 Kokila Sakthivel
Kokila Sakthivel R. J. Gana
R. J. Gana Toshitaka Shoji
Toshitaka Shoji Naoko Takenaga
Naoko Takenaga Toshifumi Dohi
Toshifumi Dohi Fateh V. Singh
Fateh V. Singh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 08 September 2023
Sec. Organic Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1217744
This article is part of the Research Topic Multicomponent Reactions (MCRs) Towards Scaffolds with Versatile Applications View all 6 articles
To prepare complicated organic molecules, straightforward, sustainable, and clean methodologies are urgently required. Thus, researchers are attempting to develop imaginative approaches. Metal-catalyzed multicomponent reactions (MCRs) offer optimal molecular diversity, high atomic efficiency, and energy savings in a single reaction step. These versatile protocols are often used to synthesize numerous natural compounds, heterocyclic molecules, and medications. Thus far, the majority of metal-catalyzed MCRs under investigation are based on metal catalysts such as copper and palladium; however, current research is focused on developing novel, environmentally friendly catalytic systems. In this regard, this study demonstrates the effectiveness of metal catalysts in MCRs. The aim of this study is to provide an overview of metal catalysts for safe application in MCRs.
The development of innovative and effective methods for the one-step synthesis of various complicated systems from basic, easily obtainable starting materials is a formidable objective in organic chemistry. Multicomponent reactions (MCRs) are chemical reactions wherein multiple initial materials combine to generate a target molecule; essentially, all or most of the initial material atoms contribute to the synthesized product (Ugi et al., 1994). Consequently, MCRs have encouraged synthetic chemists to focus on one-step synthesis of complicated organic compounds. Although MCRs have been used for more than a century, their utility is typically restricted to a small number of reactions because managing the appropriate selectivity and reactivity selectivity when using three or more components is challenging (Ugi and Steinbruckner, 1960; Bienayme and Bouzid, 1998; Groebcke et al., 1998; Kaim et al., 2005). MCRs exhibit better synthetic efficiency compared with normal multi-step reaction routes and offer other advantages, including rapid and easy operation, time and energy savings, high atom economy, and environmental friendliness (Biggs-Houck et al., 2010; Slobbe et al., 2012; Rotstein et al., 2014; Khan et al., 2015; Malinakova, 2015; Paprocki et al., 2018). MCRs are typically easy to execute experimentally and frequently without the need for dry conditions or an inert atmosphere. Using MCRs, molecules can be combined in a convergent rather than linear manner. Therefore, because all property-determining moieties are introduced simultaneously rather than sequentially, structure-activity relationships (SARs) can be quickly constructed using MCRs. In addition, more than 300 distinct scaffolds have been documented in chemical literature, and MCRs offer a tremendous amount of chemical variety (Domling and Ugi, 2000; Ganem, 2009; Domling and Huang, 2010; Domling et al., 2012; Brauch et al., 2013; Mostofi et al., 2018; Graebin et al., 2019). The regulation of selectivity is a crucial problem when various sets of reactants are used in these procedures. Nevertheless, this lack of selectivity enables the production of intricate adduct combinations that are useful in combinatorial chemistry for biological applications. However, the mechanisms behind how reactants spontaneously choose one another (innate selectivity) remain unclear, and this is possibly challenging to achieve under normal conditions. In this regard, numerous conditions can be considered, including structural, chemical, electrical, compositional, kinetic, and energetic considerations, which can affect selectivity (Ghashghaei et al., 2019). In homogeneous catalytic processes, some factors may be more significant than those in heterogeneous reactions, and vice versa. However, in most circumstances, these two important catalytic types do not offer significant distinctions, particularly for controlling product selectivity (Davis, and Suib, 1993). Transition-metal catalysts are currently being employed in multicomponent processes as catalytic platforms because they assist in the selectivity and reactivity of reactions. A single catalytic pathway can produce complex scaffolds from simple and easily accessible building blocks using metal catalysts to activate the starting components and control the selectivity of the reaction. These characteristics considerably influence the development of MCRs employing various metal catalysts (D’Souza and Muller, 2007; Sunderhaus and Martin, 2009; Cioc et al., 2014; Kakuchi, 2015; Haji, 2016).
Aryl substituted pyridines are affluented in medicinal chemistry, which have beed synthesized by the condensation of aldehydes 1, thiols 3 and malononitrile 2 in the presence of calcium oxide nanoparticles 4 (Figure 1A). When compared to homogeneous base catalysts, calcium oxide (CaO) nanoparticles are inexpensive, have a high basicity, non-corrosive, economically benign, and are simple to handle. In terms of excellent yields, fast reaction times, reusability, and minimal catalyst loading, this method proffers an advanced method for the synthesis of 2-amino-4-aryl-3,5-dicyano-6-sulfanylpyridines 5. When the reaction performed under the optimal conditions using a variety of aldehydes and thiols to evaluate the extent of this approach, the observed that in the beginning of the reaction, electron-withdrawing groups like NO2, Cl, and Br interacted quite easily with malononitrile, producing 2-amino-3,5-dicarbonitrile-6-thio-pyridines 4 in a short amount of time. Additionally, compared to unimpeded aldehydes, sterically hindered aldehydes reacted more slowly. When malononitrile, thiophenol, and benzaldehyde were used as substrates, the best results were obtained (Safaei et al., 2013).

FIGURE 1. Synthesis of (A) substituted pyridines 5 and (B) bicyclic sulfonamides 11 using Ca-catalyst via multicomponent reaction (MCR).
Later, in place of pricey and extremely hazardous noble metal catalysts, Gao and team created a straightforward calcium catalyst that may be employed for a huge synthetic applications. The high Lewis acidity of the calcium catalyst efficiently transforms precursors into reactive carbocations, enabling reversible covalent bond formation due to its potency as a Lewis acid. The aliphatic alkynes as nucleophiles in carbocation cascade reactions to synthesize bicyclic sulfonamides (Figure 1B). The reaction proceeded through the ionization of hexenol 6 in the presence of Bu4NPF6 10 and calcium(II) bis(trifluoromethanesulfonimide) 9. The team discovered that changing the substrates of the disubstituted sulfonamides had no effect. When a bulky group was introduced into the alkyne, the target product 11 was produced in excellent yield, along with complete diastereoselectivity (Gao et al., 2015).
Zhou et al. synthesized substituted pyridines via a Cu-catalyzed three-component reaction of alkynes 13, 2-[(amino)methylene]malononitriles 12 and sulfonyl azides 14 (Figure 2A). During the screening of the reaction conditions, the substituents on the pyridines were varied with respect to the solvent. When all three substrates were treated with the Cu catalyst in the presence of triethylamine (TEA) 16, an anionic intermediate D was formed. DMF at 50°C under N2 afforded 6-amino-2-iminopyridine 18 by nucleophilic vinylic substitution (SNV), followed by intramolecular cyclization of an anionic intermediate D. THF at 20°C–25°C afforded 4-amino-2-iminopyridine 17 via direct intramolecular cyclization of the anionic intermediate D (Figure 2B) (Zhou et al., 2013).
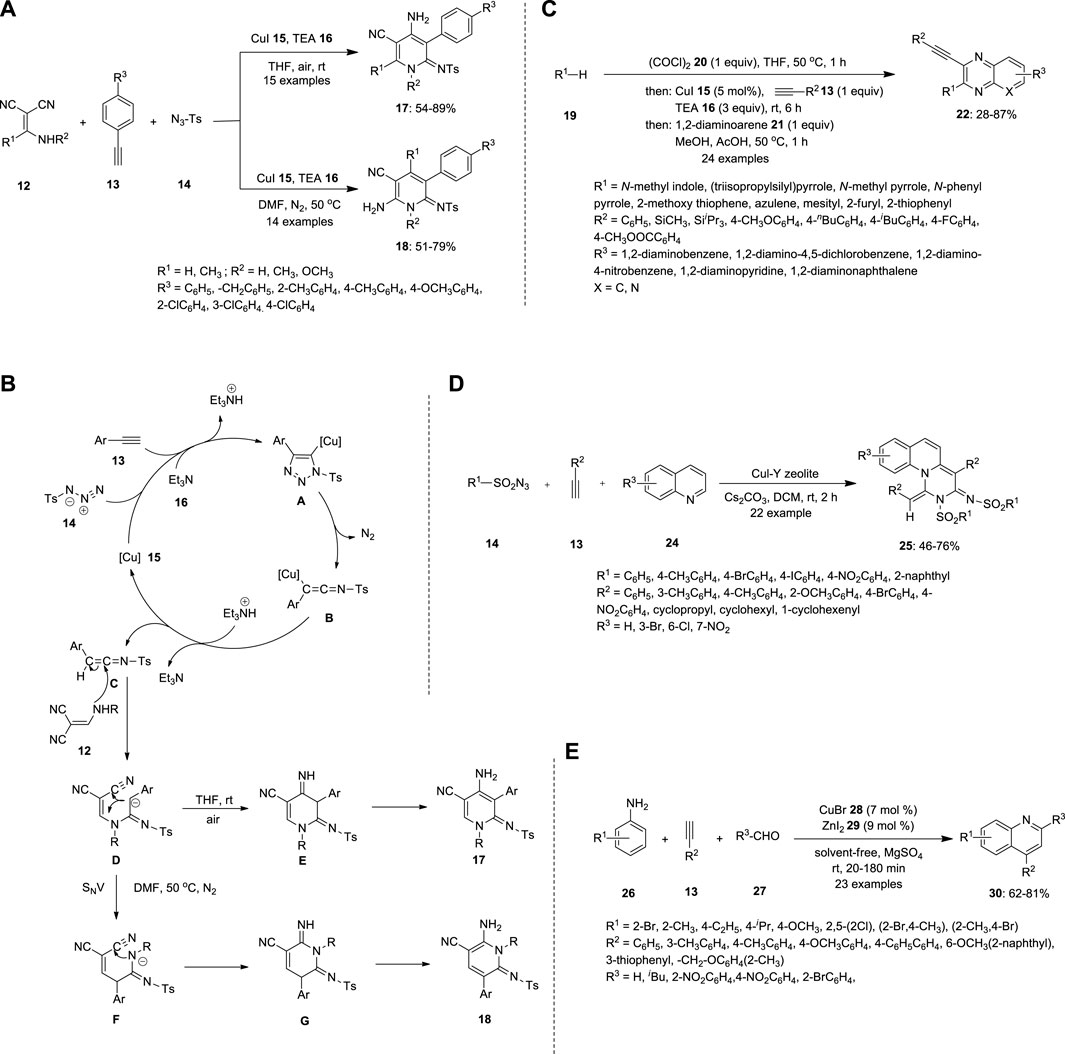
FIGURE 2. Synthesis of N-heterocyclic compounds (A) substituted pyridines 17 and 18, (B) mechanism of pyridines 17 and 18, (C) quinoxalines 22, (D) isoquinolines 25 and (E) quinolines 30 using Cu-catalyzed MCR.
Gers et al. also reported that the multicomponent reacts with the terminal alkyne 13 to produce substituted quinoxalines 22 (Figure 2C). The electron rich π-nucleophiles were reacted in presence of oxylchloride 20, thereby furnishing ynedione 23; this step is called gloxylation. Ynedione 23 further underwent Stephens−Castro coupling with 1,2-diaminoarenes 21, a cyclocondensation reaction that resulted in the formation of 2-substituted 3-ethynyl quinoxalines 22. The high fluorescence of the synthesized compounds renders them ideal sensors for protein interactions (Gers et al., 2014).
Ramanathan and Pitchumani reported a Cu-catalyzed MCR between sulfonyl azide 13 and terminal alkyne 13 (Figure 2D). The expected pyrimido [1,6-a]quinolines 25 were prepared using a tandem technique involving the ketenimine-based [3 + 2]/[2 + 2 + 2] intermolecular cycloaddition of alkyne 20, sulfonyl azide 14, and quinoline 24. In this simple, atom- and step-efficacious procedure, copper (I) promotes the cycloaddition of azide-alkyne, followed by ring-rearrangement/ketenimine formation/regio- and stereoselective termolecular cycloaddition and dehydrogenation cascade, to selectively yield pyrimido [1,6-a]quinoline E-isomer (Ramanathan and Pitchumani, 2015).
Mondal et al. synthesized quinolones 30 using a Cu catalyst by coupling various anilines 26, terminal alkynes 13 and aldehydes 27 under solvent-free conditions (Figure 2E). The expected 4-substituted, 2,4-disubstituted, and thermally labile sugar-based chiral quinolines 30 were obtained in good yields via cyclization of the C≡C bond. Terminal alkyne 13 was activated when treated with a Cu catalyst, which further formed the propargyl amine 31 intermediate through the formation of C−C and C−N bonds between aldehyde 27 and aromatic amine 26. The propargylamine 31 intermediate can be used as a CuI procatalyst, which initiates sp2 C–H activation with cyclization involving transient CuIII species. The reductive elimination step helps obtain quinolones and the CuI catalyst (Mondal et al., 2016).
Li et al. provided a selective methodology for the synthesis of indole and its derivatives throughout copper-catalyzed one-pot multicomponent cascade reactions. 1-bromo-2-(2,2- dibromovinyl) benzenes 32 were treated with aldehydes 1 and aqueous ammonia 34 in the presence of Cu catalyst in DMF at 80°C (Figure 3A). The ligands 1,4-diazabicyclo [2.2.2]octane (DABCO) and pivalic acid (PivOH) were used as additives to facilitate Cu catalysis. The resulting products also differed depending on the reaction time and substrate ratio. When the reaction was carried out in a 1:1 M ratio of aqueous ammonia to DMF for 24 h (condition A), 3-cyanoindoles 35 was formed. Whereas when the reaction was performed with a 0.5:3 M ratio of aqueous ammonia to DMF for 30 h (condition B), the produced sequence of 9H-pyrimido[4,5-b]indoles 36 disregarded the steric and electronic nature of the substituents. Similarly, pyrido[2,3-b]indole derivatives 37 were obtained under condition B using aliphatic aldehydes 33 were employed (Li et al., 2015).
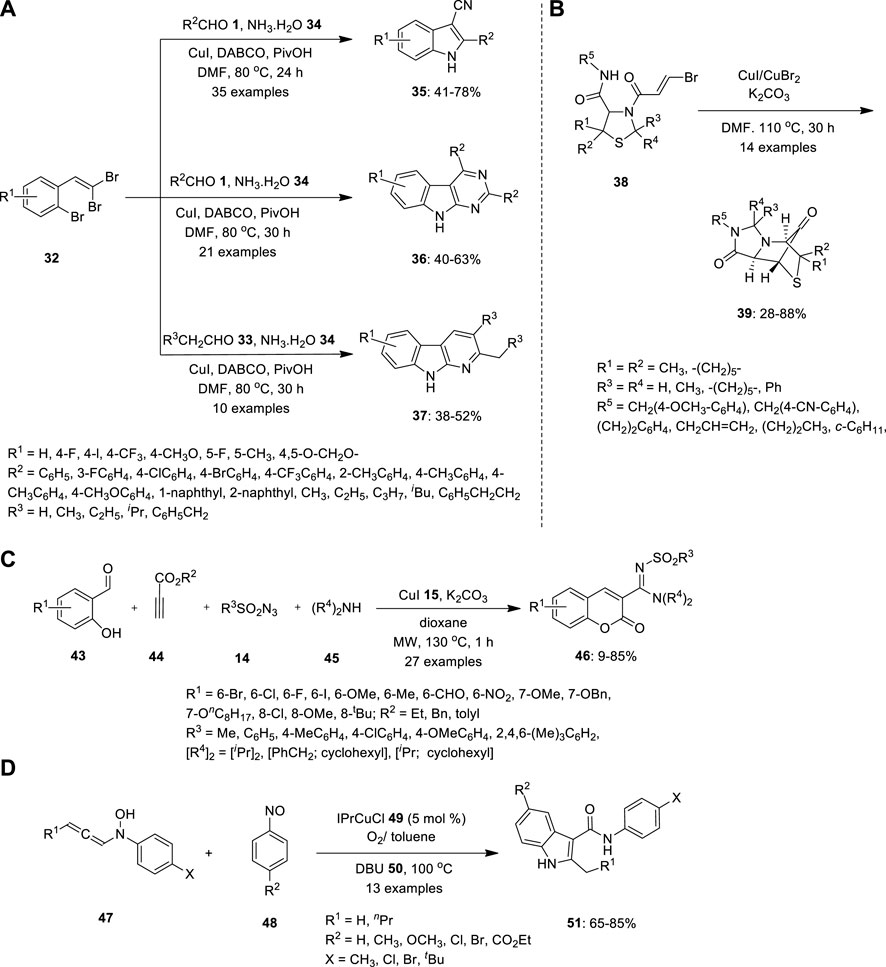
FIGURE 3. Copper catalyzed MCR for synthesis of (A) indoles 35, pyrmidoindles 36 and and pyridoindoles 37, (B) bridged N- heterocyclic compound 37, (C) 3N-sulfonylamidine and (D) functionalized indoles 51.
(Kroger et al., 2015) investigated the diastereoselective synthesis of a tricyclically annulated and bridged heterocyclic system reinforced by Cu-mediated rearrangement based on bisamides with a thiazolidine substructure. (Figure 3B). Precursors 38 were easily synthesized by a two-step synthetic dispatch known as the Asinger and Ugi reaction, using the concept of consecutive MCRs with 3-thiazolines 40, trans-bromoacrylic acid 41 and isocyanide 42. Precursor 38 further reacted with CuI/CuBr2 in the presence of K2CO3 in DMF at 110°C. Consequently, product 39 was obtained by intramolecular ring-closing reaction between the secondary amide and brominated alkene in moderate yield (Kroger et al., 2015).
Propiolate 44, salicylaldehyde 43, sulfonyl azide 14, and secondary amine 45 were coupled to form a four-component tandem copper-catalyzed 3-N-sulfonylamidine coumarin 47 (Figure 3C). Using this one-pot procedure, Murugavel et al. obtained functionalized coumarin structural frameworks in moderate-to-high yields. Thus, the cycloaddition of 44 with 14 may produce ketenimine. Nucleophilic addition of the amine 45 to the ketenimine may furnish amidine that can react with the aldehyde 44 to give cationic intermediate, which may cyclize to produce 46 by transesterification. When the reaction was conducted at room temperature, 2-imino esters and substituted phenols were formed as byproducts, which decreased the yield of the expected coumarin. The reactions were performed under microwave irradiation. Surprisingly, the expected coumarin 46 was formed as the major product, essentially dominating other by-products (Murugavel, and Punniyamurthy, 2015).
Because the reaction entails a notable structural rearrangement, the 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) 50 -mediated indole synthesis is mechanistically intriguing. In 2016, Sharma and Liu proposed process, the [3 + 2]-annulation reactions were carried out using nitrosobenzenes 48 and N-hydroxyaniline 47 in the presence of an IPrCuCl 49 additive and O2 in cold toluene (Figure 3D). The resulting isoxazolidin-5-ol derivatives were heated in (DBU) 50 in toluene. DBU can help the initial ketal’s ring open to generate intermediate, which then catalyses the ring’s rearrangement and leads the formation of ketenimine isomer, which is then converted as the O−linkage isomer. In contrast to its N−linkage isomer, O−linkage isomer is chemically reactive to undergo a 3,3-sigmatropic shift, resulting in the 3-oxo-2-arylamides and the desired product 51 with moderate to good yield (Sharma, and Liu, 2016).
Malonate 53, nitrosoarene 54, and alkene 52 undergo one-pot three-component cycloaddition under the influence of Cu(OAc)2 55 (Figure 4A). Using this technique, Wu et al., 2023 produced various isoxazolidines in good to outstanding yields. Further, they conducted mechanistic studies, which revealed that the simple production of nitrone intermediates via the Cu(OAc)2-catalyzed reaction of malonates with nitrosoarenes is an essential step in this catalytic system (Wu et al., 2023).
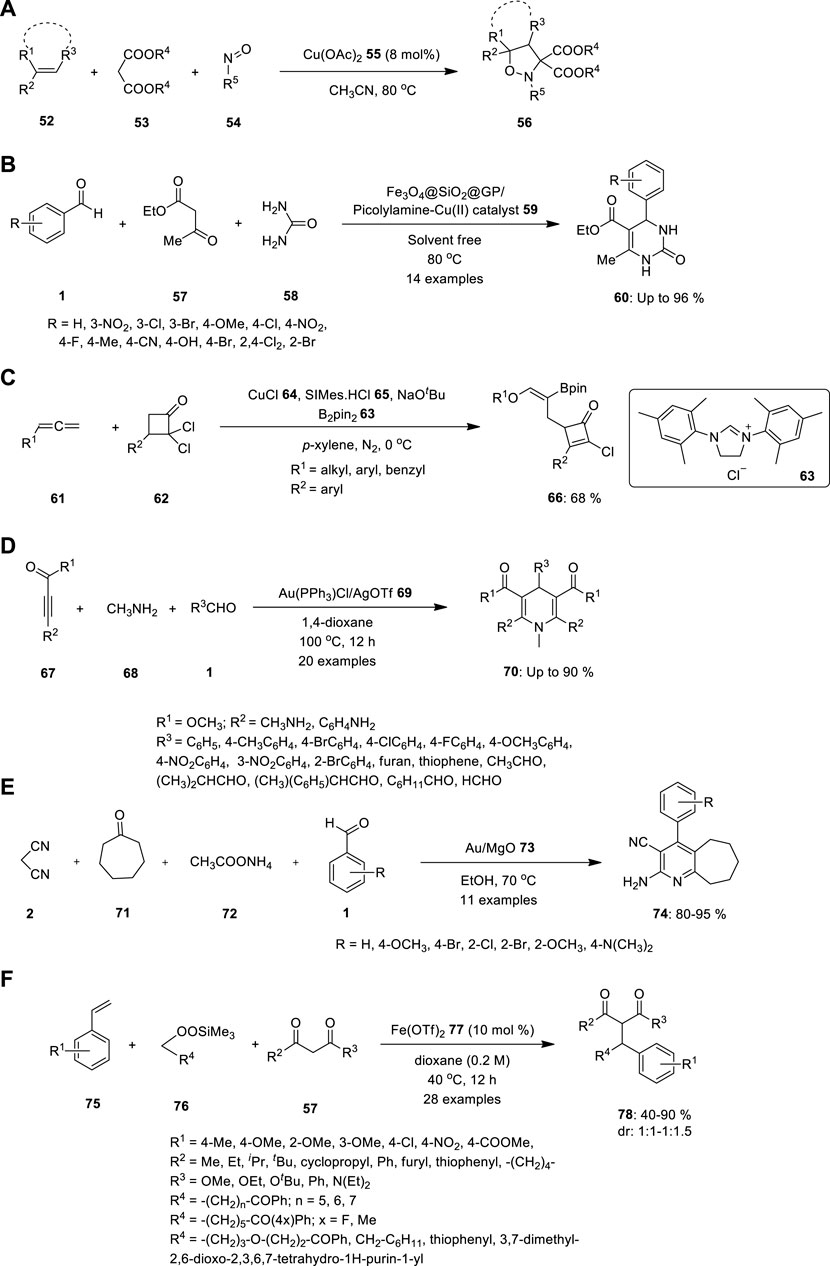
FIGURE 4. Synthesis of (A) N,O—heterocyclic compound 56, (B) 3,4-dihydropyrimidinones 60, (C) polysubstituted cyclobutenone 66 using copper catalyzed MCR, (D) N-substituted dihydropyridines 70, (E) multisubstituted pyridine compounds 74 using Au-catalyzed multicomponent reaction, (F) Dicarbofunctionalization of vinylarenes 75 in presence of catalytic Fe(OTf)2 77.
Rezayati and team reported biginelli annulation reaction catalysed by heterogeneous magnetic silica-coated picolylamine copper complex to synthesis of biologically active 3,4-dihydropyrimidinones 60. The catalyst Fe3O4@SiO2@GP/Picolylamine-Cu (II) 59 was synthesized easily using chemical attachment of the picolylamine compound on Fe3O4@SiO2@GP, followed by treatment with copper salt in ethanol under reflux conditions. The resulting catalyst system was successfully used in the Biginelli reaction through a variety of compounds such as aromatic aldehyde 1, urea 58, and ethyl acetoacetate 57 under solvent-free conditions or ethylene glycol at 80 °C and yielded the desired products with high conversions with powerful reusability (Figure 4B). The current approach was convenient and clean, and only 0.01 g of the catalyst could be used to perform the reaction. Also, the Fe3O4@SiO2@GP/Picolylamine-Cu (II) nanocatalyst 59 could be recycled by an external magnet for eight runs with only a significant loss in the product yields (Rezayati et al., 2022).
In addition, the researchers inspired by the selectivity in multicomponent reactions demonstrated by metal catalysts. To prove that, Liu and team reported copper catalyzed regio-and stereoselective three-component coupling of allenyl ethers with gem-dichlorocyclobutenones 63 and bis (pinacolato) diboron 62. The reaction yielded a variety of highly functionalized cyclobutenone products 67 tethering with an alkenylborate fragment (Figure 4C). The polysubstituted cyclobutenones also underwent diverse transformations (Liu et al., 2023).
Wang S. et al. described the one-pot MCRs of activated alkynes 67, methanamine 68, and aldehydes 1 catalysed by gold to the synthesis of bioactive N-substituted dihydropyridines (DPH) 70. The reaction provided a convergent strategy for generating a C–C bond and a C–N bond for the synthesis of N-substituted DHP derivatives in moderate to high yields (Figure 4D). Whether an electron-withdrawing or electron-donating group was introduced on the phenyl ring, the reaction proceeded without mishap and produced the corresponding N-substituted DHPs in good yields (Wang et al., 2013).
Pagadala R et al. demonstrated Au/MgO 73 to be a highly effective and reusable catalyst for multicomponent coupling reactions at 70°C (Figure 4E). The synthesis of multisubstituted pyridines 74 resulted in high yields and rapid reaction rates. Reaction proceeded through the Knoevenagel condensation of aldehyde 1 with sp3 carbon of malononitrile 2, double bond further reduced by Michael addition, which further underwent cyclization with the help of ammonium acetate 72. After a completion of reaction, the novel catalyst can be easily recovered and re-utilized without any loss of catalytic activity, as a result of its simple processing (Pagadala et al., 2014).
Recently, Xu et al. introduced a novel strategy for developing a set of carbon–carbon bonds using styryls 75 alongside 1,3 dicarbonyl substrates 57, catalyzed by the existence of catalytic Fe(OTf)2 77 under optimized reaction conditions (Figure 4F). Vinylarenes with various substituents, along with numerous alkylsilyl peroxides 76 and β-keto carbonyl substrates 57, can be employed in this process. Using the same reaction conditions, a novel benzopyran derivative 78 was created from 2-hydroxystyrene, which is an important molecule with various biological characteristics (Xu et al., 2022).
Vytla et al. reported an efficient three-component photoredox-catalyzed Petasis reaction. Under the optimized conditions using {Ir[dFCF3PPy]2(dtbpy)}PF6 81 as the photocatalyst, a wide range of sulfonamides, amides and hydrazides can efficiently undergo imine formation with various benzaldehydes 1 followed by radical α-alkylation with alkyltrifluoroborates 80 in presence of sodium hydrogen sulfate (Figure 5A). The operationally simple protocol allows rapid access to structurally diverse α-substituted secondary sulfonamides, amides and hydrazides 82 in moderate to high yields (Vytla et al., 2022).
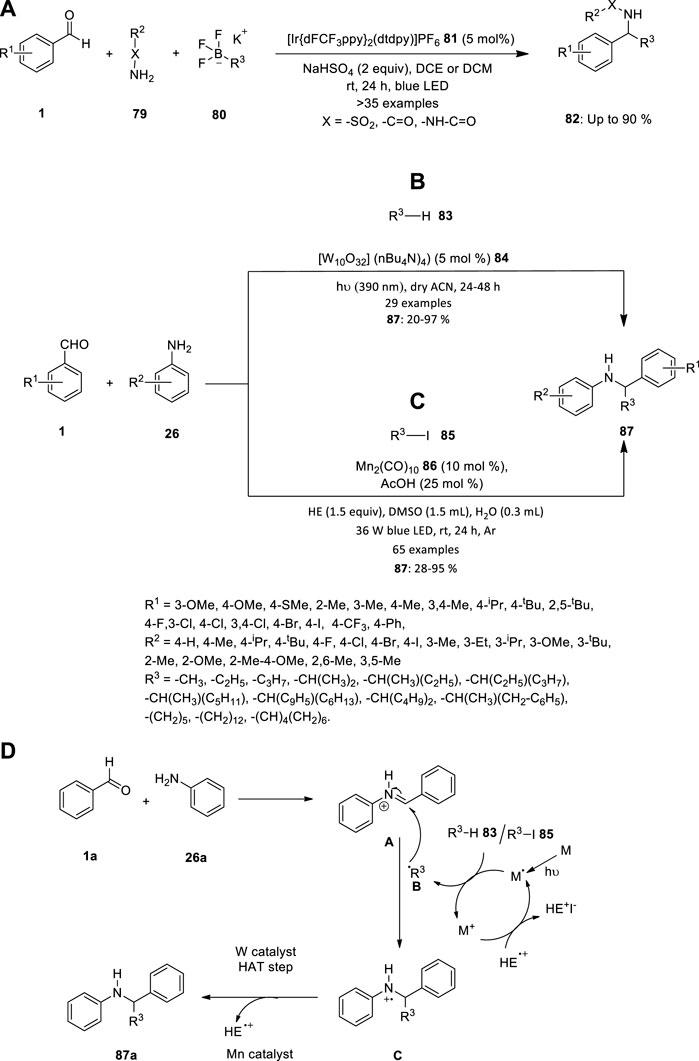
FIGURE 5. (A) Iridium catalyzed photoredox Petasis reaction, Synthesis of secondary amines using (B) TBADT 84 and (C) Mn2(CO)10 86 catalyst, (D) Proposed mechanism for the synthesis of secondary amine 89.
Due to the prevalence of secondary amines in agrochemicals, drugs, natural products, and small-molecule biological probes, there have been efforts to accelerate the synthesis of molecules with an amine functional group. The two different metal catalysts were introduced to synthesis the secondary amines in 2021. Due to the benefits that the direct functionalization of these potent C-H bonds holds, the prevalence of non-activated aliphatic C-H bonds has been the focus of ongoing research. It is clear that this discipline has advanced thanks to the discovery of photoredox catalysis, which offers a variety of solutions to problems caused by the inertness of unactivated C-H bonds. Pillitteri et al. investigated the production of C-sp3 centred radicals from aliphatic C-H bonds (Figure 5B). This is made simple by the use of polyoxometalate tetrabutylammonium decatungstate (TBADT) 84. Despite the widespread use of this photocatalyst, only a small number of radical acceptors have been used, and there aren't many reports on multicomponent reactions, primarily because of polarity match and chemoselectivity problems. Therefore, the team achieved a generally applicable method for the mild reaction conditions alkylation of in situ generated imines to produce functionalized secondary amines 87. The innate electrophilicity of imines towards radical addition is exploited in this atom-economic transition, and the starting materials' electronic characteristics are tuned to produce a wide range of products (Pillitteri et al., 2021).
Wang et al. described the three-component alkylation reactions of imines (produced in situ by condensation of benzaldehydes 1 and anilines 26) with unactivated alkyl iodides 85, catalysed by inexpensive and widely available Mn2(CO)10 86, are here as a mild, operationally straightforward method for the synthesis of secondary amines 87 (Figure 5C). This procedure is a versatile, adaptable method for the synthesis of secondary amines 87 because it is compatible with a variety of sensitive functional groups and doesn’t call for a significant amount of the alkylating reagent (Wang et al., 2021).
The mechanism depicted in Figure 5D is what causes the amination reactions to happen. When Mn catalyst is exposed to the blue LED, metal radical is produced. This metal radical takes iodine/hydrogen from R3−I 87 to produce the nucleophilic radical species B and metal cation. Radical B combines with iminium ion A (which was created in-situ by a condensation process between 26a and 27a and then protonated by AcOH) to create radical cation C. After further reducing C, a tertiary amine product 89 is produced, and this produces a radical cation called HE+, which combines with metal cation to regenerate the active catalyst radical and end the photocatalytic cycle. Whereas, while using tungsten compound used as catalyst, R3−H 85 produced radical B and hydrogen radical. Radical B combines with iminium ion A to create a radical C, which underwent hydrogen atom transfer (HAT) step to produce a tertiary amine product 89.
Zhang et al. used chiral N,N′-dioxide/Ni(OTf)2 complex 90 to develop an enantioselective three-component reaction of 2-oxo-3-ynoates 88, nitrosoarenes 54 and diazoacetates 89 (Figure 6A). This process in mild reaction conditions produces imino ketone-substituted multifunctional chiral epoxides 91 in ordinary to good yields (up to 82%), outstanding diastereo- and enantioselectivities (99% ee and up to >95/5 dr), and high Z/E ratios (up to >95/5 Z/E). Essentially, the imino group of product 91 is hydrolyzed at a low pH (<7) to afford the corresponding ketone. Zhang et al. carried out the same reaction in different orders, including nitrosoarenes 54 and diazoacetates 89, thereby producing nitrone. Subsequently, the nitrone was treated with 2-oxo-3-ynoates 88 to produce aziridine 93, which is not hydrolyzed (Zhang et al., 2020).
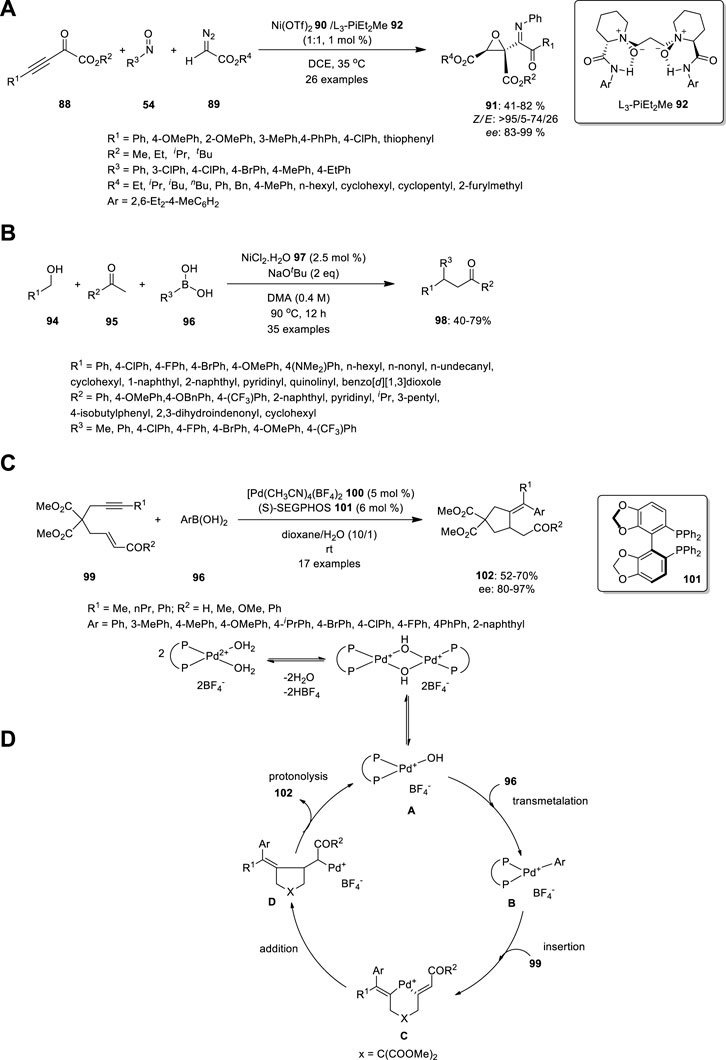
FIGURE 6. Synthesis of (A) imino ketone-substituted multifunctional chiral epoxides, (B) β,β-disubstituted ketones 98 using Ni complex, (C) An enantioselective arylative cyclization of alkyne-tethered enals or enones catalyzed by cationic Pd(II) and (D) proposed mechanism.
Recently, Balakrishnan et al. treated alcohols 94 and ketones 95 with boronic acid 96 in the presence of NiCl2 97 to produce β,β-disubstituted ketones 98 (Figure 6B). This can be achieved using hydrogen borrowing strategy, which is a cost-effective technique for the α–functionalization of ketones. The alcohol 94 reduced as aldehyde by Ni catalyst, which further underwent aldol condensation reaction with aryl methyl ketone 95 to create enone. Addition of Ni-Ph catalyst to the enone generate 1,4 addition, which further produce the target molecule 98. This methodology is tolerated by inactivated alkyl alcohols, long-alkyl chain alcohols, and sterically crowded alcohols. Methyl-substituted boronic acids reduced the yield after observing the effect of the substrate in boronic acid. Moreover, the coordination of the oxygen and sulfur atoms to the nickel center rendered thiophene and furan entirely inert (Balakrishnan et al., 2022).
Shen et al. developed an enantioselective arylative cyclization of alkyne-tethered enals or enones catalyzed by cationic Pd(II) via the carbopalladation of alkynes (Figure 6C). The acyclic allylic 2-alkynoate 99 was treated with aryl boronic acid 96 in the presence of [Pd(dppp)(H2O)2](BF4)2 103 in toluene/H2O. The expected product, (E)-2-(5-oxo-4-(1-phenylethylidene)tetrahydrofuran-3-yl) acetaldehyde 104, was obtained in 41% yield along with the uncyclized product, (Z)-4-oxobut-2-en-1-yl (E)-3-phenylbut-2-enoate 105 (48%). Subsequently, they studied the asymmetric arylative cyclization by introducing a chiral ligand to increase the yield and enantioselectivity. High enantioselectivities were observed when biphenyl or binaphthyl motifs 101 were present in the bisphosphine ligands, thereby resulting in high enantioselectivities with moderate yields, probably owing to the fierce conjugate addition side reactions. The reaction was performed in the absence of water and the yield decreased with increasing temperature (Shen et al., 2012).
The Pd hydroxo complex A is the active catalytic species, while arylboronic acids 96 form arylpalladium species B through transmetalation. Carbopalladation yields vinylpalladium intermediate C, followed by carbon-carbon double bond insertion, resulting in intermediate D. The protonolysis of the C-Pd bond generates the product and regenerates Pd(II) species A. The Pd(II) catalytic cycle is a plausible mechanism for cationic Pd(II)-catalyzed transformations, with halogen-substituted phenylboronic acids and allylic esters being substrates (Figure 6D). Both aryl-halogen bonds and allylic ester groups are cleaved first in the presence of Pd (0).
Using Pd/Cu catalysis, Semba et al. developed a convenient approach for the carboallylation of electron-deficient alkenes (Figure 7A). The yield of the alkenes was unaffected by the presence of electron-withdrawing and electron-donating groups at the para-position on the benzene ring of 112. Hence, using widely available alkenes 106, allylic carbonates 108 and organo-boron compounds 107, this approach may produce numerous carbon skeletons that are well-tolerated by numerous functional groups, including bromo, cyano, alkoxycarbonyl, and acetyl moieties (Semba et al., 2019).
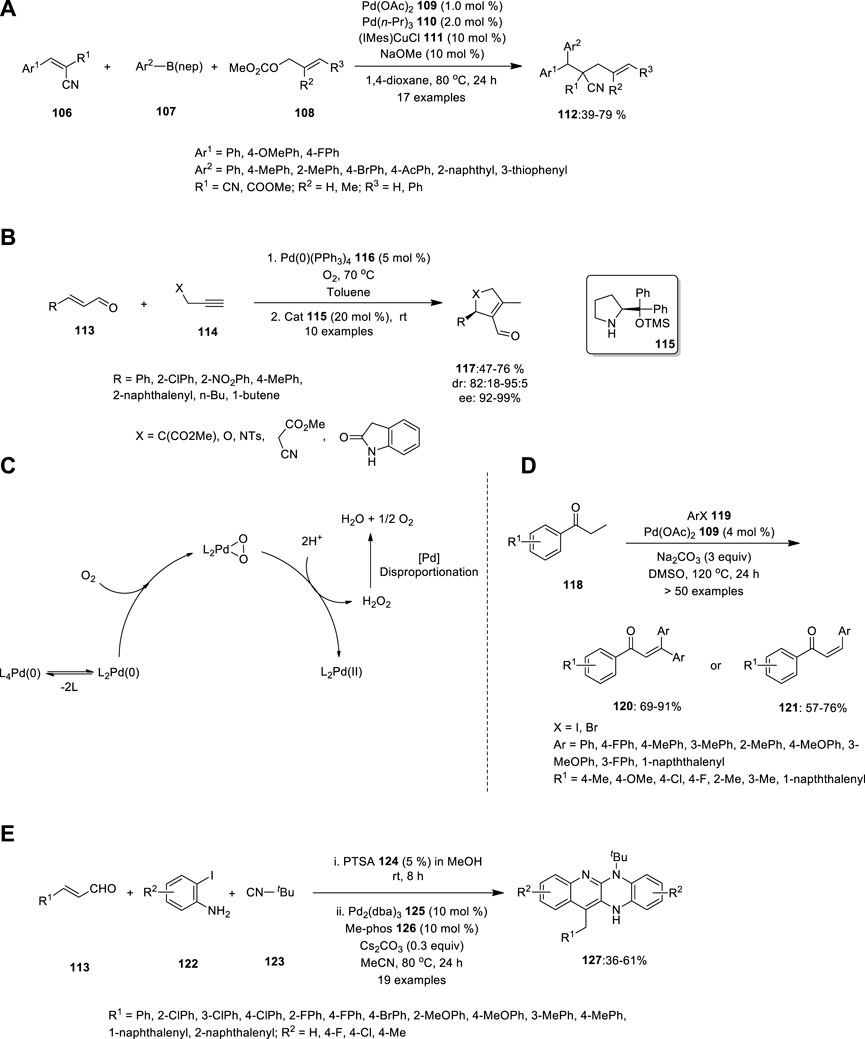
FIGURE 7. Pd—catalyzed (A) carboallylating electron-deficient alkenes 106, (B) intermolecular nucleophilic addition of unmodified aldehydes 113 reactions, (C) Oxidation of Pd (0) to Pd(II) by molecular oxygen, (D) synthesis of α,β-unsaturated carbonyl compounds 117, 120, and 121, (E) Ugi-variant one-pot synthesis of fused tetracyclic quinoline scaffolds 127.
Another chiral ligand was reported in 2014 by Santoro et al., who investigated the mechanism of the palladium and amine co-catalyzed intermolecular nucleophilic addition of unmodified aldehydes to alkynes (Figure 7B). Based on experimentations and theory, they reported that the C–C bond-forming reaction is presumably a two-step carbocyclization of enaminynes catalyzed by a Pd(II) species. In the first step, the α,β-unsaturated aldehyde 113 reacts with chiral amine 115, thereby forming a nucleophilic enamine, which further attacks the electrophilic Pd(II). This is followed by insertion into the alkyne 114 bond, which produces a syn carbopalladation intermediate. Moderate isolation yields were obtained with good enantiomeric excess ratios.
Predominantly, the catalytic cycle is connected to complete enantioselective aerobic allylic alcohol oxidation using a Pd complex with a chiral amine. Owing to the presence of PdII (PPh3)2O2 in the catalytic cycle, no other oxidizing agent is required to convert Pd(0) to Pd(II) (Figure 7C). Hence, “Pd oxidase catalysis” and “metal/organo cooperative catalysis” can be used in environmentally friendly and sustainable chemistry (Santoro et al., 2014).
Gandeepan et al. synthesized an extremely substituted α,β-unsaturated carbonyl species using a novel Pd catalyst (Figure 7D). Unlike the Heck-type coupling reaction, this reaction can produce an effective yield with low catalyst loading and desist other lavish metal oxidants or ligands. Saturated ketones, aldehydes, and esters were treated with aryl halides under optimized reaction conditions to afford α,β-unsaturated carbonyl compounds in good to excellent yields. When 2.5 equivalents of aryl iodide were employed in the reaction, monosubstituted α,β -unsaturated compounds 121 were produced; however, when 3.5 equivalents of aryl iodide were used, disubstituted α,β -unsaturated compounds 120 were formed as the major product (Gandeepan et al., 2014).
In the same year, Che et al. carried out the Ugi-variant MCR and Pd-catalyzed one-pot synthesis of bis-annulation with substrates 113, 122 and 123, and PTSA 124 in MeOH for 8 h at room temperature. The crude product was concentrated, followed by the addition of Pd2(dba)3 125, Me-phos (126 and Cs2CO3 in MeCN at 80°C for 24 h (Figure 7E). The fused tetracyclic quinoline scaffolds 127 were obtained in moderate-to-good yields. The electronic properties of the substituents on the aromatic ring of the aldehyde did not influence the reaction rate, whereas anilines with either electron-donating or electron-withdrawing groups demonstrated reactivity (Che et al., 2014).
Wang et al. reported the synthesis of chiral 3,3-disubstituted 3,4-dihydroisoquinoline and tetrahydroisoquinoline derivatives, which are important building blocks in organic synthesis for biological evaluation and ligand designing. Essentially, they carried out Pd-catalyzed enantioselective imidoylative C (sp2)−H bond activation reaction by desymmetrization of dibenzyl isocyanide 128 (Figure 9A). Dibenzyl isocyanide 128 was treated with aryl iodine 129 in the presence of the SPINOL-derived phosphoramidite ligand 130 (L*). 3,4-Dihydroisoquinolines 131 bearing a chiral quaternary carbon center were formed in high yields with good enantioselectivities. Further, the reactions were well tolerated with electronically diverse substituents at the ortho- or para-position of aryl iodides, heteroaromatic iodides, bulky groups (such as naphthalene), and substrates bearing two substituents on the phenyl ring of isocyanides (Wang et al., 2017).
In 2014, Zhu et al. synthesized symmetrical and unsymmetrical methylidenefluorene derivatives via a Pd/Cu-catalyzed MCR (Figure 9B). They developed a three-component reaction combining cyclic diphenyleneiodonium 132, alkynes 13, and boronic acid 96 to produce methylidenefluorenes 133 in a single step. The effect of the substituents was examined, and the strong electron deficiency of the substituted groups on phenylboronic acids and aryl alkynes produced the product in lower yields. By contrast, phenylboronic acids with electron-donating or electron-withdrawing substituents, terminal aryl groups, and alkyl alkynes were well tolerated under the optimized reaction conditions. Hence, numerous unsymmetrical methylidenefluorenes could be synthesized in isomeric form. The steric substituent had no influence on chemoselectivity; by contrast, the electronic effect had a significant influence on chemoselectivity. Electronically poorly substituted unsymmetrical cyclic diphenyliodoniums exhibited high chemoselectivity and vice versa (Zhu et al., 2014).
Peng et al. attempted the Pd-catalyzed MCR of propargylic carbonates 134 with isocyanides 135 (Figure 9C). They treated the propargylic carbonate 134 with isocyanide 135 in the presence of Pd(PPh3)2Cl2 136, and CsF in DMSO. Surprisingly, two different types of useful N-heterocyclic compounds, 137 and 138, were produced via the orderly insertion of isocyanides. According to a systematic investigation of the reaction conditions, the temperature and ligands can influence the selectivity for the formation of N-heterocyclic products. When they completed the reaction at room temperature, (Z)-6-imino-4,6-dihydro-1H-furo[3,4-b]pyrrol-2-amines 137 was predominantly produced, whereas when 1,1′-Bis (diphenylphosphino) ferrocene (dppf) was employed instead of PPh3 at 110°C, (E)-5-iminopyrrolones 138 was predominantly produced. Their study provides evidence for the migratory insertion of isocyanides into aryl- and alkenylpalladium species.
Proposed mechanism is given in Figure 9. According the mechanism, the reaction initiated with oxidative addition of 134 to the palladium (0) catalyst, producing the allenylpalladium A, The in situ-formed allenylpalladium species bearing multiple reaction sites allowed the successive insertion of multiple isocyanides in an orderly manner to become the crucial intermediate B. Further, the intermediate B has two possible sites C2 and C4 to get attack by H2O as a nucleophilic reagent, which determined the selectivity in the synthesis of products 138 and 137 respectively. This mechanism made sense given that compounds 137 were the kinetically preferred products and that they could be produced at room temperature (Figure 8). The thermodynamically preferred pathway for the more stable amide intermediate B, which was different from the keteniminium intermediate III, was path b (Peng et al., 2016).
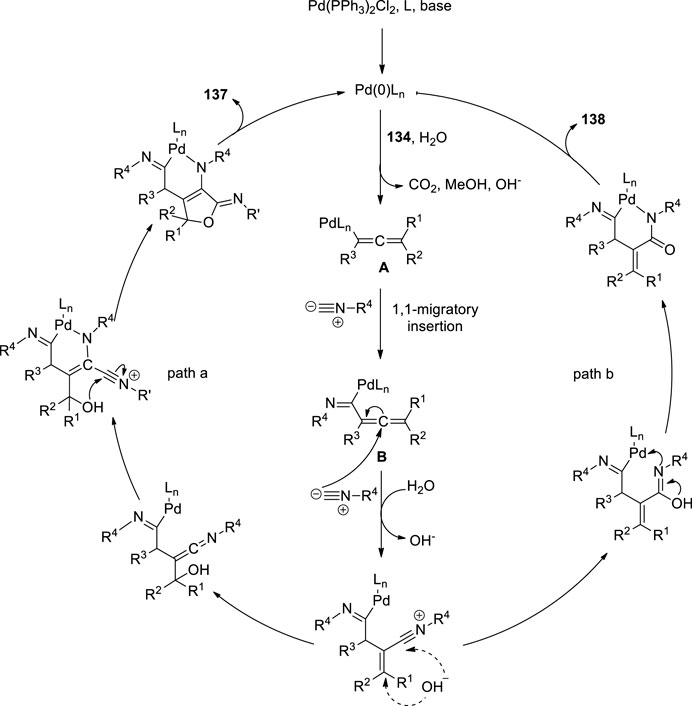
FIGURE 8. Proposed mechanism.
Lu and coworkers developed by PhI(OAc)2-mediated four-component tandem addition/cyclization/oxidation/aromatization process using simple aromatic amine 26, aldehyde 1, alkyne 13 and halide salt 139 as a precursors for the exquisite construction of functionalized 3-haloquinolines 140 (Figure 9D). The proposed methodology merging A3-coupling and redox reaction of halide salt, namely, A3-X type tandem reaction, is step economic, functional groups flexible, and potentially applicable to complex molecules under very mild condition (Lu et al., 2022).
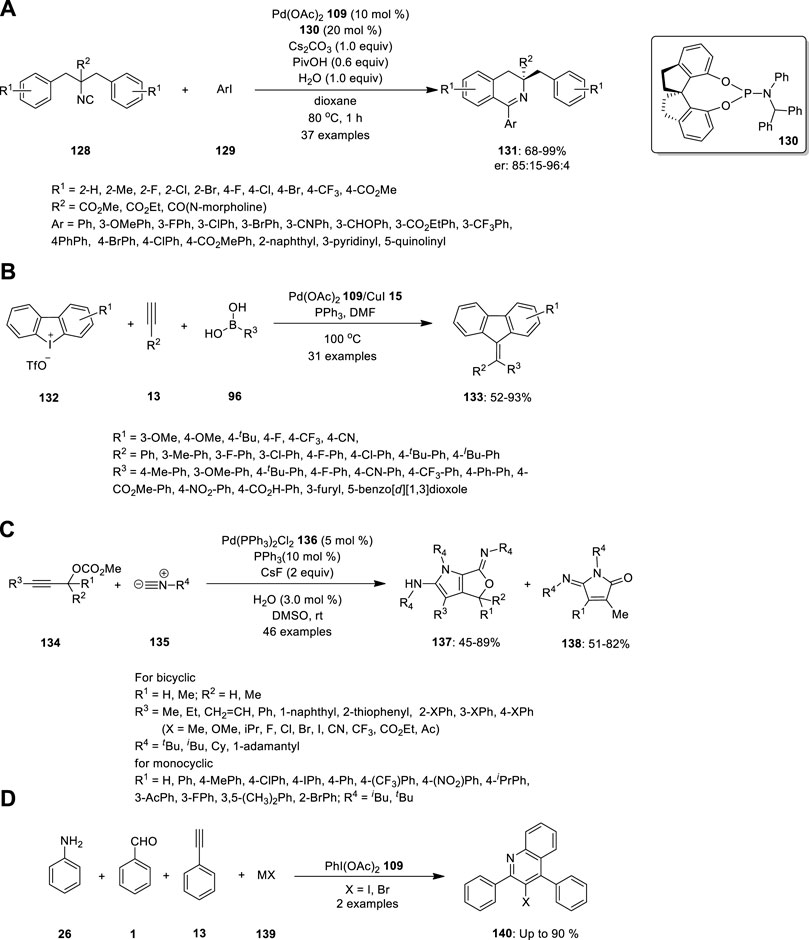
FIGURE 9. Pd-catalyzed synthesis of (A) chiral 3,3-disubstituted 3,4-dihydroisoquinoline 131, (B) methylidenefluorenes 133, (C) N-heterocyclic compounds 137 and 138, (D) functionalized 3-haloquinolines 140.
In 2019, Zhou et al. developed a novel strategy for the synthesis of isoquinoline derivative 146 using a rhodium (III)-catalyzed one-pot three-component reaction (Figure 10A). In this reaction, N-methoxybenzamide 141, α-diazoester 142 and alkyne 143 are smoothly transformed into various isoquinoline derivatives in good yields via successive O-alkylation and inert C–H activation processes in the presence of the Cp*RhIII catalyst at room temperature. When the substrate scope was investigated, both electron-withdrawing and electron-donating groups were tolerated by all substrates, including N-methoxybenzamide 141, α-diazoester 142, and alkyne 143. When asymmetric diarylalkynes were examined, two regioisomeric products were obtained in equal ratios. Subsequently, a reaction sequence was established as a plausible mechanism. N-methoxybenzamide 141 and α-diazoester 142 reacted together to produce oxoimine intermediate 147, which further combined with diaryl alkyne to produce isoquinoline derivatives 146. However, an isoquinolone resulting from changing the sequence of treatment of N-methoxybenzamide with diaryl alkyne was not observed (Zhou et al., 2019).
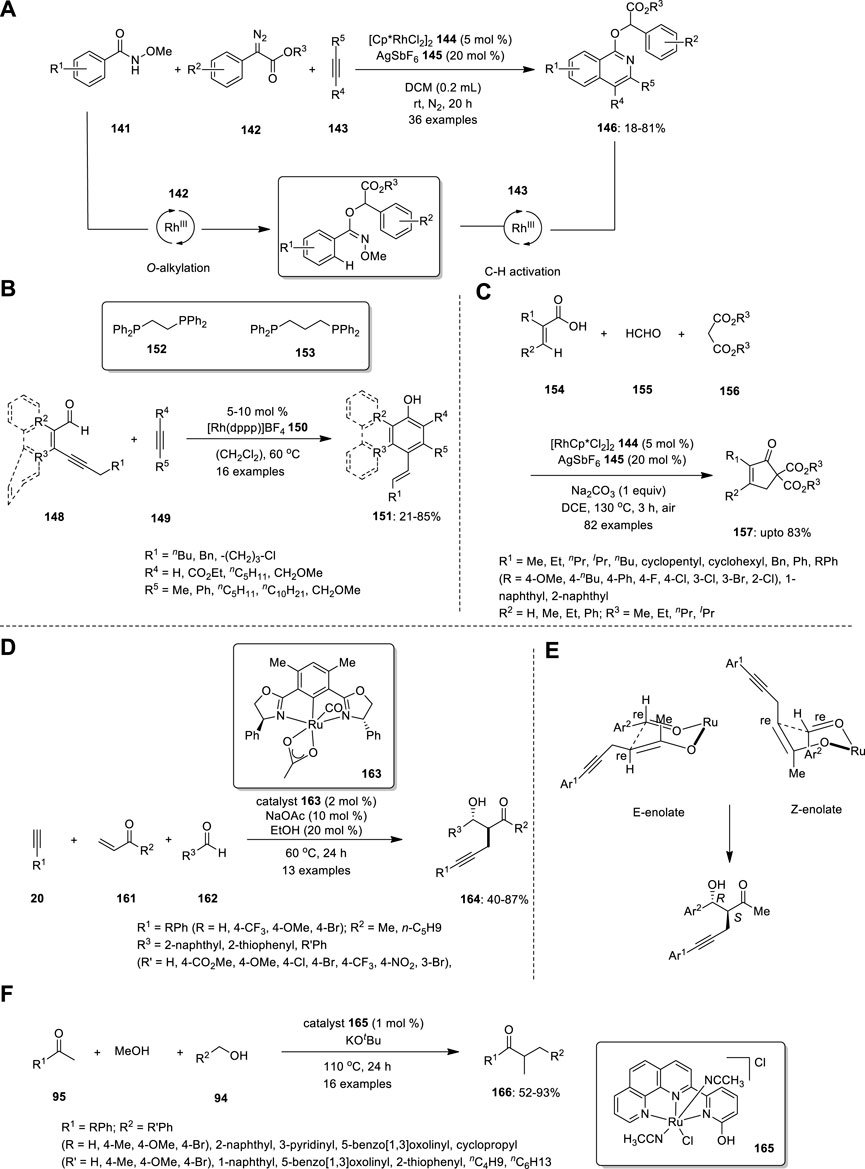
FIGURE 10. Rhodium (III)-catalyzed synthesis of (A) isoquinoline derivatives 146, (B) alcoholic derivatives 151, (C) cyclopentenones 157 and Ru—catalyzed synthesis of (D) β−hydroxyketone compounds 164, (E) Isomers of six-membered cyclic transition state, (F) α -methylated ketones 166.
Hojo and Tanaka developed a new methodology for the synthesis of phenol, naphthol, phenanthrenol, and triphenylenol derivatives from readily available conjugated alkynyl aldehydes 148 and alkynes 149 via cationic rhodium(I)/dppp complex catalysis of the aldehyde C−H bond activation/[4 + 2] annulation/aromatization cascade (Figure 10B). The effect of the ligand was examined. Evidently, in the cationic rhodium (I)/dppe complex-catalyzed [4 + 2] annulation, electron-deficient internal alkynes were more reactive, whereas the electron-rich internal alkyne 149 was more reactive when dppf was used as the ligand instead of dppe. All the internal alkynes exhibited high reactivity using a cationic rhodium(I)/dppp complex as a catalyst. The structure of 1,2-Bis(diphenylphosphino) ethane (dppe) 152 and 1,3-Bis(diphenylphosphino) propane (dppp) 153 are shown in Figure 10B. The resulting compound can react to produce pentacyclic compounds via intramolecular Friedel−Crafts alkylation. The synthesis of chrysenol derivatives is also possible by intramolecular olefin metathesis, such as the treatment of the resultant compound with 2nd generation Grubbs catalyst (Hojo, and Tanaka, 2012).
Yu et al. established an efficient Rh-catalyzed protocol for the synthesis of cyclopentenones based on a three-component reaction of acrylic acids 154, formaldehyde 155, and malonates 156 via vinylic C−H activation (Figure 10C). An MCR involving the profusion of formaldehyde 155 and malonate 156 produces multisubstituted cyclopentenones 157 via Michael addition. The substrates were reacted in the presence of rhodium catalyst, and 20 mol% AgSbF6 145 afforded the corresponding 5,5- diester cyclopentenone 157. When various alkyl halides were treated with 5,5- diester cyclopentenone and 1 equiv of Na2CO3 in MeOH, 5-alkylated cyclopentenones 158 were successfully produced. In addition, the ester group of cyclopentenone could be easily removed by decarboxylation. Moreover, this strategy can produce hexahydro-1H-inden-1-one derivative 159 when cyclohexene-1-carboxylic acid 160 is used instead of acrylic acid 154 (Yu et al., 2021).
Ubukata et al. described direct conjugate alkynylation and subsequent aldol reaction-mediated three-component coupling of commercially available alkyne,-unsaturated ketones, and aldehydes (Figure 10D). The advantage of this catalytic system is the commercial accessibility of the substrates. Using this approach, they effectively prepared a range of novel β−hydroxyketone compounds 164 with α−propargyl groups with excellent yields and good chemoselectivities. However, building complex and functionalized organic compounds from accessible starting materials in a one-pot process using this synthetic technique is challenging, although the enantioselectivity is moderate.
The absolute configuration of the main anti-isomer S (α), R (β) could be obtained by C−C bond formation between the re face of the aldehyde and the re face of the enolate in a six-membered cyclic transition state (Figure 10E). This further confirmed by X-ray diffraction analysis. Control investigations revealed that the Ru−O−enolate species produced by conjugating an alkyne to enone functions as a crucial intermediary for the subsequent aldol coupling process (Ubukata et al., 2016).
For the first time, ketones, alcohols, and methanol were successfully coupled via cooperative Ru(II)-catalyzed tandem three-component coupling (Figure 10F). Surprisingly, under mild reaction conditions, numerous functionalized acetophenones, benzyl alcohol derivatives, and methanol were successfully linked in tandem to produce the corresponding α-methylated ketones 139. The regioselective conversion of the final ketone products to acetanilide derivatives increased the synthetic usefulness of the reaction. The catalyst loading was decreased to 0.5 mol% at 85°C for the methylation of ketones using methanol as a methylating agent, and it was further decreased to 0.1 mol% at 120°C. To understand the catalytic cycle and the higher reactivity of the bifunctional catalyst, numerous kinetic experiments and DFT calculations were conducted. This tandem three-component coupling procedure is particularly appealing because of the straightforward use of air- and moisture-stable, nonphosphine-based Ru(II) catalysts (Chakrabarti et al., 2017).
Complex organic compounds are typically synthesized via multistep processes, which typically consume more time, energy, and solvent. Instead, an appropriate alternative to multistep synthesis is MCR, which is more efficient, and time- and energy-saving compared with multistep synthesis. However, MCR has a drawback in regard with its byproducts and selectivity. Thus, metallic catalysts have been introduced to enhance its selectivity. Essentially, the maximum amount of starting materials is consumed when metal catalysts are used, which limits the amount of byproducts. In this study, we summarized the current available literature on metal catalysts utilized in mMCRs for synthesizing organic compounds.
KS, RG, TS, and NT have covered the literature and collectively written the manuscript. TD and FS have given the proper plan of manuscript; finalize the contents and final editing of manuscript. All authors contributed to the article and approved the submitted version.
The review article is prepared without the support of any funding agency.
Authors are thankful to VIT Chennai for providing infrastructure for preparing the review article. TD and TS acknowledge support from the Ritsumeikan Global Innovation Research Organization (R-GIRO) project and Ritsumeikan NEXT Student Fellow program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Balakrishnan, V., Ganguly, A., and Rasappan, R. (2022). Interception of nickel hydride species and its application in multicomponent reactions. Org. Lett. 24, 4804–4809. doi:10.1021/acs.orglett.2c01862
Bienayme, H., and Bouzid, K. (1998). A new heterocyclic multicomponent reaction for the combinatorial synthesis of fused 3- aminoimidazoles. Angew. Chem. Int. Ed. 37, 2234–2237. doi:10.1002/(SICI)1521-3773(19980904)37:16<2234:AID-ANIE2234>3.0.CO;2-R
Biggs-Houck, J. E., Younai, A., and Shaw, J. T. (2010). Recent advances in multicomponent reactions for diversity- oriented synthesis. Curr. Opin. Chem. Biol. 14, 371–382. doi:10.1016/j.cbpa.2010.03.003
Brauch, S., van Berkel, S. S., and Westermann, B. (2013). Higher-order multicomponent reactions: Beyond four reactants. Chem. Soc. Rev. 42, 4948–4962. doi:10.1039/C3CS35505E
Chakrabarti, K., Maji, M., Panja, D., Paul, B., Shee, S., Das, G. K., et al. (2017). Utilization of MeOH as a C1 building block in tandem three-component coupling reaction. Org. Lett. 19, 4750–4753. doi:10.1021/acs.orglett.7b02105
Che, C., Yang, B., Jiang, X., Shao, T., Yu, Z., Tao, C., et al. (2014). Syntheses of fused tetracyclic quinolines via Ugi-variant MCR and Pd-catalyzed bis-annulation. J. Org. Chem. 79, 436–440. doi:10.1021/jo4024792
Cioc, R. C., Ruijter, E., and Orru, R. V. (2014). Multicomponent reactions: Advanced tools for sustainable organic synthesis. Green Chem. 16, 2958–2975. doi:10.1039/C4GC00013G
D'Souza, D. M., and Mueller, T. J. (2007). Multicomponent syntheses of heterocycles by transition-metal catalysis. Chem. Soc. Rev. 36, 1095–1108. doi:10.1039/B608235C
Davis, M. E., and Suib, S. L. (1993). Selectivity in catalysis ACS symposium series. Washington, DC: J. Am. Chem. Soc.
Dömling, A., and Huang, Y. (2010). Piperazine scaffolds via isocyanide-based multicomponent reactions. Synth 17, 2859–2883. doi:10.1055/s-0030-1257906
Dömling, A., and Ugi, I. (2000). Multicomponent reactions with isocyanides. Chem. Bio Chem. 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168:aid-anie3168>3.0.co;2-u
Domling, A., Wang, W., and Wang, K. (2012). Chemistry and biology of multicomponent reactions. Chem. Rev. 112, 3083–3135. doi:10.1021/cr100233r
Gandeepan, P., Rajamalli, P., and Cheng, C. H. (2014). Palladium-catalyzed dehydrogenative β-arylation of simple saturated carbonyls by aryl halides. ACS Catal. 4, 4485–4489. doi:10.1021/cs501326p
Ganem, B. (2009). Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 42, 463–472. doi:10.1021/ar800214s
Gao, S., Stopka, T., and Niggemann, M. (2015). Calcium-catalyzed dynamic multicomponent reaction. Org. Lett. 17, 5080–5083. doi:10.1021/acs.orglett.5b02593
Gers, C. F., Nordmann, J., Kumru, C., Frank, W., and Muller, T. J. (2014). Solvatochromic fluorescent 2-substituted 3-ethynyl quinoxalines: Four-component synthesis, photophysical properties, and electronic structure. J. Org. Chem. 79, 3296–3310. doi:10.1021/jo4025978
Ghashghaei, O., Seghetti, F., and Lavilla, R. (2019). Selectivity in multiple multicomponent reactions: Types and synthetic applications. Beilstein J. Org. Chem. 15, 521–534. doi:10.3762/bjoc.15.46
Graebin, C. S., Ribeiro, F. V., Rogério, K. R., and Kümmerle, A. E. (2019). Multicomponent reactions for the synthesis of bioactive compounds: A review. Curr. Org. Synth. 16, 855–899. doi:10.2174/1570179416666190718153703
Groebcke, K., Weber, L., and Mehlin, F. (1998). Synthesis of imidazo[1,2-a] annulated pyridines, pyrazines and pyrimidines by a novel three-component condensation. Synlett 6, 661–663. doi:10.1055/s-1998-1721
Haji, M. (2016). Multicomponent reactions: A simple and efficient route to heterocyclic phosphonates. Beilstein J. Org. Chem. 12, 1269–1301. doi:10.3762/bjoc.12.121
Hojo, D., and Tanaka, K. (2012). Rhodium-catalyzed C–H bond activation/[4+ 2] annulation/aromatization cascade to produce phenol, naphthol, phenanthrenol, and triphenylenol derivatives. Org. Lett. 14, 1492–1495. doi:10.1021/ol300234g
Khan, M. M., Yousuf, R., Khan, S., and Shafiullah, S. (2015). Recent advances in multicomponent reactions involving carbohydrates. RSC Adv. 5, 57883–57905. doi:10.1039/C5RA08059B
Kaïm, L. E., Grimaud, L., and Oble, J. (2005). Phenol ugi–smiles systems: Strategies for the multicomponent N-arylation of primary amines with isocyanides, aldehydes, and phenols. Angew. Chem. 117, 8175–8178. doi:10.1002/ange.200502636
Kakuchi, R. (2015). Metal-catalyzed multicomponent reactions for the synthesis of polymers. Multicomponent Sequential React. Polym. Synthesis 269, 1–15. Advances in Polymer Science. doi:10.1007/12_2014_300
Kroger, D., Franz, M., Schmidtmann, M., and Martens, J. (2015). Sequential multicomponent reactions and a cu-mediated rearrangement: Diastereoselective synthesis of tricyclic ketones. Org. Lett. 17, 5866–5869. doi:10.1021/acs.orglett.5b03057
Li, B., Guo, S., Zhang, J., Zhang, X., and Fan, X. (2015). Selective access to 3-cyano-1 H-indoles, 9 H-pyrimido [4, 5-b] indoles, or 9 H-pyrido [2, 3-b] indoles through copper-catalyzed one-pot multicomponent cascade reactions. J. Org. Chem. 80, 5444–5456. doi:10.1021/acs.joc.5b00239
Liu, S., Qian, Y. S., Xu, J. L., Xu, L., and Xu, Y. H. (2023). Copper-catalyzed regio-and stereoselective three-component coupling of allenyl ethers with gem-dichlorocyclobutenones and B2pin2. Org. Lett. 25 (24), 4498–4503. doi:10.1021/acs.orglett.3c01515
Lu, H., Qiu, Y. C., Zhao, Q., Tang, R., Chen, T., Hu, L., et al. (2022). An efficient approach for 3-haloquinoline synthesis: PhI (OAc)2-mediated A3-X type tandem annulation of amine, aldehyde, alkyne and halide salt. Tetrahedron Lett. 101, 153927. doi:10.1016/j.tetlet.2022.153927
Malinakova, H. C. (2015). Recent advances in the discovery and design of multicomponent reactions for the generation of small-molecule libraries. Rep. Org. Chem. 5, 75–90. doi:10.2147/ROC.S65115
Mondal, R. R., Khamarui, S., and Maiti, D. K. (2016). CuBr–ZnI2 combo-catalysis for mild CuI–CuIII switching and sp2 C–H activated rapid cyclization to quinolines and their sugar-based chiral analogues: A UV–vis and xps study. ACS Omega 1, 251–263. doi:10.1021/acsomega.6b00185
Mostofi, F., and Mohebat, R. (2018). Efficient synthesis of novel polyfunctionalized pyrazole derivatives via isocyanide-based three-component reaction. Polycycl. Aromat. Compd. 38, 213–218. doi:10.1080/10406638.2016.1200101
Murugavel, G., and Punniyamurthy, T. (2015). Microwave-assisted copper-catalyzed four-component tandem synthesis of 3-N-sulfonylamidine coumarins. J. Org. Chem. 80, 6291–6299. doi:10.1021/acs.joc.5b00738
Pagadala, R., Maddila, S., Moodley, V., van Zyl, W. E., and Jonnalagadda, S. B. (2014). An efficient method for the multicomponent synthesis of multisubstituted pyridines, a rapid procedure using Au/MgO as the catalyst. Tetrahedron Lett. 55 (29), 4006–4010. doi:10.1016/j.tetlet.2014.05.089
Paprocki, D., Madej, A., Koszelewski, D., Brodzka, A., and Ostaszewski, R. (2018). Multicomponent reactions accelerated by aqueous micelles. Front. Chem. 6, 502. doi:10.3389/fchem.2018.00502
Peng, J., Gao, Y., Hu, W., Gao, Y., Hu, M., Wu, W., et al. (2016). Palladium-catalyzed multicomponent reaction (MCR) of propargylic carbonates with isocyanides. Org. Lett. 18, 5924–5927. doi:10.1021/acs.orglett.6b02999
Pillitteri, S., Ranjan, P., Voskressensky, L. G., Van der Eycken, E. V., and Sharma, U. K. (2021). Alkylation of in situ generated imines via photoactivation of strong aliphatic CH bonds. Mol. Catal. 514, 111841. doi:10.1016/j.mcat.2021.111841
Ramanathan, D., and Pitchumani, K. (2015). Copper (I)-Y zeolite-catalyzed regio-and stereoselective [2+ 2+ 2] cyclotrimerization cascade: An atom-and step-economical synthesis of pyrimido [1, 6-a] quinoline. J. Org. Chem. 80, 10299–10308. doi:10.1021/acs.joc.5b01896
Rezayati, S., Kalantari, F., Ramazani, A., Sajjadifar, S., Aghahosseini, H., and Rezaei, A. (2022). Magnetic silica-coated picolylamine copper complex [fe3o4@sio2@ gp/picolylamine-cu (ii)]-catalyzed biginelli annulation reaction. Inorg. Chem. 61, 992–1010. doi:10.1021/acs.inorgchem.1c03042
Rotstein, B. H., Zaretsky, S., Rai, V., and Yudin, A. K. (2014). Small heterocycles in multicomponent reactions. Chem. Rev. 114, 8323–8359. doi:10.1021/cr400615v
Safaei, G. J., Ghasemzadeh, M. A., and Mehrabi, M. (2013). Calcium oxide nanoparticles catalyzed one-step multicomponent synthesis of highly substituted pyridines in aqueous ethanol media. Sci. Iran. 20, 549–554. doi:10.1016/j.scient.2012.12.037
Santoro, S., Deiana, L., Zhao, G. L., Lin, S., Himo, F., and Cordova, A. (2014). Mechanism of palladium/amine cocatalyzed carbocyclization of aldehydes with alkynes and its merging with “Pd Oxidase Catalysis”. ACS Catal. 4, 4474–4484. doi:10.1021/cs500905r
Semba, K., Ohta, N., and Nakao, Y. (2019). Carboallylation of electron-deficient alkenes with organoboron compounds and allylic carbonates by cooperative palladium/copper catalysis. Org. Lett. 21, 4407–4410. doi:10.1021/acs.orglett.9b00915
Sharma, P., and Liu, R. S. (2016). [3+2]-Annulations of N-hydroxy allenylamines with nitrosoarenes: One-pot synthesis of substituted indole products. Org. Lett. 18, 412–415. doi:10.1021/acs.orglett.5b03447
Shen, K., Han, X., and Lu, X. (2012). Cationic Pd (II)-catalyzed highly enantioselective arylative cyclization of alkyne-tethered enals or enones initiated by carbopalladation of alkynes with arylboronic acids. Org. Lett. 14, 1756–1759. doi:10.1021/ol3003546
Slobbe, P., Ruijter, E., and Orru, R. V. A. (2012). Recent applications of multicomponent reactions in medicinal chemistry. Med. Chem. Comm. 3, 1189–1218. doi:10.1039/C2MD20089A
Sunderhaus, J. D., and Martin, S. F. (2009). Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds. Chem. Eur. J. 15, 1300–1308. doi:10.1002/chem.200802140
Ubukata, S., Ito, J. I., Oguri, R., and Nishiyama, H. (2016). Asymmetric three-component coupling reaction of alkyne, enone, and aldehyde catalyzed by chiral phebox ruthenium catalysts. J. Org. Chem. 81, 3347–3355. doi:10.1021/acs.joc.6b00374
Ugi, I., Domling, A., and Horl, W. (1994). Multicomponent reactions in organic chemistry. Endeavor 18, 115–122. doi:10.1016/S0160-9327(05)80086-9
Ugi, I., and Steinbruckner, C. (1960). Concerning a new condensation principle. Angew. Chem. 72, 267–268. doi:10.1002/ange.19600720709
Vytla, D., Emmadi, J., Velayuthaperumal, R., Shaw, P., Cavallaro, C. L., Mathur, A., et al. (2022). Visible-light enabled one-pot three-component Petasis reaction for synthesis of α-substituted secondary sulfonamides/amides/hydrazides. Tetrahedron Lett. 106, 154055. doi:10.1016/j.tetlet.2022.154055
Wang, J., Gao, D. W., Huang, J., Tang, S., Xiong, Z., Hu, H., et al. (2017). Palladium-catalyzed enantioselective C (sp2)–H imidoylation by desymmetrization. ACS Catal. 7 (6), 3832–3836. doi:10.1021/acscatal.7b00731
Wang, S., Chen, H., Zhao, H., Cao, H., Li, Y., and Liu, Q. (2013). Gold-catalyzed multicomponent reaction: Facile strategy for the synthesis of N-substituted 1, 4-dihydropyridines by using activated alkynes, aldehydes, and methanamine. Eur. J. Org. Chem. 2013 (32), 7300–7304. doi:10.1002/ejoc.201301203
Wang, X., Zhu, B., Dong, J., Tian, H., Liu, Y., Song, H., et al. (2021). Visible-light-mediated multicomponent reaction for secondary amine synthesis. Chem. Commun. 57, 5028–5031. doi:10.1039/D1CC01560E
Wu, Y., Liu, Q., Huang, S., Zhang, C., Wei, W., and Li, X. (2023). Cu(OAc)2-catalyzed three-component cycloaddition of malonates, nitrosoarenes and alkenes: Access to isoxazolidines. Org. Biomol. Chem. Adv. Artic. 21, 3669–3674. doi:10.1039/D3OB00189J
Xu, W., Kato, T., Liu, Y., Matsumoto, A., and Maruoka, K. (2022). Fe-catalyzed dicarbofunctionalization of vinylarenes with alkylsilyl peroxides and β-keto carbonyl substrates. Org. Lett. 24, 2641–2645. doi:10.1021/acs.orglett.2c00656
Yu, S., Hong, C., Liu, Z., and Zhang, Y. (2021). Synthesis of cyclopentenones through rhodium-catalyzed C–H annulation of acrylic acids with formaldehyde and malonates. Org. Lett. 23, 5054–5059. doi:10.1021/acs.orglett.1c01569
Zhang, Y., Pan, H., Liu, W., Cao, W., and Feng, X. (2020). Asymmetric catalytic synthesis of epoxides via three-component reaction of diazoacetates, 2-Oxo-3-ynoates, and nitrosoarenes. Org. Lett. 22, 6744–6749. doi:10.1021/acs.orglett.0c02108
Zhou, C., Jiang, J., and Wang, J. (2019). Three-component synthesis of isoquinoline derivatives by a relay catalysis with a single rhodium (III) catalyst. Org. Lett. 21, 4971–4975. doi:10.1021/acs.orglett.9b01456
Zhou, F., Liu, X., Zhang, N., Liang, Y., Zhang, R., Xin, X., et al. (2013). Copper-catalyzed three-component reaction: Solvent-controlled regioselective synthesis of 4-amino-and 6-amino-2-iminopyridines. Org. Lett. 15, 5786–5789. doi:10.1021/ol4028368
Keywords: multicomponent reactions, arylation, cyclization, cycloaddition, metal, copper, palladium, catalyst
Citation: Sakthivel K, Gana RJ, Shoji T, Takenaga N, Dohi T and Singh FV (2023) Recent progress in metal assisted multicomponent reactions in organic synthesis. Front. Chem. 11:1217744. doi: 10.3389/fchem.2023.1217744
Received: 05 May 2023; Accepted: 21 August 2023;
Published: 08 September 2023.
Edited by:
Kamaldeep Paul, Thapar Institute of Engineering and Technology, IndiaReviewed by:
Satyajit Saha, Institute of Chemical Technology, IndiaCopyright © 2023 Sakthivel, Gana, Shoji, Takenaga, Dohi and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshifumi Dohi, dGQxMjAzQHBoLnJpdHN1bWVpLmFjLmpw; Fateh V. Singh, ZmF0ZWh2ZWVyLnNpbmdoQHZpdC5hYy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.