, the emission energies calculated from the S1,0 minima are equal to 541, 566, 729, and 725 nm (2.29, 2.19, 1.70, and 1.71 eV) in hexane, toluene, ACN, and DMSO, respectively. Despite the fact that these theoretical energies are considerably lower than the experimental energies, they correctly describe the experimentally recorded redshifting in the emission energy passing from non-polar to polar solvents. For example, the experimental data register a redshift of the emission in ACN with respect to toluene of approximately 0.6 eV, a value that agrees with the 0.5 eV difference based on the computational results. It is known that the functional used here tends to lower the energy to CT-type states (Cammi et al., 2000). Therefore, theoretical results predict the energetic minima of the CT character for all solvents, which are indeed in agreement with the experimental emission spectra in all solvent except hexane. Regarding the S1,90 minima of D1, they appear to be non-emissive, with the computed oscillator strength for the transition to the ground state being equal to zero. This is a consequence of the 90° rotation of the amine group, making the overlap between HOMO and LUMO insignificant (Supplementary Figure S10). For D2 (Supplementary Figure S11), the emission energies computed from S1,40 are equal to 545, 561, 715, and 710 nm (2.27, 2.21, 1.73, and 1.75 eV) in hexane, toluene, ACN, and DMSO, respectively. Again, the absolute values predicted for the emission energies are too low compared to the experimental values but correctly described the redshifting of the emission from non-polar to polar solvents.
Employing the same example as that of D1, for D2, the experimental data found a redshift of 0.6 eV from toluene to ACN, a value that agrees with the 0.5 eV difference based on the computational results.
The drastic decrease observed experimentally for the value of ΦF of D1 in polar solvents can be rationalized by the presence of the S1,90 structure, which is instead absent in D2. The S1,90 minimum corresponds, in polar solvents, to the lowest S1 minimum-energy structure (i.e., lower than the S1,0 structure), and consequently, it will be the geometry toward which the S1 population will evolve (Supplementary Table S6 for D1 and Supplementary Table S7 for D2). Considering the geometrical similarities between the S0 and S1,0 structures (Supplementary Figures S4, S9, S12), it is, however, plausible to assume that the molecule will initially decay to the S1,0 minimum. The energy barrier that separates the S1,0 and S1,90 structures was evaluated by computing the corresponding minimum energy path (MEP, Figure 5). From S1,0, an energy barrier of 0.25 and 0.24 eV was computed to reach S1,90 in ACN and DMSO, respectively. These relatively small values confirm the ability of the system to attain the S1,90 structure. Even more relevant is the fact that the S1,90 structure is separated from the ground state by a small energy gap of only 0.39 eV in polar solvents like ACN (0.40 eV in DMSO), which consequently favors the non-radiative decay back to the ground state (Figure 5A). The S1,90 structure is then able to promote the non-radiative decay of D1 in polar solvents, thus explaining the experimentally recorded low ΦF values for such environments. In non-polar solvents instead, S1,0 is the lowest S1 minimum (Supplementary Table S6) to which consequently the system will tend to evolve. The barrier from the S1,0 to S1,90 minima in non-polar solvents was computed to have values of 0.44 and 0.43 eV for hexane and toluene, respectively. The population of S1,90 in non-polar solvents is then much less probable than in polar solvents for two reasons: first, S1,90 is not the lowest S1 minimum; second, it requires to surmount a significantly higher energy barrier. Moreover, in non-polar solvents, the energy gap with the ground state at the S1,90 structure is of 1.94 and 1.76 eV in hexane and toluene, respectively, and will not allow a non-radiative decay (Figure 5B). This, together with the impossibility to emit fluorescence due to the small overlap between HOMO and LUMO orbitals, makes the contribution of the S1,90 structure to the photophysics of the D1 molecule in non-polar environments of marginal relevance. These theoretical findings support the high ΦF values experimentally observed for D1 in non-polar solvents.

FIGURE 5. Schematic representation of the S0 and S1 PESs of D1 (A, B) and D2 (C, D) in polar (ACN) and non-polar (toluene) solvents.
Finally, the absence of an S1,90 structure for D2 explains why for such a system high ΦF values are recorded in both non-polar and polar solvents (Figure 1; Table 1). The influence of the solvent polarity on the fluorescence properties of the D2 molecule is thus reduced to a redshift of the emission passing from non-polar to polar solvents (Figures 5C,D).
A large fluorosolvatochromism is observed for biphenylpyrimidine-based push–pull systems bearing dimethylamino (D1) or diphenylamino (D2) as donor groups, where the N,N-substituent (dimethyl or diphenyl) determines their emissive properties according to the polarity of the solvent. Theoretical calculations have demonstrated to be a key tool to explain this effect. In the case of D1, the presence of a twisted geometry for the lowest-energy, charge-transfer excited state (S1,90) was found to promote the non-radiative decay in polar solvents due to the lower energy of this structure and its energy proximity to the ground S0 state. In contrast, in non-polar solvents, the S1,90 structure is higher in energy and less attainable, making the non-radiative decay less likely. In the case of D2, the S1,90 structure is not a minimum due to the steric hindrance between the phenyl rings of the amine group, and consequently, the fluorescence quantum yield is maintained independently of the polarity of the solvent. These systems are of high interest as possible hole transporters in electroluminescent devices based on semiconductor materials such as perovskites, due to their stunning emissive and electronic properties.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
AC-V: formal analysis, investigation, and writing–original draft. IS-D: formal analysis, software, and writing–original draft. MD: methodology, supervision, and writing–review and editing. MV: methodology and writing–review and editing. PR: methodology and writing–review and editing. CA: methodology, supervision, and writing–review and editing. AG: formal analysis, software, and writing–original draft. AD-C: formal analysis and writing–review and editing. EO: conceptualization, software, supervision, and writing–review and editing. RG: conceptualization, project administration, supervision, and writing–review and editing. JP-P: conceptualization, funding acquisition, project administration, supervision, and writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Financial support by the MCIN/AEI of Spain (projects PID 2020-115710GBI00, PID 2021-128569NB-I00, and CEX 2019-000919-M, funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”) and the Generalitat Valenciana (IDIFEDER/2018/064, IDIFEDER/2021/064, CIPROM/2022/57, PROMETEO/2020/077, MFA/2022/017, and MFA/2022/051) are acknowledged. The MFA/2022/017 and MFA/2022/051 projects form part of the Advanced Materials programme supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by the Generalitat Valenciana. The study was also supported by CEDENNA AFB180001 and FONDECYT 1200192/1200116 projects.
AC-V thanks the “Maria de Maeztu” Programme for Units of Excellence in R&D (CEX 2019-000919-M) for a predoctoral fellowship (PRE2018-084294) funded by MCIN/AEI/10.13039/501100011033 and ESF Investing in your future. IS-D also thanks the Generalitat Valenciana for their predoctoral grant CIACIF/2021/438. AG is indebted to MCIN/AEI for granting the Juan de la Cierva (IJC2018-035123-I) fellowship funded by MCIN/AEI/10.13039/501100011033 and by “European Union NextGenerationEU/PRTR.”
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1292541/full#supplementary-material
Achelle, S., and Robin-Le Guen, F. (2017). Emission properties of diazines chromophores: structure-properties relationship. J. Photochem. Photobiol. A Chem. 348, 281–286. doi:10.1016/j.jphotochem.2017.08.060
Achelle, S., Rodríguez-López, J., and Robin-Le Guen, F. (2023). The arylvinylpyrimidine scaffold: a tunable platform for luminescent and optical materials. Org. Biomol. Chem. 21, 39–52. doi:10.1039/D2OB01841A
Al-Ahmed, Z. A., Habib, I. H. I., Khattab, R. R., Abdelhameed, R. M., El-Naggar, M., Abu Bieh, M. H., et al. (2021). Synthesis, spectrophotometric, voltammetric, and density functional theory studies of tetrahydro[3,2-b]indolocarbazoles for sensing small molecules. J. Heterocycl. Chem. 58, 127–136. doi:10.1002/jhet.4153
Anderson, R. S., Nagirimadugu, N. V., and Abelt, C. J. (2019). Fluorescence quenching of carbonyl-twisted 5-Acyl-1-dimethylaminonaphthalenes by alcohols. ACS Omega 4, 14067–14073. doi:10.1021/acsomega.9b01905
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Bouihi, F., Schmaltz, B., Mathevet, F., Kreher, D., Faure-Vincent, J., Yildirim, C., et al. (2022). D-π-A-Type pyrazolo[1,5-a]pyrimidine-based hole-transporting materials for perovskite solar cells: effect of the functionalization position. Materials 15, 7992. doi:10.3390/ma15227992
Bureš, F. (2014). Fundamental aspects of property tuning in push–pull molecules. RSC Adv. 4, 58826–58851. doi:10.1039/C4RA11264D
Cammi, R., Mennucci, B., and Tomasi, J. (2000). Fast evaluation of geometries and properties of excited molecules in solution: a tamm-dancoff model with application to 4-dimethylaminobenzonitrile. J. Phys. Chem. A 104, 5631–5637. doi:10.1021/jp000156l
Chen, W., Chen, C.-L., Zhang, Z., Chen, Y.-A., Chao, W.-C., Su, J., et al. (2017). Snapshotting the excited-state planarization of chemically locked N,N′-Disubstituted dihydrodibenzo[a,c]phenazines. J. Am. Chem. Soc. 139, 1636–1644. doi:10.1021/jacs.6b11789
Chibani, S., Laurent, A. D., Blondel, A., Mennucci, B., and Jacquemin, D. (2014). Excited-state geometries of solvated molecules: going beyond the linear-response polarizable continuum model. J. Chem. Theory Comput. 10, 1848–1851. doi:10.1021/ct5001507
Denneval, C., Achelle, S., Baudequin, C., and Robin-Le Guen, F. (2014). Prediction of photophysical properties of pyrimidine chromophores using Taguchi method. Dyes Pigments 110, 49–55. doi:10.1016/j.dyepig.2014.05.030
Dreuw, A., and Head-Gordon, M. (2004). Failure of time-dependent density functional theory for long-range charge-transfer excited states: the Zincbacteriochlorin−Bacteriochlorin and Bacteriochlorophyll−Spheroidene complexes. J. Am. Chem. Soc. 126, 4007–4016. doi:10.1021/ja039556n
Fecková, M., Kalis, I. K., Roisnel, T., Le Poul, P., Pytela, O., Klikar, M., et al. (2021). Photophysics of 9,9-dimethylacridan-substituted phenylstyrylpyrimidines exhibiting long-lived intramolecular charge-transfer fluorescence and aggregation-induced emission characteristics. Chem. – A Eur. J. 27, 1145–1159. doi:10.1002/chem.202004328
Fecková, M., Le Poul, P., Bureš, F., Robin-Le Guen, F., and Achelle, S. (2020). Nonlinear optical properties of pyrimidine chromophores. Dyes Pigments 182, 108659. doi:10.1016/j.dyepig.2020.108659
Francl, M. M., Pietro, W. J., Hehre, W. J., Binkley, J. S., Gordon, M. S., Defrees, D. J., et al. (1982). Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 77, 3654–3665. doi:10.1063/1.444267
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16. Wallingford, CT: Rev. C.01.
Galván, I. F., Martín, M. E., and Aguilar, M. A. (2010). Theoretical study of the dual fluorescence of 4-(N,N-Dimethylamino)benzonitrile in solution. J. Chem. Theory Comput. 6, 2445–2454. doi:10.1021/ct9006713
Gnida, P., Slodek, A., Chulkin, P., Vasylieva, M., Pająk, A. K., Seweryn, A., et al. (2022). Impact of blocking layer on DSSC performance based on new dye -indolo[3,2,1-jk]carbazole derivative and N719. Dyes Pigments 200, 110166. doi:10.1016/j.dyepig.2022.110166
Gómez, S., Soysal, E. N., and Worth, G. A. (2021). Micro-solvated DMABN: excited state quantum dynamics and dual fluorescence spectra. Molecules 26, 7247. doi:10.3390/molecules26237247
Grabowski, Z. R., Rotkiewicz, K., and Rettig, W. (2003). Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem. Rev. 103, 3899–4032. doi:10.1021/cr940745l
Grabowski, Z. R., Rotkiewicz, K., and Siemiarczuk, A. (1979). Dual fluorescence of donor-acceptor molecules and the twisted intramolecular charge transfer (TICT) states. J. Luminescence 18-19, 420–424. doi:10.1016/0022-2313(79)90153-4
Haberhauer, G. (2017). Planarized and twisted intramolecular charge transfer: a concept for fluorophores showing two independent rotations in excited state. Chem. – A Eur. J. 23, 9288–9296. doi:10.1002/chem.201700566
Haberhauer, G., Gleiter, R., and Burkhart, C. (2016). Planarized intramolecular charge transfer: a concept for fluorophores with both large Stokes shifts and high fluorescence quantum yields. Chem. – A Eur. J. 22, 971–978. doi:10.1002/chem.201503927
Hadad, C., Achelle, S., García-Martinez, J. C., and Rodríguez-López, J. (2011). 4-Arylvinyl-2,6-di(pyridin-2-yl)pyrimidines: synthesis and optical properties. J. Org. Chem. 76, 3837–3845. doi:10.1021/jo200204u
Henkelman, G., Uberuaga, B. P., and Jónsson, H. (2000). A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904. doi:10.1063/1.1329672
Herbich, J., Karpiuk, J., Grabowski, Z. R., Tamai, N., and Yoshihara, K. (1992). Modification of the intramolecular electron transfer by hydrogen bonding: 4-(dialkylamino) pyrimidines. J. Luminescence 54, 165–175. doi:10.1016/0022-2313(92)90005-T
Herbich, J., and Waluk, J. (1994). Excited charge transfer states in 4-aminopyrimidines, 4-(dimethylanilino)pyrimidine and 4-(dimethylamino)pyridne. Chem. Phys. 188, 247–265. doi:10.1016/0301-0104(94)00256-8
Il'ichev, Y. V., Kühnle, W., and Zachariasse, K. A. (1998). Intramolecular charge transfer in dual fluorescent 4-(dialkylamino)benzonitriles. Reaction efficiency enhancement by increasing the size of the amino and benzonitrile subunits by alkyl substituents. J. Phys. Chem. A 102, 5670–5680. doi:10.1021/jp980426o
Improta, R., Barone, V., Scalmani, G., and Frisch, M. J. (2006). A state-specific polarizable continuum model time dependent density functional theory method for excited state calculations in solution. J. Chem. Phys. 125, 054103. doi:10.1063/1.2222364
Improta, R., Scalmani, G., Frisch, M. J., and Barone, V. (2007). Toward effective and reliable fluorescence energies in solution by a new state specific polarizable continuum model time dependent density functional theory approach. J. Chem. Phys. 127, 074504. doi:10.1063/1.2757168
Itami, K., Yamazaki, D., and Yoshida, J.-I. (2004). Pyrimidine-core extended π-systems: general synthesis and interesting fluorescent properties. J. Am. Chem. Soc. 126, 15396–15397. doi:10.1021/ja044923w
Kalyagin, A., Antina, L., Ksenofontov, A., Antina, E., and Berezin, M. (2022). Solvent-dependent fluorescence properties of CH2-bis(BODIPY)s. Int. J. Mol. Sci. 23, 14402. doi:10.3390/ijms232214402
Kinoshita, M., Okamoto, Y., and Hirata, F. (2000). Peptide conformations in alcohol and water: analyses by the reference interaction site model theory. J. Am. Chem. Soc. 122, 2773–2779. doi:10.1021/ja993939x
Kozlov, O. V., Liu, X., Luponosov, Y. N., Solodukhin, A. N., Toropynina, V. Y., Min, J., et al. (2017). Triphenylamine-based push–pull molecule for photovoltaic applications: from synthesis to ultrafast device photophysics. J. Phys. Chem. C 121, 6424–6435. doi:10.1021/acs.jpcc.6b12068
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. doi:10.1103/PhysRevB.37.785
Lippert, E. (1955). Dipolmoment und Elektronenstruktur von angeregten Molekülen. Z. für Naturforsch. A 10, 541–545. doi:10.1515/zna-1955-0707
Liu, J., Chen, C., and Fang, C. (2022). Polarity-dependent twisted intramolecular charge transfer in diethylamino coumarin revealed by ultrafast spectroscopy. Chemosensors 10, 411. doi:10.3390/chemosensors10100411
Maddala, G., Gade, R., Ahemed, J., Kalvapalli, S., Simhachalam, N. B., Chetti, P., et al. (2021). Efficient, dopant free phenazine based hole transporting materials for high performance perovskite solar cells. Sol. Energy 226, 501–512. doi:10.1016/j.solener.2021.08.055
Manda, K., Kore, R., Ambapuram, M., Chetti, P., Roy, S., Jadhav, V. D., et al. (2022). Benzodithiophene-based, donor-acceptor-pi-donor-acceptor systems as hole transporting materials for efficient perovskite solar cells. Chemphotochem 6, e202200062. doi:10.1002/cptc.202200062
Mansoor, S. S., and Shafi, S. S. (2015). Oxidation of methionine by tetraethylammonium chlorochromate in non-aqueous media – a kinetic and mechanistic study. Arabian J. Chem. 8, 480–486. doi:10.1016/j.arabjc.2011.01.031
Marcus, Y. (1993). The properties of organic liquids that are relevant to their use as solvating solvents. Chem. Soc. Rev. 22, 409–416. doi:10.1039/CS9932200409
Martin, R. L. (2003). Natural transition orbitals. J. Chem. Phys. 118, 4775–4777. doi:10.1063/1.1558471
Mataga, N., Kaifu, Y., and Koizumi, M. (1955). The solvent effect on fluorescence spectrum, change of solute-solvent interaction during the lifetime of excited solute molecule. Bull. Chem. Soc. Jpn. 28, 690–691. doi:10.1246/bcsj.28.690
Nakao, K., Sasabe, H., Komatsu, R., Hayasaka, Y., Ohsawa, T., and Kido, J. (2017). Significant enhancement of blue OLED performances through molecular engineering of pyrimidine-based emitter. Adv. Opt. Mater. 5, 1600843. doi:10.1002/adom.201600843
Neese, F. (2022). Software update: the ORCA program system—version 5.0. WIREs Comput. Mol. Sci. 12, e1606. doi:10.1002/wcms.1606
Pannipara, M., Asiri, A. M., Alamry, K. A., Arshad, M. N., and El-Daly, S. A. (2014). Spectroscopic investigation, effect of solvent polarity and fluorescence quenching of a new D-π-A type chalcone derivative. J. Fluoresc. 24, 1629–1638. doi:10.1007/s10895-014-1449-1
Qian, X., Yan, R., Hang, Y., Lv, Y., Zheng, L., Xu, C., et al. (2017). Indeno[1,2-b]indole-based organic dyes with different acceptor groups for dye-sensitized solar cells. Dyes Pigments 139, 274–282. doi:10.1016/j.dyepig.2016.12.028
Qiao, W., Duan, G., Wang, J., Dong, J., Pan, B., and Mu, X. (2020). Photoinduced charge transfer in push/pull systems of two-photon absorption. ACS Omega 5, 17275–17286. doi:10.1021/acsomega.0c01482
Qin, X., Yang, X., Du, L., and Li, M. (2021). Polarity-based fluorescence probes: properties and applications. RSC Med. Chem. 12, 1826–1838. doi:10.1039/D1MD00170A
Rodríguez-Aguilar, J., Vidal, M., Pastenes, C., Aliaga, C., Rezende, M. C., and Domínguez, M. (2018). The solvatofluorochromism of 2,4,6-triarylpyrimidine derivatives. Photochem. Photobiol. 94, 1100–1108. doi:10.1111/php.12982
Ryutaro, K., Hisahiro, S., and Junji, K. (2018). Recent progress of pyrimidine derivatives for high-performance organic light-emitting devices. J. Photonics Energy, 032108. doi:10.1117/1.JPE.8.032108
Sasaki, S., Drummen, G. P. C., and Konishi, G.-I. (2016). Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry. J. Mater. Chem. C 4, 2731–2743. doi:10.1039/C5TC03933A
Segarra-Martí, J., and Coto, P. B. (2014). A theoretical study of the intramolecular charge transfer in 4-(dimethylamino)benzethyne. Phys. Chem. Chem. Phys. 16, 25642–25648. doi:10.1039/C4CP03436H
Slodek, A., Zych, D., Maroń, A., Malecki, J. G., Golba, S., Szafraniec-Gorol, G., et al. (2019). Does the length matter? - synthesis, photophysical, and theoretical study of novel quinolines based on carbazoles with different length of alkyl chain. Dyes Pigments 160, 604–613. doi:10.1016/j.dyepig.2018.08.048
Soriano-Díaz, I., Ortí, E., and Giussani, A. (2023). On the importance of equatorial metal-centered excited states in the photophysics of cyclometallated Ir(iii) complexes. Dalton Trans. 52, 10437–10447. doi:10.1039/D3DT01404E
Subuddhi, U., Haldar, S., Sankararaman, S., and Mishra, A. K. (2006). Photophysical behaviour of 1-(4-N,N-dimethylaminophenylethynyl)pyrene (DMAPEPy) in homogeneous media. Photochem. Photobiological Sci. 5, 459–466. doi:10.1039/B600009F
Sun, H., Liu, D., Wang, T., Li, P. A., Bridgmohan, C. N., Li, W., et al. (2018). Charge-separated sensitizers with enhanced intramolecular charge transfer for dye-sensitized solar cells: insight from structure-performance relationship. Org. Electron. 61, 35–45. doi:10.1016/j.orgel.2018.06.045
Tan, C.-J., Yang, C.-S., Sheng, Y.-C., Amini, H. W., and Tsai, H.-H. G. (2016). Spacer effects of donor-π spacer-acceptor sensitizers on photophysical properties in dye-sensitized solar cells. J. Phys. Chem. C 120, 21272–21284. doi:10.1021/acs.jpcc.6b07032
Tomasi, J., and Persico, M. (1994). Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem. Rev. 94, 2027–2094. doi:10.1021/cr00031a013
Venkateswararao, A., Tyagi, P., Justin Thomas, K. R., Chen, P.-W., and Ho, K.-C. (2014). Organic dyes containing indolo[2,3-b]quinoxaline as a donor: synthesis, optical and photovoltaic properties. Tetrahedron 70, 6318–6327. doi:10.1016/j.tet.2014.04.009
Verbitskiy, E. V., Cheprakova, E. M., Subbotina, J. O., Schepochkin, A. V., Slepukhin, P. A., Rusinov, G. L., et al. (2014). Synthesis, spectral and electrochemical properties of pyrimidine-containing dyes as photosensitizers for dye-sensitized solar cells. Dyes Pigments 100, 201–214. doi:10.1016/j.dyepig.2013.09.006
Verbitskiy, E. V., Kvashnin, Y. A., Bogdanov, P. I., Medvedeva, M. V., Svalova, T. S., Kozitsina, A. N., et al. (2021a). The effect of molecular structure on the efficiency of 1,4-diazine–based D–(π)–A push-pull systems for non-doped OLED applications. Dyes Pigments 187, 109124. doi:10.1016/j.dyepig.2020.109124
Verbitskiy, E. V., Schepochkin, A. V., Makarova, N. I., Dorogan, I. V., Metelitsa, A. V., Minkin, V. I., et al. (2015). Synthesis, photophysical and redox properties of the D–π–A type pyrimidine dyes bearing the 9-phenyl-9H-carbazole moiety. J. Fluoresc. 25, 763–775. doi:10.1007/s10895-015-1565-6
Verbitskiy, E. V., Steparuk, A. S., Zhilina, E. F., Emets, V. V., Grinberg, V. A., Krivogina, E. V., et al. (2021b). Pyrimidine-based push–pull systems with a new anchoring amide group for dye-sensitized solar cells. Electron. Mater. 2, 142–153. doi:10.3390/electronicmat2020012
Wong, K.-T., Hung, T. S., Lin, Y., Wu, C.-C., Lee, G.-H., Peng, S.-M., et al. (2002). Suzuki coupling approach for the synthesis of Phenylene−Pyrimidine alternating oligomers for blue light-emitting material. Org. Lett. 4, 513–516. doi:10.1021/ol017066z
Wu, C. C., Lin, Y. T., Chiang, H. H., Cho, T. Y., Chen, C. W., Wong, K. T., et al. (2002). Highly bright blue organic light-emitting devices using spirobifluorene-cored conjugated compounds. Appl. Phys. Lett. 81, 577–579. doi:10.1063/1.1493669
Yang, S., Guan, D., Yang, M., Tian, J., Chu, W., and Sun, Z. (2015). Synthesis and characterization of novel butterfly-shaped aryl-substituted indolo[2,3-a]carbazole derivatives. Tetrahedron Lett. 56, 2223–2227. doi:10.1016/j.tetlet.2015.03.058
Zachariasse, K. A., Grobys, M., Von Der Haar, T., Hebecker, A., Il'ichev, Y. V., Jiang, Y. B., et al. (1996). Intramolecular charge transfer in the excited state. Kinetics and configurational changes. J. Photochem. Photobiol. A Chem. 102, 59–70. doi:10.1016/S1010-6030(96)04368-7
Zachariasse, K. A., Grobys, M., Von Der Haar, T., Hebecker, A., Il'ichev, Y. V., Morawski, O., et al. (1997). Photoinduced intramolecular charge transfer and internal conversion in molecules with a small energy gap between S1 and S2. Dynamics and structure. J. Photochem. Photobiol. A Chem. 105, 373–383. doi:10.1016/S1010-6030(96)04601-1
Zimosz, S., Slodek, A., Gnida, P., Glinka, A., Ziółek, M., Zych, D., et al. (2022). New D−π–D−π–A systems based on phenothiazine derivatives with imidazole structures for photovoltaics. J. Phys. Chem. C 126, 8986–8999. doi:10.1021/acs.jpcc.2c01697
Zych, D., Slodek, A., and Frankowska, A. (2019). Is it worthwhile to deal with 1,3-disubstituted pyrene derivatives? – Photophysical, optical and theoretical study of substitution position effect of pyrenes containing tetrazole groups. Comput. Mater. Sci. 165, 101–113. doi:10.1016/j.commatsci.2019.04.041
Keywords: intramolecular charge transfer, donor–acceptor systems, fluorosolvatochromism, photophysical properties, theoretical calculations
Citation: Cortés-Villena A, Soriano-Díaz I, Domínguez M, Vidal M, Rojas P, Aliaga C, Giussani A, Doménech-Carbó A, Ortí E, Galian RE and Pérez-Prieto J (2023) Governing the emissive properties of 4-aminobiphenyl-2-pyrimidine push–pull systems via the restricted torsion of N,N-disubstituted amino groups. Front. Chem. 11:1292541. doi: 10.3389/fchem.2023.1292541
Received: 11 September 2023; Accepted: 23 October 2023;
Published: 10 November 2023.
Edited by:
Claudia Espro, University of Messina, ItalyReviewed by:
Kenkera Rayappa Naveen, Julius Maximilian University of Würzburg, GermanyCopyright © 2023 Cortés-Villena, Soriano-Díaz, Domínguez, Vidal, Rojas, Aliaga, Giussani, Doménech-Carbó, Ortí, Galian and Pérez-Prieto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolina Aliaga, Y2Fyb2xpbmEuYWxpYWdhQHVzYWNoLmNs; Enrique Ortí, ZW5yaXF1ZS5vcnRpQHV2LmVz; Raquel E. Galian, cmFxdWVsLmdhbGlhbkB1di5lcw==; Julia Pérez-Prieto, anVsaWEucGVyZXpAdXYuZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.