 Seokwoo Lee1,2†
Seokwoo Lee1,2† Jae Eui Shin
Jae Eui Shin Sanghee Kim
Sanghee Kim- 1College of Pharmacy, Seoul National University, Seoul, Republic of Korea
- 2College of Pharmacy, Chungnam National University, Daejeon, Republic of Korea
The formation of N-heterocycles with multiple substituents is important in organic synthesis. Herein, we report a novel method for the construction of functionalized dihydropyridinone rings through the annulation of an amide α-carbon with a tethered alkyne moiety. The reaction of the amide with the alkyne was achieved via O-silyl N,O-ketene acetal formation and silver-mediated addition. Furthermore, the developed method was applied for the total synthesis of phenanthroindolizidine and phenanthroquinolizidine alkaloids. By varying the coupling partners, a concise and collective total synthesis of these alkaloids was achieved.
1 Introduction
The construction of N-heterocycles containing multiple substituents still remains an important synthetic challenge (Vo et al., 2014; Nandakumar et al., 2015; He et al., 2021; Chen et al., 2023). We recently described the Ag(I) and Brønsted acid co-catalyzed cyclization of an enamine with a tethered alkyne moiety as a one-pot method for pyridinium formation (Scheme 1A) (Lee et al., 2023). Our proposed mechanism for the transformation involves the addition of a nucleophilic enamine to a silver(I)-complexed alkyne, followed by protonolysis of the resulting vinyl-silver species and subsequent aromatization. Based on this annulation, we envisioned that the nucleophilic addition of the amide α-carbon onto the appended alkyne would form a dihydropyridinone (Scheme 1B). To the best of our knowledge, the reactions of alkynes with amides have not been well explored, although reactions with various types of carbon nucleophiles, especially stabilized carbon nucleophiles such as malonates, β-ketoesters, and diketones, have been well explored (Dénes et al., 2010; Hack et al., 2015; Lin et al., 2021).
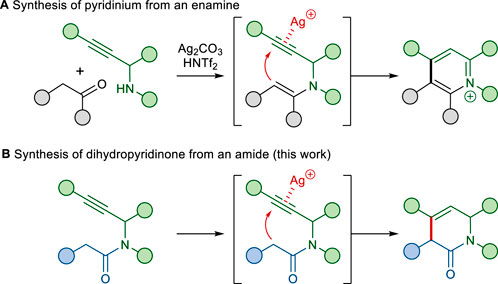
SCHEME 1. Synthesis of N-heterocycles via the annulation of an alkyne with a tethered moiety.
Herein, we report the annulation of an amide α-carbon with a tethered alkyne moiety, which is a new complementary process for the functionalization of dihydropyridinone rings. In addition, we discuss the application of this C–C bond-forming reaction for the expedient total synthesis of phenanthroindolizidine and phenanthroquinolizidine alkaloids.
2 Results and discussion
We examined the feasibility of the proposed reaction using model substrate 1 (Table 1), which was prepared in two steps from commercially available materials. Based on our previous results on pyridinium formation (Lee et al., 2023), Ag2CO3 or AgNTf2 were employed as a catalyst for alkyne activation. Without a base, no conversion of 1 occurred. The addition of a conventional base, such as an alkali metal carbonate or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), did not result in product formation (Supplementary Table S1). Despite the reported incompatibility of strong bases and Lewis-acidic metals (Yamamoto, 2000; Yamamoto et al., 2008; Lappert et al., 2009), the strong bases generally used for amide enolate generation, including potassium bis(trimethylsilyl)amide (KHMDS) and LiHMDS, were also examined. However, all of these attempts failed, and most of the starting material decomposed or was recovered (Supplementary Table S1).
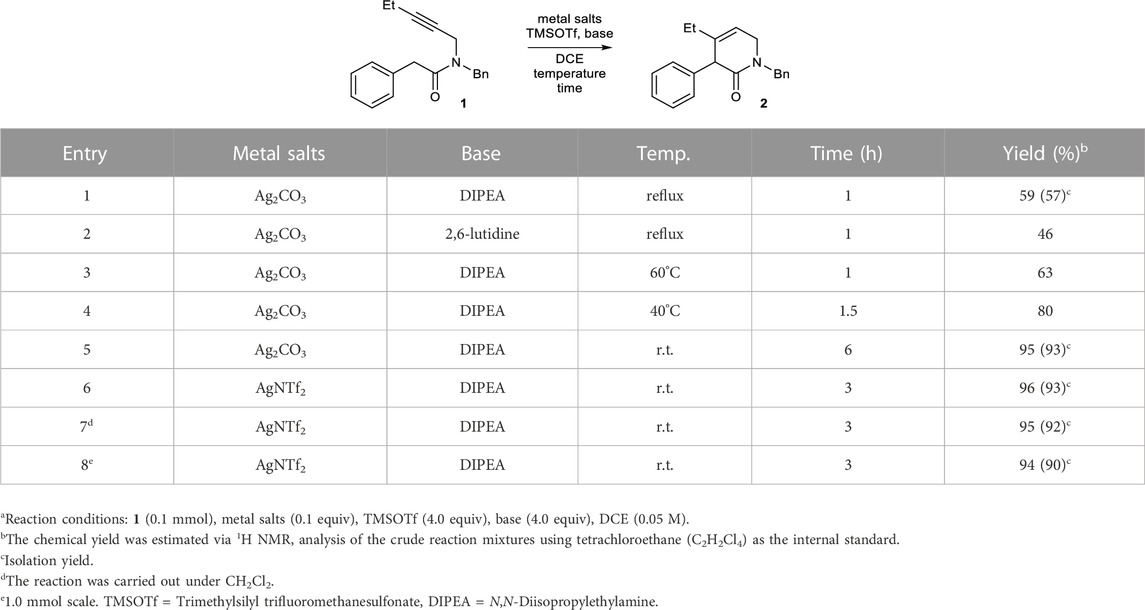
TABLE 1. Conditions for the formation of 2 from 1.a
We turned our attention to an O-silyl N,O-ketene acetal as a surrogate for the amide enolate. O-Silyl N,O-ketene acetals have been typically used for Mukaiyama-type reactions (Paris et al., 2022) and generated in situ by the treatment of an amide with a silylating agent and tertiary amine base (Kobayashi et al., 2011; Downey et al., 2015; Takeda et al., 2017). Previously, Shen and coworkers reported the gold(I)-catalyzed cyclization of alkynes with an O-silyl ketene amide or carbamate nucleophiles (Minnihan et al., 2007). However, the reaction of an alkyne with a silyl N,O-ketene acetal has not been reported.
At the outset of this study, TMSOTf was used as a silylating agent, and various amine bases were screened in the presence of 0.1 equiv of Ag2CO3 in dichloroethane (DCE) under reflux conditions. Among the tested amine bases, N,N-diisopropylethylamine (DIPEA) exhibited the best performance, affording dihydropyridinone 2 in a modest 59% yield along with a mixture of unidentifiable polar side products (Table 1, entry 1). The other sterically hindered base 2,6-lutidine also provided 2, albeit in a lower yield (46%, entry 2).
After determining the feasibility of the reaction, further screening of the reaction conditions was performed using DIPEA and TMSOTf. Under reflux conditions, the yield of compound 2 was reduced, likely due to the formation of considerable amounts of unidentified side products. A reduction in the reaction temperature led to an increase in the yield of compound 2 (entries 3–5), likely as a result of decreased formation of side products. For example, 2 was formed in an excellent yield of 95% at room temperature, although a longer reaction time was required (entry 5). When Ag2CO3 was replaced with AgNTf2, the reaction time was reduced by half (3 h), and the yield was also excellent (96%, entry 6). As in our previous study on the annulation of enamines with alkynes (Lee et al., 2023), the 5-membered heterocycles formed via 5-exo-dig cyclization were not observed under the conditions. Other silylating agents did not lead to better yields than TMSOTf (Supplementary Table S2). Several solvents were tested for this transformation. The only other effective solvent was CH2Cl2, which furnished 2 with a similar yield (95%, entry 7). Other solvents did not enable the formation of 2 (Supplementary Table S3). Under the optimal conditions, the reaction could be enlarged to a 1.0 mmol scale without a significant decrease in yield (94%, entry 8).
Based on these results, we attempted the total synthesis of phenanthroindolizidine alkaloids (Figure 1). This family of natural products exhibits a wide range of biological effects, including significant anticancer and antiviral activities (De Fatima Pereira et al., 2015; Jia et al., 2021). Therefore, these alkaloids have been the synthetic targets of numerous research groups over the past few decades (Chemler, 2009; Burtoloso et al., 2014).
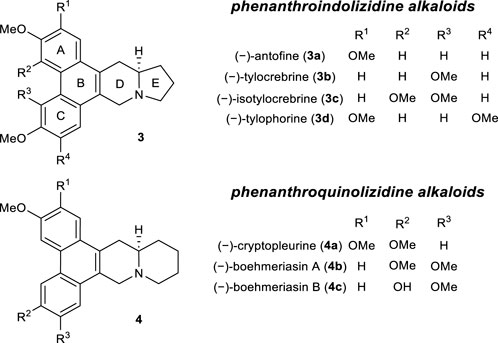
FIGURE 1. The structures of representative phenanthroindolizidine and phenanthroquinolizidine alkaloids.
Our retrosynthetic analysis, based on the above dihydropyridinone synthetic strategy, is depicted in Scheme 2. The B ring of the phenanthroindolizidine skeleton of Ⅰ could be constructed at the last stage of the synthesis via the biaryl coupling of Ⅱ. We envisioned that dihydropyridinone ring of Ⅱ could be formed by the annulation of an amide α-carbon with a tethered alkyne moiety in Ⅲ, according to the above-mentioned method. An obvious disconnection of the amide bond in Ⅲ led to the 2-alkyne-pyrrolidine Ⅳ and 2-arylacetic acid Ⅴ. Pyrrolidine derivative Ⅳ would be accessed by coupling of an aryl halide Ⅵ with the known alkyne Ⅶ. According to this retrosynthetic scheme, many members of this phenanthroindolizidine family and analogs could be synthesized by varying the two coupling partners Ⅴ and Ⅵ. Even, this scheme would permit the synthesis of phenanthroquinolizidine alkaloids if 2-alkyne-piperidine was employed instead of Ⅶ.
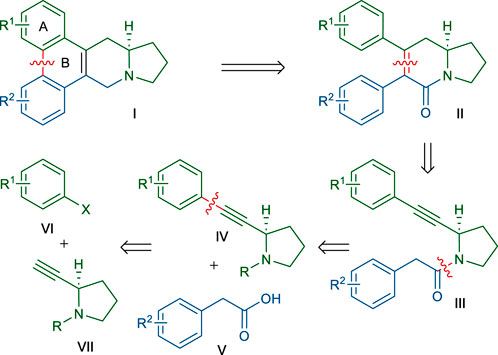
SCHEME 2. Retrosynthetic route for the synthesis of phenanthroindolizidine alkaloids.
Our synthesis began with the preparation of known alkyne 5 (Scheme 3), which is available in two steps from commercially available N-Boc-d-prolinol (Mercado-Marin et al., 2014). The Sonogashira coupling of 5 with 3,4-dimethoxy phenyl iodide afforded 6a in high yield. Removal of the N-Boc group, followed by EDCI-mediated coupling with 2-arylacetic acid 7a, generated amide 8a in a good overall yield. Application of the developed reaction conditions to 8a was successful, resulting in the formation of 3,6-dihydropyridin-2-one 9a as the major product (90%) after 1 h. Treatment with DBU at 90°C led to the isomerization of 9a to the thermodynamically more favorable 5,6-dihydropyridinone 10a (see Supplementary Material). At this stage, we envisaged that 10a could be obtained directly from the alkyne–amide cyclization. Fortunately, we found modified conditions that allowed the direct formation of 10a from 8a. At an elevated temperature of 60°C for 2 h, 10a was obtained directly from 8a in a 71% yield.
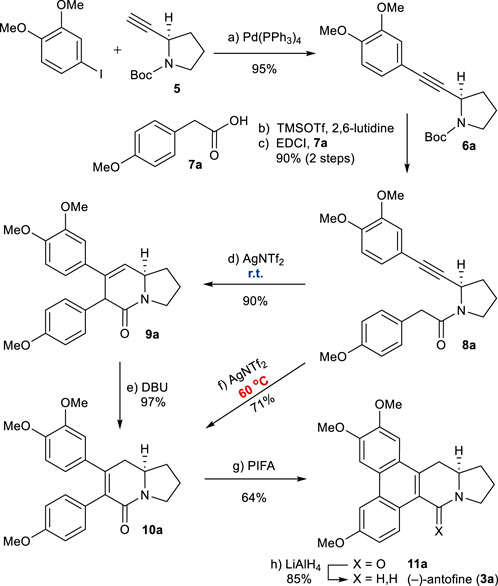
SCHEME 3. Synthesis of (−)-antofine (3a). Reagents and conditions: a) Pd(PPh3)4 (0.05 equiv), piperidine/MeCN (1:1), reflux, 3 h, 95%; b) TMSOTf (2.0 equiv), 2,6-lutidine (3.0 equiv), CH2Cl2, 0°C, 10 min; c) 7a (1.2 equiv), EDCI (1.1 equiv), DMAP (1.1 equiv), CH2Cl2, r.t., 12 h, 90% for 2 steps; d) AgNTf2 (0.1 equiv), TMSOTf (4.0 equiv), DIPEA (4.0 equiv), DCE, r.t., 1 h, 90%; e) DBU (4.0 equiv), toluene, 90°C, 30 min, 97%; f) AgNTf2 (0.1 equiv), TMSOTf (4.0 equiv), DIPEA (4.0 equiv), DCE, 60°C, 2 h, 71%; g) PIFA (1.1 equiv), BF3OEt2 (1.5 equiv), CH2Cl2, –10°C, 1 h, 64%; h) LiAlH4 (2.0 equiv), THF, reflux, 1 h, 85%. EDCI = N-Ethylcarbodiimide hydrochloride, DMAP = 4-Dimethylaminopyridine.
The oxidative biaryl coupling of 10a was accomplished with hypervalent iodine reagent phenyliodine(III) bis(trifluoroacetate) (PIFA) to give pentacyclic product 11a in a 64% yield (Kwon et al., 2015). Finally, the amide group of 11a was reduced with LiAlH4 to give (−)-antofine (3a) in an 85% yield (Iwao et al., 1983). Overall, this asymmetric total synthesis was completed in only 8 steps from N-Boc-d-prolinol with a 33% overall yield (5 steps from known 6a and a 35% overall yield).
With an established route to (−)-antofine, we pursued the total synthesis of (−)-tylocrebrine (3b), whose structure differs from that of (−)-antofine (3a) due to the presence of a methoxy group at C-5. Unlike the method for B-ring formation in 3a, radical-mediated oxidative biaryl coupling could not be used for 3b synthesis because of the regioselectivity problem. Therefore, we planned to employ palladium catalyzed C–H annulation (Ghosh et al., 2022; Thombal et al., 2022).
From intermediate 6a, (−)-tylocrebrine (3b) was readily accessible. First, 6a was coupled with 2-arylacetic acid 7b to afford 8b. The annulation of an amide with a tethered alkyne moiety in 8b under the abovementioned conditions gave 5,6-dihydropyridinone 10b directly in a 63% yield. After several trials, we found that the treatment of 10b with Pd(OAc)2 and PCy3·HBF4 as the palladium source and ligand in dioxane, respectively, led to the formation of 11b as the only detectable regioisomer (Campeau et al., 2006; Yadav et al., 2010). After the reduction of the amide group in 11b, (−)-tylocrebrine (3b) was obtained in 5 steps from 6a (Scheme 4).
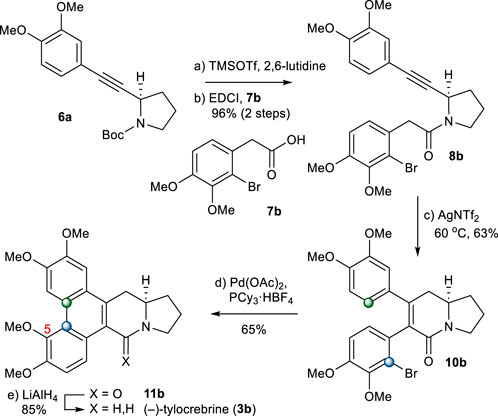
SCHEME 4. Synthesis of (−)-tylocrebrine (3b). Reagents and conditions: a) TMSOTf (2.0 equiv), 2,6-lutidine (3.0 equiv), CH2Cl2, 0°C, 10 min; b) 7b (1.2 equiv), EDCI (1.1 equiv), DMAP (1.1 equiv), CH2Cl2, r.t., 20 h, 96% for 2 steps; c) AgNTf2 (0.1 equiv), TMSOTf (4.0 equiv), DIPEA (4.0 equiv), DCE, 60°C, 2 h, 63%; d) Pd(OAc)2 (0.2 equiv), PCy3.HBF4 (0.4 equiv), K2CO3 (4.0 equiv), 1,4-dioxane, 110°C, 1 h, 65%; e) LiAlH4 (2.0 equiv), THF, reflux, 1 h, 85%.
The same chemistry was used for the total synthesis of (−)-isotylocrebrine (3c). Total synthesis of 3c started from alkyne 5. The Sonogashira coupling of 5 with 2-bromo-1-iodo-3,4-dimethoxybenzene chemoselectively afforded 6c in an excellent yield (Weijiang et al., 2013). After N-Boc group removal, 2-aryl acetic acid 7c was introduced to give amide 8c in a good yield. Under the aforementioned one-pot cyclization/isomerization conditions, 8c would not yield 5,6-dihydropyridinone 10c. Instead, 9c was formed in a high yield. Isomerization to 10c was achieved when 9c was treated with DBU in toluene at 90°C. Palladium-catalyzed B-ring formation, followed by the reduction of the amide group, provided (−)-isotylocrebrine (3c), as shown in Scheme 5.
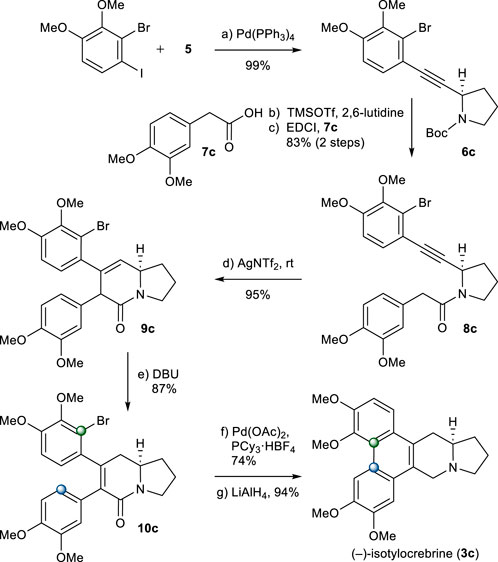
SCHEME 5. Synthesis of (−)-isotylocrebrine (3c). Reagents and conditions: a) Pd(PPh3)4 (0.05 equiv), CuI (0.1 equiv), iPr2NH, r.t., 2 h, 99%; b) TMSOTf (2.0 equiv), 2,6-lutidine (3.0 equiv), CH2Cl2, 0°C, 10 min; c) 7c (1.2 equiv), EDCI (1.1 equiv), DMAP (1.1 equiv), CH2Cl2, r.t., 12 h, 83% for 2 steps; d) AgNTf2 (0.1 equiv), TMSOTf (4.0 equiv), DIPEA (4.0 equiv), DCE, r.t., 1 h, 95%; e) DBU (4.0 equiv), toluene, 90°C, 1.5 h, 87%; f) Pd(OAc)2 (0.2 equiv), PCy3.HBF4 (0.4 equiv), K2CO3 (4.0 equiv), 1,4-dioxane, 110°C, 12 h, 74%; g) LiAlH4 (2.0 equiv), THF, reflux, 1 h, 94%.
Using the same chemistry described for the synthesis of (−)-antofine (3a), we accomplished the total synthesis of the phenanthroquinolizidine alkaloid (−)-cryptopleurine (4a), as shown in Scheme 6. A notable difference is the use of 2-alkyne-piperidine 12 in place of 5. The total synthesis of 4a was accomplished from 12 in 7 steps, using the process shown in Scheme 3. The spectra data and optical rotations of obtained alkaloids 3a, 3b, 3c, and 4a were in good agreement with those reported in the literature (Abe et al., 1995; Suzuki et al., 1995; Stærk et al., 2002; Niphakis et al., 2011).
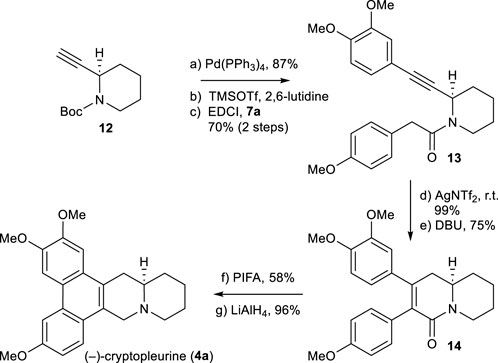
SCHEME 6. Synthesis of (−)-cryptopleurine (4a). Reagents and conditions: a) 3,4-dimethoxyiodobenzene, Pd(PPh3)4 (0.05 equiv), piperidine/MeCN (1:1), r.t., 14 h, 87%; b) TMSOTf (2.0 equiv), 2,6-lutidine (3.0 equiv), CH2Cl2, 0°C, 25 min; c) 7a (1.2 equiv), EDCI (1.1 equiv), DMAP (1.1 equiv), CH2Cl2, r.t., 12 h, 70% for 2 steps; d) AgNTf2 (0.1 equiv), TMSOTf (4.0 equiv), DIPEA (4.0 equiv), DCE, r.t., 2 h, 99%; e) DBU (4.0 equiv), toluene, 90°C, 6 h, 75%; f) PIFA (1.1 equiv), BF3OEt2 (3.0 equiv), CH2Cl2, –10°C, 30 min, 58%; g) LiAlH4 (3.0 equiv), THF, reflux, 30 min, 96%.
3 Conclusion
In conclusion, we successfully developed a new synthetic strategy for the construction of functionalized dihydropyridinone rings through the annulation of an amide α-carbon with a tethered alkyne moiety. An unexplored reaction between amide and alkyne was realized through an O-silyl N,O-ketene acetal. Our method was applied for the total synthesis of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Varying the coupling partners allowed for the culminative total synthesis of (−)-antofine (3a), (−)-tylocrebrine (3b), (−)-isotylocrebrine (3c), and (−)-cryptopleurine (4a). Further applications of this reaction for the synthesis of various functional dihydropyridinones and investigation of its extension to the total synthesis of other types of heterocyclic compounds are underway in our laboratory.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
SL: Data curation, Formal Analysis, Writing-review and editing, Investigation, Validation. JS: Data curation, Formal Analysis, Writing-review and editing, Investigation, Validation. RY: Data curation, Formal Analysis, Writing-review and editing. HY: Data curation, Formal Analysis, Writing-review and editing. SK: Supervision, Writing-original draft, Writing-review and editing, Conceptualization, Funding acquisition, Investigation, Project administration, Validation.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (RS-2023-00209322).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1267422/full#supplementary-material
References
Abe, F., Iwase, Y., Yamauchi, T., Honda, K., and Hayshi, N. (1995). Phenanthroindolizidine alkaloids from Tylophora tanakae. Phytochemistry 39, 695–699. doi:10.1016/0031-9422(95)00034-5
Burtoloso, A. C. B., Bertonha, A. F., and Rosset, I. G. (2014). Synthesis of alkaloids: Recent advances in the synthesis of phenanthroindolizidine alkaloids. Curr. Top. Med. Chem. 14, 191–199. doi:10.2174/1568026613666131213154633
Campeau, L.-C., Parisien, M., Jean, A., and Fagnou, K. (2006). Catalytic direct arylation with aryl chlorides, bromides, and iodides: Intramolecular studies leading to new intermolecular reactions. J. Am. Chem. Soc. 128, 581–590. doi:10.1021/ja055819x
Chemler, S. R. (2009). Phenanthroindolizidines and phenanthroquinolizidines: Promising alkaloids for anti-cancer therapy. Curr. Bioact. Compd. 5, 2–19. doi:10.2174/157340709787580928
Chen, W., Cao, X., and Yang, X. (2023). Transition-metal-free methods for the remote C−H bond functionalization of cyclic amines. Asian J. Org. Chem. 12, e202200547. doi:10.1002/ajoc.202200547
De Fatima Pereira, M., Rochais, C., and Dallemagne, P. (2015). Recent advances in phenanthroindolizidine and phenanthroquinolizidine derivatives with anticancer activities. Anti-Cancer Agents Med. Chem. 15, 1080–1091. doi:10.2174/1871520615666150520143600
Dénes, F., Pérez-Luna, A., and Chemla, F. (2010). Addition of metal enolate derivatives to unactivated carbon-carbon multiple bonds. Chem. Rev. 110, 2366–2447. doi:10.1021/cr800420x
Downey, C. W., Ingersoll, J. A., Glist, H. M., Dombrowski, C. M., and Barnett, A. T. (2015). One-pot silyl ketene acetal-formation Mukaiyama–Mannich additions to imines mediated by trimethylsilyl trifluoromethanesulfonate. Eur. J. Org. Chem. 2015, 7287–7291. doi:10.1002/ejoc.201500958
Ghosh, T., Biswas, D., and Bhakta, S. (2022). Palladium-catalyzed synthesis of fused carbo-and heterocycles. Chem. Asian J. 17, e202200725. doi:10.1002/asia.202200725
Hack, D., Blümel, M., Chauhan, P., Philipps, A. R., and Enders, D. (2015). Catalytic Conia-ene and related reactions. Chem. Soc. Rev. 44, 6059–6093. doi:10.1039/C5CS00097A
He, Y., Zheng, Z., Yang, J., Zhang, X., and Fan, X. (2021). Recent advances in the functionalization of saturated cyclic amines. Org. Chem. Front. 8, 4582–4606. doi:10.1039/D1QO00171J
Iwao, M., Mahalanabis, K. K., Watanabe, M., De Silva, S. O., and Snieckus, V. (1983). Directed ortho metallation of tertiary aromatic amides: A new N-heteroring annelation method and synthesis of phenanthro-quinolizidine and -indolizidine alkaloids. Tetrahedron 39, 1955–1962. doi:10.1016/S0040-4020(01)91913-1
Jia, X. H., Zhao, H. X., Du, C. L., Tang, W. Z., and Wang, X. J. (2021). Possible pharmaceutical applications can be developed from naturally occurring phenanthroindolizidine and phenanthroquinolizidine alkaloids. Phytochem. Rev. 20, 845–868. doi:10.1007/s11101-020-09723-3
Kobayashi, S., Kiyohara, H., and Yamaguchi, M. (2011). Catalytic silicon-mediated carbon−carbon bond-forming reactions of unactivated amides. J. Am. Chem. Soc. 133, 708–711. doi:10.1021/ja108764d
Kwon, Y., Song, J., Lee, H., Kim, E.-Y., Lee, K., Lee, S. K., et al. (2015). Design, synthesis, and biological activity of sulfonamide analogues of antofine and cryptopleurine as potent and orally active antitumor agents. J. Med. Chem. 58, 7749–7762. doi:10.1021/acs.jmedchem.5b00764
Lappert, M., Poer, P., Protchenko, A., and Seeber, A. (2009). Metal amide chemistry. Weinheim: Wiley VCH.
Lee, S., Yoo, H., Park, S., Yoon, R., and Kim, S. (2023). Facile one-pot synthesis of polysubstituted pyridinium salts by annulation of enamines with alkynes. Chem. Eur. J. 29, e202300059. doi:10.1002/chem.202300059
Lin, E.-Z., Xu, Y., Ji, K., and Ye, L.-W. (2021). Recent advances towards catalytic asymmetric Conia-ene-type reactions. Chin. Chem. Lett. 32, 954–962. doi:10.1016/j.cclet.2020.08.012
Mercado-Marin, E., Garcia-Reynaga, P., Romminger, S., Pimenta, E. F., Romney, D. K., Lodewyk, M. W., et al. (2014). Total synthesis and isolation of citrinalin and cyclopiamine congeners. Nature 509, 318–324. doi:10.1038/nature13273
Minnihan, E. C., Colletti, S. L., Toste, F. D., and Shen, H. C. (2007). Gold(I)-catalyzed regioselective cyclizations of silyl ketene amides and carbamates with alkynes. J. Org. Chem. 72, 6287–6289. doi:10.1021/jo071014r
Nandakumar, A., Midya, S. P., Landge, V. G., and Balaraman, E. (2015). Transition-metal-catalyzed hydrogen-transfer annulations: Access to heterocyclic scaffolds. Angew. Chem. Int. Ed. 54, 11022–11034. doi:10.1002/anie.201503247
Niphakis, M. J., and Georg, G. I. (2011). Synthesis of tylocrebrine and related phenanthroindolizidines by VOF3–mediated oxidative aryl–alkene coupling. Org. Lett. 13, 196–199. doi:10.1021/ol1023954
Paris, T. J., and Willand-Charnley, R. (2022). Modern synthesis and chemistry of stabilized ketene N, O-acetals. Synthesis 54, 1537–1550. doi:10.1055/a-1713-8481
Stærk, D., Lykkeberg, A. K., Christensen, J., Budnik, B. A., Abe, F., and Jaroszewski, J. W. (2002). In vitro cytotoxic activity of phenanthroindolizidine alkaloids from cynanchum vincetoxicum and Tylophora tanakae against drug-sensitive and multidrug-resistant cancer cells. J. Nat. Prod. 65, 1299–1302. doi:10.1021/np0106384
Suzuki, H., Aoyagi, S., and Kibayashi, C. (1995). Asymmetric total synthesis of (R)-(–)-cryptopleurine and (R)-(–)-julandine via highly enantioselective amidoalkylations with N-acylhydrazonium salts. J. Org. Chem. 60, 6114–6122. doi:10.1021/jo00124a025
Takeda, N., Futaki, E., Kobori, Y., Ueda, M., and Miyata, O. (2017). Nucleophilic arylation of N,O-ketene acetals with triaryl aluminum reagents: Access to alpha-aryl amides through an umpolung process. Angew. Chem. Int. Ed. 56, 16342–16346. doi:10.1002/anie.201708665
Thombal, R. S., Rubio, P. Y. M., Lee, D., Maiti, D., and Lee, Y. R. (2022). Modern palladium-catalyzed transformations involving C–H activation and subsequent annulation. ACS Catal. 12, 5217–5230. doi:10.1021/acscatal.2c00813
Vo, C.-V. T., and Bode, J. W. (2014). Synthesis of saturated N-heterocycles. J. Org.Chem. 79, 2809–2815. doi:10.1021/jo5001252
Weijiang, Y., and Herndon, James W. (2013). Total synthesis of (+)-antofine and (–)-cryptopleurine. Eur. J. Org. Chem. 2013, 3112–3122. doi:10.1002/ejoc.201300200
Yadav, A. K., Ila, H., and Junjappa, H. (2010). Synthesis of novel substituted phenanthrenes and polycyclic heteroarenes by Pd-catalyzed, direct, intramolecular arylation/heteroarylation. Eur. J. Org. Chem. 2010, 338–344. doi:10.1002/ejoc.200901036
Keywords: N-heterocycle, O-silyl, N,O-ketene acetal, dihydropyridinone, total synthesis, phenanthroindolizidine, phenanthroquinolizidine, alkaloid
Citation: Lee S, Shin JE, Yoon R, Yoo H and Kim S (2023) Annulation of O-silyl N,O-ketene acetals with alkynes for the synthesis of dihydropyridinones and its application in concise total synthesis of phenanthroindolizidine alkaloids. Front. Chem. 11:1267422. doi: 10.3389/fchem.2023.1267422
Received: 26 July 2023; Accepted: 28 August 2023;
Published: 21 September 2023.
Edited by:
Hyun-Joon Ha, Hankuk University of Foreign Studies, Republic of KoreaReviewed by:
Tao Shi, The Scripps Research Institute, United StatesRajendra Rohokale, University of Florida, United States
Copyright © 2023 Lee, Shin, Yoon, Yoo and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanghee Kim, cGVubmtpbUBzbnUuYWMua3I=
†These authors have contributed equally to this work